
BOOK OF ABSTRACTS Paul Ehrlich Euro-PhD Network Virtual ...
118
BOOK OF ABSTRACTS Paul Ehrlich Euro-PhD Network Virtual Meeting 2021 Zoom platform, July 26 th -28 th 2021 www.pehrlichmedchem.eu www.net4science.com medchem2021.unicz.it
Transcript of BOOK OF ABSTRACTS Paul Ehrlich Euro-PhD Network Virtual ...

BOOK OF ABSTRACTS
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
www.pehrlichmedchem.eu www.net4science.com
medchem2021.unicz.it

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
1
WELCOME PREFACE
It is a great pleasure for the Scientific and Organizing Committees to
welcome all participants to this extraordinary meeting of the Paul Ehrlich Euro-
PhD network. This edition, established formally during the local coordinators’
meeting on February 15th 2021, has been designed with the ultimate aim to keep
as much as possible united the network that physically had the occasion to meet
last time only in June 2019 in Catanzaro. In that occasion, nobody could imagine
what should happen after few months. The X edition of the Paul Ehrlich Euro-PhD
meeting, scheduled in Barcelona, has been fixed and postponed twice in 2020 and
2021 due to pandemic reasons. So, the risk of losing the texture of the network,
the contacts among all adhering Universities and the opportunity to award
brilliant PhDs with all PE requirements was really high and concrete. In order to
reduce such a risk, the Paul Ehrlich Euro-PhD community promptly reacted the
situation and to propose a virtual formula for this extraordinary edition. It is
obviously not the best choice, but the only real alternative was to skip meeting
also in 2021. In order to attract as much as possible a wide audience of
participants, our committees invested a lot in a new meeting formula, where no
keynote lectures are scheduled and only afternoon sessions are organized in three
consecutive days. The idea is to promote as much as possible the interaction
among the participants, involving them in ten selected projects, distributed in
three unedited PENP sessions, where local coordinators are actively invited to
keep high the discussion level. The traditional Paul Ehrlich Euro-PhD Awards,
named as PEEPA, are organized in two sessions and, for the first time, one of the
10 eligible PhDs will be selected for an in presence young investigator meeting
organized in February 2021 in Nantes (France). Furthermore, flash
communications from PENP and Poster contribution will enrich the program of the
three days with young speakers. Moreover, two special sessions, respectively
related to the Editor’s corner and the PE Alumni association, will complete the
programme. Finally, following the tradition of previous editions, the awarding
ceremony will close the last day and, for the first time, a special issue on a
renowned scientific journal will be officially launched.
As coordinator of the Paul Ehrlich Euro-PhD network, this meeting
represents my last activity for this community within the exciting three year

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
2
period 2019-2021. Unfortunately, the pandemic situation strongly effected the
original planning, but basing on the consistent participation of scientific
contribution reported in this book of abstract and also on additional requests
collected from some Universities to join us, I am quite satisfied of this result. I
want to emphasize that the organization of this meeting could not be done
without the precious contribution of the Scientific Board (Julio Alvarez-Builla,
Athina Geronikaki, Elias Maccioni and Serge Van Calenbergh) and the Organizing
Committee, members of my research team (Antonio Lupia, Francesco Mesiti,
Federica Moraca, Francesco Ortuso, Giulia Panzarella and Isabella Romeo), who
really worked hard to create this special event, that I hope will be reminded as a
nice virtual experience within all participants. Last but not least, I express my
gratitude to two institutions, Net4Science srl and the Life Science PhD course of
my University in Catanzaro, for hosting and promoting the PEVM2021 with all
available media channels and three additional institutions, Schrödinger software
house, Gilead Pharma company and Chemistry Europe for supporting our
initiative.
So, I wish you to enjoy the PEVM2021 and to hope to meet again in presence
as soon as possible!
Catanzaro, July 25th 2021
Stefano Alcaro
Università “Magna Græcia” di Catanzaro (Italy) Coordinator of the Paul Ehrlich Euro-PhD Network
Chair of MedChem2021 Virtual Meeting [email protected]

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
3
Commitees
SCIENTIFIC COMMITTEE ORGANIZING COMMITTEE
Stefano Alcaro Antonio Lupia Julio Alvarez-Builla Francesco Mesiti Athina Geronikaki Federica Moraca
Elias Maccioni Francesco Ortuso Serge Van Calenberg Giulia Panzarella
Isabella Romeo
Organizing Institutions
Supporting Institutions

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
4
PEVM2021 Scientific Programme MONDAY, July 26th 2021
Introduction to the Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
14:45 Welcome greetings to #PEVM2021 Stefano ALCARO, Università “Magna Græcia” di Catanzaro (Italy) and PE MedChem Euro-PhD Network Coordinator
PENP Session I - Topic: ”Cancer diseases” Chairs: Beatriz de PASCUAL-TERESA, Universidad CEU San Pablo (Spain) Luc DEMANGE, Université de Paris V Descartes (France) Rita GUEDES, Universidade de Lisboa (Portugal)
15:00 PENP-1
Pyrrolo[2’,3’:3,4]cyclohepta[1,2-d][1,2]oxazoles, a new class of anti-mitotic agents active against multiple malignant cell types
Marilia BARRECA Università di Palermo (Italy)
15:20 PENP-2 Model optimization and site-mapping of hASNS, a novel target
in the treatment of ALL 15:40
Adriana CORICELLO Università “Magna Græcia” di Catanzaro (Italy)
PENP-3 Development of Tumor-Associated Carbonic Anhydrases Inhibitors Based on Benzopyrone Scaffold
Lisa SEQUEIRA Università di Cagliari (Italy)
16:00 Coffee break and Sponsor Slideshow
PEEPA Session I – Paul Ehrlich Euro-PhD Awards Moderator: Serge VAN CALENBERGH, Ghent University (Belgium)
16:10 PEEPA-1
Molecular modeling studies on antiviral targets: Drug resistance mechanisms and rational drug design
Francesca Alessandra AMBROSIO Università “Magna Græcia” di Catanzaro (Italy)
16:25 PEEPA-2 Derivatives of Pyrazinecarboxylic Acid as Potential Antimycobacterial Active Drugs
Ghada BOUZ Charles University (Czech Republic)
16:40 PEEPA-3 Au and Ag NHC-metal complexes as effective multi-target agents in breast cancer tratment
Jessica CERAMELLA Università della Calabria (Italy)
16:55 PEEPA-4 Development of new chemical entities based on natural scaffolds with therapeutic potential towards age-related disorders Daniel CHAVARRIA
Universidade do Porto (Portugal) 17:10 PEEPA-5
Behind the allosteric inhibition of PTPRZ1, a current druggable phosphatase Bruno DI GERONIMO
Universidad San Pablo-CEU (Spain)

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
5
Flash PENP Communications (5 min) Moderator: Claudia Sissi, Università di Padova (Italy)
17:25 FC-1 Modeling Epac1 interactions with the allosteric inhibitor AM-001 by co-solvent molecular dynamics
Marianna BUFANO Università di Roma “La Sapienza” (Italy)
17:30 FC-2 Delivery for infectious diseases
Valentina DEL GENIO Università di Napoli “Federico II” (Italy)
17:35 FC-3
Discovering selective Poly (ADP-ribose) Polymerase (PARP) Inhibitors to expand the precision medicine approach
Mariagiulila NIZI Università di Perugia (Italy)
17:40 FC-4 Inhibition of ZIKA virus replication by novel inhibitors
of NS2B/NS3 complex 17:45
Michela PUXEDDU Università di Roma “La Sapienza” (Italy)
FC-5 The interaction between gab2 with sh3-domain of gbr2 as a new potential target in cancer therapy
Jessica SEBASTIANI Università di Roma “La Sapienza” (Italy)
17:50
FC-6 Development of innovative analytical tools to improve the safety of plant-based health products, application to the case of plants of the genus Tinospora used in Laos and in Europe
Kedmany SISOUKLATH Université de Paris (France)
17:55
FC-7 Proteomic contribution to the omic path for the identification of novel drugs overcoming resistance in Leishmaniasis
Lorenzo TAGLIAZUCCHI Università di Modena e Reggio Emilia (Italy)
18:15 Closing

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
6
TUESDAY, July 27th 2021 12:00 Paul Ehrlich Euro-PhD Network Coordinator Meeting (Reserved only for PE Local Coordinators)
14:45 Opening
Rita PODZUNA, Senior Director at Schrödinger – München (Germany)
PENP Session II – Topic: “Neurodegenerative diseases” Chairs: Fernanda BORGES, Universidade do Porto (Portugal) Jose Ignacio BORRELL, Ramon Llull University (Spain) Elias MACCIONI, Università di Cagliari (Italy)
15:00 PENP-4 Identification of molecular basis of praja2 and TBC1D31 interaction
Bianca FIORILLO Università di Napoli “Federico II” (Italy)
15:20 PENP-5 Flavonoid-derived acetylcholinesterase inhibitors as multitarget
drug ligands for the treatment of Alzheimer’s disease 15:40
Jorge GÓMEZ-CARPINTERO Universidad Complutese de Madrid (Spain)
PENP-6 LigAdvisor: a unified and easily accessible webserver for polypharmacology and drug design repurposing
Annachiara TINIVELLA
Università di Modena e Reggio Emilia (Italy)
16:00 Coffee break and Sponsor Slideshow
PEEPA Session II – Paul Ehrlich Euro-PhD Awards Moderator: Hanoch SENDEROWITZ, Bar-Ilan University (Israel)
16:10 PEEPA-6 Strategies against chronic kidney disease: new modulators of PTP1B and ILK
Javier GARCÍA-MARÍN Univerisdad de Alcalá (Spain)
16:25 PEEPA-7 Targeting carbonic anhydrases (CAs): rational design, synthesis Structural studies and biochemical evaluation
Francesca MANCUSO Università di Messina (Italy)
16:40 PEEPA-8 Mapping chromone-3-phenylcarboxamide pharmacophore: quid est veritas?
Francesco MESITI Università “Magna Græcia” di Catanzaro (Italy)
16:55 PEEPA-9 The search for novel histamine H3 receptor ligands in the group of piperazine derivatives
Katarzyna SZCZEPAŃSKA Jagiellonian University Medical College (Poland)
17:10 PEEPA-10 Targeting protein-protein interactions for the treatment of tumors and neurodegenerative disorders
Serena VITTORIO Università di Messina (Italy)
17:30 Editor’s Corner: a Q&A session on best practices in publishing Moderator: Rosaria GITTO, Università di Messina (Italy)
Paola BARRAJA (EJMC) Maria Laura BOLOGNESI (JMC) David PERALTA (ChemMedChem)
18:00 Closing

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
7
WEDNESDAY, July 28th 2021
14:45 Opening Michelangelo SIMONELLI, Senior Government Affairs Director at Gilead Sciences (Italy)
PENP Session III – Topic: “Infectious diseases” Chairs: Maria Paola COSTI, Università di Modena e Reggio Emilia (Italy) Athina GERONIKAKI, Aristotle University of Thessaloniki (Greece) Daniel KIKELJ, Univerza V Ljublijani (Slovenia) Oriana TABARRINI, Università di Perugia (Italy)
15:00 PENP-7 Discovery of an Effective Dual αvβ6/αvβ8 Integrin Ligand as a Herpes Simplex Virus-1 Entry Inhibitor
Vincenzo Maria D’AMORE Università di Napoli “Federico II” (Italy)
15:20 PENP-8 Amphiphilic azobenzenes: Antibacterial activities and biophysical
investigation of their interaction with bacterial membrane lipids 15:40
Antoine FRANCHE Université de Paris (France)
PENP-9 6-Methyl-7-aryl-7-deazapurine nucleosides as anti-trypanosoma cruzi agents: structure-activity relationship and in vivo efficacy
Cai LIN Ghent University (Belgium)
16:00 PENP-10 Chimeric small molecules in the search for novel anti-trypanosomatid agents
Elisa ULIASSI Università di Bologna (Italy)
16:30 Coffee break and Sponsor Slideshow
16:45- 17:15
Flash PE Poster Communications (3 min) Moderator: Agostino MARRAZZO, Università di Catania (Italy)
FC-1 Haloperidol metabolite II Valproate ester MRJF22 enantiomers as potential multifunctional agents against uveal melanoma Carla BARBARACI
Università di Catania (Italy) FC-2
Design, synthesis and biological evaluation of new 4-oxo-1,4-dihydroquinolin-3-adamtilamides derivates to develop CB2R fluorescent probes
Francesca INTRANUOVO
Università di Bari “Aldo Moro” (Italy) FC-3
Novel Proteasome Inhibitors based on γ-lactams for cancer treatment Roberta LISTRO
Università di Pavia (Italy) FC-4
New cytisine-based multitarget compounds for neurodegenerative diseases
Emmanuel OROCIO RODRÍGUEZ Universidad Complutense de Madrid (Spain)
FC-5 A novel scaffold for potent and selective inhibition of tumor-related
carbonic anhydrase isoforms IX and XII Virginia PONTECORVI
Università di Roma “La Sapienza” (Italy)
FC-6
Hybrid design, synthesis and in vitro biological evaluation of 1H-indazoles as MAO B inhibitors: effect of 1,2,4-oxadiazole bioisosteric replacement of the amide linker
Mariagrazia RULLO
Università di Bari “Aldo Moro” (Italy)

