
Chapter 2 1 CHAPTER 2 · Chapter 2 1 CHAPTER 2 1. This solvolysis reaction proceeds with loss of...
15
Chapter 2 1 CHAPTER 2 1. This solvolysis reaction proceeds with loss of the leaving group to generate a cation. The aromatic ring will stabilize that cation either by -donation from the ring or by formal resonance participation via a non-classical ion. In either case, an electron-donating substituent on the aromatic ring will stabilize the charge and, thereby, stabilize the intermediate cation. This stability will be reflected in the relative rate of the solvolysis reaction. Both OMe and Me are electron donating and therefore enhance the rate. Since OMe is a more powerful electron donating group, the effect is large. The Cl substituent is mildly electron withdrawing and, therefore, slows the relative rate by slightly destabilizing the intermediate cation. This problem is taken from J. Org. Chem., 1985, 50, 821. 2. Since the OH group in 3-pentanol is an extremely poor leaving group and iodide is a nucleophile, an S N 2 reaction is not facile. Despite the presence of the water, solvolysis of the alcohol moiety to generate a cation is somewhat slow. If an acid catalyst is added, however, protonation of the OH occurs and loss of water generates the cation. The cation is then trapped under S N 1 conditions by iodide to give 3-iodopentane. OH O I H H - H 2 O + I – H + 3. This bromide is in fact a neopentyl-like molecule [R 3 C-CH(R')X] and, therefore, very sterically hindered to nucleophilic displacement (see Sec. 2.6.A.i). Neopentyl halides react much slower with nucleophiles under S N 2 conditions than do tertiary halides (see Table 2.11 on page 105 in the text). The use of KOH in DMF promotes substitution, and the DMF does not allow facile ionization to give an S N 1 displacement. Elimination with the basic KOH dominates if the substitution reaction is too slow to compete. 4. (a) Since the phenyl ring is somewhat electron-withdrawing, the three phenoxides are less basic than the cyclohexanol anion. The OMe group is electron releasing, and this makes the availability of electron density on O - greater than on phenoxide. Similarly, the nitro group is electron withdrawing, making the electron density on O – less than in phenoxide. (b) When comparing formic acid and acetic acid, the electron releasing methyl group in acetic acid diminishes the positive character of the acidic hydrogen. It is therefore less acidic. In addition, once ionized to the carboxylate, the presence of the electron releasing methyl group slightly diminishes the adjacent positive character of the carboxyl carbon, destabilizing the anion relative to the formate anion. The smaller formate anion is probably better solvated than the acetate anion, which also contributes to enhanced acidity. Copyright © 2011 Elsevier Inc. All rights reserved.
Transcript of Chapter 2 1 CHAPTER 2 · Chapter 2 1 CHAPTER 2 1. This solvolysis reaction proceeds with loss of...

Chapter 2 1
CHAPTER 2
1. This solvolysis reaction proceeds with loss of the leaving group to generate a cation. The aromatic ring will
stabilize that cation either by -donation from the ring or by formal resonance participation via a non-classical ion.
In either case, an electron-donating substituent on the aromatic ring will stabilize the charge and, thereby, stabilize
the intermediate cation. This stability will be reflected in the relative rate of the solvolysis reaction. Both OMe and
Me are electron donating and therefore enhance the rate. Since OMe is a more powerful electron donating group,
the effect is large. The Cl substituent is mildly electron withdrawing and, therefore, slows the relative rate by
slightly destabilizing the intermediate cation.
This problem is taken from J. Org. Chem., 1985, 50, 821.
2. Since the OH group in 3-pentanol is an extremely poor leaving group and iodide is a nucleophile, an SN2
reaction is not facile. Despite the presence of the water, solvolysis of the alcohol moiety to generate a cation is
somewhat slow. If an acid catalyst is added, however, protonation of the OH occurs and loss of water generates the
cation. The cation is then trapped under SN1 conditions by iodide to give 3-iodopentane.
OH O IHH
- H2O + I–
H+
3. This bromide is in fact a neopentyl-like molecule [R3C-CH(R')X] and, therefore, very sterically hindered to
nucleophilic displacement (see Sec. 2.6.A.i). Neopentyl halides react much slower with nucleophiles under SN2
conditions than do tertiary halides (see Table 2.11 on page 105 in the text). The use of KOH in DMF promotes
substitution, and the DMF does not allow facile ionization to give an SN1 displacement. Elimination with the basic
KOH dominates if the substitution reaction is too slow to compete.
