
CagA-Dependent Downregulation of B7-H2 Expression … Downregulation of B7-H2 Expression on Gastric...
10
of June 19, 2018. This information is current as Infection Helicobacter pylori of Th17 Responses during Inhibition Expression on Gastric Mucosa and CagA-Dependent Downregulation of B7-H2 Reyes E. Yamaoka, David Y. Graham, Ellen J. Beswick and Victor Taslima T. Lina, Irina V. Pinchuk, Jennifer House, Yoshio http://www.jimmunol.org/content/191/7/3838 doi: 10.4049/jimmunol.1300524 August 2013; 2013; 191:3838-3846; Prepublished online 30 J Immunol References http://www.jimmunol.org/content/191/7/3838.full#ref-list-1 , 23 of which you can access for free at: cites 60 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved. 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on June 19, 2018 http://www.jimmunol.org/ Downloaded from by guest on June 19, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of CagA-Dependent Downregulation of B7-H2 Expression … Downregulation of B7-H2 Expression on Gastric...

of June 19, 2018.This information is current as
InfectionHelicobacter pyloriof Th17 Responses during
InhibitionExpression on Gastric Mucosa and CagA-Dependent Downregulation of B7-H2
ReyesE.Yamaoka, David Y. Graham, Ellen J. Beswick and Victor
Taslima T. Lina, Irina V. Pinchuk, Jennifer House, Yoshio
http://www.jimmunol.org/content/191/7/3838doi: 10.4049/jimmunol.1300524August 2013;
2013; 191:3838-3846; Prepublished online 30J Immunol
Referenceshttp://www.jimmunol.org/content/191/7/3838.full#ref-list-1
, 23 of which you can access for free at: cites 60 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 19, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
CagA-Dependent Downregulation of B7-H2 Expression onGastric Mucosa and Inhibition of Th17 Responses duringHelicobacter pylori Infection
Taslima T. Lina,* Irina V. Pinchuk,*,† Jennifer House,‡ Yoshio Yamaoka,x
David Y. Graham,x Ellen J. Beswick,{,1 and Victor E. Reyes*,‡,1
Gastric epithelial cells (GECs) are the primary target for Helicobacter pylori infection and may act as APCs regulating local T cell
responses. We previously reported that H. pylori infection of GECs induces the expression of the T cell coinhibitory molecule
B7-H1 on GECs. This process contributes to the hyporesponsiveness of CD4+ effector T cells and accumulation of regulatory
T cells. In the present study, we investigated the impact of H. pylori cytotoxin-associated gene A (CagA) on the modulation of the
expression of the T cell costimulator B7-H2 by GECs. B7-H2 is involved in promoting Th17 type responses. H. pylori infection
downregulates B7-H2 expression by GECs in a CagA-dependent manner. IFN-g, which is increased in the H. pylori–infected
gastric mucosa, synergizes with H. pylori in downregulating B7-H2 expression by GECs. CagA-mediated modulation of B7-H2 on
GECs involves p70 S6 kinase phosphorylation. The CagA-dependent B7-H2 downregulation in GECs correlates with a decrease in
Th17 type responses in vitro and in vivo. Furthermore, CagA-dependent modulation of Th17 responses was inversely correlated
with the H. pylori colonization levels in vivo. Our data suggest that CagA contributes to the ability of H. pylori to evade Th17-
mediated clearance by modulating expression of B7-H2 and, thus, to the establishment of the H. pylori chronic infection. The
Journal of Immunology, 2013, 191: 3838–3846.
Helicobacter pylori is a Gram-negative, spiral-shapedbacterium that infects the gastric mucosa of .50% ofthe world’s population. H. pylori infection initially
occurs in childhood and becomes persistent. This chronic infec-tion leads to gastric inflammation (1) and is the major cause ofgastritis, gastric and duodenal ulcers, as well as gastric adeno-carcinoma (2–8).The cytotoxin-associated gene A (CagA) protein is a major
virulence factor of H. pylori. Patients infected with cagA+ strainshave higher levels of inflammatory responses and are at a higherrisk of developing peptic ulcer or gastric cancer (4, 9). CagA isencoded by the cagA gene within the cag pathogenicity island,a 40-kb chromosomal region that encodes for a type IV secretionsystem (T4SS).
Gastric epithelial cells (GECs) are the primary target forH. pylori infection. After H. pylori adheres to GECs, the CagA
protein is translocated into their cytosol via a T4SS (10). Once
inside GECs, CagA becomes phosphorylated and elicits multiple
cell responses, including disruption of epithelial tight junctions,
cytoskeleton rearrangement, changes in cellular adhesion proper-
ties and polarity, as well as secretion of proinflammatory media-
tors (11, 12). Despite the marked inflammatory response within
the H. pylori–infected gastric mucosa, the host immune response
is unable to clear H. pylori, resulting in persistent infection and
development of chronic gastric inflammation (13, 14). Studies by
us and others suggested that the imbalance in CD4+ T cells
responses to H. pylori is responsible for the host’s inability to
clear the infection (15–18). Th17 cells, whose hallmark cytokine
is IL-17A, are crucial in the clearance of extracellular bacteria
(19). IL-17A is primarily associated with gastric inflammation
during H. pylori infection and, when chronically present, may
contribute to the inflammation-associated carcinogenesis (19–21).
Alternatively, IL-17A–initiated recruitment of neutrophils is crit-
ical for the clearance of the bacteria (22). Although increased IL-
17A expression is observed during chronic gastric inflammation,
the levels produced are not sufficient to clear the infection. The
mechanisms responsible for the reduced Th17 responses during
the establishment of H. pylori persistence in the gastric mucosa
remain poorly understood. Dendritic cell–mediated skewing of
T cell balance toward suppressive regulatory T cells (Tregs) has
been suggested to be important in the downregulation of Th17 and
the establishment of the H. pylori persistence (23). However, it is
not known whether and how GECs, as primary targets for H. pylori
infection, contribute to regulation of Th17 cell responses during
the establishment of the chronic infection.During H. pylori infection GECs express MHC class II and may
act as local APCs (24, 25). In addition to the recognition of MHC-
bound peptides on APCs by TCR, the outcome of APC/T cell
*Department of Microbiology and Immunology, University of Texas Medical Branch,Galveston, TX 77555; †Department of Internal Medicine, University of Texas MedicalBranch, Galveston, TX 77555; ‡Department of Pediatrics, University of Texas MedicalBranch, Galveston, TX 77555; xDepartment of Medicine, Michael E. DeBakey Vet-erans Affairs Medical Center, Baylor College of Medicine, Houston, TX 77030; and{Department of Molecular Genetics and Microbiology, University of New MexicoHealth Sciences Center, Albuquerque, NM 87131
1E.J.B. and V.E.R. share senior authorship.
Received for publication February 28, 2013. Accepted for publication July 29, 2013.
This work was supported by National Institutes of Health Grants AI68712, DK090090-01,and CA127022; the American Gastroenterological Association Research ScholarAward; National Institutes of Health Grant 1U54RR02614; the University of TexasMedical Branch Clinical and Translational Sciences Award; a University of TexasMedical Branch Sealy Center for Vaccine Development predoctoral fellowship (toT.T.L.); American Cancer Society Grant RSG-10-159-01-LIB; National Institutesof Health Grant 8UL1TR000041; and by the University of New Mexico Clinicaland Translational Science Center.
