CANCER Copyright © 2018 Enhanced preclinical antitumor ...Lan et al., Sci. Transl. Med. 10,...
16
Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018 SCIENCE TRANSLATIONAL MEDICINE | RESEARCH ARTICLE 1 of 15 CANCER Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF- Yan Lan,* Dong Zhang, Chunxiao Xu, Kenneth W. Hance, † Bo Marelli, Jin Qi, Huakui Yu, Guozhong Qin, Aroop Sircar, Vivian M. Hernández, Molly H. Jenkins, Rachel E. Fontana, Amit Deshpande, George Locke, Helen Sabzevari, ‡ Laszlo Radvanyi, Kin-Ming Lo* Antibodies targeting immune checkpoints are emerging as potent and viable cancer therapies, but not all patients re- spond to these as single agents. Concurrently targeting additional immunosuppressive pathways is a promising approach to enhance immune checkpoint blockade, and bifunctional molecules designed to target two pathways simultaneously may provide a strategic advantage over the combination of two single agents. M7824 (MSB0011359C) is a bifunctional fusion protein composed of a monoclonal antibody against programmed death ligand 1 (PD-L1) fused to the extracellular domain of human transforming growth factor– (TGF-) receptor II, which functions as a “trap” for all three TGF- isoforms. We demonstrate that M7824 efficiently, specifically, and simultaneously binds PD-L1 and TGF-. In syngeneic mouse models, M7824 suppressed tumor growth and metastasis more effectively than treatment with either an anti–PD-L1 antibody or TGF- trap alone; furthermore, M7824 extended survival and con- ferred long-term protective antitumor immunity. Mechanistically, the dual anti-immunosuppressive function of M7824 resulted in activation of both the innate and adaptive immune systems, which contributed to M7824’s anti- tumor activity. Finally, M7824 was an effective combination partner for radiotherapy or chemotherapy in mouse models. Collectively, our preclinical data demonstrate that simultaneous blockade of the PD-L1 and TGF- pathways by M7824 elicits potent and superior antitumor activity relative to monotherapies. INTRODUCTION Targeting tumor immune suppression pathways represents a paradigm shift in clinical oncology (1, 2). Programmed death 1 (PD-1) and pro- grammed death ligand 1 (PD-L1) are key components of an immuno- suppressive network that dampens T cell activity in normal physiology but can be exploited by tumors to suppress T cell–mediated antitumor immune responses (2, 3). Increased PD-L1 expression in tumors has been associated with poor clinical outcomes (2, 4–6). Antibodies di- rected against PD-1 and PD-L1 have effected striking and durable im- provements in survival for patients with melanoma (7–9), lung cancer (10–12), renal cell cancer (13), Merkel cell carcinoma (14), urothelial can- cer (15–17), head and neck cancer (18, 19), and several other indications. Despite promising clinical activity, only a minority of patients respond to anti–PD-1/PD-L1 therapies. Thus, there is an urgent need to improve objective response rates. One strategy is to combine anti– PD-L1/PD-1 with other immunotherapies (20), as exemplified by the combination of anti–PD-1 and anti–CTLA-4, which has been ap- proved for advanced melanoma (8, 21). In normal physiology, the regulatory cytokine transforming growth factor– (TGF-) functions to maintain immunological self-tolerance (22–24). However, TGF- can promote tumor progression and facili- tate tumor immune evasion through its effects on the innate and adap- tive immune systems. The three TGF- isoforms, TGF-1, TGF-2, and TGF-3 (22, 24), are highly expressed in many tumor types, and their serum concentrations correlate with poor clinical outcome (24–26). TGF- functions as an autocrine or paracrine signal within the tumor microenvironment, where it promotes tumor progression via stromal modification, angiogenesis, and induction of epithelial-mesenchymal transition (EMT) (27–29). TGF- signaling in myeloid cells is also critical in driving metastasis (30). In addition, TGF-1 can directly in- hibit T cell division and acquisition of effector function and natural killer (NK) cell function (31–35). Dual targeting of PD-L1 and TGF- represents a rational thera- peutic strategy because PD-L1 and TGF- are key pathways with in- dependent and complementary immunosuppressive functions. The respective roles of PD-L1 and TGF- in tumor cell–intrinsic and tumor cell–extrinsic pathways raise the possibility of superior anti- tumor activity compared with monotherapies. For example, T helper 1 (T H 1) cytokine production was restored most effectively when PD-1 blockade was combined with TGF- inhibition (36). Furthermore, because the up-regulation of TGF- signaling–associated genes has been linked to anti–PD-1 resistance in metastatic melanoma (37), block- ade of TGF- signaling may target mechanisms of resistance and sen- sitize tumors to anti–PD-1/PD-L1 therapies. Resistance to checkpoint blockade immunotherapy has raised sev- eral therapeutic challenges, including identifying which additional pathways to target, developing regimens to practically target two or more specific molecules, and controlling the high clinical development costs associated with deploying multiple single agents. To address these issues, we developed M7824 (MSB0011359C), a bifunctional fu- sion protein designed to simultaneously block the PD-L1 and TGF- pathways. M7824 is a fully human immunoglobulin G1 (IgG1) mono- clonal antibody against human PD-L1 fused to the extracellular do- main of human TGF- receptor II (TGF-RII), which functions as a TGF- trap. The anti–PD-L1 moiety of M7824 is based on avelumab (MSB0010718C), which has been approved for the treatment of Merkel cell carcinoma (14) and urothelial cancer (17). Here, we characterized EMD Serono Research and Development Institute Inc., Billerica, MA 01821, USA. *Corresponding author. Email: [email protected] (Y.L.); kinming.lo@emdserono. com (K.-M.L.) †Present address: GlaxoSmithKline, Collegeville, PA 19426, USA. ‡Present address: Intrexon Corporation, Germantown, MD 20876, USA. Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works by guest on April 14, 2020 http://stm.sciencemag.org/ Downloaded from
Transcript of CANCER Copyright © 2018 Enhanced preclinical antitumor ...Lan et al., Sci. Transl. Med. 10,...

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
1 of 15
C A N C E R
Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-Yan Lan,* Dong Zhang, Chunxiao Xu, Kenneth W. Hance,† Bo Marelli, Jin Qi, Huakui Yu, Guozhong Qin, Aroop Sircar, Vivian M. Hernández, Molly H. Jenkins, Rachel E. Fontana, Amit Deshpande, George Locke, Helen Sabzevari,‡ Laszlo Radvanyi, Kin-Ming Lo*
Antibodies targeting immune checkpoints are emerging as potent and viable cancer therapies, but not all patients re-spond to these as single agents. Concurrently targeting additional immunosuppressive pathways is a promising approach to enhance immune checkpoint blockade, and bifunctional molecules designed to target two pathways simultaneously may provide a strategic advantage over the combination of two single agents. M7824 (MSB0011359C) is a bifunctional fusion protein composed of a monoclonal antibody against programmed death ligand 1 (PD-L1) fused to the extracellular domain of human transforming growth factor– (TGF-) receptor II, which functions as a “trap” for all three TGF- isoforms. We demonstrate that M7824 efficiently, specifically, and simultaneously binds PD-L1 and TGF-. In syngeneic mouse models, M7824 suppressed tumor growth and metastasis more effectively than treatment with either an anti–PD-L1 antibody or TGF- trap alone; furthermore, M7824 extended survival and con-ferred long-term protective antitumor immunity. Mechanistically, the dual anti-immunosuppressive function of M7824 resulted in activation of both the innate and adaptive immune systems, which contributed to M7824’s anti-tumor activity. Finally, M7824 was an effective combination partner for radiotherapy or chemotherapy in mouse models. Collectively, our preclinical data demonstrate that simultaneous blockade of the PD-L1 and TGF- pathways by M7824 elicits potent and superior antitumor activity relative to monotherapies.
INTRODUCTIONTargeting tumor immune suppression pathways represents a paradigm shift in clinical oncology (1, 2). Programmed death 1 (PD-1) and pro-grammed death ligand 1 (PD-L1) are key components of an immuno-suppressive network that dampens T cell activity in normal physiology but can be exploited by tumors to suppress T cell–mediated antitumor immune responses (2, 3). Increased PD-L1 expression in tumors has been associated with poor clinical outcomes (2, 4–6). Antibodies di-rected against PD-1 and PD-L1 have effected striking and durable im-provements in survival for patients with melanoma (7–9), lung cancer (10–12), renal cell cancer (13), Merkel cell carcinoma (14), urothelial can-cer (15–17), head and neck cancer (18, 19), and several other indications.
Despite promising clinical activity, only a minority of patients respond to anti–PD-1/PD-L1 therapies. Thus, there is an urgent need to improve objective response rates. One strategy is to combine anti–PD-L1/PD-1 with other immunotherapies (20), as exemplified by the combination of anti–PD-1 and anti–CTLA-4, which has been ap-proved for advanced melanoma (8, 21).
In normal physiology, the regulatory cytokine transforming growth factor– (TGF-) functions to maintain immunological self-tolerance (22–24). However, TGF- can promote tumor progression and facili-tate tumor immune evasion through its effects on the innate and adap-tive immune systems. The three TGF- isoforms, TGF-1, TGF-2, and TGF-3 (22, 24), are highly expressed in many tumor types, and their serum concentrations correlate with poor clinical outcome (24–26). TGF- functions as an autocrine or paracrine signal within the tumor
microenvironment, where it promotes tumor progression via stromal modification, angiogenesis, and induction of epithelial-mesenchymal transition (EMT) (27–29). TGF- signaling in myeloid cells is also critical in driving metastasis (30). In addition, TGF-1 can directly in-hibit T cell division and acquisition of effector function and natural killer (NK) cell function (31–35).
Dual targeting of PD-L1 and TGF- represents a rational thera-peutic strategy because PD-L1 and TGF- are key pathways with in-dependent and complementary immunosuppressive functions. The respective roles of PD-L1 and TGF- in tumor cell–intrinsic and tumor cell–extrinsic pathways raise the possibility of superior anti-tumor activity compared with monotherapies. For example, T helper 1 (TH1) cytokine production was restored most effectively when PD-1 blockade was combined with TGF- inhibition (36). Furthermore, because the up-regulation of TGF- signaling–associated genes has been linked to anti–PD-1 resistance in metastatic melanoma (37), block-ade of TGF- signaling may target mechanisms of resistance and sen-sitize tumors to anti–PD-1/PD-L1 therapies.
Resistance to checkpoint blockade immunotherapy has raised sev-eral therapeutic challenges, including identifying which additional pathways to target, developing regimens to practically target two or more specific molecules, and controlling the high clinical development costs associated with deploying multiple single agents. To address these issues, we developed M7824 (MSB0011359C), a bifunctional fu-sion protein designed to simultaneously block the PD-L1 and TGF- pathways. M7824 is a fully human immunoglobulin G1 (IgG1) mono-clonal antibody against human PD-L1 fused to the extracellular do-main of human TGF- receptor II (TGF-RII), which functions as a TGF- trap. The anti–PD-L1 moiety of M7824 is based on avelumab (MSB0010718C), which has been approved for the treatment of Merkel cell carcinoma (14) and urothelial cancer (17). Here, we characterized
EMD Serono Research and Development Institute Inc., Billerica, MA 01821, USA.*Corresponding author. Email: [email protected] (Y.L.); [email protected] (K.-M.L.)†Present address: GlaxoSmithKline, Collegeville, PA 19426, USA.‡Present address: Intrexon Corporation, Germantown, MD 20876, USA.
Copyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
2 of 15
the biochemical activity of M7824 and evaluated its antitumor activity in mouse tumor models.
RESULTSM7824 is a bifunctional fusion protein designed to simultaneously block PD-L1 and TGF- pathwaysM7824 is a recombinant anti–PD-L1 antibody–TGF-RII fusion protein (Fig. 1A). The light chain of the molecule is iden-tical to the light chain of avelumab. The heavy chain of M7824 is a fusion protein composed of the heavy chain of avelumab containing three amino acid substitutions in the constant regions that result in a dif-ferent IgG1 allotype, genetically fused via a flexible (Gly4Ser)4Gly linker to the N terminus of the soluble extracellular do-main of TGF-RII (amino acids 1 to 136) (38). At the fusion junction, the C- terminal lysine residue of the antibody heavy chain was mutated to alanine to reduce potential proteolytic cleavage.
The concept behind the bifunctional M7824 molecule was to simultaneously target a receptor (PD-L1) and a soluble lig-and (TGF-) to block tumor cell–intrinsic and tumor cell–extrinsic immunosuppres-sive pathways, respectively. With this ap-proach, dosing can be based on an optimal PD-L1 target occupancy (TO) of 90 to 95%, which puts the trap moiety in vast excess of the active TGF- concentration in cancer patients (39, 40). This approach also avoids the constraints of using a bi-specific antibody to cotarget two cellular receptors, which would have different re-ceptor densities and cellular locations. The use of the natural receptor TGF-RII as a ligand trap ensures neutralization with high avidity; this is supported by a struc-tural model showing that the obligatory dimeric conformation of the receptors at the C terminus of the antibody allows bi-valent binding of the TGF- homodimer (Fig. 1B). Furthermore, targeting of the anti–PD-L1 moiety to the tumor should localize the trap moiety to the tumor mi-croenvironment, enabling it to sequester autocrine or paracrine TGF-. TGF- com-plexed with the TGF-RII moiety of M7824 cannot cause immunosuppression be-cause M7824 lacks the cytoplasmic do-main of TGF-RII, which is required to phosphorylate TGF-RI to initiate the sig-naling cascade (38). Furthermore, more than 30% of M7824 was internalized 4 hours
PD-L1 binding TGF-β binding
OD
450
nm
TGF-β1: anti−PD-L1TGF-β1: M7824
TGF-β2: M7824TGF-β2: anti−PD-L1
TGF-β3: M7824TGF-β3: anti−PD-L1
1 100 10,000
3
2
1
0
Anti−PD-L1 or M7824 (ng/ml)
A
C D
OD
450
nm
3
2
1
01 10 100TGF-β2 (ng/ml)
TGF-β2 binding
Anti−PD-L1M7824
E F Simultaneous binding
M7824 structure
PD-L1binding region
PD-L1binding region
VLVH
CLCH1
TGF-βRII
Linker
TGF-β trap
VLVH
CLCH1
TGF-βRII
Linker
CH2CH2
CH3CH3
Anti−PD-L1
M7824Trap control
0.01 1 100 10,000
MFI
600
400
200
0
Concentration (ng/ml)
OD
450
nm
1.0
0.5
01 10 1000.1 1000
Trap control or M7824 (ng/ml)
M7824Trap control
Fcγ1
TGF-βRII
TGF-β3TGF-β3
TGF-βRII
B Computer generated model
CH2
CH3
CH2
CH3
Linkers
Fig. 1. M7824 binds efficiently and specifically to PD-L1 and TGF- in vitro. (A) Structure of M7824 bifunctional fusion pro-tein. M7824 is composed of a fully human programmed death ligand 1 (PD-L1) immunoglobulin G1 (IgG1) monoclonal anti-body and a transforming growth factor– (TGF-)–neutralizing trap moiety, fused to the CH3–C terminus of the IgG via a flexible (Gly4Ser)4Gly linker. (B) Computer-generated model of an Fc1 (gray) linked by a (Gly4Ser)4Gly linker (red) to the ex tracellular domain of TGF-RII (brown). Both TGF-RII moieties can simultaneously bind a disulfide- linked TGF-3 homodimer (cyan/green) (97). (C) Binding of M7824 to PD-L1 on human embryonic kidney (HEK) 293 cells ectopically overexpressing PD-L1. Cells were in-cubated with serial dilutions of M7824, anti–PD-L1, or trap control, followed by a fluorophore-conjugated anti- human IgG sec-ondary antibody. Flow cytometry measured mean fluorescence intensity (MFI) (n = 2 technical replicates). (D) Binding of M7824 to plate-bound TGF-. Serial dilutions of anti–PD-L1 or M7824 were incubated with plate-bound TGF-1, TGF-2, or TGF-3, and binding was assessed via anti-human IgG enzyme- linked immunosorbent assay (ELISA) (n = 2 technical replicates). (E) Binding of M7824 to TGF-2 in solution. Serial dilutions of soluble human recombinant TGF-2 were incubated with plate-bound M7824 or anti–PD-L1, and binding was assessed via anti–TGF-2 ELISA (n = 2 technical replicates). (F) Simultaneous binding of M7824 to PD-L1 and TGF-1. PD-L1-Fc–coated plates were incubated with serial dilutions of M7824 or trap control, followed by biotinylated TGF-1. Binding was evaluated using streptavidin–horseradish peroxidase. Optical densities (OD) were read at 450 nm. Data are means ± SD, and nonlinear best fits are shown (n = 2 technical replicates). All experiments were repeated at least twice.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
3 of 15
after incubation with human embryonic kidney (HEK) 293 cells ecto-pically expressing PD-L1 (fig. S1); this may provide a mechanism for the clearance of M7824-bound TGF-. Finally, the cross-reactivity of the human anti–PD-L1 moiety to murine PD-L1 (41) and of the TGF-RII moiety to murine TGF- (42) allows the evaluation of M7824 in immunocompetent syngeneic tumor models.