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
8
17:30 Social Event “PE Alumni meet again” Moderators: Cosimo ALTOMARE, Università di Bari “Aldo Moro” (Italy) Federica MORACA, Università di Napoli “Federico II” (Italy) What about a PE Alumni Association? 18:00 Special Issue Launch & Awards Announcement Best PEEPA Best PENP Best POSTER Best Flash Communication
Greetings and closing remarks
Next meeting announcement

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
9
Paul Ehrlich Network Projects (PENP)

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
10
Paul Ehrlich Network Projects (PENP-1)
Pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles, a new class of anti-mitotic agents active against multiple malignant cell types
Marilia Barreca,a,d Virginia Spanò,a Roberta Rocca,c,g Alessandra Montalbano,a Maria Valeria Raimondi,a Eugenio Gaudio,d Roberta Bortolozzi,f Ruoli Bai,e Stefano Alcaro,b,c
Ernest Hamel,e Giampietro Viola,f,h Francesco Bertoni,d Paola Barrajaa
a Department of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of
Palermo, Via Archirafi 32, 90123, Palermo, Italy b Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro, Italy
cNet4Science srl, Academic Spinoff, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro, Italy dInstitute of Oncology Research, USI, Via Vincenzo Vela 6, 6500, Bellinzona, Switzerland
e Screening Technologies Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, Frederick National Laboratory for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick,
Maryland 21702, United States f Istituto di Ricerca Pediatrica IRP, Fondazione Città della Speranza, Corso Stati Uniti 4, 35127, Padova, Italy
g Dipartimento di Medicina Sperimentale e Clinica, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro, Italy
h Dipartimento di Salute della Donna e del Bambino, Laboratorio di Oncoematologia Università di Padova, Via Giustiniani 2, 35131, Padova, Italy
E-mail: [email protected]
The [1,2]oxazole moiety represents the core structure of many drug candidates with multiple
targets and is an attractive scaffold in medicinal chemistry. Diaryl[1,2]oxazoles,
naphtylcombretastin and its derivatives, incorporating the isoxazole ring, displayed potent cytotoxic
effects and inhibition of tubulin polymerization [1,2]. We incorporated the [1,2]oxazole moiety into
tricyclic systems. In this contest, we reported the synthesis and biological evaluation of
[1,2]oxazolo[5,4-e]isoindoles with in vitro and in vivo anti-tumor activity in diffuse malignant
peritoneal mesothelioma due to tubulin polymerization impairment [3].
We now report our recent results on a new class of pyrrolo[2',3':3,4]cyclohepta[1,2-d][1,2]oxazoles
for the treatment of both solid and hematological neoplasias. The new compounds showed potent
activity at nanomolar level (MG-MID 0.08–0.41 µM) in the NCI-60 human tumor cell line panel. All
compounds were further tested at the Institute of Oncology Research (IOR, Switzerland) on a
panel of lymphoma cell lines, including Idelalisib and Ibrutinib resistant clones. Five compounds
showed potent growth inhibitory effects on selected cell lines. Insight on the mechanism of action
indicated G2/M phase arrest in a concentration dependent manner and induction of apoptosis
through the mitochondrial pathway, characterized by generation of reactive oxygen species (ROS)
and activation of PARP cleavage. Inhibition of tubulin polymerization (IC50 values of 1.9–8.2 µM)
and colchicine binding to tubulin were assessed at Frederick National Laboratory for Cancer
Research. Molecular modeling studies (Unversità “Magna Grӕcia” di Catanzaro) highlighted strong
interactions with tubulin and a peculiar binding mode, characterized by the methoxybenzyl portion
placed similarly to colchicine.
In conclusion, pyrrolo[2',3':3,4]cyclohepta[1,2-d][1,2]oxazoles are a novel class of anti-mitotic
agents with anti-tumor activity in multiple cancer cell lines.
[1] Sun, C.M., et al. Bioorg. Med. Chem. Lett. 2007, 17, 1078–1081; [2] Maya, A. B. S., et al. Bioorg. Med. Chem. 2005, 13, 2097–2107; [3] Spano, V.; et al. Eur. J. Med. Chem. 2016, 124, 840–851.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
11
Paul Ehrlich Network Projects (PENP-2)
Model optimization and site-mapping of hASNS, a novel target in the treatment of ALL.
Adriana Coricello,a Antonio Lupia,b Carmen Gratteri,a Stefano Alcaro,ab and Nigel Richardsc
a Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, 88100, Catanzaro, Italy b Net4Science srl, Università “Magna Græcia” di Catanzaro, Campus Universitario “S. Venuta”, Viale Europa,
Loc. Germaneto, Catanzaro, 88100, Italy c School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK
E-mail: [email protected]
Asparagine Synthetase (ASNS) is an amidotransferase responsible for the production of L-
asparagine which plays a key role in the metabolism of tumor cells, in particular in lymphoblastic
leukemia (ALL), sarcoma and in some forms of metastatic breast cancer [1][2]. Up to date, only
one compound was identified as a potent inhibitor of human ASNS (hASNS), and the X-ray 3D
structure of hASNS was recently published [3][4]. Since the integrity of the protein is essential
when running MD simulations (MDs), homology modelling and Molecular Dynamics (MD) tools
were applied to obtain an optimized 3D model to use for the in silico analyses. hASNS features two
active sites: a glutaminase site in the N-terminal domain and an ATP binding site in the C-terminal
domain, where the synthesis of asparagine takes place. Several complexes of the protein were
built in order to mimic the behavior of the protein in different moments of the catalytic turnover and
additional MD simulations were performed. The trajectory analysis showed that the presence or
lack of a ligand within the ATP binding site strongly influences the behavior of the Gln site
residues, and vice versa. The mapping of the binding sites also revealed essential information. The
glutaminase site was hence selected for the screening of libraries of compounds aimed at the
identification of new candidate inhibitors of hASNS.
[1] Richards, N.G.J. et al., Annu Rev Biochem. 2006, 75, 629–654. [2] Radadiya, A. et al., Biochemistry 2020, 59, 3193-3200. [3] Ikeuchi, H. et al., Bioorgan Med Chem. 2012, 20, 5915–5927. [4] Zhu, W. et al., Commun Biol. 2019, 2, 345.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
12
Paul Ehrlich Network Projects (PENP-3)
Development of Tumour-Associated Carbonic Anhydrases Inhibitors Based on Benzopyrone Scaffold
Lisa Sequeira,a,b Elias Maccioni,a Fernanda Borges,c Claudiu T. Supuran,d and Eugenio Uriarteb
a Department of Life and Environmental Sciences, University of Cagliari, Cagliari, Italy b Department of Organic Chemistry, Faculty of Pharmacy of the University of Santiago de Compostela, Spain
c CIQUP/ Department of Chemistry and Biochemistry, Faculty of Sciences of the University of Porto d Dipartimento NEUROFARBA, Sezione di Scienze Farmaceutiche, Università degli Studi di Firenze, Sesto
Fiorentino, Florence, Italy
E-mail: [email protected]
Carbonic anhydrases (CAs) are a class of metallo-enzymes that catalyze the reversible hydration
of carbon dioxide into bicarbonate and a proton and are widely distributed in all living organisms.
These enzymes are involved in numerous physiological processes such as ion transport,
regulation of pH, bone resorption, and secretion of gastric, cerebrospinal fluids and pancreatic juice
[1,2]. In mammals CAs have 16 different isoforms and multiple ones are implicated in a range of
diseases, including cancer [2]. In particular, the trans-membrane CAs IX and XII are key pH
regulators that create a differential pH microenvironment within solid tumors and allow for tumor
cell survival under stressful conditions [2]. For this reason, CAs became an increasing interest to
researchers as drug targets, and, as a result, several CAs inhibitors have been designed.
However, the CAs inhibitors available are mostly unselective, leading to several side effects, thus
selectivity is mandatory [1]. Coumarins and chromones are two groups of heterocyclic compounds
commonly found in nature that show a wide range of biological activities, such as aromatase
inhibitory effect, anti-HIV, antimycotic, and antitumor activities [3]. Previous results also highlighted
the selectivity of furocoumarins towards CA IX and XII [4]. Accordingly, our project will focus its
drug design strategy on heterocycle compounds based on the coumarin and chromone scaffolds
that can inhibit selectively CA IX and XII. The results obtained so far will be presented in this
communication.
Figure 1: General structures of the compounds under investigation.
[1] Meleddu, R., et al., Acs Medicinal Chemistry Letters 2018, 9(10), 1045-1050 [2] Singh, S., et al., Molecules 2018, 23(5) [3] Abu-Hashem A. A., et al, Eur J Med Chem 2015, 90, 663-665 [4] Melis, C., et al., ACS Med. Chem. Lett. 2018, 9, 725−729
Lisa Sequeira grant was supported by Univerità degli Studi di Cagliari (funds from the Italian Ministry of Education, University and Reasearch).

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
13
Paul Ehrlich Network Projects (PENP-4)
Identification of molecular basis of praja2 and TBC1D31 interaction
Bianca Fiorillo,a Federica Moraca,a,b Emilia Pedone,c Eduard Stefan,d Antonio Feliciello,e Bruno Catalanottia
aDepartment of Pharmacy, University Federico II, 80131-Naples, Italy bNet4Science srl, Università “Magna Græcia”, 88100-Catanzaro, Italy cInstitute of Biostructures and Bioimaging, CNR, 80134-Naples, Italy.
dInstitute of Biochemistry, University of Innsbruck, 60-20-Innsbruck, Austria. eDepartment of Molecular Medicine and Medical Biotechnologies, University Federico II,80131- Naples, Italy.
E-mail: [email protected]
OFD1 is a component of the centrosome/basal body playing a key role in cilium biogenesis, and,
therefore, in the control of cell differentiation, growth and development. Mutations of the OFD1
cause ciliopathies with several implications in the development determining severe malformations
[1]. In this work, we investigated the role of the ubiquitin system in the control of OFD1 activity and
stability. In particular, we discovered that praja2 is the E3 ligase responsible for OFD1
ubiquitination, and that TBC1D31, a highly conserved protein, has a primary role to control, in the
centrosome, the TBC1D31/praja2/OFD1 molecular network necessary for the correct ciliogenesis.
In this study [2], we discovered that the C-terminal region TBC1D31941-970 (Figure 1A) acts as an
anchor for praja2, binding the praja2550-570 segment. Thus, the molecular basis of praja2550-570 and
TBC1D31941-970 interaction was further investigated. Firstly, the homology models were generated
using the I-TASSER threading approach. Then, a two-step docking procedure followed by 2 µs of
molecular dynamics (MD) simulations helped to identify the positively charged residues R957,
R959 and H960 of TBC1D31941-970 as key residues for the interaction with praja2550-570 (Figure 1B).
Microscale thermophoresis experiments validated the proposed binding mode, supporting the MD-
derived hypothesis of a specific role of residues R957, R959 and H960 of TBC1D31 in praja2
binding activity. Our findings revealed a multi-functional transduction unit at the centrosome that
links GPCR signaling to ubiquitylation and proteolysis of the ciliopathy protein OFD1, with
important implications on cilium biology and development.
Figure 1: A) Threading modelled structure of TBC1D31, with a zoom of its C-terminus. Mutated residues are highlighted in stick coloured by atom type; B) MD derived binding mode of praja2530–
570 (green cartoon) to the C-terminal region of TBC1D31 (red cartoon).
[1] Hildebrandt, F.; et al. The New England journal of medicine, 2011, 364(16), 1533–1543 [2] Senatore, E.; et al. EMBO J., 2021, 40(10):e106503

