Bryostatin 1 Affects P-Glycoprotein Phosphorylation but not...
8
Vol. 1. 1581-1587. December 1995 Clinical Cancer Research 1581 Bryostatin 1 Affects P-Glycoprotein Phosphorylation but not Function in Multidrug-resistant Human Breast Cancer Cells Stefania Scala, Bruce Dickstein, Joanna Regis, Zoltan Szallasi, Peter M Blumberg, and Susan E. Bates’ Medicine Branch, Clinical Oncology Program. Division of Cancer Treatment IS. S.. B. D., J. R., S. E. B.l, and Laboratory of Cellular Carcinogenesis and Tumor Proiiiotion, Division of Cancer Etiology lZ. S.. P. M. B.l, National Cancer Institute. NIH. Bethesda, Maryland 20892 ABSTRACT The function of P-glycoprotein (Pgp), which confers multidrug resistance by active efflux of drug, is thought to be dependent on phosphorylation. Previous studies have suggested that protein kinase C (PKC) plays an important role in Pgp phosphorylation. We report here the effects of bryostatin 1, a unique PKC activator and inhibitor, on Pgp function in a multidrug-resistant MCF-7 human breast can- cer subline which overexpresses PKC-a. Bryostatin 1 (100 nrsi) decreased Pgp phosphorylation after 24 h of treatment. In contrast, it did not affect Pgp function as demonstrated by the accumulation of [3H]vinblastine and rhodamine 123. We compared the effect of bryostatin 1 treatment on PKC-a with that of the phorbol ester 12-O-tetradecanoylphorbol- 13-acetate (200 nsi). 12-O-tetradecanoylphorbol-13-acetate caused translocation of PKC-a from the cytosol to the cell membrane after a 10-mm treatment and its down-regulation after 24 h of treatment. Likewise, bryostatin 1 (100 nsl) caused translocation, but only after longer treatment (1 h), and it caused down-regulation of PKC-a at 24 h of treat- ment. Thus, while the MCF-7TH cells overexpress the PKC-a isoform, and its down-regulation by bryostatin 1 is associated with decreased Pgp phosphorylation, these alter- ations do not modulate drug transport. We conclude that, while bryostatin 1 may be useful clinically because of its ability to inhibit PKC, it is not able to reverse Pgp-mediated multidrug resistance. INTRODUCTION The identification of clinically suitable agents able to oven- come drug resistance would have great impact for cancer treat- ment. In tumor cell lines. the best characterized mechanism of drug resistance is ‘ ‘multidrug resistance’ ‘ mediated by overex- pression of an energy-dependent membrane-bound drug effiux pump known as Pgp2. Pgp undergoes covalent modification by Received 2/6/95: revised 8/15/95: accepted 8/17/95. I To whom requests for reprints should be addressed. at NIH. Building 10/Room 12N226, 9000 Rockville Pike, Bethesda. MD 20892. Phone: (301) 402-0984; Fax: (301) 402-0172. 2 The abbreviations used are: Pgp. P-glycoprotein: PK. protein kinase: TPA, I 2-O-tetnadecanoylphonbol- I 3-acetate. rapid cycles of phosphorylation and dephosphorylation in which several different protein kinases have been implicated ( 1-6). The earliest reports suggested that phosphorylation played a role in the activity of the efflux pump (1. 7-9). Increased transport results from increased Pgp phosphorylation, whereas impaired function occurs with inhibition of phosphorylation (2. 3). PKC (2. 10-13). PKA (4. 13). and other potentially more specific kinases (3. 5. 6) have been linked to Pgp phosphoryla- tion. Chambers et a/. ( 12) reported that PKC phosphorylates Pgp at multiple sites in the linker region between the two halves of the molecule. Characterization of specific residues for PKC and PKA phosphorylation has been reported in both munine and human Pgp (11, 13). Several studies have confirmed that acti- vation of PKC by the phorbol ester TPA results in an increase in Pgp phosphorylation associated with increased drug efflux, de- creased drug accumulation, and a more resistant phenotype ( 1-3. 7. 14. 15). Conversely, PKC down-regulation by long- term TPA treatment or inhibition by staunosponne on calphostin C impairs Pgp function, resulting in decreased drug efflux and increased accumulation (7, 14, 16). Increased expression or activity of the PKC isoforms has been reported in multidrug- resistant cell lines (2, 7, 17), with a differential increase in the expression of the PKC-a isoform being the most frequent ob- servation ( 17-19). This finding has been reported for a multi- drug-resistant subline of MCF-7 human breast cancer cells (19). Bryostatin 1 . which is structurally distinct from the phorbol esters, can activate PKC in vitro but in t’is’o it can paradoxically antagonize other activators of PKC such as the tumor-promoting phonbol esters (20. 21 ). Bryostatin 1 is the best studied member of a family of more than I 3 compounds that have a macrocyclic lactone structure with varying side chains (22. 23). As the most abundant of the bryostatins isolated from the marine bryozoan Bugu/a neritina, bryostatin 1 is the first member of this class of compounds to be tested in humans. These macrocyclic lactones possess potent antineoplastic properties in human and murine tumor cell lines (22. 24-26) and in tim activity in murine B16 melanoma. The range of in vitro activities includes cytotoxicity and terminal differentiation of malignant cells (27, 28). More- over, in contrast to the phorbol esters, bryostatin I lacks tumor- promoting activity. These properties suggested that bryostatin I could be useful as an antineoplastie agent. The first Phase I clinical trial with bryostatin 1 was recently completed and demonstrated that concentrations of drug as high as 25 jig/mI could be safely achieved in humans (29). Since PKC modulation has been shown to impair Pgp function, and bryostatin 1 has been shown to be well tolerated clinically. we studied the effect of bryostatin I on a multidrug- resistant MCF-7 human breast cancer subbine which overex- presses PKC-a. We sought an agent suitable for clinical studies which could decrease Pgp phosphorylation and thereby impair its function. The results of these studies are described here. Research. on June 1, 2018. © 1995 American Association for Cancer clincancerres.aacrjournals.org Downloaded from
Transcript of Bryostatin 1 Affects P-Glycoprotein Phosphorylation but not...

Vol. 1. 1581-1587. December 1995 Clinical Cancer Research 1581
Bryostatin 1 Affects P-Glycoprotein Phosphorylation but not
Function in Multidrug-resistant Human Breast Cancer Cells
Stefania Scala, Bruce Dickstein, Joanna Regis,
Zoltan Szallasi, Peter M Blumberg,
and Susan E. Bates’
Medicine Branch, Clinical Oncology Program. Division of Cancer
Treatment IS. S.. B. D., J. R., S. E. B.l, and Laboratory of Cellular
Carcinogenesis and Tumor Proiiiotion, Division of Cancer Etiology
lZ. S.. P. M. B.l, National Cancer Institute. NIH. Bethesda, Maryland
20892
ABSTRACT
The function of P-glycoprotein (Pgp), which confers
multidrug resistance by active efflux of drug, is thought tobe dependent on phosphorylation. Previous studies have
suggested that protein kinase C (PKC) plays an important
role in Pgp phosphorylation. We report here the effects of
bryostatin 1, a unique PKC activator and inhibitor, on Pgp
function in a multidrug-resistant MCF-7 human breast can-
cer subline which overexpresses PKC-a. Bryostatin 1 (100
nrsi) decreased Pgp phosphorylation after 24 h of treatment.