M7824 binds efficiently and specifically to PD-L1 and TGF-M7824 bound ectopically expressed PD-L1 on HEK293 cells with a profile similar to that of the anti–PD-L1 antibody [half-maximal ef-fective concentration (EC50) = 48 (average of three lots) and 34 ng/ml, or 0.27 and 0.24 nM, respectively], indicat-ing that the binding of the anti–PD-L1 moiety was not affected. By contrast, no binding activity was observed when the anti–PD-L1 moiety of M7824 was mutated [anti–PD-L1(mut)/TGF- trap, hereafter referred to as the “trap control”] (Fig. 1C). In enzyme- linked immunosorbent assays (ELISAs), M7824 bound plate-bound TGF- 1 and TGF-3 (EC50 = 215.3 and 651.4 ng/ml, respectively), but not plate-bound TGF-2 (Fig. 1D), due to low intrinsic binding af-finity between TGF-2 and TGF-RII (43). M7824 bound TGF- 2 only when TGF-2 was in solution, presumably because this allowed the TGF-2 homodimer to engage in bivalent avidity binding with the oblig-atory TGF-RII dimer in M7824 (EC50 = 26.5 ng/ml) (Fig. 1E). In a two-step ELISA, M7824 captured by plate-bound PD-L1 simultaneously bound TGF-1 in solu-tion with an EC50 of 7.1 ng/ml (Fig. 1F), which was ≈30-fold lower than the EC50 of 215.3 ng/ml when TGF-1 was plate-bound (Fig. 1C).
M7824 inhibits PD-L1 and TGF- signaling in vitroIn an in vitro superantigen stimulation assay with human peripheral blood mono-nuclear cells (PBMCs), M7824, but not the trap control, enhanced T cell activity [measured by interleukin-2 (IL-2) produc-tion] in a dose-dependent manner (EC50 = 71.6 ng/ml) (Fig. 2A). In addition, M7824, but not anti–PD-L1, blocked TGF- ca-nonical signaling [half- maximal inhibi-tory concentration (IC50) = 38.3 ng/ml] in a TGF- SMAD luciferase reporter assay in transfected 4T1 cells (Fig. 2B).
M7824 showed nonlinear pharmacokinetics, depleted TGF- in vivo, achieved high PD-L1 TO, and reduced phosphorylated SMAD2 within the tumorM7824 showed nonlinear pharmacokinet-ics in mice bearing EMT-6 breast tumors
treated with single doses of M7824 (Fig. 2C). As M7824 dose increased, plasma clearance decreased. Analysis of PD-L1 TO in EMT-6 tumors showed that the maximal TO was >90%. Consistent with pharmaco-kinetics, TO remained >90% even at the last time point examined (360 hours) for the high dose cohort (Fig. 2D), demonstrating the effective tumor targeting by M7824.
TGF-1 and TGF-2 were depleted from the plasma of EMT-6 tumor–bearing mice upon M7824 treatment, even at the lowest dose tested (55 g). TGF-1 and TGF-2 concentrations began rising 24 hours after treatment at this low dose (55 g) of M7824, but at higher doses, depletion persisted throughout all time points tested (2 to 192 hours
IL-2
(pg/
ml)
6000
4000
2000
0
M7824 or trap control (ng/ml) M7824 or anti−PD-L1 (ng/ml)
TGF−
βInd
uced
sig
nal (
%)
100
50
0
200,000
150,000
100,000
50,000
0Pla
sma
M78
24 (n
g/m
L)
Hours postinjection0 100 200 300 400
M7824 pharmacokinetics
100
50
00 100 200 300 400
%P
D-L
1 ta
rget
occ
upan
cyHours postinjection
492 µg164 µg55 µg
100,000
50,000
0
6000
4000
2000
00 50 100 150 200
Hours postinjection
300
200
100
0
TGF-β1 TGF-β2 TGF-β3
PD-L1 target occupancy
TGF-
β1 (p
g/m
l)
TGF-
β2 (p
g/m
l)
TGF-
β3 (p
g/m
l)
55-µg M7824164-µg M7824492-µg M7824
NaiveIsotype control
T cell stimulation TGF-β signalingA B
C D
E TGF-β plasma concentration
0 50 100 150 200Hours postinjection
0 50 100 150 200Hours postinjection
1 10 100 1000 10000 10 100 1000
Trap controlM7824
M7824Anti−PD-L1
492 µg164 µg55 µg
Fig. 2. M7824 inhibits PD-L1 and TGF-–dependent pathways in vitro and in vivo. (A) Effect of M7824 on T cell activation in vitro. Serial dilutions of M7824 or trap control were incubated with human peripheral blood mononuclear cells (PBMCs) in the presence of the superantigen staphylococcal enterotoxin A, and supernatants were harvested after 4 days for interleukin-2 (IL-2) ELISA (n = 3 technical replicates). (B) Luciferase assay to evaluate the effect of M7824 on canonical TGF- signaling. Serial dilutions of M7824 or anti–PD-L1 were incubated with SMAD luciferase reporter–transfected 4T1 cells for 16 hours in the presence of recombinant human TGF-1 (5 ng/ml) (n = 3 technical replicates). (C and D) M7824 pharmacokinetics and PD-L1 target occupancy (TO). Jh mice were orthotopically inoculated with 0.3 × 106 EMT-6 cells (day −12) and intravenously injected with a single dose (55, 164, or 492 g) of M7824 on day 0. Plasma samples were collected at different time points between 24 and 336 hours thereafter. (C and D) M7824 concentration in plasma (C), assessed via human IgG ELISA, and percentage PD-L1 TO in tumor-infiltrating T cells (CD3+/CD45+ gate) (D), assessed via flow cytometry (n = 3 mice per time point). In (A) and (B), means ± SEM are shown; in (A) to (D), best fit lines are shown. (E) Effect of M7824 treatment on plasma concentrations of TGF- isoforms. Jh mice were orthotopically inoculated with 0.25 × 106 EMT-6 cells (day −12) and intravenously injected with a single dose of M7824 (55, 164, or 492 g) or isotype control (400 g) on day 0. Plasma was collected 2, 6, 24, 48, 72, and 192 hours after treatment or from treatment-naive mice. Mean TGF-1, TGF-2, and TGF-3 concentrations were determined using a Multiplex Discovery assay (n = 3 mice per time point). All experiments were repeated at least twice.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
4 of 15
after administration) (Fig. 2E). By contrast, TGF-3 concentration was transiently reduced at the 2-hour time point but subsequently remained at control levels through the 80-hour time point. Because the physiological concentration of TGF-3 is very low, these results may reflect low assay sensitivity rather than the ability of M7824 to deplete TGF-3 in vivo.
M7824 treatment significantly lowered phosphorylated SMAD2 (pSMAD2) in MC38 tumors relative to isotype control at 2, 4, and 48 hours after treatment (P = 0.0020, P = 0.0005, and P = 0.0071, re-spectively), as shown by anti-pSMAD2 immunohistochemistry (IHC) (fig. S2). Treatment with a TGF- receptor I (TGF- RI) kinase inhib-itor, used as a positive control, decreased pSMAD2 at 1 to 4 hours after treatment, but pSMAD2 returned to levels near those of isotype control by 48 hours. The incomplete reduction of pSMAD2 is not un-expected, given that the phosphorylation of receptor-regulated SMADs can be induced by TGF- superfamily ligands other than TGF-1 to TGF-3 (22).
The trap control exhibits antitumor activity in vivoAs an initial proof of concept for the TGF- trap moiety, we investigat-ed the antitumor activity of the trap control in a Detroit 562 human pharyngeal carcinoma xenograft model, which is dependent on TGF- for tumor growth (44). Treatment with the trap control elicited a dose- dependent reduction in tumor volume compared with the isotype control (P < 0.0001 for high dose versus equimolar isotype control; day 25 after treatment start), demonstrating the anti tumor activity of the TGF- trap molecule alone and supporting the further testing of the TGF- moiety when fused to anti–PD-L1 in M7824 (fig. S3).
M7824 treatment induces tumor regression in mouse modelsBecause M7824 is a recombinant human protein, it induces a strong immunogenic response in immunocompetent mice if dosed repeatedly for more than 6 days. Therefore, B cell–deficient mouse strains (Jh and Mt−) were used for in vivo studies to enable testing of clinically relevant repeat dosing schedules, unless otherwise indicated.
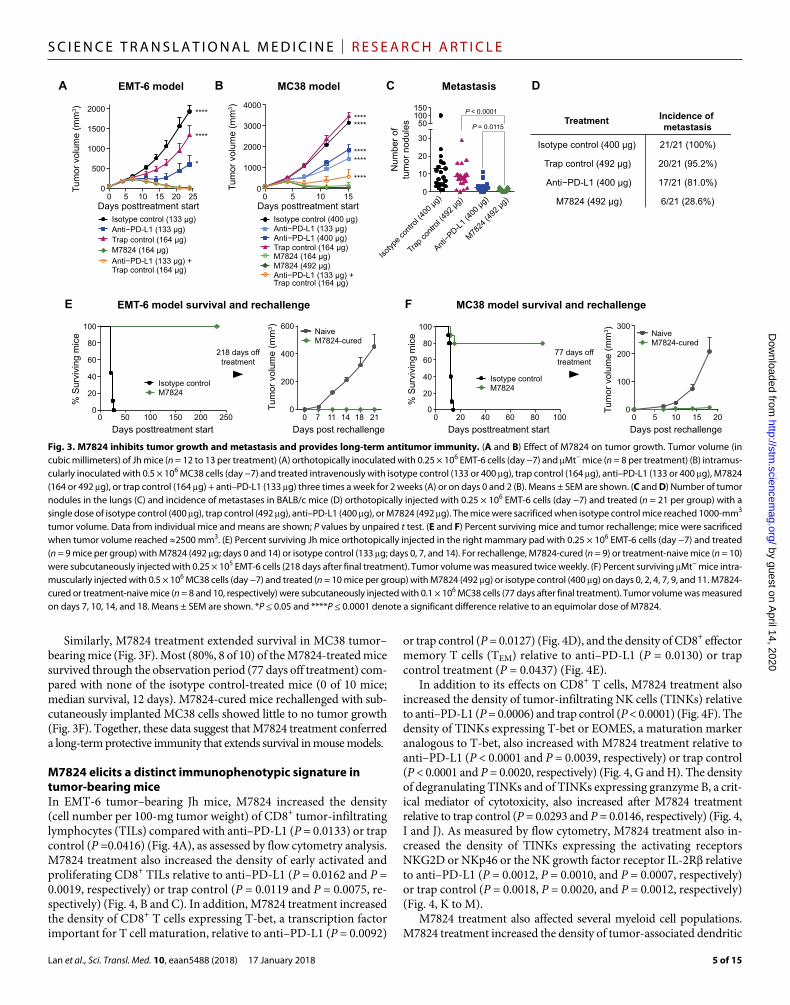
Anti–PD-L1 or trap control treatment partially inhibited tumor growth in an EMT-6 orthotopic breast tumor model. Treatment with M7824 resulted in superior tumor regression compared to treatment with an equimolar dose of either anti–PD-L1 (P = 0.0109; day 24) or the trap control (P < 0.0001; day 24) and was comparable to combina-tion therapy with anti–PD-L1 and the trap control (Fig. 3A). M7824 treatment also resulted in tumor regression in more mice (92%, 12 of 13 mice) than treatment with equimolar doses of either anti–PD-L1 (54%, 7 of 13 mice) or the trap control (8%, 1 of 13 mice) and about the same number of mice as the combination therapy with anti–PD-L1 and the trap control (100%, 13 of 13 mice) (fig. S4A). Tumor regression was rapid and pervasive upon M7824 treatment; although tumors continued to grow early after treatment initiation, possibly due to immune cell infiltration, tumors began regressing by day 11 from volumes as large as 500 mm3 (fig. S4A).
In the MC38 colorectal cancer model, the trap control alone had no antitumor activity. Although anti–PD-L1, at either dose, had statistical-ly significant antitumor activity (P < 0.0001 both doses; day 15), M7824 inhibited tumor growth more strongly than did 133 or 400 g of anti– PD-L1 (P < 0.0001 both doses; day 15) or the trap control (P < 0.0001; day 15) (Fig. 3B), suggesting a more than additive antitumor effect of M7824. Over 171 days of observation, complete tumor regression was observed in 88% (seven of eight) and 50% (four of eight) of the mice treated with 492 or 164 g of M7824, respectively, compared with
no mice (zero of eight) treated with anti–PD-L1 (133 or 400 g) or the trap control alone (164 g) and only 25% (two of eight) of mice treated with a combination of anti–PD-L1 and trap control (fig. S4B). The antitumor efficacy of M7824 was also significantly greater than that of combination therapy with anti–PD-L1 and the trap control (P < 0.0001; day 15), although the serum exposure of the trap control was about three times higher than that of M7824 (fig. S5), likely because the trap control does not bind PD-L1+ cells and internalize.
