
CHAPTER-4 Cobalt(II)- Catalyzed Dehydration of...
39
119 CHAPTER-4 Cobalt(II)- Catalyzed Dehydration of Aldoximes: A Highly Efficient practical Procedure for the Synthesis of Nitriles
Transcript of CHAPTER-4 Cobalt(II)- Catalyzed Dehydration of...

119
CHAPTER-4
Cobalt(II)- Catalyzed Dehydration
of Aldoximes: A Highly Efficient
practical Procedure for the
Synthesis of Nitriles

120
Background
4.1. Introduction
Nitriles are important synthons in organic chemical synthesis. They are key
components of range of dyes, agrochemicals (such as herbicides, insecticides, and
acaricides), pharmaceuticals, ferroelectric materials, and natural products.1-5
Nitrile group
also serves as an important intermediate structure for a multitude of possible
transformations into other functional groups.
4.2. Biological Activity of nitriles
Nitrile group serves as an important intermediate structure for a multitude of possible
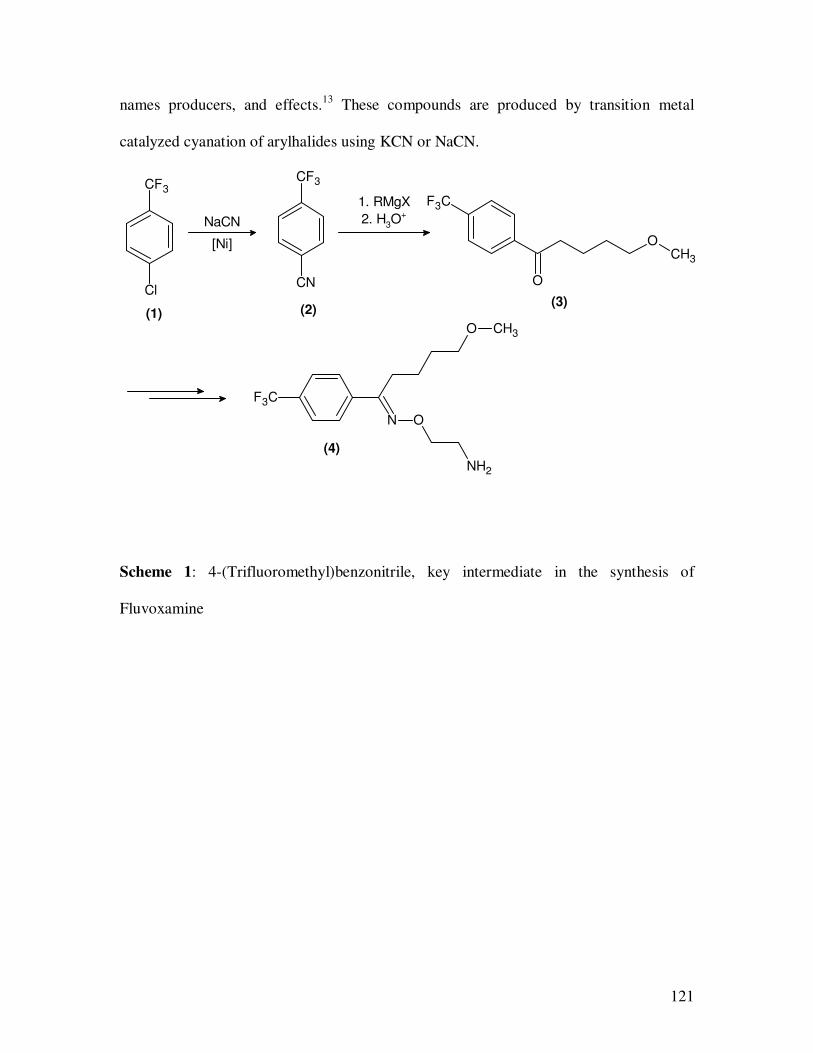
transformations into other functional groups. For example, the synthesis of Fluoxamine
(4) is shown in the Scheme 1. Here, 4-(trifluorormethyl)-benzonitrile, which is obtained
from 4-chloro-(trifluoromethyl)benzene by nickel catalyzed cyanation serves as an
intermediate.6-8
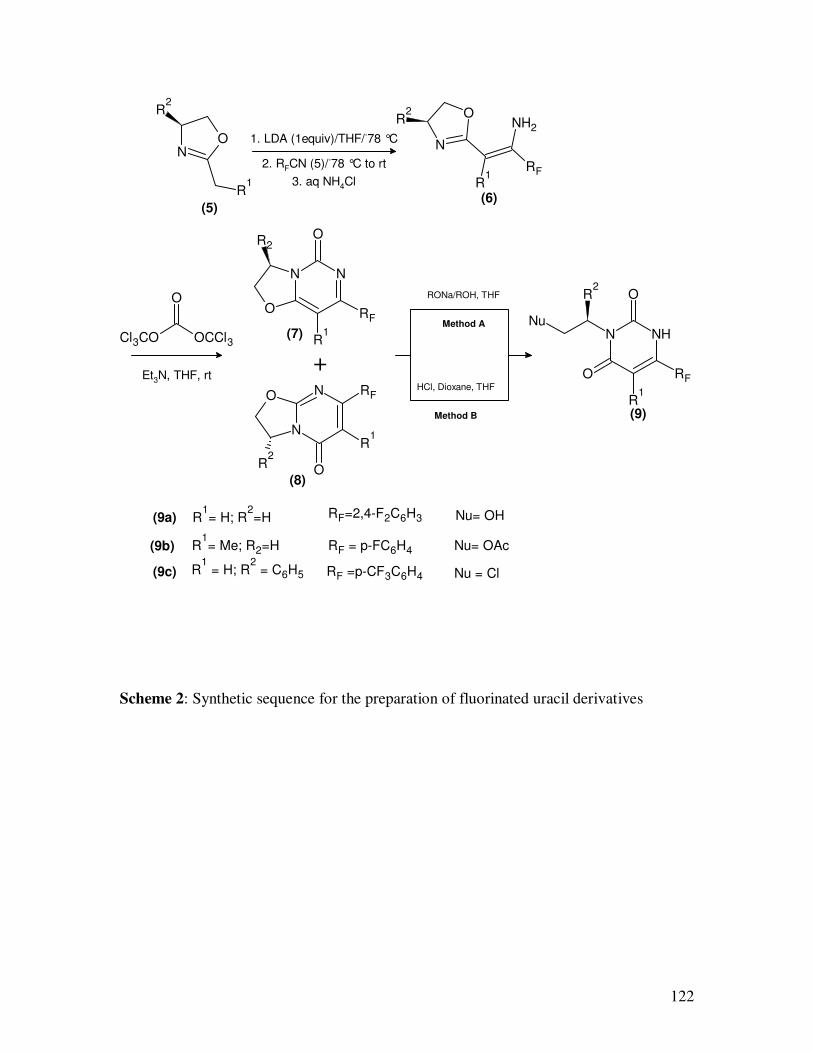
Nitriles are the starting materials in the synthesis of fluorinated uracil
derivatives (9a-9c)9 which are known for their applications as antineoplastics, antiviral,
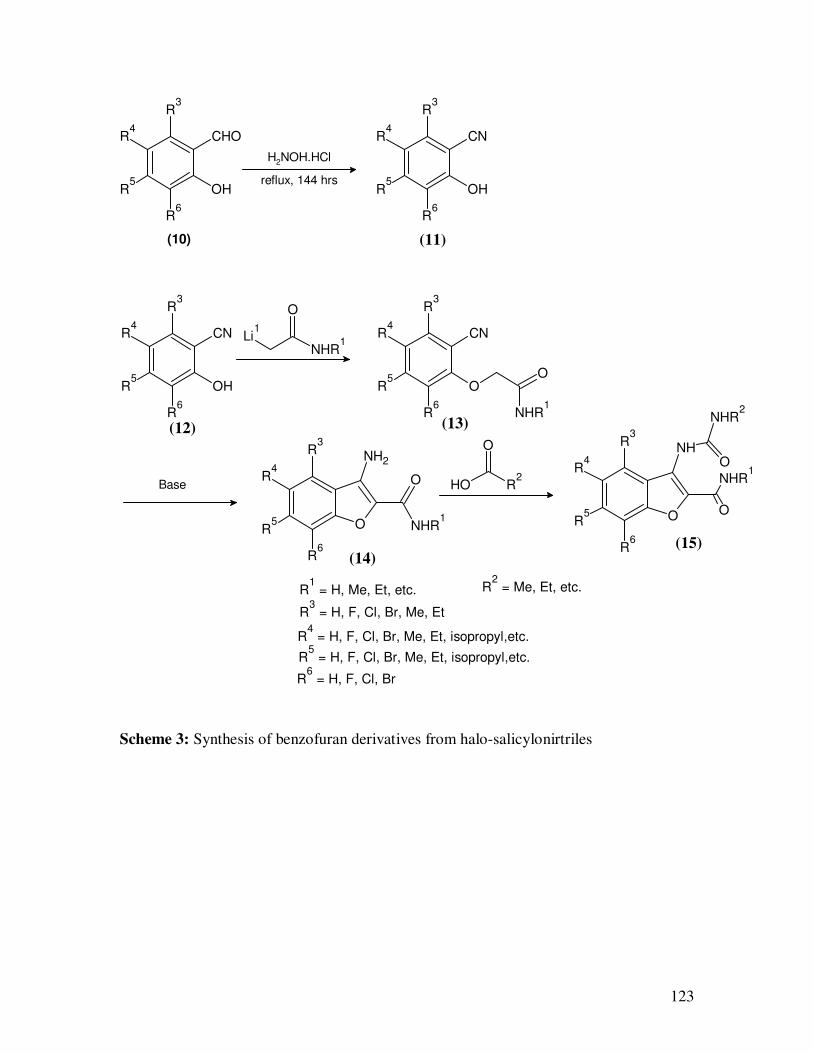
and antitumoral agents (Scheme 2). Benzofuran derivatives10-12
(16) that are potent
adenosine A2A receptor antagonists, and are useful for treating or preventing adenosine
A2A receptor mediated diseases such as motor function disorders, depression, cognitive
function disorders, and cerebral ischemia disorders, are synthesized from halo-substituted
salicylonitriles (Scheme 3).
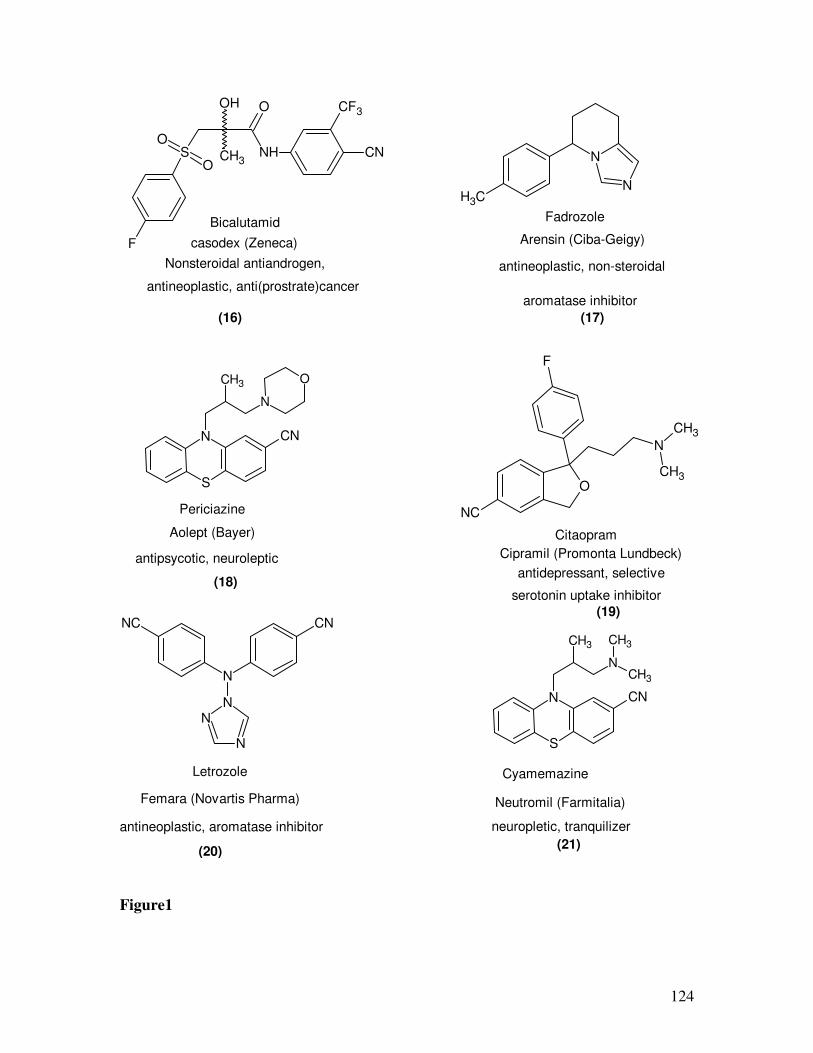
Benzonitriles themselves are also of significant interest, for example as substructures
in biologically active agents. In Figures 1, 2, selected examples of pharmaceuticals (16-
23) containing an aromatic nitrile as integral part of molecule are shown along with their
121
names producers, and effects.13
These compounds are produced by transition metal
catalyzed cyanation of arylhalides using KCN or NaCN.
Cl
CF3
NaCN
[Ni]
CN
CF3
1. RMgX
2. H3O+
F3C
OCH3
O
F3C
O CH3
N O
NH2
(1) (2)(3)
(4)
Scheme 1: 4-(Trifluoromethyl)benzonitrile, key intermediate in the synthesis of
Fluvoxamine
122
NO
R2
R1
1. LDA (1equiv)/THF/-78 °C
2. RFCN (5)/-78 °C to rt
3. aq NH4Cl
N
OR2
R1
NH2
RF
O
Cl3CO OCCl3
Et3N, THF, rt
N
O
N
O
R1
R2
RF
+
N NH
O
R1
R2
RF
Nu
O
N
NO
O
R1
RF
R2
R1= H; R
2=H
R1= Me; R2=H
Nu= OH
RONa/ROH, THF
Method A
HCl, Dioxane, THF
Method B
RF=2,4-F2C6H3
(5)(6)
(7)
(8)
(9a)
(9b) RF = p-FC6H4 Nu= OAc
(9c) R1 = H; R
2 = C6H5 RF =p-CF3C6H4 Nu = Cl
(9)
Scheme 2: Synthetic sequence for the preparation of fluorinated uracil derivatives
123
Scheme 3: Synthesis of benzofuran derivatives from halo-salicylonirtriles
OH
CHO
R3
R6
R5
R4
OH
CN
R3
R6
R5
R4
H2NOH.HCl
reflux, 144 hrs
(10) (11)
OH
CN
R3
R6
R5
R4
NHR1Li
1
O
O
CN
R3
R6
R5
R4
NHR1
O
Base
O
R3
R6
R5
R4
NHR1
O
NH2
OH R2
O
O
R6
R5
R4
NH
NHR2
R3
O
NHR1
O
R1 = H, Me, Et, etc. R
2 = Me, Et, etc.
R3 = H, F, Cl, Br, Me, Et
R4 = H, F, Cl, Br, Me, Et, isopropyl,etc.
R5 = H, F, Cl, Br, Me, Et, isopropyl,etc.
R6 = H, F, Cl, Br
(12) (13)
(14)(15)
124
Figure1
F
SO
ONH
O
CN
CF3OH
CH3
Bicalutamid
casodex (Zeneca)
Nonsteroidal antiandrogen,
antineoplastic, anti(prostrate)cancer
CH3
N
N
Fadrozole
Arensin (Ciba-Geigy)
antineoplastic, non-steroidal
aromatase inhibitor
N
S
CH3
N
O
CN
Periciazine
Aolept (Bayer)
antipsycotic, neuroleptic
O
NC
F
N
CH3
CH3
Citaopram
Cipramil (Promonta Lundbeck)
antidepressant, selective
serotonin uptake inhibitor
N
NN
N
NC CN
Letrozole
Femara (Novartis Pharma)
antineoplastic, aromatase inhibitor
N
S
CH3
NCH3
CH3
CN
Cyamemazine
Neutromil (Farmitalia)
neuropletic, tranquilizer
(16) (17)
(18)
(19)
(20)(21)

