
The Importance of Phenotype in Genotype-Phenotype Studies in

Page | 1
CentoMD®5.0_Handbook_V5_July2018
Handbook
Precautions/warnings:
For professional use only.
To support clinical diagnosis.

Page | 2
CentoMD®5.0_Handbook_V5_July2018
Contents
Introduction ................................................................................................. 3
Intended use ................................................................................................ 3
Facts and Features ........................................................................................ 4
Technologies used ......................................................................................... 5
Data acquisition and curation policy ................................................................... 7
Database curators .................................................................................. 7
Data acquisition .................................................................................... 7
Curation workflow ................................................................................. 8
Quality status ....................................................................................... 9
Variant-related information ........................................................................... 10
Genetic variants ................................................................................... 10
Variant location ................................................................................... 11
Variant type on DNA level ....................................................................... 11
Coding effect ...................................................................................... 13
Variant zygosity ................................................................................... 14
Allele frequency at CentoMD® ................................................................... 15
Publication status ................................................................................. 15
Clinical significance according to CentoMD® .................................................. 15
Information on disease and inheritance ............................................................. 19
Individual-related information on phenotype and demographics .............................. 20
Clinical statement of CENTOGENE AG ................................................................ 23
Vcf upload and annotation ............................................................................. 24
Input file............................................................................................ 24
Annotated File ..................................................................................... 25
Appendix .................................................................................................. 27
Abbreviations used in CentoMD® ................................................................ 27
Evidence-based annotation rules to determine the clinical statement ................... 27
Glossary .................................................................................................... 32

Page | 3
CentoMD®5.0_Handbook_V5_July2018
Introduction
Diagnosing a patient with a rare disease is a complex task because not all existing genetic
variants have been described or precisely annotated. Medical professionals need to obtain
all available knowledge about the detected genetic variants in a patient in order to
establish the most accurate diagnosis possible.
CentoMD® is a holistic database that combines phenotype and genotype information
gathered from genetic tests conducted at CENTOGENE AG. This means that every variant
reported in CentoMD® is linked to at least one clinically described individual analyzed at
CENTOGENE AG through a standardized workflow with accredited quality. Respectively,
CentoMD® is a growing database; newly generated data will be imported quarterly.
This handbook describes the content of CentoMD®, how this content is generated, how
clinical significance classes are defined, and how quality standards are fulfilled. The
accompanying CentoMD® user guide provides a detailed description of how to use this web
based database.
Intended use
CentoMD® is a browser based software that provides a genetic diagnosis correlating
information from a comprehensive and unique repository of genetic, biochemical (where
available) and clinical information from consented and manually curated patient data sets,
and probands of different geographical backgrounds. It will be only available to medically-
trained professionals for the evaluation of the genetic variants that have been identified
in their own patients. This enhances the validity of the genetic analytical workflow and
providing the health care professional with a result interpretation and variant assertion
recommendation in evaluating treatment options for patients with rare hereditary diseases.
The software will not allow independent review of data generated by the health care
professional. The software is designed to provide a reliable recommendation of clinical
diagnosis.

Page | 4
CentoMD®5.0_Handbook_V5_July2018
Facts and Features
CentoMD® database provides detailed information of variants detected in individuals who
were referred to genetic testing by their physicians in order to evaluate whether they are
affected by or are carriers of variants which cause rare hereditary diseases. This patient
cohort is a unique representation of the global population originating from more than 115
countries. The allele frequencies stated in CentoMD® reflect the frequency observed in this
particular worldwide cohort. For every analyzed individual, CentoMD® provides information
about the genotype-phenotype correlation based on tested clinical cases. Therefore, all
genetic variants are associated to epidemiological data and clinical information – such as
signs and symptoms of the disease – if described by the physician.
CentoMD® 5.0 contains almost 60,000 variants which are classified and curated (see Variant
quality status). In total, more than 3,400 phenotypes and more than 1.3 billion alleles have
been identified in ~180,000 analyzed individuals. The current release contains more than
13,200 HPO (Human Phenotype Ontology) terms and almost 75,000 individuals are linked
with HPO term(s).
CentoMD® 5.0 provides the following key features:
o Advanced Genotype to Phenotype module: Based on approved gene symbols given
by the users, CentoMD® provides detailed data on corresponding genetic variants and
the associated epidemiological data and clinical information following HPO
nomenclature.
o Advanced Phenotype to Genotype module: Based on HPO terms given by the users,
CentoMD® provides hints on candidate genes and related variants underlying the
phenotype of interest.
o Annotation of genetic variants contained in a single sample vcf file with CentoMD®
clinical significance
o 58% of CentoMD® classified and curated clinically relevant (CRV) and uncertain (VUS)
variants are unpublished in the literature: Users can access data of CRV/VUS which
have not been previously described in literature.
o The annotation and classification of genetic variants is strictly curated by medical
professionals: Users have access to high quality data.

Page | 5
CentoMD®5.0_Handbook_V5_July2018
o Explanation of variant classification, statistics and detailed individual-related data
are available: data can be retrieved at 4 different levels: variant rationale, positive
individuals, statistics, and individual view.
Rationale: Summary supporting the clinical significance according to the ACMG
guidelines and internal evidences. In the current release, more than 75,000
variants are linked with rationales.
Positive individuals: Detailed information on individuals tested positive for the
variant of interest.
Statistics: Statistical analyses of individuals tested positive for the variant of
interest.
Individual view: Information on individuals tested positive for the variant of
interest as well as classified and curated CRV/VUS variants associated with each
individual.
o Co-occurrences are indicated: Users can view the association of the variant of
interest with other classified and curated CRV/VUS variants in the same gene or
other genes.
o Interactive search interface: Users are given the flexibility to perform searching,
sorting, filtering and access specific data contents by simple clicks.
o Data export functions: Users can export data for activated variants into read-only
excel file.
o Users are notified via e-mail when activated variants are re-classified: Users get the
latest information on the clinical significance class of variants of interest.
Technologies used
The following validated technologies are used at CENTOGENE AG to detect changes on
genetic levels and to identify the cause of the disease:
o Sanger: Classical method of DNA sequencing, developed by Fred Sanger, using
chemically altered "dideoxy" bases to terminate newly synthesized DNA fragments
at specific bases (either A, C, T or G). These fragments are then size-separated, and
the DNA sequence can be read.
o NGS: Next-Generation Sequencing: High-throughput sequencing technology,
allowing the parallel sequencing of multiple genes, producing thousands or millions
of sequences concurrently.

Page | 6
CentoMD®5.0_Handbook_V5_July2018
o qPCR: Quantitative Polymerase Chain Reaction: Method to amplify and
simultaneously quantify a targeted DNA molecule. Used especially for detecting
large/gross and gene rearrangements.
o MLPA: Multiplex Ligation-dependent Probe Amplification: Variation of the multiplex
PCR that permits multiple targets to be amplified with only a single primer pair.
Used especially for detecting large/gross and gene rearrangements.
o CES: Clinical Exome Sequencing: Application of the next generation technology to
determine the variations of coding regions of genes which have been associated to
human disease.
o WES: Whole Exome Sequencing: Brute-force approach that involves modern day
sequencing technology and DNA sequence assembly tools to piece together all coding
portions of the genome. The sequence is then compared to a reference genome and
any differences are noted.
o WGS: Whole Genome Sequencing: modern day technology for sequencing of the
entire coding and non-coding regions of the genome.
o Other method: Used when another methodology (for example fragment length
analysis) has been employed to detect the variants.
Interpretations of the enzymatic activities and biomarker levels are provided, when
available, as supporting evidence for the relevance of the detected genetic change. For
example, for Fabry disease, which is an X-linked rare genetic lysosomal storage disease,
measurements of enzymatic activities are conducted in males, and measurements of the
biomarker levels are conducted in both males and females.
The terms used to describe the results of biochemical analyses are explained below:
o Biochemical analysis: Method to analyze enzymatic activity or levels of biomarkers
in samples obtained from patients usually suspected of being affected by a metabolic
disorder. This is a test performed via Tandem Mass Spectrometry to detect,
diagnose, and monitor diseases, disease processes, and susceptibility, and to
determine a course of treatment.
o Biomarker interpretation: Evaluation of the biomarker levels compared to the
reference interval
Normal: Biomarker levels are within the normal range (no change).

Page | 7
CentoMD®5.0_Handbook_V5_July2018
Pathological: Biomarker levels are significantly increased compared to the
normal range.
Slightly increased: Biomarker levels are only slightly increased compared to
the normal range.
o Enzyme interpretation: Evaluation of the enzyme activity compared to the reference
interval
Normal: Levels of activity are within the normal range (no change).
Pathological: Levels of activity are significantly decreased compared to the
normal range.
Slightly decreased: Levels of activity are only slightly decreased compared to
the normal range.
Data acquisition and curation policy
Curation is the process of collection, association, update and review of genetic and
phenotypic data of patients genetically analyzed at CENTOGENE AG into a structured and
standardized format. It utilizes a combination of computer-based tools and manual review
in order to assure the accuracy, efficiency and quality of the curation process.
Database curators
CentoMD® curators are biologists with strong background in human genetics. They
continuously undergo extensive training to ensure curation consistency and
standardization. They confirm that CentoMD® is error-free (items properly associated and
interpreted, no inconsistencies, and/or discrepancies against detected observations in-
house and external sources), and close the curation process by manual approval that
reviewed and curated data agree with standard procedures established in-house.
Data acquisition
Data gathering and variant curation are procedures developed and implemented in a web-
based software, that is compliant with the HGNC, HGVS and HPO nomenclatures allowing
collection of variants detected in nuclear coding, nuclear non-coding and mitochondrial
genes. The software integrates in-house sample management systems and analysis
platforms with external databases providing the curator with a comprehensive and