M7824 treatment also demonstrated superior antitumor efficacy in subcutaneous tumor models (fig. S6). In EMT-6 tumor–bearing mice, M7824 treatment significantly inhibited tumor growth relative to an equimolar dose of either the trap control (P < 0.0001; day 18) or anti–PD-L1 (P = 0.0088, every other day dosing, or P < 0.0001, twice per week dosing of M7824; day 18) and prolonged survival (fig. S6, A to C). In MC38 tumor–bearing mice, M7824 treatment (164- and 492-g doses) significantly reduced tumor volume relative to an equi-molar dose of trap control (P < 0.0001 both doses; day 19) or anti–PD-L1 (P < 0.0001 both doses; day 19) and reduced tumor weight at day 19 relative to trap control (P = 0.0002 and P < 0.0001, respectively) and anti–PD-L1 treatment (P = 0.0152 and P < 0.0001, respectively) (fig. S6, D to F). M7824 treatment showed faster treatment response kinetics and improved in vivo efficacy in the EMT-6 orthotopic tumor model (Fig. 3A and fig. S4A) relative to the subcutaneous tumor model (fig. S6, A and B). These results are encouraging, given that orthotopic models are considered more clinically relevant and better predictive models of clinical drug efficacy.
To further confirm the antitumor activity of M7824, we also per-formed experiments in wild-type, non–B cell–deficient mice. We tested the effects of single doses (164 and 492 g) of M7824, which would not induce high titers of antidrug antibodies, in the MC38 and EMT-6 tumor models. Under these more limited therapy condi-tions, the 492-g dose of M7824 still exhibited superior antitumor activity compared to anti–PD-L1 in mice bearing MC38 (P = 0.0010; day 9) (fig. S7, A and B) or EMT-6 (P < 0.0001; day 20) tumors (fig. S7, C and D), although there were no significant differences with the 164-g dose.
M7824 suppresses spontaneous metastasis in an orthotopic breast cancer mouse modelTGF- plays a key role in promoting tumor metastasis (22). In EMT-6 tumor–bearing wild-type mice, a single dose of M7824 more effec-tively reduced the number and incidence of spontaneous lung tumor metastases than did treatment with anti–PD-L1 (P = 0.0115, number of nodules) or the trap control (P < 0.0001, number of nodules) (Fig. 3, C and D). M7824 treatment also decreased the incidence and number of lung metastases in orthotopic 4T1 and Renca tumor models (fig. S8).
M7824 provides long-term protective antitumor immunity in tumor rechallenge mouse modelsM7824 treatment markedly extended survival in the EMT-6 orthotopic model relative to isotype control treatment; all M7824-treated mice (nine of nine) survived through the observation period (218 days off treatment) compared with none (zero of nine) of the isotype control-treated mice (median survival, 21 days) (Fig. 3E). When M7824-cured mice were rechallenged subcutaneously with EMT-6 cells without additional M7824 treatment, they did not develop tumors, whereas treatment-naive mice rapidly developed tumors (Fig. 3E).
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from
Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
5 of 15
Similarly, M7824 treatment extended survival in MC38 tumor–bearing mice (Fig. 3F). Most (80%, 8 of 10) of the M7824-treated mice survived through the observation period (77 days off treatment) com-pared with none of the isotype control-treated mice (0 of 10 mice; median survival, 12 days). M7824-cured mice rechallenged with sub-cutaneously implanted MC38 cells showed little to no tumor growth (Fig. 3F). Together, these data suggest that M7824 treatment conferred a long-term protective immunity that extends survival in mouse models.
M7824 elicits a distinct immunophenotypic signature in tumor-bearing miceIn EMT-6 tumor–bearing Jh mice, M7824 increased the density (cell number per 100-mg tumor weight) of CD8+ tumor-infiltrating lymphocytes (TILs) compared with anti–PD-L1 (P = 0.0133) or trap control (P =0.0416) (Fig. 4A), as assessed by flow cytometry analysis. M7824 treatment also increased the density of early activated and proliferating CD8+ TILs relative to anti–PD-L1 (P = 0.0162 and P = 0.0019, respectively) or trap control (P = 0.0119 and P = 0.0075, re-spectively) (Fig. 4, B and C). In addition, M7824 treatment increased the density of CD8+ T cells expressing T-bet, a transcription factor important for T cell maturation, relative to anti–PD-L1 (P = 0.0092)
or trap control (P = 0.0127) (Fig. 4D), and the density of CD8+ effector memory T cells (TEM) relative to anti–PD-L1 (P = 0.0130) or trap control treatment (P = 0.0437) (Fig. 4E).
In addition to its effects on CD8+ T cells, M7824 treatment also increased the density of tumor-infiltrating NK cells (TINKs) relative to anti–PD-L1 (P = 0.0006) and trap control (P < 0.0001) (Fig. 4F). The density of TINKs expressing T-bet or EOMES, a maturation marker analogous to T-bet, also increased with M7824 treatment relative to anti–PD-L1 (P < 0.0001 and P = 0.0039, respectively) or trap control (P < 0.0001 and P = 0.0020, respectively) (Fig. 4, G and H). The density of degranulating TINKs and of TINKs expressing granzyme B, a crit-ical mediator of cytotoxicity, also increased after M7824 treatment relative to trap control (P = 0.0293 and P = 0.0146, respectively) (Fig. 4, I and J). As measured by flow cytometry, M7824 treatment also in-creased the density of TINKs expressing the activating receptors NKG2D or NKp46 or the NK growth factor receptor IL-2R relative to anti–PD-L1 (P = 0.0012, P = 0.0010, and P = 0.0007, respectively) or trap control (P = 0.0018, P = 0.0020, and P = 0.0012, respectively) (Fig. 4, K to M).
M7824 treatment also affected several myeloid cell populations. M7824 treatment increased the density of tumor-associated dendritic
Isotyp
e con
trol (4
00 µg
)
Anti−P
D-L1 (4
00 µg
)
Trap c
ontro
l (492
µg)
M7824
(492
µg)
Num
ber o
ftu
mor
nod
ules
15010050
30
20
10
0
P < 0.0001
P = 0.0115
Days posttreatment start Days post rechallenge0 50 100 150 200 250
Isotype controlM7824
NaiveM7824-cured
EMT-6 model survival and rechallenge
% S
urvi
ving
mic
e
100
80
60
40
20
0 Tum
or v
olum
e (m
m3 )
218 days offtreatment
Metastasis
% S
urvi
ving
mic
e
100
80
60
40
20
00 20 40 60 80 100
Isotype controlM7824
Days posttreatment start
Tum
or v
olum
e (m
m3 ) 300
200
100
00 5 10 15 20Days post rechallenge
NaiveM7824-cured
77 days offtreatment
Isotype control (400 µg)
Anti−PD-L1 (400 µg)
Trap control (492 µg)
M7824 (492 µg)
Incidence of metastasis
21/21 (100%)
20/21 (95.2%)
17/21 (81.0%)
6/21 (28.6%)
Treatment
MC38 model survival and rechallenge
C D
E F
600
400
200
00 7 11 14 18 21
Tum
or v
olum
e (m
m3 ) 2000
1500
1000
500
00 5 10 15 20 25
Days posttreatment startIsotype control (133 µg)Anti−PD-L1 (133 µg) Trap control (164 µg) M7824 (164 µg)
Trap control (164 µg) Anti−PD-L1 (133 µg) +
Isotype control (400 µg)Anti−PD-L1 (133 µg)
Trap control (164 µg) M7824 (164 µg)
Trap control (164 µg) Anti−PD-L1 (133 µg) +
Anti−PD-L1 (400 µg)
M7824 (492 µg)
Tum
or v
olum
e (m
m3 ) 4000
3000
2000
1000
00 5 10 15
Days posttreatment start
EMT-6 model MC38 modelA B
*
****
****
****
****
********
****
Fig. 3. M7824 inhibits tumor growth and metastasis and provides long-term antitumor immunity. (A and B) Effect of M7824 on tumor growth. Tumor volume (in cubic millimeters) of Jh mice (n = 12 to 13 per treatment) (A) orthotopically inoculated with 0.25 × 106 EMT-6 cells (day −7) and Mt− mice (n = 8 per treatment) (B) intramus-cularly inoculated with 0.5 × 106 MC38 cells (day −7) and treated intravenously with isotype control (133 or 400 g), trap control (164 g), anti–PD-L1 (133 or 400 g), M7824 (164 or 492 g), or trap control (164 g) + anti–PD-L1 (133 g) three times a week for 2 weeks (A) or on days 0 and 2 (B). Means ± SEM are shown. (C and D) Number of tumor nodules in the lungs (C) and incidence of metastases in BALB/c mice (D) orthotopically injected with 0.25 × 106 EMT-6 cells (day −7) and treated (n = 21 per group) with a single dose of isotype control (400 g), trap control (492 g), anti–PD-L1 (400 g), or M7824 (492 g). The mice were sacrificed when isotype control mice reached 1000-mm3 tumor volume. Data from individual mice and means are shown; P values by unpaired t test. (E and F) Percent surviving mice and tumor rechallenge; mice were sacrificed when tumor volume reached ≈2500 mm3. (E) Percent surviving Jh mice orthotopically injected in the right mammary pad with 0.25 × 106 EMT-6 cells (day −7) and treated (n = 9 mice per group) with M7824 (492 g; days 0 and 14) or isotype control (133 g; days 0, 7, and 14). For rechallenge, M7824-cured (n = 9) or treatment-naive mice (n = 10) were subcutaneously injected with 0.25 × 105 EMT-6 cells (218 days after final treatment). Tumor volume was measured twice weekly. (F) Percent surviving Mt− mice intra-muscularly injected with 0.5 × 106 MC38 cells (day −7) and treated (n = 10 mice per group) with M7824 (492 g) or isotype control (400 g) on days 0, 2, 4, 7, 9, and 11. M7824-cured or treatment-naive mice (n = 8 and 10, respectively) were subcutaneously injected with 0.1 × 106 MC38 cells (77 days after final treatment). Tumor volume was measured on days 7, 10, 14, and 18. Means ± SEM are shown. *P ≤ 0.05 and ****P ≤ 0.0001 denote a significant difference relative to an equimolar dose of M7824.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
6 of 15
BA CD8+ tumor-infiltratinglymphocytes (TILs)
C D
CD
8+ /CD
69+ T
ILs
(×10
6 /100
-mg
tum
or)
0.000
0.010
0.004
0.008
Early activation of CD8+ TILs
0.002
0.006
CD
8+ T c
ells
(×10
6 /100
-mg
tum
or)
0.00
0.03
0.01
0.02
P = 0.0133P = 0.0416
P = 0.0092
P = 0.0162P = 0.0119
P = 0.0062
CD
8+ /Ki-6
7+ TIL
s(×
106 /1
00-m
g tu
mor
)
0.000
0.004
0.002
0.001
0.003
P = 0.0019P = 0.0075
P = 0.0003
Proliferation ofCD8+ TILs
CD
8+ /T-b
et+ T
ILs
( ×10
6 /100
-mg
tum
or)
0.000
0.008
0.004
0.002
0.006P = 0.0092
P = 0.0127P = 0.0019
T-bet+
CD8+ TILsE
GF
CD8+ TEM
cells
H I
DX
5+ cel
ls(×
106 /1
00-m
g tu
mor
)
0.00
0.06
0.04
Tumor-associated naturalkiller cells (TINKs)
0.02
CD
8+ /CD
44hi
gh/C
D62
low T
ILs
(×10
6 /100
-mg
tum
or)
0.000
0.025
0.010
0.020P = 0.0130
P = 0.0437P = 0.0067
P = 0.0006P < 0.0001
P < 0.0001
DX
5+ /CD
107a
+ cel
ls(×
106 /1
00-m
g tu
mor
)
0.000
0.008
0.004
0.002
0.006
P = 0.1173P = 0.0293
P = 0.0021
Degranulation ofTINKs
DX
5+ /T-b
et+ c
ells
(×10
6 /100
-mg
tum
or)
0.00
0.04
0.02
0.01
0.03
P < 0.0001P < 0.0001
P < 0.0001
T-bet+ TINKs J
DX
5+ /EO
ME
S+ c
ells
(×10
6 /100
-mg
tum
or)
P = 0.0039P = 0.0020
P = 0.0016
EOMES+ TINKs
0.000
0.015
0.010
0.005
0.005
0.015
LK
Granzyme B+ TINKs
M N
DX
5+ /CD
314+ c
ells
(×10
6 /100
-mg
tum
or)
0.0
0.4
0.3
NKG2D+ TINKs
0.2
DX
5+ /gra
nzym
e B
+ cel
ls(×
106 /1
00-m
g tu
mor
)
0.000
0.005
0.002
0.004
P = 0.0515P = 0.0146
P = 0.0040
P = 0.0012P = 0.0018
P = 0.0010
DX
5+ /CD
335+ c
ells
(×10
6 /100
-mg
tum
or)
0.00
0.10
0.06
0.04
0.08
P = 0.0010P = 0.0020
P = 0.0009NKp46+ TINKs
DX
5+ /CD
122+ c
ells
(×10
6 /100
-mg
tum
or)
0.0
0.4
0.2
0.1
0.3
P = 0.0007P = 0.0012
P = 0.0006
IL-2Rβ+ TINKs O
CD
11b+ /C
D11
c+ /F4/
80- c
ells
( ×10
6 /100
-mg
tum
or)
P < 0.0001P = 0.0060
P = 0.0001
Tumor-associateddendritic cells (TADCs)
0.001
0.003
0.1 0.02
0.00
0.06
0.04
0.08
0.02
QP
Mature TADCs
R S
0.0
1.5
1.0
Tumor-associatedneutrophils (TANs)
0.5
CD
86+ /C
D80
+ DC
s( ×
106 /1
00-m
g tu
mor
)
0.00
0.04
0.01
0.03P = 0.0003
P = 0.0307P = 0.0030
P = 0.0146P = 0.0260
P = 0.0870
0.0
0.6
0.2
0.4
P < 0.0001P < 0.0001
P < 0.0001
Tumor-associated monocytes
0.000
0.015
0.005
0.010P = 0.0097
P = 0.0243P = 0.0007
Tumor M1 Cells T
0.02
M1/M
2 rat
io(×
106 /1
00-m
g tu
mor
)
P = 0.0850P = 0.0602
P = 0.0053
0.0
0.6
0.4
0.2
M1/M
2 ratio
CD
11b+ /F
4/80
+ /I-A
d+ (M
1) ce
lls(×
106 /1
00-m
g tu
mor
)
CD
11b+ /L
y-6C
high
/Ly-
6G-
cells
(×10
6 /100
-mg
tum
or)
CD
11b+ /L
y-6C
low/L
y-6G
high
ce
lls (×
106 /1
00-m
g tu
mor
)
CD
11b+ /F
4/80
+ /Ly-
6C−
/PD
-1+ c
ells
(×10
6 /100
-mg
tum
or)
P = 0.0546P = 0.1125
P = 0.0028
0.000
0.020
0.015
0.010
PD1+ tumor-associatedmacrophages (TAMs)
0.005
Isotype control (400 µg) Trap control (492 µg) Anti–PD-L1 (400 µg) M7824 (492 µg) U
CD8 immunohistochemistry
Immunophenotyping analysis
Isotyp
e con
trol
Trap c
ontro
l
Anti−P
D-L1
M7824
P = 0.0452P > 0.9999
P = 0.032715
10
5
0
CD
8+ cel
ls(%
of t
otal
cel
ls)
CD8+ TIL quantification
Isotype control (400 µg) Trap control (492 µg) Anti−PD-L1 (400 µg) M7824 (492 µg)
V
Fig. 4. M7824 elicits a distinct immunophenotypic signature in EMT-6 tumor–bearing mice. (A to V) Jh mice were subcutaneously inoculated with 0.25 × 106 EMT-6 cells on day −10 (A to T) or day −11 (U and V) and treated intravenously with isotype control (400 g), trap control (492 g), anti–PD-L1 (400 g), or M7824 (492 g) on days 0, 4, 7, 11, and 13 (A to T) and sacrificed on day 14 or on days 0, 4, 7, 11, and 14 (U and V) and sacrificed on day 15. (A to T) Flow cytometry analysis of dissociated tumors (n = 8 mice per group). Abso-lute numbers of cells per 100 mg of tumor tissue were calculated for populations of (A to E) CD8+ tumor-infiltrating lymphocytes (TILs), including absolute number (A), early activation (B), proliferation (C), T-bet+ CD8+ TILs (D), and CD8+ effector memory T cells (TEM) cells (E). (F to M) Tumor-associated natural killer cells (TINKs), including absolute number (F), T-bet+ (G), EOMES+ (H), degranulation (I) of granzyme B+ (J), NKG2D+ (K), NKp46+ (L), and IL-2R+ (M) TINKs. (N to T) Myeloid cells, including absolute number (N) and mature tumor-associated dendritic cells (TADCs) (O), tumor-associated neutrophils (TANs) (P), tumor-associated monocytes (Q), M1 cells (R), M1/M2 ratio (S), and PD1+ tumor-associated macrophages (TAMs) (T). P values were determined by unpaired t test. (U and V) Representative images of anti-CD8a immunohistochemistry (IHC) of tumors (n = 5 mice per group) (U) and percentages of CD8+ cells (V) are shown. Scale bars, 100 m. P values were determined by Kruskal-Wallis test.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
7 of 15
cells (TADCs) and of mature TADCs relative to anti–PD-L1 (P < 0.0001 and P = 0.0003, respectively) and trap control (P = 0.0060 and P = 0.0307, respectively) (Fig. 4, N and O). In addition, M7824 decreased the density of tumor-associated neutrophils (TANs) and in-creased the density of tumor- associated monocytes relative to anti–PD-L1 (P = 0.0146 and P < 0.0001, respectively) or trap control (P = 0.0260 and P < 0.0001, respectively) (Fig. 4, P and Q). Finally, M7824 treatment increased the density of M1 macrophages relative to anti–PD-L1 (P = 0.0097) or trap control (P = 0.0243) (Fig. 4R) but did not significantly increase the ratio of M1 to M2 macrophages (M1/M2) relative to anti–PD-L1 or trap control (Fig. 4S). M7824 treatment sig-nificantly decreased the density of tumor- associated macrophages (TAMs) expressing PD-1, relative to iso type control, whereas anti–PD-L1 monotherapy did not (Fig. 4T); this is meaningful, given the re-duced phagocytic capacity and antitumor activity of PD-1+ TAMs (45).
