
Chapter 20 DNA Technology and Genomics. The Challenges of Studying DNA The size of the DNA poses big...
59
Chapter 20 DNA Technology and Genomics
-
Upload
brett-cuffe -
Category
Documents
-
view
221 -
download
2
Transcript of Chapter 20 DNA Technology and Genomics. The Challenges of Studying DNA The size of the DNA poses big...

Chapter 20
DNA Technology and Genomics

The Challenges of Studying DNA
The size of the DNA poses big problems.
Naturally occurring DNA is very long and particular genes may only comprise a small portion of the DNA, maybe 1/100,000 of the chromosome.
There may only be a small difference in the surrounding nucleotides.

Help in Overcoming These Challenges
To make this easier, scientists have developed methods for gene cloning.
Genes are cloned to make multiple copies of the same gene and to produce a protein product.
Cloning can be used to endow an organism with a new metabolic activity.– Pest resistance, drought resistance, etc.
We can also insert genes into organisms for medical purposes.– Insulin production by E. coli.

Most Methods for Cloning DNA Share General Features:A commonly used method employs
bacteria (E. coli) and their plasmids:– 1. The plasmid is isolated from bacteria.– 2. DNA is inserted into it.– 3. The plasmid is now recombinant (DNA
from 2 sources).– 4. The bacteria reproduces forming a
clone of the original cell.– 5. The foreign (inserted) gene is cloned
at the same time.


Restriction Enzymes
Restriction enzymes are enzymes that cut DNA molecules at a limited number of specific locations.
In nature, these enzymes help prevent a bacterial cell from foreign DNA (from phages and other organisms).
Many different restriction enzymes have been identified and isolated.

Restriction Enzymes Each restriction enzyme
is very specific and recognizes a short DNA sequence known as a restriction site.
The DNA itself is cut at specific sites within the DNA strand.
A bacterial cell will protect its own DNA from its own restriction enzymes by addition of methyl (-CH3) groups to A’s and C’s within the sequences recognized by these enzymes.

Restriction Enzymes
They recognize sequences 4-6 nucleotides in length.
Many such sequences occur by chance throughout the genome, thus a restriction enzyme will produce a numerous amount of fragments (called restriction fragments) when they are introduced to DNA.

Restriction Enzymes
All copies of a particular DNA molecule always produce the same DNA fragments when introduced to the same restriction enzymes.
Thus, a restriction enzyme cuts DNA in a reproducible way.

Restriction Enzymes The most useful RE’s cleave DNA in a
certain way and produce sticky ends. We call them sticky ends because they
combine with other DNA fragments that have been cut by the same enzyme.
These fragments usually hydrogen bond together and then are joined permanently by DNA ligase which catalyzes the formation of covalent bonds in the sugar-phosphate backbones.
This produces a stable, recombinant DNA molecule.

Restriction Enzymes
Movie

Gene Cloning in Plasmids
Genes are cloned in plasmids using a cloning vector which is the original plasmid that carries the foreign DNA into the cell.
Bacterial plasmids are commonly used because they are easy to manipulate and most of the experimentation with them can be done in vitro.

A Common Method for Cloning:
1. Isolation of plasmids from E. coli cells and DNA from human cells grown in culture.
2. Treat the 2 with the same RE producing the same sticky ends. Plasmids cut at one spot, human DNA cut at many.
3. Mix the human and plasmid fragments.