Address correspondence and reprint requests to Dr. Victor E. Reyes, Department ofPediatrics, University of Texas Medical Branch, Children’s Hospital, 301 UniversityBoulevard, Galveston, TX 77555-0366. E-mail address: [email protected]
Abbreviations used in this article: CagA, cytotoxin-associated gene A; EpCAM,epithelial cell adhesion molecule; GEC, gastric epithelial cell; PMSS1, premouseSydney strain 1; ROR, retinoic acid–related orphan receptor; SS1, Sydney strain 1;Treg, regulatory T cell; T4SS, type IV secretion system; WT, wild-type.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1300524
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

interactions depends on a second signal provided by engagementof the B7 costimulatory receptor (26). Our laboratory has previ-ously shown that GECs express the classical B7 costimulatorsB7-1 and B7-2, whose expression is increased during H. pyloriinfection (25). We also demonstrated that GECs express B7-H1,whose expression also increases during H. pylori infection andcontributes to the suppression of CD4+ effector T cell activity andupregulation of Tregs (18, 27).B7-H2 (ICOS ligand) is among the newer members of the B7
family of receptors and is known to have a costimulatory function onT cell activity upon binding to its receptor, ICOS (28). Recent studieshave implicated B7-H2/ICOS interaction in Th17 cell development,maintenance, and function (29–31). However, the role of B7-H2 inimmune responses to H. pylori is unknown. Thus, in this study weinvestigated the impact of H. pylori and its major virulence factorCagA on the modulation of B7-H2 as an important regulator ofTh17 cell responses. Our in vitro and in vivo studies showed thatH. pylori causes downregulation of B7-H2 on GECs and this effectdepends on the presence of H. pylori CagA via a process involvingp70 S6 kinase activation. CagA-dependent B7-H2 downregulationon GECs correlated with the decrease in Th17 responses and wasinversely correlated with the level of H. pylori colonization in vivo.Thus, this study points out a novel strategy used by H. pylori toimpair Th17 responses, and this impairment could contribute to theestablishment of persistent infection in the host.
Materials and MethodsAnimals
Female C57BL/6 mice were purchased from The Jackson Laboratory (BarHarbor, ME). Six- to 8-wk-old mice, which were tested negative for in-testinal Helicobacter spp., were used in the experiments. The University ofTexas Medical Branch Institutional Animal Care and Use Committee–approved protocol was followed.
Human tissue and cell lines
GECs were isolated from biopsy specimens as described previously (32).Briefly, biopsy specimens of the gastric antrum were obtained from con-senting patients undergoing gastroesophageal duodenoscopy for variousclinical indications in accordance with Institutional Review Board–approvedprotocol. Patients were considered infected when H. pylori was detected byboth rapid urease testing and histopathology, and for these studies they wereconfirmed to be infected by culture of H. pylori from biopsies. Biopsy tissuewas placed in calcium- and magnesium-free HBSS, supplemented with 5%FCS and penicillin plus streptomycin, and transported immediately to thelaboratory. The tissue was then placed in HBSS containing 0.1 mM EDTAand 0.1 mM DTT and agitated at 37˚C for 15–30 min to remove the mucus.The biopsy tissue was placed in dispase solution (2.4 U/ml; BoehringerMannheim, Mannheim, Germany) and agitated at 37˚C for 30 min witha change of fresh dispase solution after 15 min. The supernatant was col-lected and the cells were pelleted by centrifugation at 2003 g for 5 min. Thecells isolated were mostly (.90%) epithelial cells according to the mor-phology with May–Grunwald–Giemsa staining (Sigma-Aldrich, St. Louis,MO) and flow cytometry with immunofluorescence staining of anticytokeratinmAb. Human gastric carcinoma epithelial cells N87 and AGS as well asHs738.st/int human GECs were obtained from the American Type CultureCollection. HGC-27 was obtained from RIKEN (Japan) and was main-tained in RPMI 1640 with 10% FBS and 2 mM L-glutamine.
Bacterial cultures and infection of GECs
H. pylori LC11 (cagA+) and RD26 (cagA+) strains were originally isolatedfrom duodenal ulcer and peptic ulcer patients, respectively (33, 34).H. pylori strain Sydney strain 1 (SS1) and premouse SS1 (PMSS1) (35)used to infect mice were gifts from Dr. J. Pappo (Astra Research Center)and Dr. Richard Peek (Vanderbilt University), respectively. These bacterialstrains were grown on tryptic soy agar plates supplemented with 5% sheep’sblood (Becton Dickinson, San Jose, CA) or on blood agar plates with2.5 mg/ml chloramphenicol (Technova, Hollister, CA) to maintain cagA2
strains at 37˚C under microaerophilic conditions. Bacteria were transferredafter 48 h into Brucella broth containing 10% FBS for overnight. Aftercentrifugation at 3000 rpm for 10 min, bacteria were resuspended in normal
saline. The concentration of bacteria was determined by measuring theOD530 using a spectrophotometer (DU-65; BD Biosciences) and compar-ing the value to a standard curve generated by quantifying viable organ-isms from aliquots of bacteria at varying concentrations that were alsoassessed by OD and colony formation. For specific multiplicity of infec-tion, the numbers of GECs were determined using trypan blue staining, andthe required number of bacteria was added after calculation. GECs weretreated with IFN-g (100 U/ml) for 48 h, then washed and incubated anadditional day in regular medium without IFN-g before infection. WhenIFN-g needed to be neutralized, anti–IFN-g-neutralizing Ab was added atan optimal concentration (10 mg/ml). As an isotype control, mouse IgG1 kAb was used in the same concentration. B7-H2 expression was measuredafter coculture with IFN-g or H. pylori–infected GECs in the presence ofeither anti–IFN-g-neutralizing Ab or isotype control Ab.
Construction of cagA isogenic mutant
For isogenic cagA mutants, portions of the genes were amplified by PCRand the amplified fragment was inserted into the pBluescript SK(+)(Stratagene, La Jolla, CA). After mutagenesis by insertion of a chloram-phenicol resistance gene cassette (a gift from Dr. D. E. Taylor, Universityof Alberta, Edmonton, AB, Canada) in the cagA gene, the obtained plas-mids (1–2 mg) were used for inactivation of chromosomal genes by naturaltransformation as previously described (36). Correct integration of the chlor-amphenicol resistance gene cassette into the H. pylori chromosome by doublecrossover recombination was confirmed by PCR amplification followed bySouthern blot hybridization.
Abs, recombinant proteins, and cell signaling inhibitors
PE-conjugated anti-human B7-H2 (clone M1H12), PE-conjugated anti-murine B7-H2 (clone HK5.3), allophycocyanin-conjugated anti-murineepithelial cell adhesion molecule (EpCAM; clone G8.8), and PE-conjugatedretinoic acid–related orphan receptor (ROR)gt (clone AFKJS-9) were pur-chased from eBioscience, as were the isotype controls. The viability dyeeFluor 780 (eBioscience, San Diego, CA) was included in the experiments tocontrol cell viability. Human rIFN-g (Roche) was used at 100 U/ml. Neu-tralizing Abs for IFN-g included the purified functional grade anti-humanIFN-g from eBioscience. The isotype control Ab used for IFN-g studies wasfunctional grade mouse IgG1 k from eBioscience. For cell signaling inhi-bition the following inhibitors were used: CAY10512 (10 mM; CaymanChemical, Ann Arbor, MI), AG-490 (100 ng/ml; Enzo Life Sciences,Farmingdale, NY), wortmannin (100 nM; Calbiochem, Billerica, MA),and rapamycin (100 ng/ml; Calbiochem, Darmstadt, Germany).
