
Case Scenario Cv (1)
14
University of Santo Tomas THE GRADUATE SCHOOL ADVANCED MEDICAL-SURGICAL NURSING I CASE SCENARIO THE CLIENT WITH CARDIOVASCULAR ALTERATIONS Prepared by: John Henry O. Valencia, RN, RM Master of Arts in Nursing Student It is mid-morning in the medical ward where you work, and you are getting a new patient, M.A., a 60- year-old, retired businessman, is married and has 3 grown children. As you take his health history, he tells you that he began to feel palpitations and “on and off” chest discomfort about 10 days ago. He has hypertension and a 5- year history of angina pectoris. He is 5’9” and weighs 200 lb. He is sedentary and gets no routine exercise. During the past week, he has had more frequent episodes of midchest discomfort. During the week, he has also experienced increased fatigue. He states “I just feel crappy most of the time”. He tells you a cardiac catheterization done several years ago revealed 50% occlusion of the right coronary artery and 50% occlusion of the LAD artery. He also tells you that both his parents had CAD. He is taking NTG, amlodipine, metoprolol, lipitor and baby ASA.The MD writes his diagnosis as CAD, HPN. At 1AM the following day, M.A. turns on his call light. When you respond, he is talking rapidly and pointing to the bathroom. His speech pattern indicates he is short of breath. You assist him to the bathroom and note that his skin feels clammy. While sitting on the commode he vomits. The resident MD comes and evaluates M.A.’s condition. He order s Furosemide 40 mg. IV push STAT. M.A. continues to experience vomiting and diaphoresis inspite of medications and comfort measures. A STAT 12-lead ECG reveals ischemic changes indicating ACS. M.A. is ordered to be transferred to the CCU. Five days later his condition is stabilized and M.A. is taken to OR for CABG x3. When he arrives from the OR, he has a Swan-Ganz catheter in place for hemodynamic monitoring and is intubated and put on a ventilator at FiO2 70. His ABG reading is: pH 7.36; PCO2 46 mmHg; PO2 61 mmHg; SaO2 85% with a Hgb 10.3mg/dL. Hemodynamic values show that the pressures within his heart and lungs are slightly elevated and that his cardiac output is low, indicating that the heart is not effectively pumping out the blood that is returned to it or that he is experiencing a little fluid overload. M.A. receives continuous IV infusions of Nitroprusside and Dobutamine. Close monitoring and intensive management of M.A.’s condition is done. His condition improves and is transferred to the Telemetry unit. Five days later, he is now preparing for discharge. INSTRUCTIONS: Identify the learning issues (at least 7) related to pathophysiology (Focus on CV) that you can draw out from this case. Prepare to discuss these issues with the class on Tuesday, September 23, 2014. Your written answer to your learning issues will be submitted next Tuesday’s class.
-
Upload
johnhenryv -
Category
Documents
-
view
219 -
download
0
Transcript of Case Scenario Cv (1)

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 1/14
University of Santo Tomas
THE GRADUATE SCHOOL
ADVANCED MEDICAL-SURGICAL NURSING I
CASE SCENARIOTHE CLIENT WITH CARDIOVASCULAR ALTERATIONS
Prepared by: John Henry O. Valencia, RN, RM
Master of Arts in Nursing Student
It is mid-morning in the medical ward where you work, and you are getting a new patient, M.A., a 60-
year-old, retired businessman, is married and has 3 grown children. As you take his health history, he tells you
that he began to feel palpitations and “on and off” chest discomfort about 10 days ago. He has hypertension and
a 5-year history of angina pectoris. He is 5’9” and weighs 200 lb. He is sedentary and gets no routine exercise.
During the past week, he has had more frequent episodes of midchest discomfort. During the week, he has also
experienced increased fatigue. He states “I just feel crappy most of the time”. He tells you a cardiaccatheterization done several years ago revealed 50% occlusion of the right coronary artery and 50% occlusion of
the LAD artery. He also tells you that both his parents had CAD. He is taking NTG, amlodipine, metoprolol, lipitor
and baby ASA.The MD writes his diagnosis as CAD, HPN.
At 1AM the following day, M.A. turns on his call light. When you respond, he is talking rapidly and
pointing to the bathroom. His speech pattern indicates he is short of breath. You assist him to the bathroom and
note that his skin feels clammy. While sitting on the commode he vomits. The resident MD comes and evaluates
M.A.’s condition. He order s Furosemide 40 mg. IV push STAT. M.A. continues to experience vomiting and
diaphoresis inspite of medications and comfort measures. A STAT 12-lead ECG reveals ischemic changes
indicating ACS. M.A. is ordered to be transferred to the CCU.