125
Figure 2: Selected Examples for Pharmaceuticals containing Benzonitriles as the integral
part
4.3. Nitrile containing natural products
Naturally occurring nitriles comprise a small and surprisingly diverse set of
secondary metabolites.14
The structures vary from simple, long-chain alkane nitriles to
architecturally complex structures such as the calyculins, with new and more metabolites
being continually reported. Naturally occurring nitriles are known to be derived from
amino acids in plant,15
arthropods,16
bacteria17
and fungi. N-Hydroxylation and
decarboxylation of amino acids affords aldoximes that are enzymatically converted to the
corresponding nitrile. A variety of phenyl and hydroxylated phenyl acetonitriles have
been obtained from plant sources. Many of these nitriles are derived from the
corresponding glucosinolates as indicated by the dependence of nitrile-containing
metabolites on the method sample preparation. The parent phenyl acetonitrile (24) and
phenylpropanenitrile18
(25) have been isolated from Nasturium officinate seeds while the
NMeO
MeO
OMe
OMe
CN
CH3
CH3CH3
Verapamil
anti-arrhythmic and
vasodilatator
(22)
N
NHO
OHCN
Vildagliptin
anti-diabetic agent
(Novartis)(23)

126
mono and dihydroxylated (26-28) analogs19-20
were isolated from Erica scoparia and
Moringa oleifera leaf extracts respectively (Fig 3).
.
Fig 3
O
HMe
OH
H
OH
H
OH
H H
OH
CN
O
NC
R3
R2
R1
O
HMe
OH
H
OHH
OH
H
HO
R1= OH R
2= OH R
3= OCH2Ph
R1= OH R2= OMe R2= OMe
(29)(30)
Figure 4
CNCN CN
OHOH
(24)
O
HMe
OH
H
OHH
OH
H
HO
CN
OH
O
HMe
AcO
H
OHH
OH
H
HO
CN
OH
(25) (26)
(28)(27)