Page | 8
CentoMD®5.0_Handbook_V5_July2018
straightforward overview of the evidences regarding genotype-phenotype correlation
available in-house as well as external information.
The data is gathered by a combination of manual submission and data import following an
individual-oriented model where characteristics belonging to a particular individual
(patient information, clinical data, methodology and detected genetic variants) are stored
and associated together.
Curation workflow
To provide high-quality data, the curation process at CENTOGENE AG is divided in 3 phases:
variant-wise, individual-wise and warnings-wise procedures.
Curation by variant: To begin the curation process, the variant-linked information is
reviewed. This includes approval of variant nomenclature, terminology, accuracy,
consistency, record completeness.
Curation by individual: In order to start curation by individual, all variants detected in this
individual must be approved. It aims at assuring that the entries belonging to an individual
follow the rules for clinical statement closely, and that all associated data is in agreement
with the agreed guidelines. The following factors are considered as critical for the clinical
statement: variant clinical significance, patient genotype (number of clinically relevant
changes, their zygosity and location -i.e. cis vs. trans), inheritance pattern of the disorder,
the sex of the patient (for X-linked diseases), the phenotypic description, and if available
- levels of biomarkers.
Curation by warning: The database generates warnings at different levels (variant,
individual, gene, database levels) to detect errors, invalid terms and nomenclatures,
inconsistencies, and can provide hints where updates and reviews are necessary. Mostly
these warnings are due to additional evidences obtained internally (medical reports issued
at CENTOGENE AG) or detected externally (e.g. additional articles, publications and
external databases). Each warning is manually resolved.
Quarterly, all approved individuals are anonymized and then released to CentoMD®,
offering the most complete and up-to-date information possible to its users.
CentoMD® is a constantly growing and enriched database. Whenever additional evidence
provided by the medical professionals in-house or by peer-reviewed literature becomes
available, the variants are revised and re-classified accordingly. A detailed overview of the

Page | 9
CentoMD®5.0_Handbook_V5_July2018
clinical significance classes captured in CentoMD® is provided in the chapters “Variant-
related information” and “Clinical significance according to CentoMD®”.
Quality status
CentoMD® offers a dataset of variants derived from various technologies of genetic testing
and processed through a standardized workflow which follows international standards and
ensures high data quality. In CentoMD®, different types of variant and individual quality
status are indicated.
There are three types of variant quality status:
o Classified and curated (++): A variant has been assigned to a clinical significance
class based on the confirmed genotype-phenotype associations and curated by
following strictly the ACMG guidelines and internal expertise.
o Classified (+): A variant has been assigned to a clinical significance class according
to ACMG guidelines but has not yet been curated in the context of genotype-
phenotype association.
o Unclassified (0): A variant has not yet been assigned to any clinical significance class
due to the lack of genotype-phenotype associations. Further evaluation is required,
once additional information is available.
There are two types of individual quality status:
o G2P individuals (++): An individual with confirmed genotype to phenotype
association for the gene in question and linked with at least one classified and
curated variant (++) during manual curation process.
o Non-G2P individuals (+): An individual with not yet confirmed genotype to phenotype
association (unresolved individual) for the gene in question during manual curation
process. This individual could not be linked to any classified and curated variant (++)
in the respective gene. Non-G2P individuals are periodically reviewed against the
most updated knowledge in context of genotype-phenotype correlations.

Page | 10
CentoMD®5.0_Handbook_V5_July2018
Variant-related information
Genetic variants
CentoMD® includes germline and de novo genetic variants detected in all types of genes. A
collection of variants detected in nuclear coding, nuclear non-coding and mitochondrial
genes is available. The HGNC-approved gene symbols are used.
A gene is defined by a sequence of DNA that represents a basic unit of heredity, being
expressed in RNA and proteins.
o Nuclear coding: A gene located in the cell nucleus of a eukaryote that encodes for
protein.
o Nuclear non-coding: A gene located in the cell nucleus that does not encode for a
protein product.
o Mitochondrial: A gene located in the mitochondria.
In CentoMD®, each gene is linked with a transcript or reference sequence, i.e. a digital
nucleic acid sequence, assembled by scientists as a representative example of a species'
set of genes. All variant-type annotations provide mapping to genomic coordinates (genome
build hg19). Coding DNA reference sequence refers to a cDNA-derived sequence containing
the full length of all coding regions and non-coding untranslated regions.
According to the reference sequence used, the genetic variants are linked with the
corresponding location within the gene, with a particular mutation type on three different
levels: genomic/mitochondrial, cDNA, and protein, closely following the HGVS guidelines
and recommendations, for both small and gross gene rearrangements.
o Genomic DNA change: Change at gDNA level following numbering based on genomic
DNA reference sequence.
o Coding DNA change: Change at cDNA level following numbering based on coding DNA
reference sequences.
o Protein change: Change at protein level following numbering based on the amino
acid sequence, using one letter amino acid code and X for designating a translation
termination codon.

Page | 11
CentoMD®5.0_Handbook_V5_July2018
Variant location
Variant location refers to the location of the DNA change relative to the transcription
initiation site, initiation codon, polyadenylation site, or termination codon of the
corresponding gene.
o Upstream: The region located 5' (upstream) from the 5'UTR region of the gene.
o 5'UTR (5'-Untranslated Region): Sequences on the 5' end of messenger RNA (mRNA)
but not translated into protein. It extends from the transcription start site to just
before the ATG translation initiation codon. 5' UTR may contain sequences that
regulate translation efficiency or mRNA stability.
o Exon: The protein-coding DNA sequence of the gene.
o Intron: The non-coding region of a gene that interrupt the protein coding regions
(exons).
o 3'UTR (3'-Untranslated Region): Particular section of mRNA that starts with the
nucleotide immediately following the stop codon of the coding region. This region
contains transcription and translation regulating sequences.
o Downstream: The region located 3' (downstream) from the polyadenylation signal of
the gene.
For large deletions/duplications and gene rearrangements, the location is indicated by the
first and the last exon affected by the change (for example, e1_e9 stands for a large
deletion/duplication affecting exon 1 to exon 9). If, for example, only one exon is linked
with a large deletion, this indicates that particular exon is completely removed (see
mutation types below).
Please note that for mitochondrial genes, only the location exon 1 is valid.
Variant type on DNA level
The variant type describes the different types of changes that can occur in the DNA
sequence. The following types are included in CentoMD®:
o Chromosomal deletion: Loss of part of chromosome.
o Complex rearrangement: Involves the structures or number of the chromosomes, it
is referred to as chromosome mutation, or rearrangement, rearranged
chromosomes.

Page | 12
CentoMD®5.0_Handbook_V5_July2018
o Conversion: Non-reciprocal transfer of information between homologous
sequences; one DNA sequence replaces a homologous sequence such that the
sequences become identical after the conversion event.
o Deletion: A sequence change where one or more nucleotides are removed (deleted).
o Duplication: A sequence change where a copy of one or more nucleotides are
inserted directly 3`-flanking of the original copy.
o Gain of methylation: Gain of the normal DNA methylation level.
o Gene & regulatory region(s) deletion: Refers to loss of the entire gene and flanking
regions.
o Gene & regulatory region(s) duplication: Refers to the gain of the entire gene and
flanking regions.
o Gene deletion: Refers to loss of the entire gene.
o Gene duplication: Refers to gain/duplication of the entire gene.
o Gross deletion: Refers to loss of part(s) of a gene.
o Gross duplication: Refers to gain/duplication of part(s) of a gene.
o Gross inversion: Refers to 180-degree inversion of part(s) of a gene.
o Insertion/Deletion (Indel): Refers to a sequence change that includes a combination
of both insertions and deletions.
o Insertion: A sequence change where one or more nucleotides are added (inserted)
into a DNA sequence, or it may involve portions of a chromosome.
o Inversion: Chromosomal abnormality where a segment of a chromosome is rotated
180 degrees and reinserted.
o Loss of methylation: Loss of the normal DNA methylation level.
o Pathological allele (D4Z4 motif): Deletion of 3.3-kb repeats from a chromosomal
tandem repeat called D4Z4 located near the end of chromosome 4 at the 4q35-ter
location. D4Z4 contains an ORF encoding a putative homeobox protein called DUX4,
a large polymorphic repeat structure consisting of 1–100 KpnI units.
o Repeat expansion: Refers to an increase number of a genomic tandem repeated DNA
sequence.
o Retrotransposon insertion: Retrotransposons (also called transposons via RNA
intermediates) are genetic elements that can amplify themselves in a genome, and
can induce mutations by inserting near or within genes. Retrotransposon-induced

Page | 13
CentoMD®5.0_Handbook_V5_July2018
mutations are relatively stable, because the sequence at the insertion site is
retained as they transpose via the replication mechanism.
o Substitution: A sequence change where one nucleotide is replaced by one other
nucleotide. Substitutions are described using a ">" character (indicating "changes
to").
o Other/complex: Refers to all other types not included in any already mentioned
category above.
Coding effect
The coding effect describes the sequence changes at protein level. The following types are
distinguished:
o Effect unknown: The coding effect on protein level has not been analyzed. An effect
is expected but difficult to predict.
o Frameshift: A sequence change caused by deletion/insertion of nucleotides affecting
an amino acid between the first (initiation, ATG) and last codon (termination, stop),
replacing the normal C-terminal sequence with one encoded by another reading
frame.
o Increased polyglutamine tract/expanded polyQ: Portion of a protein consisting of a
sequence of several glutamine (Glu; Q) units.
o In-frame: A sequence change that does not cause a shift in the triplet reading frame.
As a result, one or more amino acids are added, deleted or replaced by one or more
other amino acids.
o Missense: A single nucleotide change that results in a codon that codes for a different
amino acid. Not all missense mutations are deleterious; some changes can have no
effect. Because of the ambiguity of missense mutations, it is often difficult to
interpret the consequences of these mutations in causing disease.
o New translation initiation site: A change affecting the translation initiation codon
(Met-1) introducing a new upstream initiation codon extending the N-terminus of the
encoded protein.
o Non-coding: The change on DNA level that has no effect on protein, or the effect of
regulatory mutations is unknown.
o Nonsense: A sequence change that results in a premature stop codon, and in a
truncated, incomplete protein product.

