
Characterization ofSerratia.spp. from Water and Sediment ...
24
Characterization of Serratia. spp. from Water and Sediment from Aquaculture Environment Nurul Ain bt. Abdul'Mattin Bachelor of Science with Honours £6 (Resource Biotechnology) N974 2011 2011
Transcript of Characterization ofSerratia.spp. from Water and Sediment ...

Characterization of Serratia spp from Water and Sediment from Aquaculture Environment
Nurul Ain bt AbdulMattin
Bachelor of Science with Honours~~ pound6 (Resource Biotechnology) N974 2011 2011
PKHIDMAT MAKLUMAT AKADIMIK Pusat Khidmat Maklumat Aka emi~ Vpound t ~L vs P A
111111111 1111 1UUJLL
DECLARATION
I hereby declare that no portion of this dissertation has been submitted in support of an application for another degree ofqualification of this or any other university or institution of higher learning
I
Nurul Ain binti Abdul Mattin Resources Biotechnology Department of Molecular Biology Faculty of Resources Science and Technology University Malaysia Sarawak
II
ACKNOWLEDGEMENT
I am heartily thankful to my superVIsor Dr Samuel Lihan whose
encouragement guidance and support from the initial to the fmallevel of this whole
research project It is an honor for me to become one of his Final Year Projects
student and I am also very grateful for his willingness to always motivate me
towards completing my project and thesis write-up
I would also like to show my gratitude to my parents and beloved family
members who have given me a lot of spiritual and fmancial supports during this
whole project I would also want to thank Kathleen a master student in
Microbiology lab for willingly helping me throughout this project This research
project would not have been successful without her help
Lastly lowe my deepest gratitude to all my friends that have been helping
me around and motivating me whenever I feel like giving up Your kindness are
very much appreciated
I
III
PU3at Khidmat MakJumat Akadem ik UNIVERSITI MALAYSIA SARAWAK
TABLE OF CONTENT
Title amp Front Cover ~
Acknowledgement Table of contents III
List of Abbreviations IV
List ofTables and Figures ~ V
Abstracts 1
Chapter 1
Introduction 2
Chapter 2
Literature Review
20 Taxonomy bull 4
21 Morphology ampIdentification 4
22 Pathophysiology ofS marcescens 5
23 Epidemiology and Outbreaks Associated with Serratia spp 5
24 Identification ofSerratia spp by biochemical test 7
25 Characterization ofSerratia spp by Molecular Method 8 I
Chapter 3
Materials and Method
30 Sampling 10
31 Isolation and identification ofSerratia spp 10
32 Biochemical tests and DNA sequencing 11
15IV
I
shy
33 Characterization by molecular method
331 (GTG)5-PCR Analysis - 15
3321 Gel Electrophoresis 17
333 Antibiotic Susceptibility Testing 18
Chapter 4
Results amp Discussion
1940 Isolationof Serratia spp
41 Confirmation and Biochemical tests ofSerratia spp 21
42 Characterization of Serratia spp 30
420 (GT G)5-PCR Analysis 30
421 Antibiotic Susceptibility Testing 34
Conclusion 39
References 40
Appendices
i
v
LIST OF ABBREVIATIONS
AB
BLAST
CYVD
DNA
EtBr
HCL
LB
MHA
MR
NaCI
PCR
PVC
RAPD
SIM
TBE
TSA
TSI
VP
Antibiotic
Basic Local Alignment Search Tool
Cucurbit Yellow Vine Disease
Deoxyribonucleic acid
Ethidium Bromide
Hydrochloric acid
Luria broth
Mueller-Hinton agar
Methyl red
So dim chloride
Polymerase Chain Reaction
Polyvinyl Chloride
Random Amplified Polymorphic DNA
Sulfide-Indo le-Motility
TrislBoratelEDT A
Trypticase soy agar
r Triple sugar iron
Voges-Proskauer
VI
-
LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