14
A Common Method for Cloning:
4. Add DNA ligase to permanently fuse the sticky ends of the plasmid and human DNA.
5. Mix the recombinant plasmids with bacteria.
6. Plate the bacteria out on selective media to isolate the recombinants.
14
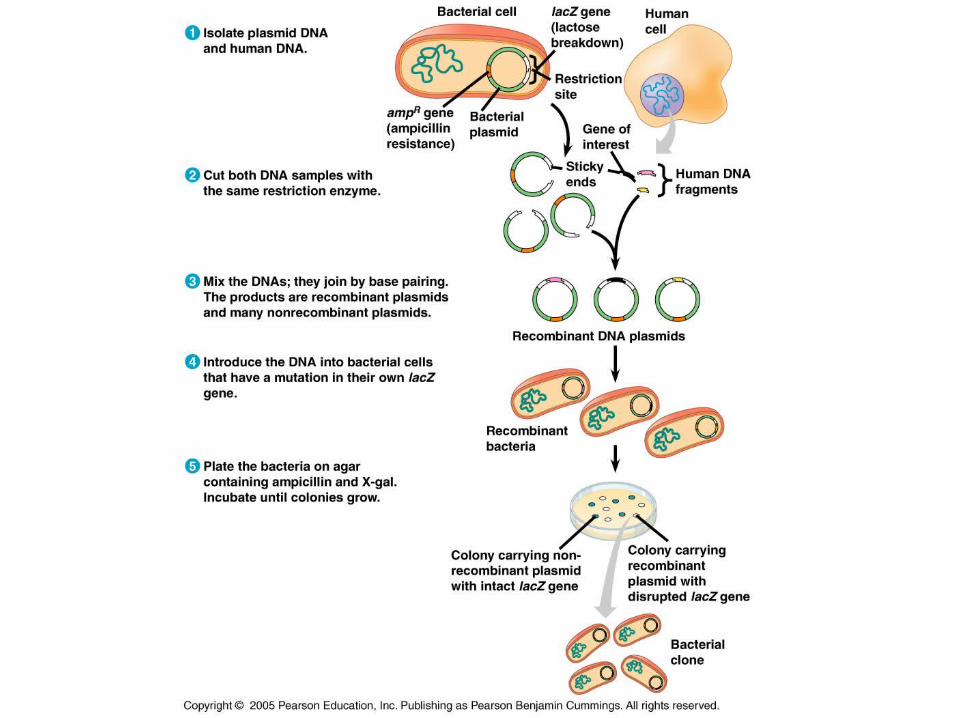

Recognizing the Clone
Originally, the bacteria lacked a gene conferring resistance to something, say an antibiotic.
When the bacterium was plated on growth medium containing the antibiotic, they would die.
Those bacteria containing the antibiotic resistance gene are now able to survive on the medium containing antibiotic.

Gene Cloning
Movie

Problems with Cloned DNA and Expression in the BacteriaTwo different types of cells (prokaryotic
vs. eukaryotic) pose problems for gene expression.
The presence of introns in eukaryotic transcripts and no way for the prokaryotes to splice them out.

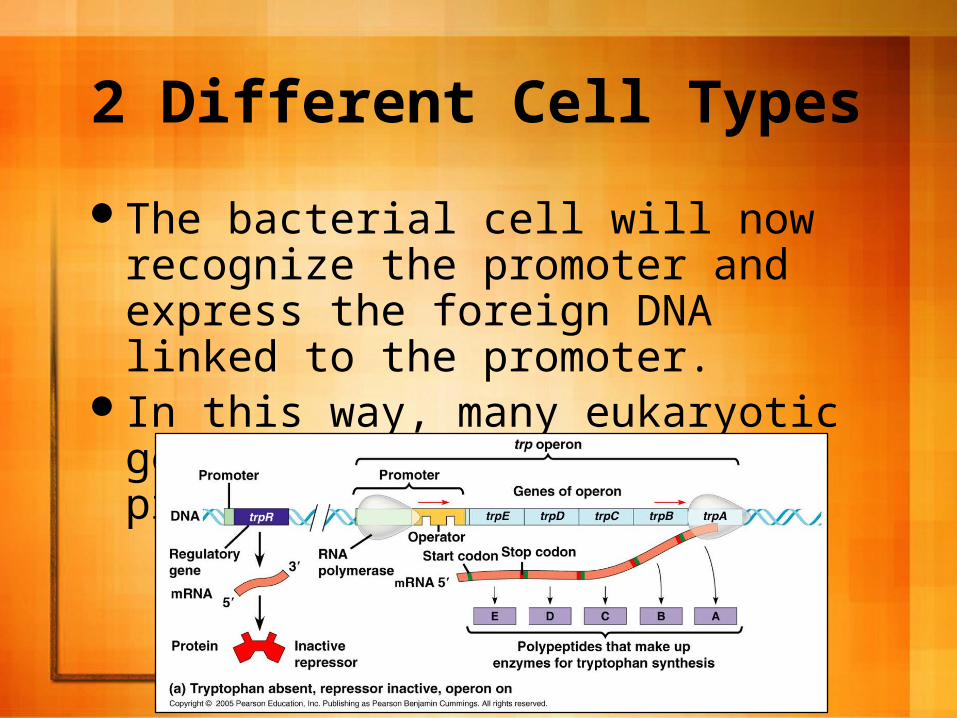
2 Different Cell Types
To overcome this, scientists use expression vectors that have highly active prokaryotic promoters just upstream from the gene to be expressed.
21
2 Different Cell Types
The bacterial cell will now recognize the promoter and express the foreign DNA linked to the promoter.
In this way, many eukaryotic genes are expressed in prokaryotic cells.
21