Page | 14
CentoMD®5.0_Handbook_V5_July2018
o Silent: A sequence change that does not result in a change of amino acid and
functional change of the protein product.
o Splicing mutation: A sequence change that affects the splicing process (i.e. intron
removal and exons joining). Splice-site mutations occur within genes in the
noncoding regions (introns) just next to the coding regions (exons). Splice-site
mutations can eliminate an existing donor or acceptor site, which will cause an exon
to be skipped and possibly result in a frameshift.
o Start loss: A sequence change in the ATG start codon that prevents the original start
translation site from being used. This kind of mutation may eliminate gene function.
o New translation termination codon: A sequence change that affects the translation
termination codon (Ter/*) introducing a new downstream termination codon,
extending the C-terminus of the encoded protein.
Variant zygosity
Zygosity indicates if a variant is detected on one chromosome or on both chromosomes and
therefore describes the degree of similarity of the alleles for a trait in an organism.
The following zygosities are included in CentoMD®:
o Heterozygous (Het): Gene locus when cells contain two different alleles of a gene.
o Homozygous (Hom): Gene locus when identical alleles of the gene are present on
both homologous chromosomes.
o Hemizygous (Hem): Used for alleles detected in genes located on X-chromosome for
male cases.
For the mitochondrial variants, the zygosity must be read as the degree of heteroplasmy,
i.e. as a mixture of more than one type of mitochondrial DNA (mDNA) within a
cell/individual. In those cases, where a variant in mDNA is responsible for a disease, the
larger the proportion of mutant mitochondria, the more likely the person will show
symptoms of the disease.
Two degrees of heteroplasmy are included:
o Heteroplasmic: The cell has some mitochondria that have a mutation in the mDNA
and some that do not.
o Homoplasmic: The cell has a uniform collection of mDNA: either completely normal
mDNA or completely mutant mDNA.

Page | 15
CentoMD®5.0_Handbook_V5_July2018
Allele frequency at CentoMD®
This term indicates the number of observations of the allele of interest at a particular locus
in CENTOGENE AG-unique population, expressed as decimal.
Publication status
The publication status indicates if the identified variant has previously been published in
the literature or not. For published variants PubMed identifier (PMID) is indicated.
Additionally, the Single Nucleotide Polymorphism Database (dbSNP) ID is provided, if
available. The dbSNP is an archive of genetic variations within and across different species
developed and hosted by the National Center for Biotechnology Information (NCBI) in
collaboration with the National Human Genome Research Institute (NHGRI) and available
to the public.
Clinical significance according to CentoMD®
In CentoMD®, based on the likelihood to predispose to or to cause the observed
phenotype/disease, the detected genetic variants are classified into one of the three
groups: clinically relevant variants (CRV), clinically irrelevant variants (CIV) and variants of
unknown significance (VUS)/uncertain variants/predicted uncertain variants. The CRVs
include the following classes: pathogenic, likely pathogenic, risk factor, modifier and
premutation. The CIVs involve neutral, likely neutral, disease-associated polymorphisms,
Centogene (likely) neutral - published as (likely) pathogenic and mutable normal
(intermediate) (see Figure 1).
For classified and curated variants (++) the classification of genetic germline variants is
done according to the ACMG guidelines which define five classes: pathogenic, likely
pathogenic, uncertain significance, likely neutral and neutral (class 1-5; Richards et al.
(2015), Genet. Med., doi:10.1038/gim2015.30). In addition to the 5 classes specified by
ACMG, CentoMD® also annotates variants as risk factors, modifiers, premutations, disease-
associated polymorphisms, mutable normal (intermediate) and the CentoMD-specific
clinical significance class Centogene (likely) neutral-published as (likely) pathogenic.
Additionally, some modifications of the ACMG guidelines are applied. These modifications
arise from our continuously growing internal expertise in the field of molecular diagnostics
and are represented mainly by new evidences regarding internal observed frequencies,

Page | 16
CentoMD®5.0_Handbook_V5_July2018
segregation data, genotype-phenotype correlation, co-occurrence, enzymatic and
biomarker levels. The adjustments to the ACMG recommendations are specified below.
Figure 1: Classification of genetic variants in CentoMD®. The classification rules determining the clinical significance of a genetic variant are provided in the text. CG: CENTOGENE
Classification as pathogenic is additionally assigned to:
Loss of function (LOF) variants which are associated with pathologically decreased
biochemical levels/ activities.
Non-LOF variants which are associated with pathologically decreased biochemical
levels/ activities and where sufficient clinical information of the associated
individual clearly supports the presence of the metabolic disease.
Classification as likely pathogenic is additionally assigned to:
All variants
Clinically relevant variants(CRV)
Pathogenic
Likely pathogenic
Risk factor
Modifier
Premutation
Variant of unknownsignificance (VUS)
Clinically irrelevant variants(CIV)
CG (likely) neutral – published as(likely) pathogenic
Disease-associated polymorphism
Likely neutral
Neutral
Mutable normal (intermediate)

Page | 17
CentoMD®5.0_Handbook_V5_July2018
LOF-variants detected in the genes related to metabolic disorders with no
biochemical evidences.
Non-LOF-variants found in an individual for whom pathological biochemical data is
supporting but insufficient clinical information was provided to confirm the presence
of the disease.
Risk factors and modifiers are classified based on their distinct manner to influence the
presence or the severity of the disease. To be included in the sub-class of risk factors,
variant should be reported as altering the risk for a disease by influencing function of other
proteins. A modifier is a variant that operates through influencing gene expression and
affects severity of the phenotype but alone is not sufficient to cause the disease.
Premutation is a repeat expansion variant in a range that may not result in the clinical
manifestation of the associated disease in the carrying individual, but that may result in
the manifestation of the disease in the offspring due to potential repeat instability.
A variant is classified as uncertain, when available information is not sufficient to state
pathogenicity. For example, in case of metabolic disorders, novel variants, which are non-
LOF and additionally are associated with inconclusive biochemical data, are annotated as
uncertain.
Variants are classified as neutral and likely neutral based on their high frequency in
population(s), no observed impact on disease presence/severity/susceptibility, or non-
segregation and/or co-occurrence detected, etc.
A mutable normal (intermediate) variant is meiotically unstable and not convincingly
associated with an abnormal phenotype. Because of the instability of alleles in the mutable
normal range, an asymptomatic individual with a mutable normal allele may be predisposed
to having a child with an expanded allele.
The class of disease-associated polymorphism includes variants related to complex,
multigenic disorders with no clear Mendelian inheritance. In order to be classified as
disease-associated polymorphism, variants must have a maximum frequency of 5% in public
databases and the association should be replicated by at least 2 independent studies or in
1 study with functional evidence.
When the internal evidence regarding the clinical significance of a variant is inconsistent
compared to other external sources, the class “CENTOGENE (likely) neutral - published as
(likely) pathogenic” is used in order to emphasize the importance of this observation. This
class of clinical significance is used only to genetic variants of high penetrance. The variants

Page | 18
CentoMD®5.0_Handbook_V5_July2018
associated with this clinical significance class are reclassified based on internal evidences
only. When a variant is reclassified based on external available information, the correct
clinical class is neutral/likely neutral, following strictly the corresponding definitions (see
schematic representation below, Figure 2).
Internal evidences refer to at least one of the following criteria:
Is the DNA change found at Centogene at a frequency above the reported incidence
of its associated disease?
Is the DNA change identified in healthy/ asymptomatic individuals?
Does the DNA change not segregate with the disease in our identified families, or
among independent individuals?
Does the DNA change co-occur with deleterious variants (in the same gene or other
genes) in screened individuals?
Does any other evidence support a likely neutral pathogenicity (like enzymatic
activities or biomarker levels)?
Figure 2: Schematic representation of reclassification of pathogenic variants
Classification of (+) genetic germline variants (not curated, automatically classified), five
ACMG criteria (BA1- stand alone, BP6, PVS1, PM2, and PP5; for more information please

Page | 19
CentoMD®5.0_Handbook_V5_July2018
see
https://www.acmg.net/docs/standards_guidelines_for_the_interpretation_of_sequence_
variants.pdf) are applied. Variants with allele frequency higher than 5% in any of the
following database are automated classified as predicted neutral: gnomAD, ExAc, ESP,
1000Genome or CentoMD.
Rare variants where PVS1 applies (null variants) and / or reputated sources have been
already reported as pathogenic, are automated classified as predicted pathogenic.
Rare variants where conflicting criteria identified, or not enough other evidences at hand,
are classified automatically as predicted uncertain.
Variant re-evaluation and re-classification is a key feature of CentoMD® and performed
regularly in the light of literature, publicly available clinical databases and most important,
based on CENTOGENE AG’s own continuously growing and improving proprietary
information.
Information on disease and inheritance
Every genetic disorder which has been suggested or suspected by the physician is described
according to the Online Mendelian Inheritance in Man® (OMIM®) catalog. OMIM® was
developed for the world-wide-web by NCBI and contains a list of human genes and genetic
diseases with links to other relevant resources (http://www.ncbi.nlm.nih.gov/omim).
Every entry in OMIM® has a unique identifier, which is also captured in CentoMD®.
Each genetic disorder is linked with the observed mode of inheritance (MOI). MOI is defined
by the manner in which a particular genetic trait or disorder is passed from one generation
to the next. The following MOIs are included in CentoMD®:
o Autosomal dominant (AD): The pattern of inheritance in which an affected individual
has one copy of a mutant gene and one copy of normal gene on a pair of autosomal
chromosomes.
o Autosomal recessive (AR): The pattern of inheritance in which both copies of an
autosomal gene must be abnormal for a genetic condition or disease to occur.
o Digenic (Di): The pattern of inheritance that is similar to recessive inheritance,
except that the trait only develops when mutations are found in one copy of each of
the two independent genes simultaneously.
o Imprinting/Epigenetic (Imp/Epi): The pattern of inheritance by mechanisms not
directly involving nucleotide sequences, but paramutations and parental imprinting.

Page | 20
CentoMD®5.0_Handbook_V5_July2018
o Mitochondrial (Mito): The pattern of inheritance of a trait encoded in the
mitochondrial genome.
o Multifactorial (MF): The pattern of inheritance caused by the interplay between
genetic factors and environmental factors.
o Pseudoautosomal dominant (P-AD): The inheritance pattern seen with genes in the
pseudoautosomal region of the X and Y chromosome that can exchange regularly
between the two sex chromosomes. Alleles for genes in the pseudoautosomal region
can show male-to-male transmission, and therefore mimic autosomal inheritance,
because they can cross over from the X to the Y chromosome during male
gametogenesis and be passed on from a father to his male offspring.
o X-linked (X): The mode of inheritance of a trait encoded in the X chromosome.
o Y-linked (Y): The pattern of inheritance that may result from a mutant gene located
on the Y chromosome. By definition, only males are affected.
o Unknown (?): This mode of inheritance is selected for genes not yet associated with
any pathological condition or disease, therefore no pattern of inheritance has been
observed.
Individual-related information on phenotype and demographics
All patient data in CentoMD® are fully anonymized. The following epidemiological and
clinical data are reported for individuals associated with classified and curated CRV and/or
VUS in CentoMD®:
o Random patient ID: Unique identifier assigned to each consented individual in
CentoMD®.
o Finding: Indicates if a variant is related to the indication for testing. Primary findings
are variants related to the indication for testing. Secondary (incidental) findings are
derived from whole exome sequencing (WES) and are pathogenic or likely pathogenic
variants identified in 59 genes recommended by ACMG for reporting of secondary
findings in clinical exome and genome sequencing (Genetics in Medicine, 2017).
Secondary findings are unrelated to the indication for testing.
o OMIM® disease: A number/identifier given by OMIM® to phenotype/disease. For
example, OMIM® disease 230800 stands for Gaucher disease, type I.