127
The co-occurrence21
of the nitrolosides (Fig. 4), (benzyl nitrile (29 with
ehretiosides 30) is further support for the biosynthesis of nitrilsides from phenyalanine or
tyrosine by hydrogenation. A new nitrile glycoside, niaziridine (31), (Figure 5) isolated
from the pods of Moringa Oleifera was able to increase the bioactivity of commonly used
antibiotics such as rifampicine, tetracycline, and ampicillin against Gram (+) and Gram (-
) bacteria.22-23
Niaziridine also enhances activity of antifungal drugs against C. albicans
and increases the absorption of antibiotics through gastro-intestinal membrane.
Figure 5: Niaziridine isolated from Moringa Oleifera
Organic compounds possessing a cyano group occur in nature, including compound (32),
which has antibiotic activity and compound (33) which is an antiviral agent isolated from
a Verongida sponge (Figure 6).
Figure 6: Naturally occurring compound containing cyano-function
CN
OH
O
O
OH
OH
NC
CH3
H
CH3
H
CH3CH3
(32)
(33)
O
HMe
OH
H
OH
H
OH
H H
CN
OH
O
(31)

128
4.4. Applications of nitriles in Material science
2,4-Dihyroxybenzonitrile (28) is the starting material in the preparation of a electro
optical materials24
( Scheme 4). Pelzl et.al., in their report, presented a series of banana-
shaped compounds (33), C6O, C8O, C9O, and C12O, which exhibit a cyano substituent at
4-position of the central core. NMR studies, X-ray investigations, microscopical and
electro-optical measurements had shown that all members of the series not only form the
chiral B2 phase characteristic of a bent molecular shape but also SmA and SmC phases.
The electro-optical studies on B2 phase proved that an antielectric ground state could be
switched into ferroelectric states.
Scheme 4: Synthesis of electro-optical materials C6O, C8O, C9O, and C12O
CHO
OHOH
NH2OH.HClCHO
OHOH
1) Ac2O
2) KOH OHOH
CN
CN
OHOHR N
COOH +DCC/DMAP
R
N
OCN
OO
R
N
O
R= OC 6H13, OC 8H17, OC 9H19, OC 12H25
(34) (35)(36)
(37) (38)
(39)

129
4.5. Nitriles as intermediates for Tetrazole Derivatives
One of the most important applications of nitriles is that they serve as
intermediates in the synthesis of tetrazoles. Tetrazoles are a class of heterocycles with a
wide range of applications, which are currently receiving considerable attention.25
This
functional group has a role in coordination chemistry as a ligand,26,27
as well as in various
materials sciences applications including photography and specialty explosives.
Tetrazoles readily tolerate a wide range of chemical environments and new uses for this
unique family of heterocycles continue to emerge in both materials science, and
pharmaceutical applications.
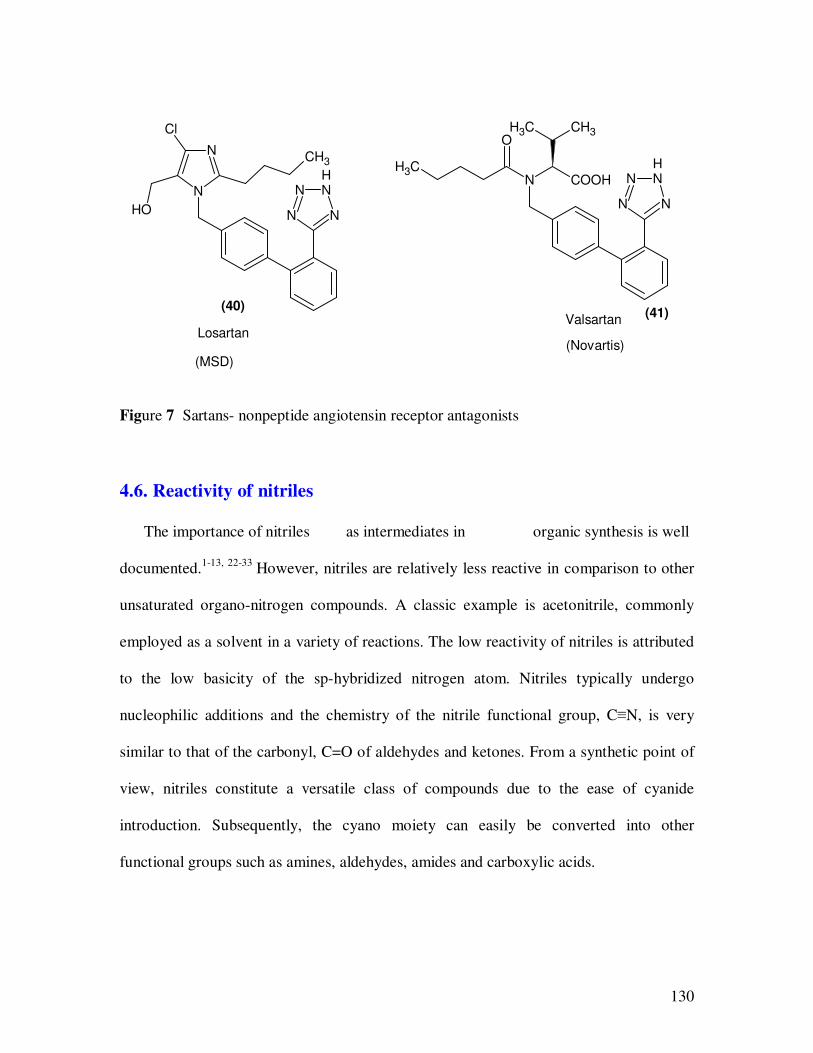
The 5-(4’-methyl-1,1’-biphenyl-2-yl)-tetrazole subunit has been used as a
carboxylic acid mimic in the class of so called sartan derivatives (40,41 in Figure 4).
Angiotensin II (AII) is the octapeptide responsible for the peripheral effects of the rennin-
angiotensin system28-33
which include the regulation of blood pressure and volume
homeostasis. Losartan was the first nonpeptide angiotensin receptor antagonist to appear
on the market followed by Valsartan (Figure 7). The 5-(4’-methyl-1,1’-biphenyl- 2-yl)-
1H-tetrazole subunit has become ubiquitous in the most potent and bioavailable
antagonists disclosed to date.
130
Figure 7 Sartans- nonpeptide angiotensin receptor antagonists
4.6. Reactivity of nitriles
The importance of nitriles as intermediates in organic synthesis is well
documented.1-13, 22-33
However, nitriles are relatively less reactive in comparison to other
unsaturated organo-nitrogen compounds. A classic example is acetonitrile, commonly
employed as a solvent in a variety of reactions. The low reactivity of nitriles is attributed
to the low basicity of the sp-hybridized nitrogen atom. Nitriles typically undergo
nucleophilic additions and the chemistry of the nitrile functional group, C≡N, is very
similar to that of the carbonyl, C=O of aldehydes and ketones. From a synthetic point of
view, nitriles constitute a versatile class of compounds due to the ease of cyanide
introduction. Subsequently, the cyano moiety can easily be converted into other
functional groups such as amines, aldehydes, amides and carboxylic acids.
N
N NH
N
N
N
OH
Cl
CH3
N
N NH
N
N COOH
CH3 CH3
CH3
O
Losartan
(MSD)
Valsartan
(Novartis)
(40)(41)

131
4.7. Synthesis of Nitriles
The development of new methods for the synthesis of nitriles is important in organic
chemistry, since nitriles are useful as intermediates for the reparation of amines,
tetrazoles and other functional groups. The synthetic methods for the preparation of
nitriles can be related mainly to four reaction types: addition, substitution, elimination,
ammoxidation, and conversion of other nitriles. The important methods currently
employed for the purpose are as follows.
1. Addition of HCN
2. Cyanation of aryl halides with Copper(I) Cyanide
3. Ammoxidation toluene derivatives
4. Transition Metal catalyzed Cyantion
5. Oxidation of primary amines
6. Dehydration of aldoximes
4.7.1. Preparation of nitriles by addition of hydrogen cyanide
A variety of processes for the introduction of cyanide functionality into aromatic
compounds have been described.34
including the addition of HCN in the presence of
dicobalt octacarbonyl,35
nickel catalysts,36-37
and palladium catalysts.38
4.7.2. Preparation of alkyl nitriles
One of the most general methods for the preparation of nitriles is a direct
nucleophilic substitution of alkyl halides with inorganic cyanides.34
The classical