PKHIDMAT MAKLUMAT AKADIMIK Pusat Khidmat Maklumat Aka emi~ Vpound t ~L vs P A
111111111 1111 1UUJLL
DECLARATION
I hereby declare that no portion of this dissertation has been submitted in support of an application for another degree ofqualification of this or any other university or institution of higher learning
I
Nurul Ain binti Abdul Mattin Resources Biotechnology Department of Molecular Biology Faculty of Resources Science and Technology University Malaysia Sarawak
II
ACKNOWLEDGEMENT
I am heartily thankful to my superVIsor Dr Samuel Lihan whose
encouragement guidance and support from the initial to the fmallevel of this whole
research project It is an honor for me to become one of his Final Year Projects
student and I am also very grateful for his willingness to always motivate me
towards completing my project and thesis write-up
I would also like to show my gratitude to my parents and beloved family
members who have given me a lot of spiritual and fmancial supports during this
whole project I would also want to thank Kathleen a master student in
Microbiology lab for willingly helping me throughout this project This research
project would not have been successful without her help
Lastly lowe my deepest gratitude to all my friends that have been helping
me around and motivating me whenever I feel like giving up Your kindness are
very much appreciated
I
III
PU3at Khidmat MakJumat Akadem ik UNIVERSITI MALAYSIA SARAWAK
TABLE OF CONTENT
Title amp Front Cover ~
Acknowledgement Table of contents III
List of Abbreviations IV
List ofTables and Figures ~ V
Abstracts 1
Chapter 1
Introduction 2
Chapter 2
Literature Review
20 Taxonomy bull 4
21 Morphology ampIdentification 4
22 Pathophysiology ofS marcescens 5
23 Epidemiology and Outbreaks Associated with Serratia spp 5
24 Identification ofSerratia spp by biochemical test 7
25 Characterization ofSerratia spp by Molecular Method 8 I
Chapter 3
Materials and Method
30 Sampling 10
31 Isolation and identification ofSerratia spp 10
32 Biochemical tests and DNA sequencing 11
15IV
I
shy
33 Characterization by molecular method
331 (GTG)5-PCR Analysis - 15
3321 Gel Electrophoresis 17
333 Antibiotic Susceptibility Testing 18
Chapter 4
Results amp Discussion
1940 Isolationof Serratia spp
41 Confirmation and Biochemical tests ofSerratia spp 21
42 Characterization of Serratia spp 30
420 (GT G)5-PCR Analysis 30
421 Antibiotic Susceptibility Testing 34
Conclusion 39
References 40
Appendices
i
v
LIST OF ABBREVIATIONS
AB
BLAST
CYVD
DNA
EtBr
HCL
LB
MHA
MR
NaCI
PCR
PVC
RAPD
SIM
TBE
TSA
TSI
VP
Antibiotic
Basic Local Alignment Search Tool
Cucurbit Yellow Vine Disease
Deoxyribonucleic acid
Ethidium Bromide
Hydrochloric acid
Luria broth
Mueller-Hinton agar
Methyl red
So dim chloride
Polymerase Chain Reaction
Polyvinyl Chloride
Random Amplified Polymorphic DNA
Sulfide-Indo le-Motility
TrislBoratelEDT A
Trypticase soy agar
r Triple sugar iron
Voges-Proskauer
VI
-
LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

ACKNOWLEDGEMENT
I am heartily thankful to my superVIsor Dr Samuel Lihan whose
encouragement guidance and support from the initial to the fmallevel of this whole
research project It is an honor for me to become one of his Final Year Projects
student and I am also very grateful for his willingness to always motivate me
towards completing my project and thesis write-up
I would also like to show my gratitude to my parents and beloved family
members who have given me a lot of spiritual and fmancial supports during this
whole project I would also want to thank Kathleen a master student in
Microbiology lab for willingly helping me throughout this project This research
project would not have been successful without her help
Lastly lowe my deepest gratitude to all my friends that have been helping
me around and motivating me whenever I feel like giving up Your kindness are
very much appreciated
I
III
PU3at Khidmat MakJumat Akadem ik UNIVERSITI MALAYSIA SARAWAK
TABLE OF CONTENT
Title amp Front Cover ~
Acknowledgement Table of contents III
List of Abbreviations IV
List ofTables and Figures ~ V
Abstracts 1
Chapter 1
Introduction 2
Chapter 2
Literature Review
20 Taxonomy bull 4
21 Morphology ampIdentification 4
22 Pathophysiology ofS marcescens 5
23 Epidemiology and Outbreaks Associated with Serratia spp 5
24 Identification ofSerratia spp by biochemical test 7
25 Characterization ofSerratia spp by Molecular Method 8 I
Chapter 3
Materials and Method
30 Sampling 10
31 Isolation and identification ofSerratia spp 10
32 Biochemical tests and DNA sequencing 11
15IV
I
shy
33 Characterization by molecular method
331 (GTG)5-PCR Analysis - 15
3321 Gel Electrophoresis 17
333 Antibiotic Susceptibility Testing 18
Chapter 4
Results amp Discussion
1940 Isolationof Serratia spp
41 Confirmation and Biochemical tests ofSerratia spp 21
42 Characterization of Serratia spp 30
420 (GT G)5-PCR Analysis 30
421 Antibiotic Susceptibility Testing 34
Conclusion 39
References 40
Appendices
i
v
LIST OF ABBREVIATIONS
AB
BLAST
CYVD
DNA
EtBr
HCL
LB
MHA
MR
NaCI
PCR
PVC
RAPD
SIM
TBE
TSA
TSI
VP
Antibiotic
Basic Local Alignment Search Tool
Cucurbit Yellow Vine Disease
Deoxyribonucleic acid
Ethidium Bromide
Hydrochloric acid
Luria broth
Mueller-Hinton agar
Methyl red
So dim chloride
Polymerase Chain Reaction
Polyvinyl Chloride
Random Amplified Polymorphic DNA
Sulfide-Indo le-Motility
TrislBoratelEDT A
Trypticase soy agar
r Triple sugar iron
Voges-Proskauer
VI
-
LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