Page | 21
CentoMD®5.0_Handbook_V5_July2018
o MOI: Mode of inheritance: It is the manner in which a particular genetic trait or disorder
is passed from one generation to the next.
o Anonymized random family number (ARFN): Unique family number used to keep all
family members together when relationship links are provided.
o Pedigree: Indicates the connection/relation among individuals by blood, marriage, or
adoption in relation to the index patient. Based on the ARFN and the relationships within
one family, it is possible to reconstruct the family trees accordingly. In each family, the
index patient is indicated. The index patient represents the affected individual through
whom the family with a genetic disorder is first diagnosed.
o Sex: Indicates the biological state of the individual of being male, female, intersex,
unknown sex (when no information was provided or a prenatal case was analyzed).
o Age: Age at diagnosis. It is calculated as date of sample entry at CENTOGENE AG minus
date of birth, and is expressed in years. For patients referred to CENTOGENE AG several
times, the date of the first order entry is used by default to calculate the age at
diagnosis.
o Country: Country of sample origin. It indicates the area of the world the patient is
coming from. The basis for this information is the country from which the sample has
been sent to CENTOGENE AG. If physician provides information about the ethnicity of
the patient (e.g. Canadian citizen of German origin), then this (in this case Germany) is
the country selected in this situation.
o Region: Continental region the sample is coming from.
o Clinical information (HPO terms): Description of features and characteristics that the
corresponding physician has provided as supporting evidence of the presence of a
particular disease translated into the vocabulary defined by the HPO
(http://www.human-phenotype-ontology.org/).
Sometimes it is not possible to describe the clinical picture accurately, because the
details are not given by the physician or only general assumptions have been made.
Such cases are documented in CentoMD® in the following manner:
No info/unknown: when no clinical information has been provided;
Healthy/asymptomatic: when the physician has explicitly indicated that the
person is healthy, asymptomatic, or not affected;

Page | 22
CentoMD®5.0_Handbook_V5_July2018
Suspected/affected: when only very general statements are provided by the
physician (e.g. “patient suffering from Fabry disease” or “clinical features of
Marfan”).
o Variant zygosity: Indication if the variant is detected on one chromosome or on both
chromosomes.
o Total number of variants: Total number of detected variants for this case (clinically
relevant; clinically irrelevant) on this particular gene. For example, “10 (1 ; 9)” is to be
interpreted as follows: the total number of variants that were identified in this
proband/patient for this particular gene is 10, one of them is clinically relevant, while
9 are clinically irrelevant variants.
o Genotype: Genetic constitution of a case with respect to the number of alleles and their
clinical significance for this particular gene.
o Enzyme interpretation: Interpretation of the enzyme activity compared to the
reference interval.
o Biomarker interpretation: Interpretation of biomarker levels compared to the reference
interval.
o Clinical statement: The finding or the conclusion of the molecular genetic test
conducted at CENTOGENE AG.
o Sample type: Type of sample sent to CENTOGENE AG for testing. It includes DNA, Blood,
dry blood spot (DBS) or other (e.g. amniotic fluid).
o Age at onset: Refers to the age at which an individual acquires, develops, or first
experiences a condition or symptoms of a disease or disorder.
o Carrier testing: Indicates if the individual was interested in performing a carrier
screening when the presence of specific genetic variant was detected already in other
family members.
o Consanguineous parents: Refers to the marriage between two genetically related
persons.
o Family history: Indicates the presence or the absence of a particular disorder or
symptomatology in blood relatives of a patient.
o Detailed family history: Detailed description of disorders from which direct blood
relatives of the patient have suffered.

Page | 23
CentoMD®5.0_Handbook_V5_July2018
Clinical statement of CENTOGENE AG
The clinical statement is the finding or the conclusion of the molecular genetic test
conducted at CENTOGENE AG. The clinical statement may confirm or disprove the
suspected diagnosis, or serve to elucidate the genetic cause of an uncertain or questionable
condition or disease. When deriving the clinical statement, the following factors are
considered:
o Mode of inheritance of the disorder
o Patient’s genotype
o Clinical significance of all identified genetic variants
o Clinical data provided, if available
o Additionally, sex and/or biochemical evidences, if applicable
The evidence-based rules determining the clinical statement are summarized in Table 1
and Figure 3. The following clinical statements are used in CentoMD®:
o Affected: Individual with confirmed diagnosis at genetic level.
o Probably affected: Fabry male patients carrying an uncertain variant associated with
pathological enzymatic levels, but biomarker levels are within normal range.
o At least carrier: Individual with clinical suspicion most likely confirmed at genetic
level. It includes individuals carrying in trans a pathogenic variant with an uncertain
variant in case of autosomal recessive mode of inheritance, or Fabry females
carrying uncertain variants.
o Probably carrier: Individual carrying an uncertain variant in the context of autosomal
recessive disorders or X-linked disorders (in this last situation it applies only to
female individuals).
o Carrier: Individual who inherited one mutated allele at genetic level in case of
autosomal recessive mode of inheritance, or female in case of X-linked mode of
inheritance.
o Increased risk of developing the disease: Individual with confirmed susceptibility at
genetic level to develop a particular medical condition.
o Increased risk of having affected offspring: Individual who carries a premutation
variant and who may not be clinically affected of the disease himself, but who has
a higher risk of having an affected offspring due to potential repeat instability.

Page | 24
CentoMD®5.0_Handbook_V5_July2018
o Not determined: Individual carrying uncertain variant(s) where clear statement on
either presence or susceptibility to develop a particular disease was not possible.
o Unaffected: Indicates an individual where the susceptibility of the disease was not
confirmed at genetic level.
For example, for an autosomal dominant disorder where the patient’s genotype is
heterozygote, meaning he carries one clinical relevant variant, the expected clinical
statement is either “Affected” or “Increased risk of developing the disease” (according to
the provided clinical information).
Vcf upload and annotation
This functionality refers to annotation of genetic variants detected within genes confirmed to be
associated or cause human diseases / conditions, according to CentoMD®. A single vcf- file is
uploaded, and the genetic variants are subjected to two different approaches:
i) Variants which are identified in CentoMD® are annotated and classified according to the
current version, following the 5 standard ACMG classes (pathogenic, likely pathogenic,
uncertain, likely neutral and neutral) for classified and curated variants (++) and
predicted pathogenic, predicted uncertain and predicted neutral for classified variants
(+).
ii) Variants that are not yet identified in CentoMD® are subjected to on the fly
annotation using Variant Effect Predictor (VEP, see
https://www.ensembl.org/info/docs/tools/vep/index.html) and automatically
classified using the process described above for classified variants (+).
Input file
The input file must be in Variant Call Format (VCF). CentoMD® supports VCF v.4.1 and later
on hg19 genome assembly. For specification see https://samtools.github.io/hts-
specs/VCFv4.1.pdf.
The input file should contain the 8 fixed mandatory columns ('#CHROM', 'POS', 'ID', 'REF',
'ALT', 'QUAL', 'FILTER', 'INFO'), followed by a 'FORMAT' column and then at least one column
containing sample-specific genotype data. The genotype (GT) at every site is mandatory.
If it is not found, the respective variant call will be excluded. Acceptable formats are values

Page | 25
CentoMD®5.0_Handbook_V5_July2018
separated by forward slash (e.g., 0/1) or pipe (e.g., 0|1). The sample in the 10th column
is considered the "active sample". That is, if the file contains additional columns with
genotype data of additional samples or for the same sample but made from additional
variant callers, they will be ignored. The maximum allowed file size is 100MB. Bigger files
will not be accepted. The function does not support multi-sample vcf files.
Annotated File
Once the annotation of the uploaded vcf file is complete the annotated file is created for
download by the user. The annotated file will be provided as a csv file. It contains the
following information:
- Genomic position: The 1-based position of the variation on the given sequence on genome
build hg19
- Ref: The reference base (or bases in the case of an indel) at the given position on the given
reference sequence.
- Alt: The list of alternative alleles at this position.
- Gene: A gene is defined by a sequence of DNA that represents a basic unit of
heredity, being expressed in RNA and proteins. In CentoMD® the HGNC-approved
gene symbols are used. Every gene-variant combination is represented by one entry.
In case a variant maps on more than one gene it will be included more than once.
- Transcript: Coding DNA reference sequence refers to a cDNA-derived sequence
containing the full length of all coding regions and non-coding untranslated regions.
CentoMD® uses RefSeq transcripts (genome build hg19) and only one transcript per
Gene-variant combination will be included. When several transcripts are available
for a single gene the selection of the transcript displayed is done as follows: 1.
Transcript where the variant has the highest impact, 2. Longest transcript, 3.
Transcript with the most number of exons, 4. Length of genomic locus, 5. Transcript
where the variant falls within an exon rather than the transcript where the variant
falls into a non-coding region.
- Coding DNA change
- genomic DNA change
- protein change
- location
- variant type on DNA level

