
CLIA Webinar Slides - Food and Drug Administration · Welcome to today’s FDA/CDRH Webinar Thank...
29
Welcome to today’s FDA/CDRH Webinar Thank you for your patience while we register all of today’s participants. If you have not connected to the audio portion of the webinar, please do so now: U.S. Callers Dial: 800-369-2039; International Callers Dial: 1-517-308-9315 Passcode: 5179628
Transcript of CLIA Webinar Slides - Food and Drug Administration · Welcome to today’s FDA/CDRH Webinar Thank...

Welcome to today’s FDA/CDRH Webinar
Thank you for your patience while we register all of today’s participants.
If you have not connected to the audio portion of the
webinar, please do so now: U.S. Callers Dial: 800-369-2039; International Callers
Dial: 1-517-308-9315 Passcode: 5179628

Clinical Laboratory Improvement Amendments of 1998 (CLIA)
Waiver Applications Draft Guidances Peter Tobin, Ph.D.
Chemist Division of Program Operations and Management
Office of In Vitro Diagnostics and Radiological Health Center for Devices and Radiological Health
January 8, 2018

3
This Webinar Covers Two Complementary Draft Guidances for CLIA Waiver Applications:
• Select Updates for Recommendations for Clinical Laboratory
Improvement Amendments of 1988 (CLIA) Waiver Applications for
Manufacturers of In Vitro Diagnostic Devices
– Referred to as: Draft “Sec V” Guidance
• Recommendations for Dual 510(k) and CLIA Waiver by Application
Studies
– Referred to as: Draft “Dual” Guidance
www.fda.gov

4
21st Century Cures Requires an Update to Sec V. of the CLIA Waiver Guidance
• Sec. 3057, CLIA Waiver Improvements, requires FDA to publish guidance that:
(1) revises “Section V. Demonstrating Insignificant Risk of an Erroneous
Result – Accuracy” of the guidance entitled “Recommendations for Clinical
Laboratory Improvement Amendments of 1988 (CLIA) Waiver Applications for
Manufacturers of In Vitro Diagnostic Devices” and dated January 30, 2008;
and
(2) includes the appropriate use of comparable performance between a
waived user and a moderately complex laboratory user to demonstrate
accuracy.
www.fda.gov

5
The Draft Guidances Address Two CLIA Waiver Pathways
Stepwise CLIA Waiver by Application (2008 CW Guidance & Draft Sec V)
Dual 510(k) and CLIA Waiver by Application (Draft Dual Guidance)
Marketing Submission (PMA, 510(k), De Novo
CR: Moderate
Pre-Submission Dual Submission (Combined 510(k) and CW)
CLIA Waiver by Regulation or Clearance for Home or OTC use
Clearance /Approval of test type listed in 42 CFR 493.15(c), or Clearance/Approval for home or Over-the-Counter (OTC) use
CLIA Record (CR): Waived
CLIA Waiver by Application (CW) Pre-Submission

6
Outline
• Background
– Recent CLIA Waiver Program Improvements
• Draft Section V Guidance
• Draft Dual Guidance
• Recommendations for Semi-Quantitative Tests
www.fda.gov

7
The CLIA Waiver Program Has Improved Dramatically
• In the last few years, CLIA waiver program improvements
have addressed stakeholder concerns with CLIA waiver
review times, approval rates, study design flexibility, and
transparency
www.fda.gov

