
Bridging scales: Ab initio molecular dynamics and kinetic Monte Carlo simulations
28
Bridging scales: Ab initio molecular dynamics and kinetic Monte Carlo simulations Karsten Reuter Fritz-Haber-Institut, Berlin
-
Upload
jillian-lara -
Category
Documents
-
view
36 -
download
3
description
Bridging scales: Ab initio molecular dynamics and kinetic Monte Carlo simulations. Karsten Reuter Fritz-Haber-Institut, Berlin. Multiscale modeling. Ab initio atomistic thermodynamics and statistical mechanics of surface properties and functions K. Reuter, C. Stampfl and M. Scheffler, - PowerPoint PPT Presentation
Transcript of Bridging scales: Ab initio molecular dynamics and kinetic Monte Carlo simulations

Bridging scales:
Ab initio molecular dynamics and
kinetic Monte Carlo simulations
Karsten Reuter
Fritz-Haber-Institut, Berlin

Multiscale modeling
Ab initio atomistic thermodynamics and statistical mechanics of surface properties and functions
K. Reuter, C. Stampfl and M. Scheffler, in: Handbook of Materials Modeling, Part A. Methods,
(Ed.) Sidney Yip, Springer (Berlin, 2005).http://www.fhi-berlin.mpg.de/th/paper.html

I. Straightforward time-evolution:Ab initio molecular dynamics
Understanding Molecular Simulation,D. Frenkel and B. Smit, Academic Press (2002)

)())](()1
[()()(2)( 42 terrorttrVm
ttrtrttr
MD: Numerical integration of Newton’s equation
),,,( 21
..
Nrii rrrVrmi
e.g. Verlet algorithm
Alternative algorithms: - velocity Verlet- leapfrog- predictor-corrector
- Runge-Kutta…
Solid-state: Δt ~ 10-15 s

Keeping everything: expensive, limited time scale
Example:
O2 dissociation at Al(111)
Total time of trajectory: 0.5 psTime step: 2.5 fs (200 steps)
CPU cost: 45 days on 1 Compaq ES45 processor
J. Behler et al., Phys. Rev. Lett. 94, 036104 (2005)

II. First-principles kinetic Monte Carlo simulations:rare events and long time scales
IPAM tutorial by Kristen Fichthornhttp://www.ipam.ucla.edu/publications/matut/matut_5907.ppt

First-principles kinetic Monte Carlo simulations
A
BMolecularDynamics
ΔEA ΔEBrA→B
rB→A
kineticMonte CarloN
t
B
A
equilibriumMonte Carlo
A
B
< N >
EB
EA
TS
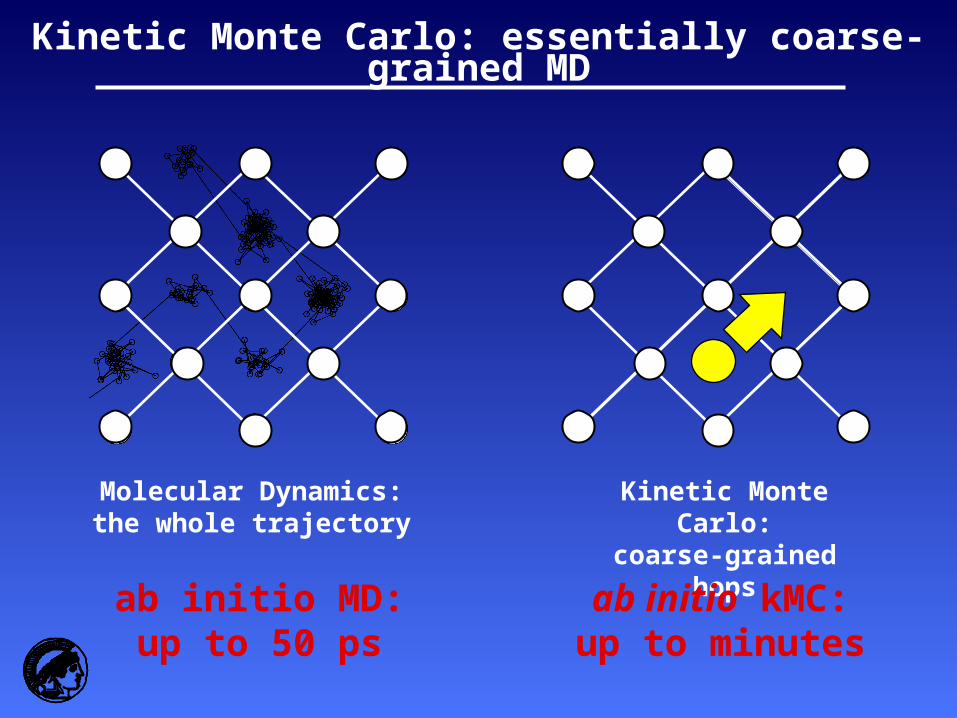
Molecular Dynamics:the whole trajectory
Kinetic Monte Carlo:coarse-grained hops
ab initio MD:up to 50 ps
ab initio kMC:up to minutes
Kinetic Monte Carlo: essentially coarse-grained MD