Page | 26
CentoMD®5.0_Handbook_V5_July2018
- coding effect
- clinical significance according to CentoMD® or the predicted clinical significance
- HGMD accession number
- ClinVar classification, comma separated if more than one classification exists for the
variant
Page | 27
CentoMD®5.0_Handbook_V5_July2018
Appendix
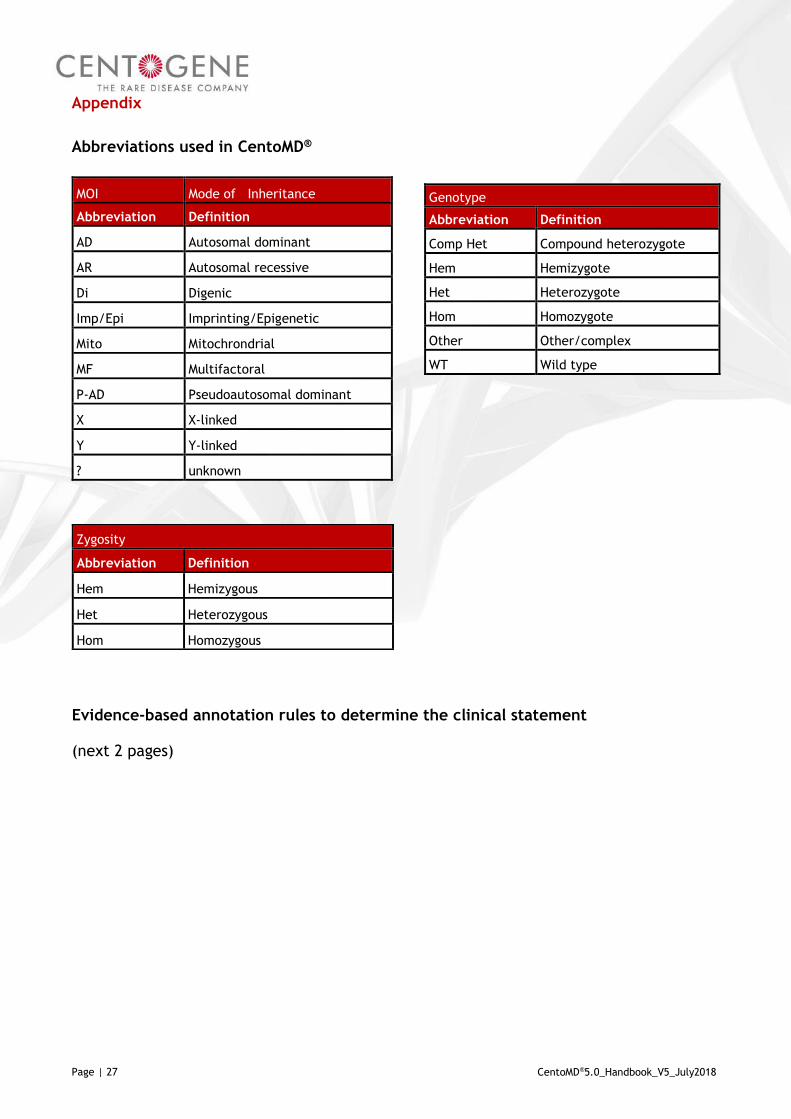
Abbreviations used in CentoMD®
Evidence-based annotation rules to determine the clinical statement
(next 2 pages)
MOI Mode of Inheritance
Abbreviation Definition
AD Autosomal dominant
AR Autosomal recessive
Di Digenic
Imp/Epi Imprinting/Epigenetic
Mito Mitochrondrial
MF Multifactoral
P-AD Pseudoautosomal dominant
X X-linked
Y Y-linked
? unknown
Genotype
Abbreviation Definition
Comp Het Compound heterozygote
Hem Hemizygote
Het Heterozygote
Hom Homozygote
Other Other/complex
WT Wild type
Zygosity
Abbreviation Definition
Hem Hemizygous
Het Heterozygous
Hom Homozygous

Page | 28
CentoMD®5.0_Handbook_V5_July2018
Genotype1)
MOI2)
Significance3)
Significance 23)
CI4)
Clinical statement
AD
AR
X-linked7)
Path5)
VUS6)
Path5)
VUS6)
- + ?
Hom/
Hem
x
x
x
increased risk
x
x
x
affected
x
x
x affected/increased risk
x
x
x
not determined
x
x
x
not determined
x
x
x not determined
x
x
x
increased risk
x
x
x
affected
x
x
x affected
x
x
x
not determined
x
x
x
not determined
x
x
x not determined
x x
x
increased risk
x x
x
affected
x x
x affected/increased risk
x
x
x
not determined
x
x
x
not determined
x
x
x not determined
Het
x
x
x
increased risk
x
x
x
affected
x
x
x affected/increased risk
x
x
x
not determined
x
x
x
not determined
x
x
x not determined
x
x
x
carrier
x
x
x
carrier
x
x
x carrier
x
x
x
probably carrier
x
x
x
probably carrier
x
x
x probably carrier
x x
x
carrier
x x
x
carrier
x x
x carrier
x
x
x
not determined
x
x
x
not determined
x
x
x not determined

Page | 29
CentoMD®5.0_Handbook_V5_July2018
Table 2: Evidence-based annotation rules to determine the clinical statement at CentoMD®. See Figure 3 for further illustration of the decision process.
1) The most often detected annotation classes are included. The wild type genotype is excluded. For wild type the clinical statement is “Unaffected”. 2) Mode of Inheritance. 3) Indicates the clinical significance of the identified variant. 4) Clinical information:
- indicates the absence of signs and symptoms of the disease (i.e. healthy/unaffected); + indicates the presence of signs and symptoms of the disease: ? indicates that no clinical information was provided.
5) Refers to a variant annotated as pathogenic, likely pathogenic, modifier or risk factor. 6) Uncertain variant. 7) Two X-linked diseases (i.e. Fabry disease and Hunter disease) do not follow these definitions closely, as additional information is available and used as a decision factor when selecting the finding. For these two diseases, please see the decision trees presented in Figure 4.
Comp Het
x
x
x
x
increased risk
x
x
x
x
affected
x
x
x
x affected/increased risk
x
x
x x
increased risk
x
x
x
x
affected
x
x
x
x affected/increased risk
x
x
x x
not determined
x
x
x
x
not determined
x
x
x
x not determined
x
x
x
x
increased risk
x
x
x
x
affected
x
x
x
x affected
x
x
x x
at least carrier
x
x
x
x
at least carrier
x
x
x
x at least carrier
x
x
x x
not determined
x
x
x
x
not determined
x
x
x
x not determined
x x
x
x
increased risk
x x
x
x
affected
x x
x
x affected
x x
x x
at least carrier
x x
x
x
affected
x x
x
x at least carrier
x
x
x x
not determined
x
x
x
x
not determined
x
x
x
x not determined

Page | 30
CentoMD®5.0_Handbook_V5_July2018
Figure 3: Decision trees that illustrate the evidence-based annotation rules which determine the clinical statement at CentoMD® The decision levels illustrated are: MOI – Genotype – Clinical significance (variant effect) – Clinical information – Clinical statement (the
caption of Table 1 also applies to this figure).

Page | 31
CentoMD®5.0_Handbook_V5_July2018
Figure 4: Decision trees that illustrate the evidence-based annotation rules which determine the clinical statement for Fabry and Hunter disease. The decision levels illustrated are: MOI – Genotype – Clinical significance (variant effect) – Clinical information – Clinical
statement (the caption of Table 1 also applies to this figure)
Fabry disease Hunter/MPS2 disease

Page | 32
CentoMD®5.0_Handbook_V5_July2018
Glossary
Term Explanation
Allele One of two (or more) forms of a gene/genetic locus.
Allele frequency at CentoMD®
Indicates the number of observations of the allele of interest at a particular locus in CentoMD-unique population, expressed as decimal.
Biochemical analysis
Method to analyze enzymatic activity or levels of biomarkers in samples obtained from patients, usually suspected being affected by a metabolic disorder. This is a test performed to detect, diagnose and monitor diseases, disease processes, susceptibility and determine a course of treatment.
Data File Upload-VCF
Variant Call Format: the format of a text file used in bioinformatics for storing gene sequence variations.
Degree of heteroplasmy
Mixture of more than one type of mitochondrial DNA (mDNA) within a cell/individual. In those cases where a mutant mDNA is responsible for a disease, the larger the proportion of mutant mitochondria, the more likely the person will show symptoms of the disease.
Degree of heteroplasmy-Heteroplasmic
Cell has some mitochondria that have a mutation in the mDNA and some that do not.
Degree of heteroplasmy-Homoplasmic
Cell has a uniform collection of mDNA: either completely normal mDNA or completely mutant mDNA.
Disease
Particular abnormal, pathological condition that affects part or all of an organism. It is often construed as a medical condition associated with specific symptoms and signs.
Disease name
Name of a disease according to Online Mendelian Inheritance in Man (OMIM) database.
Gene
Sequence of DNA that represents a basic unit of heredity, being expressed in RNA and proteins.
Gene symbol A unique abbreviation for the gene name assigned by the HUGO Gene Nomenclature Committee (HGNC).
Gene-cDNA
DNA that is synthesized from a messenger RNA template; the single-stranded form is often used as a probe in physical mapping.
Gene-mDNA
An extranuclear double-stranded DNA found exclusively in mitochondria that in most eukaryotes is a circular molecule and is maternally inherited.
Gene-Mitochondrial A gene located in the mitochondria.
Gene-Nuclear coding
A gene located in the cell nucleus of a eukaryote that encodes for protein.
Gene-Nuclear non-coding
A gene located in the cell nucleus that does not encode for a protein product.
Genotype to Phenotype module
Tool allowing search initiation using approved gene symbols. The corresponding genetic variants are associated with epidemiological data and clinical information following the HPO nomenclature.
Genotype to Phenotype module-Gene statistics-Screened individuals
Indicates the total number of individuals screened at genetic level.