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
14
Paul Ehrlich Network Projects (PENP-5)
Flavonoid-derived acetylcholinesterase inhibitors as multitarget drug ligands for the treatment of Alzheimer's disease
Jorge Gómez-Carpintero1, Juan Francisco González1, Sagrario Martín-Aragón2, Paloma Bermejo2, Paula Moyano,3 Javier del Pino,3 José Carlos Menéndez1
1. Department of Chemistry in Pharmaceutical Sciences (Organic and Medicinal Chemistry Unit),
Faculty of Pharmacy, Universidad Complutense, Madrid 2. Department of Pharmacology, Pharmacognosy and Botanics, Faculty of Pharmacy, Universidad
Complutense, Madrid. 3. Department of Pharmacology and Toxicology, Faculty of Veterinary Medicine, Universidad
Complutense, Madrid.
E-mail: [email protected]
Alzheimer´s disease (AD), is the world´s leading cause of neurodegenerative disorders with a fast-growing incidence (the cases reported in 2016 doubled the ones in 1990). Hence, AD stands as a worldwide challenge for all health systems, as the absence of an effective drug prevents its treatment. It is widely recognized that AD has a multifactorial nature, with many pathological pathways that contribute to the development of the disease. In this context, multitarget drug ligands (MTDLs) emerge as an interesting approach to the treatment of AD.1 Acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) are two enzymes that have a close relationship with the development of AD, as they contribute to reduce the levels of acetylcholine (Ach) in the brain, and both are validated AD pharmacological targets. 2,3 Alternatively, oxidative stress is a common hallmark in a wide range of neurodegenerative disorders and also has an important role in the development and progression of AD.4 In this communication we present the synthesis and pharmacological evaluation of a series of hybrid compounds that are designed to act as MTDLs for the treatment of Alzheimer´s disease. These compounds possess a common flavonoid scaffold (naringenin or hesperitin) merged with the well-known AChE and BuChE inhibitors tacrine and donepezil. Thus, the compounds were designed to possess a strong antioxidant activity together with AChE and BuChE inhibitory activity, making them promising agents for the treatment of AD. [1] (a) Maramai, S.; Benchekroun, M.; Gabr, M. T.; Yahiaoui, S. Multitarget therapeutic strategies for Alzheimer’s disease: Review on emerging target combinations. BioMed Res. Int. 2020, Article ID 5120230. (b) González JF, Alcántara AR, Doadrio AL, Sánchez-Montero JM. Developments with multi-target drugs for Alzheimer's disease: an overview of the current discovery approaches. Expert Opin. Drug Discov. 14, 879-891 (2018). [2] Santos, M. A., Chand, K. & Chaves, S. Recent progress in repositioning Alzheimer’s disease drugs based on a multitarget strategy. Future Med. Chem. 8, 2113–2142 (2016). [3] Hampbel, H.; Mesulam. M. M.; Cuello, A. C.; Farlow, M. R.; Giacobini, E. et al. Brain 141, 1917-1933 (2018). [4] Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s
Dis. 57, 1105-1121 (2017).

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
15
Paul Ehrlich Network Projects (PENP-6)
LigAdvisor: a unified and easily accessible webserver for polypharmacology and drug repurposing
Annachiara Tinivella,a,b Luca Pinzib and Giulio Rastellib
a Department of Life Science, University of Modena and Reggio Emilia, Via Giuseppe Campi 103, 41125-Modena, Italy
b Clinical and Experimental Medicine PhD Program, University of Modena and Reggio Emilia, Via Giuseppe Campi 287, 41125-Modena, Italy
E-mail: [email protected]
Recently, a considerable amount of interest in drug discovery has been directed towards the often
challenging areas of polypharmacology (design of multi-target agents) and drug repurposing
(identification of new uses for known molecules) [1, 2]. Therefore, the exploitation of ever-evolving
data-driven approaches has become pivotal, enabling use of the vast amount of biological,
chemical and clinical data currently available [2].
We herein present LigAdvisor (https://ligadvisor.unimore.it/), a user-friendly webserver
designed to assist also non-experts in computer-aided drug design [3]. LigAdvisor is designed as
an easily accessible platform, which integrates information reported in multiple databases of
pharmaceutical interest (DrugBank, Protein Data Bank, UniProt, ClinicalTrials.gov, Therapeutic
Target Database) with ligand-based similarity calculations. Indeed, >30,000 unique ligands,
350,000 clinical data records, >9,000 target-disease associations and 18,000 targets are currently
reported in the core dataset.
As shown in Figure 1, users can perform both ligand- and target-oriented queries on
LigAdvisor, and either explore existing molecules or upload their own. Remarkably, LigAdvisor first-
of-its-kind webserver implements multi-ligand and multi-target queries, able to efficiently guide
users in retrieving the most suitable candidates for a wide range of drug discovery-related
applications, such as polypharmacology, drug repositioning, target profiling and target fishing
campaigns.
LigAdvisor is regularly updated, and is available at https://ligadvisor.unimore.it/) [3].
Figure 1: Schematic features of LigAdvisor and its implemented search types.
[1] Anighoro, A.; Bajorath, J.; Rastelli, G., J. Med. Chem 2014, 57, 7874–7887 [2] March-Vila, E. et al., Front Pharmacol. 2017, 8, 298. [3] Pinzi, L.; Tinivella, A.; Gagliardelli, L.; Beneventano, D.; Rastelli, G., Nucleic Acids Res. 2021, 1, doi:10.1093/nar/gkab385.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
16
Paul Ehrlich Network Projects (PENP-7)
Discovery of an Effective Dual αvβ6/αvβ8 Integrin Ligand as a Herpes Simplex Virus-1 Entry Inhibitor
Vincenzo Maria D’Amore,a Stefano Tomassi,a Francesco Saverio Di Leva,a Salvatore Di Maro,b Tatiana Gianni,c Giovanna Muscod, Horst Kessler,e Luciana Marinellia
a Dipartimento di Farmacia, Università degli Studi di Napoli Federico II, Via D. Montesano 49, 80131 Napoli, Italy; b Dipartimento di Scienze e Tecnologie Ambientali Biologiche e Farmaceutiche, Università degli Studi della Campania "Luigi Vanvitelli", Via A. Vivaldi 43, 81100, Caserta, Italy; c Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, 40126 Bologna, Italy d Biomolecular NMR Unit c/o IRCCS S. Raffaele, Via Olgettina 58, 20132 Milano, Italy; e Klinik für Nuklearmedizin and TranslaTUM, Central Institute for Translational Cancer Research, Technische Universität München, Ismaninger Str. 22, 81675, München, Germany.
E-mail: [email protected]
Herpes simplex virus (HSV) are widespread human pathogens which commonly causes recurrent
infections of the skin, mouth, lips, eyes, and genitals. The HSV cell entry-fusion is a multistep
process orchestrated by four essential glycoproteins, gD, gH/gL, and gB [1], which exploits the
nectin-1 and HVEM (herpesvirus entry mediator) receptors to penetrate host cells. In addition to
these, αvβ6 and αvβ8 Arg-Gly-Asp (RGD) integrins has recently come to the limelight as
interchangeable co-receptors for the cellular penetration of HSV-1. In fact, a consistent drop in the
infectivity of this virus has been obtained by contemporary inhibiting αvβ6 and αvβ8 either by cell
exposure to subtype-selective monoclonal antibodies (mAbs) or through siRNA transfection. [2] In
this work, we focused on a more affordable pharmaceutical approach, based on the design of
small RGD-containing cyclic pentapeptides. We started this campaign from our recently developed
αvβ6-selective peptide [RGD-Chg-E]-CONH2 (1) [3], which was submitted to a systematic N-
methylation with the aim to increase its affinity also toward αvβ8. Thus, a small library of N-
methylated derivatives of 1 was synthesized and one of them, namely [RGD-Chg-(NMe)E]-CONH2
(6), resulted in a potent dual αvβ6/αvβ8 binder. Extensive in cell evaluations demonstrated the
capability of 6 to effectively impair HSV-1 cellular penetration through an integrin-dependent
mechanism, prompting its further development as a new anti-HSV agent. Furthermore, a
NMR/molecular modeling combined approach was employed to rationalize the renewed selectivity
profile of 6 and to provide novel valuable hints for the design of RGD integrin ligands with the
desired subtype specificity.
[1] Campadelli-Fiume, G.; Collins-McMillen, D.; Gianni, T.; Yurochko, A. D. Annu Rev Virol. 2016, 3, 215-236. [2] Gianni, T.; Massaro, R.; Campadelli-Fiume, G. PNAS, 2015, 112, 3901-10 [3] Di Leva, F. S.; Tomassi, S.; Di Maro, S.; Reichart, F.; Notni, J.; Dangi, A.; Marelli, U. K.; Brancaccio, D.; Merlino, F.; Wester, H. J.; Novellino, E.; Kessler, H.; Marinelli, L. Angew. Chemie - Int. Ed. 2018, No. 57, 14645–14649. [4] Tomassi, S.; D’Amore, V. M.; Di Leva, F. S.; Vannini, A.; Quilici, G.; Weinmüller, M.; Reichart, F.; Amato, J.; Romano, B.; Izzo, A. A.; Di Maro, S.; Novellino, E.; Musco, G.; Gianni, T.; Kessler, H.; Marinelli, L. J. Med. Chem. 2021, 64, 6972–6984.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
17
Paul Ehrlich Network Projects (PENP-8)
Amphiphilic azobenzenes: Antibacterial activities and biophysical investigation of their interaction with bacterial membrane lipids
Antoine Franche,a Antoine Fayeulle,b Laurence Lins,c Isabelle Pezron,b Magali Deleuc and Estelle Léonardd
aFaculty of Pharmacy, Université de Paris, 4 Avenue de l’observatoire 75006 Paris, France bSorbonne University, Université de technologie de Compiègne, Centre de recherche de Royallieu, 60203
Compiègne Cedex, France cTERRA Research Center, Laboratory of Molecular Biophysics at Interfaces, Gembloux Agro-Bio Tech,
Université de Liège, Passage des Déportés, 2, 5030 Gembloux, Belgium dESCOM, 1 allée du Réseau Jean-Marie Buckmaster, 60200 Compiègne, France
E-mail: [email protected]
With the emergence of multi-drug resistant bacteria and hospital-acquired infections, there is an
urgent need to develop new antibiotics. Azo compounds are known to have antibacterial activities
since long [1]. Cationic amphiphilic azo compounds are reported to interact with bacterial
membrane [2]. Here, we report the synthesis, physico-chemical characterizations, and
antimicrobial activity assays of four Azo compounds that differ in their alkyl chain length (Figure 1).
The molecular mechanism of their antibacterial activity was investigated by complementary in vitro
and in silico biophysical studies. The compounds with alkyl chain lengths of four or six carbons
showed a low MIC50 against Escherichia coli and Bacillus subtilis. Our investigations into the
mechanism of their action revealed that phosphatidylethanolamine in the bacterial plasma
membrane plays an important role in their antibacterial activity.
Figure 1: Synthesis of the cationic azo compounds
[1] Banaszak-Leonard, E., Fayeulle, A., Franche, A. et al. J. Iran.Chem. Soc., 2021 [2] Salta, J.; Benhamou, I.; Herzog, I, Fridman M.. Chem. Eur. J. 2017, 23, 12724 – 12728