In a separate set of flow cytometry experiments, the proportions of mature dendritic cells (DCs) and CD8+ T cells–expressing EOMES positively correlated with M7824 dosage and dosing frequency in the EMT-6 tumor model (fig. S9). IHC analysis of EMT-6 tumor–bearing Jh mice also revealed an increase in the percentage of CD8+ TILs with M7824 treatment relative to anti–PD-L1 (Fig. 4, U and V).
We also analyzed immunophenotypic changes in the MC38 model. In MC38 tumor–bearing wild-type mice, M7824 treatment increased the proportions of CD8+ TILs and CD8+ TILs–expressing T-bet relative to anti–PD-L1 and trap control (fig. S10, A and B). M7824 also increased the proportion of CD8+ TILs specific for p15E (an endogenous retroviral antigen expressed in MC38 tumor cells), of CD8+ TEM, and of TINKs relative to trap control (fig. S10, C to E). M7824 treatment also decreased the proportions of TANs relative to anti–PD-L1 and of tumor-associated myeloid-derived suppressor cells relative to anti–PD-L1 or trap control (fig. S10, F and G).
M7824 promotes the expression of immune cell signature genesTo examine changes in gene expression induced by M7824 treatment, we analyzed EMT-6 tumor tissue by RNA sequencing (RNA-seq) (Fig. 5A). Compared to isotype control treatment, M7824 resulted in differential expression (defined as P < 0.05 and a fold change of >1.5:1) of 3,605 of 39,012 genes evaluated. Anti–PD-L1 resulted in a similarly high number (4064) of differentially expressed genes, whereas trap control treatment resulted in 720 differentially expressed genes. These findings suggest that most of the gene expression changes induced by M7824 were mediated by its anti–PD-L1 moiety. Pathways up-regulated by M7824 treatment relative to isotype control include interferon- (IFN-) response and extracellular matrix remodeling.
To further evaluate the effects of M7824 treatment on the immune infiltrate, we quantified the expression of gene signatures associated with distinct immune cell types (Fig. 5, B to E) and IFN- and IFN- responses (Fig. 5, F and G). The genes comprising each immune sig-nature are listed in table S1; immune cell signatures are based on pub-lished lists (46, 47), and IFN response genes are from the Broad Institute’s hallmark gene sets (48). M7824 treatment increased signature scores, defined as the mean log2(fold change) among all genes in the signature, for T cells, NK cells, macrophages, and DCs relative to anti–PD-L1 (P < 0.0001, P < 0.0001, P = 0.0207, and P = 0.0010, respectively) or the trap control (P = 0.0042, P = 0.0019, P = 0.0008, and P < 0.0001, respec-tively) (Fig. 5, B to E). M7824 also increased the expression of IFN- and IFN- response signatures relative to anti–PD-L1 (P = 0.0007 and P < 0.0001, respectively) or the trap control (P = 0.0301 and P = 0.0004, respectively) (Fig. 5, F and G).
Analysis of individual genes revealed that M7824 treatment in-creased the expression of granzyme A (GzmA), granzyme B (GzmB), and perforin 1 (Prf1), which are critical mediators of CD8+ T cell and NK cell cytotoxicity (49), relative to anti–PD-L1 (P = 0.0045, P < 0.0001, and P < 0.0001, respectively) and trap control (P = 0.0195, P = 0.0432, and P = 0.0268, respectively) (Fig. 5, H to J, and heat map inset in Fig. 5A).
CD8+ T cells and NK cells mediate the antitumor activity of M7824In a series of antibody-based immune cell depletion studies in MC38 tumor–bearing mice, we found that the antitumor efficacy of M7824 was abrogated by the depletion of CD8+ T cells with an anti-CD8 (2.43) antibody and by the depletion of NK cells with an anti-NK (ASGM1) antibody (Fig. 6A). By contrast, CD4+ T cell depletion with anti-CD4 (GK1.5) did not hinder the antitumor efficacy of M7824 (Fig. 6A and fig. S11A).
Because M7824 contains a fully human IgG1, it has the potential to mediate antibody-dependent cellular cytotoxicity (ADCC) against PD-L1–expressing tumor cells as a secondary mechanism of tumor growth inhibition. However, we observed no difference in antitumor activity between MC38 tumor–bearing mice treated with M7824 and those treated with an aglycosylated M7824 (through mutation to remove the N-glycan in the CH2 region), which was confirmed to be devoid of effector function in vitro (Fig. 6, B and C, and fig. S11B). These findings suggest that ADCC did not play a major role in the antitumor activity of M7824 and are consistent with the weaker ADCC activity of M7824 in vitro compared to anti–PD-L1 (Fig. 6B).
M7824 treatment reduces -SMA expression in mouse tumorsIncreased TGF- activity induces the expression of -smooth muscle actin (-SMA), a marker of cancer-associated fibroblasts (CAFs), which can contribute to drug resistance and are emerging as immuno therapy targets (50, 51). To evaluate whether M7824 treatment altered -SMA expression, we performed -SMA IHC on EMT-6 tumor sections (Fig. 7). M7824 treatment significantly reduced -SMA expression relative to isotype control or anti–PD-L1 treatment (P = 0.0080 and P = 0.0197, respectively) but not to the trap control (Fig. 7, B and D), suggesting that the trap moiety was likely the primary mediator of reduced -SMA expression after M7824 treatment.
We next used picrosirius red staining to investigate the effect of M7824 treatment on the deposition of collagen, a fibrous protein produced by CAFs. Although quantitative analysis did not show significant reduction in picrosirius red staining with M7824 treat-ment relative to isotype control (Fig. 7E), collagen deposition seen in the IHC images suggests that there may be morphological differences between control and M7824-treated tumors (Fig. 7C). Specifically, the collagen network in isotype control-treated tumors appears to have thicker fibers that are densely arranged, compared with the less structured collagen network structure seen in M7824-treated tumors. A similar reduction in collagen networks was observed af-ter trap control or anti–PD-L1 treatment relative to isotype control (Fig. 7C), possibly resulting from reduced CAF activity.
M7824 is an effective combination partner for radiotherapy or chemotherapy in mouse modelsPrevious studies have shown that radiation and chemotherapy can in-duce TGF- (52, 53) and enhance PD-L1 expression (54–56), suggesting
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
8 of 15
that M7824 may be effective in combination therapy regi-mens. In MC38 tumor– bearing mice, treatment with a single low dose of M7824 or local fractionated radiation moderately reduced tumor volume and tumor weight compared with isotype control treatment (Fig. 8, A and B). However, combination therapy with M7824 and radia-tion reduced tumor volume and tumor weight relative to M7824 (P < 0.0001 and P = 0.0014, respectively; day 10) or radiation therapy alone (P < 0.0001 and P < 0.0001; days 10 and 14, respectively) (Fig. 8, A and B, and fig. S12A). Enzyme-linked immunospot (ELISpot) analysis showed that mice treated with the combination therapy had an increased frequency of p15E-reactive, IFN-–producing CD8+ T cells compared with M7824 (5.3-fold; P < 0.0001) or radiation therapy alone (6.7-fold; P < 0.0001), whereas mice treated with either monotherapy did not have a sig-nificant induction relative to isotype control (Fig. 8C).
Localized radiation therapy can elicit anticancer ef-fects at distal sites, a phenomenon known as the abscopal effect. To test whether M7824 can potentiate the abscopal effect of radiation therapy, C57BL/6 mice were inoculated with MC38 cells at two sites to generate a primary, intra-muscular MC38 tumor in the thigh and a secondary, sub-cutaneous MC38 tumor on the opposite flank (Fig. 8D). Localized fractionated radiation was applied only to the primary tumor. As expected, combination therapy with radiation and a single dose of M7824 reduced primary tumor volume relative to M7824 or radiation alone (P < 0.0001 for both; day 14) (Fig. 8E and fig. S12B). However, combination therapy also reduced secondary tumor volume relative to M7824 or radiation alone (P = 0.0066 and P = 0.0006, respectively; day 14) (Fig. 8F and fig. S12C), in-dicating that M7824, together with radiation, induced an
****
Macrophages
A
B C D
Isotype controlIsotype controlIsotype control Trap controlTrap controlTrap control Anti−PD-L1Anti−PD-L1Anti−PD-L1 M7824M7824M7824
M78
24 v
s. a
nti−
PD
-L1
M78
24 v
s. a
nti−
PD
-L1
M78
24 v
s. a
nti−
PD
-L1
M78
24 v
s. tr
ap c
ontro
lM
7824
vs.
trap
con
trol
M78
24 v
s. tr
ap c
ontro
lM
7824
vs.
isot
ype
cont
rol
M78
24 v
s. is
otyp
e co
ntro
lM
7824
vs.
isot
ype
cont
rol
Ant
i−P
D-L
1 vs
. iso
type
con
trol
Ant
i−P
D-L
1 vs
. iso
type
con
trol
Ant
i−P
D-L
1 vs
. iso
type
con
trol
Trap
con
trol v
s. is
otyp
e co
ntro
lTr
ap c
ontro
l vs.
isot
ype
cont
rol
Trap
con
trol v
s. is
otyp
e co
ntro
l
Up-
regu
late
dU
p-re
gula
ted
Up-
regu
late
dD
own-
regu
late
dD
own-
regu
late
dD
own-
regu
late
dN
o si
gnifi
cant
cha
nge
No
sign
ifica
nt c
hang
eN
o si
gnifi
cant
cha
nge
Sig
natu
re s
core
0.0
−0.4
0.4
DCs
NK cellsT cells
Sig
natu
re s
core
Sig
natu
re s
core
Sig
natu
re s
core
0.2
−0.2
0.6
0.0
−0.4
0.4
0.2
−0.2
0.0
−1.0
1.0
0.5
−0.5
0.0
−1.0
1.0
0.5
−0.5
E F
****
**
****
****
**
***
*
***
****
******
Sig
natu
re s
core
0.0
−0.4
0.4
0.2
−0.2
−0.6
Sig
natu
re s
core
0.0
−0.4
0.4
0.2
−0.2
−0.6
IFN-α response IFN-γ response
GzmAGzmAGzmA
GzmBGzmBGzmB
Prf1Prf1Prf1
Log 2 (
fold
cha
nge)
Log 2 (
fold
cha
nge)
Log 2 (
fold
cha
nge)
222
111
000
−1−1−1
−2−2−2
G
Immune signature scores
****
****
***
****
***
*
H I J
Isotyp
e con
trol
Trap c
ontro
l
Anti−P
D-L1
M7824
4
0
1
3
2
2.5
2.0
1.5
1.0
0.5
0.0
2.0
1.5
1.0
0.5
0.0
GzmA Prf1
Gra
nzym
e A
(TP
M)
Gra
nzym
e B
(TP
M)
Per
forin
(TP
M)
Gene expression analysis
Isotyp
e con
trol
Trap c
ontro
l
Anti−P
D-L1
M7824
Isotyp
e con
trol
Trap c
ontro
l
Anti−P
D-L1
M7824
Dif
fere
nti
al g
ene
exp
ress
ion
GzmB
****
*
**
****
*
****
****
*
****
Fig. 5. M7824 promotes gene expression associated with innate and adaptive immune cells. RNA sequencing (RNA-seq) analysis of tumor tissue from BALB/c mice (n = 10 mice per group) that were ortho-topically inoculated with 0.25 × 106 EMT-6 cells on day −20 and treat-ed intravenously with isotype control (400 g), trap control (492 g), anti–PD-L1 (400 g), or M7824 (492 g) on days 0, 1, and 2 and sacri-ficed on day 6. (A) Heat map of gene expression changes for all signifi-cantly differentially expressed genes (defined as P < 0.05 and a fold change of >1.5) from RNA-seq analysis. The colors in each box repre-sent the log2(fold change) in the expression of a gene after treatment relative to the median isotype control; rows represent individual genes, and columns represent individual mice. The five columns on the left of the heat map show significance of up-regulation or down- regulation of genes in treatment comparisons. Inset shows magnified heat map of expression of GzmA (granzyme A), GzmB (granzyme B), and Prf1 (perforin 1). (B to G) Gene expression signatures associated with T cells (B), natural killer (NK) cells (C), macrophages (D), dendritic cells (DCs) (E), and interferon- (IFN-) (F) and IFN- (G) responses. Signature scores [defined as the mean log2(fold change) among all genes in the signature] are presented as box plots. P values were generated with the ROAST method (96). (H to J) Gene expression [transcript parts per million (TPM)] was evaluated for GzmA (H), GzmB (I), and Prf1 (J). False discovery rate–corrected P values were determined by DESeq2 (98). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 denote a significant difference relative to the M7824 treatment.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
9 of 15
abscopal effect. Neither radiation alone nor a single low dose of M7824 alone inhibited secondary tumor growth relative to vehicle treatment. Neither anti–PD-L1 nor trap control significantly poten-tiated the abscopal effect of radiation in the MC38 model (fig. S13).