PU3at Khidmat MakJumat Akadem ik UNIVERSITI MALAYSIA SARAWAK
TABLE OF CONTENT
Title amp Front Cover ~
Acknowledgement Table of contents III
List of Abbreviations IV
List ofTables and Figures ~ V
Abstracts 1
Chapter 1
Introduction 2
Chapter 2
Literature Review
20 Taxonomy bull 4
21 Morphology ampIdentification 4
22 Pathophysiology ofS marcescens 5
23 Epidemiology and Outbreaks Associated with Serratia spp 5
24 Identification ofSerratia spp by biochemical test 7
25 Characterization ofSerratia spp by Molecular Method 8 I
Chapter 3
Materials and Method
30 Sampling 10
31 Isolation and identification ofSerratia spp 10
32 Biochemical tests and DNA sequencing 11
15IV
I
shy
33 Characterization by molecular method
331 (GTG)5-PCR Analysis - 15
3321 Gel Electrophoresis 17
333 Antibiotic Susceptibility Testing 18
Chapter 4
Results amp Discussion
1940 Isolationof Serratia spp
41 Confirmation and Biochemical tests ofSerratia spp 21
42 Characterization of Serratia spp 30
420 (GT G)5-PCR Analysis 30
421 Antibiotic Susceptibility Testing 34
Conclusion 39
References 40
Appendices
i
v
LIST OF ABBREVIATIONS
AB
BLAST
CYVD
DNA
EtBr
HCL
LB
MHA
MR
NaCI
PCR
PVC
RAPD
SIM
TBE
TSA
TSI
VP
Antibiotic
Basic Local Alignment Search Tool
Cucurbit Yellow Vine Disease
Deoxyribonucleic acid
Ethidium Bromide
Hydrochloric acid
Luria broth
Mueller-Hinton agar
Methyl red
So dim chloride
Polymerase Chain Reaction
Polyvinyl Chloride
Random Amplified Polymorphic DNA
Sulfide-Indo le-Motility
TrislBoratelEDT A
Trypticase soy agar
r Triple sugar iron
Voges-Proskauer
VI
-
LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

I
shy
33 Characterization by molecular method
331 (GTG)5-PCR Analysis - 15
3321 Gel Electrophoresis 17
333 Antibiotic Susceptibility Testing 18
Chapter 4
Results amp Discussion
1940 Isolationof Serratia spp
41 Confirmation and Biochemical tests ofSerratia spp 21
42 Characterization of Serratia spp 30
420 (GT G)5-PCR Analysis 30
421 Antibiotic Susceptibility Testing 34
Conclusion 39
References 40
Appendices
i
v
LIST OF ABBREVIATIONS
AB
BLAST
CYVD
DNA
EtBr
HCL
LB
MHA
MR
NaCI
PCR
PVC
RAPD
SIM
TBE
TSA
TSI
VP
Antibiotic
Basic Local Alignment Search Tool
Cucurbit Yellow Vine Disease
Deoxyribonucleic acid
Ethidium Bromide
Hydrochloric acid
Luria broth
Mueller-Hinton agar
Methyl red
So dim chloride
Polymerase Chain Reaction
Polyvinyl Chloride
Random Amplified Polymorphic DNA
Sulfide-Indo le-Motility
TrislBoratelEDT A
Trypticase soy agar
r Triple sugar iron
Voges-Proskauer
VI
-
LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

LIST OF ABBREVIATIONS
AB
BLAST
CYVD
DNA
EtBr
HCL
LB
MHA
MR
NaCI
PCR
PVC
RAPD
SIM
TBE
TSA
TSI
VP
Antibiotic
Basic Local Alignment Search Tool
Cucurbit Yellow Vine Disease
Deoxyribonucleic acid
Ethidium Bromide
Hydrochloric acid
Luria broth
Mueller-Hinton agar
Methyl red
So dim chloride
Polymerase Chain Reaction
Polyvinyl Chloride
Random Amplified Polymorphic DNA
Sulfide-Indo le-Motility
TrislBoratelEDT A
Trypticase soy agar
r Triple sugar iron
Voges-Proskauer
VI
-
LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