Five days later his condition is stabilized and M.A. is taken to OR for CABG x3. When he arrives from
the OR, he has a Swan-Ganz catheter in place for hemodynamic monitoring and is intubated and put on a
ventilator at FiO2 70. His ABG reading is: pH 7.36; PCO2 46 mmHg; PO2 61 mmHg; SaO2 85% with a Hgb
10.3mg/dL. Hemodynamic values show that the pressures within his heart and lungs are slightly elevated and
that his cardiac output is low, indicating that the heart is not effectively pumping out the blood that is returned to it
or that he is experiencing a little fluid overload. M.A. receives continuous IV infusions of Nitroprusside and
Dobutamine. Close monitoring and intensive management of M.A.’s condition is done. His condition improves
and is transferred to the Telemetry unit. Five days later, he is now preparing for discharge.
INSTRUCTIONS:
Identify the learning issues (at least 7) related to pathophysiology (Focus on CV) that you can draw out from
this case. Prepare to discuss these issues with the class on Tuesday, September 23, 2014. Your written answer
to your learning issues will be submitted next Tuesday’s class.

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 2/14
Page 2 of 14
OLD AGE (60 YEARS OLD), OBESITY (97KGS OR 200LBS) AND SEDENTARY LIFESTYLE
Nonspecific Injury to endothelial lining
Desquamation of endothelial lining
Activation of tissue inflammatory response
Release of pro-inflammatory cytokines
(MCP-1, TNF-α, Interleukins, CRP and Serum Amyloid A)
Interleukin - 6
Leukocyte andEndothelial cell
activation
Hepatic Acute-phase reactants
production
Increased LiverEnzymes (SGPT)
MonocyteChemoreactant Protein-1
Leukocyte-Endothelium binding
Migration to site ofinflammation
Mononuclearphagocyte activation
IncreasedWBC count
Complete BloodCount
Interleukin-18 and 10
Expression ofinterferon-γ
Endothelialadhesion molecule
activation
Intercellular Adhesion Molecule(ICAM) Formation
Vascular Cell Adhesion Molecule
(VCAM)
E-selectinFormation
Monocyte Adhesion
Leukocyte diapedesisinto extravascular space
Adhesion of molecules
Increase vonWillebrand Factor
Developing fatty StreakOxidative Stress
LDL Oxidation
Oxidized LDL attracts monocytes andmacrophages to the site
Plaques begin to form from cells whichembedded into the endothelium
Blood Tests(Cardiac Enzymes
Test)
COMPREHENSIVE
PATHOPHYSIOLOGIC BASIS OF
CLIENT’s CONDITION
8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 3/14
Page 3 of 14
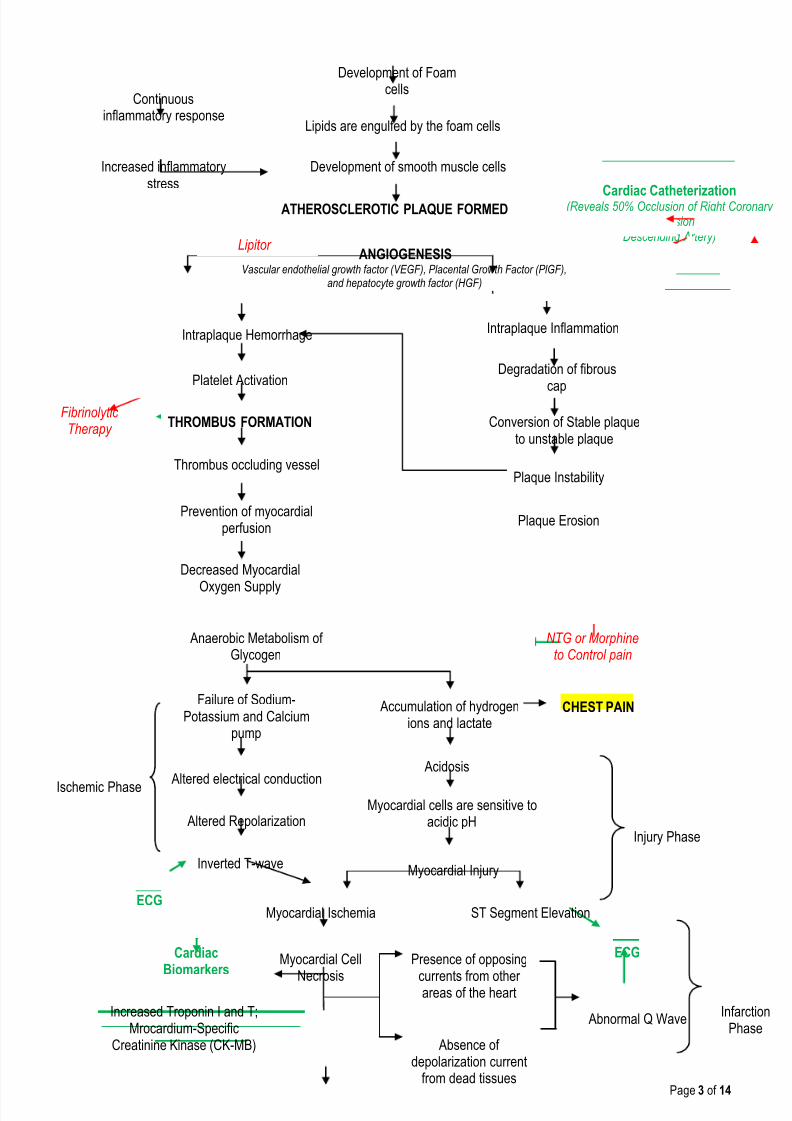
Development of Foamcells
Lipids are engulfed by the foam cells
Development of smooth muscle cells
ATHEROSCLEROTIC PLAQUE FORMED
Increased inflammatorystress
Continuousinflammatory response
ANGIOGENESISVascular endothelial growth factor (VEGF), Placental Growth Factor (PlGF),
and hepatocyte growth factor (HGF)
Intraplaque Hemorrhage
Conversion of Stable plaqueto unstable plaque
Intraplaque Inflammation