In contrast, it did not affect Pgp function as demonstratedby the accumulation of [3H]vinblastine and rhodamine 123.
We compared the effect of bryostatin 1 treatment on PKC-awith that of the phorbol ester 12-O-tetradecanoylphorbol-
13-acetate (200 nsi). 12-O-tetradecanoylphorbol-13-acetate
caused translocation of PKC-a from the cytosol to the cell
membrane after a 10-mm treatment and its down-regulationafter 24 h of treatment. Likewise, bryostatin 1 (100 nsl)
caused translocation, but only after longer treatment ( 1 h),
and it caused down-regulation of PKC-a at 24 h of treat-ment. Thus, while the MCF-7TH cells overexpress thePKC-a isoform, and its down-regulation by bryostatin 1 isassociated with decreased Pgp phosphorylation, these alter-ations do not modulate drug transport. We conclude that,
while bryostatin 1 may be useful clinically because of itsability to inhibit PKC, it is not able to reverse Pgp-mediated
multidrug resistance.
INTRODUCTION
The identification of clinically suitable agents able to oven-
come drug resistance would have great impact for cancer treat-
ment. In tumor cell lines. the best characterized mechanism of
drug resistance is ‘ ‘multidrug resistance’ ‘ mediated by overex-
pression of an energy-dependent membrane-bound drug effiux
pump known as Pgp2. Pgp undergoes covalent modification by
Received 2/6/95: revised 8/15/95: accepted 8/17/95.
I To whom requests for reprints should be addressed. at NIH. Building10/Room 12N226, 9000 Rockville Pike, Bethesda. MD 20892. Phone:
(301) 402-0984; Fax: (301) 402-0172.
2 The abbreviations used are: Pgp. P-glycoprotein: PK. protein kinase:TPA, I 2-O-tetnadecanoylphonbol- I 3-acetate.
rapid cycles of phosphorylation and dephosphorylation in which
several different protein kinases have been implicated ( 1-6).
The earliest reports suggested that phosphorylation played a role
in the activity of the efflux pump ( 1. 7-9). Increased transport
results from increased Pgp phosphorylation, whereas impaired
function occurs with inhibition of phosphorylation (2. 3).
PKC (2. 10-13). PKA (4. 13). and other potentially more
specific kinases (3. 5. 6) have been linked to Pgp phosphoryla-
tion. Chambers et a/. ( 12) reported that PKC phosphorylates Pgp
at multiple sites in the linker region between the two halves of
the molecule. Characterization of specific residues for PKC and
PKA phosphorylation has been reported in both munine and
human Pgp (11, 13). Several studies have confirmed that acti-
vation of PKC by the phorbol ester TPA results in an increase in
Pgp phosphorylation associated with increased drug efflux, de-
creased drug accumulation, and a more resistant phenotype
( 1-3. 7. 14. 15). Conversely, PKC down-regulation by long-
term TPA treatment or inhibition by staunosponne on calphostin
C impairs Pgp function, resulting in decreased drug efflux and
increased accumulation (7, 14, 16). Increased expression or
activity of the PKC isoforms has been reported in multidrug-
resistant cell lines (2, 7, 17), with a differential increase in the
expression of the PKC-a isoform being the most frequent ob-
servation ( 17-19). This finding has been reported for a multi-
drug-resistant subline of MCF-7 human breast cancer cells (19).
Bryostatin 1 . which is structurally distinct from the phorbol
esters, can activate PKC in vitro but in t’is’o it can paradoxically
antagonize other activators of PKC such as the tumor-promoting
phonbol esters (20. 2 1 ). Bryostatin 1 is the best studied member
of a family of more than I 3 compounds that have a macrocyclic
lactone structure with varying side chains (22. 23). As the most
abundant of the bryostatins isolated from the marine bryozoan
Bugu/a neritina, bryostatin 1 is the first member of this class of
compounds to be tested in humans. These macrocyclic lactones
possess potent antineoplastic properties in human and murine
tumor cell lines (22. 24-26) and in tim activity in murine B16
melanoma. The range of in vitro activities includes cytotoxicity
and terminal differentiation of malignant cells (27, 28). More-
over, in contrast to the phorbol esters, bryostatin I lacks tumor-
promoting activity. These properties suggested that bryostatin I
could be useful as an antineoplastie agent. The first Phase I
clinical trial with bryostatin 1 was recently completed and
demonstrated that concentrations of drug as high as 25 jig/mI
could be safely achieved in humans (29).
Since PKC modulation has been shown to impair Pgp
function, and bryostatin 1 has been shown to be well tolerated
clinically. we studied the effect of bryostatin I on a multidrug-
resistant MCF-7 human breast cancer subbine which overex-
presses PKC-a. We sought an agent suitable for clinical studies
which could decrease Pgp phosphorylation and thereby impair
its function. The results of these studies are described here.
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
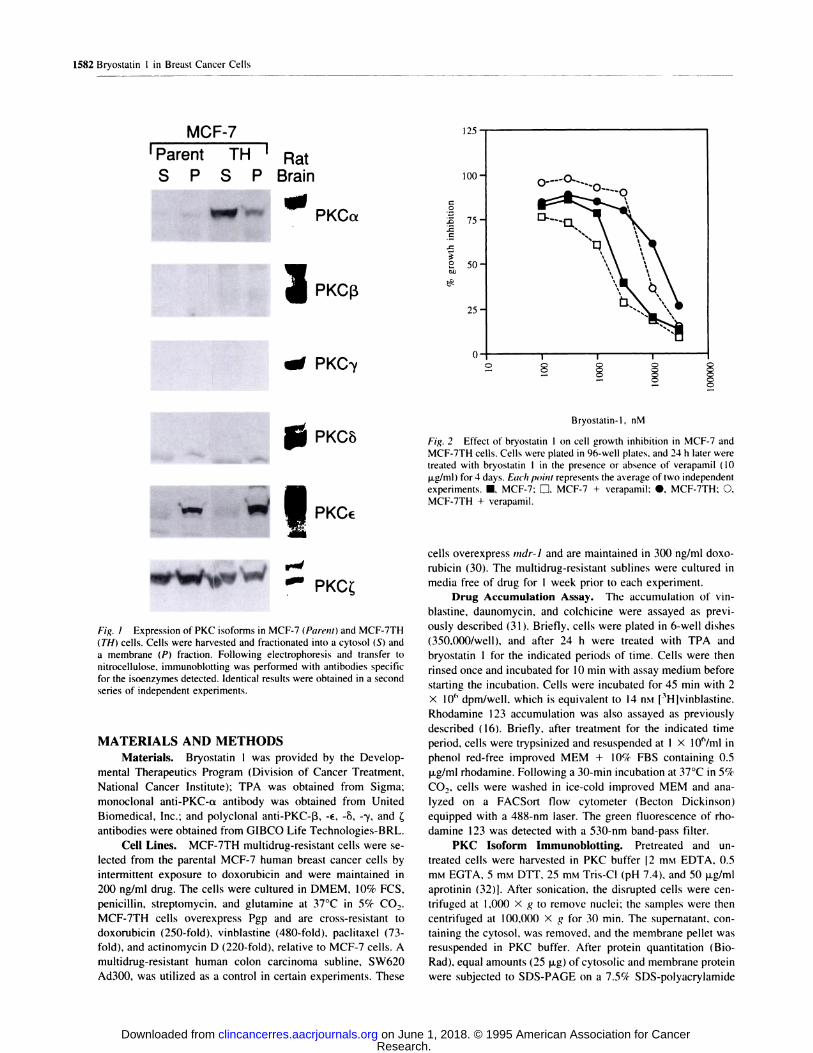
MCF-7
‘Parent TH I Rat
S P S PBrain
� - PKCa
I� PKC’y
100�
75.