4. (a) Since the phenyl ring is somewhat electron-withdrawing, the three phenoxides are less basic than the
cyclohexanol anion. The OMe group is electron releasing, and this makes the availability of electron density on O-
greater than on phenoxide. Similarly, the nitro group is electron withdrawing, making the electron density on O–
less than in phenoxide.
(b) When comparing formic acid and acetic acid, the electron releasing methyl group in acetic acid diminishes
the positive character of the acidic hydrogen. It is therefore less acidic. In addition, once ionized to the
carboxylate, the presence of the electron releasing methyl group slightly diminishes the adjacent positive character
of the carboxyl carbon, destabilizing the anion relative to the formate anion. The smaller formate anion is probably
better solvated than the acetate anion, which also contributes to enhanced acidity.
Copyright © 2011 Elsevier Inc. All rights reserved.

2 Organic Synthesis Solutions Manual
ortho -Methoxyphenol is more acidic than para-methoxyphenol due to the "ortho effect." The proximity of the
OMe in the ortho derivative allows internal hydrogen bonding with the O-H moiety, making that bond more
polarized and more acidic. Such internal hydrogen bonding is not possible in the para derivative, although
intermolecular hydrogen bonding may occur.
Acetic acid is more acidic in water, although THF is a stronger base. Ionization is much easier in water than in
THF, and water can better stabilize the ionic products. This makes the ionization (loss of H+) easier, enhancing
acidity relative to THF where ionization is less efficient.
5.
OMe
N(CH2Ph)2
Et
O
N
Et
H
O OSiMe3
O
Cl
MeOOMe
O
O
Cl NHi-Pr
ONEt2
OMe
OMe
MeO
O
O
N
HPh
Me
Me(CH2)9
ON
O
HN
O
O
O
OO MeO
see J. Org. Chem., 1999, 64, 4980 see J. Org. Chem., 1999, 64, 7586 see Tetrahedron Lett., 2000, 41, 733
see Tetrahedron Lett., 2000, 41, 1975see J. Am. Chem. Soc., 1994, 116, 9921 see J. Org. Chem., 1999, 64, 2450
see J. Org. Chem., 1999, 64, 3736 see J. Am. Chem. Soc., 1994, 116, 9921
(a)(b)
(c)
(d) (e) (f)
(g) (h)
6. Taken from Stock, L.M., Aromatic Substitution Reactions, Prentice-Hall, Englewood Cliffs, N.J., 1968, p. 91.
This reaction proceeds via a benzyne intermediate (X). Addition of NH2 to either carbon of the triple bond leads to
the two products shown and the 14C label on both carbons relative to the ipso carbon bearing the amine unit.
NH2Cl
+
NH2
**
**
H2N
* = 14C X
H2O
7. This cyclohexane derivative exists in two chair forms, A and B. An E2 reaction cannot occur from A because
Copyright © 2011 Elsevier Inc. All rights reserved.

Chapter 2 3
the bromine is equatorial. A trans-diaxial relationship between the bromine and any -hydrogen(s) is a
requirement. Only when the bromine is axial in conformation B is one -hydrogen is axial. This means that the E2
reaction can give only one alkene with the double bond toward the methyl group rather than the ethyl group. The
stereochemistry in the halide makes this a regiospecific elimination.
Me
Br
MeEt
Br
Me
Me
H
Et
H
H
Br
MeMe
EtH
A B
Me
Me
Et
8. See Tetrahedron Lett., 2000, 41, 3411. The tertiary allylic alcohol reacts with the acidic resin (H+) to give the
oxonium ion. There are two pathways. An SN2' displacement of water by the primary alcohol unit leads to the
product directly. This is the mechanism presented by the authors of this paper. An alternative mechanism would
be ionization to an allylic tertiary cation followed by cyclization and loss of a proton, as shown in the diagram.