8
CLIA Waiver Approvals Have Increased Significantly
www.fda.gov
FY08 FY09 FY10 FY11 FY12 FY13 FY14 FY15 FY16
MDUFA III
FY08 FY09 FY10 FY11 FY12 FY13 FY14 FY15 FY16 12 7 5 5 6 3 15 14 10 5 3 2 3 2 2 11 8 8
Received Approved
Approved 80% in FY2016
Approved 42% in FY2008
MDUFA II
MDUFA = Medical Device User Fee Amendments
9
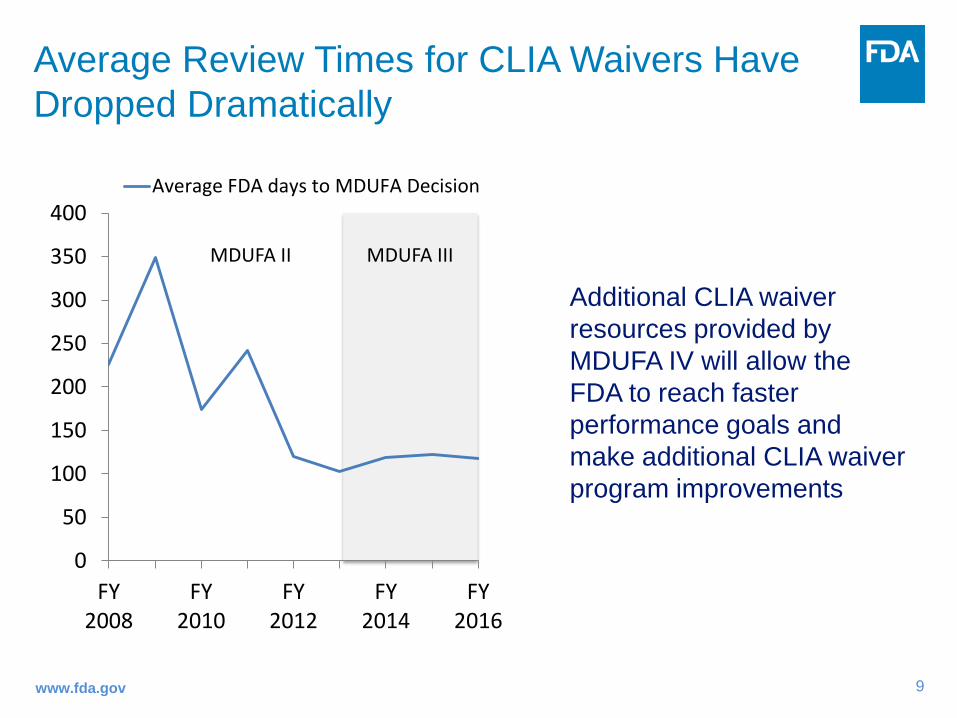
Average Review Times for CLIA Waivers Have Dropped Dramatically
www.fda.gov
MDUFA III
Additional CLIA waiver resources provided by MDUFA IV will allow the FDA to reach faster performance goals and make additional CLIA waiver program improvements
0
50
100
150
200
250
300
350
400
FY2008
FY2010
FY2012
FY2014
FY2016
Average FDA days to MDUFA Decision
MDUFA II

10
CLIA Waiver Decision Summaries Demonstrate Flexibility in Study Designs Supporting CLIA Waiver Approvals and Increase Transparency in the CLIA Waiver Review Process
https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHTransparency/ucm578178.htm

11
Draft Sec V. Guidance
• Describes how manufacturers may leverage existing accuracy data from a marketing submission (e.g., 510(k), PMA) and conduct less costly agreement studies that compare performance of the test between a waived user and a moderately complex laboratory user to demonstrate accuracy
• Follows least burdensome principles and references recognized consensus standards for study designs
www.fda.gov

12
CLIA Statutory Criteria for Waiver CLIA, 42 U.S.C. 263a(d)(3) Examinations and Procedures, as modified by the Food and Drug Administration Modernization Act of 1997 (FDAMA):
“The examinations and procedures [that may be performed by a laboratory with a Certificate of Waiver]… are laboratory examinations and procedures that have been approved by the Food and Drug Administration for home use or that, as determined by the Secretary, are simple laboratory examinations and procedures that have an insignificant risk of an erroneous result, including those that − A) employ methodologies that are so simple and accurate as to render the
likelihood of erroneous results by the user negligible, or B) the Secretary has determined pose no unreasonable risk of harm to the
patient if performed incorrectly.”
www.fda.gov

