
CENTER FOR BIOLOGICAL SEQUENCE ANALYSIS Probabilistic modeling and molecular phylogeny Anders Gorm...
32
CENTER FOR BIOLOGICAL SEQUENCE ANALYSIS CENTER FOR BIOLOGICAL SEQUENCE ANALYSIS Probabilistic modeling and Probabilistic modeling and molecular phylogeny molecular phylogeny Anders Gorm Pedersen Anders Gorm Pedersen Molecular Evolution Group Molecular Evolution Group Center for Biological Sequence Analysis Center for Biological Sequence Analysis Technical University of Denmark (DTU) Technical University of Denmark (DTU)
-
date post
19-Dec-2015 -
Category
Documents
-
view
228 -
download
1
Transcript of CENTER FOR BIOLOGICAL SEQUENCE ANALYSIS Probabilistic modeling and molecular phylogeny Anders Gorm...

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS Probabilistic modeling and Probabilistic modeling and
molecular phylogenymolecular phylogeny
Anders Gorm PedersenAnders Gorm Pedersen
Molecular Evolution GroupMolecular Evolution GroupCenter for Biological Sequence AnalysisCenter for Biological Sequence AnalysisTechnical University of Denmark (DTU)Technical University of Denmark (DTU)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
What is a model?
Mathematical models are:Mathematical models are:
• IncomprehensibleIncomprehensible
• UselessUseless
• No fun at allNo fun at all

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
What is a model?
Mathematical models are:Mathematical models are:
• IncomprehensibleIncomprehensible
• UselessUseless
• No fun at allNo fun at all

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
What is a model?
• Model = hypothesis !!!Model = hypothesis !!!
• Hypothesis (as used in most biological research): Hypothesis (as used in most biological research): – Precisely stated, but qualitativePrecisely stated, but qualitative– Allows you to make qualitative predictionsAllows you to make qualitative predictions
• Arithmetic model: Arithmetic model: – Mathematically explicit (parameters)Mathematically explicit (parameters)
– Allows you to make quantitative predictionsAllows you to make quantitative predictions……

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
The Scientific Method
ObservationObservationof dataof data
ModelModel of how system of how system worksworks
Prediction(s) about Prediction(s) about system behavior system behavior
(simulation)(simulation)
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
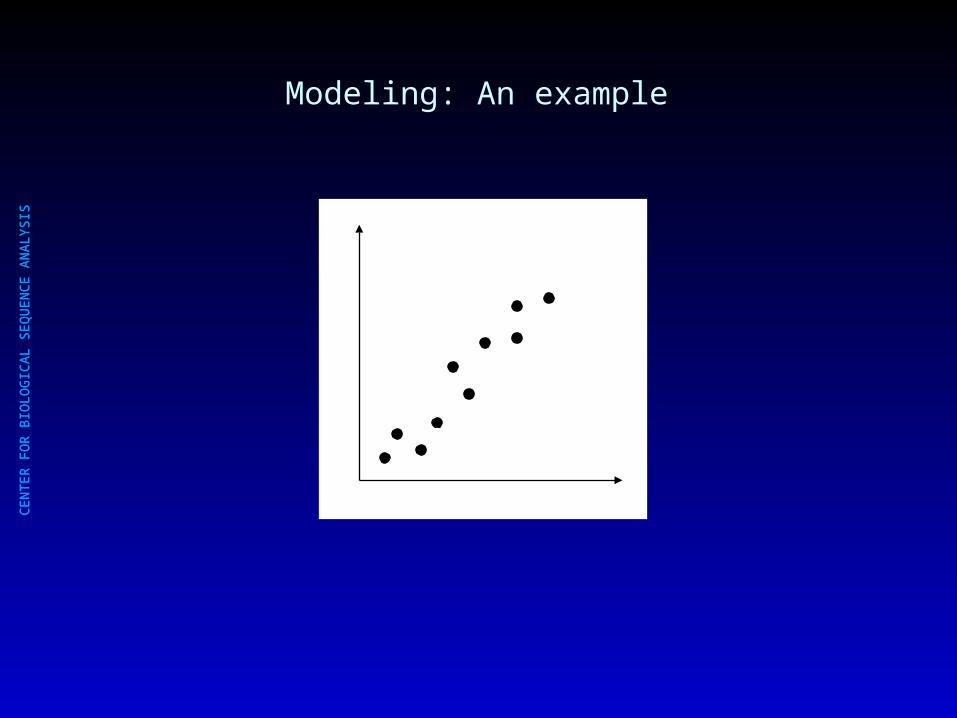
Modeling: An example