Degradation of fibrous
cap
Plaque Instability
Platelet Activation
Plaque Erosion
THROMBUS FORMATION
Thrombus occluding vessel
Prevention of myocardialperfusion
Decreased Myocardial
Oxygen Supply
Anaerobic Metabolism ofGlycogen
Failure of Sodium-Potassium and Calcium
pump
Accumulation of hydrogenions and lactate
Acidosis
Altered electrical conduction
Altered Repolarization
Inverted T-wave
Ischemic PhaseMyocardial cells are sensitive to
acidic pH
Myocardial Injury
ST Segment ElevationMyocardial Ischemia
Myocardial Cell
Necrosis
Absence ofdepolarization current
from dead tissues
Presence of opposing
currents from otherareas of the heart
Abnormal Q Wave
Injury Phase
InfarctionPhase
ECG
ECG
CHEST PAIN
Cardiac Catheterization(Reveals 50% Occlusion of Right Coronary Artery and 50% Occlusion of Left Anterior
Descending Artery)
Increased Troponin I and T;Mrocardium-Specific
Creatinine Kinase (CK-MB)
CardiacBiomarkers
NTG or Morphineto Control pain
FibrinolyticTherapy
Lipitor
8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 4/14
Page 4 of 14
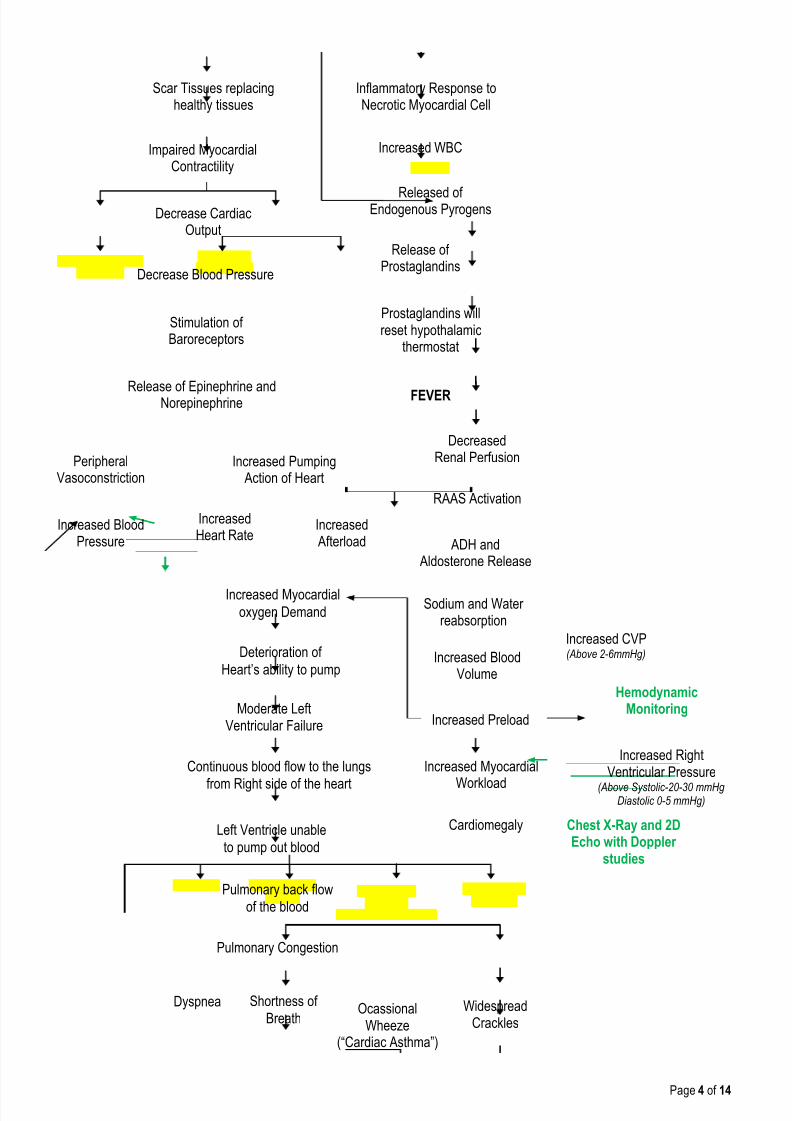
Inflammatory Response toNecrotic Myocardial Cell
Impaired MyocardialContractility
Increased WBC
Released ofEndogenous Pyrogens
Release ofProstaglandins
Prostaglandins willreset hypothalamic
thermostat
FEVER
Scar Tissues replacinghealthy tissues
Decrease CardiacOutput
Decrease Blood Pressure
Release of Epinephrine andNorepinephrine
Increased BloodPressure
IncreasedHeart Rate
Increased Afterload
Stimulation ofBaroreceptors
PeripheralVasoconstriction
Increased Pumping Action of Heart
DecreasedRenal Perfusion
RAAS Activation
Increased Myocardialoxygen Demand
ADH and Aldosterone Release
Sodium and Waterreabsorption
Increased BloodVolume
Increased Preload
Increased Myocardial
Workload
Cardiomegaly Chest X-Ray and 2DEcho with Doppler
studies
Deterioration of
Heart’s ability to pump
Moderate LeftVentricular Failure
Continuous blood flow to the lungs
from Right side of the heart
Left Ventricle unable
to pump out blood
Pulmonary back flow
of the blood
Pulmonary Congestion
Dyspnea Shortness of
BreathWidespread
CracklesOcassional
Wheeze(“Cardiac Asthma”)
Increased RightVentricular Pressure
(Above Systolic-20-30 mmHgDiastolic 0-5 mmHg)
HemodynamicMonitoring
Increased CVP(Above 2-6mmHg)

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 5/14
Page 5 of 14
Fluids leak intoalveolar space
Lymphatic system drains excessinterstitial fluid volume
Additional fluid in the pleural spacedrains into titer lymph nodes
Fluid moves from the interstitial spacein the alveolar walls
Damaged to alveolarepithelium
Further fluid accumulation in thealveolar space
Alveolar Edema
Impaired gas exchange
H oxemiaHypercarbia
Decrease lung complianceDecrease oxygen diffusion
Use of Accessory Muscle Bradypnea
Poor cerebral
oxygenation
tered Level ofonsciousness
Agitation /
Restlesness
Poor Peripheraloxygenation