GEC/T cell coculture
For GEC/T cell coculture experiments, naive CD4+ T cells were isolatedfrom peripheral blood as previously described (17). Briefly, heparinizedvenous blood samples were collected from healthy volunteers negative forH. pylori (Institutional Review Board–approved protocol 06-122 at theUniversity of Texas Medical Branch). PBMCs were prepared from col-lected blood by density gradient centrifugation over Ficoll-Paque Plus.Naive CD4+ T cells were isolated from the peripheral blood by negativeselection using a MACS naive CD4+ T cell isolation kit II (MiltenyiBiotec, Bergisch Gladbach, Germany). GECs where exposed to H. pyloristrains for 8 h, and then supernatants from H. pylori–treated cultures werefiltered to remove bacteria, and GECs were washed twice with PBS toremove attached bacteria and replaced with filtered supernatants until 24 h.CD4+ T cells were preactivated for 1 h with anti-CD3/CD28 beads (Invi-trogen) according to the manufacturer’s instructions and added to each wellat 3:1 T cell/GEC ratio and then incubated at 37˚C with 5% CO2 for 2 d.
Western blot analysis
Western blot analysis was performed as previously described (37).
Infection of mice, processing of stomach, and detection ofH. pylori in murine stomach
Experimental animal groups were inoculated by orogastric gavage with 108
colony-forming units (in 100 ml PBS) of H. pylori SS1 or PMSS1 strainsthree times during a week. The placebo group was inoculated with PBS.Four weeks later, animal serum was collected, mice were euthanized, andstomach was removed. Stomachs were dissected longitudinally in two tofour pieces and used for analysis of the H. pylori load, histopathology, RT-PCR, and flow cytometry analysis. For histopathology analysis one lon-gitudinal strip of stomach was placed in 10% normal buffered formalin for24 h at 4˚C, transferred into 70% ethanol solution, and stored at 4˚C untilneeded. Tissue was then embedded in paraffin and processed by H&E
The Journal of Immunology 3839
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
staining. Stomach tissue dissociation and enzymatic digestion was per-formed using a gentleMACS dissociator (Miltenyi Biotec, Auburn, CA)according to the manufacturer’s instructions. Briefly, DTT/EDTA treat-ment was performed similarly as described above for human tissue. Cellswere then washed twice with HBSS followed by treating with collagenase
I, II, and IV (100 U/ml). After the first round of dissociation, cells wereincubated with enzymatic mixture at 37˚C for 30 min. Tissue was thensubjected to the second round of dissociation and treated with DNAse (10mg/ml; Worthington Biochemical) at 37˚C for 15 min. Finally, tissue wasprocessed using the dissociator and washed twice with HBSS. The digested
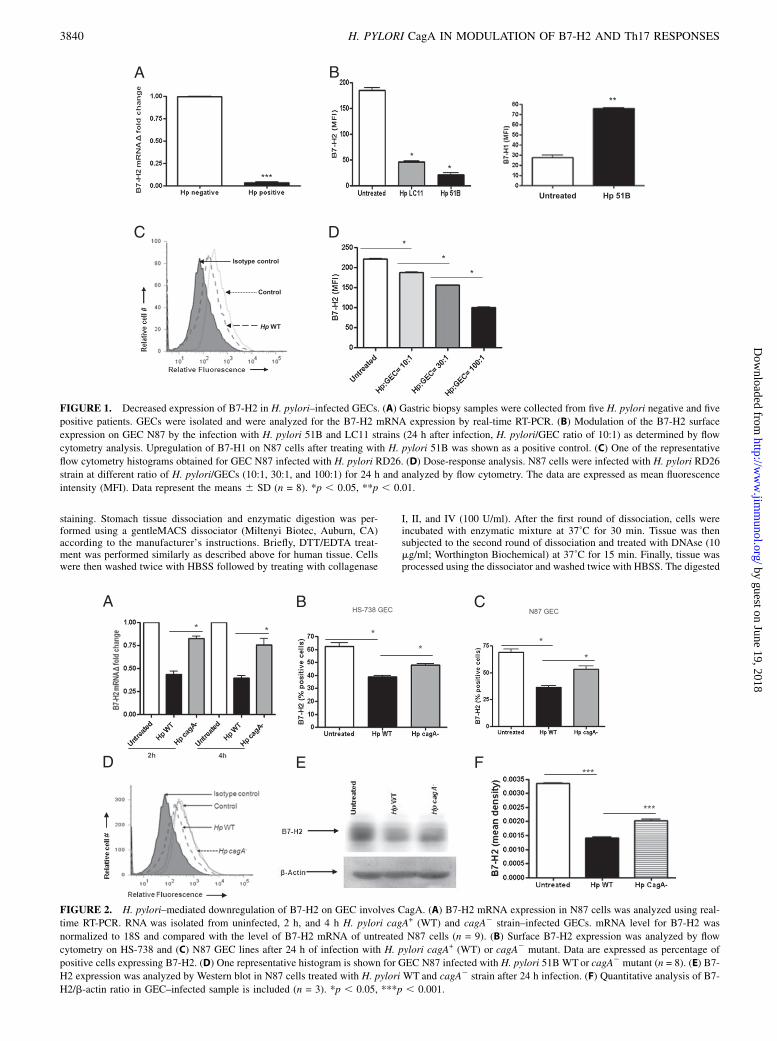
FIGURE 1. Decreased expression of B7-H2 in H. pylori–infected GECs. (A) Gastric biopsy samples were collected from five H. pylori negative and five
positive patients. GECs were isolated and were analyzed for the B7-H2 mRNA expression by real-time RT-PCR. (B) Modulation of the B7-H2 surface
expression on GEC N87 by the infection with H. pylori 51B and LC11 strains (24 h after infection, H. pylori/GEC ratio of 10:1) as determined by flow
cytometry analysis. Upregulation of B7-H1 on N87 cells after treating with H. pylori 51B was shown as a positive control. (C) One of the representative
flow cytometry histograms obtained for GEC N87 infected with H. pylori RD26. (D) Dose-response analysis. N87 cells were infected with H. pylori RD26
strain at different ratio of H. pylori/GECs (10:1, 30:1, and 100:1) for 24 h and analyzed by flow cytometry. The data are expressed as mean fluorescence
intensity (MFI). Data represent the means 6 SD (n = 8). *p , 0.05, **p , 0.01.
FIGURE 2. H. pylori–mediated downregulation of B7-H2 on GEC involves CagA. (A) B7-H2 mRNA expression in N87 cells was analyzed using real-
time RT-PCR. RNA was isolated from uninfected, 2 h, and 4 h H. pylori cagA+ (WT) and cagA2 strain–infected GECs. mRNA level for B7-H2 was
normalized to 18S and compared with the level of B7-H2 mRNA of untreated N87 cells (n = 9). (B) Surface B7-H2 expression was analyzed by flow
cytometry on HS-738 and (C) N87 GEC lines after 24 h of infection with H. pylori cagA+ (WT) or cagA2 mutant. Data are expressed as percentage of
positive cells expressing B7-H2. (D) One representative histogram is shown for GEC N87 infected with H. pylori 51B WTor cagA2 mutant (n = 8). (E) B7-
H2 expression was analyzed by Western blot in N87 cells treated with H. pylori WT and cagA2 strain after 24 h infection. (F) Quantitative analysis of B7-
H2/b-actin ratio in GEC–infected sample is included (n = 3). *p , 0.05, ***p , 0.001.
3840 H. PYLORI CagA IN MODULATION OF B7-H2 AND Th17 RESPONSES
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

tissue was passed through a 40-mm cell strainer. Recovered cells werecounted and used for immunostaining followed by flow cytometry analysis.
Murine gastric tissue was homogenized and DNA was extracted usinga DNeasy blood and tissue kit (Qiagen, Valencia, CA) followed by a pu-rification of the DNA according to the manufacturer’s protocol. ExtractedDNA was used for the detection of H. pylori by real-time PCR usinga protocol originally described by Roussel et al. (38). A primer/probe set16SHP229BP for 16S gene was used for the quantification of the H. pyloribacterial load. To determine bacterial load, standard curves were generatedby PCR of serial dilutions of extracted H. pylori DNA. Quantification ofH. pylori in murine gastric mucosa and determination of absolute genomecopy number were calculated according to the method described by Rousselet al. (38). Murine GADPH gene amplification was used to control the equalloading of total DNA used in the PCR reaction.