132
conditions involve heating a halide with a cyanide salt in aqueous alcohol solution or in
aprotic polar solvents such as DMSO (Kolbe nitrile synthesis) (Scheme 5).39
In analogy,
the use of metal thiocyanates such as KSCN in a nucleophilic substitution with organic
halides is a general procedure to introduce the thiocyanate group into a molecule.40
R X + NaCNDMSO
90-160 °CR CN
Scheme 5: Nucleophilic substitution of alkyl halide
4.7.3. Preparation of nitriles from nitroalkanes
A convenient protocol for the synthesis of optically active aldoximes and nitriles
starting from chiral nitroalkanes was reported by Carreira et al.41
In this method, the
optically active nitro-compound was treated with benzyl bromide, KOH and nBu4NI
followed by the addition of SOCl2 to afford the nitrile directly, in relative good yields
without loss of optical activity (Scheme 6).
CH3
NO2
CH3
CN
i. BnBr, KOH, n-Bu4NI, THF
ii. SOCl2, Et3N, THF
75 °C
Scheme 6: One pot conversion of optically active nitro-alkane to nitrile

133
4.7.4. Preparation of nitriles from hydrazones
Several procedures have been documented for the preparation of nitriles from
hydrazones, including oxidative cleavage of dimethylhydrazone of aldehydes with
magnesium monoperoxyphthalate hexahydrate (MMPP)34,42
and microwave-assisted
solvent-free oxidative cleavage using oxone with wet alumina.43
A convenient procedure
to form nitriles under mildly basic conditions is the treatment of dimethylhydrazones with
excess of methyl iodide followed by reaction with DBU (Scheme 7).44
NN
OMe
CH3
i. MeI, THF, 6h
DNU, 0 °C, 3h
CH3
CN
Scheme 7: Synthesis of nitrile from hydrozone of aldehyde
4.7.5. Preparation of nitrile by diazotization
Nitriles can also be prepared on laboratory scale as well as on industrial scale by
diazotization of anilines and subsequent Sandmeyer reaction.45-48
4.7.6. Cyanation of Aryl Halides
Most often nitriles are synthesized by Rosenmund-von Braun reaction49-54
from aryl
halides on laboratory scale as well as on industrial scale. Unfortunately several problems
limit the generality of these classic methods. A main drawback of the Rosenmund-von

134
Braun and the Sandmeyer reactions is the use of stoichiometric amounts of copper(I)
cyanide as cyanating agent. When used in industrial scale, the stoichiometric amounts of
copper salts also present a significant waste disposal problem. The other disadvantages
of Rosenmund-van Braun reactions are the high temperature (150-280ºC) and the low
reactivity of aryl chlorides and bromides. In general, the use of expensive aryl iodides is
required.
CH3
BrCuCN
DMF
CH3
CN
Scheme 8: van Braun reaction
4.7.8. Ammoxidation of Toluene Derivatives
On ton scale the method of choice in industries is the ammoxidation of toluene
derivatives. In this process, toluene derivatives are reacted with oxygen and ammonia at
300-550º in the presence of heterogeneous fixed bed catalysts.55,56
Since, ammoxidation
requires high temperature and high pressure, and also a large quantity of ammonia, the
process is has very narrow scope. Hence the method is limited to the preparation of
products such as benzonitrile, and chlorobenzonitrile.57-59
Ammoxidation of 4-nitrotolune
catalyzed by rutile or alumina supported vanadium phosphate (VOHPO4) and vanadium
oxide (V2O5) is shown in the Scheme 9. In this procedure, the supported V2O5 and
VOHPO4 (with alumina and titanium oxide) were prepared solid-solid wetting method.
Appropriate quantities of vanadium phosphate (VPO) precursors and supporting

135
materials were mixed thoroughly and ground in an agate mortar and then transferred to a
grinding machine where the powders electrically mixed thoroughly. The resulting solid
mixture was pelletised, crushed and sieved to required particle size
Scheme 9: Ammoxidation of Toluene Derivatives
(1.0-1.25 mm) and then calcined at 450ºC for 3 h. Clearly, this method is not applicable
for the substrates containing functional groups.
4.7.9. Oxidation of Primary Amines
An alternative method for the preparation of nitriles is the oxidation of primary
amines. A plethora of oxidizing agents for such transformations documented in the
literature demonstrates the importance of with which the functional group transformation
has been addressed.60
Although several procedures that use stoichiometric amount of
reagents have been known,61-64
only a few catalytic methods have been reported. A
number of protocols using ruthenium complexes as catalysts, and dioxygen,65-68
iodosylbenzene69
and persulfate ions70
as oxidation agents have been reported.
Yamakuchi and co-workers71,72
have shown that Ru/Al2O3 and Ru/Fe2O3 were more
active for aerobic oxidation of benzylamine. However, these oxidation methodologies
CH3
NO2
+ NH3 + 3/2 O2
supported V2O5
350 °C
CN
NO2

136
have very limited substrate scope as applied only to benzyl amine. Very recently, Wang
and co-workers73
have reported an improved catalytic system that consists of Ru/Co3O4.
The Co3O4 supported ruthenium catalyst was found to exhibit the best catalytic
performance for the aerobic oxidation (Scheme 10). However, these
HH
RNH2
O2
Ru/Al2O3
100 °C+ R-CN O2+
Scheme 10: Ruthenium Catalyzed Oxidation of Primary Amines
methods involve tedious work-up procedures and often the reaction is accompanied by
side products such as imines and secondary amines. Furthermore, the aerobic oxidation
method is incompatible with substrates containing sensitive functional groups.
4.7.10. Transition Metal catalyzed Cyanation of Aryl halides
A useful alternative for the preparation of benzonitriles is the transition metal
catalyzed cyanation of aryl –X compounds(X= Cl, Br, I, OTf, etc). Buchwald et.al.,
reported copper catalyzed domino exchange-cyanation of aryl halides.74
In this method,
the aryl halides are cyanated with NaCN in the presence 1,2-diamines and KI at 100ºC
(Scheme 11). However, the most common catalysts for coupling of aryl halides or
triflates with cyanide are the transition metal complexes of platinum group, particularly
palladium and nickel complexes.

137
Scheme 11: Copper catalyzed cyanation aryl halides
4.7.11. Palladium Catalyzed Cyanation of aryl halides
Palladium catalysts tolerate a wide range of functional groups and are less sensitive to air
and humidity. Palladium catalyzed cyanation of aryl halides with KCN as the cyanating
agent was introduces by Takagi74,75
and co-workers in 1973. Since then, the palladium
catalyzed cyanation methodology has grown into a common and powerful process to
obtain substituted benzonitriles.76
Recent examples of this transformation in areas as
diverse as process chemistry,77
medicinal chemistry,78
and ligand synthesis79
are
indicative of the importance benzonitriles as end products or as synthetic intermediates
which can be converted to a multitude of different functional groups. For example in the
preparation of 2-((1H-Pyrrolo[2,3-b]pyridine-4-yl)methylamino)-5-fluoronicotinic Acid80
4-Cyano-7-azaindole serves as an intermediate which in turn is obtained by cyanation of
4-Chloro-7-azaindole using Zn(CN)2 as cyanating agent (Scheme 12).
Ar Br
10 Mol % CuI, 20 Mol % KI
1 equiv ligand
1.2 equiv NaCN
Toluene 110-130 °C, 24 h
Ar - CN
Me(H)NN(H)Me
Ligand=

138
N NH
Cl
N NH
CNZn/Zn(CN)2
Pd2dba
3/dppf
DMAC/120 °C
LAH/THF
40 °C
N NH
NH2
+N
F COOH
Cl
NaHCO3
1-pentanol/130 °C
N NH
NH2
N
F COOH
N
NH
NH
Scheme 12: Palladium catalyzed cyanation of arylhalides with Zn(CN)2
From nearly as early as the discovery of the reaction it was known that palladium-
catalyzed cyanation reactions are sensitive to cyanide. While cyanide is necessary for the
reaction, it has been proposed that an excess of cyanide can sequester the catalyst,
rendering it inactive. This is one of the reasons for the widespread use of zinc cyanide in
these reactions.81-84
However, even reactions using Zn(CN)2 can be unreliable, suggesting
that they may also suffer from the problem of high levels of cyanide in solution
sequestering the catalyst.98
This reaction is very sensitive to traces of oxygen, which can
cause catalyst poisoning. Recently, potassiumhexacyanoferrate (K4[Fe(CN)6]) was
introduced as a nontoxic cyanide source for palladium catalyzed cyanations85-87
For
example, Weissman and co-workers have developed a method for the cyanation of aryl
bromides using potassiumferrocyanaide as cyanating agent. The reaction (Scheme 13) is
catalyzed by palladium under ligand free condition in DMAC at 120ºC.100
An efficient
application of potassiumhexacyanoferrate (Scheme 14) as cyanating agent in the concise