Pallor Sweaty coolperiphery
Reducedcapillaryreturn
Increased work
of breathing
Exhaustion
Fatigue
Decreased PO2 (PO2 61mmHg)
Increased Pulmonary Artery
Pressure(Above 25mmHg)
andPulmonary Artery Wedge Pressure(PAWP) (Above 4-12mmHg)
Decreased SaO2 (SaO2 85%)
HemodynamicMonitoring
Arterial Blood GasAnalysis

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 6/14
Page 6 of 14
Identify the learning issues (at least 7) related to pathophysiology (Focus on CV) that you can draw out
from this case.
1. STAGES OF PATHOPHYSIOLOGIC BASIS OF CORONARY ARTERY DISEASE
Coronary artery disease (CAD) also known as atherosclerotic heart disease, coronary heart
disease, or ischemic heart disease (IHD),is the most common type of heart disease and cause of heart
attacks.The disease is caused by plaque building up along the inner walls of thearteries of the heart,
which narrows the arteries and reduces blood flow to the heart.
Atherogenesis in humans typically occurs over a period of many years, usually many decades.
Growth of atherosclerotic plaques probably does not occur in a smooth, linear fashion but
discontinuously, with periods of relative quiescence punctuated by periods of rapid evolution. It
undergoes different steps; Initiation, Leukocyte recruitment and Foam Cell formation.
A.
INITIATION
Studies of human atherosclerosis suggests that the "fatty streak" represents the initial lesion of
atherosclerosis. These early lesions most often seem to arise from focal increases in the content of
lipoproteins within regions of the intima. Our patient being a smoker without exercise and with a
susceptible age predisposes him to atherosclerotic plaque formation which is heralded by the Fatty
streak formation from lipoprotein accumulation. This accumulation of lipoprotein particles may not result
simply from increased permeability, or "leakiness," of the overlying endothelium. Rather, the lipoproteins
may collect in the intima of arteries because they bind to constituents of the extracellular matrix,
increasing the residence time of the lipid-rich particles within the arterial wall.
Lipoproteins that accumulate in the extracellular space of the intima of arteries often associate
with glycosaminoglycans of the arterial extracellular matrix, an interaction that may slow the egress of
these lipid-rich particles from the intima. Lipoprotein particles in the extracellular space of the intima,
particularly those retained by binding to matrix macromolecules, may undergo oxidative modifications.
Considerable evidence supports a pathogenic role for products of oxidized lipoproteins in
atherogenesis. Lipoproteins sequestered from plasma antioxidants in the extracellular space of the
intima become particularly susceptible to oxidative modification, giving rise to hydroperoxides,
lysophospholipids, oxysterols, and aldehydic breakdown products of fatty acids and phospholipids.