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
y = ax + b
Simple 2-parameter modelSimple 2-parameter model
Modeling: An example

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
y = ax + b
Predictions based on Predictions based on model
Modeling: An example
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
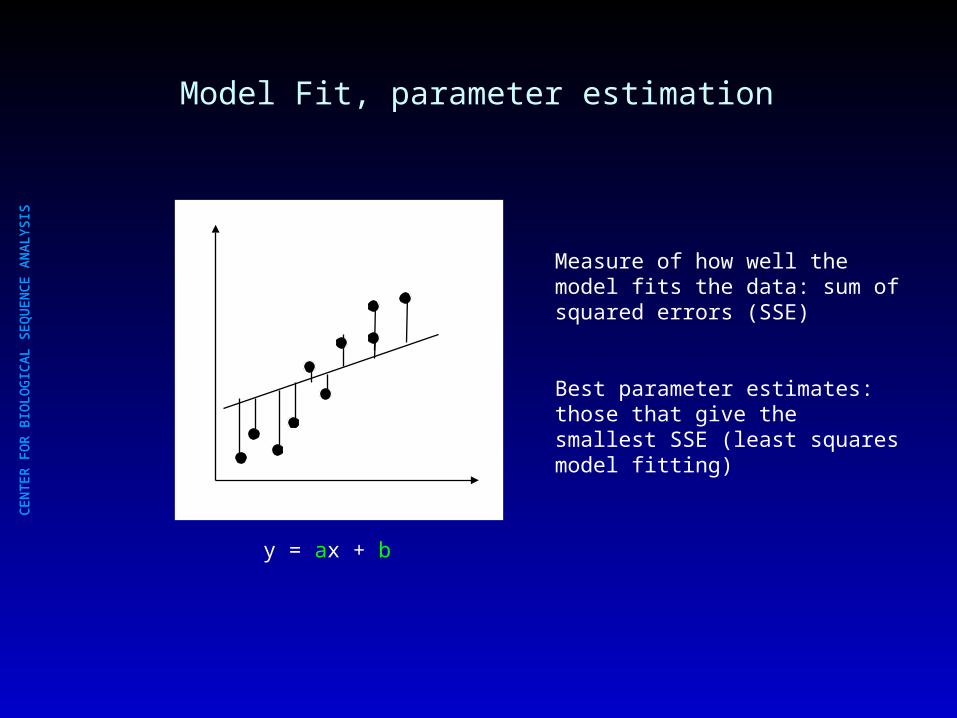
Model Fit, parameter estimation
y = ax + b
Measure of how well the model fits the data: sum of squared errors (SSE)
Best parameter estimates: those that give the smallest SSE (least squares model fitting)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Model Fit , parameter estimation
y = 1.24x - 0.56
Measure of fit between model and data: sum of squared errors (SSE)
Best parameter estimates: those that give the smallest SSE (least squares)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Probabilistic modeling applied to phylogeny
• Observed data: multiple alignment of sequencesObserved data: multiple alignment of sequences
H.sapiens globinH.sapiens globin A G G G A T T C AA G G G A T T C A
M.musculus globinM.musculus globin A C G G T T T - AA C G G T T T - A
R.rattus globinR.rattus globin A C G G A T T - AA C G G A T T - A
• Probabilistic model parameters (simplest case):Probabilistic model parameters (simplest case):– Tree topology and branch lengthsTree topology and branch lengths– Nucleotide-nucleotide substitution rates (or substitution probabilities)Nucleotide-nucleotide substitution rates (or substitution probabilities)– Nucleotide frequenciesπ: πNucleotide frequenciesπ: πAA, π, πCC, π, πGG, π, πTT
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
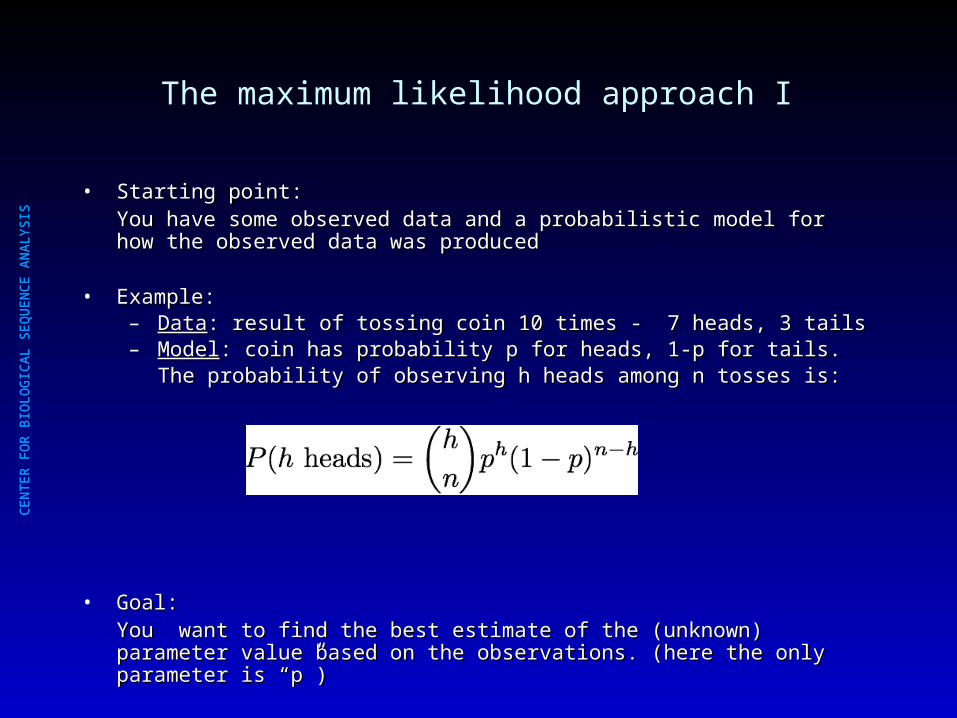
The maximum likelihood approach I
• Starting point:Starting point:You have some observed data and a probabilistic model for how the observed You have some observed data and a probabilistic model for how the observed data was produceddata was produced
• Example: Example: – DataData: result of tossing coin 10 times - 7 heads, 3 tails: result of tossing coin 10 times - 7 heads, 3 tails– ModelModel: coin has probability p for heads, 1-p for tails. : coin has probability p for heads, 1-p for tails.
The probability of observing h heads among n tosses is:The probability of observing h heads among n tosses is:
• Goal:Goal:You want to find the best estimate of the (unknown) parameter value based on You want to find the best estimate of the (unknown) parameter value based on the observations. (here the only parameter is “p”) the observations. (here the only parameter is “p”)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
The maximum likelihood approach II
Likelihood = Probability (Data | Model)
Maximum likelihood: Best estimate is the set of parameter values which gives the highest possible likelihood.