13
Accuracy is a Widely Used and Generally Understood Term in Clinical Laboratory Science
• For example, the Clinical Laboratory Standard Institute in EP21 defines accuracy as “the closeness of an agreement between a test result and the accepted reference value”
• The more accurate a test is, the less error there is, and therefore, the more likely the test is to have an “insignificant risk of erroneous result”
• Inaccurate tests may lead to incorrect patient management decisions and inappropriate public health responses
www.fda.gov

14
Why is it Important to Demonstrate Accuracy in Order for a Test to be CLIA Waived?
So that CLIA-waived laboratories don’t have to!
Under CLIA, for a non-waived test:
• The responsibility for delivering accurate results to patients is broadly shared between the test manufacturer and laboratories that choose to perform the test
• The data provided by the manufacturer to support FDA clearance/approval is only part of the data supporting that the test is accurate
• Non-waived laboratories complete additional verification or validation before performing a test for patients

15
Why is it Important to Demonstrate Accuracy in Order for a Test to be CLIA Waived? Unlike non-waived laboratories, CLIA-waived laboratories do not:
1. Verify or establish the accuracy and other performance characteristics of a cleared or approved test
2. Verify or establish that the test’s reference intervals (normal values) are appropriate for the laboratory's patient population
3. Perform proficiency testing 4. Operate within a quality system
• Validation of the accuracy of a test performed by the manufacturer to support CLIA waiver saves over 180,000 CLIA-waived laboratories from having to perform comparable validation themselves
16
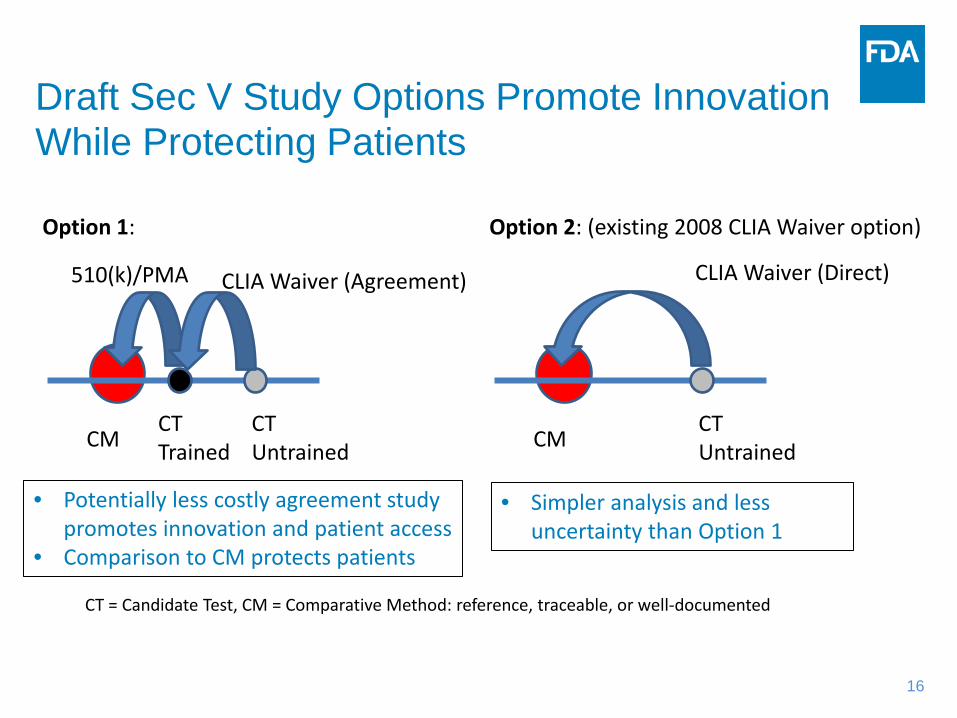
Draft Sec V Study Options Promote Innovation While Protecting Patients
CT = Candidate Test, CM = Comparative Method: reference, traceable, or well-documented
• Potentially less costly agreement study promotes innovation and patient access
• Comparison to CM protects patients
CM CT Trained
CT Untrained
510(k)/PMA CLIA Waiver (Agreement)
Option 1:
CM CT Untrained
CLIA Waiver (Direct)
Option 2: (existing 2008 CLIA Waiver option)
• Simpler analysis and less uncertainty than Option 1