The Presence of Introns
Prokaryotes lack splicing machinery, and the long eukaryotic gene is often prevented from being expressed in the bacteria.
Scientists use cDNA to circumvent the problem. – cDNA contains only the exons.

cDNA Synthesis
cDNA is synthesized from mRNA extracted from the cells.
Retroviruses make reverse transcriptase and this is used to make single stranded DNA molecules from the mRNA.
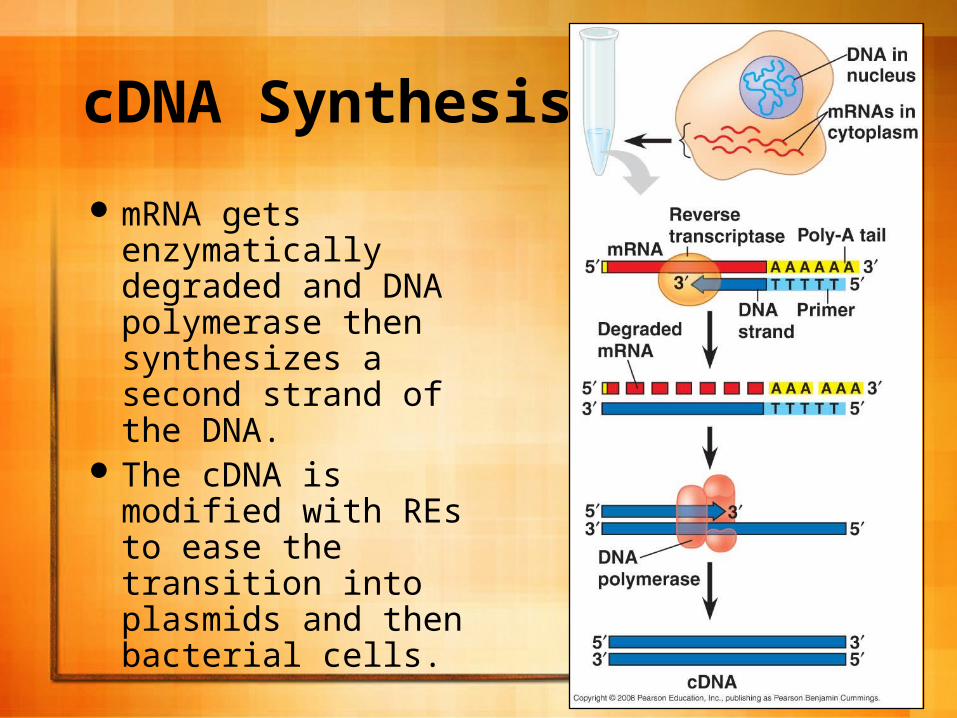
24
cDNA Synthesis
mRNA gets enzymatically degraded and DNA polymerase then synthesizes a second strand of the DNA.
The cDNA is modified with REs to ease the transition into plasmids and then bacterial cells.
25
cDNA Synthesis
Movie
25

cDNA Synthesis
Scientists can overcome the differences between prokaryotes and eukaryotes using expression vectors.
cDNA is used and will be expressed by the bacterial cell as long as the expression vector contains the bacterial promoter and other control elements necessary for transcription and translation.

Other Ways to Get Around Compatibility Problems:
Scientists often use eukaryotic yeasts and single-celled fungi which are as easy to grow as bacteria and also contain plasmids.
Scientists have also made recombinant plasmids that combine yeast and bacterial DNA that can replicate in either type of cell.