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum likelihood: coin tossing example
DataData: result of tossing coin : result of tossing coin 10 times - 7 heads, 3 tails10 times - 7 heads, 3 tails
ModelModel: coin has probability p : coin has probability p for heads, 1-p for tails. for heads, 1-p for tails.

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Probabilistic modeling applied to phylogeny
• Observed data: multiple alignment of sequencesObserved data: multiple alignment of sequences
H.sapiens globinH.sapiens globin A G G G A T T C AA G G G A T T C A
M.musculus globinM.musculus globin A C G G T T T - AA C G G T T T - A
R.rattus globinR.rattus globin A C G G A T T - AA C G G A T T - A
• Probabilistic model parameters (simplest case):Probabilistic model parameters (simplest case):– Tree topology and branch lengthsTree topology and branch lengths– Nucleotide-nucleotide substitution rates (or substitution probabilities)Nucleotide-nucleotide substitution rates (or substitution probabilities)– Nucleotide frequenciesπ: πNucleotide frequenciesπ: πAA, π, πCC, π, πGG, π, πTT

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Other models of nucleotide substitution

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
General Time Reversible Model
Time-reversibility:
The amount of change from state x to y is equal to the amount of change from y to x
πA x PAG = πG x PGA => πA x πG x a = πG x πA x a

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
More elaborate models of evolution
• Codon-codon substitution rates Codon-codon substitution rates
(64 x 64 matrix of codon substitution rates)(64 x 64 matrix of codon substitution rates)
• Different mutation rates at different sites in the geneDifferent mutation rates at different sites in the gene
(the “gamma-distribution” of mutation rates)(the “gamma-distribution” of mutation rates)
• Molecular clocks Molecular clocks
(same mutation rate on all branches of the tree). (same mutation rate on all branches of the tree).
• Different substitution matrices on different branches of the treeDifferent substitution matrices on different branches of the tree
(e.g., strong selection on branch leading to specific group of animals)(e.g., strong selection on branch leading to specific group of animals)
• Etc., etc.Etc., etc.