139
total synthesis of esermethole and physostigmine, powerful inhibitors of acetyl- and
butyryl-cholinesterase, was developed by Zhu etal.87
Br
R
0.1 mol % Pd(OAc)2
0.22 equiv K4[Fe(CN)6]
CN
R
Scheme 13: Cyanation of aryl bromide using potassiumferrocyanide
The intermediate, 3-alkyl-3-cyanomethyl-2-oxindole was synthesized by a palladium-
catalyzed domino Heck-cyanation reaction. Efforts have also been made to use other
cyanide sources such as thiocyanates and TMSCN in the palladium catalyzed reactions.
As a compliment to the classic cyanation of aryl halides using cyanide sources and
transition metal catalyst, Liebeskind et al., reported the palladium-catalyzed cross-
coupling of thiocyanates with boronic acids in the presence of copper(I) thiophene-2-
carboxylate (CuTC).89
R1
B(OH)2
Pd cat, CuTC
dioxane, 100 °C+R2SCN R
1CN
Scheme 14: Nitrile synthesis by palladium-catalyzed, copper(I)-mediated coupling of
boronic acid with thiocyanates
4.8. Objectives of Present Research
A number of methods as explained in the previous sections of this chapter could be
used in the synthesis of nitrile. Recently, palladium-catalyzed cyanation of aryl halides
garnered wide attention mainly for two reasons: 91) it is highly efficient and versatile (2)

140
it tolerates a variety of functional groups. However, palladium catalysts are costly and the
reagents used in these methods are toxic. Moreover, these methods are not applicable in
the synthesis of alkyl nitriles. Dehydration of aldoximes to nitriles is a viable alternative
and requires stoichiometric amounts of metal or non-metal reagents. Having succeeded in
using a cheap and widely available Co-catalyst, a project was set out to develop a cobalt-
catalyzed method for the purpose. The main objective of the present research was to
develop a method for the dehydration of salicylaldoximes. The existing fewer methods
are time consuming (existing methods 1-2 days) and involve tedious work-up procedures
leading to poor yields.
4.9. Results and Discussion
The nitriles can be prepared conveniently by dehydration of aldoximes. In the recent
past, there had been several reports describing the dehydration of aldoximes with the use
of stoichiometric amounts of certain main group and transition metal complexes.90-93
There are now a number of efficient procedures available for the catalytic conversion of
aldoximes into nitriles and advances have been made in the way of low catalyst loadings,
short reaction time,94
and the use of microwave,95
and flash vacuum pyrolysis
technology.96
Unfortunately, the use of high cost and commercially unavailable catalysts,
high power microwave and very high temperature for pyrolysis make these methods
unattractive. Moreover, these methods are not suitable for preparation of salicylonitriles.
The compounds prepared from salicylonitriles find their applications as superoxide
inhibitors, ferrielectric liquid crystal dopants96
antipicornaviral, anti-inflammatory and
anti-asthma agents, and fibrinogen antagonists.97-98
Salicylonitriles, including 2,4-

141
dihydroxybenzonitrile, a precursor to the potent, less toxic form of desferrithiocin known
as 4'-hydroxydesazadesferrithiocin, 2-hydroxy-3-methoxybenzonitrile, and halo-
substituted salicylonitriles, the intermediates for the benzofuran derivatives, remain a
synthetic challenge as the catalyst and the reagents should display tolerance to hydroxyl
group. For instance, 3-bromo-5-chloro-2-hydroxybenzonitrile, an important intermediate
in the synthesis of benzofuran derivatives known for their potent adenosine A2A receptor
antagonist activity97-99
and BLT1, BLT2 activities,97-99
is currently prepared in three steps.
Salicylonitrile (2-cyanophenol) serves as starting material) in the synthesis of
benzofuro[3,2-c]isoquinolinone. It is known to be an important poly(ADP-
ribose)polymerase-1 (PARP-1) inhibitor (Scheme 15).
OR
O
COOR
Br
NC
OH
acetone, K2CO
3 or
CH3CN, (CH
3CH
2)3N
NH
O
O
R= Me, Et
Scheme 15: Synthesis of benzofuro[3,2-c]isoquinolinone
At present, various dehydrating reagents, including thiophosphoric diamide, thionyl
diimidazole, trichloromethyl chloroformate, etc., are used in the dehydration of
salicylaldoximes to salicylonitriles. Most of these reagents, however, are corrosive,
toxic, expensive or commercially not available. These methods require fairly high
pressure and elevated temperature, and the hydroxyl groups of salicylaldoximes must be
protected before dehydration.97-99
Thus the development of a catalytic method that is

142
compatible with sensitive functional groups, and avoids harsh reaction conditions would
be an interesting target, and beneficial from the commercial point of view.
Having succeeded in developing a cobalt(II)-catalyzed versatile method for the
Friedel-crafts acylation of electron rich aromatics, we set out a project to study the effect
of cobalt(II) species on dehydration of aldoximes to nitriles. Toward developing a
cobalt-catalyzed method for dehydration, we first examined the catalytic activity of
Co(II) chloride, Co(II) acetate, Co(II) TPP, and Co(II) acetylacetonate in the conversion
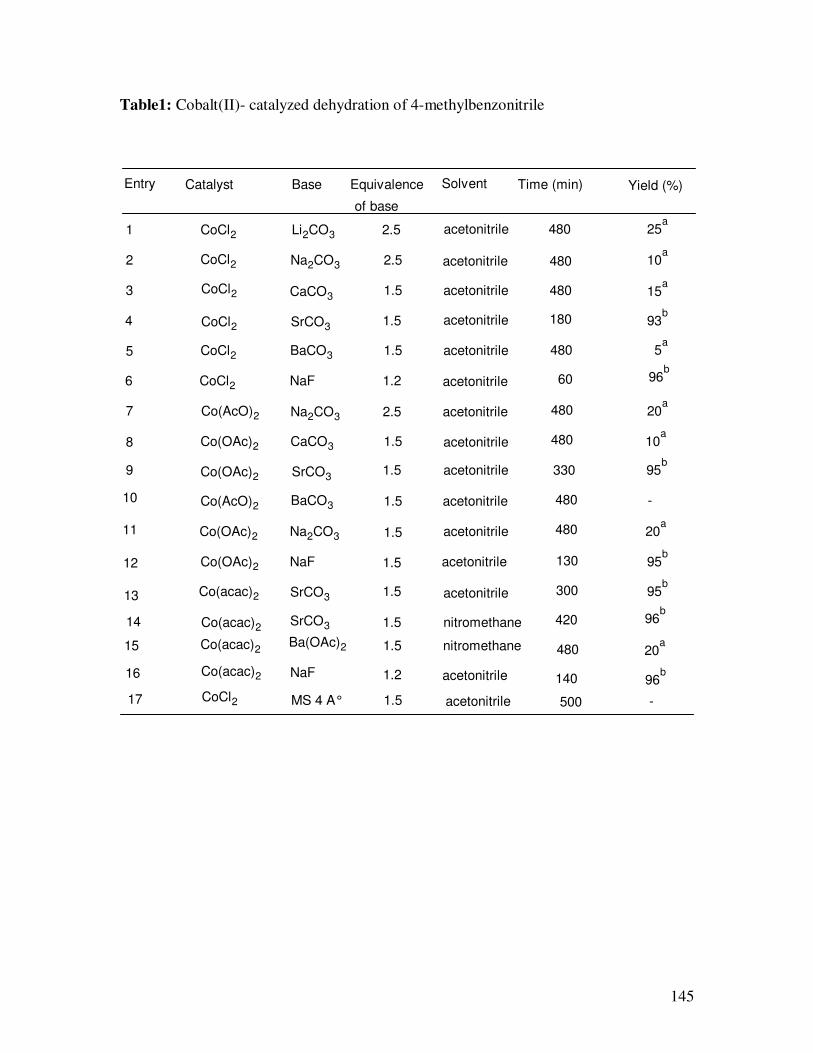
of 4-methylbenzaldoxime to 4-methylbenzonitrile (Table 1) in the presence of various
inorganic bases such as fluorides, carbonates and acetates of various alkali and alkaline
earth metals in acetonitrile. Whereas no significant conversion was noticed when
cobalt(II) TPP catalyst was used, all other catalysts have been found to be effective in the
conversion of various aldoximes to nitriles in the presence of NaF or SrCO3. Among the
three active catalysts, cobalt(II) chloride emerged as the best suitable for the conversion.
While cobalt(II) chloride catalyzed dehydration in the presence of NaF was complete
within 1 h (entry 7), cobalt(II) acetate and cobalt(II) acetylacetonate catalyzed reaction
took more than 2 h (entries 12 and 16). No conversion of aldoximes in the absence of
catalyst or the base was observed even after 24 h of stirring at 80º C. Among the bases
used, NaF gave the best results. On contrary to the reported ruthenium catalyzed
dehydration [20] of oximes, the use of molecular sieves had not shown any significant
results (entry 17). Furthermore, the usual side product, amide, associated with
dehydration of aldoximes to nitriles was not noticed in this reaction.
Then, in order to achieve optimum yields, different solvents were tested. While
cobalt(II) acetylacetonate catalyzed the transformation both in nitromethane and