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
18
Paul Ehrlich Network Projects (PENP-9)
6-Methyl-7-aryl-7-deazapurine nucleosides as anti-trypanosoma cruzi agents: structure-activity relationship and in vivo efficacy
Cai Lin,a Ludmila Ferreira de Almeida Fiuza,b Camila Cardoso Santos,b Daniela Ferreira
Nunes,c Otacílio Cruz Moreira,c Jakob Bouton,a Izet Karalic,a Louis Maes,d Guy Caljon,d
Fabian Hulpia,a,e* Maria de Nazaré C. Soeiro,b* and Serge Van Calenbergha,*
a Laboratory for Medicinal Chemistry (Campus Heymans), Ghent University, Ottergemsesteenweg 460, B-9000, Gent, Belgium. b Laboratório de Biologia Celular, Instituto Oswaldo Cruz (FIOCRUZ), Fundação Oswaldo Cruz, Rio de Janeiro, Avenida Brasil 4365, Manguinhos, RJ, Brazil. c Plataforma de PCR em Tempo Real RPT09A-Laboratório de Biologia Molecular e Doenças Endêmicas, Instituto Oswaldo Cruz (FIOCRUZ), Fundação Oswaldo Cruz, Rio de Janeiro, Avenida Brasil 4365, Manguinhos, RJ, Brazil. d Laboratory of Microbiology, Parasitology and Hygiene, University of Antwerp, Universiteitsplein 1 (S7), B-2610, Wilrijk, Belgium. d Janssen Pharmaceutica NV Turnhoutseweg 30, 2340 Beerse, Belgium.
E-mail: [email protected]
Chagas disease is a tropical infectious disease resulting in progressive organ-damage and
currently lacks efficient treatment and vaccine options.[1] The causative pathogen, Trypanosoma
cruzi, requires uptake and processing of preformed purines from the host because it cannot
synthesize these de novo,[2] instigating the evaluation of modified purine nucleosides as potential
trypanocides. By modifying the pyrimidine part of a previously identified 7-aryl-7-deazapurine
nucleoside,[3] we found that substitution of a 6-methyl for a 6-amino group allows retaining T. cruzi
amastigote growth inhibitory activity but confers improved selectivity towards mammalian cells. By
keeping the 6-methyl group unaltered, and introducing different 7-aryl groups, we identified several
analogues with submicromolar antitrypanosomal activity. The 7-(4-chlorophenyl) analogue 14,
which was stable in microsomes, was evaluated in an acute mouse model. Oral administration of
25 mg/kg b.i.d. suppressed peak parasitemia and protected mice from infection-related mortality,
gave similar reductions as the reference drug of blood parasite loads determined by qPCR, but as
benznidazole failed to induce sterile cure in the short time period of drug exposure (5 days).
[1] M. De Rycker, B. Baragana, S. L. Duce, I. H. Gilbert, Nature 2018, 559, 498-506.
[2] M. Berg, P. Van der Veken, A. Goeminne, A. Haemers, K. Augustyns, Curr Med Chem 2010, 17, 2456-
2481.
[3] F. Hulpia, K. Van Hecke, C. Franca da Silva, D. da Gama Jaen Batista, L. Maes, G. Caljon, C. S. M. de
Nazare, S. Van Calenbergh, J Med Chem 2018, 61, 9287-9300.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
19
Paul Ehrlich Network Projects (PENP-10)
Chimeric small molecules in the search for novel anti-trypanosomatid agents
Elisa Uliassi,a J. Carlos Menéndez,b,* and Maria Laura Bolognesia,*
aDepartment of Pharmacy and Biotechnology, Alma Mater Studiorum – University of Bologna, Via Belmeloro 6, 40126 Bologna, Italy.
bUnidad de Química Orgánica y Farmacéutica, Departamento de Química en Ciencias Farmacéuticas, Facultad de Farmacia, Universidad Complutense, 28040 Madrid, Spain.
E-mail: [email protected]
Trypanosomatid infections (Leishmania spp. and Trypanosoma spp.) are a major obstacle to global
health and economic development. Current pharmacotherapy is unsatisfactory; hence, new drugs
are urgently required.
A collaborative project between the University of Bologna and Universidad Complutense was set
with the aim of developing new chimeric small molecules. These heterobifunctional compounds,
consisting of two protein-binding moieties, have demonstrated potential to tackle many questions in
biomedical research, as well as translation potential [1]. In this project, bivalency has been
exploited for a polypharmacological modality and a targeted delivery in the field of Trypanosomatid
diseases. First, we developed a series of chimera by combining the naphthoquinone and coumarin
frameworks to obtain dual inhibitors of glyceraldehyde-3-phosphate dehydrogenase/trypanothione
reductase with promising anti-trypanosomal profile [2]. A second series of chimera was based on
the privileged anti-Leishmania quinoline scaffold, which was linked to different polyamines at C-4,
with the objective of targeting parasite mitochondrion [3]. Further decoration of the quinoline
scaffold considered the introduction at C-2 of a styryl. Evaluation of the anti-Leishmania profile in
promastigote and amastigote forms revealed the mitochondrial involvement in the leishmanicidal
mechanism of action.
[1] Rossi, M.; Bolognesi, M. L. La Chimica & L'Industria 2019, 3, 26-30.
[2] Uliassi, E.; Fiorani, G.; Krauth-Siegel, R. L.; Bergamini, C.; Fato, R.; Bianchini, G.; Menéndez, J.C.;
Molina, M. T.; López-Montero, E.; Falchi, F.; Cavalli, A.; Gul, S.; Kuzikov, M.; Ellinger, B.; Witt, G.; Moraes,
C. B.; Freitas-Junior, L. H.; Borsari, C.; Costi, M. P.; Bolognesi, M. L. European Journal of Medicinal
Chemistry 2017, 141, 138-148.
[3] Staderini, M.; Piquero, M.; Abengózar, M.Á.; Nacher-Vazquez, M.; Romanelli, G.; Lopez-Alvarado, P.; Rivas, L.; Bolognesi, M.L.; Menéndez, J.C. European Journal of Medicinal Chemistry 2019, 171, 38-53.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
20
Paul Ehrlich Euro-PhD Awards (PEEPA)

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
21
Paul Ehrlich Euro-PhD Awards (PEEPA-1)
Molecular modeling studies on antiviral targets: drug resistance mechanisms and rational drug design
Francesca Alessandra Ambrosio,a Anna Artese,a,b and Stefano Alcaroa,b
a Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Campus “S. Venuta”, Viale Europa, 88100-Catanzaro, Italy.
b Net4Science Academic Spin-Off, Università “Magna Græcia” di Catanzaro, Campus “S. Venuta”, Viale Europa, 88100 Catanzaro, Italy.
E-mail: [email protected]
HIV-1 reverse transcriptase (RT) and integrase (IN) are essential enzymes for the virus replication,
since the first catalyzes the conversion of virus single-stranded RNA genome to double stranded
DNA and the second the integration of the viral DNA into the host cell-genome. In order to
elucidate the role of the novel IN mutation N155H alone and in presence of a secondary mutations
pathway, associated to Dolutegravir resistance, Induced Fit Docking and Molecular Dynamics
simulations were performed to rationalize the drug resistance profile associated to Dolutegravir in
the presence of the analyzed mutations [1]. By a combined shape- and structure-based virtual
screening approach, a new series of lead compounds towards HIV-1 RT were identified. In
particular, an indoline pyrrolidine, an indonyl piperazine and an indolyl indolinone derivatives were
identified as novel non-nucleoside RT inhibitors in the low micromolar range [2]. With the aim to
exploit a coumarin-based scaffold acting as inhibitor of IN and RT-associated Ribonuclease H
(RNase-H), modeling studies were applied to evaluate the theoretical binding affinity of the
synthesized compounds on both HIV-1 IN and RT RNase-H active sites. The computational results
were confirmed by biological assays.
Figure 1: 3D representation of HIV-1 A) Integrase and B) Reverse Transcriptase enzymes.
[1] Malet, I.; Ambrosio, F.A.; Subra, F.; Herrmann, B.; Leh, H.; Bouger, M.C.; Artese, A.; Katlama, C.; Talarico, C.; Romeo, I.; Alcaro, S.; Costa, G.; Deprez, E.; Calvez, V.; Marcelin, A.G.; Delelis,O., Journal of Antimicrobial Chemotherapy, 2018, 73, 1158-1166. [2] Costa, G.; Rocca, R.; Corona, A.; Grandi, N.; Moraca, F.; Romeo, I.; Talarico, C.; Gagliardi, M.G.; Ambrosio, F.A.; Ortuso, F.; Alcaro, S.; Distinto, S.; Maccioni, E.; Tramontano, E.; Artese, A., European Journal of Medicinal Chemistry, 2019, 161, 1-10. [3] Esposito F.; Ambrosio, F.A.; Maleddu, R.; Costa, G.; Rocca, R.; Maccioni, E.; Catalano, R.; Romeo, I.; Eleftheriou, P.; Karia, D.C.; Tsirides, P.; Godvani, N.; Pandya, H.; Corona A.; Alcaro, S.; Artese, A.; Geronikaki, A.; Tramontano, E., European Journal of Medicinal Chemistry, 2019, 182: 111617.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
22
Paul Ehrlich Euro-PhD Awards (PEEPA-2)
Derivatives of Pyrazinecarboxylic Acid as Potential Antimycobacterial Active Drugs
Ghada Bouz, Martin Doležal, and Jan Zitko
Department of Pharmaceutical Chemistry and Pharmaceutical Analysis, Charles University, Akademika Heyrovského 1203, Hradec Králové, Czech Republic
E-mail: [email protected]
Despite the established treatments, tuberculosis remains an alarming threat to public health
according to WHO [1]. Novel agents are needed to overcome the increasing rates of resistance
and perhaps achieve eradication. As part of our long-term research on pyrazine derivatives, we
focused during my PhD study on the design, synthesis, and biological evaluation of compounds
belonging to six structural types: 3-(phenylcarbamoyl)pyrazine-2-carboxylic acids 1, 3-
aminopyrazine-2-carboxamides 2, ureidopyrazines 3, N-(pyrazin-2-yl)benzenesulfonamides 4, N-
substituted quinoxaline-2-carboxamides 5, and hybrid compounds combining pyrazinamide and 4-
aminosalicylic acid 6. All prepared compounds were screened in vitro against six mycobacterial
strains (with the main focus on the virulent Mtb H37Rv), eight bacterial strains, and eight fungal
stems, along with in vitro cytotoxicity evaluation in HepG2 liver cancer cells. Additional testings
such as in vivo activity and toxicity screenings, stability experiments, and mechanism of action
determination (computational and experimetal) were performed for the most promising compounds.
General structural types and the best in vitro Mtb actitivies (expressed as MIC) for each structural
group are presented in Figure 1. Most important findings along with structure-activity-relationsinps
will be discussed during the presentation.
Figure 1: The general structures and best in vitro activities against Mtb H37 expressed as MIC.
[1] World Health Organization, Global Tuberculosis, Report 2020. www.who.int/tb/publications/global_report/en/ accessed: 04.01.2021

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
23
Paul Ehrlich Euro-PhD Awards (PEEPA-3)
Au and Ag NHC-metal complexes as effective multi-target agents in breast cancer treatment
Jessica Ceramella,a Domenico Iacopetta,a Annaluisa Mariconda,b Camillo Rosano,c Marco Sirignano,d Carmela Saturnino,b Pasquale Longo,d and Maria Stefania Sinicropia
a Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende (CS), Italy. b Department of Science, University of Basilicata, Viale dell’Ateneo Lucano 10, 85100 Potenza, Italy.
c Proteomics and Mass Spectrometry Unit, IRCCS Policlinico San Martino, L. go Rosanna Benzi 10, 16132, Genova, Italy.
d Department of Biology and Chemistry, University of Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano, Italy.
E-mail: [email protected]
Recently, the use of N-heterocyclic carbenes (NHCs), as ligands for the design of new metal
complexes, represents an expanding field due to the wide applications in medicinal chemistry.
Indeed, silver complexes are promising antimicrobial agents, while the gold ones have been
proved as effective anticancer drugs [1].
Our recent results reported the good anticancer activity of some silver and gold complexes with
NHC ligands, mostly towards breast cancer cell lines [2,3]. Considering these evidences, new Au
and Ag NHC complexes were designed and synthesized in order to improve their
pharmacokinetics and pharmacodynamics properties. These complexes exerted an interesting
anticancer activity towards the breast cancer MDA-MB-231 and MCF-7 cell lines. In addition, in
silico and in vitro studies demonstrated that they target the human topoisomerases I and II and
actin polymerization and depolymerization reactions, leading to cancer cells death by apoptosis.
Our outcomes highlight the multi-target activity of these complexes, a desired feature in the fight
against cancer.
Figure 1. Multi-target action of Au and Ag NHC complexes.
[1] Patil, S.A.; Hoagland, A.P.; Patil, S.A.; Bugarin, A., Future Med Chem 2020, 12, 2239-2275. [2] Iacopetta, D.; Mariconda, A.; Saturnino, C.; Caruso, A.; Palma, G.; Ceramella, J.; Muià, N.; Perri, M.; Sinicropi, M.S.; Caroleo, M.C.; Longo, P., ChemMedChem 2017, 12, 2054-2065. [3] Iacopetta, D.; Rosano, C.; Sirignano, M.; Mariconda, A.; Ceramella, J.; Ponassi, M.; Saturnino, C.; Sinicropi, M.S.; Longo, P., Pharmaceuticals 2020, 13, 91.
Human Topoisomerase I
Actin polymerization (Au and Ag NHC)
and depolymerization (Ag NHC)
Human Topoisomerase II Multi-target action