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Different rates at different sites: the gamma distribution

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Computing the probability of one column in an alignment given tree topology and other parameters
A C G G A T T C AA C G G A T T C AA C G G T T T - AA C G G T T T - AA A G G A T T - AA A G G A T T - AA G G G T T T - AA G G G T T T - A
AA
C
C A
G
Columns in alignment contain homologous nucleotides
Assume tree topology, branch lengths, and other parameters are given. Assume ancestral states were A and A. Start computation at any internal or external node.
Pr = πC PCA(t1) PAC(t2) PAA(t3) PAG(t4) PAA(t5)
t4
t5
t3t1
t2

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Computing the probability of an entire alignment given tree topology and other parameters
• Probability must be summed over all possible combinations of ancestral nucleotides.
(Here we have two internal nodes giving 16 possible combinations)
• Probability of individual columns are multiplied to give the overall probability of the alignment, i.e., the likelihood of the model.
• Often the log of the probability is used (log likelihood)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Maximum likelihood phylogeny: heuristic tree search• Data: Data:
– sequence alignmentsequence alignment
• Model parameters: Model parameters: – nucleotide frequencies, nucleotide substitution rates, tree topology, branch lengths.nucleotide frequencies, nucleotide substitution rates, tree topology, branch lengths.
Choose random initial values for all parameters, compute likelihood
Change parameter values slightly in a direction so likelihood improves
Repeat until maximum found
Results:(1) ML estimate of tree topology (2) ML estimate of branch lengths(3) ML estimate of other model parameters(4) Measure of how well model fits data (likelihood).

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Model Selection?
y = 1.24x - 0.56
Measure of fit between model and data (e.g., SSE, likelihood, etc.)
How do we compare different types of models?

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Over-fitting: More parameters always result in a better fit to the data, but not necessarily in a better description
y = ax + b
2 parameter modelGood description, poor fit
y = ax6+bx5+cx4+dx3+ex2+fx+g
7 parameter modelPoor description, good fit

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Selecting the best model: the likelihood ratio test
• The fit of two alternative models can be compared using the ratio of their likelihoods:The fit of two alternative models can be compared using the ratio of their likelihoods:
LR =LR = P(Data P(Data | | M1) = L,M1M1) = L,M1
P(Data | M2) L,M2P(Data | M2) L,M2
• Note that LR > 1 if model 1 has the highest likelihoodNote that LR > 1 if model 1 has the highest likelihood
• For For nested modelsnested models it can be shown that it can be shown that
Δ Δ = 2*ln(LR) = 2* (lnL,M1 - lnL,M2)= 2*ln(LR) = 2* (lnL,M1 - lnL,M2)
follows a Χfollows a Χ22 distribution with degrees of freedom equal to the number of extra parameters in the distribution with degrees of freedom equal to the number of extra parameters in the most complicated model.most complicated model.
This makes it possible to perform stringent statistical tests to determine which model (hypothesis) This makes it possible to perform stringent statistical tests to determine which model (hypothesis) best describes the databest describes the data

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Asking biological questions in a likelihood ratio testing framework
• Fit two alternative, nested models to the data.Fit two alternative, nested models to the data.
• Record optimized likelihood and number of free parameters for each fitted model.Record optimized likelihood and number of free parameters for each fitted model.
• Test if alternative (parameter-rich) model is Test if alternative (parameter-rich) model is significantlysignificantly better than null-model, given better than null-model, given number of additional parameters (nnumber of additional parameters (nextraextra):):
• Compute Δ = 2 x (lnLCompute Δ = 2 x (lnLAlternative Alternative - lnL- lnLNullNull) )
• Compare Δ to Compare Δ to ΧΧ22 distribution with n distribution with nextraextra degrees of freedom degrees of freedom
• Depending on models compared, different biological questions can be addressed Depending on models compared, different biological questions can be addressed (presence of molecular clock, presence of positive selection, difference in mutation (presence of molecular clock, presence of positive selection, difference in mutation rates among sites or branches, etc.)rates among sites or branches, etc.)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Likelihood ratio test example: Which model fits best: JC or K2P?
A C G T
A - α α α
C α - α α
G α α - α
T α α α -
Jukes and Cantor model (JC):
All nucleotides have same frequency All substitutions have same rate
Kimura 2 parameter model (K2P):
All nucleotides have same frequency Transitions and transversions have different rate
A C G T
A - β α β
C β - β α
G α β - β
T β α β -
=> K2P has one extra parameter compared to JC