50’
25’
0� �- C
- C
1582 Bryostatin I in Breast Cancer Cells
...
. PKC�I
I PKC#{128}
.. PKC�
Fig. I Expression of PKC isoforms in MCF-7 (Parent) and MCF-7TH
(TI-f) cells. Cells were harvested and fractionated into a cytosol (S) and
a membrane (P) fraction. Following electrophoresis and transfer to
nitrocellulose, immunoblotting was performed with antibodies specific
for the isoenzymes detected. Identical results were obtained in a second
series of independent experiments.
MATERIALS AND METHODS
Materials. Bryostatin 1 was provided by the Develop-
mental Therapeutics Program (Division of Cancer Treatment,
National Cancer Institute); TPA was obtained from Sigma;
monoelonal anti-PKC-a antibody was obtained from United
Biomedical, Inc.; and polycbonal anti-PKC-�3, -#{128},-�, -“y, and �
antibodies were obtained from GIBCO Life Technobogies-BRL.
Cell Lines. MCF-7TH multidrug-resistant cells were Se-
leeted from the parental MCF-7 human breast cancer cells by
intermittent exposure to doxorubicin and were maintained in
200 ng/ml drug. The cells were cultured in DMEM, 10% FCS.
penicillin, streptomycin, and glutamine at 37#{176}Cin 5% CO2.
MCF-7TH cells overexpress Pgp and are cross-resistant to
doxorubicin (250-fold), vinblastine (480-fold), paclitaxel (73-
fold), and actinomycin D (220-fold), relative to MCF-7 cells. A
multidrug-resistant human colon carcinoma subline, SW620
Ad300, was utilized as a control in certain experiments. These
0
.0
.00
.0
SC
Bryostatin-l, nM
Fig. 2 Effect of bryostatin 1 on cell growth inhibition in MCF-7 and
MCF-7TH cells. Cells were plated in 96-well plates. and 24 h later weretreated with bryostatin 1 in the presence or absence of verapamil (10
pg/mb) for 4 days. Each point represents the average of two independent
experiments. �, MCF-7: El. MCF-7 + verapamil: #{149},MCF-7TH: 0,MCF-7TH + verapamil.
cells overexpress mndr-I and are maintained in 300 ng/ml doxo-
rubicin (30). The multidrug-resistant sublines were cultured in
media free of drug for I week prior to each experiment.
Drug Accumulation Assay. The accumulation of yin-
blastine, daunornycin. and colehicine were assayed as previ-
ously described (31). Briefly, cells were plated in 6-well dishes
(350,000/well), and after 24 h were treated with TPA and
bryostatin 1 for the indicated periods of time. Cells were then
rinsed once and incubated for 10 mm with assay medium before
starting the incubation. Cells were incubated for 45 mm with 2
x l0� dpm/well. which is equivalent to 14 nsi [3Hlvinblastmne.
Rhodamine I 23 accumulation was also assayed as previously
described ( I 6). Briefly. after treatment for the indicated time
period, cells were trypsinized and resuspended at 1 X 106/ml in
phenol red-free improved MEM + 10% FBS containing 0.5
jig/mb rhodamine. Following a 30-mm incubation at 37#{176}Cin 5%
CO,. cells were washed in ice-cold improved MEM and ana-
byzed on a FACSort flow cytometer (Becton Dickinson)
equipped with a 488-nm laser. The green fluorescence of rho-
damine 123 was detected with a 530-nm band-pass filter.
PKC Isoform Immunoblotting. Pretreated and un-
treated cells were harvested in PKC buffer [2 mM EDTA. 0.5
mM EGTA, 5 mM DTT, 25 msi Tris-CI (pH 7.4), and 50 jig/mI
aprotinin (32)1. After sonication. the disrupted cells were cen-
trifuged at 1,000 X g to remove nuclei; the samples were then
centrifuged at 100.000 X g for 30 mm. The supernatant. con-
taming the cytosol, was removed, and the membrane pellet was
resuspended in PKC bufThr. After protein quantitation (Bio-
Rad), equal amounts (25 jig) of cytosolic and membrane protein
were subjected to SDS-PAGE on a 7.5% SDS-polyaerylamide
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
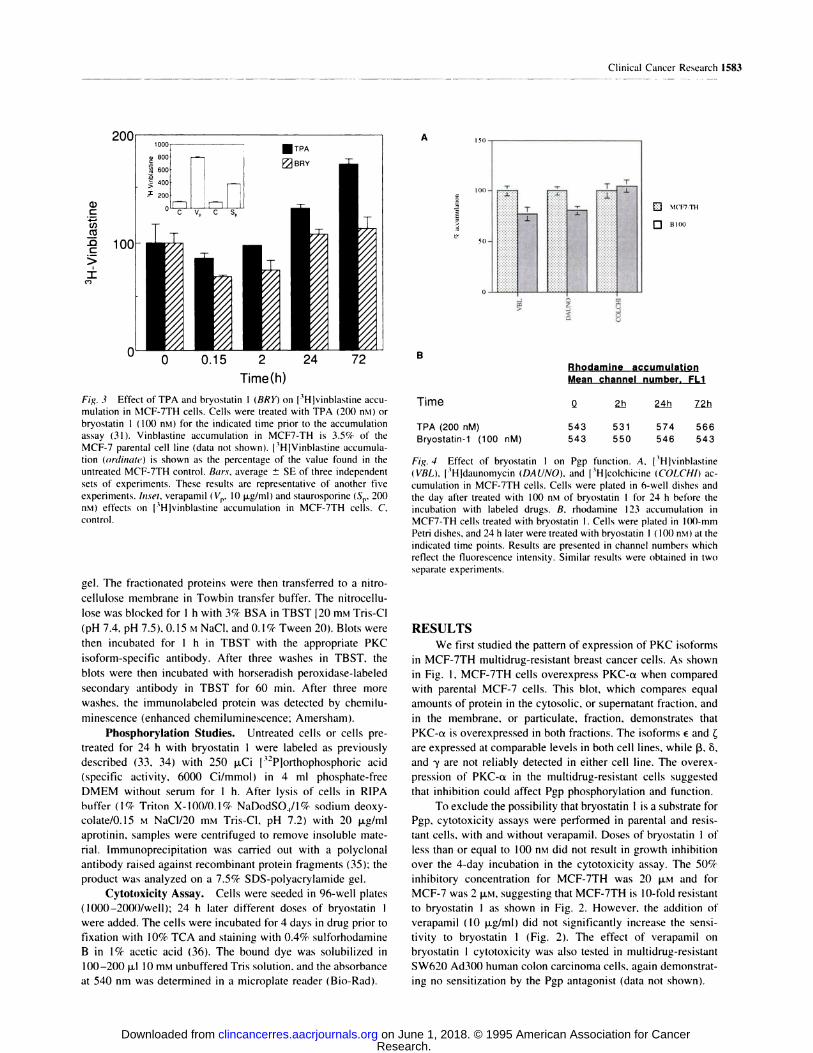
C/)
C
>
IC,)
A
B
00 BlOt)
0 0.15
Time(h)Rhodamine accumulationMean channel number. FL1
Time Q 2.ti 2�4�h ZZb
TPA (200 nM)
Bryostatin-1 (100 nM)
543 531 574 566
543 550 546 543
Clinical Cancer Research 1583
Fig. 3 Effect of TPA and bryostatin 1 (BRY) on [3Hlvinblastine accu-
mulation in MCF-7TH cells. Cells were treated with TPA (200 nsi) or
bryostatin 1 (100 n�i) for the indicated time prior to the accumulation
assay (31). Vinblastine accumulation in MCF7-TH is 3.5% of the
MCF-7 parental cell line (data not shown). 13H]Vinblastine accumula-tion (ordinate) is shown as the percentage of the value found in the
untreated MCF-7TH control. Bars, average ± SE of three independent
sets of experiments. These results are representative of another five
experiments. Inset, verapamil (Vu. 10 jig/nil) and staurosporinc (Se. 2(X)
nM) effects on l3H]vinblastine accumulation in MCF-7TH cells. C.