HOOH + H+
H2OOH
OHO
H
– H2O
– H+
– H2O
O
SN2'
vinylogous SN1-like
Tetrahedron Lett., 2000, 41, 3411
9. This reaction proceeds via a mercury-stabilized secondary cation. In this paper, a detergent (sodium dodecyl
sulfate) was added and it had an important effect. The cation formed in the reaction can react with water (from the
aqueous solvent) to give an alcohol after reductive cleavage of the C-Hg bond with sodium borohydride. The ether
is formed by attack of octanol on the cation. There is a large excess of water, however, so the alcohol is the major
product. Increasing the proportion of octanol leads to increased amounts of ether. Even when 10 equivalents of
octanol are used, water is present in a large excess. In fact, water and octanol are close in nucleophilic strength.
The detergent leads to enhanced local concentrations of octanol that can overcome the bulk concentration effects of
the excess water, leading to more ether product.
Taken from J. Org. Chem., 1987, 52, 5039.
10. This problem was taken from J. Org. Chem., 1987, 52, 260; 1984, 47, 4855.
(a) Product A is the usual product of reaction of an alkene with chlorine. Formation of chloronium ion X was
Copyright © 2011 Elsevier Inc. All rights reserved.

4 Organic Synthesis Solutions Manual
followed by addition of chloride ion to give A, as shown. Product B arises by a cationic mechanism that involves
participation of the aromatic ring. If chloronium ion X opens to form cation Y, the benzene ring attacks the
positive charge to form the bridged cation Z. If chloride ion attacks the three-membered ring, as shown, B results.
Cl
ClMeO
Cl
MeO
Cl
Cl
MeO
Y
Cl
MeO
ClX
Cl—Cl
B
MeO
Cl
Cl
MeO
A
Z
(b) If the OMe group, which is electron-releasing, is replaced with Cl, which is electron-withdrawing, product B
should be more difficult to form. Cation Z is stabilized by the electron releasing OMe group, but an electron
withdrawing Cl would destabilize this intermediate, making formation of Z more difficult and, thereby, formation
of B.
11. See Tetrahedron Lett., 1997, 38, 3469. When drawn in a form that approximates the three-dimensional shape it
is clear that the cyclobutanone unit flattens the tricyclic system. Protonation of the glycol unit is followed by acid-
catalyzed addition of methanol to the ketone. A Grob-like fragmentation leads to formation of the methyl ester unit
and a cation. This cation traps water and with proton transfers loses ethylene glycol to form the ketone unit found
in the final product.
Copyright © 2011 Elsevier Inc. All rights reserved.
Chapter 2 5
O
O
O
H
HH
O
OO
H
H
OO
OH
RT
HH
OMe
OOH
OH
H
CO2Me
O
H
H
OMe
OOH
O
OOH
O
OH
H
HH
OMe
H
H
OMe
O
O
H
H
OMe
OOH
O
OH2
+ H+
+ MeOH;H+ transfer
– H+
+ H2O
–H+;+H+
– ethylene glycol–H+
MeOH , 1N HCl
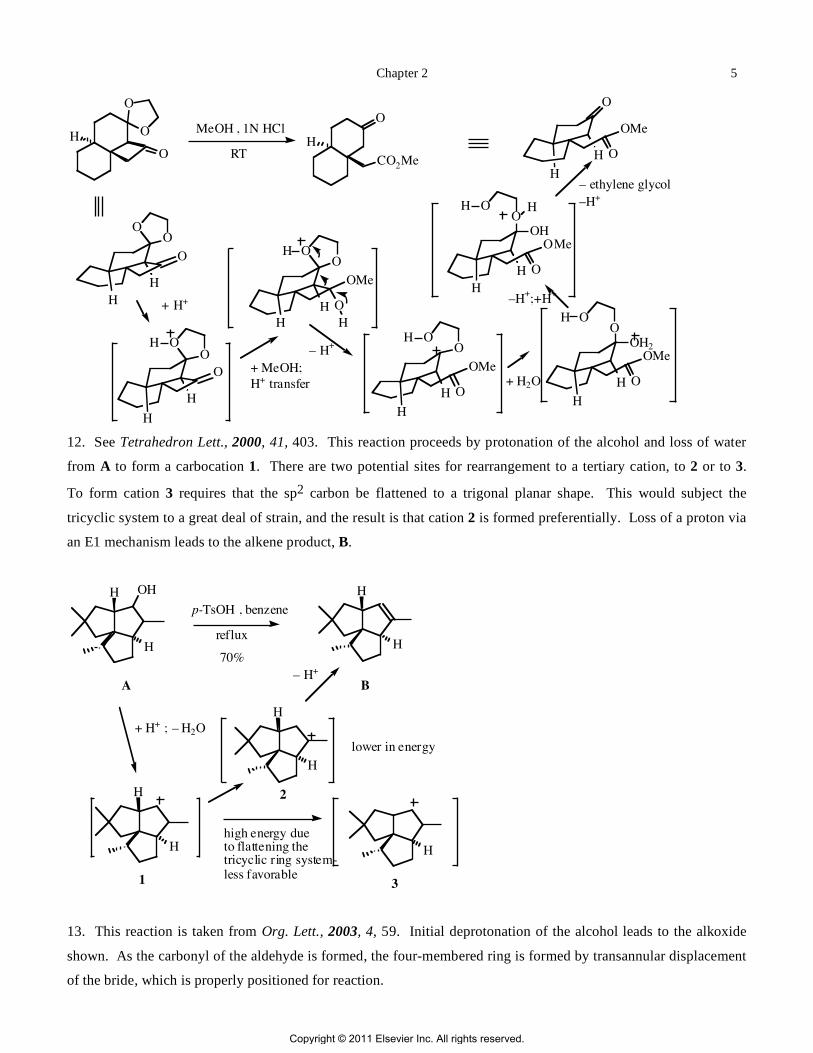