17
When is Option 1 Appropriate?
1. The accuracy of the candidate test when performed by trained operators was demonstrated through comparison to an appropriate comparative method in the premarket submission (510(k), PMA, De Novo, etc.), and
2. The premarket study subjects/samples were representative of the intended use population at CLIA waived sites.
Valid statistical methods may then be used to estimate the accuracy of the test when performed by untrained operators using a combined analysis of the data from both studies
www.fda.gov

18
When is Option 1 Not Appropriate?
CT = Candidate Test, CM = Comparative Method: reference, traceable, or well-documented
CM CT Trained
CT Untrained
510(k)/PMA CLIA Waiver (Agreement)
?
Predicate Trained
Unknown Relationship to True Value – Can’t determine if the candidate test performed by untrained users has an insignificant risk of erroneous results
When data are not available to support accuracy of the test with trained operators compared to an appropriate comparative method

19
When is Option 2 Appropriate?
• Option 2 is always accepted and is also used in the Dual approach
• Option 2 is recommended for sponsors who want the best study to determine accuracy with less uncertainty and simpler analysis then Option 1
www.fda.gov

20
Draft Dual Guidance
• Describes study designs for generating data to support both 510(k) clearance and
CLIA Waived categorization
• Intended to assist manufacturers in using the optional Dual 510(k) and CLIA
Waiver by Application pathway (Dual pathway), formalized and piloted at the
request of industry groups in MDUFA III
• FDA believes the Dual pathway is in many instances the least burdensome and
fastest approach for manufacturers to obtain a CLIA waived categorization in
addition to 510(k) clearance for new In Vitro Diagnostic (IVD) devices
www.fda.gov

21
Historically, Separate 510(k) and CLIA Waiver Studies Have Been Conducted in Different Clinical Settings
www.fda.gov
Two Step: Direct (Existing 2008 and Draft Sec V: Option 2)
510(k) Candidate Test by Trained Users vs. Comparative Method or Predicate
CLIA Waiver by Application
Candidate Test by Untrained Users vs. Comparative Method
• In this two step approach, manufacturers conduct separate comparison and reproducibility studies, in different clinical settings, first to support 510(k) clearance and later to support CLIA Waiver by Application.

22
The Draft Dual Guidance Describes a More Efficient Single Study Approach for New IVDs
www.fda.gov
Two Step: Agreement (Draft Sec V: Option 1)
Two Step: Direct (Existing 2008 and Draft Sec V: Option 2)
Dual Study (Draft Dual)
510(k) Candidate Test (CT) by Trained Users vs. Comparative Method (CM)
CT by Trained Users vs. CM or Predicate
CLIA Waiver by Application
CT by Untrained Users vs. CT by Trained Users
CT by Untrained Users vs. CM
CT by Untrained Users vs. CM

23
Recommendations for Semi-Quantitative Tests Are a New Feature of the Draft Guidances
• The appropriate design of the studies and data analysis is
strongly influenced by whether the candidate test is quantitative,
qualitative or semi-quantitative
• For the purpose of these guidances, a semi-quantitative test is a
test with a few ordinal categories (e.g., negative, trace, +, ++,
+++) in which the order of categories together with the
definitions of these categories contain information used during
the interpretation of the test results
www.fda.gov