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
24
Paul Ehrlich Euro-PhD Awards (PEEPA-4)
Development of new chemical entities based on natural scaffolds with therapeutic potential towards age-related disorders
Daniel Chavarria,a Carlos Fernandes,a Sofia Benfeito,a Pedro Soares a, Catia Soares,a Fernando Cagide,a Jorge Garrido,b Vera Silva,c Eva Gil-Martins,c Renata Silva,c Ophelie Da Silva,d Xavier Brazzolotto,d Florian Nachon,d José Dias,d Fernando Remião,c Paulo J.
Oliveira,e and Fernanda Borgesa
aCIQUP/Depart.of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007 Porto,
Portugal b CIQUP/Depart. of Chemical Engineering, School of Engineering (ISEP), Polytechnic of Porto, 4200-072
Porto, Portugal c UCIBIO-REQUIMTE, Laboratory of Toxicology, Department of Biological Sciences, Faculty of Pharmacy,
University of Porto, 4050-313 Porto, Portugal d Département de Toxicologie et Risques Chimiques, Institut de Recherche Biomédicale des Armées,91223
Brétigny-sur-Orge, France e CNC—Center for Neuroscience and Cell Biology, University of Coimbra, UC Biotech, Biocant Park, 3060-
197 Cantanhede, Portugal.
E-mail: [email protected]
Neurodegenerative diseases represent a set of progressive and incurable age-related
disorders whose clinical symptoms are commonly associated with the decline of cognitive and/or
motor functions. The selective neuronal loss and consequent neurotransmitter depletion are
pathological hallmarks of NDs. Other factors, such as oxidative stress and mitochondrial
dysfunction, also contribute to neurodegeneration. Based on these findings, inhibitors of
cholinesterases (ChEs) and monoamine oxidases (MAOs) were developed to increase brain
acetylcholine and dopamine levels. However, current single-target treatments for NDs are only
palliative and fail to modify the disease progression [1].
Ferulic acid (FA), caffeic acid (CA) and piperine are considered privileged structures useful
for central nervous system (CNS) drug discovery programs due to their relevant antioxidant or
MAO inhibition properties [2, 3]. With the aim of developing new chemical entities able to target
oxidative stress and/or neurotransmitter depletion, libraries based on FA, CA and piperine were
developed [4-6]. From the data obtained so far we highlight: 1) the bioisosteric OH to SH
replacement on the FA scaffold as a strategy to improve its antioxidant profile [4]; 2) the
development of a small library of piperine derivatives as innovative hMAO-B inhibitors [5]; 3) the
development of innovative piperine- and CA-based mitochondria-targeted agents with antioxidant
activity and inhibitory activities towards ChEs and MAOs [6].
This project was supported by the Foundation for Science and Technology (FCT) and FEDER/COMPETE (Grants UIDB/00081/2020,
PTDC/MED-QUI/29164/2017, POCI-01-0145-FEDER-29164, PTDC/BIA-MOL/28607/2017, POCI-01-0145-FEDER-028607). D.
Chavarria, S. Benfeito and F. Cagide grants are supported by FCT and FEDER/COMPETE.
[1] M. G. Savelieff et al., Chem. Rev., 2019, 119 (2), 1221-1322. [2] S. Ojha et al., Drug Des. Devel. Ther., 2015, 9 5499-5510. [3] S. A. Lee et al., Chem. Pharm. Bull. (Tokyo), 2005, 53 (7), 832-835. [4] D. Chavarria et al., Org. Biomol. Chem., 2019, 17 (44), 9646-9654. [5] D. Chavarria et al., Eur. J. Med. Chem., 2020, 185 111770. [6] D. Chavarria et al., Antioxidants, 2021, 10 (2), 329.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
25
Paul Ehrlich Euro-PhD Awards (PEEPA-5)
Behind the allosteric inhibition of PTPRZ1, a current druggable phosphatase
Bruno Di Geronimo,a Miryam Pastor, Claire Coderch,b and Beatriz de Pascual-Teresa.b
aExperimental Therapeutics Programme, Spanish National Cancer Research Center (CNIO), Calle de Melchor Fernández Almagro, 3, 28029 Madrid, Spain.
bUniversidad San Pablo CEU, Division of Chemistry and Biochemistry, Calle de Julián Romea, 23, 28003 Madrid, Spain.
E-mail: [email protected]
Protein tyrosine phosphatases (PTP) are well-known phosphatases and interesting to investigate
as potential drug targets because of their relationship with several diseases such as diabetes,
obesity, cancer and neurological disorders1. Selective modulation of phosphatases is a challenging
objective due to the high sequence and structural homology. Moreover, potent inhibition of PTPs is
also a complex process due to its intrinsic activity. The allosteric inhibition has become an
interesting strategy to avoid multiple PTPs activity modulation as well as activity modulation.
Among all PTPs, we focused our attention on PTPRZ1 due to its interesting pharmacological
properties as it is implicated in the neurological protection against drug abuse as well as in cancer
disease. The most recent PTPRZ1 holo structure was deposited (PDB code 5H08)2 in a
“superopen” conformation within the ligand 7WL (Figure 1). This conformation was only previously
described in its close homologue PTPRG. By using both “superopen” conformations and molecular
modelling techniques we have explored the different plausible binding mode of our published and
potent molecule 10a3. In this work, we highlighted that presumably our molecules are able to
interact in an allosteric binding mode inside PTPRZ1, occupying the hydrophobic pocket and
interacting thought the arc of the WPD-loop.
Figure 1: 2D structure of 7WL and 10a showing in red the similar moieties. Superimposition of the crystal structure 5H08 with 7WL (green) and the docking result of 10a (blue).
[1] Barr, A. J. Protein Tyrosine Phosphatases as Drug Targets: Strategies and Challenges of Inhibitor Development. Future Med Chem 2010, 2 (10), 1563–1576. [2] Fujikawa, A. Targeting PTPRZ Inhibits Stem Cell-Like Properties and Tumorigenicity in Glioblastoma Cells. Scientific Rep 2017, 7 (1), 5609. [3] Pastor, M. Development of Inhibitors of Receptor Protein Tyrosine Phosphatase Z1 (PTPRZ1) as Candidates for CNS Disorders. Eur J Med Chem 2017, 144, 318–329.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
26
Paul Ehrlich Euro-PhD Awards (PEEPA-6)
Strategies against chronic kidney disease: new modulators of PTP1B and ILK
Javier García-Marín,a Mercedes Griera ,b Sergio de Frutos,b Manuel Rodriguez-Puyol,b Ramón Alajarín,a Diego Rodriguez-Puyol,b Juan J. Vaqueroa
a Department of Organic and Inorganic Chemistry, University of Alcalá, Ctra. Colmenar Viejo, km. 9100, 28034-Alcalá de Henares, Spain.
b Department of System Biology, University of Alcalá, Ctra. Colmenar Viejo, km. 9100, 28034-Alcalá de Henares, Spain.
c Department of Medicine, University of Alcalá, Ctra. Colmenar Viejo, km. 9100, 28034-Alcalá de Henares, Spain.
E-mail: [email protected]
Chronic kidney disease (CKD) is the non–transmissible global cause of death that raised the most
within the past 20 years. It increases the risk of all–cause mortality and may progress to end–stage
of renal failure. There is no effective therapy for CKD and current approaches do not prevent its
progression.
The main cause of CKD is diabetes Mellitus type 2 (DM2), as such, new antidiabetic drugs with
improved profiles seems of a very interest to fight against this disease. In this regard, our research
group has identified a serie of pyrrolo[1,2-a]quinoxalines as potent inhibitors of Protein Tyrosine
Phosphatase 1B (PTP1B), a validated target against DM2 [1]. After the synthesis, molecular
modelling, in vitro and phenotypic assays have confirmed the utility of these compounds for further
development as promising drugs against PTP1B.
On the other hand, searching for a validated target for CKD is a need nowadays, because there is
no pharmacological therapy approved. In this context, we turn our attention in the Integrin Linked
Kinase (ILK), a protein which has been related with CKD [2]. After studying its protein-protein
interaction with α-parvin and thanks to computational approaches, a small set of peptides were
prepared as chemical probes to target this interaction. Its evaluation has showed that they bind to
ILK, partially altering its union with α-parvin with very interesting properties in phenotypic models,
thus opening new opportunities for drug development.
[1] García-Marín, J.; Griera, M.; Sánchez-Alonso, P.; Di Geronimo, B.; Mendicuti, F.; Rodríguez-Puyol, M.; Alajarín, R.; de Pascual-Teresa, B.; Vaquero, J. J.; Rodríguez-Puyol, D. ChemMedChem 2020, 15, 1788. [2] de Frutos, S; Luengo, A; Garcia-Jerez, A; Hatem-Vaquero, M; Griera, M; O'Valle, F;; Rodriguez-Puyol, D; Calleros, L. Biochim. Biophys. Acta, 2019, 1865(6):1284-1297.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
27
Paul Ehrlich Euro-PhD Awards (PEEPA-7)
Targeting carbonic anhydrases (CAs): rational design, synthesis, structural studies and biochemical evaluation
Francesca Mancuso,a Laura De Luca,a Andrea Angelib, Anna Di Fiore,c Giuseppina De Simone,c Milan Vrabel,d Clemente Capasso,e Claudiu T. Supuran, b and Rosaria Gitto,a
aChiBioFarAm Department, University of Messina, Viale Palatucci, I-98168, Messina, Italy. bNEUROFARBA Department, University of Florence, Via U. Shiff 6, I-50019, Florence, Italy.
cIstituto di Biostrutture e Bioimmagini, CNR, Via Mezzocannone, I-80134, Napoli, Italy. dIOCB, Czech Academy of Sciences, Flemingovo nám. 2, 16610, Prague, Czech Republic.
eDepartment of Biology, Agriculture and Food Sciences, CNR, I-80134 Napoli, Italy.
E-mail: [email protected]
Carbonic anhydrases (CAs, E.C. 4.2.1.1.) are ubiquitous enzymes catalyzing the reversible
hydration of CO2 to HCO3- and H+. This simple reaction controls pH homeostasis, transport of
CO2/HCO3-, respiration and a multitude of biosynthetic reactions in distinct organisms. Thereby,
several CAs became well-established targets for designing modulators endowed with therapeutic
application. Fifteen CAs belonging to α–class have been identified in humans (hCAs I–IV, VA and
VB, VI–XIV), some of them are implicated in a multitude of pathological processes, (epilepsy and
others neuropathic pain, glaucoma, obesity, cancer, etc.).
We addressed our interest toward classical and non-classical human CA inhibitors (hCAIs)
possessing aryl-sulfonamide and coumarin nucleus respectively. The designed compounds were
tested against selected hCAs, thus revealing that some compounds demonstrated nanomolar
affinity and high selectivity toward the druggable hCA VII, IX and XII. By means of structural and
computational studies we deciphered the binding pose of the most intriguing compounds within
hCA catalytic site [1-4].
Our attention has been also focused on the prokaryotic CAs as innovative target for the
development of novel chemotherapeutic agents able to overcome the global threat of antimicrobial
drug resistance [5-6]. By applying different drug design strategies, we designed and synthesized
potent and selective inhibitors against the α, β and γ classes of the pathogenic species Vibrio
Cholerae (VchCAα, β, γ). Interestingly, some of them combined high affinity toward VchCAs with
surprising selectivity over the human off–target isoforms. Thus, they could represent the starting
point for the identification of novel anti–infective agents characterized by a peculiar mechanism of
action.
[1] L. De Luca, F. Mancuso, S. Ferro, et al., Eur. J. Med. Chem. 2018, 143, 276–282. [2] M. Buemi, A. Di Fiore, L. De Luca, et al., Eur. J. Med. Chem. 2019, 163, 443-452. [3] F.Mancuso, A. Di Fiore, L. De Luca, et al., ACS Med. Chem. Lett. 2020, 11, 1000–1005. [4] F. Mancuso, L. De Luca, A. Angeli, et al., J. Enzyme Inhib. Med. Chem. 2020, 1442–1449. [5] R. Gitto, L. De Luca, F. Mancuso, et al., J. Enzyme Inhib. Med. Chem. 2019, 34, 1186-1192. [6] F.Mancuso, L. De Luca, A. Angeli, et al., ACS Med. Chem. Lett. 2020, 11, 2294-2299.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
28
Paul Ehrlich Euro-PhD Awards (PEEPA-8)
Mapping chromone-3-phenylcarboxamide pharmacophore: quid est veritas?
Francesco Mesitia, b, c, Alexandra Gasparc*, Daniel Chavarriac, Annalisa Marucaa, b, Roberta Roccab,e, Eva-Gil Martinsd, Sandra Barreirod, Renata Silvad, Carlos Fernandesc, Sheraz
Gulf,g, Oliver Keminerf,g, Stefano Alcaroa, and Fernanda Borgesc*.
a Dipartimento di “Scienze della Salute”, Università “Magna Græcia”, Campus Salvatore Venuta, 88100, Catanzaro, Italy.
b Net4Science srl, Università "Magna Græcia", Campus Salvatore Venuta, 88100, Catanzaro, Italy. c CIQUP/Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007,
Porto, Portugal. d UCIBIO-REQUIMTE, Laboratory of Toxicology, Department of Biological Sciences, Faculty of Pharmacy,
University of Porto, 4050-313, Porto, Portugal. e Dipartimento di Medicina Sperimentale e Clinica, Università “Magna Græcia” di Catanzaro, Campus “S.
Venuta”, Viale Europa, Loc. Germaneto, 88100, Catanzaro, Italy. f Fraunhofer Institute for Translational Medicine and Pharmacology, 22525 Hamburg, Germany.
g Fraunhofer Cluster of Excellence Immune-Mediated Diseases CIMD, Hamburg Site, 22525 Hamburg, Germany.
E-mail: [email protected]
Chromone-3-phenylcarboxamides (Crom-1 and Crom-2) are potent and selective inhibitors of
human monoamine oxidase B [1]. In the presented study, new derivatives aimed to map the
benzopyran chemical features were synthetized and screened against hMAO-A and hMAO-B. SAR
data supported by molecular docking studies provide a rationale for the contribution of the
heterocycle’s rigidity, carbonyl group and benzopyran heteroatom for enzymatic inhibition. Notably,
N-(3-chlorophenyl)-4H-thiochromone-3-carboxamide (31) (Figure 1) exhibited an improved
pharmacological profile when compared to Crom-1 and Crom-2, displaying potent and selective
hMAO-B inhibition (hMAO-B IC50=1.52±0.15 nM) with no obvious ADME-toxicity liability.
Additionally, for quinolone derivatives the tautomerism relevance on MAO-B inhibition was also
explored and the data showed that prototropic tautomerism markedly influences the biological
activity. Therefore, to understand the attained results the unequivocal characterization of the
quinolone tautomers was performed using 1H-15N HSQC and 1H-15N HMBC. Overall, our data
provided robust SAR and STR of benzopyran-based compounds concerning their bioactivity and
cytotoxicity.
Figure 1. Scaffold mapping and biological properties of compound 31.
[1]. Reis, J.; Cagide, F.; Chavarria, D.; Silva, T.; Fernandes, C.; Gaspar, A.; Uriarte, E.; Remiao, F.; Alcaro, S.; Ortuso, F.; Borges, F., Discovery of new chemical entities for old targets: insights on the lead optimization of chromone-based monoamine oxidase B (MAO-B) inhibitors. J. Med. Chem. 2016, 59, 5879-5893.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
29
Paul Ehrlich Euro-PhD Awards (PEEPA-9)
The search for novel histamine H3 receptor ligands in the group of piperazine derivatives
Katarzyna Szczepańska,a Kamil J. Kuder,a Tadeusz Karcz,a Steffen Pockes,b Magdalena Kotańska,c Bassem Sadek,d Sigurd Elz,b Holger Stark,e and Katarzyna Kieć-Kononowicza
a Department of Technology and Biotechnology of Drugs, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9, Kraków 30-688, Poland
b Institute of Pharmacy, Faculty of Chemistry and Pharmacy, University of Regensburg, Universitatsstraße 31, D-93053 Regensburg, Germany
c Department of Pharmacological Screening, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9, Kraków, 30-688, Poland
d Department of Pharmacology & Therapeutics, College of Medicine & Health Sciences, United Arab Emirates University, P.O. Box 17666, Al Ain, United Arab Emirate e Institute of Pharmaceutical and Medicinal Chemistry, Heinrich Heine University Düsseldorf, Universitaetsstr.
1, 40225 Duesseldorf, Germany
E-mail: [email protected]
Proteins belonging to G-protein coupled receptors (GPCR) superfamily are one of the most often
explored therapeutic target in drug design and discovery process. Undoubtedly, one of the GPCR
receptors – histamine H3 receptor (H3R) serves as an interesting research object. This relatively
new biological target was discovered in the early eighties of the last century and its importance in
the pathogenesis of central and peripheral nervous system diseases has not been fully explained
yet [1].
The overall aim of this work was to obtain a series of novel, active and selective, histamine H3R
ligands, whilst maintaining a favorable physicochemical properties and ADMET parameters.
As a result of this work, a total of eighty new piperazine derivatives were synthesized [2-4].
Structure–activity studies allowed for the establishment of the 4-pyridylpiperazine moiety as a new
bioisosteric piperidine replacement in H3R ligands. The results of the in vitro tests proved this
scaffold being a crucial element for high hH3R affinity. Global analysis of collected data referring to
influence of alkyl linker length, allowed for the selection of three methylene homologues, due to
their highest H3R affinity values among all described 4-pyridylpiperazine derivatives. Interestingly,
benzophenone derivative (KSK63) showed the highest affinity among all tested compounds (hH3R
Ki = 3.12 nM). Moreover, the most promising compounds exhibited anticonvulsant activity in the
maximal electroshock-induced seizure (MES) model in mice. Additional, pro-cognitive properties of
compound KSK19 were confirmed in the passive avoidance test. Consequently, KSK19 has been
chosen as a new lead structure and therefore proceeded to further studies, including its potential
anti-obesity activity. Animals fed with high-fat diet and treated with KSK19 showed significantly less
weight gain, in comparison with the control group.
We are pleased to acknowledge the generous support of the National Science Center, Poland granted on the basis of decision No.
2020/36/C/NZ7/00284.
[1] Szczepańska, K.; Kuder, K.J.; Kieć-Kononowicz, K. Curr Med Chem. 2018, 10, 279-290. [2] Szczepańska, K.; Karcz, T.; Mogilski, S.; et al. Eur J Med Chem. 2018, 152, 223-234. [3] Szczepańska, K.; Karcz, T.; Kotańska, M.; et al. Bioorg Med Chem. 2018, 26, 6056-6066. [4] Szczepańska, K.; Karcz, T.; Siwek, A.; et al. Bioorg Chem. 2019, 91, 103071.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
30
Paul Ehrlich Euro-PhD Awards (PEEPA-10)
Targeting protein-protein interactions for the treatment of tumors and neurodegenerative disorders.
Serena Vittorio,a Thierry Langer,b Ugo Perricone,c Salvador Ventura,d Rosaria Gitto,a and Laura De Lucaa
aDepartment of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, Viale Palatucci 13, I-98168, Messina, Italy. bDepartment of Pharmaceutical Chemistry, University of Vienna, Althanstrasse 14, 1090-Vienna, Austria. c Molecular Informatics Unit, Fondazione Ri.MED, Via Filippo Marini
14, 90138-Palermo, Italy. d Institut de Biotecnologia i de Biomedicina and Departament de Bioquímica i Biologia Molecular, Universitat Autònoma de Barcelona, 08193-Bellaterra, Barcelona, Spain
E-mail: [email protected]
Protein-Protein interactions (PPIs) are implicated in several biological processes such as cellular
growth, DNA replication, transcriptional activation, translation and transmembrane signal
transduction. PPIs are often dysregulated in diseases thus representing promising targets for
pharmacological intervention. Despite the hurdles associated with the modulation of PPIs by small
molecules, mainly due to the large and flat protein interfaces, research efforts led to the discovery
of low-molecular weight compounds able to inhibit PPIs [1]. During my PhD, my research was
focused on two different PPIs: the aggregation of α-synuclein (α-syn) involved in neurological
disorders, and MUC-CIN85 implicated in cancer progression.
α-syn is a presynaptic protein whose abnormal aggregation plays a central role in the
pathogenesis of Parkinson’s disease (PD). PD is the second most prevalent neurodegenerative
disorder characterized by the loss of dopaminergic neurons in the substantia nigra of the brain. To
date no effective therapies are available for the cure of PD and, therefore, there is an increasing
interest in the development of new therapeutic tools. In the last decades, the modulation of α-syn
aggregation proved to be a promising disease-modifying strategy for reducing or halting the
neurodegenerative process [2]. Within this context, the aim of my research was to identify new α-
syn aggregation inhibitors by using in silico approaches [3].
MUC1 is a transmembrane glycoprotein extensively glycosylated in physiological conditions and
under-glycosylated in tumors. The hypo-glycosylation increases the accessibility of the protein
backbone promoting the instauration of new protein-protein interactions that are characteristic of
cancer cells. CIN85 is a multifunctional adaptor protein that interacts with the tumor form of MUC1
inducing the formation of metastasis [4]. The scope of my research was to exploit computational
methods to gain useful structural insights for the design of MUC1-CIN85 PPI inhibitors [5, 6].
[1] Mabonga, L.; Kappo, A. P.; Biophys Rev 2019, 11, 559-581.
[2] Oliveri, V.; Eur J Med Chem 2019, 167, 10-36.
[3] Vittorio, S.; Adornato, I.; Gitto, R.; Peña-Díaz, S.; Ventura, S.; De Luca, L.; J Enzyme Inhib Med Chem
2021, 35, 1727-1735.
[4] Cascio, S.; Farkas, A. M.; Hughey, R. P.; Finn, O. J., Oncotarget 2013, 4, 1686-1697.
[5] Vittorio, S.; Seidel, T.; Garon, A.; Gitto, R.; Langer, T.; De Luca, L; Int J Mol Sci 2021, 22, 534.
[6] Gulotta, M.R.; Vittorio, S.; Gitto, R; Perricone, U.; De Luca, L; Int J Mol Sci 2021, 22, 2208.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
31
Flash PENP Communications (FC)