Considerable evidence supports the presence of such oxidation products in atherosclerotic lesions.
B.
LEUKOCYTE RECRUITMENT
Accumulation of leukocytes characterizes the formation of early atherosclerotic lesions. Thus,
from its very inception, atherogenesis involves elements of inflammation, a process that now provides a
unifying theme in the pathogenesis of this disease. The inflammatory cell types typically found in the
evolving atheroma include monocyte-derived macrophages and lymphocytes. A number of adhesion
molecules or receptors for leukocytes expressed on the surface of the arterial endothelial cell probably
participate in the recruitment of leukocytes to the nascent atheroma. Constituents of oxidatively modified
low-density lipoprotein can augment the expression of leukocyte adhesion molecules. This example
illustrates how the accumulation of lipoproteins in the arterial intima may link mechanistically with
leukocyte recruitment, a key event in lesion formation.

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 7/14
Page 7 of 14
Once captured on the surface of the arterial endothelial cell by adhesion receptors, the
monocytes and lymphocytes penetrate the endothelial layer and take up residence in the intima. In
addition to products of modified lipoproteins, cytokines (protein mediators of inflammation) can regulate
the expression of adhesion molecules involved in leukocyte recruitment. For example, interleukin 1 (IL-
1) or tumor necrosis factor (TNF-) induce or augment the expression of leukocyte adhesion molecules
on endothelial cells. Because products of lipoprotein oxidation can induce cytokine release fromvascular wall cells, this pathway may provide an additional link between arterial accumulation of
lipoproteins and leukocyte recruitment. Chemoattractant cytokines such as monocyte chemoattractant
protein 1 appear to direct the migration of leukocytes into the arterial wall.
C. FOAM-CELL FORMATION
Once resident within the intima, the mononuclear phagocytes mature into macrophages and
become lipid-laden foam cells, a conversion that requires the uptake of lipoprotein particles by receptor-
mediated endocytosis. One might suppose that the well-recognized "classic" receptor for LDL mediates
this lipid uptake; however, humans or animals lacking effective LDL receptors due to genetic alterations(e.g., familial hypercholesterolemia) have abundant arterial lesions and extraarterial xanthomata rich in
macrophage-derived foam cells. In addition, the exogenous cholesterol suppresses expression of the
LDL receptor; thus, the level of this cell-surface receptor for LDL decreases under conditions of
cholesterol excess. Candidates for alternative receptors that can mediate lipid loading of foam cells
include a growing number of macrophage "scavenger" receptors, which preferentially endocytose
modified lipoproteins, and other receptors for oxidized LDL or very low-density lipoprotein (VLDL).
Monocyte attachment to the endothelium, migration into the intima, and maturation to form lipid-laden
macrophages thus represent key steps in the formation of the fatty streak, the precursor of fully formed
atherosclerotic plaques.
D. ARTERIAL REMODELING DURING ATHEROGENESIS
During the initial part of the life history of an atheroma, growth is often outward, preserving the
caliber of the lumen. This phenomenon of "compensatory enlargement" accounts in part for the
tendency of coronary arteriography to underestimate the degree of atherosclerosis. Rupture of the
plaque's fibrous cap causes thrombosis. Physical disruption of the atherosclerotic plaque commonly
causes arterial thrombosis by allowing blood coagulant factors to contact thrombogenic collagen found
in the arterial extracellular matrix and tissue factor produced by macrophage-derived foam cells in the
lipid core of lesions. In this manner, sites of plaque rupture form the nidus for thrombi. The normal artery
wall has several fibrinolytic or antithrombotic mechanisms that tend to resist thrombosis and lyse clots
that begin to form in situ. Such antithrombotic or thrombolytic molecules include thrombomodulin,
tissue- and urokinase-type plasminogen activators, heparan sulfate proteoglycans, prostacyclin, and
nitric oxide. When the clot overwhelms the endogenous fibrinolytic mechanisms, it may propagate and
lead to arterial occlusion.
The consequences of this occlusion depend on the degree of existing collateral vessels. In a
patient with chronic multivessel occlusive coronary artery disease (CAD), collateral channels have often
formed. In such circumstances, even a total arterial occlusion may not lead to myocardial infarction (MI),
or it may produce an unexpectedly modest or a non-ST-segment elevation infarct because of collateral
flow. In a patient with less advanced disease and without substantial stenotic lesions to provide a
stimulus for collateral vessel formation, sudden plaque rupture and arterial occlusion commonly
produces an ST-segment elevation infarction. These are the types of patients who may present with MI
or sudden death as a first manifestation of coronary atherosclerosis. In some cases, the thrombus may

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 8/14
Page 8 of 14
lyse or organize into a mural thrombus without occluding the vessel. Such instances may be clinically
silent.
The subsequent thrombin-induced fibrosis and healing causes a fibroproliferative response
that can lead to a more fibrous lesion that can produce an eccentric plaque that causes a
hemodynamically significant stenosis. In this way, a nonocclusive mural thrombus, even if clinically
silent or causing unstable angina rather than infarction, can provoke a healing response that canpromote lesion fibrosis and luminal encroachment.