Flow cytometry
Flow cytometry was performed as we described previously (17).
Real-time RT-PCR
Real-time RT-PCR was performed according to the a two-step real-timeRT-PCR protocol (Applied Biosystems, Foster City, CA). The appropri-ate Assays-on-Demand gene expression assay primers/probe mix (AppliedBiosystems) for human 18S and gene of interest (a 203 mix of unlabeledPCR primers and TaqMan MGB probe, FAM dye–labeled) and 2 ml cDNAwas added to the PCR reaction step. The reactions were carried out in 20ml final volume using a Bio-Rad Q5 real-time PCR machine according tothe following protocol: 2 min at 50˚C, 10 min at 95˚C (1 cycle) and 15 s at95˚C, and 1 min at 60˚C (40 cycles). Triplicate cycle threshold values wereanalyzed in Microsoft Excel using the comparative CT (DDCT) method asdescribed by the manufacturer (Applied Biosystems).
Bio-Plex
The levels of total and phosphorylated cell signaling proteins in N87 cellsinfected with H. pylori strains—IL-17A from T cell/GEC coculture andIL-17, IL-21, IL-22, IL-23, and IL-6 from murine serum collected fromH. pylori–infected mice—were measured using a Luminex array (Millipore,Billerica, MA) according the manufacturer’s instructions. Samples wereanalyzed using Bio-Plex Manager software (Bio-Rad).
Statistical analysis
The results were expressed as the means 6 SD of data obtained from atleast three independent experiments done with triplicate sets in each ex-periment, unless otherwise indicated. Differences between means wereevaluated by ANOVA using a Student t test for multiple comparisons. Thep values , 0.05 were considered statistically significant.
ResultsH. pylori downregulates B7-H2 expression on GECs
To assess B7-H2 expression during H. pylori infection, mRNAlevels were examined in GECs isolated from gastric mucosa biopsysamples from H. pylori–infected and uninfected individuals usingreal-time RT-PCR. B7-H2 mRNA expression was significantly de-creased in GECs from H. pylori+ subjects when compared withGECs from uninfected controls (Fig. 1A). To determine whetherH. pylori directly induced a reduction of B7-H2 by GECs, a humanGEC line (N87) was infected with H. pylori 51B, LC-11, or RD26strains. A significant decrease in surface B7-H2 expression on GECswas observed at 24 h after infection with all H. pylori strains (Fig.1B, 1C). Fig 1B shows differential regulation of B7-H1 and B7-H2by N87 cells infected with H. pylori 51B. This effect was dose-dependent (Fig. 1D). Similar results were observed when other hu-man GEC lines (e.g., AGS, HGC-27, and HS-738 cells) were infected(not shown). Thus, our results indicate that H. pylori infectiondownregulates B7-H2 expression on human GECs in a dose-dependent manner.
Downregulation of B7-H2 expression depends on the presenceof H. pylori CagA
Because CagA is an importantH. pylori virulence factor capable ofeliciting multiple host cell responses (39), we sought to determine
whether downregulation of B7-H2 is influenced by H. pylori CagA.Infection of GECs with H. pylori 51B wild-type (WT) strain (Fig.2A) led to as much as 50% decrease of B7-H2 mRNA as comparedwith uninfected controls at the time points examined (2, 4 h). Incontrast, the H. pylori cagA2 mutant had a very limited effect onB7-H2 expression compared with the controls. These data wereconfirmed at the protein level. In contrast to the cagA+ H. pyloristrain, infection with the cagA2 mutant had a minor effect on thereduction of B7-H2 surface expression by GECs (Fig. 2B–D).Western blot analysis of N87 cells treated under the same conditionsprovided an independent approach to validate decreased B7-H2protein levels in cells infected with H. pylori WT as comparedwith cells infected with cagA2 strain (Fig. 2E, 2F). These resultssuggested that the major H. pylori virulence factor CagA is in-volved in the B7-H2 downregulation on GECs.
IFN-g and H. pylori have synergistic effects on B7-H2downregulation
IFN-g is produced within the H. pylori–infected gastric mucosa(40), and previously we showed a synergistic effect of IFN-g andH. pylori on B7-H1 upregulation on GECs (18). Thus, we ex-amined whether IFN-g could modulate H. pylori–mediated B7-H2downregulation on GECs. N87 cells treated with either IFN-g orH. pylori alone showed significant decreases in B7-H2 expression.However, treatment with both IFN-g and H. pylori resulted incomplete abrogation of B7-H2 expression. B7-H2 expression de-creased after culturing GECs with IFN-g, and decreased expres-sion was more prominent when GECs were pretreated with IFN-gprior to infection with H. pylori (Fig. 3). Blocking the IFN-g withneutralizing anti–IFN-g mAb prevented the decrease in the levelsof B7-H2 expression. Our results clearly showed a synergisticeffect of IFN-g and H. pylori in reduced B7-H2 expression byGECs. Interestingly, the synergism of IFN-g and H. pylori in de-creasing B7-H2 expression was less obvious when cells wereinfected with a cagA2 strain (Fig. 3). This result supports a keyrole of CagA in B7-H2 downregulation during H. pylori infection.
FIGURE 3. IFN-g synergizes H. pylori–mediated B7-H2 downregula-
tion on gastric epithelium. N87 cells were treated with IFN-g (100 U/ml)
for 48 h. Cells treated with IFN-g were washed and cultured with medium
for 24 h before they were either infected with H. pylori 51B WT and
cagA2 strain in the presence or absence of IFN-g–neutralizing Ab. B7-H2
expression on N87 GECs was measured by flow cytometry after 24 h.
Graph represents the mean 6 SD (n = 8). *p , 0.05.
The Journal of Immunology 3841
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

B7-H2 downregulation involve activation of mTOR/p70 S6kinase
Previous studies showed that H. pylori CagA protein can activateNF-kB, MAPK, STAT3, and PI3K pathways (41–44). To under-stand the underlying mechanisms regulating B7-H2 decreasedexpression during H. pylori infection, we first globally analyzedpathways activated in H. pylori–infected GECs using a Luminexcell signaling array. Our data demonstrated that, in addition to NF-kB and STAT3 pathways, H. pylori infection of GECs also leadsto the activation of mTOR/p70 S6 kinase within the first 5 min ofinfection (Fig. 4A, 4B). In contrast, no significant phosphorylationof p70 S6 kinase was observed in GECs infected with cagA2
strain (Fig. 4C, 4D). Thus, we examined the role of these path-ways in H. pylori–mediated downregulation of B7-H2 by usingspecific inhibitors. We observed that downregulation of B7-H2 by
the cagA+ H. pylori strain was blocked in the presence of rapa-mycin, a p70 S6 kinase/mTOR–specific inhibitor (Fig. 4E). In con-trast, inhibition of PI3K, STAT3, and NF-kB pathways withpharmacological inhibitors did not affect H. pylori–mediateddownregulation of B7-H2 expression (Fig. 4F). These results sug-gest that p70 S6 kinase is a key signaling pathway in H. pylori–mediated downregulation of B7-H2 on GECs.