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
32
Flash PENP Communications (FC-1)
Investigating the AM-001 binding site to EPAC1 protein using co-solvent molecular dynamics
Marianna Bufano,a Frank Lezoualc’h,b Romano Silvestri,a and Antonio Colucciaa
a Department of Chemistry and Technologies of Drug, Sapienza University of Rome, Laboratory affiliated to Istituto Pasteur Italia – Fondazione Cenci Bolognetti, Piazzale Aldo Moro 5, I-00185 Roma, Italy
b Université de Toulouse - Paul Sabatier, 31432 Toulouse, Cedex 04, France
E-mail: [email protected]
The exchange proteins activated by cAMP (EPAC) are implicated in a large variety of physiological
processes and they are as considered promising targets for a wide range of therapeutic
applications [1]. EPAC1 is considered as a novel protein target for the treatment of various cardiac
diseases [2-3]. In that context, we recently characterized a selective EPAC1 antagonist named
AM-001[2]. This compound was featured by a non-competitive mechanism of action but the
localization of its allosteric site to EPAC1 structure has yet to be investigated. Therefore, we
performed cosolvent molecular dynamics to identify a suitable allosteric binding site. The CMD led
to the identification of a series of suitable pockets then studied by docking and classical molecular
dynamics. This approach led us to the identification of a suitable allosteric binding pocket for AM-
001. As a model validation, we also evaluated the binding poses of the available AM-001
analogues, with a different biological activity. Finally, the complex EPAC1 with AM-001 bound at
the putative allosteric site was further refined by molecular dynamics. The principal component
analysis led us to identify the protein motion that resulted in an inactive like conformation upon the
allosteric inhibitor binding (Figure 1).
Figure 1: Porcupine plot of the top two eigenvectors. Right panel: eigenvector 1, left panel: eigenvector 2. Epac1 is reported as tube: CNBD and DEP green; REM orange; CDC25-HD blue and RA red. AM-001 is reported as cyan stick. AM-001 is reported as cyan stick. The yellow and grey arrows attached to each α-carbon atoms indicate the direction of the movement; the size of each arrow shows the magnitude of the corresponding movement. [1] Robichaux, W.GA.; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919-1053 [2] Laudette, M.; A, Sainte-Marie, Y.; Solari, A.; Fazal, L.; Sicard P.; Silvestri, R.; Mialet-Perez, J.; Pons, S.; Ghaleh, B.; Blondeau, J.P.; Lezoualc'h, F. Identification of a pharmacological inhibitor of Epac1 that protects the heart against acute and chronic models of cardiac stress. Cardiovasc. Res. 2019, 115, 1766-1777 [3] Bouvet, M.; Lezoualc’h, F. The Epac1 Protein: Pharmacological Modulators, Cardiac Signalosome and
Pathophysiology. Cells. 2019, 8, 1543-1561.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
33
Flash PENP Communications (FC-2)
Delivery for infectious diseases
Valentina Del Genioa, Annarita Falangab, Massimiliano Galdieroc, Stefania Galdieroa
a Department of Pharmacy, University of Naples "Federico II", Via Domenico Montesano 49, 80131 Naples,
Italy. b Department of Agricultural Sciences, University of Naples Federico II, Via Università 100, 80055, Portici,
Italy. c Department of Experimental Medicine, Second University of Naples, Via de Crecchio 7, Naples 80138,
Italy.
E-mail: [email protected]
Nanotechnology is widely used to develop innovative carriers, which ultimately enable the
obtainment of targeted delivery systems of drugs with increased efficacy and reduced toxicity.
Nanotechnology will undoubtedly lead a breakthrough also in the biomedical field relative to
infectious diseases [1]. Enveloped viruses are characterized by the presence of one or more
glycoproteins on their external surface, which are involved in the infection process. In particular,
conformational changes of viral glycoproteins represent a crucial step that govern the fusion of viral
and cellular membranes in the entry of enveloped viruses [2,3]. Antiviral peptides are able to inhibit
viral attachment and penetration, competing for receptor sites or interfering with the numerous
conformational modifications of surface glycoproteins that are required for viral fusion. Peptides
mimicking domains of viral glycoproteins are apt to interfere with the fusion event, we have
developed a peptide sequence with a high potential to inhibit the entry of Herpes simplex virus type
1 and we developed a strategy, similarly to other viruses, to deliver these sequences directly to the
membrane through cholesterol conjugation in order to potently block fusion [4]. The peptide
conjugated to polyethylenglycol and cholesterol interacts with viral and cell membranes thanks to
the presence of cholesterol and blocks the conformational rearrangements of the glycoprotein B
[5,6]. Our results show that the most efficient nanosystem is able to self-assemble into looser
aggregates and bind extensively to membranes where fusion takes place increasing the local
peptide concentration.
[1] Chakravarty, Malobika, and Amisha Vora. Drug delivery and translational research. Nanotechnology-based antiviral therapeutics, 2020, 1-40. [2] Falanga, Annarita, et al. Protein and peptide letters. Membrane fusion and fission: enveloped viruses, 16.7 2009: 751-759. [3] Harrison, Stephen C. Virology. Viral membrane fusion, 2015, 479: 498-507. [4] Porotto, Matteo, et al. Journal of virology. Viral entry inhibitors targeted to the membrane site of action, 2010,84.13: 6760. [5] Pessi, Antonello. Journal of Peptide Science. Cholesterol‐conjugated peptide antivirals: a path to a rapid response to emerging viral diseases, 2015, 21.5: 379-386. [6] Augusto, Marcelo T., et al. Molecules. Antiviral lipopeptide-cell membrane interaction is influenced by PEG linker length, 2017, 22.7: 1190.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
34
Flash PENP Communications (FC-3)
Discovering selective Poly (ADP-ribose) Polymerase (PARP) Inhibitors to expand the precision medicine approach
Maria Giulia Nizi,a Mirko M. Maksimainen,b Sudarshan Murthy,b Serena Massari,a Albert Galera-Prat,b Sven T. Sowa,b Alaviuhkola, J.; b Lari Lehtiö,b and Oriana Tabarrinia
a Department of Pharmaceutical Sciences, University of Perugia, Via del Liceo,1, 06123, Italy
b Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7, 20230, Finland
E-mail: [email protected]
Among the most recent anti-cancer agents that reached the market there are the poly-(ADP-ribose) polymerases (PARPs) inhibitors (PARPIs). PARP is a class of 17 enzymes that post-translationally modify proteins. In particular, using nicotinamide adenine dinucleotide (NAD+) as a co-substrate, they catalyze the covalent attachment of ADP-ribose in form of monomer or linear and branched polymer (MARylation and PARylation, respectively) to the target protein, while releasing nicotinamide as a side product. Since 2014, four PARP inhibitors were approved as a treatment or maintenance therapy. Originally developed for the treatment of BRCA 1/2 mutated advanced ovarian cancer, where the synthetic lethality mechanism was generated in the cells, their potentiality in other cancers such as breast, prostate, pancreatic or lung is emerging, highlighting a wider therapeutic use [1]. The approved drugs act mainly on PARP1 and 2, but they also recognize other PARPs thus working as pan-inhibitors; it is not still clear if this property is desirable or not. In this context, our groups have been involved in recent years in the identification of new chemotypes as PARPIs selective for only one subfamily as valid approach to better understand their role and hopefully to identify new anti-cancer agents [2,3]. In particular, starting from two hits identified by screening commercial libraries against a large panel of PARPs, and through iterative medicinal chemistry cycles, we have developed a series of analogues that, based on a very minor structural modifications, are able to reach one or a narrow subgroup of PARPs. Co-crystallographic studies allowed to understand the key interactions responsible for their selective inhibition. The design, the biological and structural characterization will be the object of the presentation.
[1] Sachdev E, Tabatabai R, Roy V, Rimel BJ, Mita MM, Target Oncol 2019, 14, 657-679.
[2] Nkizinkiko Y, Desantis J, Koivunen J, Haikarainen T, Murthy S, Sancineto L, Massari S, Ianni F, Obaji E,
Loza MI, Pihlajaniemi T, Brea J, Tabarrini O, Lehtiö L.Sci. Rep. 2018, 8, 1680-1689.
[3] Murthy S, Desantis J, Verheugd P, Maksimainen MM, Venkannagari H, Massari S, Ashok Y, Obaji E,
Nkizinkinko Y, Lüscher B, Tabarrini O, Lehtiö L. Eur. J. Med. Chem. 2018, 156, 93-102.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
35
Flash PENP Communications (FC-4)
Inhibition of ZIKA virus replication by novel inhibitors of NS2B/NS3 complex
Michela Puxeddu,a Giuseppe La Regina,a Antonio Coluccia,a Marianna Nalli,a Jin-Ching Lee,b and Romano Silvestri.a
a Laboratory Affiliated to Institute Pasteur Italy − Cenci Bolognetti Foundation, Department of Drug Chemistry and Technologies, Sapienza University of Rome, I00185 Rome, Italy
b Department of Biotechnology, College of Life Science, College of Medicine, and Drug Development and Value Creation Research Center, Kaohsiung Medical University, Kaohsiung 807, Taiwan.
E-mail: [email protected]
Zika virus (ZIKV) is an RNA virus of the Flaviviridae family, which is responsible for a condition
known as Zika fever. While initially, the infection was endemic only in Africa and Asia, now it is
spread all over the world. ZIKV is a viral ailment transmitted by Aedes mosquitoes, mainly located
in the equatorial zone. The symptoms are generally mild. Nevertheless, during pregnancy this
infection can cause the birth of infants with microcephaly or other inborn malformation. The NS2B /
NS3 viral protease complex is implicated in virus replication and immune system escape, so that
there is growing interest in the design of new ZIKV inhibitors with this target. Preliminary studies
with molecular models provided insights into the molecular determinants responsible for its high
affinity toward the target enzyme. On these bases, we have designed and synthesized 14 new
potential allosteric inhibitors of the NS2B / NS3 complex, characterized by an indole with a benzyl
or benzoyl group in position 3 [1].
Two of these new compounds, showed strong activity in both enzymatic and cellular assays. As a
proof of concept, the most promising compound was evaluated in a mouse animal model. (Figure
1). Not only the compound has significantly reduced NS2B / NS3 synthesis and viral replication,
but also it prevented the mice from a life-threatening infection showing a powerful reduction of the
brain damages produced by the viral infection. These results pave the way to new ZIKV drug
candidates able to cross the blood-brain barrier to reach the neural cells.
Figure 1: Proposed binding mode and biological evaluation of the derivative.
[1] A. Coluccia et al. ACS Med. Chem. Lett. 2020, 11, 10, 1869–1874.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
36
Flash PENP Communications (FC-5)
The interaction between GAB2 with SH3-Domain of GBR2 as a new potential target in cancer therapy
Jessica Sebastiani,a* Antonio Coluccia,a Giuseppe La Regina, a Stefano Gianni,b Romano Silvestria
aLaboratory affiliated to Istituto Pasteur Italia — Fondazione Cenci Bolognetti, Dipartimento di Chimica e Tecnologie del Farmaco, Sapienza Università di Roma, Piazzale Aldo Moro 5, 00185, Rome, Italy
bIstituto Pasteur — Fondazione Cenci Bolognetti, Dipartimento di Scienze Biochimiche “A. Rossi Fanelli” and Istituto di Biologia e Patologia Molecolari del CNR, Sapienza Università di Roma, 00185 Rome, Italy
E-mail: [email protected]
Gab2 is a scaffolding protein that plays a key role in cellular proliferation, migration ad
differentiation. Indeed, it is overexpressed in many types of cancer’s tissues as breast gastric, lung,
and colorectal. The interaction between Gab2 and the C-terminal SH3 domain of Gbr2 represents
one of the first steps of activation of the Ras/Erk, an important proliferation-signaling pathway, thus
being a valuable potential anticancer drug target. Therefore, a virtual screening study allowed us to
identify seven potential inhibitor molecules that were tested through kinetic and equilibrium binding
experiments. From the results obtained, the only compound that showed inhibitory activity against
the above interaction is AN-465-J137-985 [1]. Molecular docking studies on Gbr’s SH3-domain of
this derivative provided insights into the molecular determinants responsible for its high affinity
toward the target. The superimposition of the AN-465-J137-985 proposed binding and Gab2 core
highlighted that three aromatic rings were superimposable with Pro512 and Val513 side chains,
besides the Arg515 backbone is similar to hydrophobic contact observed for the substrate (Figure
1). To further investigate the effect of AN-465-J137-985 we treated A549 and H1299, lung cancer
cell lines, with increasing doses of AN-465-J137-985. The compound has been shown to
significantly inhibit the growth of both cancer cell lines. In particular, it has lethal dose values of 50
(LD50) of about 5 and 7 µM for H1299 and A549, respectively. In this context, our studies will
focus on the synthesis of new Gab 2 inhibitors starting from lead compound AN-465-J137-985. We
will try to improve pharmacokinetic properties by trying to alternately insert groups of various
groups of various kinds on the aromatic rings.
Figure 1: (A) Binding mode for derivative AN-465-J137-985. (B) Superimposition of AN-465-J137-985 proposed binding and Gab2a core binding.
[1] Malagrinò, F. et al. Targeting the Interaction between the SH3 Domain of Grb2 and Gab2. Cells, 2020, 9, e2435.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
37
Flash PENP Communications (FC-6)
Development of innovative analytical tools to improve the safety of plant-based health products, application to the case of plants of the
genus Tinospora used in Laos and in Europe.
Kedmany Sisouklath
Faculty of Pharmacy, University of Paris, 4 avenue de l’Observatoire, 75006 Paris, France
E-mail: [email protected]
Tinospora crispa (T.c.) (Menispermaceae) is a medicinal plant widely used all over the world,
including Southeast Asia and French overseas territories. All parts of the plant, especially the
stems, are used to maintain good health and treat various diseases such as appetite disturbance,
fevers, malaria, some other tropical diseases, diabetes, hypertension (Ahmad, Jantan, and Bukhari
2016). However, several cases of toxic hepatitis have been attributed to the consumption of T.c.
stems as reported by the laboratory in references [2] and [3]. We hypothesized that toxicity could
be linked to the presence of furanoditerpenoid lactones, considering the close structural similarity
observed between borapetosides present in stems and furanoditerpenoids like 8-Epidiosbulbin E
acetate (EEA) and teucrin A isolated from Dioscorea bulbifera (Druckova and Marnett 2006; Lin et
al. 2016) and Teucrium chamaedrys (Stickel, Patsenker, and Schuppan 2005), respectively, both
responsible for human hepatotoxicity. As evidenced by analytical studies, the latter compounds
underwent cytochrome P450 metabolic activation of the furan ring leading to the formation of an
enedial moiety, whose quenching by GSH and/or protein induces direct or indirect toxicity toward
hepatocytes (Li, Peng, and Zheng n.d.). In order to elucidate relationships between the presence of
borapetosides in T.c. stems and observed hepatotoxicity, and more generally to assess the safety
of their consumption, several experiments have been carried out to date, including i) qualitative
and quantitative analyses of the furanoditerpenoids present in the stem of T.c. collected in Laos, ii)
chemical epoxidation reactions by dimethyldioxirane, and results will be presented herein.
[1] Ahmad, W.; Jantan, I.; Bukhari, S. N., Frontiers in pharmacology 2016, 7, 59
[2] Cachet, X.; Langrand, J.; Riffault-Valois, L.; Bouzidi, C.; Colas, C.; Dugay, A.; Michel, S.; Boucaud-Maitre,
D., Scientific reports 2018, 8(1), 1-11
[3] Druckova, A.; Marnett, L. J., Chemical research in toxicology 2006, 19(10), 1330-1340
[4] Langrand, J.; Regnault, H.; Cachet, X.; Bouzidi, C.; Villa, A. F.; Serfaty, L.;Garnier, R.; Michel,
S., Phytomedicine 2014, 21(8-9), 1120-1123.
[5] Li, H.; Peng, Y.; Zheng, J. Advances in Molecular Toxicology. Vol 10: Metabolic Activation and Toxicities
of Furanoterpenoids. Elsevier B.V, 2016, pp. 55-97
[6] Lin, D.; Li, W.; Peng, Y.; Jiang, C.; Xu, Y.; Gao, H.; Zheng, J., Chem Res Toxicol 2016, 29, 359−366
[7] Stickel, F.; Patsenker, E.; Schuppan, D., Journal of Hepatology 2005, 43, 901–910