Such a sequence of events may convert a "vulnerable" atheroma with a thin fibrous cap that is
prone to rupture into a more "stable" fibrous plaque with a reinforced cap. Angioplasty of unstable
coronary lesions may "stabilize" the lesions by a similar mechanism, producing a wound followed by
healing.
E. MYOCARDIAL ISCHEMIA AS CAUSE OF PATIENTS CHEST PAIN
Central to an understanding of the pathophysiology of myocardial ischemia is the concept of
myocardial supply and demand. In normal conditions, for any given level of a demand for oxygen, themyocardium will control the supply of oxygen-rich blood to prevent underperfusion of myocytes and the
subsequent development of ischemia and infarction. The major determinants of myocardial oxygen
demand are heart rate, myocardial contractility, and myocardial wall tension (stress).The normal
coronary circulation is dominated and controlled by the heart's requirements for oxygen. This need is
met by the ability of the coronary vascular bed to vary its resistance (and, therefore, blood flow)
considerably while the myocardium extracts a high and relatively fixed percentage of oxygen. By
reducing the lumen of the coronary arteries, atherosclerosis limits appropriate increases in perfusion
when the demand for flow is augmented, as occurs during exertion or excitement.
During episodes of inadequate perfusion caused by coronary atherosclerosis, myocardial
tissue oxygen tension falls and may cause transient disturbances of the mechanical, biochemical, andelectrical functions of the myocardium. Coronary atherosclerosis is a focal process that usually causes
nonuniform ischemia. During ischemia, regional disturbances of ventricular contractility cause
segmental hypokinesia, akinesia, or, in severe cases, bulging (dyskinesia), which can reduce
myocardial pump function.

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 9/14
Page 9 of 14
CAD
CLINICAL MANIFESTATIONS
CHRONIC STABLE ANGINA PECTORIS UNSTABLE ANGINA PECTORIS (Preinfarction)
Chest pain or discomfort provoked by EXERTION or
EMOTIONAL STRESS. Relieved by REST or
NITROGLYCERIN.
Characteristics
substernal chest pain, pressure, heaviness or
discomfort
pain may be mild or severe
gradual buildup of discomfort and subsequent
gradual fading
numbness or weakness in arms, wrists, or
hands
diaphoresis
tachycardia
increased BP
Location
Behind middle or upper third of sternum
+ Levine Sign
Radiation
Radiates to neck, jaw, shoulders, arms, hands,
and posterior intracapsular area
Duration
2-15 minutes (after stopping activities)
1 minute after NITROGLYCERINE
Other Precipitating Factor
Exposure to hot or cold weather
Eating heavy meal
Coitus (increase workload of the heart, increase
oxygen demand)
Chest pain occurring at REST, no OXYGEN
DEMAND is placed on the heart, but an ACUTE
LACK of BLOOD FLOW to the HEART occurs
because of:
Coronary artery spasm
Presence of an enlarge plaque
Hemorrhage / ulceration of a complicated lesion
Critical narrowing of the vessel lumen occurs
A change in FREQUENCY, DURATION, and
INTENSITY of stable angina symptoms Pain lasts longer than 10 MINUTES
Pain UNRELIEVED by rest or Nitroglycerine
Mimics S&S of MI
CAN CAUSE SUDDEN DEATH OR RESULT IN
MI.
SILENT ISCHEMIA
Absence of chest pain with documented evidence of
an imbalance between myocardial oxygen supply
and demand (ST DEPRESSION of 1mm or more). CIRCADIAN EVENT (occurs during the first few
hours after awakening due to an increase in
sympathetic nervous system activity) o Increase heart rate
o Increase BP
o Increase coronary vessel tone
o Increase blood viscosity
2. Significant clinical presentation of the disease presented by the patient:

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 10/14
Page 10 of 14
3. Possible Complications of Acute Coronary Syndrome.
The complications of acute coronary syndromes depend on how much, how long, and where a
coronary artery is blocked. If the blockage affects a large amount of heart muscle, the heart will notpump effectively. If the blockage shuts off blood flow to the electrical system of the heart, the heart
rhythm may be affected.
Pumping problems:
In a heart attack, part of the heart muscle dies. Dead tissue, and the scar tissue that eventually
replaces it, does not contract. The scar tissue sometimes even expands or bulges when the rest of
the heart contracts. Consequently, there is less muscle to pump blood. If enough muscle dies, the
heart's pumping ability may be so reduced that the heart cannot meet the body's need for blood
and oxygen. Heart failure, low blood pressure, or both develop. If more than half of the heart tissueis damaged or dies, the heart generally cannot function, and severe disability or death is likely.
Drugs such as beta-blockers and especially angiotensin-converting enzyme (ACE) inhibitors
can reduce the extent of the abnormal areas by reducing the workload of and the stress on the
heart. Thus, these drugs help the heart maintain its shape and function more normally.