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Likelihood ratio test example: Which model fits best: JC or K2P?
Starting point: set of DNA sequences, fit JC and K2P models to data, record likelihoods
JC: lnL = -2034.3K2P: lnL = -2031.2
K2P has better fit than JC: lnLK2P has better fit than JC: lnLK2PK2P > lnL > lnLJCJC
Test whether K2P is Test whether K2P is significantlysignificantly better better
Δ= 2 x (lnLΔ= 2 x (lnLAlternative Alternative - lnL- lnLNullNull) = 2 x (-2031.2 - -2034.3) = 6.2) = 2 x (-2031.2 - -2034.3) = 6.2
Degrees of freedom = 1 Degrees of freedom = 1 (K2P has one extra parameter compared to JC) (K2P has one extra parameter compared to JC)

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Δ = 2 x (lnLΔ = 2 x (lnLAlternative Alternative - lnL- lnLNullNull) = 6.2 Degrees of freedom = 1) = 6.2 Degrees of freedom = 1
Critical value (5% level) = 3.8415Statistic = 6.2
=> 1% < p < 5%=> Difference is significant=> K2P is significantly better description than JC
Likelihood ratio test example: Which model fits best: JC or K2P?
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
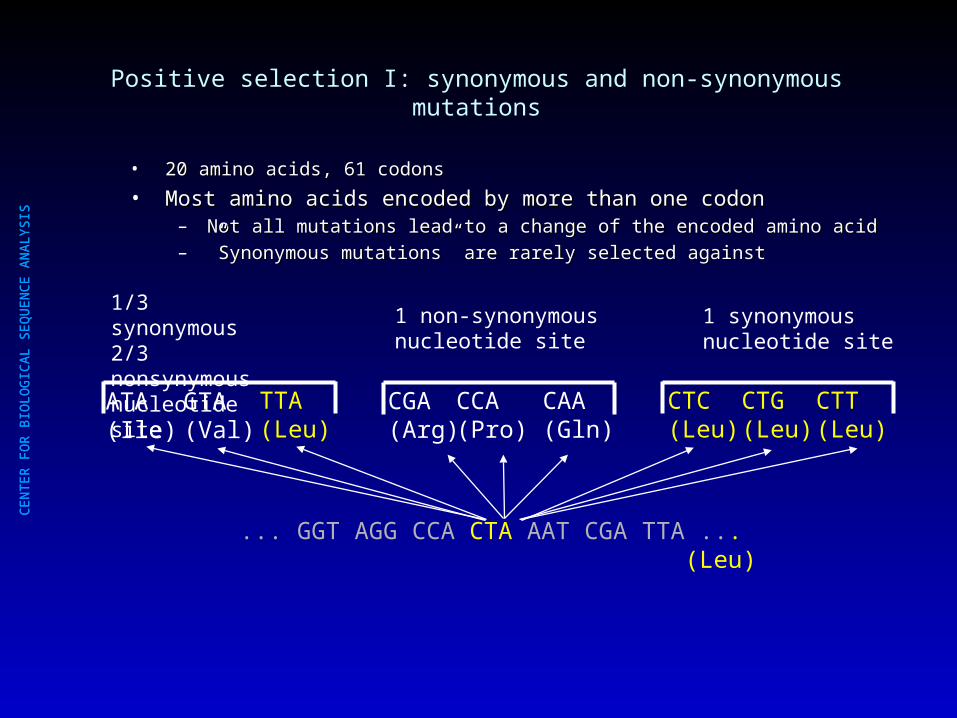
Positive selection I: synonymous and non-synonymous mutations
• 20 amino acids, 61 codons 20 amino acids, 61 codons
• Most amino acids encoded by more than one codonMost amino acids encoded by more than one codon– Not all mutations lead to a change of the encoded amino acidNot all mutations lead to a change of the encoded amino acid– ””Synonymous mutations” are rarely selected againstSynonymous mutations” are rarely selected against
... GGT AGG CCA CTA AAT CGA TTA ... (Leu)
CTC(Leu)
CTG(Leu)
CTT(Leu)
CAA(Gln)
CCA(Pro)
CGA(Arg)
ATA(Ile)
GTA(Val)
TTA(Leu)
1 non-synonymous nucleotide site
1 synonymous nucleotide site
1/3 synonymous2/3 nonsynymousnucleotide site

CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
Positive selection II: non-synonymous and synonymous mutation rates contain information about selective pressure
• dN: rate of non-synonymous mutations dN: rate of non-synonymous mutations per non-synonymous siteper non-synonymous site
• dS: rate of synonymous mutations dS: rate of synonymous mutations per synonymous siteper synonymous site
• Recall: Evolution is a two-step process:Recall: Evolution is a two-step process:
(1) Mutation (random)(1) Mutation (random)
(2) Selection (non-random)(2) Selection (non-random)
• Randomly occurring mutations will lead to dN/dS=1.Randomly occurring mutations will lead to dN/dS=1.
• Significant deviations from this most likely caused by subsequent selection.Significant deviations from this most likely caused by subsequent selection.
• dN/dS < 1dN/dS < 1: Higher rate of synonymous mutations: : Higher rate of synonymous mutations: negative (purifying) selectionnegative (purifying) selection
• dN/dS > 1dN/dS > 1: Higher rate of non-synonymous mutations: : Higher rate of non-synonymous mutations: positive selectionpositive selection
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
CE
NT
ER
FO
R B
IOLO
GIC
AL
SE
QU
EN
CE
AN
ALY
SIS
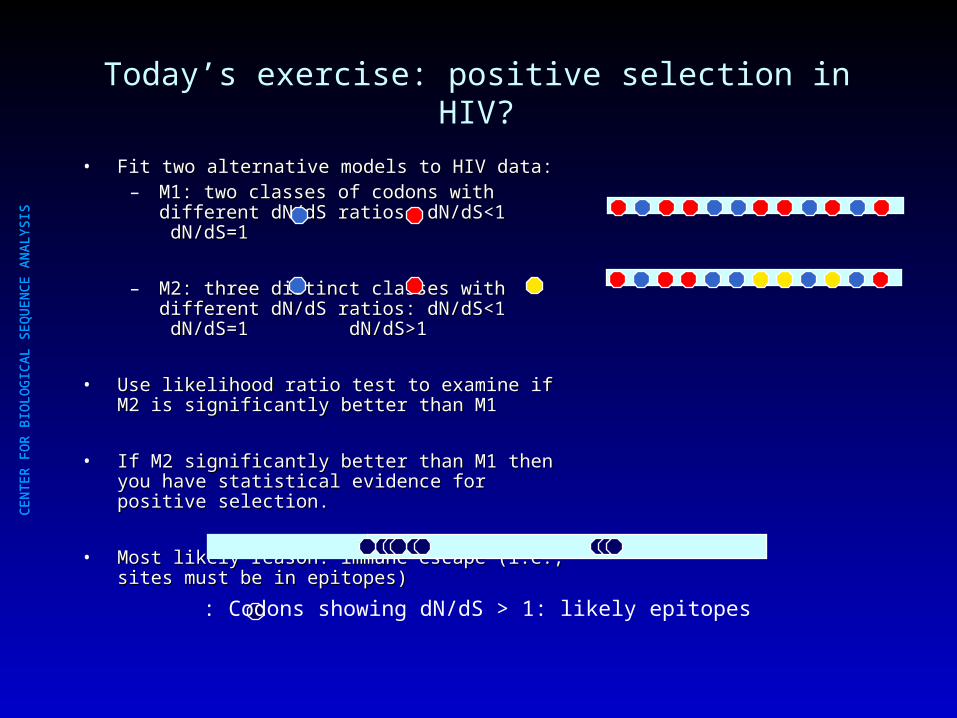
Today’s exercise: positive selection in HIV?
• Fit two alternative models to HIV data:Fit two alternative models to HIV data:– M1: two classes of codons with different dN/dS M1: two classes of codons with different dN/dS
ratios: dN/dS<1 dN/dS=1ratios: dN/dS<1 dN/dS=1
– M2: three distinct classes with different dN/dS M2: three distinct classes with different dN/dS ratios: dN/dS<1 dN/dS=1 dN/dS>1ratios: dN/dS<1 dN/dS=1 dN/dS>1
• Use likelihood ratio test to examine if M2 is significantly Use likelihood ratio test to examine if M2 is significantly better than M1better than M1
• If M2 significantly better than M1 then you have If M2 significantly better than M1 then you have statistical evidence for positive selection. statistical evidence for positive selection.
• Most likely reason: immune escape (i.e., sites must be Most likely reason: immune escape (i.e., sites must be in epitopes)in epitopes)
: Codons showing dN/dS > 1: likely epitopes


















