
Clinical trials with reference to GCP Guidelines Certificate course in Research Ethics Review...
38
Clinical trials with Clinical trials with reference to GCP reference to GCP Guidelines Guidelines Certificate course in Research Ethics Certificate course in Research Ethics Review (CCRER) Review (CCRER) 25 25 th th April 2008 April 2008 Priyadarshani Galappatthy Priyadarshani Galappatthy MD,MRCP MD,MRCP Department of Pharmacology Department of Pharmacology
-
Upload
emil-knight -
Category
Documents
-
view
216 -
download
2
Transcript of Clinical trials with reference to GCP Guidelines Certificate course in Research Ethics Review...

Clinical trials with reference Clinical trials with reference to GCP Guidelinesto GCP Guidelines
Certificate course in Research Ethics Review Certificate course in Research Ethics Review (CCRER)(CCRER)
2525thth April 2008 April 2008Priyadarshani Galappatthy Priyadarshani Galappatthy MD,MRCP MD,MRCP
Department of PharmacologyDepartment of Pharmacology

22
OverviewOverview
Definition and phases of clinical trialsDefinition and phases of clinical trials Principles of GCP guidelinesPrinciples of GCP guidelines Responsibilities, functions, procedures Responsibilities, functions, procedures
for ERC/IRB in GCP guidelinesfor ERC/IRB in GCP guidelines Monitoring and auditing Monitoring and auditing Documents for clinical trialsDocuments for clinical trials Approvals for a clinical trial in SLApprovals for a clinical trial in SL

33
Clinical ResearchClinical Research
Broad definition:Broad definition:• Medical research in human subjectsMedical research in human subjects
Narrow definition:Narrow definition:• The gamut of activities related to testing The gamut of activities related to testing
and development of diagnostic, and development of diagnostic, prophylactic, therapeutic and cosmetic prophylactic, therapeutic and cosmetic modalities in human subjectsmodalities in human subjects

44
Clinical Research in Clinical Research in Drug DevelopmentDrug Development
Defines efficacy, safety, PK, PD, PE – hence Defines efficacy, safety, PK, PD, PE – hence transforms a chemical substance to a drugtransforms a chemical substance to a drug
Involves multiple stakeholdersInvolves multiple stakeholders Every aspect is subject to regulatory reviewEvery aspect is subject to regulatory review Governed by specific laws, rights, & legal Governed by specific laws, rights, & legal
implicationsimplications Involves risks and benefits for patientsInvolves risks and benefits for patients Holds ethical implications and penaltiesHolds ethical implications and penalties Has liabilities for investigators and ethics Has liabilities for investigators and ethics
committeescommittees

55
Phases of Clinical Research (1)Phases of Clinical Research (1) Phase 0Phase 0
Very early human dosing Very early human dosing Helps determine critical pharmacokinetic Helps determine critical pharmacokinetic
factorsfactors Increasingly used for early decision-makingIncreasingly used for early decision-making
Phase 1Phase 1 Series of healthy volunteer studiesSeries of healthy volunteer studies Terminology often used for all volunteer Terminology often used for all volunteer
studiesstudies Establishes safety, PK, and healthy PD in Establishes safety, PK, and healthy PD in
humanshumans Conducted in specialized unitsConducted in specialized units

66
Phases of Clinical Research (2)Phases of Clinical Research (2) Phase 2Phase 2
• Proof of concept (PoC) and dose range finding Proof of concept (PoC) and dose range finding (DRF) studies in patients(DRF) studies in patients
• Successful PoC transforms valueSuccessful PoC transforms value Phase 3Phase 3
• Definitive studies to confirm efficacy & safetyDefinitive studies to confirm efficacy & safety• Establishes level of less frequent side-effectsEstablishes level of less frequent side-effects• Often unaffordable for smaller companiesOften unaffordable for smaller companies
Phase 4Phase 4• Post marketing surveillance (PMS) Post marketing surveillance (PMS) • Establishes long-term benefit risk ratioEstablishes long-term benefit risk ratio• Establishes place in therapyEstablishes place in therapy

77
How to ensure clinical trials are How to ensure clinical trials are conducted ethically and according conducted ethically and according
to regulatory requirements ?to regulatory requirements ?
GuidelinesGuidelines BenchmarksBenchmarks

88

99
Good Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects.
Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles in the Declaration of Helsinki, and that the clinical trial data are credible.
Guideline was developed with consideration of the current good clinical practices of the European Union, Japan, United States, Australia, Canada, the Nordic countries and the WHO.
GCP: The Code of ConductGCP: The Code of Conduct