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
38
Flash PENP Communications (FC-7)
Proteomic contribution to the omic path for the identification of novel drugs overcoming resistance in Leishmaniasis.
Lorenzo Tagliazucchi,a Greta Fiorini,a Ana Perea-Martínez,b Raquel García-Hernández,b
José Ignacio Manzano,b Francisco Gamarro,b Maria Paola Costi a
a Università di Modena e Reggio Emilia, Dipartimento di Scienze della Vita, b Instituto de Parasitología y Biomedicina “López-Neyra”, IPBLN-CSIC, Granada (Spain).
E-mail: [email protected]
As the world is now facing increasing treatment failure against leishmaniasis, deeper investigation
on the molecular mechanisms responsible for drug resistance and therapeutic failure is needed.
The currently few available drugs, such as Sb complexes, paromomycin and miltefosine, show
severe side effects and develop different resistances, so innovative molecules should be
considered [1]. The aim of our research in the frame of the Spanish Grant RTI2018-097210-B-100
(IPBLN-CSIC) is to study the modulation of infected human monocytes by L. infantum clinical
isolates from patients with therapeutic failure, using proteomics approaches that can be further
integrated with the transcriptomics studies to achieve information for novel drug discovery studies.
A THP-1 cell line was infected with different clinical isolates of L. infantum lines from therapeutic
failure patients with leishmaniasis. The samples were divided in two groups: a drug resistant group,
and a cluster of strains isolated from immunocompromised patients with therapeutic failure and
without drug resistance phenotype. Samples were digested and analyzed in a UHPLC-Orbitrap Q-
Ex™, then Progenesis QIP™ and Mascot Matrix™ were used for peptide quantification. A
differential analysis between non-infected THP-1 cells and the human cell line infected with heat
inactivated L. infantum promastigotes assessed a baseline reference. The comparison between
samples generated a list of differentially expressed proteins (DEPs). A network enrichment
analysis process was applied through different freely accessible bioinformatic tools (STRING,
Panther, Reactome, others). This work led to the identification of relevant biological process
associated with drug resistance/therapeutic failure mechanisms. A comparison between
proteomics and transcriptomics datasets identified at the IPBLN-CSIC, among other results,
evidenced two proteins/transcripts with the same expression trend: Transferrin receptor protein 1
(TRFC), involved in Fe2+ homeostasis, and Nucleoside diphosphate kinase (NDK3), responsible for
apoptotic process. Both processes are observed in Leishmania infection [2,3]. The expected
results is to reconcile the proteomic results with transcriptomic data achievements as founding
concepts to identify new protein targets involved in Leishmania drug resistance and therapeutic
failure mechanisms. We aim to set up drug design programs of new molecules and from
repurposing studies and to offer a drug combination therapy to avoid drug resistance and
therapeutic failure phenomena.
This work was supported by the Spanish Grant RTI2018-097210-B-100 (to Francisco Gamarro) and
agreement n° 603240 NMTrypI - New Medicine for Trypanosomatidic Infections (to M. Paola Costi).
[1] Ponte-Sucre, A. et al. 2017. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl Trop Dis 11,
e0006052. [2] Das, N.K., et al. 2009. Leishmania donovani depletes labile iron pool to exploit iron uptake capacity of macrophage for its
intracellular growth. Cellular Microbiology 11, 83–94. [3] Moreira, D.S. et al. 2016. Involvement of nucleoside diphosphate kinase b and
elongation factor 2 in Leishmania braziliensis antimony resistance phenotype. Parasites Vectors 9, 641.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
39
Poster Communications (PC)