LIST OF TABLES AND FIGURES Page
Table 1 PCR reaction set-up for 16SrRNA 14
Table 2 PCR conditions for amplification of 16srRNA using 27fand 519r primers 15
Table 3 PCR reaction set-up for (GTG)s-PCR analysis 16
Table 4 PCR conditions for (GTG)s-PCR analysis 17
Table 5 Table of biochemical tests reaction 27
Table 6 Isolate codes and their species representatives 28
Table 7 Zone of inhibition (cm) of different types of antibiotics 35
Table 8 Antibiotic Resistance patterns and MAR (Multiple Antibiotic Resistance) Index 36
Table 9 Resistant Percentages () of the Types of Antibiotics Tested 37
Figure 1 Green-grey colonies of Serratia spp on PaIcam selective agar 19
Figure 2 Positive result ofTSI test 21
Figure 3 Positive result of catalase test 22
Figure 4 Positive result of SIM motility test 23
Figure 5 Gram negative with a cocci-shaped bacterium 24
Figure 6 (Left) Negative MR test (Right) Positive VP test 25
Figure 7 Agarose gel electrophoresis (15) of (GTG)s PCR of the Serratia spp 30
Figure 8 Dendogram of all isolates showing the degree of relatedness between each species typed 32
Figure 9 Antibiotic susceptibility testing of a sample 34
VII
Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

Characterization ofSerratia spp from Water and Sediment Samples FromAquaculture Environment
Nurul Ain bt Abdul Mattin
Resource Biotechnology Programme
Faculty of Resource Science and Technology
Universiti Malaysia Sarawak
Abstract
Serratia spp are classified under Enterobacteriaceae and the pathogenic species Serratia marcescens is notoriously
associated with nosocomial infection in patients In this study Serratia spp were isolated from water and sediment samples The sampling was carried out at a pond in Samariang and 7U1 Mile Kuching Further identification and
confmnation tests were carried out using TSI test citrate utilization test catalase test urease test MRVP test gram
staining and DNA sequencing (GTG)s-PCR was conducted to analyze the genetic diversity of species isolated II isolates of the Serratia spp were characterized by antibiotic susceptibility testing and DNA profiling using (GTG)sshy
PCR In antibiotic susceptibility testing the antibiotics used were ampicillin vancomycin bacitracin nitrofurantoin norfloxacin kanamycin erythromycin chloramphcnicol streptomycin tetracycline nalidixic acid sulphamethoxazoletrimethoprim gentamycin and carbenicillin All isolates were resistant to vancomycin bacitracin nitrofurantoin erythromycin and chloramphenicol The phylogenetic tree constructed for DNA fingerprinting using
(GTG)5-PCR technique indicates that Serratia spp isolated from the water and sediment samples were genetically
diverse and the isolates showed multiple-resistance to the antibiotics being tcsted
Key words Serratia spp biochemical tests DNA sequencing antibiotic susceptibility DNA fmgerprinting
Abstrak
Serratia spp dikategorikan di dalam kumpulan Enterobakter dan kewujudan strain pathogen Serratia marcescens biasanya bertanggungjawab di dalam jangkitan nosokomial di kalangan pesakit Di dalam kajian yang telah dilakukan ini spesis Serratia spp telah dipencilkan daripada persekitaran air dan mendapan tanah Persampelan telah dijalankan
di tasik berdekatanSwvariang dan Batu 7 Seterusnya pengesahan spesis tersebut telah dijalankan dengan menggunakan kaedah-kaedah biokimia seperti ujian TSI ujian pemanfaatan sitrat ujian katalase ujian urease ujian MRVP pewamaan gram dan teknik penjujukan DNA Kaedah penjujukan DNA dengan menggunakan (GTG)s-PCR berperanan untuk menguji diversiti genetik daripada pencilan yang telah dilakukan II pencilan Serratia spp juga telah
dicirikan menggunakan ujian ketahanan antibiotik dan pemprofilan DNA pula dijalankn menggunakan teknik (GTG)sshyPCR Antibiotik yang telah digunakan di dalam ujian ketahanan antibiotic adalah ampisilin vankomisin basitrasin nitrofurantoin norfloksasin kanamisin eritromisin k1oramfenikol streptomisin tetrasikl in asid nalidiksik
sulfametoksazoVtrimetoprim gentamisin dan karbenisilin Kesemua pencilan pula bertahan terhadap vankomisin basitasin nitrofurantoin eritromisin dan kloramfenikoL Pokok filogenetik yg telah dibuat daripada cap jari DNA menggunakan kaedah (GTG)s-PCR menunjukkan bahawa Serratia spp yang tclah dipencilkan daripada komponen air
dan mendapan mempunyai kepeJbagaian genetik yang luas dan pencilan tersebut menunjukkan kadar ketahanan antibiotik yang berbagai terhadap antibiotik yang diuji
Kata kunci Serralia spp ujian biokirnia penjujukan DNA ketahanan antibiotik cap jari DNA
1
CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