12. See Tetrahedron Lett., 2000, 41, 403. This reaction proceeds by protonation of the alcohol and loss of water
from A to form a carbocation 1. There are two potential sites for rearrangement to a tertiary cation, to 2 or to 3.
To form cation 3 requires that the sp2 carbon be flattened to a trigonal planar shape. This would subject the
tricyclic system to a great deal of strain, and the result is that cation 2 is formed preferentially. Loss of a proton via
an E1 mechanism leads to the alkene product, B.
H OH
H
H
H
p-TsOH , benzene
reflux
70%
A B
H
H
H
H
Hhigh energy dueto flattening thetricyclic ring system-less favorable1
2
3
lower in energy
– H+
+ H+ ; – H2O
13. This reaction is taken from Org. Lett., 2003, 4, 59. Initial deprotonation of the alcohol leads to the alkoxide
shown. As the carbonyl of the aldehyde is formed, the four-membered ring is formed by transannular displacement
of the bride, which is properly positioned for reaction.
Copyright © 2011 Elsevier Inc. All rights reserved.

6 Organic Synthesis Solutions Manual
O
Br
O
H
O
CHO
O
Br
OH
H
KH
14. Initial attack of hydroxide leads to opening of the oxazolone ring, and the nitrogen attacks the epoxide, forming
the indolizidine ring and generating an alkoxide. Protonation to give the alcohol is followed by a second attack of
hydroxide on the carbonate ester. Carbonic acid is formed as the alkoxide is released in an acyl substitution
reaction. The carbonic acid decomposes to water and carbon dioxide and protonation by ethanol gives the final
product.
N
H
OBn
OH
OO
HO
NO
O
OBn
O
OH
HO
NO
O
OBn
O OH
N
H
OBn
OH
O
HO O
OH
HO
O
OH
N
H
OBn
OH
OH
EtOH
N
H
OBn
O
OO
HO
N
H
OBn
OH
O
EtOH
see J. Org. Chem., 2000, 65, 9129
–
15. (a) This is the Hofmann elimination reaction (Sec. 2.9.C.i) and demands a syn transition state (an eclipsed
conformation for reaction). Because of the requirement for an eclipsed conformation (where the leaving group on
the -carbon and the "base" can be in close enough contact) the lowest energy eclipsed conformation will lead to
the major product. In this case, the lowest energy transition state is A rather than B, and ethene is the major alkene
product, not 4-methyl-2-pentene. The i-Pr Me interaction in B destabilizes that conformation relative to the
H H interactions in A. In both A and B, the NR3 H interactions are about the same.
Copyright © 2011 Elsevier Inc. All rights reserved.

Chapter 2 7
NEt2C4H9
HH
H
H H
NEt3
HMe
H
MeH
A B
(b) Taken from J. Org. Chem., 2004, 69, 7616. The strongly electronegative fluorine atoms withdraw electron
density, making the C=C unit very electrophilic. This inductive effect makes the alkene carbon more susceptible to
attack than the epoxide carbon, despite the fact that there is modest steric hindrance. Such attack drives the SN2'
reaction to the allylic alcohol shown, as a mixture of E and Z isomers.