CagA+ H. pylori infection reduces induction of Th17 by humangastric epithelium
Because B7-H2 has been implicated in Th17 cell differentiation,we examined whether the CagA-dependent B7-H2 downregulationimpaired the capacity of GECs to induce Th17 cell differentiationfrom naive CD4+ T cells. Our results showed that there is a smallinduction of RORg expressing CD4+ T cells when naive CD4+
FIGURE 4. Activation of mTOR/p70 S6 kinase involved in the H. pylori–mediated downregulation of B7-H2 expression. N87 cells were incubated with
H. pylori 51B WT and cagA2 strain. Cells were lysed after 5, 15, 30, 45, and 60 min. Cell lysates from GECs exposed to H. pylori 51B WT and cagA2
strain were analyzed for (A, C) phosphorylated and (B, D) total p70 S6 kinase using Luminex bead arrays, respectively. (E) GECs were treated with
rapamycin (100 ng/ml), an inhibitor of mTOR/p70 S6 kinase pathway, or (F) with wortmannin (100 nM), AG-490 (100 ng/ml), and CAY10512 (10 mM)
inhibitors of PI3K, STAT3, and NF-kB pathways, respectively, for 1 h and then were infected with H. pylori. B7-H2 expression was analyzed using flow
cytometry 24 h later. Data are expressed as a percentage of positive cells. Results represents as the means 6 SD (n = 8). *p , 0.05, **p , 0.01.
3842 H. PYLORI CagA IN MODULATION OF B7-H2 AND Th17 RESPONSES
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

T cells were cocultured with N87 cells infected with H. pylori WT,but this induction was significantly increased when cells wereinfected with a cagA2 strain (Fig. 5A). The presence of Th17 wasfurther confirmed by measuring IL-17A in coculture supernatantsby a Luminex array (Fig. 5B). Analysis of mRNA levels inparallel cultures confirmed these findings by showing increasedRORg and IL-17A mRNA levels in CD4+ T cells (Fig. 5C, 5D).These data demonstrate the critical role of CagA in maintaininglow-level Th17 responses, and this may contribute to H. pyloriimmune evasion.
Downregulation of B7-H2 on GECs during murine H. pyloriinfection depends on CagA and correlates with the decrease ofTh17 responses and increase in H. pylori colonization
To understand the relevance of our observations in vitro to theH. pylori–associated immunopathophysiology, we used a mousemodel of H. pylori infection. Mice infected with H. pylori PMSS1,which contains a functional T4SS, had downregulated B7-H2 ex-pression by gastric epithelial (EpCAM+) cells. Interestingly, infec-tion of mice with H. pylori SS1 strain in which the T4SS is defectiveand cannot deliver CagA in GECs (45) resulted in upregulation ofB7-H2 (Fig. 6A, 6B). No significant difference was observed ingastric mucosal inflammation in mice infected with either strain atweek 4. However, the H. pylori load was drastically different be-tween the mice infected with SS1 and PMSS1 strains. Mice infectedwith PMSS1 strain had .100-fold higher bacterial burden whencompared with those infected with SS1 strain (Fig. 6C). Further-more, analysis of serum cytokine profile demonstrated that in con-trast to the mice infected with SS1 strain, which had significantserum levels of IL-17 and IL-21 (p , 0.05), mice infected withPMSS1 strain failed to upregulate proinflammatory Th17 cytokines(Fig. 7). Serum levels of IL-6, IL-22, and IL-23 were also increasedsignificantly inH. pylori SS1–infected mice (Fig. 7). Taken together,our in vivo data correlate with our in vitro findings and suggest thatCagA-mediated downregulation of B7-H2 might be involved in theprevention of the Th17-mediated clearance of the H. pylori duringonset of the infection.
DiscussionDuring H. pylori infection the host mounts an immune response,but this response is insufficient to clear the infection, leading tothe establishment of a persistent infection and development ofchronic inflammation. Infiltration of CD4+ T cells into the gastricmucosa is among the major factors contributing to the ongoinginflammation. At the same time, these cells are required for theimmunization-induced protective responses (46–48). Recent datasuggested that Th17 type responses are required for the clearanceof the bacteria (49). The exact mechanisms implicated in theH. pylori–mediated escape of host immunity to prevent clearanceof H. pylori remain far from understood. In this study, we dem-onstrated that H. pylori infection of GECs leads to a decrease ofB7-H2, which is a positive costimulatory ligand, and that thisprocess might be important in H. pylori–mediated escape of Th17-mediated bacterial clearance.Costimulatory interactions between B7 family ligands and their
receptors play important roles in the growth, development, anddifferentiation of T cells. Recent data demonstrated that inter-actions between B7-H2 on APCs with its putative receptor ICOSon T and B cells regulate adaptive immune responses (30, 50).Stimulation of ICOS was demonstrated to be critical for the de-velopment of human IL-17–producing CD4+ T cells (31). Fur-thermore, Bauquet et al. (29) demonstrated that ICOS was criticalfor maintaining effector memory Th17 cells. B7-H22/2 knockoutmice also were noted to have lower Th17 responses to chlamydialinfection than do WT mice (51). The blockade of B7-H2/ICOSsignaling inhibited Th1 and Th17 cells responses in chronic in-flammatory conditions such as rheumatoid arthritis (30). Takentogether, these findings implicate B7-H2/ICOS signaling as animportant mediator in the activation of Th17 cells in inflamma-tion. Because expression of ICOS ligand, B7-H2, was previouslyreported on intestinal epithelial cells (52), and owing to its im-portance in activation of Th17 responses, we measured the ex-pression of B7-H2 in human biopsy samples and found thatepithelial cells in gastric biopsies isolated from H. pylori–infectedpatients had decreased levels of B7-H2 expression compared with
FIGURE 5. H. pylori–mediated Th17 devel-
opment from activated naive T cells in coculture
with N87. N87 cells were treated with either
media, with H. pylori WT, or with H. pylori
cagA2 strain (H. pylori/GEC ratio of 10:1) for
8 h. After treatment, the GECs were washed
and cocultured with preactivated human CD4+
T cells (3:1 T cell/GEC ratio) for 2 d and were
analyzed for (A) RORg expression in T cells by
flow cytometry, (B) IL-17A production in su-
pernatant by Luminex array, (C) RORg mRNA
levels, and (D) IL-17A mRNA levels in T cells
by real-time PCR. mRNA was normalized to
18S and compared with untreated cells (n = 6).
*p , 0.05, **p , 0.01.
The Journal of Immunology 3843
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

uninfected biopsy samples. B7-H2 expression on colonic andairway epithelium was previously noted (52). However, little isknown about the role of B7-H2 costimulation in the responsesassociated with bacterial immunopathogenesis and clearance. Inthe present study, we demonstrated for the first time, to our knowl-edge, that H. pylori significantly downregulated B7-H2 expression ingastric mucosa, particularly on GECs.Because CagA has been shown to play an important role in
H. pylori–mediated pathogenesis and immune evasion mechanisms(53), we sought to investigate its role in the observed B7-H2 down-
regulation. In this study using a CagA isogenic mutant and theircorresponding parental strains, we showed that CagA plays acrucial role in downregulating B7-H2 expression on GECs.Compared with the untreated cells, H. pylori cagA2 strains alwaysshowed some downregulation of B7-H2, suggesting that othercomponents of H. pylori might also have an influence in down-regulating B7-H2 expression, but they are less effective than theWT CagA+ strains. Our in vivo data using C57BL/6 mice alsoshowed that H. pylori mediated transfer of CagA via a T4SSsignificantly downregulates B7-H2 expression in the GECs in the
FIGURE 6. H. pylori–mediated downregulation of B7-H2 on GECs in vivo involves CagA function and is inversely correlated with bacterial clearance.