Combination treatment with M7824 and the chemotherapies oxaliplatin and 5-fluorouracil (Ox/5-FU) reduced tumor volume and tumor weight more effectively than M7824 monotherapy (P < 0.0001 and P = 0.0007, respectively; day 18) or Ox/5-FU (P < 0.0001 and P = 0.0065, respectively; day 18) (Fig. 8, G and H, and fig. S14). Combination treatment with M7824 and Ox/5-FU also increased the frequency of p15E- reactive, IFN-–producing CD8+ T cells compared with M7824 (2.9-fold; P = 0.0003) or Ox/5-FU therapy alone (3.4-fold; P = 0.0002), neither of which had a significant effect relative to iso-type control (Fig. 8I).
DISCUSSIONTGF- can play both tumor-promoting and tumor-suppressive roles, de-pending on the tumor type and stage, genetic/epigenetic changes in the TGF- pathway, and the larger microenvironmental context (22, 27, 57). This duality has called into question the targeting of TGF- as a therapeutic strategy in cancer (57). However, during malignant pro-gression, certain tumors become resistant to the cytostatic effects of TGF- and instead subvert the pathway to promote EMT (58). Tumor cells also often lose direct responsiveness to TGF- because TGF-R is mutated, deleted, or down-regulated (59, 60). Under such conditions, TGF- instead acts directly on immune cells, inhibiting their anti-tumor activity and driving the transition of TGF- from a tumor-
suppressive to a tumor-promoting role (31, 32).
The bifunctional nature of M7824 allows the targeting of both TGF- and PD-1/PD-L1 immunosuppressive path-ways simultaneously. Although the anti-tumor activity of the trap control and anti–PD-L1 monotherapies varied be-tween tumor models, M7824 treatment demonstrated superior antitumor ac-tivity relative to monotherapies, even in models in which trap control or anti– PD-L1 treatment alone did not trigger substantial antitumor responses. Some potential limitations to our study are that, due to the immunogenicity of M7824 in mice, the number of doses that can be administered to wild-type mice is limited and that, to allow repeated dosing beyond 1 week, we had to use B cell–deficient mouse strains. The fact that we observed robust antitumor activity of M7824 in both wild-type and B cell–deficient mice suggests that B cell–deficient mice are appropriate hosts for MC38 and EMT-6 tumor models.
The superior efficacy of M7824 could be mediated by related but nonredun-dant functions of the TGF- and PD-1/PD-L1 pathways. In our studies, deple-tion of CD8+ T cells or NK cells eliminat-
ed the antitumor activity of M7824, suggesting major roles for these two cell types in the antitumor effects of M7824. Immunopheno-typic and RNA-seq analyses suggest that M7824 promoted the antitu-mor effects of CD8+ T cells and NK cells by enhancing their cytotoxic activity, con sistent with previous reports of enhanced cytotoxic activ-ity of these cells after individual blockade of PD-L1/PD-1 or TGF- (34, 35, 61–65). These results align with previous findings that TGF- signaling in T cells directly regulates their expression of Prf1, GzmA, and GzmB, among other genes involved in cytotoxicity (34). TGF- also inhibits granzyme expression in NK cells (65, 66).
Despite the importance of NK cells to M7824’s antitumor activity, ADCC, which is primarily mediated by NK cells, did not contribute significantly. This can be partly explained by our in vitro data showing that M7824 has reduced ADCC relative to anti–PD-L1, presumably because the Fc in the antibody fusion protein is less accessible for Fc gamma receptor engagement. Because TGF- suppresses the expression of NK cell–activating receptors such as NKp46 (67) and NKG2D (68), NK cells relieved of TGF-–mediated immunosuppres-sion may contribute to the antitumor activity of M7824 through en-hanced spontaneous killing and antigen presentation by DCs. This therapeutic enhancement of NK cell effector function may also con-tribute to the decreased metastasis seen in M7824-treated mice, con-sistent with a study showing that the inhibition of TGF- signaling promoted NK cell–mediated suppression of metastasis (35). Our finding that M7824 treatment increases the number of NK cells ex-pressing activating receptors supports this hypothesis.
The antitumor effects of M7824 may also involve several myeloid cell populations. PD-L1, in addition to being expressed in cancer
Tum
or v
olum
e (m
m3 ) 3000
2000
1000
0
Isotype control (400 µg)M7824 (492 µg)M7824 + anti-CD4M7824 + anti-CD8M7824 + anti-CD4/CD8M7824 + anti-ASGM1
Days0 10 30 4020
Days7 140 21
A B
Tum
or v
olum
e (m
m3 ) 2500
2000
1000
0
500
1500
Isotype control (133 µg)M7824 (55 µg)Aglycosylated M7824 (55 µg)M7824 (164 µg)Aglycosylated M7824 (164 µg)
27
Aglycosylated M7824M7824
Isotype controlAnti−PD-L1
Log concentration (ng/ml)−1 0 1 2 3
% L
ysis
0
−20
20
40
60
80
CImmune cell depletion
********************
ADCC in vitro ADCC in vivo
Fig. 6. Innate and adaptive immunity, but not ADCC, contributes to M7824 antitumor activity. (A) Mt− mice were injected intramuscularly with 0.5 × 106 MC38 cells on day −7 and treated (n = 10 mice per group) on day 0 with isotype control (400 g), M7824 (492 g), or M7824 (492 g) dosed with anti-murine CD4 (GK1.5; 100 g), anti-murine CD8 (2.43; 100 g), anti-murine CD4 + anti-murine CD8 (100 g each), or anti-murine asialo GM1 (ASGM1; 50 l). All in-jections were intravenous, except for ASGM1, which was intraperitoneal. Anti-murine antibodies were injected on day 0, and M7824 and isotype control were injected on days 1, 3, and 5. Tumor volumes (in cubic millimeters) were measured twice weekly and are presented as mean ± SEM. (B) Effect of M7824 on PBMC-mediated antibody-dependent cellular cytotoxicity (ADCC) against tumor cells. PBMCs from a human donor were cultured with 51Cr-labeled A431 human epidermoid carcinoma cells for 4 hours in the presence of different concentrations of M7824, anti–PD-L1, aglycosylated M7824, or isotype control. 51Cr release from labeled target cells was measured with a Wallac MicroBeta Trilux Liquid Scintillation Counter, and the percent lysis was determined. Data were fit to a four-parameter dose-response curve. The data shown are representative of five different human donors. (C) Mt− mice were injected intramuscularly with 0.5 × 106 MC38 cells on day −7 and treated (n = 15 mice per group) on day 0 with isotype control (133 g), M7824 (55 or 164 g), or aglycosylated M7824 (55 or 164 g) on days 0, 2, and 4. Tumor volumes (in cubic millimeters) were measured twice weekly and are presented as mean ± SEM. ****P ≤ 0.0001 denotes a significant difference relative to M7824 treatment.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
10 of 15
cells, is highly expressed in antigen-presenting cells in the tumor microenvironment (69). PD-L1 blockade in myeloid DCs is crucial in enhancing their activation of T cells, suggesting an important role for myeloid cells in the mechanism of action of anti–PD-L1 thera-pies (70). We found that M7824 treatment increased the density of tumor-associated DCs and M1 macrophages, reduced the density of neutrophils, and increased the proportion of mature splenic DCs. Roles for DCs and macrophages are further supported by our RNA-seq data showing that M7824 treatment increased the expression of genes associated with these cell types. Given that TAM phagocytic potency can be enhanced by PD-1 blockade and negatively correlates with their expression of PD-1 (45, 71), the decreased number of PD-1–expressing TAMs after M7824 treatment may also contribute to the antitumor effect of M7824.
Furthermore, the decreased incidence and number of metastases seen with M7824 treatment compared with monotherapies are con-sistent with the known role of TGF- in promoting the EMT (22) and with studies suggesting a link between PD-L1 and the EMT (72–74). A recent publication has shown that M7824 reverts the EMT of human lung cancer cells (75). In addition, the reduced -SMA expression and changes in collagen deposition observed after M7824 or trap con-trol treatment are consistent with previous studies showing that the blockade of TGF- signaling reduces tumor stroma content, expression of extracellular matrix proteins, and fibroblast activation (76–79). Given the role of CAFs in promoting metastasis, it is possible that
M7824 treatment reduces metastasis, in part, through inhibitory ef-fects on CAFs.
Neutralization of TGF- with M7824 can potentially increase therapeutic efficacy and reduce the safety concerns associated with other therapies targeting the TGF- pathway, such as anti–TGF- antibodies, anti–TGF- receptor antibodies, and TGF-RI kinase inhibitors (80–82). In a phase 1 study of an anti–TGF-RII human IgG1 monoclonal antibody (LY3022859), dose escalation beyond 25 mg (flat dose) was considered unsafe because of uncontrolled cytokine release, despite prophylactic treatment (83). In addition, in a phase 1 study of the TGF- antibody fresolimumab (GC1008), treatment- emergent cutaneous lesions with characteristics of keratoacanthomas (KAs) were observed, as well as a single case of squamous cell carcino-ma (84). Although these KAs were benign and reversible after treat-ment completion, they underscored the importance of monitoring adverse events with TGF- inhibition therapy. In a phase 1 clinical trial of M7824, a KA was observed in one patient treated at the highest dose (20 mg/kg). Notably, this lesion regressed with treatment com-pletion. Furthermore, M7824 treatment did not cause enhanced cytokine release (85). Given the favorable tolerability of M7824 in cancer patients (85), we believe that this bifunctional fusion protein is an attractive candidate for inclusion in combination therapy regimens. We have confirmed this in our murine models, in which combining M7824 with radiotherapy or chemotherapy enhanced antitumor activity.
Picrosirius red
α-SMA
Pro
porti
on α
-SM
A+ p
ixel
s 0.5
0.4
0.3
0.2
0.1
0.0
0.0
0.10
0.05
0.15
Anti–P
D-L1
Trap c
ontro
l
M7824
P = 0.0080
P = 0.0197
Isotype control (400 µg) Trap control (492 µg) Anti–PD-L1 (400 µg) M7824 (492 µg) P
icro
siri
us
red
α -S
MA
C
B
P = 0.1705
AH
&E
Isotyp
e con
trol
Anti–P
D-L1
Trap c
ontro
l
M7824
D
E
Pro
porti
on c
olla
gen+ p
ixel
s
Isotyp
e con
trol
Fig. 7. M7824 reduces the expression of -SMA and collagen in tumors. (A to C) Jh mice were subcutaneously inoculated with 0.25 × 106 EMT-6 cells on day −11 and treated (n = 5 mice per group) intravenously with isotype control (400 g), trap control (492 g), anti–PD-L1 (400 g), or M7824 (492 g) on days 0, 4, 7, 11, and 14. Mice were sacrificed on day 15. Representative images of hematoxylin and eosin (H&E) staining (A), anti–-SMA IHC (B), or picrosirius red staining (C) for collagen on formalin- fixed paraffin-embedded tumor sections. For quantification, the numbers of -SMA+ pixels (D) or collagen+ pixels (E) were determined for multiple regions of interest (ROIs) per tumor and normalized to ROI area; each symbol represents the proportion of positive pixels for a single tumor (n = 5 tumors per group). P values were determined by Kruskal-Wallis one-way analysis of variance (ANOVA). Scale bars, 100 m. by guest on A
pril 14, 2020http://stm
.sciencemag.org/
Dow
nloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
11 of 15
Combination with M7824 may be particularly beneficial in radiation therapy regimens in which resistance to treat-ment is often associated with abnormal TGF- signaling and up-regulated PD-L1 expression. Both TGF- signaling in-hibitors (86–88) and anti–PD-1/PD-L1 (55, 56) have separately been shown to increase sensitivity to radiation therapy in cancer models. In addition, TGF- and PD-1 blocking antibodies in combination with radiation treatment led to an increase in complete regressions and a decrease in tumor recurrence in the 4T1 murine breast cancer model relative to radiation combined with either antibody alone (89). These results suggest that the combina-tion of radiation therapy with M7824 may be beneficial. Radiation-induced fibrosis and activation of fibroblasts also have been implicated in mechanisms of resistance (90), as well as therapy-induced injury to normal tissue (91, 92). In our models, M7824 treatment enhanced the efficacy of radiation therapy and the abscopal effect. Our results suggest that M7824 treatment may be particularly effective in combi-nation with therapies that increase TGF- signaling, such as radiation therapy, by addressing resistance mechanisms and re-ducing fibrosis- related toxicities. M7824 therefore has the potential to improve not only upon the impressive clinical efficacy of anti–PD-1 or anti–PD-L1 agents but also upon com binatorial approaches involving anti–PD-1 or anti–PD-L1 inhibitors, which are often plagued by serious tolerability concerns (20, 93).
Finally, the demonstrated capacity of M7824 to efficiently and specifically de-plete all three TGF- isoforms allays con-cerns that a TGF-RII–based trap may not effectively impair TGF-2–dependent sig-naling, which occurs in some tumor types despite TGF-1 being the predominant form in the tumor microenvironment (57, 94). Our results contrast with previous find-ings showing that soluble TGF-RII binds TGF-1 and TGF-3 but not TGF-2 (43). These differences can be explained by the stoichiometry of TGF- binding to the TGF-RII extra cellular domain (one dimeric ligand can bind two receptor molecules). When TGF- RII is in a dimeric configuration, as occurs in the M7824 molecule, binding avidity to the TGF- homodimer is several orders of magni-tude higher than the intrinsic binding affinity (95).