The crucial ingredients to a kMC simulation
i) Elementary processes
Fixed process list vs. „on-the-fly“ kMC
Lattice vs. off-lattice kMC
ii) Process rates
PES from density-functional theory
Transition state theory
x
xOcus
CObr

Flowchart of a kinetic Monte-Carlo simulation
determine all possible processes i for given configuration of your
system and build a list.
Get all rates (i)
Get two random numbers 1 , 2 [0,1[
Calculate R = i (i)
and find process “k”:
k k-1 (i) 1 R (i)
i=1 i =1
Execute process number “k”, i.e. update configuration
update clock
t t – ln(2)/R
START
END
0
1
1 Rk

III. Getting the rates…
Chemical kinetics,J.K. Laidler, Harper & Row 3rd ed. (New York, 1987)
Methods for finding saddle points and minimum energy paths,G. Henkelman, G. Jóhannesson, and H. Jónsson,in “Progress on theoretical chemistry & physics”,S.D. Schwartz (Ed.), Kluwer (Amsterdam, 2000)
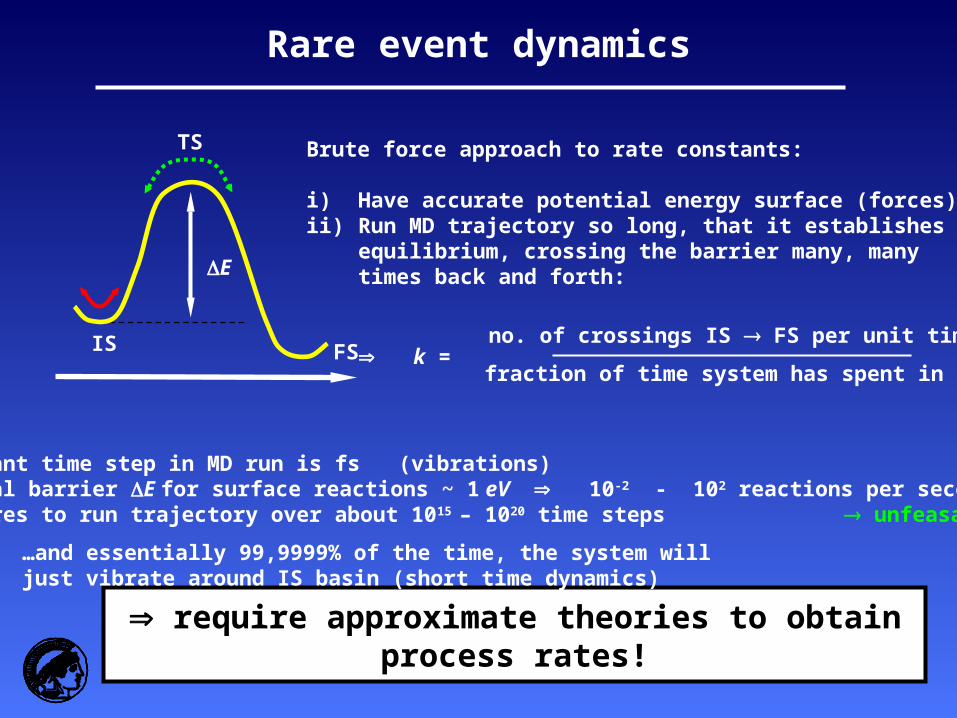
Rare event dynamics
E
IS
TS
FS
Brute force approach to rate constants:
i) Have accurate potential energy surface (forces)ii) Run MD trajectory so long, that it establishes
equilibrium, crossing the barrier many, manytimes back and forth:
k =no. of crossings IS FS per unit time
fraction of time system has spent in IS
require approximate theories to obtain process rates!
Yet: - Relevant time step in MD run is fs (vibrations) - Typical barrier E for surface reactions ~ 1 eV 10-2 - 102 reactions per second (TOF!) - Requires to run trajectory over about 1015 – 1020 time steps unfeasable…
…and essentially 99,9999% of the time, the system willjust vibrate around IS basin (short time dynamics)