The damaged heart may enlarge, partly to compensate for the decrease in pumping ability (a
larger heart beats more forcefully). Enlargement of the heart makes abnormal heart rhythms more
likely.
Rhythm problems:
Abnormal heart rhythms (arrhythmias) occur in more than 90% of people who have had a heart
attack. These abnormal rhythms may occur because the heart attack damaged part of the heart's
electrical system. Sometimes there is a problem with the part of the heart that triggers the
heartbeat, so heart rate may be too slow. Other problems can cause the heart to beat rapidly or
irregularly. Sometimes the signal to beat is not conducted from one part of the heart to the other,
and the heartbeat may slow or stop.
In addition, areas of heart muscle that have poor blood flow but that have not died can be very
irritable. This irritability can cause heart rhythm problems, such as ventricular tachycardia orventricular fibrillation. These rhythm problems greatly interfere with the heart's pumping ability and
may cause the heart to stop beating (cardiac arrest). A loss of consciousness or death can result.
These rhythm disturbances are a particular problem in people who have an imbalance in blood
chemicals, such as a low potassium level.
Other problems:
Pericarditis (inflammation of the membranes enveloping the heart) may develop in the first day
or two after a heart attack or about 10 days to 2 months later. Pericarditis is more common in
people who have not had the blocked artery opened by percutaneous coronary intervention (PCI)or coronary artery bypass grafting (CABG). People seldom notice symptoms of early developing

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 11/14
Page 11 of 14
Acute CoronarySyndromes
Rupture, fissuring, orulceration
Exposing
Highly thrombogenicplaque constituents
Underlyingsubendothelial
basement membrane
Hemorrhage into thecore of plaques
Expansion of plaquevolume
Worsening of theluminal occlusion
Plaque rupture
Intrinsic factors
• Large atheromatous core
• Thickness of the fibrous cap
Extrinsic factors
• Adrenergic stimulation
• Intense emotional stress
collagen
synthesis
collagen
degradation
pericarditis because their heart attack symptoms are more prominent. However, pericarditis
produces a scratchy rhythmic sound that can sometimes be heard through a stethoscope 2 to 3
days after a heart attack. Sometimes, the inflammation causes a small amount of fluid to collect in
the space between the two layers of the pericardium (pericardial effusion). Later developing
pericarditis is usually called Dressler (post-myocardial infarction) syndrome. This syndrome causes
fever, pericardial effusion, inflammation of the membranes covering the lungs, pleural effusion(extra fluid in the space between the two layers of the pleura), and joint pain.
Other complications after a heart attack include malfunction of the mitral valve,rupture of the
heart muscle, a bulge in the wall of the ventricle (ventricular aneurysm), blood clots (emboli), and
low blood pressure (hypotension). Nervousness and depression are common after a heart attack.
Depression after a heart attack may be significant and may persist.
4. Triggering factors for Myocardial infarction from Acute Coronary Syndrome
Role of Acute Plaque Changes
In most patients, unstable angina, infarction, and many cases of SCD all occur because of
abrupt plaque change followed by thrombosis. Hence the term acute coronary syndrome.
A. Events that trigger the abrupt plaque changes
Rupture reflects the inability of a plaque to withstand mechanical stresses.