CBA
Isotype control (133 µg)Radiation (3.6 Gy)
Radiation + M7824M7824 (55 µg)
Tum
or v
olum
e (m
m3 )
Days0 6 84 10
2000
1500
1000
0
500
Isotyp
e con
trol (1
33 µg
)
Radiat
ion (3
.6 Gy)
Radiat
ion +
M7824
M7824
(55 µ
g)
Tum
or w
eigh
t (m
g)
Isotyp
e con
trol (1
33 µg
)
Radiat
ion (3
.6 Gy)
Radiat
ion +
M7824
M7824
(55 µ
g)2
4000
3000
1000
0
2000
P < 0.0001P = 0.0014
P < 0.0001
IFN
-γ+ s
pots
per
5 ×
105 C
D8+ T
cel
ls
350
300100
50
0
400P < 0.0001P < 0.0001
OVAP15E
P < 0.0001
Primary tumor (i.m.)
Secondary tumor (s.c.)
Radiationtherapy
Primary tumor Secondary tumor
Tum
or V
olum
e (m
m3 )
Tum
or V
olum
e (m
m3 )
Days posttreatment start Days posttreatment start
2000
1500
1000
0
500
2500
0 3 7 119 14 0 3 7 119 14
600
400
200
0
Radiation + M7824M7824 (164 µg)Radiation (5 Gy) Isotype control (400 µg)
Abscopal effect:Experimental design
FED
G H I
Tum
or v
olum
e (m
m3 )
Days
Isotype control (400 µg)
0 5 10 15 20
2500
2000
1500
1000
500
0
M7824 (492 µg)
Ox/5-FU + M7824Ox/5-FU (5 mg/kg)
4000
3000
1000
0
2000
Isotyp
e con
trol (4
00 µg
)
M7824
(492
µg)
Ox/5-F
U + M78
24
Ox/5-F
U (5 m
g/kg)
Tum
or w
eigh
t (m
g) 400
300
200
100
0
IFN
-γ+ s
pots
per
5 ×
105 C
D8+ T
cel
ls
Isotyp
e con
trol (4
00 µg
)
M7824
(492
µg)
Ox/5-F
U + M78
24
Ox/5-F
U (5 m
g/kg)
P = 0.0065P < 0.0001
P = 0.0007 OVAP15E
P < 0.0001P < 0.0001
P < 0.0001
Radiation therapy
Chemotherapy
********
****
****
****
****
****
****
****
*********
Fig. 8. Combining M7824 with radiation or chemotherapy enhances antitumor efficacy. (A to C) Mt−mice were inoculated intramuscularly (i.m.) with 0.5 × 106 MC38 cells (day −8) and treated (n = 10 mice per group) with isotype control (133 g, intravenously; day 2), radiation [3.6 Gray (Gy)/day; days 0 to 3], M7824 (55 g, intravenously; day 2), or M7824 + radiation. (A) Tumor volumes, measured twice weekly. (B) Tumor weight (day 14). (C) ELISpot of IFN-–producing, p15E-responsive CD8+ T cells (day 14). Spleens were harvested (n = 5 mice), and CD8+ T cells were isolated. Antigen- presenting cells derived from naive splenocytes were pulsed with the KPSWFTTL (p15E) peptide or the irrelevant SIINFEKL (OVA) peptide, irradiated, and cultured with isolated CD8+ T cells. (D to F) C57BL/6 mice were inoculated intramuscular-ly (day −7) with 0.5 × 106 MC38 cells in the right thigh (primary tumor) and subcutaneously (s.c.) with 1 × 106 MC38 cells in the left flank (secondary tumor) and treated (n = 6 mice per group) with isotype control (400 g; days 0, 2, and 4), ra-diation (5 Gy/day; days 0 to 3), M7824 (164 g; day 0), or M7824 + radiation. Radiation was applied only to the primary tumor, as shown in (D). (E) Primary tumor volumes and (F) secondary tumor volumes were measured twice weekly. (G to I) Mt− mice were subcutaneously inoculated with 1 × 106 MC38 cells (day −7) and treated (n = 10 mice per group) with isotype control (400 g; days 3, 6, 9, 12, and 17), M7824 (492 g, intravenously; days 3, 6, 9, 12, and 17), oxaliplatin (Ox)/5-fluorouracil (5-FU) (5 mg/kg, intraperitoneally, and 60 mg/kg, intravenously; day 0), or M7824 + Ox/5-FU. (G) Tumor volumes were measured twice weekly. (H) Tumor weight (day 18). (I) ELISpot of IFN-–producing, p15E-responsive CD8+ T cells (n = 5 mice). Tumor volumes are presented as mean ± SEM. **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001 denote a significant difference relative to radiation + M7824 treatment or Ox/5-FU + M7824 treatment. Tumor weights are shown for individual mice, with lines representing the means, and they were compared by unpaired t test. IFN- ELISpot data are means ± SEM of three replicates and were evaluated by two-way ANOVA.
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
12 of 15
In conclusion, M7824 elicited potent and superior antitumor ac-tivity in preclinical models via the simultaneous blockade of two key immune suppression pathways. Targeting two pathways using a bi-functional molecule such as M7824 has strategic advantages relative to the traditional approach of combining two single agents. Bifunctional molecules can be combined more practically with additional thera-pies to target three or even more pathways to enhance clinical ben-efit. In light of these highly encouraging preclinical observations in mice, M7824 has entered clinical development. On the basis of its safety profile and early evidence of clinical activity from phase 1 dose- escalation studies (85), M7824 is being investigated in phase 1 expan-sion studies in patients with advanced solid tumors (NCT02517398 and NCT02699515).
MATERIALS AND METHODSStudy designThis study was designed to evaluate the preclinical efficacy of M7824 in multiple tumor types. This objective was addressed by (i) deter-mining whether M7824 could bind simultaneously to PD-L1 and TGF- and inhibit both pathways in vitro, (ii) evaluating differences in tumor growth and metastasis between treatment groups in mouse tumor models, (iii) assessing whether M7824 could extend survival and provide protective antitumor immunity in vivo, (iv) evaluating the innate and adaptive immunophenotypic signature of the spleen and tumor tissue between treatment groups, and (v) determining whether M7824 was effective when combined with radiotherapy or chemotherapy in mouse tumor models.
In animal studies, all treatment and control groups had about 10 mice per group (specified in the figure legends) and were ran-domized according to tumor volume at the start of treatment. The primary end points were survival and tumor size. Body weight was measured twice weekly, and mice were euthanized when tumor volume exceeded 12.5% of their body weight (≈2500 mm3). All procedures were performed in accordance with institutional proto-cols approved by the Institutional Animal Care and Use Committee of EMD Serono Research and Development Institute. All in vitro and in vivo results are representative of two to five independent experiments.
Statistical analysesStatistical analyses were performed using GraphPad Prism Software, versions 5.0 to 7.0. Tumor volume data are presented graphically as means ± SEM. For other in vivo studies, symbols represent data from individual mice; the horizontal line across each treatment group rep-resents the mean. To assess differences in tumor volumes between treatment groups, tumor volume data were log-transformed, and two-way analysis of variance (ANOVA) was performed, followed by Tukey’s multiple comparison test. A Kaplan-Meier plot was generated to show survival by treatment group, and significance was assessed by log-rank (Mantel-Cox) test. An unpaired t test with Welch’s cor-rection was performed to assess differences in tumor weights be-tween treatment groups. Two-way ANOVA with Tukey’s multiple comparison tests was used for ELISpot analyses and comparing anti-pSMAD staining between groups at different time points. An unpaired, two-tailed t test was used to compare treatment groups in immunophenotyping analyses. For anti-CD8a IHC of tumors, P values were determined by Kruskal-Wallis test. For in vitro analyses, nonlinear regression was performed, and EC50 or IC50 values were determined.
For RNA-seq analysis, signature score P values were determined with the ROAST method (96).
For all other Materials and Methods, see the Supplementary Materials.
SUPPLEMENTARY MATERIALSwww.sciencetranslationalmedicine.org/cgi/content/full/10/424/eaan5488/DC1Materials and MethodsFig. S1. M7824 has a similar internalization rate as anti–PD-L1Fig. S2. M7824 lowers pSMAD2 in MC38 tumor–bearing mice.Fig. S3. The trap control exhibits antitumor activity in vivo.Fig. S4. M7824 inhibits tumor growth in individual mice in syngeneic tumor models.Fig. S5. The trap control has about three times higher serum exposure than M7824.Fig. S6. M7824 inhibits tumor growth in subcutaneous tumor models.Fig. S7. M7824 inhibits tumor growth in wild-type mice.Fig. S8. M7824 inhibits incidence of lung metastases relative to isotype control.Fig. S9. M7824 increases mature DCs and EOMES-expressing CD8+ T cells in a dosage- and dosing frequency-dependent manner.Fig. S10. M7824 induces an immunophenotypic signature in MC38 tumor–bearing wild-type mice.Fig. S11. Innate and adaptive immunity, but not ADCC, contributes to M7824 antitumor activity.Fig. S12. Combining M7824 with radiation enhances antitumor efficacy in individual mice.Fig. S13. Neither anti–PD-L1 nor trap control significantly potentiated the abscopal effect of radiation.Fig. S14. Combining M7824 with chemotherapy enhances antitumor efficacy in individual mice.Table S1. Gene lists of immune signatures from differentially expressed gene analysis.References (99–105)
REFERENCES AND NOTES 1. I. Mellman, G. Coukos, G. Dranoff, Cancer immunotherapy comes of age. Nature 480,
480–489 (2011). 2. D. M. Pardoll, The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev.
Cancer 12, 252–264 (2012). 3. L. Zitvogel, G. Kroemer, Targeting PD-1/PD-L1 interactions for cancer immunotherapy.
Oncoimmunology 1, 1223–1225 (2012). 4. D. F. McDermott, M. B. Atkins, PD-1 as a potential target in cancer therapy. Cancer Med. 2,
662–673 (2013). 5. J. Hamanishi, M. Mandai, M. Iwasaki, T. Okazaki, Y. Tanaka, K. Yamaguchi, T. Higuchi,
H. Yagi, K. Takakura, N. Minato, T. Honjo, S. Fujii, Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 3360–3365 (2007).
6. Y. Qing, Q. Li, T. Ren, W. Xia, Y. Peng, G. L. Liu, H. Luo, Y. X. Yang, X. Y. Dai, S. F. Zhou, D. Wang, Upregulation of PD-L1 and APE1 is associated with tumorigenesis and poor prognosis of gastric cancer. Drug Des. Devel. Ther. 9, 901–909 (2015).
7. J. S. Weber, S. P. D’Angelo, D. Minor, F. S. Hodi, R. Gutzmer, B. Neyns, C. Hoeller, N. I. Khushalani, W. H. Miller Jr., C. D. Lao, G. P. Linette, L. Thomas, P. Lorigan, K. F. Grossmann, J. C. Hassel, M. Maio, M. Sznol, P. A. Ascierto, P. Mohr, B. Chmielowski, A. Bryce, I. M. Svane, J.-J. Grob, A. M. Krackhardt, C. Horak, A. Lambert, A. S. Yang, J. Larkin, Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 16, 375–384 (2015).
8. M. A. Postow, J. Chesney, A. C. Pavlick, C. Robert, K. Grossmann, D. McDermott, G. P. Linette, N. Meyer, J. K. Giguere, S. S. Agarwala, M. Shaheen, M. S. Ernstoff, D. Minor, A. K. Salama, M. Taylor, P. A. Ott, L. M. Rollin, C. Horak, P. Gagnier, J. D. Wolchok, F. S. Hodi, Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 372, 2006–2017 (2015).
9. C. Robert, J. Schachter, G. V. Long, A. Arance, J. J. Grob, L. Mortier, A. Daud, M. S. Carlino, C. McNeil, M. Lotem, J. Larkin, P. Lorigan, B. Neyns, C. U. Blank, O. Hamid, C. Mateus, R. Shapira-Frommer, M. Kosh, H. Zhou, N. Ibrahim, S. Ebbinghaus, A. Ribas, Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
10. H. Borghaei, L. Paz-Ares, L. Horn, D. R. Spigel, M. Steins, N. E. Ready, L. Q. Chow, E. E. Vokes, E. Felip, E. Holgado, F. Barlesi, M. Kohlhaufl, O. Arrieta, M. A. Burgio, J. Fayette, H. Lena, E. Poddubskaya, D. E. Gerber, S. N. Gettinger, C. M. Rudin, N. Rizvi, L. Crino, G. R. Blumenschein Jr., S. J. Antonia, C. Dorange, C. T. Harbison, F. Graf Finckenstein, J. R. Brahmer, Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
11. R. S. Herbst, P. Baas, D.-W. Kim, E. Felip, J. L. Pérez-Gracia, J.-Y. Han, J. Molina, J.-H. Kim, C. D. Arvis, M.-J. Ahn, M. Majem, M. J. Fidler, G. De Castro, M. Garrido, G. M. Lubiniecki,
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
13 of 15
Y. Shentu, E. Im, M. Dolled-Filhart, E. B. Garon, Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 387, 1540–1550 (2016).
12. A. Rittmeyer, F. Barlesi, D. Waterkamp, K. Park, F. Ciardiello, J. von Pawel, S. M. Gadgeel, T. Hida, D. M. Kowalski, M. C. Dols, D. L. Cortinovis, J. Leach, J. Polikoff, C. Barrios, F. Kabbinavar, O. A. Frontera, F. De Marinis, H. Turna, J. S. Lee, M. Ballinger, M. Kowanetz, P. He, D. S. Chen, A. Sandler, D. R. Gandara, Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265 (2017).
13. R. J. Motzer, B. Escudier, D. F. McDermott, S. George, H. J. Hammers, S. Srinivas, S. S. Tykodi, J. A. Sosman, G. Procopio, E. R. Plimack, D. Castellano, T. K. Choueiri, H. Gurney, F. Donskov, P. Bono, J. Wagstaff, T. C. Gauler, T. Ueda, Y. Tomita, F. A. Schutz, C. Kollmannsberger, J. Larkin, A. Ravaud, J. S. Simon, L.-A. Xu, I. M. Waxman, P. Sharma; CheckMate 025 Investigators, Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813 (2015).
14. H. L. Kaufman, J. Russell, O. Hamid, S. Bhatia, P. Terheyden, S. P. D’Angelo, K. C. Shih, C. Lebbé, G. P. Linette, M. Milella, I. Brownell, K. D. Lewis, J. H. Lorch, K. Chin, L. Mahnke, A. von Heydebreck, J.-M. Cuillerot, P. Nghiem, Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 17, 1374–1385 (2016).