Transition state (activated complex) theory I
Assumptions:
i) Reaction system passes the barrier only once (no re-crossings)
ii) Energy distribution of reactant DOF is Boltzmann-like (many collisions compared to reaction eventsyield equilibrium between activated complex and IS,except with respect to the reaction coordinate)
iii) Passage over barrier is the motion of only one DOF,the reaction coordinate, which is independent of allother motions of the activated complex (no concertedmotions)
iv) Passage over barrier is a classical event (no tunneling)
( Eyring, Evans, Polanyi, ~1935 )
IS FS
eq.
X
X
kTST = [ ( ) e S/k ] e-E/kTkThISFS
Derivation: see e.g.K.J. Laidler, Chemical kinetics,
Harper & Row, New York (1987)

Transition state (activated complex) theory II
problem reduces to locating transition states,i.e. saddle points in high dimensional PES
- Attempt frequency/preexponential factor ko
TST = e S/k
~ 1013 sec-1 ~ 1
- In harmonic TST, koTST is given by the harmonic normal modes at the IS and TS
(as explained in talk by Christian Ratsch on Tuesday)
kTh
- If assumptions i)-iv) are fulfilled, kTST is exact. In general, kTST is an upper limit to the real rate
k = fdyn kTST
In principle, one can compute so-called dynamical corrections. In contrast to liquid & gas phase, fdyn ~ 1 for solid-state processes ( TST is a rather good approximation)

i) Grid method:
- Compute PES on a (regular) grid- Scales like: (no. of points) dim
- e.g. 59 ~ 2 million grid points
often unfeasable
ii) Drag method:
- Choose appropriate reaction coordinate q- Constrain q and relax all other DOF- Move from IS to FS
highly dependent on good reaction coordinate hysteresis!
IS
FS
TSx
x
xx
x
x
x
x
x
x
x
x
x
Transition state search algorithms I: grid and drag

Transition state search algorithms II: ridge
- Initialize with straight line interpolation and choose max-energy point Ro
- Create two replicas slightly displaced from Ro on either side of the ridge (side-step)- Displace replicas along gradient (downhill- step)- Find max-energy point Ri along connecting line between two replicas
- Sequentially decrease displacements in downhill- and side-steps when approaching TS
works nicely on well-defined ridges difficult to optimize the displacements
for a given system then poor performance (many force
evaluations required)
I.V. Ionova and E.A. Carter,J. Chem. Phys. 98, 6377 (1993)
IS
FS
TSx
x
x
x
x
x
xx
xRo
xx
xxxxxx
R1
R2

- Initialize with several images {Ri} along a straight-line interpolation- Minimize
S(R1, …, RN) = i E(Ri) + i k/2 (Ri+1 - Ri )2
- Problem:- elastic band cuts corners- images tend to slide down towards low-energy IS/FS regions, leaving few images for relevant TS region
- Solution:- only spring force component parallel to path (no corner cutting)- only true force component perpendicular to path (no down-sliding)
widely applied workhorse has problems, if energy varies largely along path,
but very little perpendicular to it (kinky PES)
G. Mills and H. Jónsson,Phys. Rev. Lett. 72, 1124 (1994)
Transition state search algorithms III: nudged elastic band
IS
FS
TSx
x
xx x
xx
x
x
x
F true
F ||spring
x

Transition state search algorithms IV: dimer
- Initialize by putting dimer at an extremum of a high temperature MD-run- Rotate dimer to minimize energy ( direction of lowest frequency normal mode)- Move dimer along projected gradient perpen- dicular to dimer axis works without any information about FS
G. Henkelman and H. Jónsson,J. Chem. Phys. 111, 7010 (1999)
IS
FS
TSx
x
x
Generally:
- performance scaling with DOF not really known- not good for rough PES- high CPU cost - no algorithm is fool-proof (still lots of room for new ideas)
F
F R