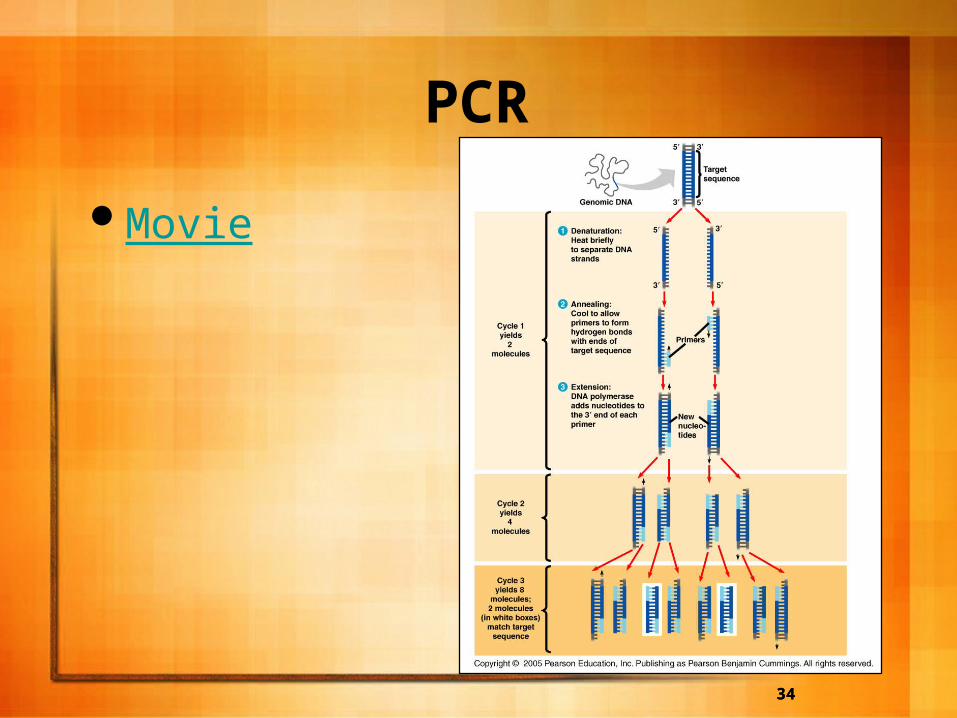
PCR
Sometimes scientists want to prepare a large quantity of DNA when only a small amount is present.
To get around this, PCR is used and can quickly generate a large amount of DNA from a small amount.

PCR A 3-step cycle brings about a chain reaction
that produces exponential growth of identical DNA molecules.
A double stranded piece of of DNA is obtained.– A. The solution containing the piece of DNA is
heated so as to denature the DNA and separate it into single strands.
– B. DNA primers (short, single stranded DNA molecules) are added to the mixture and it is allowed to cool so the primers anneal to the cDNA strands.
– C. The heat-stable DNA polymerase adds nucleotides to the primers in the standard 5’-->3’ direction synthesizing the target sequence.


PCR
What makes this process so useful is its specificity. If a target segment is identified and a primer made to it, then only a small amount is really necessary from the start.
It is easy to see how quickly a large amount of DNA can be made: 1, 2, 4, 8, 16, …..

PCR
The primer will only replicate the target segment because this is all they are able to bind to. After just a few cycles, a very large amount of the target segment will be identified.

PCR
DNA cloning in cells remains the best way to prepare a large quantity of a gene or DNA segment. PCR can’t be used to obtain a large quantity of gene because occasional errors in PCR replication impose limits on the number of good copies that can be made.
Often times though, enough of a specific DNA fragment can be made to insert it into a vector and clone it.

Gel Electrophoresis To study DNA,
scientists often use gel electrophoresis.
Agarose gel is often used to separate DNA fragments based on size, charge, etc., that have been treated with a restriction enzyme.
The fibers in the gel separate out the fragments; smaller fragments migrate further than the larger fragments.

Gel Electrophoresis
The negatively charged DNA fragments migrate toward the positive pole of the electrophoresis box.

Restriction Fragment Analysis
This is a powerful tool scientists use to analyze differences in the nucleotide sequences of DNA molecules.
When the researchers began analyzing the restriction fragments of non-coding DNA from individuals, they began noticing small nucleotide differences on homologous chromosomes.

Restriction Fragment Analysis
Treating the DNA with restriction enzymes and then running the samples through a gel enable researchers to produce banding patterns characteristic of the starting molecule and the restriction enzyme(s) used to treat the DNA.
You will use this analysis to examine your bacterial chromosome for certain genes.