control.
gel. The fractionated proteins were then transferred to a nitro-
cellulose membrane in Towbin transfer buffer. The nitroceblu-
lose was blocked for I h with 3% BSA in TBST [20 hiM Tris-Cl
(pH 7.4, pH 7.5). 0.15 si NaCI, and 0.1% Tween 20). Blots were
then incubated for I h in TBST with the appropriate PKC
isoform-specific antibody. After three washes in TBST. the
blots were then incubated with horseradish peroxidase-labeled
secondary antibody in TBST for 60 mm. After three more
washes, the immunolabeled protein was detected by chernilu-
mineseence (enhanced chernilurninescence; Amersharn).
Phosphorylation Studies. Untreated cells or cells pre-
treated for 24 h with bryostatin 1 were labeled as previously
described (33. 34) with 250 jiCi I32Plorthophosphonic acid
(specific activity, 6000 Ci/rnrnol) in 4 ml phosphate-free
DMEM without serum for 1 h. After lysis of cells in RIPA
buffer (1% Triton X-l00/0.l% NaDodSO.4/I% sodium deoxy-
colate/0.15 M NaC1/20 m�vi Tris-CI, pH 7.2) with 20 jig/mb
aprotinin, samples were centrifuged to remove insoluble mate-
rial. lrnrnunoprecipitation was carried out with a polyclonal
antibody raised against recombinant protein fragments (35): the
product was analyzed on a 7.5% SDS-polyacrylamide gel.
Cytotoxicity Assay. Cells were seeded in 96-well plates
( 1000-2000/well); 24 h later different doses of bryostatin I
were added. The cells were incubated for 4 days in drug prior to
fixation with 10% TCA and staining with 0.4% sulforhodamine
B in 1% acetic acid (36). The bound dye was solubilized in
100-200 jil 10 mM unbuffered Tnis solution. and the absorbance
at 540 nrn was determined in a microplate reader (Bio-Rad).
Fig. 4 Effect of bryostatin I on Pgp function. A. l3Hlvinhlastine
(VBL), l3Hldaunomycin (DAUNO). and l3Hlcolchicine (COLCHI) ac-
cumulation in MCF-7TH cells. Cells were plated in 6-well dishes and
the day after treated with 100 n�i of bryostatin 1 for 24 h hefcre the
incubation with labeled drugs. B. rhodamine 123 accumulation in
MCF7-TH cells treated with bryostatin I. Cells were plated in 100-mm
Petni dishes, and 24 h later were treated with bryostatin 1 (1(X) nM) at the
indicated time points. Results are presented in channel numbers which
reflect the fluorescence intensity. Similar results were obtained in two
separate experiments.
RESULTS
We first studied the pattern of expression of PKC isoforms
in MCF-7TH multidrug-resistant breast cancer cells. As shown
in Fig. 1 , MCF-7TH cells overexpress PKC-ct when compared
with parental MCF-7 cells. This blot, which compares equal
amounts of protein in the cytosobic. or supernatant fraction, and
in the membrane. or particulate, fraction, demonstrates that
PKC-ot is overexpressed in both fractions. The isoforms #{128}and �
are expressed at comparable levels in both cell lines. while �, �,
and -y are not reliably detected in either cell line. The overex-
pression of PKC-a in the multidrug-resistant cells suggested
that inhibition could affect Pgp phosphorylation and function.
To exclude the possibility that bryostatin I is a substrate for
Pgp. cytotoxicity assays were performed in parental and resis-
tant cells, with and without verapamil. Doses of bryostatin I of
less than or equal to 100 nM did not result in growth inhibition
over the 4-day incubation in the cytotoxicity assay. The 50%
inhibitory concentration for MCF-7TH was 20 jisi and for
MCF-7 was 2 jiM, suggesting that MCF-7TH is 10-fold resistant
to bryostatin I as shown in Fig. 2. However, the addition of
venapamil (10 jig/mI) did not significantly increase the sensi-
tivity to bryostatin I (Fig. 2). The effect of verapamil on
bryostatin 1 cytotoxicity was also tested in multidrug-resistant
SW620 Ad300 human colon carcinoma cells. again demonstrat-
ing no sensitization by the Pgp antagonist (data not shown).
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
100
e� 80
� � 60lace
� .� 40
� 20
C
Control 10” lh 24h �
Is pH� pl� � p’ �
TPA � -_ __ � - - PKC a
Bryo � � - � � - - PKC a
TPA .� -PKC#{128}
Bryo . - -PKCe
TPA ‘�ZW� � __� - � - � - PKC �
Bryo -� - � � - � - - - PKC �
1584 Bryostatin 1 in Breast Cancer Cells
MCF-7TH Cells
I BryostatinI I
Control 10 pM 100 nM TPA
�#{248}..