Page | 33
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
HGVS
Human Genome Variant Society that promotes: i) collection, documentation and distribution of genomic variation information and associated clinical variations; ii) guidelines and recommendations for mutation and gene nomenclature (http://www.hgvs.org/).
HGVS nomenclature
Standardized system recommended by HGVS to describe and document variant sequences.
HPO term
Phenotypic description of individuals provided by medical experts and translated into the vocabulary defined by the HPO.
Individuals-Analyzed individuals
Indicates the screened individuals at genetic level, under General statistics
Individuals-G2P individuals
Indicates the screened and consented individuals where the manual curation confirmed the genotype-phenotype correlation.
Individuals-Non-G2P individuals
Indicates the screened and consented individuals where the manual curation could not confirm any genotype-phenotype correlation. These individuals are periodically reviewed in context of genotype-phenotype correlations.
Manual curation
Manual review of submitted entries to identify typing, mis-selection and omission errors. This process ensures that all collected items are properly documented, associated and interpreted.
Mutation
Rare difference and permanent change in a DNA sequence or gene at a given locus. In medical genetics it is often used to indicate a disease-causing allele.
OMIM
Online Mendelian Inheritance in Man: Database which contains a list of human genes and genetic diseases with links to other relevant resources, developed for the world-wide-web by NCBI (http://www.ncbi.nlm.nih.gov/omim).
Phenotype to Genotype module
Tool allowing search initiation using valid HPO terminology. Using the population of similar cases sharing the HPO terms, hints on the potential candidate genes explaining the particular phenotype are provided.
Phenotype to Genotype module-Search for similar cases-Candidate genes
Represent the most potential genes associated with a particular combination of HPO terms (phenotype). Candidate genes linked with similar cases that are within 25% of the highest similarity score are displayed.
Phenotype to Genotype module-Search for similar cases-Case ID Random patient ID referring to a consented case.
Phenotype to Genotype module-Search for similar cases-HPO ID Unique HPO identifier for the attributed HPO term.
Phenotype to Genotype module-Search for similar cases-HPO name
Phenotypic description of individuals provided by medical experts and translated into the vocabulary defined by the HPO.
Phenotype to Genotype module-Search for similar cases-P-value
Defines the likeliness of obtaining the corresponding similarity score or higher by accident. The p-value is calculated by comparing individuals with random symptoms and their similarity scores. The p-value reasons over the similarity score distribution. The higher the p-value, the more likely it is to obtain the corresponding similarity score by accident. The p-value ranges from 0 to 1, where 0 is best.
Page | 34
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Phenotype to Genotype module-Search for similar cases-Shared HPO terms
Indication how many HPO terms of a case analyzed at CENTOGENE match the HPO terms provided by the users.
Phenotype to Genotype module-Search for similar cases-Similarity score
Phenotypic semantic similarity measure based on the HPO. The similarity score of two patients is a formal measure of their resemblance with respect to their standardized symptoms. The score is calculated by a pairwise comparison between each symptom of the two patients. The higher the score, the more similar the patients.
Phenotype to Genotype module-Similar cases
The cases analyzed at CENTOGENE which match the HPO terms provided by the user. Displayed are all cases that have a similarity score of 1 or higher.
Positive individual Indicates an individual carrying a particular genetic variant.
Statistics-Carrier Individual who has only one copy of a genetic variant for a recessive disease.
Statistics-Case
Indicates an individual where the diagnosis was confirmed by genetic testing at CENTOGENE.
Statistics-Geographical region
Indicates the area of the world the patient is coming from. The basis for this information is the region where the patient lives.
Statistics-Wildtype Represents a person carrying only normal genetic variations.
Variant A sequence variation in a gene.
Variant-Alt The list of alternative alleles at this position
Variant-cDNA change
Change at cDNA level following numbering based on coding DNA reference sequences.
Variant-Classified and curated
A variant which has been assigned to a clinical significance class based on confirmed genotype-phenotype association and curated by strictly following the curation workflow.
Variant-Clinical significance according to CentoMD®
Indicates the likelihood of this variant to predispose to or to cause the disorder.
Variant-Clinical significance -Disease associated polymorphism (DP)
Variant reported to be significantly associated with a phenotype/disease.
Variant-Clinical significance-CENTOGENE (likely) neutral - published as (likely) pathogenic
Variant published consistently in literature as (likely) pathogenic but re-classified as (likely) neutral based on internal evidences (observed allele frequency, family segregation studies, co-occurrence with other deleterious genetic variants, etc.). This class is used only to genetic variants of high penetrance.
Variant-Clinical significance-Likely neutral
Variant reported to be likely neutral, prediction software indicates a probably not pathological effect, and or high frequency in population observed. This classification class is equivalent to "likely benign".
Variant-Clinical significance-Likely pathogenic
Variant with probable pathogenicity, or the effect on the protein function is predicted to be likely deleterious (>90% probability to cause the disease).
Variant-Clinical significance-Modifier Variant that can alter the expression of another gene in the phenotype of an individual.
Variant-Clinical significance-Mutable normal (intermediate)
Variant that is meiotically unstable and not convincingly associated with an abnormal phenotype. Because of the instability of alleles in the mutable normal range, an

Page | 35
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
asymptomatic individual with a mutable normal allele may be predisposed to having a child with an expanded allele.
Variant-Clinical significance-Neutral
Variant reported not to influence the disease risk of the individual, or predicted to be neutral based on the high frequency in population, no effect on protein or regulatory regions. This classification class is equivalent to "benign".
Variant-Clinical significance-Pathogenic Variant that is known to cause the phenotype/disease.
Variant-Clinical significance-Pathological D4Z4 allele
Large, polymorphic repeat structure associated with a rough and inverse relationship between clinical severity and the residual repeat size, with the smallest repeats causing the most severe phenotype.
Variant-Clinical significance-Predicted neutral
Variant predicted not to influence the disease risk of the individual, or predicted to be neutral based on the high frequency in population, no effect on protein or regulatory regions. This classification class is equivalent to predicted "benign".
Variant-Clinical significance-Predicted pathogenic
Variant that has been automatically predicted to cause a phenotype/disease based on 5 ACMG criteria (BA1, BP6, PVS1, PM2 and PP5).
Variant-Clinical significance-Predicted uncertain
Variant with predicted unknown or questionable impact on a particular clinical phenotype.
Variant-Clinical significance-Premutation
A repeat expansion variant in a range that may not result in the clinical manifestation of the associated disease in the carrying individual, but that may result in the manifestation of the disease in the offspring due to potential repeat instability.
Variant-Clinical significance-Risk factor
Variant reported to be associated with the phenotype/disease and influencing the function(s) of the protein.
Variant-Clinical significance-Secondary mitochondrial mutation
The primary molecular defect resides in a nuclear gene, which leads to secondary mDNA abnormalities, such as loss of mDNA copy number or multiple mDNA deletions.
Variant-Clinical significance-Uncertain (VUS)
Variant with unknown or questionable impact on a particular clinical phenotype.
Variant-Coding effect Describes the impact of the observed DNA change on protein level.
Variant-Coding effect-Effect unknown
The coding effect on protein level has not been analyzed. An effect is expected but difficult to predict.
Variant-Coding effect-Extension
Affect either the first (start, translation initiation, N-terminus, ATG) or last codon (translation termination, stop) and as a consequence extend the protein sequence N- or C-terminally with one or more amino acids.
Variant-Coding effect-Frameshift
A sequence change caused by deletion/insertion of nucleotides affecting an amino acid between the first (initiation, ATG) and last codon (termination, stop), replacing the normal C-terminal sequence with one encoded by another reading frame.
Variant-Coding effect-Increased polyglutamine tract/expanded polyQ
Portion of a protein consisting of a sequence of several glutamine (Glu; Q) units.
Page | 36
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Coding effect-In-frame
A sequence change that does not cause a shift in the triplet reading frame. As a result one or more amino acids are replaced by one or more other amino acids.
Variant-Coding effect-Missense
A single nucleotide change that results in a codon that codes for a different amino acid. Not all missense mutations are deleterious, some changes can have no effect. Because of the ambiguity of missense mutations, it is often difficult to interpret the consequences of these mutations in causing disease.
Variant-Coding effect-New translation initiation codon
A sequence change that creates a new ATG start codon upstream of the original start translation site. If the new ATG is close enough to the original one (so that it is within the processed transcript and downstream of a ribosome-binding site) and in frame, it will be used to initiate translation, adding amino acids to the amino terminus of the original protein.
Variant-Coding effect-New translation initiation site
A sequence change affecting the translation initiation codon (Met-1) introducing a new upstream initiation codon extending the N-terminus of the encoded protein.
Variant-Coding effect-New translation termination codon
A sequence change that affects the translation termination codon (Ter/*) introducing a new downstream termination codon extending the C-terminus of the encoded protein.
Variant-Coding effect-Non-coding
The change on DNA level produces no effect on protein, or the effect of regulatory mutations is unknown.
Variant-Coding effect-Nonsense
A sequence change that results in a premature stop codon, and in a truncated, incomplete protein product.
Variant-Coding effect-Silent
A sequence change that results in a codon that codes for the same amino acid and without any functional change in the protein product.
Variant-Coding effect-Splicing mutation
A sequence change that affects the splicing process (i.e. intron removal and exons joining). Splice-site mutations occur within genes in the noncoding regions (introns) just next to the coding regions (exons). Splice site mutations can eliminate an existing donor or acceptor site, which will cause an exon to be skipped and possibly result in a frameshift.
Variant-Coding effect-Start loss
A sequence change in the ATG start codon that prevents the original start translation site from being used. This kind of mutation may eliminate gene function.
Variant-dbSNP
The Single Nucleotide Polymorphism Database (dbSNP) is a free public archive for genetic variation within and across different species developed and hosted by the National Center for Biotechnology Information (NCBI) in collaboration with the National Human Genome Research Institute (NHGRI).
Variant-gDNA change
Change at genomic DNA level following numbering based on genomic DNA reference sequence.
Variant-genomic position
The 1-based position of the variation on the given sequence on genome build hg19
Variant-Individual Represents a unique individual who was tested for a certain disease, condition or carrier status at CENTOGENE.

Page | 37
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Individual view-Quality status-G2P individual
Individual with confirmed genotype-phenotype association. At database level a G2P individual (++) is linked to at least one classified and curated variant (++).
Variant-Individual view-Quality status-Non-G2P individual
Individual with not yet confirmed genotype-phenotype association. At database level a Non-G2P individual (+) is linked to at least one classified variant (+).
Variant-Location
The location of the DNA change relative to the transcriptional initiation site, initiation codon, polyadenylation site or termination codon of the corresponding gene.
Variant-Location-3'UTR
3'-Untranslated Region: Particular section of messenger RNA that starts with the nucleotide immediately following the stop codon of the coding region. This region contains transcription and translation regulating sequences.
Variant-Location-5'UTR
5'-Untranslated Region: Sequences on the 5' end of messenger RNA but not translated into protein. It extends from the transcription start site to just before the ATG translation initiation codon. 5' UTR may contain sequences that regulate translation efficiency or messenger RNA stability.
Variant-Location-Downstream
The region located 3' (downstream) from the polyadenylation signal of the gene.
Variant-Location-Exon The protein-coding DNA sequence of the gene.
Variant-Location-Intron
The non-coding region of a gene that interrupt the protein coding regions (exons).
Variant-Location-Upstream
The region located 5' (upstream) from the 5'UTR region of the gene.
Variant-PMID
PubMed-Index for MEDLINE, PubMed identifier or PubMed unique identifier is a unique number assigned to each PubMed record.
Variant-Positive individuals
Indicates how many times a particular variant was observed at CENTOGENE in comparison to the total number of analyzed individuals for a particular gene, expressed as fraction.
Variant-Positive individuals (%)
Indicates how many times a particular variant was observed at CENTOGENE in comparison to the total number of analyzed individuals for a particular gene, expressed as percent (%).
Variant-Positive individuals-Age at onset
Refers to the age at which an individual acquires, develops or first experience a condition or symptoms of a disorder.
Variant-Positive individuals-ARFN
Anonymized random family number: Family unique number used to keep all members together when relationship links are provided.
Variant-Positive individuals-Biomarker interpretation
Evaluation of the biomarker levels compared to the reference interval.
Variant-Positive individuals-Biomarker interpretation-Normal Biomarker levels are within the normal range (no change).
Variant-Positive individuals-Biomarker interpretation-Pathological
Biomarker levels are significantly increased compared to the normal range.