1010
ICH GCP OrganizationICH GCP Organization
Glossary and PrinciplesGlossary and Principles ResponsibilitiesResponsibilities
• ERC/IRB ERC/IRB • InvestigatorsInvestigators• SponsorSponsor
Trial protocol and protocol amendmentsTrial protocol and protocol amendments Investigators’ brochureInvestigators’ brochure Document inventory and locationDocument inventory and location

1111

1212
Essential Elements of GCPEssential Elements of GCP
Autonomy and informed consentAutonomy and informed consent Ethical oversightEthical oversight Benefit-risk evaluationBenefit-risk evaluation Documentation and monitoringDocumentation and monitoring Drug product integrity & accountabilityDrug product integrity & accountability Fidelity of dataFidelity of data Qualified and trained staffQualified and trained staff Audits & InspectionsAudits & Inspections

1313
Autonomy & Informed ConsentAutonomy & Informed Consent
Enshrined as Respect for Persons - One of Enshrined as Respect for Persons - One of the 3 Basic Ethical Principles in the the 3 Basic Ethical Principles in the Belmont Report of 1979 and in the Basic Belmont Report of 1979 and in the Basic Principles of the Declaration of Helsinki.Principles of the Declaration of Helsinki.
Requires the documentation of consent:Requires the documentation of consent:• Involves a consent procedure, materials, Involves a consent procedure, materials,
discussiondiscussion• Written where possible, else witnessedWritten where possible, else witnessed• Includes autonomy to refuse and withdrawIncludes autonomy to refuse and withdraw• A healthy refusal rate is expectedA healthy refusal rate is expected

1414
Ethical OversightEthical Oversight
Aspects pertaining to the Ethical Aspects pertaining to the Ethical Review committee/ Institutional Review committee/ Institutional review Board (ERC/IRB) in GCP review Board (ERC/IRB) in GCP guidelinesguidelines
ResponsibilitiesResponsibilities Composition, functions and Composition, functions and
operationsoperations ProceduresProcedures

1515
Responsibilities of IRB/IEC (1) Safeguard the rights, safety, and well-being of all
trial subjects. Special attention paid to vulnerable subjects. The IRB/IEC should obtain the following documents:
• trial protocol/amendments, • written informed consent forms consent form updates, • subject recruitment procedures (e.g. advertisements), • written information to be provided to subjects, • Investigator's Brochure (IB), • available safety information, • information about payments and compensation to subjects, • the investigator’s current curriculum vitae and/or other
documentation evidencing qualifications, • any other documents that the IRB/IEC may need to fulfil its
responsibilities.

1616
Responsibilities of ERC/IRB (2)Responsibilities of ERC/IRB (2) Consider the qualifications of the
investigator/s Conduct continuing review of each ongoing
trial, at least once per year. If the protocol indicates that prior consent
of the trial subject is not possible, the IRB/IEC should determine that the protocol addresses ethical concerns and meets regulatory requirements
Review amount and method of payment to subjects to assure that coercion or undue influence on the subjects is prevented.
Ensure that information regarding payment to subjects, is in the informed consent form

1717
Responsibilities of ERC/IRB- (3)Responsibilities of ERC/IRB- (3)
The IRB/IEC should review a proposed clinical trial within a reasonable time
Document IRB/ERC views in writing, identifying the trial, the documents reviewed and the dates for the following • approval/favourable opinion • modifications required prior to its
approval/favourable opinion • disapproval / negative opinion• termination/suspension of any prior
approval/favourable opinion.

1818
Composition, Functions and Operations of ERC (1)
Consist of a reasonable number of members, who collectively have the qualifications and experience to review and evaluate the science, medical aspects, and ethics of the proposed trial.
It is recommended that the IRB/IEC should include: • At least five members. • At least one member whose primary area of interest is in a
non-scientific area. • At least one member who is independent of the
institution/trial site. A list of IRB/IEC members and their qualifications
should be maintained. An IRB/IEC may invite non-members with expertise
in special areas for assistance

1919
Composition, Functions and Operations of ERC (2)
The IRB/IEC should function according to written operating procedures
Maintain written records of its activities and minutes Comply with GCP and regulatory requirements. Make decisions at meetings at which a quorum, as
stipulated in operating procedures, is present. Only members who participate in the IRB/IEC review
and discussion should vote/provide their opinion and/or advise.
The investigator may provide information on the trial, but should not participate in the deliberations of the IRB/IEC or in the vote/opinion of the IRB/IEC.

2020
Procedures in ERC/IRB (1) Document in writing, and follow its
procedures, including • composition and the authority under
which it is established. • Schedule, notify conducting its meetings. • Conduct initial and continuing review of
trials. • Provide expedited review and approval of
amendments/revisions in ongoing trials that have the approval of the IRB/IEC.
• Specify that no subject should be admitted to a trial before the IRB/IEC issues its written approval of the trial.