�llUnUControl � pM 100 nM � TPA
Bryostatin
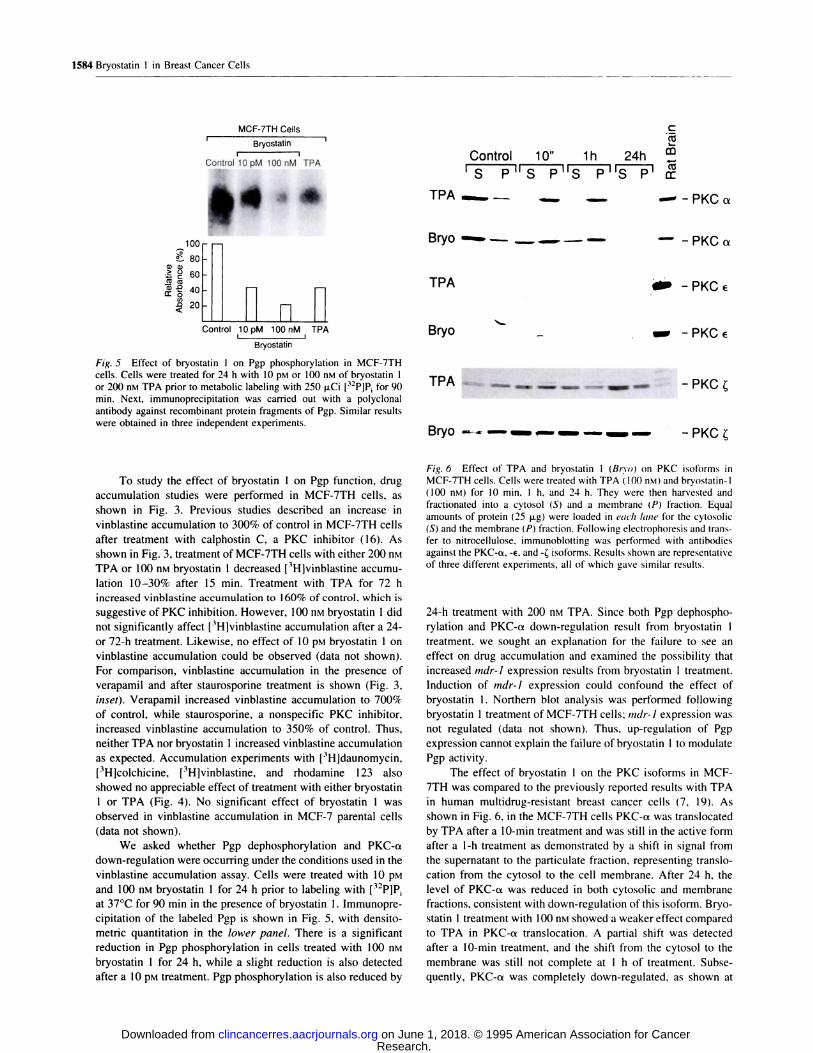
Fig. 5 Effect of bryostatin I on Pgp phosphorylation in MCF-7TH
cells. Cells were treated for 24 h with 10 �M or 100 nsi of bryostatin 1
or 200 nM TPA prior to metabolic labeling with 250 p.Ci [32P1P for 90mm. Next, immunoprecipitation was carried out with a polyclonal
antibody against recombinant protein fragments of Pgp. Similar results
were obtained in three independent experiments.
To study the effect of bryostatin I on Pgp function, drug
accumulation studies were performed in MCF-7TH cells, as
shown in Fig. 3. Previous studies described an increase in
vinblastine accumulation to 300% of control in MCF-7TH cells
after treatment with calphostin C, a PKC inhibitor ( 16). As
shown in Fig. 3, treatment of MCF-7TH cells with either 200 nst
TPA or 100 nM bryostatin 1 decreased [3Hjvinblastine aecurnu-
lation 10-30% after 15 mm. Treatment with TPA for 72 h
increased vinblastine accumulation to 160% ofeontrol, which is
suggestive of PKC inhibition. However, 100 nrvt bryostatin 1 did
not significantly affect [3Hlvinblastine accumulation after a 24-
or 72-h treatment. Likewise, no effect of 10 �M bryostatin I on
vinblastine accumulation could be observed (data not shown).
For comparison, vinblastine accumulation in the presence of
verapamil and after staurosporine treatment is shown (Fig. 3,
inset). Verapamil increased vinblastine accumulation to 700%
of control, while staurosporine, a nonspecific PKC inhibitor,
increased vinblastine accumulation to 350% of control. Thus,
neither TPA nor bryostatin I increased vinblastine accumulation
as expected. Accumulation experiments with [3Hldaunomyein,
[3Hjeolehieine, [3H]vinblastine, and rhodamine 123 also
showed no appreciable effect of treatment with either bryostatin
1 or TPA (Fig. 4). No significant effect of bryostatin 1 was
observed in vinblastine accumulation in MCF-7 parental cells
(data not shown).
We asked whether Pgp dephosphorylation and PKC-a
down-regulation were occurring under the conditions used in the
vinblastine accumulation assay. Cells were treated with 10 �M
and 100 nM bryostatin I for 24 h prior to labeling with [32P]P1
at 37#{176}Cfor 90 mm in the presence of bryostatin 1. Immunopre-
cipitation of the labeled Pgp is shown in Fig. 5, with densito-
metric quantitation in the lower panel. There is a significant
reduction in Pgp phosphorylation in cells treated with 100 nrvt
bryostatin 1 for 24 h, while a slight reduction is also detected
after a 10 �M treatment. Pgp phosphorylation is also reduced by
Fig. 6 Effect of TPA and bryostatin I (Brvo) on PKC isoforms inMCF-7TH cells. Cells were treated with TPA ( 100 nM) and bnyostatin-l
(100 nM) for 10 mm, 1 h, and 24 h. They were then harvested and
fractionated into a cytosol (S) and a membrane (P) fraction. Equal
amounts of protein (25 jig) were loaded in each lane for the cytosolic
(S) and the membrane (P) fraction. Following electrophoresis and trans-
fen to nitnocellulose, immunoblotting was performed with antibodies
against the PKC-a, -#{128}, and -� isoforms. Results shown are representative
of three different experiments, all of which gave similar results.
24-h treatment with 200 nM TPA. Since both Pgp dephospho-
rylation and PKC-cs down-regulation result from bryostatin 1
treatment. we sought an explanation for the failure to see an
effect on drug accumulation and examined the possibility that
increased mdr-1 expression results from bryostatin I treatment.
Induction of mdr-1 expression could confound the effect of
bryostatin 1 . Northern blot analysis was performed following
bryostatin I treatment of MCF-7TH cells; ,i,dr-I expression was
not regulated (data not shown). Thus. up-regulation of Pgp
expression cannot explain the failure of bryostatin 1 to modulate
Pgp activity.
The effect of bryostatin 1 on the PKC isoforms in MCF-
7TH was compared to the previously reported results with TPA
in human multidrug-resistant breast cancer cells (7, 19). As
shown in Fig. 6, in the MCF-7TH cells PKC-a was translocated
by TPA after a 10-mm treatment and was still in the active form
after a 1-h treatment as demonstrated by a shift in signal from
the supernatant to the particulate fraction, representing transbo-
cation from the cytosol to the cell membrane. After 24 h. the
level of PKC-a was reduced in both cytosolie and membrane
fractions, consistent with down-regulation of this isoform. Bryo-
statin 1 treatment with I 00 rust showed a weaker effect compared
to TPA in PKC-a translocation. A partial shift was detected
after a 10-mm treatment, and the shift from the cytosol to the
membrane was still not complete at 1 h of treatment. Subse-
quently, PKC-ct was completely down-regulated, as shown at
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
MCF-7
I � � 100 � C .01
Is P S P S P�S
MCF-7TH .�
100 I
PSPSP’c�
..