143
acetonitrile, however, acetonitrile was the suitable solvent for the other two catalysts,
cobalt(II) chloride and cobalt(II) acetate. In contrast, no significant conversion was
observed in DMF, dioxane, or dichloromethane. Several examples of cobalt(II) chloride
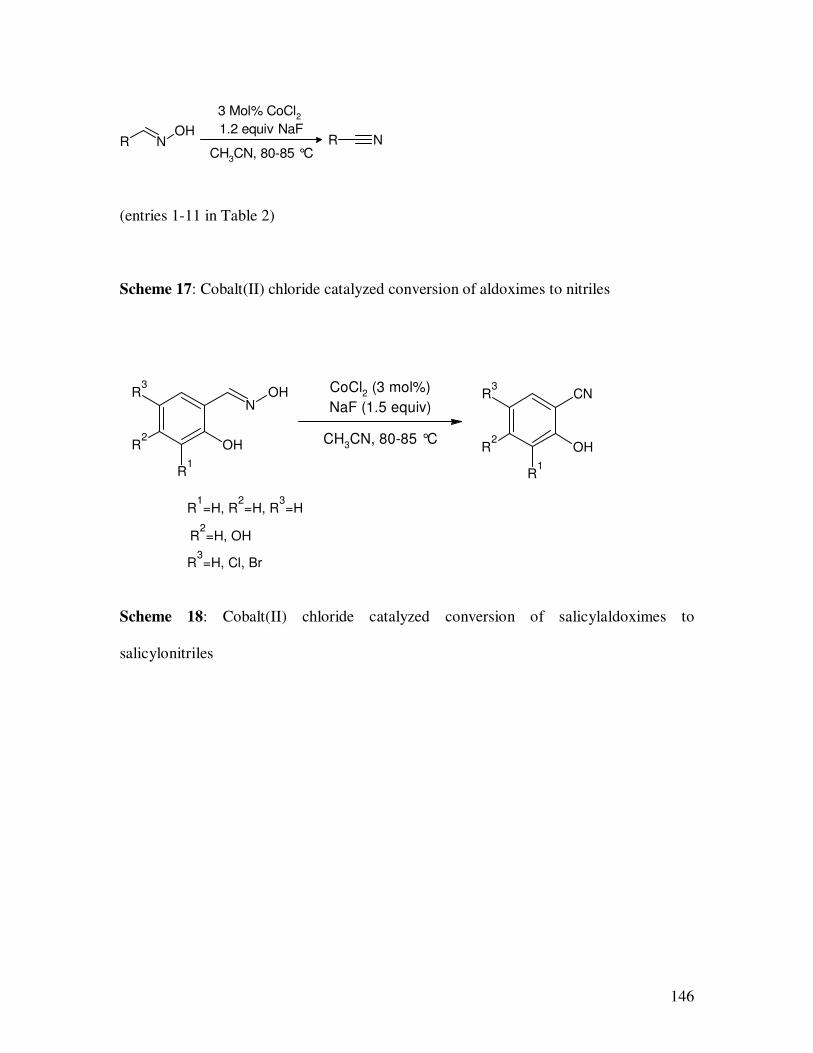
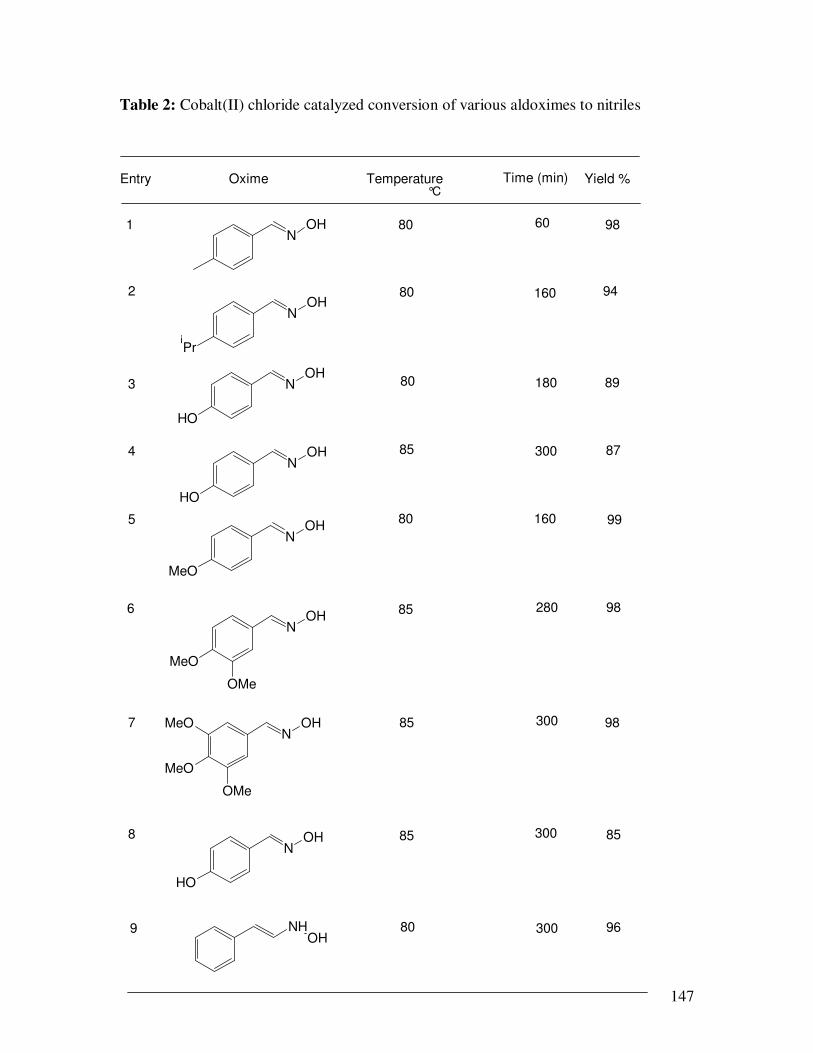
catalyzed dehydration of aldoximes to nitriles were shown in Table 2. In all cases, the
reactions proceeded smoothly in the presence of NaF. The dehydration of trans-
cinnamaldoxime also proceeded smoothly with stereochemical retention of the double
bond, and no trace of polymer was observed.
The success of this protocol, however, relies largely on the fact that it could be very
conveniently extended to the dehydration of salicylaldoximes. Currently no single
catalytic methodology, which could be applied to the dehydration of a variety of
aldoximes including substituted salicylaldoximes, is found in the literature. As
salicylaldoximes contain sensitive –OH group at C-2, we once again examined the
catalytic activity of CoCl2, Co(ACO)2, and Co(acac)2 using salicylaldoxime as test
substrate. Gratifyingly, CoCl2/NaF/acetonitrile emerged as the general and efficient
catalytic system for the conversion of salicylaldoximes into salicylonitriles. The
conversion of all salicylaldoximes (Table 2, entries 12-18,), including 3-, 5-
dichlorosalicylaladoxime (entry 16), 3-bromo-5-chlorosalicylaladoxime (entry 17), 2,4-
dihydroxybenzaldoxime (entry 18) into corresponding salicylonitriles was achieved
elegantly in the presence of NaF in acetonitrile in short reaction time (within 8 h) under
mild reaction condition when compared with existing methods [13, 14]. It is noteworthy

144
Scheme 16: Cobalt(II)-catalyzed dehydration of 4-methylbenzaldoxime to
4-methylbenzonitrile
that SrCO3 was also found to be a suitable base for the conversion of many aromatic
aldoximes including salicylaldoxime, 5-chlorosalicylaladoxime, and 5-
bromosalicylaldoxime, albeit with longer reaction time. However, the dehydration of 2,4-
dihydroxybenzaldoxime, 3,5-dichlorosalicylaldoxime, and 3-bromo-5-
chlorosalicylaldoxime proceeded smoothly only in the presence of NaF. Surprisingly, that
SrCO3 was also found to be a suitable base for the conversion of many aromatic
aldoximes including salicylaldoxime, 5-chlorosalicylaladoxime, and 5-
bromosalicylaldoxime, albeit with longer reaction time. However, the dehydration of 2,4-
dihydroxybenzaldoxime, 3,5-dichlorosalicylaldoxime, and 3-bromo-5-
chlorosalicylaldoxime proceeded smoothly only in the presence of NaF. Surprisingly,
Me
NOH 3 mol% Co(II)
Inorganic Base
Solvent, 80 °CMe N
145
Table1: Cobalt(II)- catalyzed dehydration of 4-methylbenzonitrile
Entry Catalyst Base Equivalence
of base
Solvent Time (min)
1 CoCl2 Li2CO3 2.5 acetonitrile 480 25a
2 CoCl2 Na2CO3 2.5 acetonitrile 480 10a
3 CoCl2 CaCO3 1.5 acetonitrile 480 15a
4 CoCl2 SrCO3 1.5 acetonitrile 93b
5 CoCl2 BaCO3 1.5 acetonitrile
6 CoCl2 NaF 1.2 acetonitrile
7 Co(AcO)2 Na2CO3 2.5 acetonitrile 20a
8 Co(OAc)2 CaCO3 1.5 acetonitrile 10a
9 Co(OAc)2 SrCO31.5 acetonitrile 95
b
Co(AcO)2 BaCO3 1.5 acetonitrile -
Co(OAc)2 Na2CO3 1.5 acetonitrile 20a
Co(OAc)2 NaF 1.5 acetonitrile 95b
Co(acac)2 SrCO3 1.5 acetonitrile 95b
Co(acac)2SrCO3 1.5 nitromethane
Co(acac)2Ba(OAc)2 1.5 nitromethane
Co(acac)2 NaF 1.2 acetonitrile
10
11
12
13
16
14
15
Yield (%)
180
480
60
5a
480
480
330
480
480
130
300
420 96b
480
140 96b
20a
96b
17 CoCl2 MS 4 A° 1.5 acetonitrile 500 -
146
(entries 1-11 in Table 2)
Scheme 17: Cobalt(II) chloride catalyzed conversion of aldoximes to nitriles
Scheme 18: Cobalt(II) chloride catalyzed conversion of salicylaldoximes to
salicylonitriles
R NOH
R N
3 Mol% CoCl2
1.2 equiv NaF
CH3CN, 80-85 °C
CN
OH
R1
R2
R3
OH
R1
R2
R3
NOH
R1=H, R
2=H, R
3=H
R2=H, OH
R3=H, Cl, Br
CoCl2 (3 mol%)
NaF (1.5 equiv)
CH3CN, 80-85 °C
147
Table 2: Cobalt(II) chloride catalyzed conversion of various aldoximes to nitriles
Entry Oxime Temperature °C
Time (min) Yield %
1N
OH 80 60 98
NOH
iPr
2 80 160 94
3 NOH
HO
80 180 89
4N
OH
HO
85 300 87
5
NOH
MeO
80 160 99
6
NOH
MeO
OMe
85 98
7N
OH
MeO
OMe
MeO 85 300 98
280
NOH
HO
8 85 300 85
9 NHOH
80 300 96