CHAPTER
INTRODUCTION
Serratia spp are a Gram negative facultatively anaerobic and rod-shaped
bacteria Serratia spp is a genus under the family group of Enterobacteriaceae There are
6 species of Serratia spp namely S orodijera S rnbidaea S liquejaciens S
plymuthica S jonticola and the pathogenic strain of Serratia spp is known as S
marcescens S marcescens is associated with nosocomial infection in humans Infection
of the pathogenic strain of Serratia spp may cause some serious health complications
including meningitis and arthritis Serratia spp usually infect the bloodstream lower
respiratory and urinary tract Patients may experience fever chills and other serious
complications like endocarditis renal failure pneumonia chronic obstructive pulmonary
and urinary tract disease and many others
Serratia spp have become a considerable concern in causing chronic diseases due
to their wide distribution in the environments Based on a IS-month bacterial infection
surveillance a number of 732 babies were admitted to the neonatal care unit and S
marcescens had been reported to have been isolated in 153 babies (Smith et al 1984)
These alarming high rate of infection among neonatal had led to the research of Serratia
spp
Ongoing research on Serratia spp is still being conducted in order to know more
about how infectious it is to human beings and the best prevention and treatment method
Early identification of the presence of Serratia spp was done and several biochemical
tests was conducted to confirmed the presence of the bacteria from water and soil sample
2
Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

Molecular identification of Serratia spp also was conducted using peR analysis and ~
antibiotic susceptibility testing was also conducted on the isolates DNA fingerprinting
was used to further characterize the Serratia spp strains isolated The significant of this
study was to detect the presence of Serratia spp that could indicate the presence of
pathogenic strain ofS marcescens
Hence the objectives of this study were
I) to isolate Serratia spp from water and sediment samples
2) to identify the isolated Serratia sppusing biochemical tests and DNA sequencing
3) to characterize the isolated strains using molecular DNA fingerprinting
3
CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

CHAPTER 2
LITERATURE REVIEW
20 Taxonomy
The genus Serratia is categorized under the tribe Klebsiella and large family of
Enterobacteriaceae The 6 known species of Serratia are S orodifora S rubidaea S
Liquefaciens S plymuthica S fonticola and S marcescens S marcescens were first
discovered by a Venetian pharmacist in 1819 where he discovered a red discolouration
occurring in Polenta in Padua city (Sehdev amp Donnenberg 1998) Later in 1923 he
named the bacteria as Serratia in honor of an Italian physicist who invented steamboat
Serrafino Serrati The word marcescens in Latin means decaying Therefore the word
has been used together due to the rapid red color deterioration
21 Morphology and Identification
Serratia spp is a Gram negative rod-shaped bacterium It is a facultative anaerobe
and it is motie Serratia spp produces red pigments called prodiginines It is a highly
mucoid colonies when grown on agar Based on the 2nd edition of Bergeys manual of
Determinative Bacteriology Serratia spp falls under the family Enterobacteriaceae
lactose negative group It cannot ferment lactose and is indole-negative It gives a
negative result upon urease test and H2S production Serratia spp is a motile organism
4
II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

II 1 hiJI IJN It
and citrate-positive It utilizes malonate and produces DNAse gelatinase lipase ~
lecithinase chitinase and esterase enzymes
22 Pathophysiology ofS marcescens
Infection by Serratia spp is responsible for the nosocomial infections of the
following sites bloodstream lower respiratory tract urinary tract surgical wounds and
epithelial tissues of adults The infection can also cause meningitis and arthritis in
pediatric ward patients Those addicted to heroin drug infection of the pathogenic strain
of S marcescens may lead to other serious complications including endocarditis and
osteomyelitis
Infection of S marcescens reported may also cause 2 types of pneumonia in the
year of 1968-1980 nine patients experienced acute haemorrhagic bronchopneumonia
and seven more had diffuse neutropenic vasculitis (Goldstein et ai 1982)
23 Epidemiology and Outbreaks Associated with Serratia spp
In October 1999 7 patients had been reported to have been infected with S
marcescens in a hospital located in Ontario Canada Among them 5 patients suffered ~
bull r
from bacteremia while 2 of them suffered from wound infections Among 5 of them who
showed presence ofbacteria in their blood 2 of them died later (Bonnie et ai 2001)
There was also an outbreak occurring from September 1998 to June 1999 which
involved 24 patients admitted in the bone marrow transplant and oncology unit 14 of
them developed very serious infection due to the presence of Serratia marcescens in their
5
I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