2121
Procedures in ERC/IRB- (2)• Specify that the investigator should promptly
report to the IRB/IEC: Deviations from, or changes of, the protocol. Changes increasing the risk to subjects and/or affecting
significantly the conduct of the trial All adverse drug reactions (ADRs) that are both serious
and unexpected. New information that may affect adversely the safety
of the subjects or the conduct of the trial.
• Ensure that the IRB/IEC promptly notify in writing the investigator/institution concerning:
Its trial-related decisions/opinions. The reasons for its decisions/opinions. Procedures for appeal of its decisions/opinions.

2222
Qualified & Trained StaffQualified & Trained Staff Applies to:Applies to:
• Investigators and other site staffInvestigators and other site staff• Monitors, auditors, and sponsor’s administrative staffMonitors, auditors, and sponsor’s administrative staff• Statisticians and data managersStatisticians and data managers• Independent data monitoring committeeIndependent data monitoring committee
CVs of investigators, co-investigators, and CVs of investigators, co-investigators, and sponsor’s staff must be on filesponsor’s staff must be on file
Investigators & site staff should be trained on Investigators & site staff should be trained on protocol & procedures through investigators’ protocol & procedures through investigators’ meetings and site initiation meetingsmeetings and site initiation meetings
Training files of sponsors’ staff to be maintainedTraining files of sponsors’ staff to be maintained

2323
Benefit-risk EvaluationBenefit-risk Evaluation
Applies to: Sponsor, regulatory authority, Applies to: Sponsor, regulatory authority, investigators, ethics committeesinvestigators, ethics committees
Based on: Prior information available to Based on: Prior information available to sponsor, data submitted to regulatory sponsor, data submitted to regulatory authority, investigators brochure, authority, investigators brochure, emerging safety reportsemerging safety reports
Currently a qualitative exercise – outcome Currently a qualitative exercise – outcome may vary based on the evaluator’s may vary based on the evaluator’s perspective and individual sensitivitiesperspective and individual sensitivities

2424
Safety Reporting
All serious adverse events (SAEs) should be reported immediately to the sponsor
The investigator should also comply with the applicable regulatory requirements related to the reporting of unexpected serious adverse drug reactions to the regulatory authorities and the IRB/IEC.
Adverse events and/or laboratory abnormalities identified should be reported within the time periods specified in the protocol.

2525
Documentation and MonitoringDocumentation and Monitoring
The long-term nature of clinical The long-term nature of clinical developmentdevelopment
Sponsor’s responsibilities:Sponsor’s responsibilities:• Securing direct access for sponsor and Securing direct access for sponsor and
regulatorsregulators• Monitoring and audits to verify protection of Monitoring and audits to verify protection of
subjects’ rights, data accuracy, and compliance subjects’ rights, data accuracy, and compliance to protocol, GCP, & regulationsto protocol, GCP, & regulations
Direct access monitoringDirect access monitoring• Source data verification, consent verification,Source data verification, consent verification,
drug accountability, training & information, drug accountability, training & information, communication of deviations through communication of deviations through monitoring reportsmonitoring reports

2626
Monitoring
The purposes of trial monitoring are to verify that: • The rights and well-being of human subjects
are protected. • The reported trial data are accurate, complete,
and verifiable from source documents. • The conduct of the trial is in compliance with
the currently approved protocol /amendments, with GCP, and with the applicable regulatory requirements.

2727
Product Integrity & AccountabilityProduct Integrity & Accountability
Manufacturing & packaging under GMPManufacturing & packaging under GMP Appropriate labelingAppropriate labeling Transport under appropriate conditionsTransport under appropriate conditions Secure and appropriate storageSecure and appropriate storage Accountability of dispensing and returnsAccountability of dispensing and returns Compliance monitoringCompliance monitoring

2828
Audits & InspectionsAudits & Inspections
DefinitionDefinition Auditors and inspectorsAuditors and inspectors The audit/inspection processThe audit/inspection process
• NotificationNotification• Presentation of credentialsPresentation of credentials• Hospitality and accessHospitality and access• Audit report, observations, & corrective Audit report, observations, & corrective
action planaction plan• Termination and notification of terminationTermination and notification of termination
Regulatory access to audit reportsRegulatory access to audit reports

2929
Audit A systematic and independent examination of trial
related activities and documents to determine whether the evaluated trial related activities were conducted, and the data were recorded, analyzed and accurately reported according to the protocol, sponsor's standard operating procedures (SOPs), Good Clinical Practice (GCP), and the applicable regulatory requirements.
If or when sponsors perform audits, as part of implementing quality assurance, they should consider: • Purpose • Selection and Qualification of Auditors
The sponsor should appoint individuals, who are independent of the clinical trials/systems, to conduct audits.
The sponsor should ensure that the auditors are qualified by training and experience to conduct audits properly. An auditor’s qualifications should be documented.