Restriction Fragment Length Polymorphisms
When non-coding regions of DNA were treated with restriction enzymes and banded, scientists discovered differences in non-coding regions on homologous chromosomes.
These were given the name restriction fragment length polymorphisms.

Restriction Fragment Length Polymorphisms
These serve as genetic markers of non-coding DNA that appears near a particular locus in a genome.
There are many RFLP variants within a population.
RFLP data is often used in crime investigations because the likelihood that two individuals will have the same banding patterns are minuscule at best.

Scientists use a Southern Blot to Analyze RFLPs or Regular DNA.
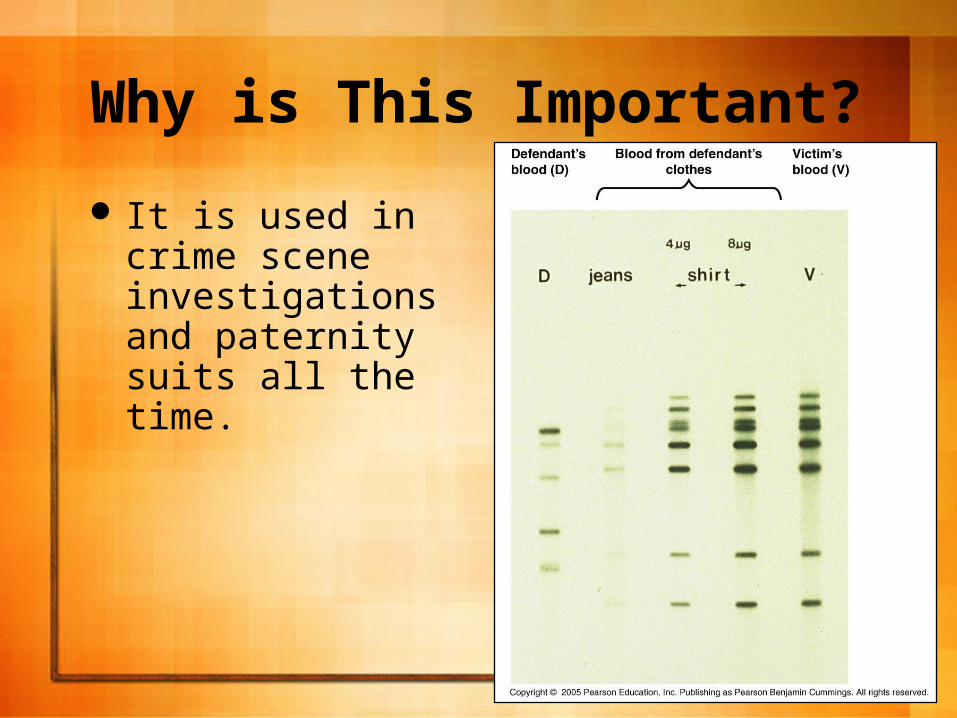
Why is This Important?
It is used in crime scene investigations and paternity suits all the time.

Why is This Important?
Scientists use the information to determine if a person has a particular disease.

Why is This Important?
Scientist can also use RFLP information to determine the likelihood of inheriting a certain genetic disease.

HGP Project
Was started in 1990, and was completed in 2003. The goal was to map the entire human genome.
It proceeded in 3 stages:– Making a genetic (linkage) map.
Using genetic markers to determine the order of genes.
– Making a physical map.Determining the distance between genes.
– DNA sequencing.Determining the exact nucleotide sequence of the
genes.

DNA Sequencing
Became the biggest drawback. There wasn’t a fast way to sequence the genes. The initial challenge was to develop new technologies to quickly sequence the DNA.
Frederick Sanger developed the first method to sequence the genes called the dideoxy chain-termination method.

The Dideoxy Chain-Termination Method
A set of complementary DNA strands are synthesized from an original DNA strand.
Each strand starts with the same primer and ends with a modified nucleotide that is fluorescently labeled and lacks a hydroxyl group; it is called a dideoxyribonucleotide.
The dideoxyribonucleotide terminates the growing DNA strand because it lacks a 3’-OH group.

The Dideoxy Chain-Termination Method
In the newly synthesized strands, each nucleotide position along the original sequence is represented by strands ending at that point with the complementary ddNTP.
Each type of ddNTP, tagged with a distinct fluorescent label, identifies the ending nucleotides of the new strands, ultimately revealing the sequence of the DNA.