(A) C57BL/6 mice were challenged with H. pylori strain PMSS1, which express functional T4SS and can deliver CagA, or with H. pylori SS1, which does
not. Gastric mononuclear cells were isolated 4 wk after H. pylori challenge using enzymatic digestion, and expression of B7-H2 and epithelial cell marker
EpCAM was analyzed by flow cytometry. (B) Level of the B7-H2–expressing epithelial cells (EpCam+) in the gastric mucosa from the cells was measured
by flow cytometry. (C) Infection rate was determined by quantification of H. pylori genome copy per half of stomach based on the analysis of H. pylori 16S
gene amplification by real-time PCR. Each datum point represents a single mouse tested in quadruplicate. Average bars of infection rate were calculated
from five mice per group and are demonstrated as means 6 SD. *p , 0.05.
FIGURE 7. In vivo infection with H. pylori expressing functional T4SS and that can deliver CagA fails to upregulate Th17 type responses. C57BL/6
mice were challenged with H. pylori strain PMSS1 or with H. pylori SS1. Blood was collected 4 wk after H. pylori challenge, and cytokine profile was
analyzed using a Luminex bead array. Data are represented as means 6 SD (n = 12). *p , 0.05.
3844 H. PYLORI CagA IN MODULATION OF B7-H2 AND Th17 RESPONSES
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

murine gastric mucosa. These in vitro and in vivo data reveal anovel mechanism whereby H. pylori uses one of its importantvirulence factors, CagA, to create a favorable environment for itspersistence via suppression of the positive costimulators requiredfor an efficient effector T cell response.Because cytokines play an important role in regulating immune
function, and IFN-g has been detected in H. pylori–infected gas-tric tissues in both humans and mice (54), we also investigatedwhether IFN-g has any role in B7-H2 expression. Our resultsshowed that IFN-g synergizes with the effect of H. pylori cagA+
strains in downregulating B7-H2 expression by GECs. Severalstudies showed induction of B7-2, B7-H1, and B7-DC in differentclassical APCs by IFN-g (55, 56). Stanciu et al. (57) showed asynergistic effect of respiratory syncytial virus and IFN-g in theupregulation of B7-H1 and B7-DC in respiratory epithelial cells.Additionally, their study showed that treatment of respiratory syn-cytial virus infected cells with IFN-g causes downregulation ofB7-H2 and B7-H3 expression. Previous findings from our groupshowed that IFN-g and H. pylori synergize in B7-H1 upregulation(18). Thus, the synergistic effect of IFN-g and H. pylori in B7-H2downregulation could result from an IFN-g–mediated increase inexpression by GEC receptors that are used by H. pylori (32).CagA interacts with several intracellular components of signal
transduction and activates NF-kB, MAPK, STAT3, and PI3K/Aktpathways (41–44). Previous reports have highlighted the fact thatH. pylori activates STAT3 to modulate host immune responses(43). Although our results showed activation of these pathwaysby CagA-expressing H. pylori strains, our data imply that CagA-mediated activation of the NF-kB, STAT3, and PI3K pathways isnot required for the H. pylori–mediated downregulation of B7-H2on GECs. Furthermore, to our knowledge, our data demonstratefor the first time that CagA contributes to the H. pylori–mediatedactivation of the mTOR/p70 S6 kinase pathway. Serine/threonineprotein kinase mTOR acts in a signaling pathway downstream fromPI3K/Akt and regulates the activation of the p70 S6 kinase, whichis required for translational regulation of ribosomal proteins (58).Using specific cell signaling inhibitors we showed that H. pyloriuses CagA to manipulate B7-H2 expression by activating p70 S6kinase pathway to prevent GECs from providing positive costim-ulation needed for protective Th17 cells.Previously our group showed that H. pylori uses its CagA and
VacA proteins to induce TGF-b production from GECs, whichcauses inhibition of CD4+ effector T cell proliferation and in-duction of Tregs (17). In this study, using in vitro GEC/T cellcocultures we showed that H. pylori uses CagA cytotoxin todownregulate Th17 cell type responses. A significant downregu-lation of Th17 cell transcription factor RORgt and IL-17A wasobserved when GECs were exposed to the H. pylori strainsexpressing CagA, but not in presence of the cagA2 mutant. Ourin vivo data with the H. pylori mouse model support this in vitroobservation. We have further shown that H. pylori CagA-mediatedB7-H2 downregulation correlates with a decrease in Th17 typeresponses detected in murine serum and an increase in H. pyloricolonization of the gastric mucosa. Although our data showedincreased H. pylori bacterial load and decreased Th17 type re-sponse in PMSS1-infected mice compared with those infectedwith the SS1 strain, we did not observe severe inflammatory changesin any of the H. pylori–infected mice. This might be becauseH. pylori infection in the mouse model results mostly in lympho-cytic gastritis, which does not progress to severe inflammation. Anoptimum induction of chronic gastritis can be achieved using Heli-cobacter felis as a model (59). Th17 cells have been suggested tohave dual roles in both infection control on the one hand and pre-neoplastic changes on the other hand. Several studies showed that
protective immunity against H. pylori infection requires a strongTh17 response (60). Our study showed reduced Th17 cell cyto-kines and increased bacterial load in the PMSS1-infected micecorrelate with results from another recent study (49). Horvathet al. (49) showed that mice lacking IL-23 when infected withH. pylori showed reduced IL-17 production and increased bacte-rial load in their stomachs. Another study suggested that ICOS-induced signaling is essential for IL-21–mediated regulation ofIL-23R expression in differentiated Th17 cells and for IL-23–driven expansion of Th17 cells (29). Our study also supports theimportance of B7-H2/ICOS signaling in Th17 cell developmentbecause the downregulation of B7-H2 expression in the gastricmucosa of PMSS1 H. pylori–infected mice correlates with de-creased Th17 type responses.Our data suggest a novel CagA-dependent mechanism, which
involves downregulation of B7-H2 on GECs, a primary target forH. pylori infection, used by the bacteria to avoid a Th17 cell–mediated clearance. Thus, our in vitro and in vivo studies suggestthat H. pylori use the T4SS to downregulate Th17 cell responses,which are critical for clearing the pathogen. Therefore, the H.pylori CagA delivery system may be an important target in vaccinedevelopment for achieving acceptable levels of immune protectionand for designing a therapeutic strategy to treat patients infectedwith this prevalent and deadly human pathogen.
AcknowledgmentsWe thank technical assistant Yu Lin for excellent assistance in performing
the animal work.
DisclosuresThe authors have no financial conflicts of interest.
References1. Marshall, B. J. 1994. Helicobacter pylori. Am. J. Gastroenterol. 89(8, Suppl.):
S116–S128.2. Parsonnet, J., G. D. Friedman, D. P. Vandersteen, Y. Chang, J. H. Vogelman,
N. Orentreich, and R. K. Sibley. 1991. Helicobacter pylori infection and the riskof gastric carcinoma. N. Engl. J. Med. 325: 1127–1131.
3. Huang, J. Q., S. Sridhar, Y. Chen, and R. H. Hunt. 1998. Meta-analysis of therelationship between Helicobacter pylori seropositivity and gastric cancer.Gastroenterology 114: 1169–1179.
4. Uemura, N., S. Okamoto, S. Yamamoto, N. Matsumura, S. Yamaguchi,M. Yamakido, K. Taniyama, N. Sasaki, and R. J. Schlemper. 2001. Helicobacterpylori infection and the development of gastric cancer. N. Engl. J. Med. 345:784–789.
5. Parsonnet, J., S. Hansen, L. Rodriguez, A. B. Gelb, R. A. Warnke, E. Jellum,N. Orentreich, J. H. Vogelman, and G. D. Friedman. 1994. Helicobacter pyloriinfection and gastric lymphoma. N. Engl. J. Med. 330: 1267–1271.
6. Rauws, E. A., and G. N. Tytgat. 1990. Cure of duodenal ulcer associated witheradication of Helicobacter pylori. Lancet 335: 1233–1235.