PhOF
F
Li
Ph
OH
F
FBu E + Z2nd step of
workup included
(c) This is an SN2 reaction, and inspection of the Walden inversion transition state shows that these two neutral
reactants produce a charged transition state where a positive charge builds on the nitrogen (not on carbon) and a
negative charge builds on iodine (see A). Separation of these two charges (water promotes separation
C
C3H9
HH
Et3N I C
R
RR
X I
A B
+ +
of charges) favors formation of the final products since the carbon is transferred to the positive nitrogen, with
iodide as the counter-ion. This contrasts with transition state B, the "normal" Walden inversion that arises when a
charged nucleophile attacks a neutral substrate. Water as a solvent will separate charges and promote ionization.
Separation of charges in B will slow the reaction since the incoming charged nucleophile is separated from the
developing charge on the central carbon. In A, however, separation of positive and negative charges favors product
formation. For this reason, the reaction of an amine and a halide to give an ammonium halide is faster in aqueous
media than in non-aqueous media (which cannot separate charges).
(d) The proximity of the ammonium group to the carboxyl carbon is the key to this answer. The aminopropanoic
acid has the amino group two carbons away, whereas the amino group is four carbons away in aminopentanoic
acid. Both inductive effects and field effects are strongest when the ammonium groups are close. For this reason,
amino propanoic acid is more acidic than amine pentanoic acid.
(e) The nitro group in 4-nitrophenol is electron withdrawing and, therefore, stabilizes the charge in the phenoxide
product. No such stabilization is possible with phenol. The electronic effects also weaken the
O—H bond, enhancing acidity.
16. 3-Bromo-4-methylhexane reacts with hydroxide (a base for an E2 reaction) by removing Ha. The orientation
of the molecule does not matter because the important feature is the anti-relationship of the Br and Ha. When Ha is
Copyright © 2011 Elsevier Inc. All rights reserved.

8 Organic Synthesis Solutions Manual
removed, the transition state for the E2 reaction will retain the stereochemical relationship of the groups. Since the
two ethyl groups are on the same side in the anti-orientation, they will be on the same side in the transition state,
and this will lead to cis-3-hexene as the major product, with the two ethyl groups still on the same side. If this
reaction were carried out under E1 conditions, ionization of the bromine would lead to planar carbocation C, and
removal of Ha could occur from either face since the bond to the carbon bearing Ha is free to rotate. This leads to
scrambling of the stereochemistry and a mixture of cis- and trans-alkenes.
Ha
Br Me
H
A
B
Ha
Br Me
H
OH
MeH
Ha
MeH
65
43
21
65
43
21
rotation about this bondleads to a mixture ofcis- and trans-alkenes
17. See J. Org. Chem., 1997, 62, 641.
18. See Synthesis, 1996, 219. Since NBS is a source of bromine, reaction with the alkene unit generates a
bromonium ion. The oxygen of the alcohol is fixed on the bottom of the molecule relative to the Br, and properly
positioned to open the bromonium ion to form the ether unit. This places the bromine on the top of the molecule.