I
blood Serratia spp were isolated from several sources including the control buttons of
intravenous infusion pump and urine jug assigned to a patient without evidence of S
marcescens carriage (Knowles et at 2000)
In Zurich (April 1998-May 1999) S marcescens pathogen had been isolated from
bottles of liquid theophylline Eleven out of 20 neonated had been infected and the
colonization of pathogen occurred within 24 hours after delivery Isolates were obtained
from stool and gastric aspirate specimens (Fleisch et at 2002)
In 2000 an outbreak in a number of Scotland NICUs (neonatal intensive care
units) had been reported This outbreak occurred over 6 week period where 12 babies
were being admitted at the Glasgow Royal Maternity Hospital (GRMH) and 5 more
babies at the Queen Mothers Hospital (QMH) Among those admitted in GRMH 3
babies suffered from septicaemia and 2 of them actually died After subsequent
investigations the outbreak pathogenic strain of S marcescens had been proved to have
been successfully isolated from laryngoscope blade and an expressed breast milk sample
(Jones et at 2000)
In 2006 at the Farabi Hospital of Karadeniz Technical University in Trabzon
Turkey 3 of 9 neonates admitted were dead due to the presence ofS marcescens in their
blood After investigation the pathogenic bacteria were found in one hand-washing
sample and two breast milk samples (Bayramoqluet at 2011)
6
24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

24 Identification of Serratia spp by biochemical test
Serratia spp is a motile organism and can grow in temperatures ranging from 5shy
40degC and pH levels ranging from 5 to 9 It can be distinguished by other types of
Enterobacteriaceae by its ability to hydrolyze casein which allows it to produce
extracellular metaUoproteinases S marcescens is able to degrade tryptophan and citrate
Degradation of tryptophan may yield pyruvic acid as by-product which later be used by
S marcescens in their metabolic processes Degradation of citrate on the other hand will
yield carbon which is used as a C source
Methyl red test is used to determine whether the bacteria is undergoing mixed-
acid fermentation or not In the case of S marcescens the result of methyl red test is
supposed to be negative Other than that S marcescens is able to ferment lactose
producing lactic acid as the end-product TSI reaction were acid-butt with no production
of hydrogen sulfide The bacteria is motile and did not give positive result for indole test
MR test were negative and the opposite positive result in VP test
In spirit blue lipase test S marcescens give positive result in which the presence
of clearing zone surrounding the sample This differential test determines whether the
organism produ~es the secreted lipase enzymes S marcescens also are able to digest
gelatin Indication of positive result is shown by the liquefaction of the media upon
refrigeration
S marcescens gives positive result upon catalase test in which visible bubbles
appear upon contact with hydrogen peroxide Negative result is obtained with oxidase
test
7
shy
25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

25 Characterization ofSerratia spp by Molecular Method
Based on article by Kur et al (1995) molecular methods that had been used in the
epidemiological study of Serratia spp includes the conventional PCR This method had
successfully detected 40 clinical strains of the pathogenic strain of S marcescens using a
primer that amplifY the spacer regions between the 16S and 23S genes in the prokaryotic
rDNA loci A combination of biotyping and RAPD-PCR method had also been
conducted in the isolates from the nasocomial infection of pediatric patients This method
shows the clonal variations available in the sample isolated (Enciso-Moreno et ai 2004)
A genotyping study using rep-PCR of the strain S marcescens that causes CYVD
(cucurbit yellow vine disease) showed the banding pattern were identical to the strains
genotyped using the DNA-DNA hybridization technique (Zhang et aI 2003) In other
study rep-pcr using the primer (GTG)s also had been done in genotyping analysis in 2
premature infants that suffered from the S marcescens infection and the result showed
that 3 out of 5 sample of patients involved were genetically-related ( Campbell et ai
1998)
In Spain 38 isolates from different oak species that has been reported as a J
I
causative agents of causing cankers in oak trees from different locations was analyzed by
sequencing using 16SrDNA and rep-PCR fingerprinting method The sequencing result
showed that 34 out of 38 isolates were from Brenneria spp and 4 of the isolates were
from Serratia spp Dendogram obtained showed that Serratia spp also are
phylogenetic ally close to the Brenneria spp (Gallego et ai 2008)
8
A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

A different study carried out in Korea proved that RAPD-PCR ERIC-PCR and ~
Rep-PCR were a reliable method in genotyping studies of the strain S marcescens All
banding patterns from the 24 samples obtained from patients admitted at the Changbuk
National University Hospital were identical in the same epidemic strain and unidentical
in non-epidemic strain ( Shin 2003)
9
CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