7. Wu, X. C., P. Andrews, V. W. Chen, and F. D. Groves. 2009. Incidence ofextranodal non-Hodgkin lymphomas among whites, blacks, and Asians/PacificIslanders in the United States: anatomic site and histology differences. CancerEpidemiol. 33: 337–346.
8. Coghlan, J. G., D. Gilligan, H. Humphries, D. McKenna, C. Dooley, E. Sweeney,C. Keane, and C. O’Morain. 1987. Campylobacter pylori and recurrence ofduodenal ulcers: a 12-month follow-up study. Lancet 330: 1109–1111.
9. Blaser, M. J., and J. E. Crabtree. 1996. CagA and the outcome of Helicobacterpylori infection. Am. J. Clin. Pathol. 106: 565–567.
10. Asahi, M., T. Azuma, S. Ito, Y. Ito, H. Suto, Y. Nagai, M. Tsubokawa,Y. Tohyama, S. Maeda, M. Omata, et al. 2000. Helicobacter pylori CagA proteincan be tyrosine phosphorylated in gastric epithelial cells. J. Exp. Med. 191: 593–602.
11. Bourzac, K. M., and K. Guillemin. 2005. Helicobacter pylori-host cell inter-actions mediated by type IV secretion. Cell. Microbiol. 7: 911–919.
12. Hatakeyama, M. 2004. Oncogenic mechanisms of the Helicobacter pylori CagAprotein. Nat. Rev. Cancer 4: 688–694.
13. Beswick, E. J., D. A. Bland, G. Suarez, C. A. Barrera, X. Fan, and V. E. Reyes.2005. Helicobacter pylori binds to CD74 on gastric epithelial cells and stim-ulates interleukin-8 production. Infect. Immun. 73: 2736–2743.
14. Muller, A., M. Oertli, and I. C. Arnold. 2011. H. pylori exploits and manipulatesinnate and adaptive immune cell signaling pathways to establish persistent in-fection. Cell Commun. Signal. 9: 25.
The Journal of Immunology 3845
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

15. Harris, P. R., S. W. Wright, C. Serrano, F. Riera, I. Duarte, J. Torres, A. Pena,A. Rollan, P. Viviani, E. Guiraldes, et al. 2008. Helicobacter pylori gastritis inchildren is associated with a regulatory T-cell response. Gastroenterology 134:491–499.
16. Mitchell, P., C. Germain, P. L. Fiori, W. Khamri, G. R. Foster, S. Ghosh,R. I. Lechler, K. B. Bamford, and G. Lombardi. 2007. Chronic exposure toHelicobacter pylori impairs dendritic cell function and inhibits Th1 develop-ment. Infect. Immun. 75: 810–819.
17. Beswick, E. J., I. V. Pinchuk, R. B. Earley, D. A. Schmitt, and V. E. Reyes. 2011.Role of gastric epithelial cell-derived transforming growth factor b in reducedCD4+ T cell proliferation and development of regulatory T cells during Heli-cobacter pylori infection. Infect. Immun. 79: 2737–2745.
18. Das, S., G. Suarez, E. J. Beswick, J. C. Sierra, D. Y. Graham, and V. E. Reyes.2006. Expression of B7-H1 on gastric epithelial cells: its potential role in regulatingT cells during Helicobacter pylori infection. J. Immunol. 176: 3000–3009.
19. Kabir, S. 2011. The role of interleukin-17 in the Helicobacter pylori inducedinfection and immunity. Helicobacter 16: 1–8.
20. Luzza, F., T. Parrello, G. Monteleone, L. Sebkova, M. Romano, R. Zarrilli,M. Imeneo, and F. Pallone. 2000. Up-regulation of IL-17 is associated withbioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa.J. Immunol. 165: 5332–5337.
21. Resende, C., A. Thiel, J. C. Machado, and A. Ristimaki. 2011. Gastric cancer:basic aspects. Helicobacter 16(Suppl. 1): 38–44.
22. DeLyria, E. S., R. W. Redline, and T. G. Blanchard. 2009. Vaccination of miceagainst H pylori induces a strong Th-17 response and immunity that is neutrophildependent. Gastroenterology 136: 247–256.
23. Kao, J. Y., M. Zhang, M. J. Miller, J. C. Mills, B. Wang, M. Liu, K. A. Eaton,W. Zou, B. E. Berndt, T. S. Cole, et al. 2010. Helicobacter pylori immune escapeis mediated by dendritic cell-induced Treg skewing and Th17 suppression inmice. Gastroenterology 138: 1046–1054.
24. Ishii, N., M. Chiba, M. Iizuka, H. Watanabe, T. Ishioka, and O. Masamune. 1992.Expression of MHC class II antigens (HLA-DR, -DP, and -DQ) on human gastricepithelium. Gastroenterol. Jpn. 27: 23–28.
25. Ye, G., C. Barrera, X. Fan, W. K. Gourley, S. E. Crowe, P. B. Ernst, andV. E. Reyes. 1997. Expression of B7-1 and B7-2 costimulatory molecules byhuman gastric epithelial cells: potential role in CD4+ T cell activation duringHelicobacter pylori infection. J. Clin. Invest. 99: 1628–1636.
26. Lenschow, D. J., T. L. Walunas, and J. A. Bluestone. 1996. CD28/B7 system ofT cell costimulation. Annu. Rev. Immunol. 14: 233–258.
27. Beswick, E. J., I. V. Pinchuk, S. Das, D. W. Powell, and V. E. Reyes. 2007.Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cellsafter Helicobacter pylori exposure promotes development of CD4+ CD25+
FoxP3+ regulatory T cells. Infect. Immun. 75: 4334–4341.28. Aicher, A., M. Hayden-Ledbetter, W. A. Brady, A. Pezzutto, G. Richter,
D. Magaletti, S. Buckwalter, J. A. Ledbetter, and E. A. Clark. 2000. Charac-terization of human inducible costimulator ligand expression and function. J.Immunol. 164: 4689–4696.
29. Bauquet, A. T., H. Jin, A. M. Paterson, M. Mitsdoerffer, I. C. Ho, A. H. Sharpe,and V. K. Kuchroo. 2009. The costimulatory molecule ICOS regulates the ex-pression of c-Maf and IL-21 in the development of follicular T helper cells andTH-17 cells. Nat. Immunol. 10: 167–175.
30. Frey, O., J. Meisel, A. Hutloff, K. Bonhagen, L. Bruns, R. A. Kroczek,L. Morawietz, and T. Kamradt. 2010. Inducible costimulator (ICOS) blockadeinhibits accumulation of polyfunctional T helper 1/T helper 17 cells and miti-gates autoimmune arthritis. Ann. Rheum. Dis. 69: 1495–1501.
31. Paulos, C. M., C. Carpenito, G. Plesa, M. M. Suhoski, A. Varela-Rohena,T. N. Golovina, R. G. Carroll, J. L. Riley, and C. H. June. 2010. The induciblecostimulator (ICOS) is critical for the development of human TH17 cells. Sci.Transl. Med. 2: 55ra78.
32. Fan, X., S. E. Crowe, S. Behar, H. Gunasena, G. Ye, H. Haeberle, N. Van Houten,W. K. Gourley, P. B. Ernst, and V. E. Reyes. 1998. The effect of class II majorhistocompatibility complex expression on adherence of Helicobacter pylori and in-duction of apoptosis in gastric epithelial cells: a mechanism for T helper celltype 1-mediated damage. J. Exp. Med. 187: 1659–1669.
33. Crowe, S. E., L. Alvarez, M. Dytoc, R. H. Hunt, M. Muller, P. Sherman, J. Patel,Y. Jin, and P. B. Ernst. 1995. Expression of interleukin 8 and CD54 by humangastric epithelium after Helicobacter pylori infection in vitro. Gastroenterology108: 65–74.