HO
OOH
BrNBS , CH2Cl2
–25°C RT
Br
O
Br
H
Br2 from NBS – H+
H
HH
H
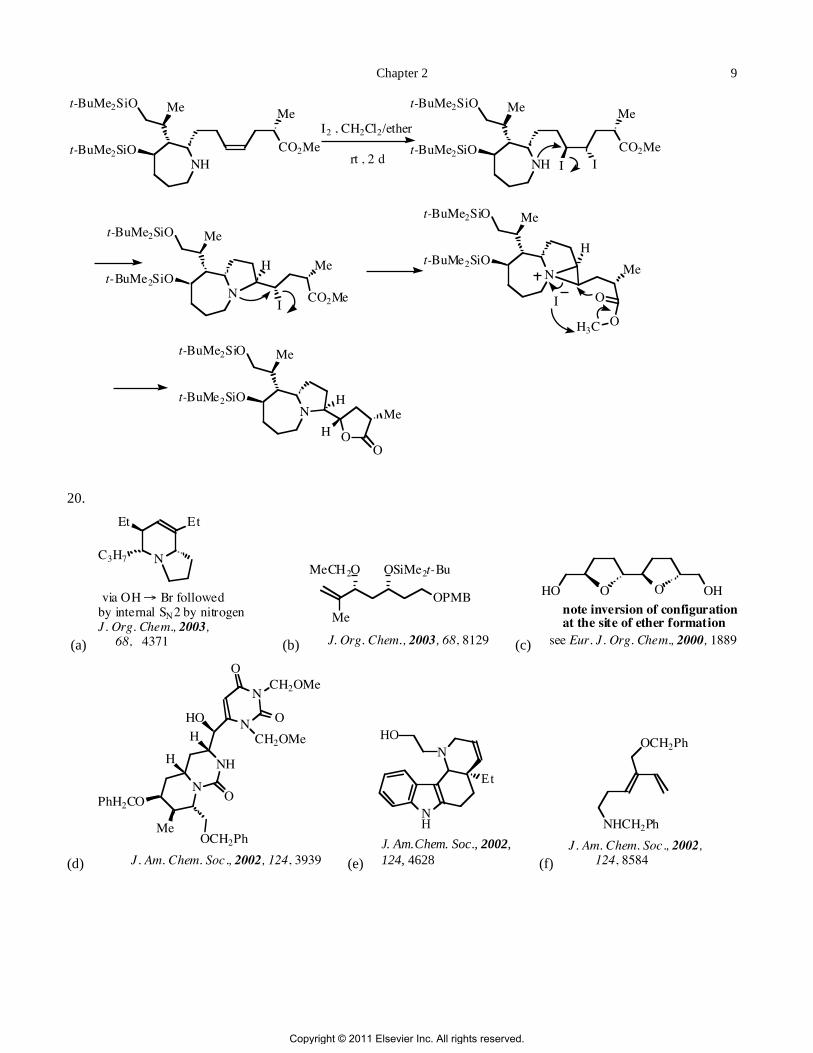
19. This sequence is taken from Org. Lett., 2003, 5, 3361. The alkene unit reacts with iodine to give the diiodide
in situ, and the proximal iodide is displaced by the amine in an internal SN2 reaction to give the bicyclic amine. A
second internal SN2 reaction displaces the remaining iodide to form an aziridinium iodide. the nucleophilic iodide
ion attacks the methyl group of the ester, displacing the carboxylate group, and the electron flow is such that the
oxygen opens the aziridinium ion to form the lactone unit in the product.
Copyright © 2011 Elsevier Inc. All rights reserved.
Chapter 2 9
NHCO2Me
Me
t-BuMe2SiO
t-BuMe2SiO Me
Nt-BuMe2SiO
t-BuMe2SiO Me
OO
MeH
H
N CO2Me
Met-BuMe2SiO
t-BuMe2SiO Me
I
HN Me
t-BuMe2SiO
t-BuMe2SiO Me
H
O
OH3C
NHCO2Me
Me
t-BuMe2SiO
t-BuMe2SiO Me
I I
I
I2 , CH2Cl2/ether
rt , 2 d
20.
(a)
N
EtEt
C3H7
via OH Br followedby internal SN2 by nitrogenJ. Org. Chem., 2003, 68, 4371 (b)
OPMB
Me
MeCH2O OSiMe2t-Bu
J. Org. Chem., 2003, 68, 8129 (c)
O OHO OH
note inversion of configurationat the site of ether formation
see Eur. J. Org. Chem., 2000, 1889
(d)
N
NH
N
N
PhH2CO
Me
H
H
O
OCH2Ph
OCH2OMe
O
CH2OMe
HO
J. Am. Chem. Soc., 2002, 124, 3939 (e)
NH
N
Et
HO
J. Am.Chem. Soc., 2002,
124, 4628 (f)
OCH2Ph
NHCH2Ph
J. Am. Chem. Soc., 2002, 124, 8584
Copyright © 2011 Elsevier Inc. All rights reserved.