Page | 38
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Positive individuals-Biomarker interpretation-Slightly decreased
Biomarker levels are only slightly decreased compared to the normal range.
Variant-Positive individuals-Biomarker interpretation-Slightly increased
Biomarker levels are only slightly increased compared to the normal range.
Variant-Positive individuals-Carrier testing
Indicates if the individual was interested in performing a carrier screening when the presence of specific genetic variant was detected already in other family members.
Variant-Positive individuals-Clinical information (HPO terms)
Description of features and characteristics that the corresponding physician has provided as supporting evidence of the presence of a particular disease translated into the vocabulary defined by the HPO.
Variant-Positive individuals-Clinical information (HPO terms)-Healthy/asymptomatic
Selected when the physician has explicitly indicated that the person is healthy, asymptomatic or not affected.
Variant-Positive individuals-Clinical information (HPO terms)-No info/unknown Selected when no clinical information has been provided.
Variant-Positive individuals-Clinical information (HPO terms)-Suspected/affected
Selected when only very general statements are provided by the physician (e.g. "patient is suffering from Breast Cancer" or "clinical features of Parkinson").
Variant-Positive individuals-Clinical statement of CENTOGENE
Refers to the finding or the conclusion of the molecular genetic test conducted at CENTOGENE. The clinical statement may confirm or disprove the suspected diagnosis, or serve to elucidate the genetic cause of an uncertain or questionable condition or disease.
Variant-Positive individuals-Clinical statement-Affected Individual with confirmed diagnosis at genetic level.
Variant-Positive individuals-Clinical statement-At least carrier
Individual with clinical suspicion most likely confirmed at genetic level. It includes individuals carrying in trans a pathogenic variant with an uncertain variant in case of autosomal recessive mode of inheritance, of Fabry females carrying uncertain variants.
Variant-Positive individuals-Clinical statement-Carrier
Individual who inherited one mutated allele at genetic level in case of autosomal recessive mode of inheritance, or female in case of X-linked mode of inheritance.
Variant-Positive individuals-Clinical statement-Increased risk of developing the disease
Individual with confirmed susceptibility at genetic level to develop a particular medical condition.
Variant-Positive individuals-Clinical statement-Increased risk of having affected offspring
Individual who carries a premutation variant and who may not be clinically affected of the disease himself, but who has a higher risk of having an affected offspring due to potential repeat instability.
Variant-Positive individuals-Clinical statement-Not determined
Individual carrying uncertain variant(s) where clear statement on either presence or susceptibility to develop a particular disease was not possible.
Variant-Positive individuals-Clinical statement-Probably affected
Fabry male patients carrying an uncertain variant associated with pathological enzymatic levels, but biomarker levels within normal range.
Page | 39
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Positive individuals-Clinical statement-Probably carrier
Individual carrying an uncertain variant in the context of autosomal recessive disorders or X-linked disorders (in this last situation it applies only to female individuals).
Variant-Positive individuals-Clinical statement-Unaffected
Indicates an individual where the susceptibility or presence of the disease was not confirmed at genetic level.
Variant-Positive individuals-Clinically irrelevant variant (CIV)
Variants which do not cause or influence the presence or severity of the disease. It includes variants of the following significance: neutral, likely neutral, disease-associated polymorphism, CENTOGENE (likely) neutral - published as (likely) pathogenic.
Variant-Positive individuals-Clinically relevant variant (CRV)
Variants which do cause or influence the presence or the severity of the disease. It includes variants of the following significance: likely pathogenic, pathogenic, risk factor, modifier.
Variant-Positive individuals-Consanguineous parents Refers to the marriage between two genetically related persons.
Variant-Positive individuals-Co-occurrence, other genes
Indicates the presence of other clinical relevant variant(s) or uncertain variant (s) in other genes than the gene of interest.
Variant-Positive individuals-Co-occurrence, same gene
Indicates the presence of other clinically relevant variant(s) or uncertain variant (s) in the gene of interest.
Variant-Positive individuals-Country
Indicates the area of the world the patient is coming from. The basis for this information is the country where the patient lives. If physician provides information about the ethnicity of the patient (e.g. Canadian citizen of German origin), then this (in this case Germany) is the item selected in this situation.
Variant-Positive individuals-Detailed family history
Detailed description of disorders from which direct blood relatives of the patient have suffered.
Variant-Positive individuals-Enzyme interpretation
Evaluation of the enzyme activity compared to the reference interval.
Variant-Positive individuals-Enzyme interpretation-Normal
Levels of enzyme activity are within the normal range (no change).
Variant-Positive individuals-Enzyme interpretation-Pathological
Levels of enzyme activity are significantly decreased compared to the normal range.
Variant-Positive individuals-Enzyme interpretation-Slightly decreased
Levels of enzyme activity are only slightly decreased compared to the normal range.
Variant-Positive individuals-Enzyme interpretation-Slightly increased
Levels of enzyme activity are only slightly increased compared to the normal range.
Variant-Positive individuals-Family history
Indicates the presence or the absence of a particular disorder or symptomatology in blood relatives of a patient.
Variant-Positive individuals-Finding
Indicates if a variant is related or unrelated to the indication for testing.
Variant-Positive individuals-Finding-Primary Variant related to the indication for testing.
Variant-Positive individuals-Finding-Secondary
Variant unrelated to the indication for testing (incidental finding).
Variant-Positive individuals-Genotype
Represents the genetic constitution of an individual with respect to the number of alleles and their clinical significance identified for a particular gene.

Page | 40
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Positive individuals-Genotype-Compound heterozygote
An individual carrying two different, heterozygous, in trans, uncertain or clinically relevant alleles (likely pathogenic, pathogenic, risk factor, modifier) at a given locus.
Variant-Positive individuals-Genotype-Hemizygote
A male individual carrying one uncertain or clinically relevant allele (pathogenic, likely pathogenic, risk factor, modifier) located on X-chromosome.
Variant-Positive individuals-Genotype-Heterozygote
An individual carrying one uncertain or clinically relevant allele (pathogenic, likely pathogenic, risk factor, modifier).
Variant-Positive individuals-Genotype-Homozygote
An individual carrying two identical, uncertain or clinically relevant alleles (pathogenic, likely pathogenic, risk factor, modifier) at one locus.
Variant-Positive individuals-Genotype-Other/complex
An individual carrying uncertain or clinically relevant alleles (pathogenic, likely pathogenic, risk factor, modifier) in other combinations than described above (e.g. two alleles located in cis, three heterozygous mutations, one homozygous and one heterozygous, etc.).
Variant-Positive individuals-Genotype-Wild type
An individual carrying clinically irrelevant alleles (neutral, likely neutral, disease-associated polymorphism, CENTOGENE (likely) neutral - published as (likely) pathogenic).
Variant-Positive individuals-Mode of Inheritance (MOI)
The manner in which a particular genetic trait or disorder is passed from one generation to the next.
Variant-Positive individuals-MOI-Autosomal dominant (AD)
The pattern of inheritance in which an affected individual has one copy of a mutant gene and one normal gene on a pair of autosomal chromosomes.
Variant-Positive individuals-MOI-Autosomal recessive (AR)
The pattern of inheritance in which both copies of an autosomal gene must be abnormal for a genetic condition or disease to occur.
Variant-Positive individuals-MOI-Digenic (Di)
The pattern of inheritance that is similar to recessive inheritance, except that the trait only develops when mutations are found in one copy of each of the two independent genes simultaneously.
Variant-Positive individuals-MOI-Imprinting/Epigenetic (Imp/Epi)
The pattern of inheritance by mechanisms not directly involving nucleotide sequences, but paramutations and parental imprinting.
Variant-Positive individuals-MOI-Mitochondrial (Mito)
The pattern of inheritance of a trait encoded in the mitochondrial genome.
Variant-Positive individuals-MOI-Multifactorial (MF)
The pattern of inheritance caused by the interplay between genetic factors and environmental factors.
Variant-Positive individuals-MOI-Pseudoautosomal dominant (P-AD)
The inheritance pattern seen with genes in the pseudoautosomal region of the X and Y chromosome that can exchange regularly between the two sex chromosomes. Alleles for genes in the pseudoautosomal region can show male-to-male transmission, and therefore mimic autosomal inheritance, because they can cross over from the X to the Y chromosomes during male gametogenesis and be passed on from a father to his male offspring.
Variant-Positive individuals-MOI-Unknown (?)
This mode of inheritance is selected for genes not yet being associated with any pathological condition or disease, and therefore no pattern of inheritance observed.
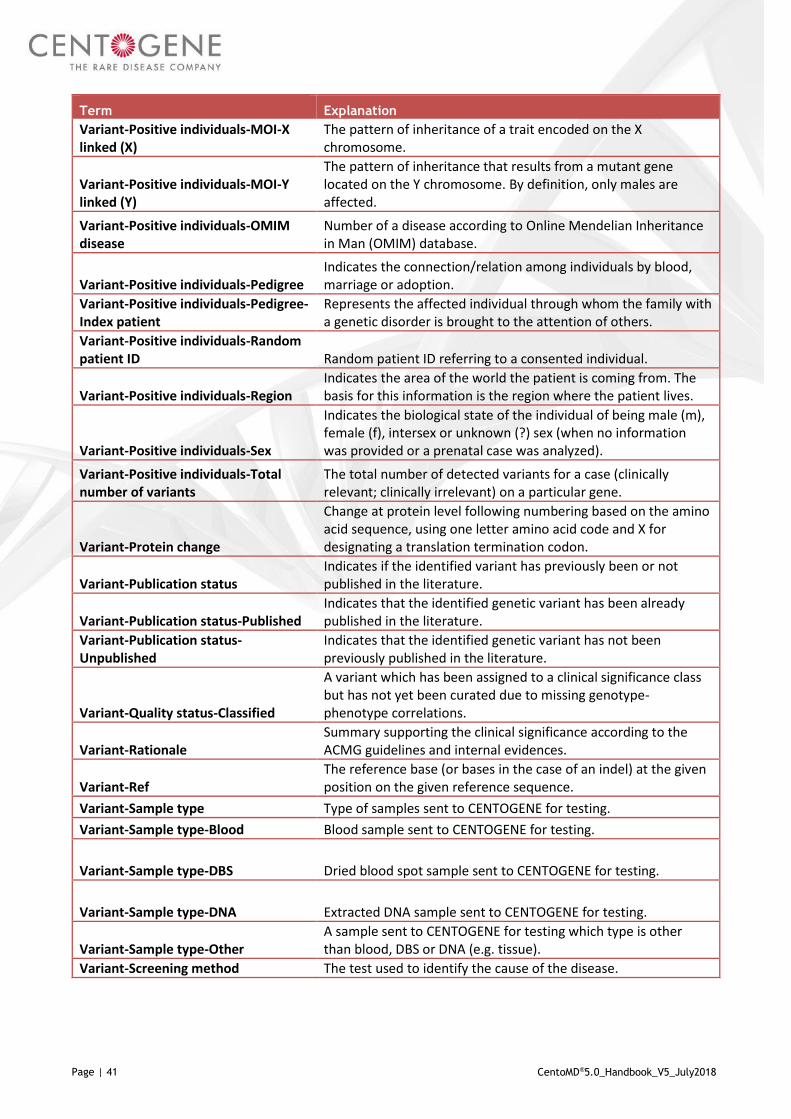
Page | 41
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Positive individuals-MOI-X linked (X)
The pattern of inheritance of a trait encoded on the X chromosome.
Variant-Positive individuals-MOI-Y linked (Y)
The pattern of inheritance that results from a mutant gene located on the Y chromosome. By definition, only males are affected.
Variant-Positive individuals-OMIM disease
Number of a disease according to Online Mendelian Inheritance in Man (OMIM) database.
Variant-Positive individuals-Pedigree
Indicates the connection/relation among individuals by blood, marriage or adoption.
Variant-Positive individuals-Pedigree-Index patient
Represents the affected individual through whom the family with a genetic disorder is brought to the attention of others.
Variant-Positive individuals-Random patient ID Random patient ID referring to a consented individual.
Variant-Positive individuals-Region
Indicates the area of the world the patient is coming from. The basis for this information is the region where the patient lives.
Variant-Positive individuals-Sex
Indicates the biological state of the individual of being male (m), female (f), intersex or unknown (?) sex (when no information was provided or a prenatal case was analyzed).
Variant-Positive individuals-Total number of variants
The total number of detected variants for a case (clinically relevant; clinically irrelevant) on a particular gene.
Variant-Protein change
Change at protein level following numbering based on the amino acid sequence, using one letter amino acid code and X for designating a translation termination codon.
Variant-Publication status Indicates if the identified variant has previously been or not published in the literature.
Variant-Publication status-Published
Indicates that the identified genetic variant has been already published in the literature.
Variant-Publication status-Unpublished
Indicates that the identified genetic variant has not been previously published in the literature.
Variant-Quality status-Classified
A variant which has been assigned to a clinical significance class but has not yet been curated due to missing genotype-phenotype correlations.
Variant-Rationale
Summary supporting the clinical significance according to the ACMG guidelines and internal evidences.
Variant-Ref The reference base (or bases in the case of an indel) at the given position on the given reference sequence.
Variant-Sample type Type of samples sent to CENTOGENE for testing.
Variant-Sample type-Blood Blood sample sent to CENTOGENE for testing.
Variant-Sample type-DBS Dried blood spot sample sent to CENTOGENE for testing.
Variant-Sample type-DNA Extracted DNA sample sent to CENTOGENE for testing.
Variant-Sample type-Other A sample sent to CENTOGENE for testing which type is other than blood, DBS or DNA (e.g. tissue).
Variant-Screening method The test used to identify the cause of the disease.