CHAPTER 3
MATERIALS AND METHOD
30 Sampling
Water (50ml) and soil (25g) samples were taken from Samariang and 7th Mile
aquaculture pond The sediments were collected using a PVC pipe These samples were
transported to the laboratory in ice container Samples brought to the lab were then
immediately processed pH of the water samples were obtained and recorded using a pH
meter
31 Isolation and identification ofSerratia spp
310 Isolation using Palcam Selective agar
225ml of Fraser broth were used for enrichment An equal amount of samples
from 3 random area for each water and soil samples were added into the broth and mixed
1 ml of sample from Fraser broth was then added into a 9ml of saline-containing tubes
(100 ml distilled water in 085g NaCl) yielding a total of 10ml solution Serial dilution of
101 102 103 and 104 were prepared using the standard technique
1 00 ~l of each dilution tube was then spread out onto Palcam selective agar
Then the plate were incubated for 24 hours at 29degC After incubation each colony were
10
first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

first being streaked and incubated onto non-selective slant TSA agar to get pure culture ~
for biochemical confirmation tests
32 Biochemical tests
320 TSI test
The agar was first prepared Then a colony of the bacterial isolate was then
stabbed into the center of the tubes containing the medium agar using a sterile needle
After that the needle was streaked back and forth onto the surface of the slanted medium
and be incubated for not more than 24 hours The TSI agar contains three sugars
(dextrose lactose and sucrose) phenol red to detect carbohydrate fermentation and
ferrous ammonium sulphate for detection of hydrogen sulfite production The caps were
closed loosely upon incubation to permit free exchange of air so as to enhance the
alkaline condition of the slant
321 Citrate utilization test
After prepanng the Simmons citrate medium bacterial colonies from the
cultured slant agar were picked by a sterile wire loop and streaked onto the medium The
streaked plate were then incubated overnight 322 Catalase test
Bacterial colony were cultured onto a non-selective TSA agar and incubated for
24 hours After cultivation 2 or 3 drops of catalase (hydrogen peroxide) reagent were
added onto the culture in the plate Formation of bubbles upon addition were observed
and record ed
11
323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

323 Urease test
After preparing the urease agar the isolates were streaked onto the medium by a
wire loop The plate were incubated for 24 hours and the color changes of the medium
were observed
324 81M test
Motility test was carried out in this study to determine whether isolated bacteria
was motile or not Medium used was the BBLTM 81M motility test medium A colony
from the pure culture was stabbed through the center of the jelly-like consistency of the
medium The tubes containing the cultured medium were incubated for 24 hours After
incubation the appearance of the agar were observed and analyzed
325 MRVP test
The isolates were first cultured into sterile LB broth After cultivation the culture
was divided into two sterile bottles to perform the MR (methyl red) and VP (vogesshy
proskauer) test
F or methyl red (MR) test 2 or 3 drops of methyl red reagent were added into the
bottle and cdlorlchanges were observed
Meanwhile voges-proskauer (VP) test were conducted using Barrits reagent A
and B A few drops of each reagent were added subsequently and the color changes were
observed
12
326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

326 Gram Staining
This test detennined whether the bacterium is either Gram positive or Gram
negative indicated by the colour of the stain obsetved under the microscope
First the cultured isolates were streaked and fixed onto a clean glass slide using a
Bunsen burner flame The smear was then drained with a few drops of the primary stain
crystal violet for 1 minute This primary stain renders all the bacteria unifonnly into
violet colour Then the excess stain were washed out under rwming tap water The smear
were then treated with a few drops of a mordant iodine for 1 minute The slides were
then washed under running water before being decolourized in alcohol for only about a
few seconds Prolonged exposure to alcohol may cause over-decolorization Then the
slides were washed under rwming tap water before treating them with the red
counterstain safranin for 1 minute Tills stain gives the colour red in Gram staining
Excess safranin were then washed out and the slide were let to air-dried before
observation of the bacterial morphology under the microscope
327 DNA Sequencing
I 01
DNA sequencing was carried out by amplifying 16SrRNA of the bacterial isolates
using universal primers 27f and 519r (Hutter et al 2003) The primers sequence were
27f (5-AGAGTTTGATCMTGGCTCAG-3)
519r (5-GWATTACCGCGGCKGCTG-3)
13
The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

The PCR reaction set-up used consists of the following (Table 1)
Table 1 PCR reaction set-up for 16SrRNA
peR components
Taq Polymerase (2U)
27fand 519rprimers
MgCh
dNTPmix
Distilled H2O
Buffer (1 Ox)
Template DNA
Total
Vohime
04 )11
1)11
3 )11
1)11
46 )11
5 )11
10 )11
25 III
14
- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