148
Entry Oxime Temperature °C
Time (min) Yield %
10 85 240 94
N
NOH
11 85 380 86
12 80 240 99
13
NOH
OH
14 80 98
15N
OH
OH
Cl 85 240 96
240
NOH
OH
Cl
Cl
1685 92
17 80 360 94
SN OH
NOH
OH
OMe
80 360 98
NOH
OH
Br
NOH
OH
Cl
Br
360

149
Table 2 continued
Entry Oxime Temperature °C
Time (min) Yield %
NHOH
1980 300
20
NOH
OHOH
18 80 360 94
96
NOH
CH3
21
CH3
CH3
NOH
80
80 160 97
120 89

150
4.10. Mechanism
The mechanism for the formation of nitrile from aldoxime could be postulated as
depicted in the Scheme 19. (i) First, the oxime coordinates with coordinatively
unsaturated cobalt through a dative bond to form an intermediate, (I). The intermediate
(II), could be formed from (I) by insertion of cobalt between N-O, bond of the oxime. In
other words, cobalt could undergo oxidative addition to give the intermediate (II) similar
R NOH
R
N O
H
Co2+
L2
Solvent
R
N
CoL2H
OH
R N
β−Η Elimination
Solvent
Co2+
L2
(I) (II)
CoL2
OH
HCo
2+L2 + OH2
Reductive
elimination
Solvent
Insertion of cobalt
(III)
Scheme 19: Proposed mechanism for the cobalt-catalyzed dehydration of aldoximes
to the insertion of palladium across N-O bond, as recently proposed for the palladium-
catalyzed dehydration of aldoximes. Intermediate (II) on β-H elimination yields nitrile,
and coordinatively saturated cobalt species (III), which on reductive elimination of water
molecule regenerates Co2+
. The support for the formation of cobalt-hydride could be
found in a recent literature. Park and co-workers proposed a pathway for the formation of
hollow FCC Co nanoparticles from oleylamine. In the proposed mechanism, CoO is

151
inserted between C-N bond of the amine, which on β-H elimination gives cooridinatively
saturated cobalt-hydride species. We also suggest that the inorganic base is involved in
the abstraction of β-H and then in the formation of cobalt-hydride. Further mechanistic
investigations of these catalytic processes as well as further studies on the scope of this
methodology in the dehydration of other aldoximes are currently in progress.
References
1. Friederich, K.; Wallenfels, K. In The Chemistry of the Cyano Group; Rappaport,
Z., Ed.; Wiley-Interscience Publishers: New York, 1970 p 67.
2. Fatiadi, A. J. In preparation and synthetic applications of cyano compounds;
Patai, S., Rappaport, Z., Eds.; Wiley: New York, 1983; p 1057.
3. Miller, J. S.; Manson, J. L. Acc. Chem. Res. 2001, 34, 563-570.
4. Fustero, S.; Salavert, E.; Sanz-Cervera, J. F.; Piera, J.; Asensio, A. J. Chem. Soc.
Chem. Commun. 2003, 844-845.
5. Ozaki, S. Med. Res. Rev., 1996, 16, 51-86.
6. Hugl, H. (Bayer AG); presentation at the conference ‘‘50 Years of Catalysis
Research in Rostock’’, July 1st –3
rd 2002, Rostock.
7. Rock, M. H.; Merhold, A. (Bayer AG), WO 98/37058,1998.
8. Rock, M. H.; Merhold, A. (Bayer AG), US 6162942, 2000.
9. Fustero, S.; Salavert, E.; Sanz-Cervera, J. F.; Piera, J.; Asensio, A. J. Chem. Soc.
Chem. Commun., 2003, 844-845., and references cited therein.
10. Mukund, S. C.; Mukund, K. G.; Joseph, C. US patent 6875882, 2005.
11. Shirai, M.; Shiba, K.; Furuya, T. EP1270550, 2004.

152
12. Nakamura, T.; Shiohara, H.; Terao, Y.; Nakayama, S, ; Miyazawa,
K.; Ohnota, H. E. P. 171023, 2005, and references cited therein.
13. Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical substances:
syntheses, patents and applications, 4th Ed. Georg Thiem Verlag, Stuttgart, New
York, 2001, 214-142, 488-489, 553, 825-826, 1154, 1598-1599.
14. Fleming, F. F. Nat. Pod. Rep. 1999, 16, 597-606.
15. Mahadevan, S. Ann. Rev. Plant. Physiol. 1973, 24, 69.
16. Duffy, S. S. in Cyanide in Biology, Academic Press, London, 1981, pp. 385-414.
17. Legras, J. L.; Ghuzel, G.; Arnaud, A,; Galzy, P. World J. Micobiol. Biotechnol.,
1990, 6, 83.
18. Gould, S. J.; He, W.; Cone, M. C. J. Nat. Prod. 1993, 56, 1239.
19. Ballester, A.; Verwey, A.; Overeem, J. C. Phytochemistry, 1975, 14, 1667.
20. Faizi, S.; Siddiqui, B. S.; Saleem, R.; Siddiqui, S.; Aftab, K. J. Nat. Prod. 1994,
57, 1256.
21. Simpol, L. R.; Otsuka, H.; Ohtani, K.; Kasai, R.; Yamasaki, S. Phytochemistry,
1994, 36, 91.
22. Kirtikar, K. R.; Basu, B. D. in Indian Medicinal plants 1975, vol. 1,
Ed.2, 675-683, (M/S Singh, B.; Singh, M. P. New Cannaught Place,
Dehra Dun.
23. Khanujia, S.P. S.; Arya, J. S.; Santha, R.; Kumar, T.; Saikia, D.; Kaur, H.; Singh,
M.; Gupta, S. C.; Shasany, A. K.; Darokar, M. P.; Srivstava, K. S.; Gupta, M. M.;
Varma, S. C.; Pal. A. US Patent 6858588, 2005.

153
24. Ina, W.; Diele, S.; Eremin. A.; Pelzl, G.; Grande, S.; Kovalenko, L.; Pancenko,
N.; Weissflog, W. J. Mater. Chem., 2001, 11, 1642-1650 and references cited
therein.
25. Butler, R. N. in Comprehensive Heterocyclic Chemistry II; A. R. Katritzky, C. W.
Rees, E. F. V. Scriven, Eds., Pergamon Press: Oxford, 1996, Vol. 4, p. 621, 905.
26. Lin, P.; Clegg, W.; Harrington, R. W.; Henderson, R. A. Dalton Trans. 2005,
2388.
27. Gupta, A. K.; Rim, C. Y.; Oh, C. H. Synlett 2004, 12, 2227.
28. Wittenberger, S. J.; Donner B. J. J. Org. Chem. 1993, 58, 4139.
29. Lusina, M.; Cindric, T.; Tamaic, J.; Peko, M.; Pozaic, L.; Musulin,
N. Int. J. Pharm.2005, 291, 127.
30. Duncia, J. V.; Carini, D. J.; Chiu, A. T.; Johnson, A. L.; Price, W. A.; Wong, P.
C.; Wexler, R. R.; Timmermans, P. B. M. W. Med. Res. Rev. 1992, 12, 141.
31. Smith, R. D.; Duncia, J. V.; Lee, R. J.; Christ, D. D.; Chiu, A. T.; Carini, D. J.;
Herblin, W. F.; Timmermans, P. B. M. W.; Wexler, R. R. Methods Neurosci.
1993, 13, 258.
32. Buehlmayer, P.; Criscione, L.; Fuhrer, W.; Fuhret, P.; de Gasparo, M.; Stutz, S.;
Whitebread, S. J. Med. Chem. 1991, 34, 3105.
33. Carini, D. J.; Duncia, J. V. Adv. Med. Chem. 1993, 2, 153.
34. Sharifi, A.; Mohsenzadeh, F.; Mojtahedi, M. M.; Saidi, M. R.;
Balalai, S. Synth.Commun. 2001, 31, 431
35. Justribo, V.; Colombo, M. I., Tetrahedron Lett. 2003, 8023-8024.