Triggers may be intrinsic or extrinsic
B. Integrity of the Plaque Fibrous caps are continuously remodeling
8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 12/14
Page 12 of 14
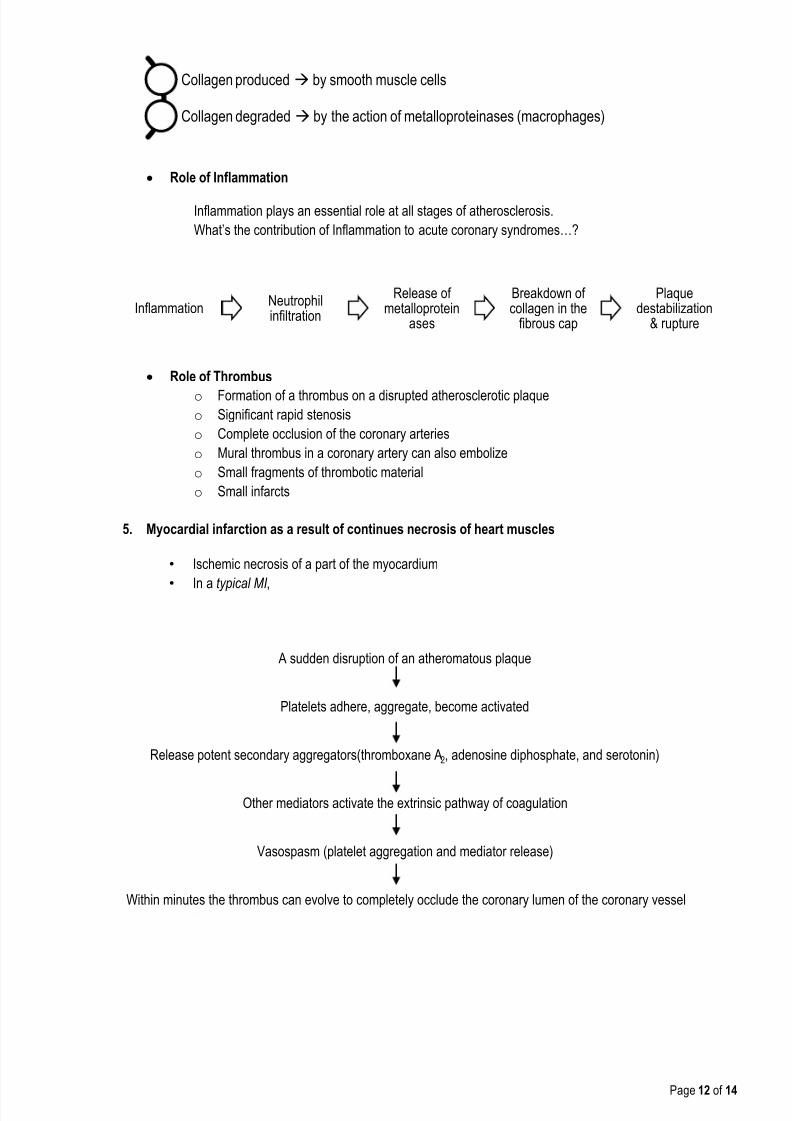
Collagen produced by smooth muscle cells
Collagen degraded by the action of metalloproteinases (macrophages)
InflammationNeutrophilinfiltration
Release ofmetalloprotein
ases
Breakdown ofcollagen in the
fibrous cap
Plaquedestabilization
& rupture
Within minutes the thrombus can evolve to completely occlude the coronary lumen of the coronary vessel
Vasospasm (platelet aggregation and mediator release)
Other mediators activate the extrinsic pathway of coagulation
Release potent secondary aggregators(thromboxane A2, adenosine diphosphate, and serotonin)
Platelets adhere, aggregate, become activated
A sudden disruption of an atheromatous plaque
Role of Inflammation
Inflammation plays an essential role at all stages of atherosclerosis.
What’s the contribution of Inflammation to acute coronary syndromes…?
Role of Thrombus
o Formation of a thrombus on a disrupted atherosclerotic plaqueo Significant rapid stenosis
o Complete occlusion of the coronary arteries
o Mural thrombus in a coronary artery can also embolize
o Small fragments of thrombotic material
o Small infarcts
5. Myocardial infarction as a result of continues necrosis of heart muscles
• Ischemic necrosis of a part of the myocardium
•
In a typical MI ,

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 13/14
Page 13 of 14
Ischemia Death of myocardium
Electrical instability of themyocardium
Arrhythmias (ventricularfibrillation)
Reduction in thecontractility of the
myocardiumReduction in the ejectionfraction & increase in end
systolic volume &pressure (Heart failure)
Or a fatal mechanicalfailure
6.
Role of Ischemia in development of Myocardial Infarction and significant signs and symptoms.
Please see the above comprehensive pathophysiologic basis of the client’s condition for furtherexplanation.
7. Likelihood that signs & symptoms represent an ACS secondary to CAD
Normal Normal Elevated cardiac TnI,TnT or CK-MB
CardiacMarker
T wave flattening orinversion in leads withdominant R wave
Normal EKG
Fixed Q waves
Abnormal ST segmentsor T waves notdocumented to be new
New or presumablynew, transient STsegment deviation(≥0.05mV) or T waveinversion (≥0.2mV)with symptoms
EKG
Chest discomfortreproduced bypalpation or respiration
Extracardiac vasculardisease
Transient MR,hypotension,diaphoresis, pulmonaryedema or rales
Exam
Probable ischemicsymptoms in absence
of the intermediatelikelihoodcharacteristics
Recent cocaine use
Chest or left arm painor discomfort as chief
symptom Age > 70
Male gender
Diabetes mellitus
Chest or left arm painor discomfort as chief
symptom reproducingprior documentedangina
Known history of CAD,including MI
History
Low Intermediate High Feature

8/11/2019 Case Scenario Cv (1)
http://slidepdf.com/reader/full/case-scenario-cv-1 14/14
References:
McPhee SJ, Hammer GD, et al. Pathophysiology of Disease: an Introduction to Clinical
Medicine 6th
Edition; 2013; 26:985-998
Huether S, McCance K, Parkinson C, et al. Understanding Pathophysiology 5th
edition; 2012;
22:782-892
Carol JR, Mattson JH, Porth JB, et al. Essentials of Pathophysiology: Concepts of Altered
Health States; 2014; 18:865-872
Corwin EJ, West BJ, Lilly LS, et al. Pathophysiology of Heart Disease: A Collaborative Project
of Medical Students and Faculty 5th
Edition; 2013; 8:345-356


