S
PKC#{128}
Clinical Cancer Research 1585
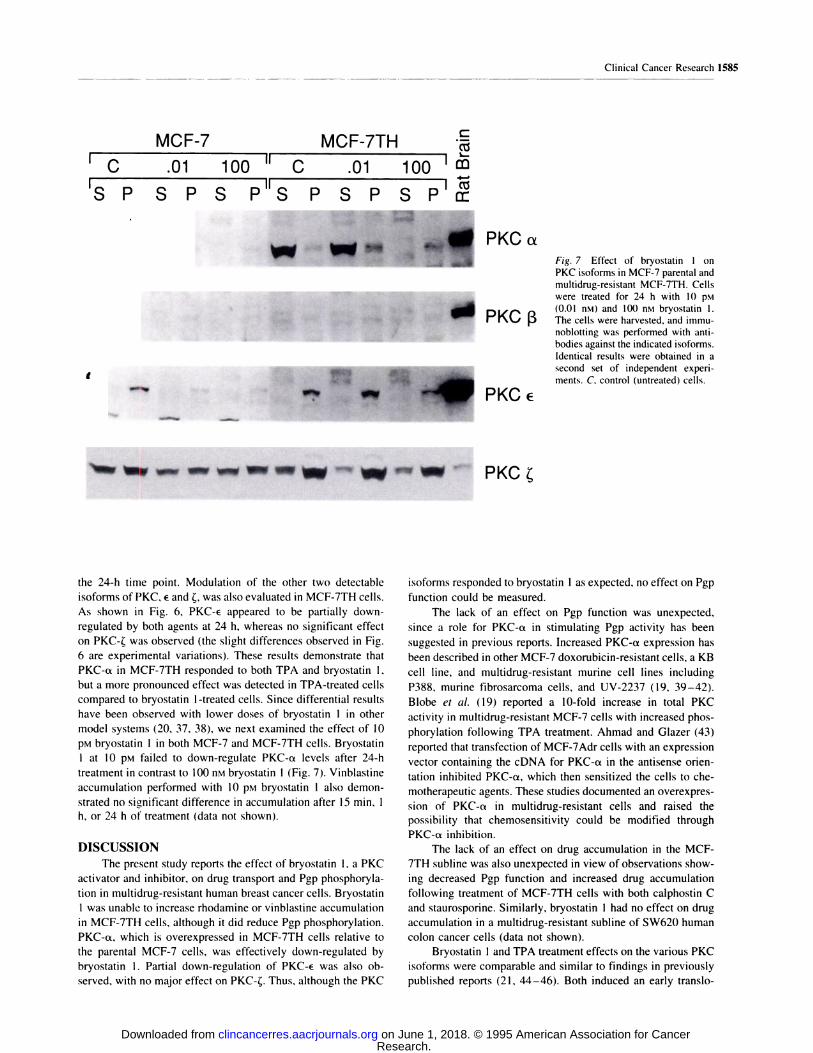
Fig. 7 Effect of bryostatin 1 onPKC isofonms in MCF-7 parental and
multidrug-resistant MCF-7TH. Cells
were treated for 24 h with 10 �M
0 “ (0.01 nM) and 100 flM bryostatin 1.,,� . . l’\Li 13 The cells were harvested, and immu-
noblotting was performed with anti-
bodies against the indicated isoforms.
Identical results were obtained in a
second set of independent experi-
ments. C. control (untreated) cells.
� � � PKC�
the 24-h time point. Modulation of the other two detectable
isoforms of PKC, #{128}and �, was also evaluated in MCF-7TH cells.
As shown in Fig. 6, PKC-#{128} appeared to be partially down-
regulated by both agents at 24 h, whereas no significant effect
on PKC-� was observed (the slight differences observed in Fig.
6 are experimental variations). These results demonstrate that
PKC-a in MCF-7TH responded to both TPA and bryostatin I,
but a more pronounced effect was detected in TPA-treated cells
compared to bryostatin 1-treated cells. Since differential results
have been observed with bower doses of bryostatin 1 in other
model systems (20. 37, 38), we next examined the effect of 10
�M bryostatin 1 in both MCF-7 and MCF-7TH cells. Bryostatin
I at 10 �M failed to down-regulate PKC-a levels after 24-h
treatment in contrast to 100 nM bryostatin 1 (Fig. 7). Vinblastine
accumulation performed with 10 �M bryostatin 1 also demon-
strated no significant difference in accumulation after 15 mm, 1
h, or 24 h of treatment (data not shown).
DISCUSSION
The present study reports the effect of bryostatin I . a PKC
activator and inhibitor, on drug transport and Pgp phosphoryla-
tion in multidrug-resistant human breast cancer cells. Bryostatin
I was unable to increase rhodamine or vinbbastine accumulation
in MCF-7TH cells, although it did reduce Pgp phosphorylation.
PKC-a, which is overexpressed in MCF-7TH cells relative to
the parental MCF-7 cells, was effectively down-regulated by
bryostatin 1 . Partial down-regulation of PKC-#{128} was also ob-
served, with no major effect on PKC-�. Thus. although the PKC
isoforms responded to bryostatin I as expected, no effect on Pgp
function could be measured.
The lack of an effect on Pgp function was unexpected,
since a role for PKC-a in stimulating Pgp activity has been
suggested in previous reports. Increased PKC-a expression has
been described in other MCF-7 doxorubiein-nesistant cells, a KB
cell line, and multidrug-resistant murine cell lines including
P388, murine fibrosarcoma cells, and UV-2237 (19, 39-42).
Bbobe et al. ( 19) reported a 10-fold increase in total PKC
activity in multidrug-resistant MCF-7 cells with increased phos-
phorylation following TPA treatment. Ahmad and Glazer (43)
reported that transfection of MCF-7Adr cells with an expression
vector containing the eDNA for PKC-a in the antisense orien-
tation inhibited PKC-a, which then sensitized the cells to ehe-
motherapeutic agents. These studies documented an overexpres-
sion of PKC-rt in multidrug-resistant cells and raised the
possibility that ehemosensitivity could be modified through
PKC-a inhibition.
The lack of an effect on drug accumulation in the MCF-
7TH subline was also unexpected in view ofobservations show-
ing decreased Pgp function and increased drug accumulation
following treatment of MCF-7TH cells with both ealphostin C
and staurosporine. Similarly, bryostatin 1 had no effect on drug
accumulation in a multidrug-resistant subline of SW620 human
colon cancer cells (data not shown).
Bryostatin I and TPA treatment effects on the various PKC
isoforms were comparable and similar to findings in previously
published reports (21, 44-46). Both induced an early translo-
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

1586 Bryostatin 1 in Breast Cancer Cells
3 z. Szallasi et a!. , submitted for publication.
cation of PKC-a from the cytosol to the membrane in MCF-
7TH cells, although bryostatin 1 activation of PKC-a was less
pronounced than that of TPA and both resulted in down-regu-
lation by 24 h. Identical results were observed in a multidrug-
resistant human colon carcinoma subline (data not shown)
which does not overexpress PKC-a compared to parental cells
as do the multidrug-resistant MCF-7TH cells reported here.
TPA and bryostatin 1 induced a partial down-regulation of
PKC-#{128}, and had no obvious effect on PKC-�. This was also
consistent with previous reports showing partial down-regula-
tion of PKC-#{128}in response to TPA or bryostatin 1 treatment of
NIH3T3 cells, mouse keratinocytes, and SH-SY5Y neuroblas-
toma cells (21, 46),� while neither transloeation nor down-
regulation of PKC-� was observed in NIH3T3 mouse fibroblast
cells, platelets, and murine epidermis (21, 47-49).
The present results contrast with previous observations
showing that decreased Pgp phosphorylation is associated with
decreased drug transport (2, 3, 16, 33). Decreased Pgp phos-
phorylation and impaired vinblastine transport were seen after
sodium butyrate treatment of SW620 human colon cancer cells,
although colehicine transport was preserved (33). Similar find-
ings were observed in SW62OAd300 cells treated with calphos-
tin C, a PKC inhibitor which impaired vinblastine while inereas-
ing verapamil transport (1 6). We now report a decrease in Pgp
phosphorylation following bryostatin 1 treatment accompanied
by unaffected vinblastine, rhodamine, and daunomycin trans-
port. These discrepancies may be explained by a differential
effect of these agents on distinct sites of phosphorylation by
several kinases which may include but are not limited to the
PKC family. These data suggest that kinases other than PKC
may be more important in regulating Pgp. Taken together, these
data suggest that Pgp phosphorylation modulates drug binding
and affinity and resulting transport, rather than having a direct
effect on Pgp function.