15. J. E. Rosenberg, J. Hoffman-Censits, T. Powles, M. S. van der Heijden, A. V. Balar, A. Necchi, N. Dawson, P. H. O’Donnell, A. Balmanoukian, Y. Loriot, S. Srinivas, M. M. Retz, P. Grivas, R. W. Joseph, M. D. Galsky, M. T. Fleming, D. P. Petrylak, J. L. Perez-Gracia, H. A. Burris, D. Castellano, C. Canil, J. Bellmunt, D. Bajorin, D. Nickles, R. Bourgon, G. M. Frampton, N. Cui, S. Mariathasan, O. Abidoye, G. D. Fine, R. Dreicer, Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 387, 1909–1920 (2016).
16. P. Sharma, M. Retz, A. Siefker-Radtke, A. Baron, A. Necchi, J. Bedke, E. R. Plimack, D. Vaena, M.-O. Grimm, S. Bracarda, J. A. Arranz, S. Pal, C. Ohyama, A. Saci, X. Qu, A. Lambert, S. Krishnan, A. Azrilevich, M. D. Galsky, Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 18, 312–322 (2017).
17. M. S. Farina, K. T. Lundgren, J. Bellmunt, Immunotherapy in urothelial cancer: Recent results and future perspectives. Drugs 77, 1077–1089 (2017).
18. R. L. Ferris, G. Blumenschein Jr., J. Fayette, J. Guigay, A. D. Colevas, L. Licitra, K. Harrington, S. Kasper, E. E. Vokes, C. Even, F. Worden, N. F. Saba, L. C. Iglesias Docampo, R. Haddad, T. Rordorf, N. Kiyota, M. Tahara, M. Monga, M. Lynch, W. J. Geese, J. Kopit, J. W. Shaw, M. L. Gillison, Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 375, 1856–1867 (2016).
19. J. Bauml, T. Y. Seiwert, D. G. Pfister, F. Worden, S. V. Liu, J. Gilbert, N. F. Saba, J. Weiss, L. Wirth, A. Sukari, H. Kang, M. K. Gibson, E. Massarelli, S. Powell, A. Meister, X. Shu, J. D. Cheng, R. Haddad, Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: Results from a single-arm, phase II study. J. Clin. Oncol. 35, 1542–1549 (2017).
20. I. Melero, D. M. Berman, M. A. Aznar, A. J. Korman, J. L. Perez Gracia, J. Haanen, Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 15, 457–472 (2015).
21. J. Larkin, V. Chiarion-Sileni, R. Gonzalez, J. J. Grob, C. L. Cowey, C. D. Lao, D. Schadendorf, R. Dummer, M. Smylie, P. Rutkowski, P. F. Ferrucci, A. Hill, J. Wagstaff, M. S. Carlino, J. B. Haanen, M. Maio, I. Marquez-Rodas, G. A. McArthur, P. A. Ascierto, G. V. Long, M. K. Callahan, M. A. Postow, K. Grossmann, M. Sznol, B. Dreno, L. Bastholt, A. Yang, L. M. Rollin, C. Horak, F. S. Hodi, J. D. Wolchok, Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
22. R. J. Akhurst, A. Hata, Targeting the TGF signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811 (2012).
23. M. O. Li, R. A. Flavell, Contextual regulation of inflammation: A duet by transforming growth factor- and interleukin-10. Immunity 28, 468–476 (2008).
24. S. H. Wrzesinski, Y. Y. Wan, R. A. Flavell, Transforming growth factor- and the immune response: Implications for anticancer therapy. Clin. Cancer Res. 13, 5262–5270 (2007).
25. L. J. A. C. Hawinkels, H. W. Verspaget, W. van Duijn, J. M. van der Zon, K. Zuidwijk, F. J. G. M. Kubben, J. H. Verheijen, D. W. Hommes, C. B. H. W. Lamers, C. F. M. Sier, Tissue level, activation and cellular localisation of TGF-1 and association with survival in gastric cancer patients. Br. J. Cancer 97, 398–404 (2007).
26. D. R. Principe, J. A. Doll, J. Bauer, B. Jung, H. G. Munshi, L. Bartholin, B. Pasche, C. Lee, P. J. Grippo, TGF-: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 106, djt369 (2014).
27. J. J. Lebrun, The dual role of TGF in human cancer: From tumor suppression to cancer metastasis. ISRN Mol. Biol. 2012, 381428 (2012).
28. C. Neuzillet, A. Tijeras-Raballand, R. Cohen, J. Cros, S. Faivre, E. Raymond, A. de Gramont, Targeting the TGF pathway for cancer therapy. Pharmacol. Ther. 147, 22–31 (2015).
29. J. M. Yingling, K. L. Blanchard, J. S. Sawyer, Development of TGF- signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 3, 1011–1022 (2004).
30. Y. Pang, S. K. Gara, B. R. Achyut, Z. Li, H. H. Yan, C.-P. Day, J. M. Weiss, G. Trinchieri, J. C. Morris, L. Yang, TGF- signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 3, 936–951 (2013).
31. D. Gate, M. Danielpour, J. Rodriguez Jr., G.-B. Kim, R. Levy, S. Bannykh, J. J. Breunig, S. M. Kaech, R. A. Flavell, T. Town, T-cell TGF- signaling abrogation restricts medulloblastoma progression. Proc. Natl. Acad. Sci. U.S.A. 111, E3458–E3466 (2014).
32. M. O. Li, S. Sanjabi, R. A. Flavell, Transforming growth factor- controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471 (2006).
33. M. O. Li, Y. Y. Wan, R. A. Flavell, T cell-produced transforming growth factor-1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591 (2007).
34. D. A. Thomas, J. Massague, TGF- directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005).
35. S. Viel, A. Marcais, F. S. Guimaraes, R. Loftus, J. Rabilloud, M. Grau, S. Degouve, S. Djebali, A. Sanlaville, E. Charrier, J. Bienvenu, J. C. Marie, C. Caux, J. Marvel, L. Town, N. D. Huntington, L. Bartholin, D. Finlay, M. J. Smyth, T. Walzer, TGF- inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 9, ra19 (2016).
36. S. L. Topalian, C. G. Drake, D. M. Pardoll, Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 24, 207–212 (2012).
37. W. Hugo, J. M. Zaretsky, L. Sun, C. Song, B. H. Moreno, S. Hu-Lieskovan, B. Berent-Maoz, J. Pang, B. Chmielowski, G. Cherry, E. Seja, S. Lomeli, X. Kong, M. C. Kelley, J. A. Sosman, D. B. Johnson, A. Ribas, R. S. Lo, Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 168, 542 (2017).
38. H. Y. Lin, X.-F. Wang, E. Ng-Eaton, R. A. Weinberg, H. F. Lodish, Expression cloning of the TGF- type II receptor, a functional transmembrane serine/threonine kinase. Cell 68, 775–785 (1992).
39. K. Krasagakis, D. Tholke, B. Farthmann, J. Eberle, U. Mansmann, C. E. Orfanos, Elevated plasma levels of transforming growth factor (TGF)-1 and TGF-2 in patients with disseminated malignant melanoma. Br. J. Cancer 77, 1492–1494 (1998).
40. F. Kong, R. L. Jirtle, D. H. Huang, R. W. Clough, M. S. Anscher, Plasma transforming growth factor-beta1 level before radiotherapy correlates with long term outcome of patients with lung carcinoma. Cancer 86, 1712–1719 (1999).
41. A. J. Vandeveer, J. K. Fallon, R. Tighe, H. Sabzevari, J. Schlom, J. W. Greiner, Systemic immunotherapy of non-muscle invasive mouse bladder cancer with avelumab, an anti–PD-L1 immune checkpoint inhibitor. Cancer Immunol. Res. 4, 452–462 (2016).
42. J. Won, H. Kim, E. J. Park, Y. Hong, S.-J. Kim, Y. Yun, Tumorigenicity of mouse thymoma is suppressed by soluble type II transforming growth factor receptor therapy. Cancer Res. 59, 1273–1277 (1999).
43. H. Y. Lin, A. Moustakas, P. Knaus, R. G. Wells, Y. I. Henis, H. F. Lodish, The soluble exoplasmic domain of the type II transforming growth factor (TGF)- receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF- ligands. J. Biol. Chem. 270, 2747–2754 (1995).
44. L. A. Van Aarsen, D. R. Leone, S. Ho, B. M. Dolinski, P. E. McCoon, D. J. LePage, R. Kelly, G. Heaney, P. Rayhorn, C. Reid, K. J. Simon, G. S. Horan, N. Tao, H. A. Gardner, M. M. Skelly, A. M. Gown, G. J. Thomas, P. H. Weinreb, S. E. Fawell, S. M. Violette, Antibody-mediated blockade of integrin v6 inhibits tumor progression in vivo by a transforming growth factor-–regulated mechanism. Cancer Res. 68, 561–570 (2008).
45. S. R. Gordon, R. L. Maute, B. W. Dulken, G. Hutter, B. M. George, M. N. McCracken, R. Gupta, J. M. Tsai, R. Sinha, D. Corey, A. M. Ring, A. J. Connolly, I. L. Weissman, PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
46. M. Angelova, P. Charoentong, H. Hackl, M. L. Fischer, R. Snajder, A. M. Krogsdam, M. J. Waldner, G. Bindea, B. Mlecnik, J. Galon, Z. Trajanoski, Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 16, 64 (2015).
47. G. Bindea, B. Mlecnik, M. Tosolini, A. Kirilovsky, M. Waldner, A. C. Obenauf, H. Angell, T. Fredriksen, L. Lafontaine, A. Berger, P. Bruneval, W. H. Fridman, C. Becker, F. Pages, M. R. Speicher, Z. Trajanoski, J. Galon, Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795 (2013).
48. A. Liberzon, C. Birger, H. Thorvaldsdottir, M. Ghandi, J. P. Mesirov, P. Tamayo, The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
49. J. Lieberman, The ABCs of granule-mediated cytotoxicity: New weapons in the arsenal. Nat. Rev. Immunol. 3, 361–370 (2003).
50. A. Calon, D. V. F. Tauriello, E. Batlle, TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 25, 15–22 (2014).
51. S. Kakarla, X.-T. Song, S. Gottschalk, Cancer-associated fibroblasts as targets for immunotherapy. Immunotherapy 4, 1129–1138 (2012).
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
14 of 15
52. H. C. Dancea, M. M. Shareef, M. M. Ahmed, Role of radiation-induced TGF-beta signaling in cancer therapy. Mol. Cell. Pharmacol. 1, 44–56 (2009).
53. S. Biswas, M. Guix, C. Rinehart, T. C. Dugger, A. Chytil, H. L. Moses, M. L. Freeman, C. L. Arteaga, Inhibition of TGF- with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Invest. 117, 1305–1313 (2007).
54. M. Luo, L. Fu, The effect of chemotherapy on programmed cell death 1/programmed cell death 1 ligand axis: Some chemotherapeutical drugs may finally work through immune response. Oncotarget 7, 29794–29803 (2016).
55. S. J. Dovedi, A. L. Adlard, G. Lipowska-Bhalla, C. McKenna, S. Jones, E. J. Cheadle, I. J. Stratford, E. Poon, M. Morrow, R. Stewart, H. Jones, R. W. Wilkinson, J. Honeychurch, T. M. Illidge, Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 74, 5458–5468 (2014).
56. L. Deng, H. Liang, B. Burnette, M. Beckett, T. Darga, R. R. Weichselbaum, Y.-X. Fu, Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Invest. 124, 687–695 (2014).
57. B. Bierie, H. L. Moses, Tumour microenvironment: TGF: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 6, 506–520 (2006).
58. J. Massague, TGF in cancer. Cell 134, 215–230 (2008). 59. L. Yang, Y. Pang, H. L. Moses, TGF- and immune cells: An important regulatory axis in the
tumor microenvironment and progression. Trends Immunol. 31, 220–227 (2010). 60. M. Yu, P. Trobridge, Y. Wang, S. Kanngurn, S. M. Morris, S. Knoblaugh, W. M. Grady,
Inactivation of TGF- signaling and loss of PTEN cooperate to induce colon cancer in vivo. Oncogene 33, 1538–1547 (2014).
61. A. Beldi-Ferchiou, M. Lambert, S. Dogniaux, F. Vely, E. Vivier, D. Olive, S. Dupuy, F. Levasseur, D. Zucman, C. Lebbe, D. Sene, C. Hivroz, S. Caillat-Zucman, PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 7, 72961–72977 (2016).
62. D. M. Benson Jr., C. E. Bakan, A. Mishra, C. C. Hofmeister, Y. Efebera, B. Becknell, R. A. Baiocchi, J. Zhang, J. Yu, M. K. Smith, C. N. Greenfield, P. Porcu, S. M. Devine, R. Rotem-Yehudar, G. Lozanski, J. C. Byrd, M. A. Caligiuri, The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 116, 2286–2294 (2010).
63. X. L. Iraolagoitia, R. G. Spallanzani, N. I. Torres, R. E. Araya, A. Ziblat, C. I. Domaica, J. M. Sierra, S. Y. Nuñez, F. Secchiari, T. F. Gajewski, N. W. Zwirner, M. B. Fuertes, NK cells restrain spontaneous antitumor CD8+ T cell priming through PD-1/PD-L1 interactions with dendritic cells. J. Immunol. 197, 953–961 (2016).
64. K. E. Pauken, E. J. Wherry, Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276 (2015).
65. E. B. Wilson, J. J. El-Jawhari, A. L. Neilson, G. D. Hall, A. A. Melcher, J. L. Meade, G. P. Cook, Human tumour immune evasion via TGF- blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLOS ONE 6, e22842 (2011).
66. R. Trotta, J. Dal Col, J. Yu, D. Ciarlariello, B. Thomas, X. Zhang, J. Allard II, M. Wei, H. Mao, J. C. Byrd, D. Perrotti, M. A. Caligiuri, TGF- utilizes SMAD3 to inhibit CD16-mediated IFN- production and antibody-dependent cellular cytotoxicity in human NK cells. J. Immunol. 181, 3784–3792 (2008).
67. R. H. Rouce, H. Shaim, T. Sekine, G. Weber, B. Ballard, S. Ku, C. Barese, V. Murali, M.-F. Wu, H. Liu, E. J. Shpall, C. M. Bollard, K. R. Rabin, K. Rezvani, The TGF-/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia 30, 800–811 (2016).
68. R. Castriconi, C. Cantoni, M. Della Chiesa, M. Vitale, E. Marcenaro, R. Conte, R. Biassoni, C. Bottino, L. Moretta, A. Moretta, Transforming growth factor 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 100, 4120–4125 (2003).
69. W. Zou, J. D. Wolchok, L. Chen, PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Trans. Med. 8, 328rv4 (2016).
70. T. J. Curiel, S. Wei, H. Dong, X. Alvarez, P. Cheng, P. Mottram, R. Krzysiek, K. L. Knutson, B. Daniel, M. C. Zimmermann, O. David, M. Burow, A. Gordon, N. Dhurandhar, L. Myers, R. Berggren, A. Hemminki, R. D. Alvarez, D. Emilie, D. T. Curiel, L. Chen, W. Zou, Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 9, 562–567 (2003).