10 Organic Synthesis Solutions Manual
(g)
THPOBnO
J. Org. Chem., 2003, 68, 6905 (h)
N
CO2t-Bu
Br
J. Org. Chem., 2003, 68, 6279 (i)
C12H25 OCH2OCH2CH2OMe
OH
J. Am. Chem. Soc., 2004, 126, 36
(j)
NC
OCH2OMe
Org. Lett. 2002, 4, 937 (k)
O
O
O
O
BuOH
J. Org. Chem., 2003, 68, 4039 (l)
OO
J. Nat. Prod., 2002, 65, 909
(m)
O
OH
H
HBr
J. Org. Chem., 2004, 69, 1744 (n)
O
CN
CO2MePhH2CO
HOH
J. Org. Chem.,2003, 68, 7422 (o)
N
OO
Me
OMe HO
J. Org. Chem., 2004, 69, 2191
(p)
MeO
NMe
HEt
J. Chem. Soc., Perkin Trans. 1, 2001, 2398 (q)
ON3AcO
AcO OAc
HOOMe
Org. Lett. 2001, 3, 3353 (r)
O
Me3SiO
O
Org. Lett., 2004 6, 2961
(s)
N
H
HN
H
O
Me2N
HN
J. Org. Chem., 2002, 67, 7147 (t)
OH
O
CO2Me
I
HO
H
MeO2C
OH
H
I
Org. Lett. 2003, 5 , 4385 (u)
Br
HO
Tetrahedron Lett., 2000, 41, 2573
Copyright © 2011 Elsevier Inc. All rights reserved.
Chapter 2 11
(v)
OMe
MeO
Me
Me
Me
OH
see Tetrahedron Lett., 2000, 41, 1151 (w)
Me
see J. Org. Chem., 1993, 58, 2186 (x)
Nt-BuO2C
I
J. Org. Chem., 2002, 67, 6181
(y)
N
OMeOMeO
J. Am. Chem. Soc., 2001, 123, 3214 (z)
BnO
TBSO SiMe3
J. Am. Chem. Soc., 2005,
127, 5596 (aa)
MeO2C CO2Me
HH
HNNH
O OChem. Commun., 2004, 2404
(bb)
Ph CH3
OOHNHS
O
p-Tol
Org. Lett., 2003, 5, 3855
21. (a) The transition state is shown in brackets. Hydroxide removes the -hydrogen only when that hydrogen is in
a conformation that places it anti to the leaving group (here, bromine). In this transition state, the relative positions
of the groups are fixed and this is translated to the alkene, where a single product is formed. The reaction is,
therefore, stereospecific.
Me
HEt
BrH
Ph Ph
Me
BrH
H
EtBr
H
Ph
EtMe
H
Br
H
Ph
EtMe
H–OH
–HBr
Ph
HEt
Merotate
(b) The major product is trans-1,2-diphenyl-1-propene. (c) This is an E2 reaction.
22.
O
HO
MeO CO2H
Ph
HO
PhMeOAc
NC
Me(a) (b) (c) (d) (e)
(a) The most acidic hydrogen is the phenolic hydrogen (pKa 10) vs. pKa 17 for the primary alcohol.
Copyright © 2011 Elsevier Inc. All rights reserved.

12 Organic Synthesis Solutions Manual
Deprotonation gives the phenoxide, which reacts with iodomethane to give the anisole derivative shown.
(b) Two equivalents of base deprotonate both the carboxyl first (most acidic) and the alcohol second (least acidic).
Of the two anions, the carboxyl anion is resonance stabilized and less nucleophilic than the alkoxide, where the
charge resides almost entirely on oxygen. Since the alkoxide oxygen is more nucleophilic, it will react
preferentially with one equivalent of allyl bromide to give the ether shown.
(c) There are two leaving groups in this molecule that can be displaced by the nucleophilic cyanide in an SN2
reaction. The mesylate group is a much better leaving group than acetate. For this reason, one expects the
cyanoacetate product shown to predominate rather than the alternative cyanomesylate.
(d) The dianion shown has a carbon nucleophile and an oxygen nucleophile. The carbon nucleophile is more
nucleophilic, despite the fact that it is resonance stabilized by the adjacent phenyls. The product will therefore be
the methylated derivative shown. Since the nucleophilic strength of the carbanion is diminished by resonance,
some alkoxide may be formed.
(e) In this case, the primary iodide is treated with base. Normally, primary iodides undergo elimination slowly,
and substitution predominates when a nucleophilic base is used. In this case, however, DBU is a non-nucleophilic
base, and reaction will give the alkene shown as the major product.
(f) The product is that shown. Initial formation of the enolate anion and quenching with PhSeCl generated the
phenylselenide. Oxidation with hydrogen peroxide gave the selenoxide in situ, which eliminated spontaneously to
give the alkene unit in the conjugated lactam product.