- I~
P CR conditions for 16srRNA amplification of bacterial isolates were as follows
(Table 2)
Table 2 PCR co nditions for amplification of 16srRNA using 27fand 519r primers
Steps
Initial Denaturati on
Denaturation
Temperature
95degC
94 degc
Time
10 min
30 sec
Annealing 55degC amp 58 degc respectively 1 min
Extension
Final Extension
72degC
72degC
1 min 30 sec
20 min
2 6
c y c I e s
T he PCR products were then purified using Qiagen DNA purification kit The
pure DN A (elution of 200 d) was sent to First Base Sdn Bhd for DNA sequencing
services
33 Characterization by Molecular Method
331 (GTG)s-PCR Analysis
The (GTG)s-PCR was carried out according to the method described by Matsheka
et al (2006) The primer used was (GTG)s The sequence ofthe primer is shown below
(GTG)s 5-GTG GTG GTG GTG GTG-3
15
The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17

The PCR reaction set-up used consists of the following (Table 3)
Table 3 PCR reaction set-up of (GTG)5-PCR
peR components Volume
Taq Polymerase (2U) 03 III
(GTG)5 primer 1III
MgCh 3111
dNTP 081l1
Distilled H2O 99 III
Buffer (lax)
5 III
Template DNA 5 III
Total
I
25 III
16
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17
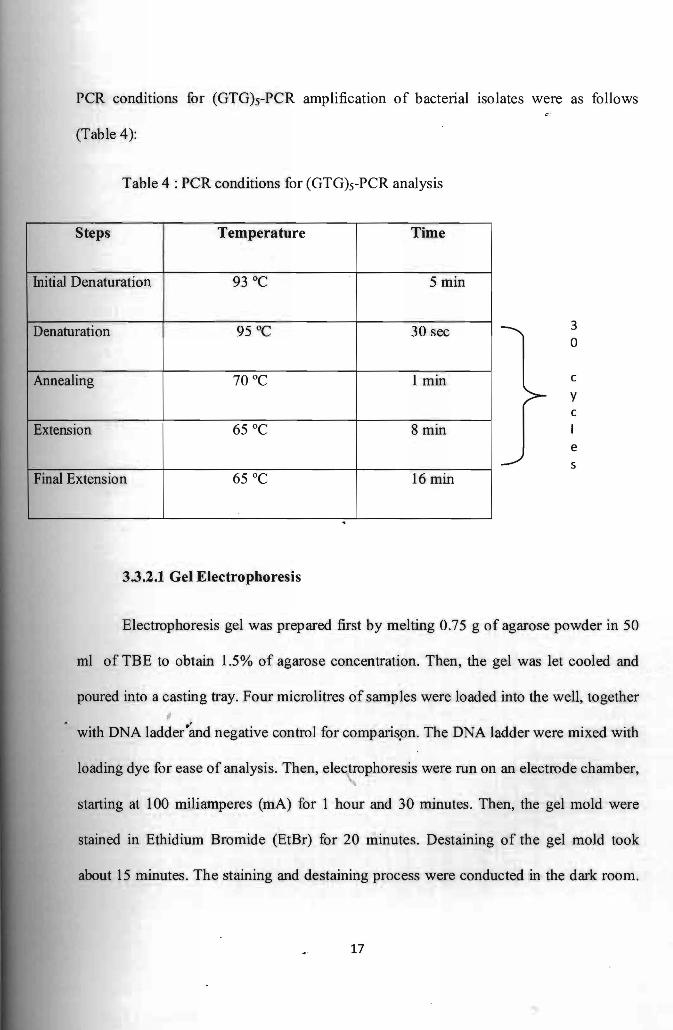
PCR conditions for (GTG)s-PCR amplification of bacterial isolates were as follows
(Table 4)
Table 4 PCR conditions for (GTG)s-PCR analysis
Steps I Temperature Time I I
Initial Denaturation 5 min 93degC
Denaturation 95degC 30 sec
Annealing 70degC 1 min
Extension 65degC 8 min
Final Extension 65degC 16 min
3 o
c y c
e s
3321 Gel Electrophoresis
Electrophoresis gel was prepared first by melting 075 g of agarose powder in 50
ml of TBE to obtain 15 of agarose concentration Then the gel was let cooled and
poured into a casting tray Four microlitres of samples were loaded into the well together ~
with DNA ladder~~d negative control for comparison The DNA ladder were mixed with
loading dye for ease of analysis Then electrophoresis were run on an electrode chamber
starting at 100 miliamperes (rnA) for 1 hour and 30 minutes Then the gel mold were
stained in Ethidium Bromide (EtBr) for 20 minutes Destaining of the gel mold took
about 15 minutes The staining and de staining process were conducted in the dark room
17


