Our results show that bryostatin 1 down-regulates PKC-a
and reduces Pgp phosphorylation in MCF-7TH multidrug-resis-
tant cells overexpressing PKC-a, without an effect on drug
transport. Although bryostatin 1 appears to be clinically well
tolerated and may have a role as an antineoplastie agent, based
on these observations, its use clinically to modulate Pgp-rnedi-
ated multidrug resistance cannot be recommended.
REFERENCES
1. Hamada, H., Hagiwara, K-I., Nakajima, T., and Tsuruo, T. Phos-
phorylation of the Mr 170,000 to 180,000 glycoprotein specific to
multidrug-nesistant tumor cells: effects of venapamil, tnifluopenazine,
and phorbol esters. Cancer Res., 47: 2860-2865, 1987.
2. Chambers, T. C., Chalikonda, I., and Eibon, G. Correlation of protein
kinase C translocation, P-glyeoprotein phosphorylation and reduced
drug accumulation in multidrug resistant human KB cells. Biochem.
Biophys. Res. Commun., 169: 253-259, 1990.
3. Ma, L., Marquardt, D., Takemoto, L., and Center, M. S. Analysis of
P-glycopnotein phosphorylation in HL6O cells isolated for resistance to
vincristine. J. Biol. Chem., 266: 5593-5599, 1991.
4. Mellado, W., and Honwitz, S. B. Phosphorylation of the multidrug
resistance associated glycoprotein. Biochemistry. 26: 6900-6904.1987.
5. Staats, J., Marquardt, D., and Center, M. S. Characterization of a
membrane-associated protein kinase of multidrug-nesistant HL-60 cells
which phosphorylates P-glycopnotein. J. Biol. Chem., 265: 4084-4090.
1990.
6. Sampson, K. E., MeCroskey, M. C., and Abraham, I. Identification
of A 170 kDa membrane kinase with increased activity in KB-Vl
multidrug resistant cells. J. Cell. Biochem., 52: 384-395, 1993.
7. Fine, R. L., Patel. J., and Chabner, B. A. Phorbol esters induce
multidrug resistance in human breast cancer cells. Proc. NatI. Acad. Sci.
USA, 85: 582-586, 1988.
8. Center, M. S. Mechanisms regulating cell resistance to Adniamycin.
Evidence that drug accumulation in resistant cells is modulated by
phosphorylation of a plasma membrane glycoprotein. Biochem. Phan-
macol., 34: 1471-1476, 1985.
9. Center, M. S. Evidence that Adniamycin resistance in Chinese ham-
ster lung cells is regulated by phosphorylation of a plasma membrane
glycoprotein. Bioehem. Biophys. Res. Commun., 1/5: 159-166, 1983.
10. Chambers, T. C., Zheng, B., and Kuo, J. F. Regulation by phorbol
ester and protein kinase C inhibitors, and by a protein phosphatase
inhibitor (okadaic acid), of P-glycoprotein phosphorylation and relation-
ship to drug accumulation in multidrug-nesistant human KB cells. Mol.
Pharmacol., 41: 1008-1015, 1992.
I 1. Chambers, T., PohI, J., and Kuo, J. F. Phosphorylation by protein
kinase C and cyclic AMP-dependent protein kinase of synthetic peptides
derived from linker region of human P-glycoprotein. Biochem. J.. 299:
309-315, 1994.
12. Chambers, T. C., PohI, J., Raynor, R. L., and Kuo, J. F. Identifica-
tion of specific sites in human P-glycoprotein phosphonylated by protein
kinase C. Comm., 1994.
13. Orr, G. A., Han, E., Browne, P. C., Nieves, E., O’Connor, B. M..
Yang, C., and Honwitz, S. B. Identification of major phosphorylation
domain of munine ,;idrlb P-glycopnotein. J. Biol. Chem., 25: 25054-
25062, 1993.
14. Chambers, T. C., McAvoy, E. M., Jacobs, J. W., and Eilon, Ci.
Protein kinase C phosphorylates P-glycoprotein in multidrug resistanthuman KB carcinoma cells. J. Biol. Chem., 265: 7679-7686, 1990.
15. Yang, C-P. H., Cohen, D.. Greenberger, L. M., Hsu, S. I-H., and
Horwitz, S. B. Differential transport properties of two mdr gene prod-
ucts are distinguished by progesterone. J. Biol. Chem., 265: 10282-
10288, 1990.
16. Bates, S. E., Lee, J. S., Dickstein, B., Spolyar, M., and Fojo, A. T.
Differential modulation of P-glycoprotein transport by protein kinase
inhibition. Biochemistry, 32: 9156-9164, 1993.
17. Lee, S. A., Karaszkiewicz, J. W., and Anderson. W. B. Elevated
level of nuclear protein kinase C in multidrug-nesistant MCF-7 human
breast carcinoma cells. Cancer Res., 52: 3750-3759, 1992.
18. Yu, G., Ahmad, S., Aquino. A., Fairchild, C. R., Trepel. J. B..
Ohno. S., Suzuki, K., Tsunuo, T., Cowan, K. H., and Glazer, R. I.Transfection with protein kinase C confers increased multidrug resis-
tance to MCF-7 cells expressing P-glycopnotein. Cancer Commun., 3:
181-189, 1991.
19. Blobe, G. C., Sachs, C. W., Khan, W. A., Fabbno, D., Stabel, S.,
Wetsel, W. C., Obeid. L. M., Fine, R. L., and Hannun, Y. A. Selective
regulation of expression of protein kinase C (PKC) isoenzymes in
multidrug-nesistant MCF-7 cells. J. Biol. Chem., 268: 658-664, 1993.
20. Gschwendt, M., Furstenberger, G., Rose-John, S., Rogers, M.,
Kittstein, W., Pettit, G. R., Herald, C. L., and Marks, K. Bryostatin I, an
activator of protein kinase C. mimics as well as inhibits biological
effects of the phorbob ester TPA in vito and in vitro. Carcinogenesis. 9:
555-562, 1988.
21. Szallasi, Z., Smith, C. B., Pettit, Ci. R., and Blumbeng, P. M.
Differential regulation of protein kinase C isozymes by bynostatin 1 and
phonbol 12-mynistate 13-acetate in NIH 3T3 fibroblasts. J. Biol. Chem.,
269: 2118-2124, 1994.
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

Clinical Cancer Research 1587
22. Reddi, 0. S. Antineoplastic components of marine animals. Nature
(Lond.), 227: 962, 1970.
23. Blumberg. P. M. Protein kinase C as the receptor for the phorbol
ester tumor promoters: sixth Rhoads memorial award lecture. Cancer
Res.. 48: 1-8. 1988.
24. Schuchter, L. M., Esa, A. H., May. W. S., Laulis. M. K.. Pettit,
G. R.. and Hess, A. D. Successful treatment of munine melanoma with
bryostatin I. Cancer Res., 5/: 682-687, 1991.
25. Hornung, R. L., Pearson, J. W., Beckwith, M., and Longo, D. L.
Preclinical evaluation of bryostatin as anticancer agent against several
murine tumor cell lines: in vitro versus in vito activity. Cancer Res., 52:
101-107. 1992.