Page | 42
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Screening method-CES
Clinical Exome Sequencing: application of the next generation technology to determine the variations of coding regions of genes which have been associated to human disease.
Variant-Screening method-MLPA
Multiplex Ligation-dependent Probe Amplification: Variation of the multiplex PCR that permits multiple targets to be amplified with only a single primer pair. Used especially for detecting large/gross and gene rearrangements.
Variant-Screening method-NGS
Next-Generation Sequencing: High-throughput sequencing technology, allowing the parallel sequencing of multiple genes, producing thousands or millions of sequences concurrently.
Variant-Screening method-Other method
Refers to other methodology (like fragment length) used to detect the variants.
Variant-Screening method-qPCR
Quantitative Polymerase Chain Reaction: Method to amplify and simultaneously quantify a targeted DNA molecule. Used especially to detect large/gross and gene rearrangements.
Variant-Screening method-Sanger
Classical method of DNA sequencing, developed by Fred Sanger, using chemically altered dideoxy bases to terminate newly synthesized DNA fragments at specific bases (either A, C, T or G). These fragments are then size-separated, and the DNA sequence can be read.
Variant-Screening method-WES
Whole Exome Sequencing: application of the next-generation technology to determine the variations of all coding regions, or exons, of known genes.
Variant-Screening method-WGS
Whole Genome Sequencing: modern day technology for sequencing of the entire coding and non-coding regions of the genome.
Variant-Statistics-Age at diagnosis
Is calculated as date of sample entry at CENTOGENE minus date of birth, and is expressed in years. For patients referred to CENTOGENE several times, the date of the first order entry is used by default to calculate the age at diagnosis.
Variant-Statistics-Clinical information distribution-Frequency in cases
Indication how many times a particular variant in cases with particular phenotype (HPO term) was observed in comparison to the total number of analyzed cases for a particular variant.
Variant-Transcript used in CentoMD®
The transcript that is used at CENTOGENE/CentoMD® as a reference sequence.
Variant-Transcript/Reference Sequence
Digital nucleic acid sequence, assembled by scientists as a representative example of a species' set of genes. Coding DNA reference sequence refers to a cDNA-derived sequence containing the full length of all coding regions and non-coding untranslated regions.
Variant-Type of variant on DNA level Different types of change than can occur in the DNA sequence.
Variant-Type of variant on DNA level-Chromosomal deletion Refers to loss of parts of chromosomes.
Variant-Type of variant on DNA level-Complex rearrangement
Involves the structures or number of the chromosomes, it is referred to as chromosome mutation, or rearrangement, rearranged chromosomes.
Variant-Type of variant on DNA level-Conversion
Non-reciprocal transfer of information between homologous sequences; one DNA sequence replaces a homologous sequence

Page | 43
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
such that the sequences become identical after the conversion event.
Variant-Type of variant on DNA level-Deletion
A sequence change where one or more nucleotides are removed (deleted).
Variant-Type of variant on DNA level-Duplication
A sequence change where a copy of one or more nucleotides are inserted directly 3’-flancking of the original copy.
Variant-Type of variant on DNA level-Gain of methylation Gain of the normal DNA methylation level.
Variant-Type of variant on DNA level-Gene & regulatory region(s) deletion Refers to loss of the entire gene and flanking regions.
Variant-Type of variant on DNA level-Gene & regulatory region(s) duplication Refers to the gain of the entire gene and flanking regions.
Variant-Type of variant on DNA level-Gene deletion Refers to loss of the entire gene.
Variant-Type of variant on DNA level-Gene duplication Refers to gain/duplication of the entire gene.
Variant-Type of variant on DNA level-Gross deletion Refers to loss of parts of a gene.
Variant-Type of variant on DNA level-Gross duplication Refers to gain/duplication of part(s) of a gene.
Variant-Type of variant on DNA level-Gross inversion Refers to 180 degree inversion of part(s) of a gene.
Variant-Type of variant on DNA level-Insertion
Genetic mutation where one or more nucleotides are added (inserted) into a DNA sequence or it may involve portions of a chromosome.
Variant-Type of variant on DNA level-Insertion/Deletion (Indel)
Refers to the mutation class that includes a combination of both insertions and deletions.
Variant-Type of variant on DNA level-Inversion
Chromosomal abnormality where a segment of a chromosome is rotated 180 degrees and reinserted.
Variant-Type of variant on DNA level-Loss of methylation Loss of the normal DNA methylation level.
Variant-Type of variant on DNA level-Other/complex
Refers to all other types not included in any category under Type of variant on DNA level.
Variant-Type of variant on DNA level-Pathological allele (D4Z4 motif)
Deletion of 3.3-kb repeats from a chromosomal tandem repeat called D4Z4 located near the end of chromosome 4 at the 4q35-ter location. D4Z4 contains an ORF encoding a putative homeobox protein called DUX4, a large polymorphic repeat structure consisting of 1-100 KpnI units.
Variant-Type of variant on DNA level-Repeat expansion
Refers to an increase number of repeats of a genomic tandemly repeated DNA sequence.
Variant-Type of variant on DNA level-Retrotransposon insertion
Retrotransposons (also called transposons via RNA intermediates) are genetic elements that can amplify themselves in a genome, and can induce mutations by inserting near or within genes. Retrotransposon-induced mutations are relatively stable, because the sequence at the insertion site is retained as they transpose via the replication mechanism.

Page | 44
CentoMD®5.0_Handbook_V5_July2018
Term Explanation
Variant-Type of variant on DNA level-Substitution
A sequence change where one nucleotide is replaced by one other nucleotide. Substitutions are described using a ">"-character (indicating "changes to").
Variant-Unclassified A variant which has not yet been assigned to any clinical significance class due to the lack of information.
Variant-Zygosity
Indicates if a variant is detected on one chromosome or on both chromosomes. Describes the degree of similarity of the alleles for a trait in an organism.
Variant-Zygosity-Hemizygous (Hem) Used for alleles detected in genes located on X-chromosome for male cases.
Variant-Zygosity-Het/Hom/Hem Ratio indicating the number of individuals relative to variant zygosity.
Variant-Zygosity-Heterozygous (Het) Gene locus when cells contain two different alleles of a gene.
Variant-Zygosity-Homozygous (Hom) Gene locus when identical alleles of the gene are present on both homologous chromosomes.


