154
36. Arthur, P.; England, D. C.; Pratt, B. C.; Whitman, G. M.; J. Am. Chem. Soc.
1954, 76, 5364.
37. RajanBabu, T. V.; Casalnuovo, A. L. J. Am. Chem. Soc. 1992, 114, 6265.
38. Jackson, W. R.; Perlmutter, P.; Smallridge, A. J. Tetrahedron Lett. 1988, 29,
1983.
39. Elmes, P. S.; Jackson, W. R. J. Am. Chem. Soc. 1979, 101, 6128.
40. Kim, D. W.; Song, C. E.; Chi, D. Y. J. Org. Chem. 2003, 68, 4281.
41. Reeves, W. P.; White, M. R.; Hilbrich, R. G.; Biegert, L. L. Synth.
Commun. 1976, 6, 509.
42. Czekelius, C.; Carreira, E. M. Angew. Chem. 2005, 117, 618.
43. Fernández, R.; Gasch, C.; Lassaletta, J. M.; Llera, J. M.; Vázquez, J. Tetrahedron
Lett. 1993, 34, 141.
44. Ramalingam, T.; Reddy, B. V. S.; Srinivas, R.; Yadav, J. S. Synth. Commun.
2000, 30, 4507.
45. Moore, J. S.; Stupp, S. I. J. Org. Chem. 1990, 55, 3374.
46. Sandmeyer, T. Ber. Dtsch. Chem. Ges. 1885, 18, 1946-1948.
47. Sandmeyer, T. Ber. Dtsch. Chem. Ges. 1885, 18, 1492-1496.
48. Sandmeyer, T. Ber. Dtsch. Chem. Ges. 1885, 17, 2650-2653.
49. Rosenmund, K. W.; Struck, E. Chem. Ber. 1919, 52, 1749.
50. Pongratz, A. Monatsh. Chem. 1927, 48, 585.
51. von Braun, J.; Manz, G. Liebigs. Ann. Chem. 1931, 488, 111.
52. Lindley, J. Tetrahedron. 1984, 40, 1433-1448.
53. Wu, J. X.; Beck, B.; Ren, R.X. Tetrahedron Lett. 2002, 43, 387.

155
54. Ellis, G. P.; Romney-Alexander, T. M. Chem. Rev. 1987, 87, 779-794.
55. Mowry, D. F. Chem. Rev. 1947, 42, 1433.
56. Stevenson, A. C. Ind. Engg. Chem. 1949, 41, 1846-1851.
57. Denton, W. I.; Bishop, R. P.; Caldwell, h. P.; Chapman, H. D. Ind. Engg. Chem.
1950, 42, 796-800.
58. Martin, A.; Kalevaru, N. V.; Lucke, B.; Sans, J. Green. Chem. 2002, 4, 481-485.
59. Martin, A.; Wolf, G. U.; Steinike, U, Luke, B. J. Chem. Soc. Faraday. Trans.
1998, 94, 2227-2233.
60. Martin, A.; Lucke, B. Catal. Today. 1996, 32, 279-283.
61. Chen, F. E.; Huang, Y. Y.; Dai, H. F.; Lu, L.; Huo, M. Synthesis, 2003, 17,
2629-2631 and the references cited therein.
62. George, M. V.; Balachandran, K. S. Chem. Rev. 1975, 491.
63. Capdevielle, P.; Lavigne, A.; Maumy, M. Synthesis 1989, 453.
64. Lee, J. B.; Parkin, C.; Shaw, M. J.; Hampson, N. A.; Mac Donald,
K. I. Tetrahedron. 1973, 29, 751.
65. Belew, J. S.; Garza, C.; Mathieson, J. W. J. Chem. Soc. 1970, 634.
66. Mori, K.; Yamakuchi, Y.; Mizugaki, T.; Ebitani, K.; Kaneda, K. J. Chem. Soc.
Chem. Commun. 2001, 416.
67. Bailey, A. J.; James, B. R. J. Chem. Soc. Chem. Commun. 1996, 2343.
68. Cenini, S. Porta, F.; Pizzotia, M. J. Mol. Catal. 1982, 15, 297.
69. Tang, R.; Diamond, S. E.; Neary, N.; Mares, F.; J. Chem. Soc. Chem. Commun.
1978, 562.
70. Porta, F.; Crotti, C. Cenini, S. J. Mol. Catal. 1989, 50, 333.

156
71. Green, G, Griffith, W. P. Hollinshead, D. M.; Ley, S. V.; Schroder, M. J. Chem..
Soc. Perkin Trans. 1 1984, 681.
72. Yamaguchi, K.; Mizuno, N. Angew. Chem. Int. Ed. 2003, 42, 1480- 1483 and
references cited therein.
73. Kotani, K.; Koike, T.; Yamakuchi, K.; Mizuno, N. Green Chem. 2006, 8, 735.
74. Li, F.; Chen, C.; Zhang, Q.; Wang, Y. Green Chem. 2008, 10, 553-562.
75. Zanon, J.; Klapars, A.; Buchwald, S. L. J. Am. Chem. Soc. 2003,
125, 2890-2890, and references cited therein.
76. Takagi, K.; Okamoto, T.; Sakakibara, Y.; Oka, S. Chem. Lett. 1973, 471-474.
77. Tagaki, K. In Handbook of Organopalladium Chemistry for Organic Synthesis;
Negishi, E., Ed.; J. Wiley & Sons: Hoboken, NJ, 2002; Vol. 1, pp 657-672.
78. For an excellent review see: Sundermeier, M.; Zapf, A.; Beller, M., Eur. J. Inorg.
Chem. 2003, 3513-3526.
79. Wang, X.; Zhi, B.; Baum, J.; Chen, y.; Crockett, R.; Huang, L.; Eisenberg, S.; Ng,
J.; Larsen, R.; Martinelli, M.; Reider, P.; J. Org. Chem. 2006, 71, 4021-4023.
80. Qiao, J. X.; Cheng, X.; Modi, D. P.; Rossi, K. a.; Luettgen, J. M.; Knabb, R. M.;
Jadhav, P. K.; Wexler, R. R.; Bioorg. Med. Chem. Lett. 2005, 15, 29-35.
81. Veauthier, J. M.; Carlson, C. N.; Collis, G. E.; Kiplinger, J. L.; John, K. D.
Synthesis 2005, 2683-2686.
82. Jin, F.; Conflone, P. N. Tetrahedron Lett. 2000, 41, 3271-3273.
83. Littke, A.; Soumeillant, M.; Kaltenbach, R. F.; Cherney, R. J.; Tarby, C. M.;
Kiau, S. Org. Lett. 2007, 9, 1711-1714 and references cited therein.

157
84. Marcantonio, K. M.; Frey, L. F.; Liu, Y.; Chen, Y.; strine, J.; Phenix, P.; Wallace,
D. J.; Chen, C. Org. Lett. 2004, 6, 3723-3725.
85. Schareina, T.; Zapf, A.; Beller, M. J. Chem. Soc. Chem. Commun. 2004, 1388.
86. Weissman, S. A.; Zewge, D.; Chen, C. J. Org. Chem. 2005, 70, 1508.
87. Grossman, O.; Gelman, D. Org. Lett. 2006, 8, 1189.
88. Pinto, A.; Jia, Y.; Neuville, L.; Zhu, J. Chemistry Eu. J. 2006, 13, 961-967.
89. Liesbkind, L. S.; Zhang, Z. Org. Lett. 2007, 8, 4331-4333.
90. Sakamoto, H.; Mori, H.; Takizawa, M.; Kikugawa, Y., Synthesis 1991, 750.
91. Meshram, H. M., Synthesis 1992, 943.
92. Iranpoor, N.; Zeyni Zadaeh, Synth. Commun. 1999, 29, 2747.
93. Barman, D. C.; Thakut, A. J.; Prajapathi, D.; Sandhu, J. S., Chem. Lett., 2000, 29,
1196.
94. S.H. Yang, S. Chang, Org. Lett. 3 (2001) 4209.
95. A. Hegedus, A. Cwik, Z. Hell, Z. Horvath, A. Esek, M. Uzsoki, Green.
Chem. 4 (2002) 618.
96. J.A. Campbell, G. McDougald, H. McNab, L.V.C. Rees, R.G. Tyas, Synthesis
(2007) 3179.
97. W. Ina M. Siegmar, E. Alexei, P. Gerhard, G. Siegbert, K. Laura, P. Natella,
W. Wolfgang, J. Mater. Chem., 11 (2001) 1642.
98. S.C. Mukund, K. G. Mukund, C. Joseph, US patent (2005) 6875882.
99. M. Shirai, K. Shiba, T. Furuya, . (2004) EP1270550.


