26. Mohammad, R. M., Al-Katib, A., Pettit, G. R., and Sensenbrenner,
L. L. Successful treatment of human Waldenstrom’s macroglobulinemia
with combination biological and chemotherapy agents. Cancer Res., 54:
165-168, 1994.
27. Knaft, A. S.. Smith, J. B., and Benkow, R. L. Bnyostatin. an activator
of the calcium phospholipid-dependent protein kinase, blocks phorbol
ester. Proc. NatI. Acad. Sci. USA, 83: 1334-1338, 1986.
28. Hennings. H.. Blumbeng. P. M., Pettit, G. R., Herald, C. L., Shores,
R., and Yuspa. S. H. Bnyostatin 1, an activator of protein kinase C,inhibits tumor promotion by phorbol esters in SENCAR mouse skin.
Carcinogenesis. 8: 1343-1346, 1987.
29. Philip, P. A., Thavasu, P., Carmichael, J.. Stuart, N. S., Rockett, H.,
Talbot. D. C.. Ganesan, T.. Pettit. G. R., and BaIkwill, F. Phase I study
of bryostatin 1 : assessment of interleukin 6 and tumor necrosis factor
alpha induction in vito. J. NatI. Cancer Inst., 85: 1812-1818, 1993.
30. Lai. G-M.. Chen, Y-N., Mickley. L. A.. Fojo. A. T., and Bates, S. E.
P-glycoprotein expression and schedule dependence of Adniamycin
cytotoxicity in human colon carcinoma cell lines. Int. J. Cancer, 49:
696-703. 1991.
3 1 . Fojo, A., Akyiama, S., Gottesman, M. M., and Pastan, I. Reduceddrug accumulation in multiply drug-resistant human KB carcinoma cell
lines. Cancer Res., 45. 3002-3007, 1985.
32. Leach, K. L., Powers, E. A., McGuire, J. C., Dong, L., Kiley, S. C.,
and Jaken, S. Monoclonal antibodies specific for Type 3 PKC recog-
nized distinct domains of PKC and inhibit in vitro functional activity. J.
Biol. Chem.. 263: 13223-13230, 1988.
33. Bates, S. E., Cunnier, S. J., Alvarez, M., and Fojo, A. T. Modulationof P-glycopnotein phosphorylation and drug transport by sodium bu-
tynate. Biochemistry. 31: 6366-6372. 1992.
34. Richert. N. D.. Aldwin, L., Nitecki, D., et al. Stability and covalent
modification of P-glycopnotein in multidrug-resistant KB cells. Bio-
chemistry, 27: 7607-7613, 1988.
35. Tanaka, S.. Cunrier, S. J., Bnuggemann, E. P.. Ueda, K.. Germann,
U. A., Pastan, I., and Gottesman, M. M. Use of recombinant P-glyco-
protein fragments to produce antibodies to the multidnug transporter.
Biochem. Biophys. Res. Commun., 166: 180-186, 1990.
36. Skehan, P., Stoneng, R., Scudiero, D., Monks, A.. McMahon, J.,
Vistica, D., Warren, J. T., Bokesch, H., Kenney, S., and Boyd, M. R.
New colorimetnic cytotoxicty assay for anticancer-drug screening. J.
NatI. Cancer Inst., 82: 1 107-1 1 12, 1990.
37. Basu, A.. and Lazo, J. S. Sensitization of human cervical carcinoma
cells to cis-diamminedichloroplatinum(II) by bryostatin 1 . Cancer Ret.,
52: 3119-3124, 1992.
38. Warren, B. S., Kamano, Y., Pettit, G. R., and Blumberg. P. M.
Mimicry of bryostatin 1 induced phosphorylation patterns in HL-60
cells by high phonbol ester concentrations. Cancer Res., 48: 5984-5988,
1988.
39. Posada, J. A.. MeKeegan, E. M.. Worthington. K. F.. Monin, M. J..
Jaken, S., and Tnitton, T. R. Human multidrug resistant KB cells
overexpress protein kinase C: involvement in drug resistance. Cancer
Commun., 1: 285-292, 1989.
40. Aquino. A., Warren, B. S., Omichinski, J., Hartman, K. D., and
Glazer, R. I. Protein kinase C- is present in Adniamycin resistant HL-60leukemia cells. Biochem. Biophys. Res. Commun., 166: 723-728. 1990.
41. Gollapudi, S., Patel, K., Jam, V., and Gupta. S. Protein kinase C
isoforms in multidnug resistant P388/ADR cells; a possible role in
daunonubicin transport. Cancer Lett., 62: 69-75, 1992.
42. O’Bnian, C. A., Fan, D., Ward, N., Dong, Z., Iwamoto, L., Gupta,
K. P.. Earnest, L. E., and Fidler, I. J. Transient enhancement of multi-
drug resistance by the bile acid deoxycholate in munine fibrosancoma
cells in vitro. Biochem. Pharmacol., 41: 797-806, 1991.
43. Ahmad, S., and Glazer, R. I. Expression of the antisense eDNA for
protein kinase C attenuates resistance in doxonubicin-resistant MCF-7
breast carcinoma cells. Mol. Pharmacol., 43: 858-862, 1994.
44. Isakov, N., Galnon, D., Mustelin, T., Pettit, G. R., and Altman, A.
Inhibition of phorbol ester-induced T cell proliferation by bryostatin is
associated with rapid degradation of protein kinase C. J. Immunol., /50:1195-1204, 1993.
45. Kennedy, M. J., Prestigiacomo, L. J., Tyler, G., May, W. S., and
Davidson, N. E. Differential effects of bryostatin I and phorbol ester on
human breast cancer cell lines. Cancer Res., 52: 1278-1283, 1992.
46. Jalava. A., Lintunen, M., and Heikkila, J. Protein kinase Ca but not
protein kinase C#{128}is differentially down-regulated by bryostatin I and
tetnadecanoyl phorbol I 3-acetate in SH-SY5Y human neuroblastoma
cells. Biochem. Biophys. Res. Commun.. 2: 472-478. 1993.
47. Grabarek, J., and Ware, J. A. Protein kinase C activation without
membrane contact in platelets stimulated by bryostatin. J. Biol. Chem..
268: 5543-5549. 1993.
48. Gschwendt, M., Liebenspenger, H., Kittstein, W., and Marks, F.
Protein kinase C and N in murine epidermis. TPA induces down-
regulation of PKCn but not PKC. FEBS Lett.. 307: 151-155, 1992.
49. Ozawa, K., Szallasi, Z., Kazanietz, G., Blumberg, P. M.. Mischak,
H., Mushinski, J. F., and Beaven, M. A. CA2�-dependent and Ca2�-independent isozymes of protein kinase C mediate exocytosis in anti-
gen-stimulated rat basophilic RBL-2H3 cells. J. Biol. Chem., 268:
1749-1756, 1993.
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

1995;1:1581-1587. Clin Cancer Res S Scala, B Dickstein, J Regis, et al. function in multidrug-resistant human breast cancer cells.Bryostatin 1 affects P-glycoprotein phosphorylation but not
Updated version
http://clincancerres.aacrjournals.org/content/1/12/1581
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/1/12/1581To request permission to re-use all or part of this article, use this link
Research. on June 1, 2018. © 1995 American Association for Cancerclincancerres.aacrjournals.org Downloaded from