34. Lu, H., J. Y. Wu, E. J. Beswick, T. Ohno, S. Odenbreit, R. Haas, V. E. Reyes,M. Kita, D. Y. Graham, and Y. Yamaoka. 2007. Functional and intracellularsignaling differences associated with the Helicobacter pylori AlpAB adhesinfrom Western and East Asian strains. J. Biol. Chem. 282: 6242–6254.
35. Arnold, I. C., J. Y. Lee, M. R. Amieva, A. Roers, R. A. Flavell, T. Sparwasser,and A. Muller. 2011. Tolerance rather than immunity protects from Helicobacterpylori-induced gastric preneoplasia. Gastroenterology 140: 199–209.
36. Heuermann, D., and R. Haas. 1998. A stable shuttle vector system for efficientgenetic complementation of Helicobacter pylori strains by transformation andconjugation. Mol. Gen. Genet. 257: 519–528.
37. Saada, J. I., I. V. Pinchuk, C. A. Barrera, P. A. Adegboyega, G. Suarez,R. C. Mifflin, J. F. Di Mari, V. E. Reyes, and D. W. Powell. 2006. Subepithelialmyofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J.Immunol. 177: 5968–5979.
38. Roussel, Y., A. Harris, M. H. Lee, and M. Wilks. 2007. Novel methods ofquantitative real-time PCR data analysis in a murine Helicobacter pylori vaccinemodel. Vaccine 25: 2919–2929.
39. Backert, S., and M. Selbach. 2008. Role of type IV secretion in Helicobacterpylori pathogenesis. Cell. Microbiol. 10: 1573–1581.
40. Shimada, M., T. Ando, R. M. Peek, O. Watanabe, K. Ishiguro, O. Maeda,D. Ishikawa, M. Hasegawa, K. Ina, N. Ohmiya, et al. 2008. Helicobacter pyloriinfection upregulates interleukin-18 production from gastric epithelial cells. Eur.J. Gastroenterol. Hepatol. 20: 1144–1150.
41. Churin, Y., L. Al-Ghoul, O. Kepp, T. F. Meyer, W. Birchmeier, and M. Naumann.2003. Helicobacter pylori CagA protein targets the c-Met receptor and enhancesthe motogenic response. J. Cell Biol. 161: 249–255.
42. Higashi, H., A. Nakaya, R. Tsutsumi, K. Yokoyama, Y. Fujii, S. Ishikawa,M. Higuchi, A. Takahashi, Y. Kurashima, Y. Teishikata, et al. 2004. Helicobacterpylori CagA induces Ras-independent morphogenetic response through SHP-2recruitment and activation. J. Biol. Chem. 279: 17205–17216.
43. Lee, K. S., A. Kalantzis, C. B. Jackson, L. O’Connor, N. Murata-Kamiya,M. Hatakeyama, L. M. Judd, A. S. Giraud, and T. R. Menheniott. 2012. Heli-cobacter pylori CagA triggers expression of the bactericidal lectin REG3g viagastric STAT3 activation. PLoS ONE 7: e30786.
44. Li, S. P., X. J. Chen, A. H. Sun, J. F. Zhao, and J. Yan. 2010. CagA+ Helicobaterpylori induces Akt1 phosphorylation and inhibits transcription of p21(WAF1/CIP1) and p27(KIP1) via PI3K/Akt1 pathway. Biomed. Environ. Sci. 23: 273–278.
45. Crabtree, J. E., R. L. Ferrero, and J. G. Kusters. 2002. The mouse colonizingHelicobacter pylori strain SS1 may lack a functional cag pathogenicity island.Helicobacter 7: 139–140.
46. Smythies, L. E., K. B. Waites, J. R. Lindsey, P. R. Harris, P. Ghiara, andP. D. Smith. 2000. Helicobacter pylori-induced mucosal inflammation is Th1mediated and exacerbated in IL-4, but not IFN-g, gene-deficient mice. J.Immunol. 165: 1022–1029.
47. Eaton, K. A., M. Mefford, and T. Thevenot. 2001. The role of T cell subsets andcytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J.Immunol. 166: 7456–7461.
48. Mohammadi, M., J. Nedrud, R. Redline, N. Lycke, and S. J. Czinn. 1997. MurineCD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis andTH2 cells reduce bacterial load. Gastroenterology 113: 1848–1857.
49. Horvath, D. J., Jr., M. K. Washington, V. A. Cope, and H. M. Algood. 2012. IL-23 contributes to control of chronic Helicobacter pylori infection and the de-velopment of T helper responses in a mouse model. Front. Immunol. 3: 56.
50. Burmeister, Y., T. Lischke, A. C. Dahler, H. W. Mages, K. P. Lam, A. J. Coyle,R. A. Kroczek, and A. Hutloff. 2008. ICOS controls the pool size of effector-memory and regulatory T cells. J. Immunol. 180: 774–782.
51. Kadkhoda, K., S. Wang, A. G. Joyee, Y. Fan, J. Yang, and X. Yang. 2010. Th1cytokine responses fail to effectively control Chlamydia lung infection in ICOSligand knockout mice. J. Immunol. 184: 3780–3788.
52. Nakazawa, A., I. Dotan, J. Brimnes, M. Allez, L. Shao, F. Tsushima, M. Azuma,and L. Mayer. 2004. The expression and function of costimulatory moleculesB7H and B7-H1 on colonic epithelial cells. Gastroenterology 126: 1347–1357.
53. Kusters, J. G., A. H. van Vliet, and E. J. Kuipers. 2006. Pathogenesis of Heli-cobacter pylori infection. Clin. Microbiol. Rev. 19: 449–490.
54. Karttunen, R., T. Karttunen, H. P. Ekre, and T. T. MacDonald. 1995. Interferon gand interleukin 4 secreting cells in the gastric antrum in Helicobacter pyloripositive and negative gastritis. Gut 36: 341–345.
55. Kim, J., A. C. Myers, L. Chen, D. M. Pardoll, Q. A. Truong-Tran, A. P. Lane,J. F. McDyer, L. Fortuno, and R. P. Schleimer. 2005. Constitutive and inducibleexpression of B7 family of ligands by human airway epithelial cells. Am. J.Respir. Cell Mol. Biol. 33: 280–289.
56. Morgado, P., Y. C. Ong, J. C. Boothroyd, and M. B. Lodoen. 2011. Toxoplasmagondii induces B7-2 expression through activation of JNK signal transduction.Infect. Immun. 79: 4401–4412.
57. Stanciu, L. A., C. M. Bellettato, V. Laza-Stanca, A. J. Coyle, A. Papi, andS. L. Johnston. 2006. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epi-thelial cells and regulation by respiratory syncytial virus and type 1 and 2cytokines. J. Infect. Dis. 193: 404–412.
58. Fingar, D. C., C. J. Richardson, A. R. Tee, L. Cheatham, C. Tsou, and J. Blenis.2004. mTOR controls cell cycle progression through its cell growth effectorsS6K1 and 4E-BP1/eukaryotic translation initiation factor 4E.Mol. Cell. Biol. 24:200–216.
59. O’Rourke, J. L., and A. Lee. 2003. Animal models of Helicobacter pylori in-fection and disease. Microbes Infect. 5: 741–748.
60. Czinn, S. J., and T. Blanchard. 2011. Vaccinating against Helicobacter pyloriinfection. Nat. Rev.Gastroenterol. Hepatol. 8: 133–140.
3846 H. PYLORI CagA IN MODULATION OF B7-H2 AND Th17 RESPONSES
by guest on June 19, 2018http://w
ww
.jimm
unol.org/D
ownloaded from


