N
O
CO2t-Bu
OSiPh2t-Bu
J. Am. Chem. Soc. 2002, 124, 14826
23. The product is the less substituted alkene (4,6-dimethylhept-2-ene, C) via syn elimination of an intermediate
sulfoxide. The syn elimination demands an eclipsed transition state, and the two pertinent eclipsed conformations
where a -hydrogen can be removed are A and B. From these Newman projections, A (for removal of Ha) is less
sterically hindered due to decreased torsion strain than is B (for removal of Hb). For this reason, A will have a
higher population at a lower energy and will account for the major product, C. Note that C is shown as a mixture
of cis and trans isomers, despite the fact that the starting iodide contained a stereogenic center. Removal of the two
Ha's in A will lead to the isomeric mixture of alkenes, C.
Copyright © 2011 Elsevier Inc. All rights reserved.

Chapter 2 13
PhS
O
S
Ha
Hb
O Ph
HCHb(Me)C5H11H
•
Ha
Me
Ha
PhS
O
CHHaMeH
•
Hb
Me
C5H11A B
HbC
24. (a) The Fischer projection for the starting material represents a chiral, non-racemic bromide. Once the
bromine and -hydrogen are in the proper anti conformation, the base removes the hydrogen and expels the
bromine. The other groups are fixed in this transition state, leading to trans-2-phenyl-3-methyl-pent-2-ene.
CHO C CMeBr Br
a b
(a) CBr4 , PPh3 (b) BuLi (c) H2O
(b) This cyclohexane derivative exists as an equilibrium mixture of A and B. For an E2 reaction, the bromine
and a -hydrogen must be trans and diaxial. In A, the bromine is equatorial, so there is no chance for elimination.
In B, the axial bromine is axial to two axial hydrogens. Elimination will therefore lead to a mixture of two
regioisomeric products, C and D.
BrMeEt
Br
MeEt
Br
Me
Et
HH
A B
MeEt
C
MeEt
D
(c) In this example, the cyclohexane bromide also exists as A and B, and A cannot react via an E2 reaction
because the bromine is equatorial. In B, however, only the hydrogen attached to the methyl-bearing carbon is trans
and diaxial with the bromine, so there is but one product, C.
BrEtMe
BrEt
MeBr
Et
Me
HH
A B
EtMe
C
25.
(a) CHOBr Br
C CMea b
(a) CBr4 , PPh3 (b) BuLi (c) H2O
Copyright © 2011 Elsevier Inc. All rights reserved.

14 Organic Synthesis Solutions Manual
(b)
NO2NO2
Cl
NHAc
Cl
NHAc
Cl
SO3H
NH2
Cl
SO3H
NH2
Cl
a b c d
e f(a) HNO3/H2SO4 (b) Cl2/AlCl3 , heat (c) H2 , Pd/C (see chap. 4, sec. 4.8.D) (d) Ac2O(e) SO3/H2SO4 (f) aq acid
(c)
ClCl
NO2
Cl
NH2
Cl
OH
a b c d
(a) Cl2 , AlCl3 (b) HNO3 , H2SO4 (c) H2 , Pt (d) NaNO2 , HCl ; H2O (reflux)
+ ortho(separate)
(d)
OH Br CN OOH
ONEt2
a b c d
(a) PBr3 (b) NaCN , DMF (c) 6N HCl , heat (d) 1. SOCl2 2. Et2NH
(e)
NO2NO2
Br
NH2
Br
OH
Br
a b c d
(a) HNO3 , H2SO4 (b) Br2 , AlCl3 (c) H2 , Ni (d) NaNO2 , HCl ; H2O (reflux)
(f)
O
OH
CN
OH
CN
a b
(a) 1. NaCN , THF 2. aq acid (b) 1. PhCO2Na , PPh3 , DEAD 2. saponify
(g)
OH OEta b(a) aq H2SO4 (b) 1. NaH , THF 2. EtI
(h) OH Et
a(a) 1. NaH 2. CS2 3. MeI 4. 200°C
Copyright © 2011 Elsevier Inc. All rights reserved.

Chapter 2 15
(i)
OHBr CN
CO2H
O
NMe2
a
b c
d e
(a) POCl3 , pyridine (b) HBr , h (c) NaCN , DMF (d) H3O+ , heat (e) i. SOCl2 i.. HNMe2
Copyright © 2011 Elsevier Inc. All rights reserved.


