24
• For example, consider a semi-quantitative test for Albumin with 4 categories (bins), each covering a concentration range:
Neg 1+ 2+ 3+
Near Cutoff Region 2
Near Cutoff Region 3
Near Cutoff Region 1
• Even when comparing a semi-quantitative test to itself: – Some level of disagreement is expected for samples in the near
cutoff regions
– The extent of disagreement will increase with increased numbers of samples in the near cutoff regions
Why is a Quantitative Comparative Method Recommended for Semi-Quantitative Tests?
25
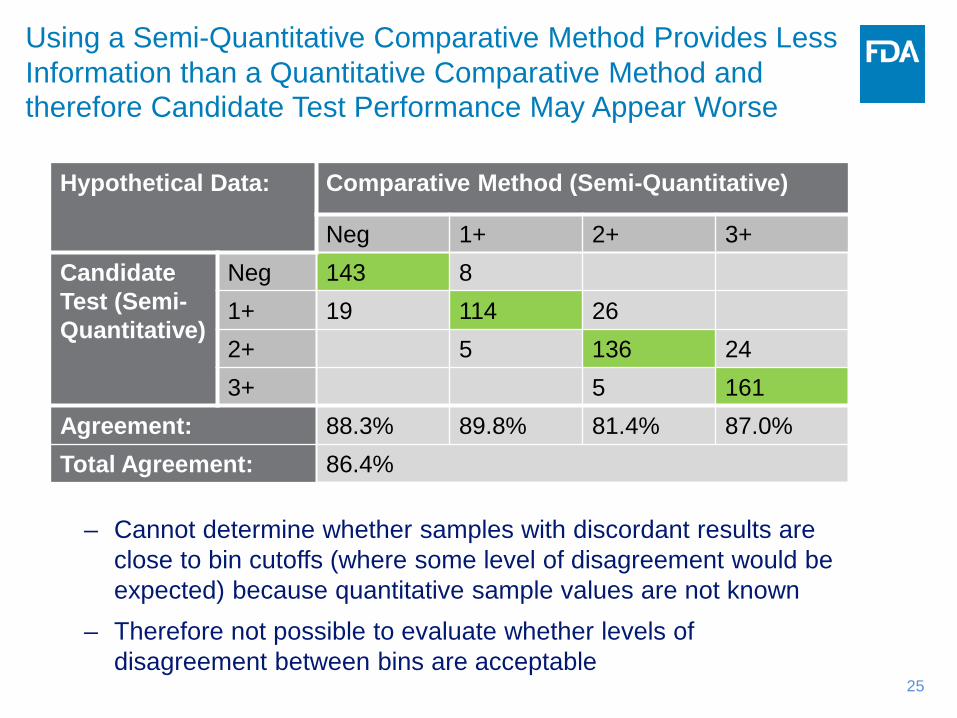
Hypothetical Data: Comparative Method (Semi-Quantitative)
Neg 1+ 2+ 3+ Candidate Test (Semi-Quantitative)
Neg 143 8 1+ 19 114 26 2+ 5 136 24 3+ 5 161
Agreement: 88.3% 89.8% 81.4% 87.0% Total Agreement: 86.4%
– Cannot determine whether samples with discordant results are close to bin cutoffs (where some level of disagreement would be expected) because quantitative sample values are not known
– Therefore not possible to evaluate whether levels of disagreement between bins are acceptable
Using a Semi-Quantitative Comparative Method Provides Less Information than a Quantitative Comparative Method and therefore Candidate Test Performance May Appear Worse

26
– Can determine whether samples are close to candidate test bin cutoffs (where some level of disagreement would be expected)
– Allows separate evaluation of agreement in 7 regions: • The 3 near cutoff regions where some disagreement is expected (grey
regions below)
• The 4 regions near the center of the bins where near 100% agreement is expected (white regions below)
Using a Quantitative Traceable Comparative Method is Advantageous
CM provides Analyte Concentration Traceable to References of Higher Order
3+ 1+ 2+ Neg
Near Cutoff Region 2
Near Cutoff Region 3 (108-123)
Near Cutoff Region 1
CT
CM 118