IV. Identifying the processes…
Extending the time scale in atomistic simulation of materials, A.F. Voter, F. Montalenti and T.C. Germann,
Annu. Rev. Mater. Res. 32, 321 (2002)

Diffusion at metal surfaces: surprises…
Hopping mechanism
Ag(100) E = 0.45 eVAu(100) E = 0.83 eV
B.D. Yu and M. Scheffler, Phys. Rev. B 56, R15569 (1997)
Exchange mechanism
Ag(100) E = 0.73 eVAu(100) E = 0.65 eV
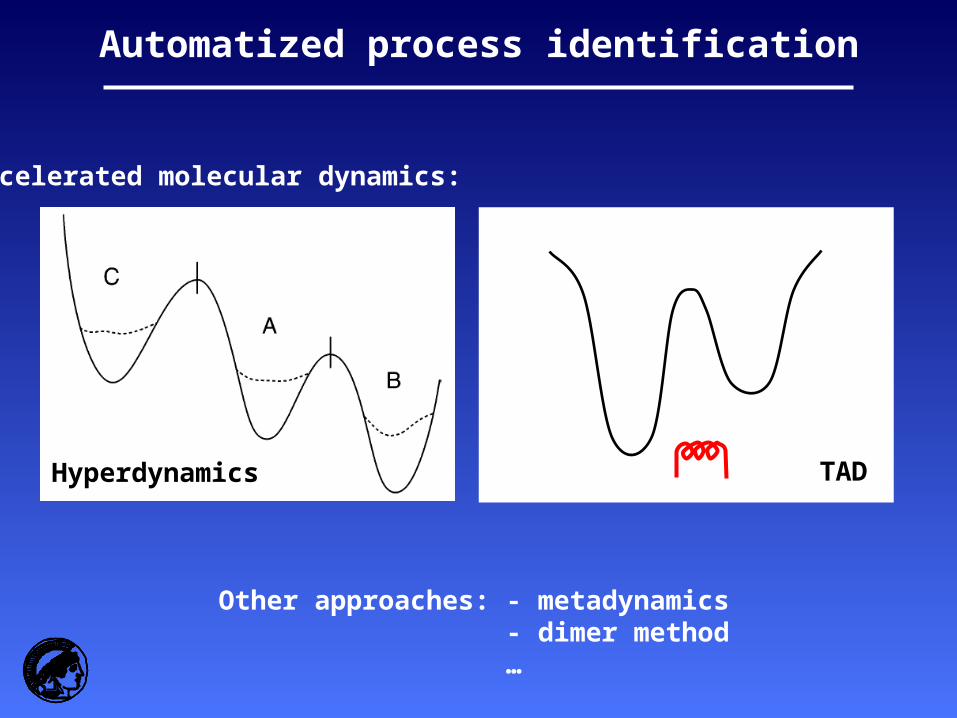
Hyperdynamics
Automatized process identification
TAD
Accelerated molecular dynamics:
Other approaches: - metadynamics- dimer method…

V. If it works, it works:First-principles kMC simulations for oxidation catalysis

cus site bridge site
RuO
A materials gap resolved: CO oxidation at Ru(0001) vs. RuO2(110)
H. Over and M. Muhler, Prog. Surf. Sci. 72, 3 (2004)
K. Reuter et al., Chem. Phys. Lett. 352, 311 (2002)