3 Main Steps of the Dideoxy Method
1. The fragment of DNA to be sequenced is denatured into single strands and incubated in a test tube with a primer known to base pair with the known 3’ end of the template stand, DNA polymerase, A,T,C,&G and the 4 ddNTP’s which are tagged with a specific fluorescent molecule.

3 Main Steps of the Dideoxy Method
2. Synthesis of the new strand starts at the 3’ end of the primer and continues until a ddNTP is added at random. This prevents further elongation. Eventually, a labeled set of strands of various lengths is generated

3 Main Steps of the Dideoxy Method
3. The labeled strands in the mixture are separated by passage through a polyacrylamide gel in a capillary tube; the shorter the strands move through faster than the larger ones.
A fluorescent detector can sense the color of each tag as the strands come through. Strands that differ in as little as 1 nucleotide can be distinguished.

The Dideoxy Method
The color of the fluorescent tag on each strand indicates the identity of each nucleotide at its end, and the results can be printed out on a spectrogram and the sequence, which is complementary to the template strand, can then be read from the bottom to top.


Whole Genome Shotgun Approach
In 1992, Craig Venter developed the shotgun method which greatly sped up the process of gene sequencing.
The process uses random DNA fragments and powerful computers to analyze the data.
The idea was initially met with skepticism.

Whole Genome Shotgun Approach
What is essentially happening is the piece of DNA is incubated with a variety of restriction enzymes and then cloned in plasmid or phage vectors to produce large numbers of these fragments.
Then, each fragment is sequenced and a computer program analyzes the fragments relative to each other to determine the sequence.
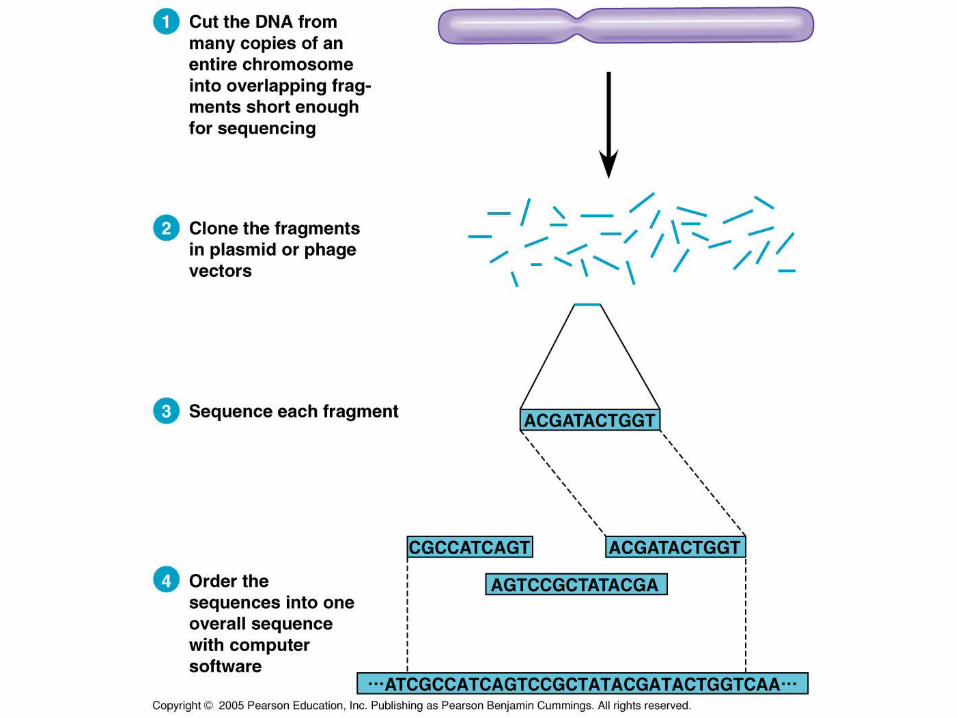

Whole Genome Shotgun Approach
His approach sequenced nearly the entire genome in about 3 years.
Using the Sanger method, the HGP took nearly 11 years.
Originally, Venter and his colleagues were accused to stealing the information made available by the HGP to sequence the genome, but it is generally understood now that the Shotgun approach is a good method.


