27
The % Coefficient of Variation (CV) of the Quantitative Traceable Comparative Method can be used to establish Allowable Total Error for a Semi-Quantitative Candidate Test
• Establishing Allowable Total Error for the difference between comparative method and candidate test
– Consider a quantitative test with acceptable level of % CV and % bias close to 0
– If one performs 2 measurements of the same sample with the quantitative device:
• there will be some difference in these two values because of random measurement errors.
– One can construct boundaries based on % CV that 95% of the differences between two repeated measurements are inside of these boundaries
– This method is used to determine near cutoff concentration ranges with acceptable variability
28
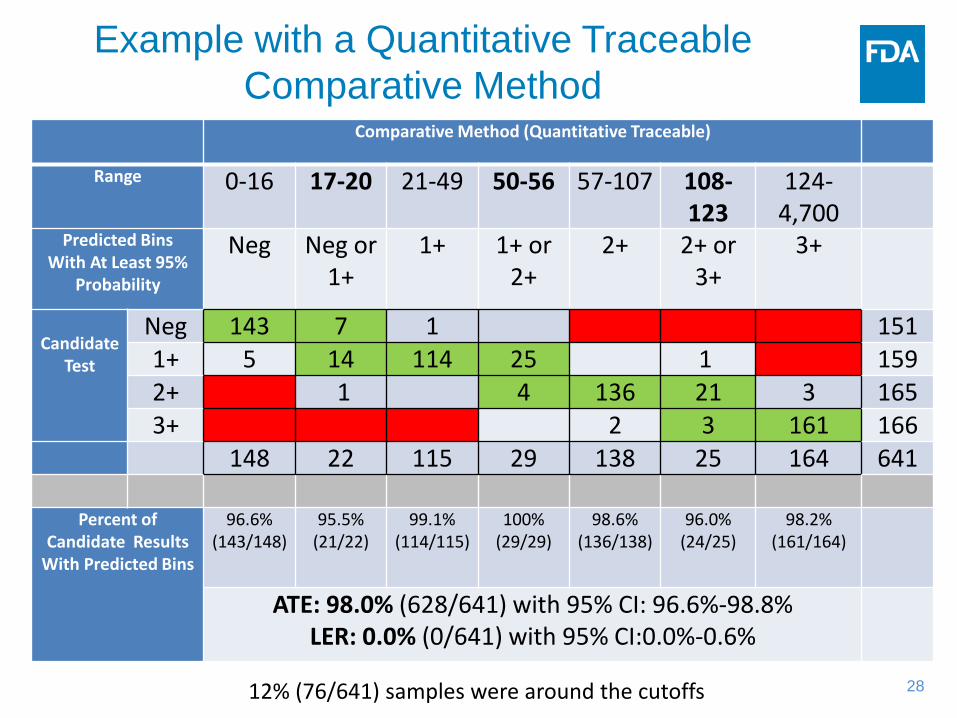
Example with a Quantitative Traceable Comparative Method
Comparative Method (Quantitative Traceable)
Range 0-16 17-20 21-49 50-56 57-107 108-123
124-4,700
Predicted Bins With At Least 95%
Probability
Neg Neg or 1+
1+ 1+ or 2+
2+ 2+ or 3+
3+
Candidate
Test
Neg 143 7 1 151 1+ 5 14 114 25 1 159 2+ 1 4 136 21 3 165 3+ 2 3 161 166
148 22 115 29 138 25 164 641
Percent of Candidate Results
With Predicted Bins
96.6% (143/148)
95.5% (21/22)
99.1% (114/115)
100% (29/29)
98.6% (136/138)
96.0% (24/25)
98.2% (161/164)
ATE: 98.0% (628/641) with 95% CI: 96.6%-98.8% LER: 0.0% (0/641) with 95% CI:0.0%-0.6%
12% (76/641) samples were around the cutoffs

Questions? [email protected]
Slide Presentation, Transcript and
Webinar Recording will be available at: http://www.fda.gov/training/cdrhlearn
Under the Heading: In Vitro Diagnostics


