kMC events for CO oxidation over RuO2(110)
Adsorption: CO - unimolecular, O2 – dissociativeno barrierrate given by impingement r ~ So p/(2mkT)
Desorption: CO – 1st order, O2 – 2nd orderout of DFT adsorption well (= barrier)prefactor from detailed balance
Diffusion: hops to nearest neighbor sitessite and element specificbarrier from DFT (TST)prefactor 1012 s-1 (generic)
Reaction: site specificimmediate desorption, no readsorptionbarrier from DFT (TST)prefactor from detailed balance
26 elementary processes considered
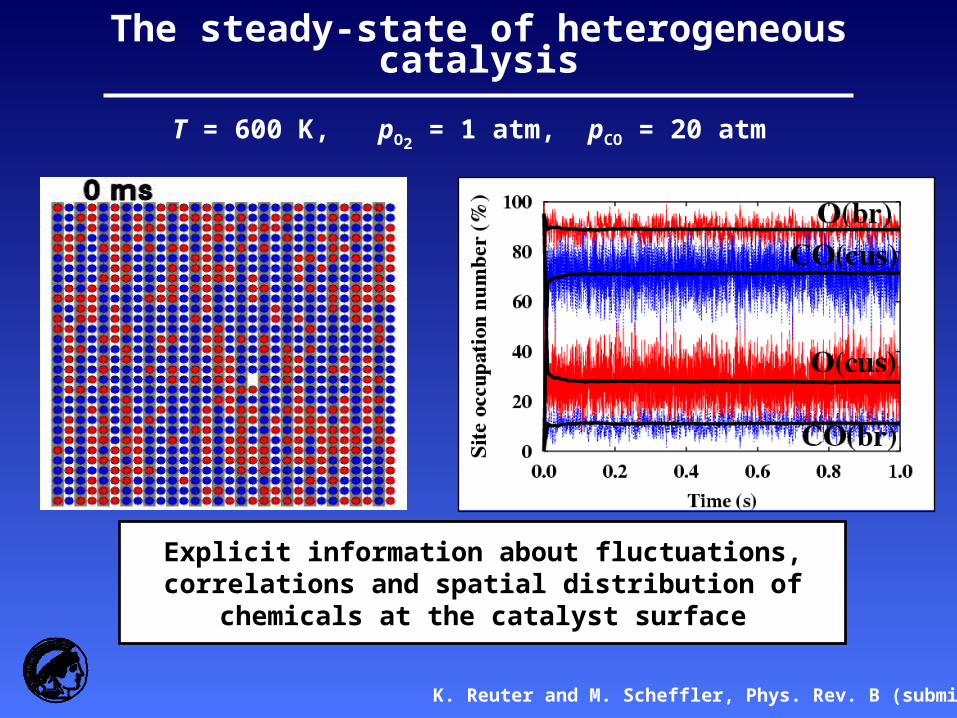
The steady-state of heterogeneous catalysis
T = 600 K, pO2 = 1 atm, pCO = 20 atm
K. Reuter and M. Scheffler, Phys. Rev. B (submitted)
Explicit information about fluctuations, correlations and spatial distribution of chemicals at the catalyst surface

A ( pCO , pO2 )-map of catalytic activity
COCObrbr/-/-
600 K pO (atm)2
1
COCObrbr/CO/COcuscus
OObrbr/O/Ocuscus
p CO (
atm
)
1
10-5
105
10-5 10+510-15
(CO/O
)br / (
CO/O)cus
10-10
OObrbr/ -/ -
COCObrbr/CO/COcuscus
OObrbr/O/Ocuscus
4.03.02.0
1.0
-1.0-2.0-3.0
0.0
pO (atm)2
10-10 110-5 10+5
log(TOF)
p CO (
atm
)
1
10-5
105
K. Reuter, D. Frenkel and M. Scheffler, Phys. Rev. Lett. 93, 116105 (2004)

pC
O (
atm
)
10-30 110-20 10-10
10-20
10-10
1 COCObrbr/CO/COcuscus
COCObrbr/-/-
- / -- / - OObrbr/O/Ocuscus
T = 350 KpCO = 10-10 atmT = 350 KpO2
= 10-10 atm
…and how about experiment?
J. Wang, C.Y. Fan, K. Jacobi, and G. Ertl,J. Phys. Chem. B 106, 3422 (2002)
pO (atm)2
-9.0
-4.0-5.0-6.0-7.0
-10.0-11.0
-8.0
log(TOF)
350 K

Ab initio MD and kMC simulations
Ab initio kinetic Monte Carlo simulations:
- coarse-grained time evolution (rare event dynamics)
- efficient treatment of statistical interplay of a larger
number of elementary processes
- time scales given by process rates, often seconds or longer
- process list (process identification, lattice models)
- accuracy of rates (DFT-TST), high CPU cost
- low speed-up, if very fast processes present
Ab initio molecular dynamics:
- fully dynamics of the system- straightforward, easy to implement
- times scales up to ~ns- acceleration techniques under development


















![A hybrid multiscale kinetic Monte Carlo methodweb.mit.edu/braatzgroup/A_hybrid_multiscale... · chastic algorithm, the kinetic Monte Carlo (KMC) [13,14] method, has been used to study](https://static.fdocuments.us/doc/165x107/5fc699ca8173b279fb4fe49a/a-hybrid-multiscale-kinetic-monte-carlo-chastic-algorithm-the-kinetic-monte-carlo.jpg)