3030
Quality Assurance and Quality Control
The sponsor is responsible for implementing and maintaining quality assurance with written SOPs to ensure that trials are conducted, data are generated, documented and reported in compliance with the protocol, GCP, and regulatory requirements.
A sponsor may transfer any or all of the sponsor's trial-related duties and functions to a CRO, but the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor.

3131
Contract Research Organization (CRO)
The CRO should implement quality assurance and quality control.
Any trial-related duty and function that is transferred to and assumed by a CRO should be specified in writing.
All references to a sponsor in GCP guideline also apply to a CRO to the extent that a CRO has assumed the trial related duties and functions
3232
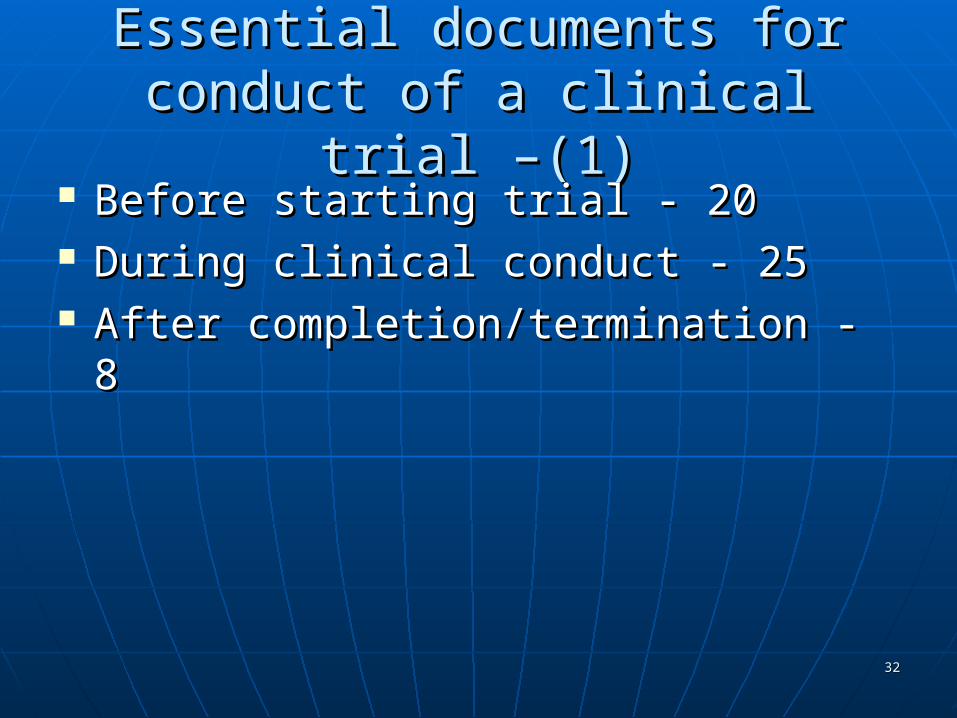
Essential documents for conduct of Essential documents for conduct of a clinical trial –(1)a clinical trial –(1)
Before starting trial - 20Before starting trial - 20 During clinical conduct - 25During clinical conduct - 25 After completion/termination - 8After completion/termination - 8

3333
Essential documents for conduct of Essential documents for conduct of a clinical trial –(2)a clinical trial –(2)

3434
Essential documents for conduct of Essential documents for conduct of a clinical trial – (3)a clinical trial – (3)

3535
Essential documents for conduct of a Essential documents for conduct of a clinical trial – (4)clinical trial – (4)

3636
GCP: The Code of ConductGCP: The Code of Conduct
A A standardstandard for the design, conduct, for the design, conduct, performance, monitoring, auditing, performance, monitoring, auditing, recording, analyses, and reporting recording, analyses, and reporting of clinical trials that provides of clinical trials that provides assuranceassurance that the data and that the data and reported reported results are credible and results are credible and accurateaccurate, and that the , and that the rights, rights, integrity, and confidentiality of trial integrity, and confidentiality of trial subjects are protectedsubjects are protected..

3737
Approvals for a clinical trial in SLApprovals for a clinical trial in SL
ERC/IRBERC/IRB Hospital authorities/ERCHospital authorities/ERC Drug regulatory authority – for Drug regulatory authority – for
registration and import of trial drugregistration and import of trial drug Sri Lanka Clinical trials registrySri Lanka Clinical trials registry

3838








![GCP & Go in 2015 [GCP編]](https://static.fdocuments.us/doc/165x107/58737f5a1a28ab272d8b474d/gcp-go-in-2015-gcp.jpg)