71. L. Shen, Y. Gao, Y. Liu, B. Zhang, Q. Liu, J. Wu, L. Fan, Q. Ou, W. Zhang, L. Shao, PD-1/PD-L pathway inhibits M.tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci. Rep. 6, 38362 (2016).
72. A. Alsuliman, D. Colak, O. Al-Harazi, H. Fitwi, A. Tulbah, T. Al-Tweigeri, M. Al-Alwan, H. Ghebeh, Bidirectional crosstalk between PD-L1 expression and epithelial to mesenchymal transition: Significance in claudin-low breast cancer cells. Mol. Cancer 14, 149 (2015).
73. S. Tsutsumi, H. Saeki, Y. Nakashima, S. Ito, E. Oki, M. Morita, Y. Oda, S. Okano, Y. Maehara, Programmed death-ligand 1 expression at tumor invasive front is associated with epithelial-mesenchymal transition and poor prognosis in esophageal squamous cell carcinoma. Cancer Sci. 108, 1119–1127 (2017).
74. C.-Y. Ock, S. Kim, B. Keam, M. Kim, T. M. Kim, J.-H. Kim, Y. K. Jeon, J.-S. Lee, S. K. Kwon, J. H. Hah, T.-K. Kwon, D.-W. Kim, H.-G. Wu, M.-W. Sung, D. S. Heo, PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 7, 15901–15914 (2016).
75. J. M. David, C. Dominguez, K. K. McCampbell, J. L. Gulley, J. Schlom, C. Palena, A novel bifunctional anti-PD-L1/TGF- trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology 6, e1349589 (2017).
76. A. Mazzocca, E. Fransvea, F. Dituri, L. Lupo, S. Antonaci, G. Giannelli, Down-regulation of connective tissue growth factor by inhibition of transforming growth factor blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 51, 523–534 (2010).
77. S. Medicherla, L. Li, J. Y. Ma, A. M. Kapoun, N. J. Gaspar, Y.-W. Liu, R. Mangadu, G. O’Young, A. A. Protter, G. F. Schreiner, D. H. Wong, L. S. Higgins, Antitumor activity of TGF- inhibitor is dependent on the microenvironment. Anticancer Res 27, 4149–4157 (2007).
78. R. A. Evans, Y. C. Tian, R. Steadman, A. O. Phillips, TGF-1-mediated fibroblast-myofibroblast terminal differentiation—The role of Smad proteins. Exp. Cell Res. 282, 90–100 (2003).
79. C. Guido, D. Whitaker-Menezes, C. Capparelli, R. Balliet, Z. Lin, R. G. Pestell, A. Howell, S. Aquila, S. Ando, U. Martinez-Outschoorn, F. Sotgia, M. P. Lisanti, Metabolic reprogramming of cancer-associated fibroblasts by TGF- drives tumor growth: Connecting TGF- signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 11, 3019–3035 (2012).
80. M. J. Anderton, H. R. Mellor, A. Bell, C. Sadler, M. Pass, S. Powell, S. J. Steele, R. R. Roberts, A. Heier, Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 39, 916–924 (2011).
81. K. Garber, Companies waver in efforts to target transforming growth factor beta in cancer. J. Natl. Cancer Inst. 101, 1664–1667 (2009).
82. A. B. Roberts, N. S. Roche, T. S. Winokur, J. K. Burmester, M. B. Sporn, Role of transforming growth factor-beta in maintenance of function of cultured neonatal cardiac myocytes. Autocrine action and reversal of damaging effects of interleukin-1. J. Clin. Invest. 90, 2056–2062 (1992).
83. A. W. Tolcher, J. D. Berlin, J. Cosaert, J. Kauh, E. Chan, S. A. Piha-Paul, A. Amaya, S. Tang, K. Driscoll, R. Kimbung, S. R. Kambhampati, I. Gueorguieva, D. S. Hong, A phase 1 study of anti-TGF receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 79, 673–680 (2017).
84. M. E. Lacouture, J. C. Morris, D. P. Lawrence, A. R. Tan, T. E. Olencki, G. I. Shapiro, B. J. Dezube, J. A. Berzofsky, F. J. Hsu, J. Guitart, Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 64, 437–446 (2015).
85. J. L. Gulley, C. R. Heery, J. Schlom, R. A. Madan, L. Cao, E. Lamping, J. L. Marte, L. M. Cordes, O. Christensen, C. Helwig, J. Strauss, Preliminary results from a phase 1 trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF-, in advanced solid tumors. J. Clin. Oncol. 35, 3006 (2017).
86. F. Bouquet, A. Pal, K. A. Pilones, S. Demaria, B. Hann, R. J. Akhurst, J. S. Babb, S. M. Lonning, J. K. DeWyngaert, S. C. Formenti, M. H. Barcellos-Hoff, TGF1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 17, 6754–6765 (2011).
87. M. E. Hardee, A. E. Marciscano, C. M. Medina-Ramirez, D. Zagzag, A. Narayana, S. M. Lonning, M. H. Barcellos-Hoff, Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-. Cancer Res. 72, 4119–4129 (2012).
88. M. Zhang, S. Kleber, M. Rohrich, C. Timke, N. Han, J. Tuettenberg, A. Martin-Villalba, J. Debus, P. Peschke, U. Wirkner, M. Lahn, P. E. Huber, Blockade of TGF- signaling by the TGFR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 71, 7155–7167 (2011).
89. C. Vanpouille-Box, J. M. Diamond, K. A. Pilones, J. Zavadil, J. S. Babb, S. C. Formenti, M. H. Barcellos-Hoff, S. Demaria, TGF is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 75, 2232–2242 (2015).
90. H. E. Barker, J. T. E. Paget, A. A. Khan, K. J. Harrington, The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015).
91. H. B. Stone, C. N. Coleman, M. S. Anscher, W. H. McBride, Effects of radiation on normal tissue: Consequences and mechanisms. Lancet Oncol. 4, 529–536 (2003).
92. J. M. Straub, J. New, C. D. Hamilton, C. Lominska, Y. Shnayder, S. M. Thomas, Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 141, 1985–1994 (2015).
93. M. K. Callahan, M. A. Postow, J. D. Wolchok, CTLA-4 and PD-1 pathway blockade: Combinations in the clinic. Front. Oncol. 4, 385 (2014).
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

Lan et al., Sci. Transl. Med. 10, eaan5488 (2018) 17 January 2018
S C I E N C E T R A N S L A T I O N A L M E D I C I N E | R E S E A R C H A R T I C L E
15 of 15
94. H. Friess, Y. Yamanaka, M. Buchler, M. Ebert, H. G. Beger, L. I. Gold, M. Korc, Enhanced expression of transforming growth factor isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 105, 1846–1856 (1993).
95. G. De Crescenzo, P. L. Pham, Y. Durocher, M. D. O’Connor-McCourt, Transforming growth factor-beta (TGF-) binding to the extracellular domain of the type II TGF- receptor: Receptor capture on a biosensor surface using a new coiled-coil capture system demonstrates that avidity contributes significantly to high affinity binding. J. Mol. Biol. 328, 1173–1183 (2003).
96. D. Wu, E. Lim, F. Vaillant, M.-L. Asselin-Labat, J. E. Visvader, G. K. Smyth, ROAST: Rotation gene set tests for complex microarray experiments. Bioinformatics 26, 2176–2182 (2010).
97. P. J. Hart, S. Deep, A. B. Taylor, Z. Shu, C. S. Hinck, A. P. Hinck, Crystal structure of the human TR2 ectodomain--TGF-3 complex. Nat. Struct. Biol. 9, 203–208 (2002).
98. M. I. Love, W. Huber, S. Anders, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
99. D. Moiani, M. Salvalaglio, C. Cavallotti, A. Bujacz, I. Redzynia, G. Bujacz, F. Dinon, P. Pengo, G. Fassina, Structural characterization of a Protein A mimetic peptide dendrimer bound to human IgG. J. Phys. Chem. B 113, 16268–16275 (2009).
100. B. Langmead, S. L. Salzberg, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
101. B. Li, C. N. Dewey, RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
102. R. Kolde, pheatmap: Pretty Heatmaps. R package version 1.0.8 (2015); https://cran.r-project.org/web/packages/pheatmap/index.html.
103. H. R. Koene, M. Kleijer, J. Algra, D. Roos, A. E. von dem Borne, M. de Haas, FcgRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell FcRIIIa, independently of the FcRIIIa-48L/R/H phenotype. Blood 90, 1109–1114 (1997).
104. J. Wu, J. C. Edberg, P. B. Redecha, V. Bansal, P. M. Guyre, K. Coleman, J. E. Salmon, R. P. Kimberly, A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Invest. 100, 1059–1070 (1997).
105. J. C. Yang, D. Perry-Lalley, The envelope protein of an endogenous murine retrovirus is a tumor-associated T-cell antigen for multiple murine tumors. J. Immunother. 23, 177–183 (2000).
Acknowledgments: We thank the study teams at EMD Serono (Billerica, MA) and Merck KGaA (Darmstadt, Germany). We thank H. Wang, B. Brunkhorst, F. Jiang, X. Xu, N. Zizlsperger,
M. Soloviev, P. A. Rolfe, and C. Wilm for technical contributions. We also thank Scripps Research Institute for the gift of the MC38 cell line. Funding: This study was supported by EMD Serono. Author contributions: Y.L. designed the overall in vivo pharmacology study and oversaw the execution of in vivo experiments and data analysis. D.Z. led the discovery project team and designed, executed, and oversaw the in vitro pharmacology studies. C.X. led the execution of in vivo experiments and immune phenotype study. K.W.H. designed in vivo studies and led the execution of experiments and data analysis. J.Q. performed in vivo studies and flow cytometry analysis. B.M., H.Y., and G.Q. performed in vivo studies. A.S. performed the structural biology analysis and linker design. V.M.H. and M.H.J. assisted in data analysis and manuscript preparation. R.E.F. and A.D. designed, performed, and analyzed IHC studies. G.L. analyzed the RNA-seq data. H.S. previously led the discovery and development program and provided scientific/immunological rationale for the combination of PD-L1 blockade and TGF- depletion. L.R. provided the overall scientific critique and guidance. K.-M.L. conceived, designed, and created the bifunctional molecule and provided the concept of the dual targeting. All authors contributed to the drafting and critical revision of the manuscript and approved the final version of the manuscript. Competing interests: Y.L., D.Z., C.X., B.M., J.Q., H.Y., G.Q., A.S., V.M.H., M.H.J., R.E.F., A.D., G.L., L.R., and K.-M.L. are all employees of EMD Serono. K.W.H. is an employee of GlaxoSmithKline. H.S. is an employee of Intrexon Corporation. K.-M.L. is the inventor on Patent Cooperation Treaty publication WO 2015/118115 and US 2015/0225483 held by applicant Merck Patent GmbH covering M7824, its methods of making, and its methods of use. Data and materials availability: The accession number for the mouse RNA-seq data is GSE107801. Requests for materials should be addressed to Y.L. or K.-M.L. and will be provided by and at EMD Serono’s sole discretion pending scientific review and the completion of a material transfer agreement with EMD Serono.
Submitted 2 May 2017Resubmitted 4 October 2017Accepted 13 November 2017Published 17 January 201810.1126/scitranslmed.aan5488
Citation: Y. Lan, D. Zhang, C. Xu, K. W. Hance, B. Marelli, J. Qi, H. Yu, G. Qin, A. Sircar, V. M. Hernández, M. H. Jenkins, R. E. Fontana, A. Deshpande, G. Locke, H. Sabzevari, L. Radvanyi, K.-M. Lo, Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-. Sci. Transl. Med. 10, eaan5488 (2018).
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from

βsimultaneously targeting PD-L1 and TGF-Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein
Radvanyi and Kin-Ming LoVivian M. Hernández, Molly H. Jenkins, Rachel E. Fontana, Amit Deshpande, George Locke, Helen Sabzevari, Laszlo Yan Lan, Dong Zhang, Chunxiao Xu, Kenneth W. Hance, Bo Marelli, Jin Qi, Huakui Yu, Guozhong Qin, Aroop Sircar,
DOI: 10.1126/scitranslmed.aan5488, eaan5488.10Sci Transl Med
promising results in multiple mouse models, alone and in combination with other treatment modalities.effective at restoring antitumor immunity than interventions targeting one pathway at a time, and it showedtargets an immune checkpoint and a different immunosuppressive pathway. This bifunctional protein was more
. designed a bifunctional protein called M7824, which simultaneouslyet alimmune checkpoint inhibition, Lan However, this type of treatment is not always sufficient to ensure protective immunity. To reinforce the effects ofcancer-associated immunosuppression and improve patients' own immune responses against the disease.
Inhibitors of immune checkpoints are increasingly used for cancer immunotherapy because they decreaseDoubling up against cancer
ARTICLE TOOLS http://stm.sciencemag.org/content/10/424/eaan5488
MATERIALSSUPPLEMENTARY http://stm.sciencemag.org/content/suppl/2018/01/12/10.424.eaan5488.DC1
CONTENTRELATED
http://stm.sciencemag.org/content/scitransmed/12/536/eaay8456.fullhttp://stm.sciencemag.org/content/scitransmed/12/525/eaay4860.fullhttp://stke.sciencemag.org/content/sigtrans/11/561/eaau0344.fullhttp://stke.sciencemag.org/content/sigtrans/12/596/eaat7527.fullhttp://stm.sciencemag.org/content/scitransmed/10/429/eaan3682.fullhttp://science.sciencemag.org/content/sci/359/6377/801.fullhttp://science.sciencemag.org/content/sci/359/6377/770.fullhttp://science.sciencemag.org/content/sci/359/6377/745.fullhttp://science.sciencemag.org/content/sci/359/6375/582.fullhttp://science.sciencemag.org/content/sci/359/6375/516.fullhttp://stm.sciencemag.org/content/scitransmed/9/385/eaak9679.fullhttp://stm.sciencemag.org/content/scitransmed/9/385/eaak9670.fullhttp://stm.sciencemag.org/content/scitransmed/9/393/eaal4922.fullhttp://stm.sciencemag.org/content/scitransmed/9/410/eaal4291.full
REFERENCES
http://stm.sciencemag.org/content/10/424/eaan5488#BIBLThis article cites 103 articles, 20 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS. is aScience Translational MedicineScience, 1200 New York Avenue NW, Washington, DC 20005. The title
(ISSN 1946-6242) is published by the American Association for the Advancement ofScience Translational Medicine
of Science. No claim to original U.S. Government WorksCopyright © 2018 The Authors, some rights reserved; exclusive licensee American Association for the Advancement
by guest on April 14, 2020
http://stm.sciencem
ag.org/D
ownloaded from