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
40
Poster Communications (PC_1)
Synthesis and structure-activity relationship of matrix metalloproteinase-13 (MMP-13) inhibitors
Lourdes Acosta1, José María Zapico1, Irene Ortín1, Claire Coderch1, Miryam Pastor1, Laura Marquez1, Beatriz de Pascual-Teresa1, Ana María Ramos1
1Departamento de Química y Bioquímica, Facultad de Farmacia, Universidad San Pablo-CEU, CEU Universities, Urbanización Montepríncipe, 28925, Alcorcón, Madrid, Spain.
E-mail: [email protected]
MMP-13 belongs to the family of matrix metalloproteinases (MMPs) which are zinc-dependent
endopeptidases responsible for the degradation of the extracellular matrix.[1] MMP-13 participates
in type II collagen degradation.[2] Its deregulation is involved in the appearance of osteoarthritis
(OA),[3] a degenerative disease of the joint cartilage for which there are only symptomatic
treatments.[4] All above-mentioned support the use of MMP-13 inhibitors as novel therapies to
prevent the progression of this disease.
Members of MMP family mainly differ in the size of the hydrophobic S1´pocket. This pocket is
larger in MMP-13 than in other MMPs, thus it can be used as strategy for the development of
selective inhibitors of this enzyme. [2, 5]
In a previous study, we synthesized polybrominated benzotriazoles, which interact selectively with
the S1´pocket of MMP-13. [6] Pursuing an improvement in solubility and pharmacokinetic
properties, in the present work we have expanded this research to new compounds modifying
benzotriazole for phthalimido core. Moreover, a different pattern of bromine substitution was used
to evaluate its influence in the binding affinity to the hydrophobic pocket. Finally, the influence on
selectivity was assessed replacing the hydroxamic acid by a weaker ZBGs such as carboxylic acid.
All synthesized compounds were docked in MMP-13 and MMP-2 3D structures to predict and
explain the interactions established with the Ω loop.
Financial support from RTI2018-093539-B-I00 (MICIU/FEDER, UE) is kindly acknowledged. L.A is supported
by PEJ-2020-AI/BMD-17635 (CAM research support).
[1] Page-McCaw, A.; Ewald, A. J.; Werb, Z. Nature Reviews Molecular Cell Biology 2007, 8 (3), 221-233. [2] Xie, X.; Wan, R.; Liu, Z. ChemMedChem, 2017, 12, 1-13. [3] Roach, H.; Yamada, N.; Cheung, K.; Tilley, S.; Clarke, N.; Oreffo, R.; Kokubun, S.; Bronner, F. Arthritis & Rheumatology, 2005, 52, 3110-3124. [4] González-Rodríguez, M.; Fernández-Romero, A.; Rabasco, A. Journal of Drug Delivery Science Technology, 2017, 42, 94-106. [5] Verma, R.; Hansch, C. Bioorganic and Medicinal Chemistry, 2007, 15, 2223-2268. [6] Pastor, M.; Zapico, J.; Coderch, C.; Maslyk, M.; Panchuk, R.; de Pascual-Teresa, B.; Ramos, A. Organic and Biomolecular Chemistry, 2019, 17, 916-929.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
41
Poster Communications (PC_2) The influence of chitosan on in situ gelling blend powders for wound
healing
Chiara Amante,a,b Chiara De Soricellis,a,b Giovanni Falcone,a,b Paola Russo,a Rita Patrizia Aquino,a Pasquale Del Gaudioa
a Department of Pharmacy, University of Salerno, Fisciano, 84084-Salerno, Italy b PhD Program in Drug Discovery and Development, University of Salerno, Fisciano, 84084-Salerno, Italy
E-mai: [email protected]
The healing of the chronic wound can be altered by pathogens, therefore avoiding the infections of
the wound site is an essential requisite of innovative wound dressing [1]. In this work, the proposal
is the development of an innovative formulation based on natural polymer blend powders able to
absorb the excess of exudate and became gel directly on the wound site. Specifically,
alginate/pectin/chitosan particles loaded with doxycycline to enhance the antibacterial power of the
formulation have been manufactured. Particularly attention on the chitosan amount has been made
to evaluate its influence on the properties of the particles and drug release when used as a wound
care formulation. Doxycycline has been used as an antimicrobial model drug for its wide
antibacterial spectrum against Gram-positive and Gram-negative bacteria [2] and for its ability to
inhibit host matrix metalloproteinases (MMPs), aberrantly expressed in chronic wounds [3].
Powders, produced by spray drying, were analyzed in terms of yield, morphology, particle size, and
fluid uptake ability. The encapsulation process demonstrated good yield and encapsulation
efficiency (e.e.) depending on the relative amount of chitosan into the feed. Fluid uptake studies
showed that despite all the powders absorbed a great amount of simulated wound fluid in less than
5 minutes, the formulations with a higher concentration of chitosan were able to absorb a greater
quantity of it. Moreover, in vitro tests were conducted. The experiment of drug release in vitro
showed a burst effect in the first hours (4 h) followed by a prolonged release till 24h. The diffusion
test against Staphylococcus aureus showed an antimicrobial effect increased thanks to the
bacteriostatic effect of chitosan. Moreover, zymography studies have shown that doxycycline
loaded particles were able to increase drug activity against MMPs, with good activity against MMP-
9 even at 0.5 μg/mL over 72h. Such results suggest that spray drying technology has permitted the
production of in situ gelling powders that could be a promising wound dressing.
Figure 1. Representative SEM image of APC particles and zymograms of pure doxycycline and APC loaded with the drug.
[1] Gopinath, D., et al., Biomaterials, 2004. 25(10): p. 1911-7. [2] Golub, L.M., et al., 1995. 22(2): p. 100-109. [3] García, R.A., et al., Mol Pharmacol, 2005. 67(4): p. 1128-36.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
42
Poster Communications (PC_3)
UHPLC-HRMS analysis of non-psychoactive Cannabis sativa L. extracts and assessment of their in vitro antiproliferative activity
Lisa Anceschi,a Alessandro Codeluppi,a Virginia Brighenti,a Lorenzo Corsi,a Federica Pellatia
a Department of Life Sciences, University of Modena and Reggio Emilia, Via G. Campi 103-287,
41125 Modena, Italy.
E-mail: [email protected]
Cannabis sativa L. is an herbaceous plant belonging to the Cannabinaceae family. Cannabinoids
are mainly synthesized in glandular trichomes. Among them, the most representative compounds
are cannabidiolic acid (CBDA), ∆9-tetrahydrocannabinolic acid (∆9-THCA), and cannabigerolic acid
(CBGA). These native acidic cannabinoids undergo a spontaneous decarboxylation under the
action of light and heat, leading to the formation of their neutral counterparts. Fiber-type C. sativa
(hemp) is characterized by a high content of CBD and CBG, and a level of psychoactive ∆9-THC
lower than 0.2%. Recently, the interest in non-psychoactive C. sativa extracts is increased due to
many biological activities related to cannabinoids and other compounds [1-3].
This study was focused on the possible role and application of non-psychoactive Cannabis sativa
L. extracts as antiproliferative agents. The compounds present in different extracts from non-
psychoactive C. sativa varieties were characterized by means of ultra high-performance liquid
chromatography coupled with high-resolution mass spectrometry (UHPLC-HRMS) and the
complete quantitative analysis was performed using HPLC-UV, following a previously validated
method [4]. The antiproliferative activity of the extracts was assessed on human cancer cells of
endodermal (HT29), mesodermal (K562) and ectodermal (U87MG) origin. The K562 chronic
myelogenous leukemia cell line was the most sensitive to the treatment and the CBD-type extract
was the one that provided the lowest IC50 value. The cytofluorimetric analysis of K562 treated cells
revealed that the antiproliferative effect was mainly due to the induction of apoptosis. The
mechanism of cell death did not involve the main pro- and anti-apoptotic markers such as p53, Bcl-
2 and Bcl-xl. Moreover, caspase 3 and 7 seemed to be involved in the mechanism of apoptosis.
Finally, dose-response curves were built for the associations of the CBD-type extract at 5 µg/mL
and pure CBD at 5 µM with anticancer drugs currently used in therapy. The dose-response curves
did not show a significant decrease of the IC50 value for imatinib and doxorubicin both in
association with the extract and with pure CBD. Differently, vincristine associated with the CBD-
type extract showed a 10 times higher efficacy then the vincristine alone. Since the
pharmacological activity of vincristine is related to its microtubule-destabilizing properties, the
CBD-rich extract and pure CBD might act on the same target/s, thus enhancing the antiproliferative
activity of this anticancer drug.
[1] Pellati, F.; Borgonetti, V.; Brighenti, et al. Biomed. Res. Int., 2018, vol. 2018.
[2] Corsi, L.; Pellati, F.; Brighenti, V.; et al. Curr. Bioact. Compd., 2019, vol. 15, 201–210. [3] Iseppi, R.; Brighenti, V.; Licata, M.; et al. Molecules, 2019, vol. 24(12). [4] Brighenti, V.; Pellati, F.; Steinbach, M.; et al. J Pharm Biomed Anal., 2017, vol. 143, 228-236.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
43
Poster Communications (PC_4)
Design of a fully automated system for the synthesis of pharmacologically relevant building blocks exploiting immobilized
Rhodotorula Rubra and new solvent systems
Francesca Annunziata,a Alessandra Guaglio,a Paola Conti,a Raffaella Gandolfia and Lucia Tamborinia
a Department of Pharmaceutical Sciences, University of Milan, Via Mangiagalli 25, 20133 Milan, Italy
E-mail: [email protected]
Rhodotorula, a widespread yeast genus, can be easily found in soil, water, air, milk, and fruit juice.
In particular, R. rubra displayed the ability to selectively reduce prochiral ketones with an
outstanding enantiomeric excess [1,2].
The aim of our work was the exploitation of a bioreactor made with immobilized whole cells of R.
rubra to efficiently produce a key building block for the synthesis of the antidepressant drug
duloxetine, i.e., (S)-3-hydroxy-3-(thiophen-2-yl)propanenitrile, (S)-1 (Scheme 1). A choline chloride-
glucose natural deep eutectic solvent (NADES) was employed with a dual function, as a co-solvent
and as a source of glucose, fundamental for the cofactor regeneration. To develop a fully
automated protocol for the production of (S)-1, an in-line purification procedure has been designed.
Firstly, an in-line extraction of the desired product was performed with a flow stream of ethyl
acetate followed by a liquid/liquid separation. A polymer supported benzylamine packed in a glass
column and connected to the organic stream allowed the removal of the unreacted ketone
thereafter.
The optimized protocol allowed the obtainment of compound (S)-1 in only 60 minutes with >90%
conversion and >99% e.e. The protocol resulted to be versatile and was successfully used for the
enantioselective reduction of different β-ketonitriles.
Scheme 1: Schematic set-up for the flow synthesis of duloxetine intermediate (S)-1.
[1] Wirth F.; Goldani L.Z., Interdisciplinar perspectives on infectious diseases 2012, 2012, 465717. [2] Facchetti G.; Gandolfi R.; Fusè M.; Zerla D.; Cesarotti E.; Pellizzoni M.; Rimoldi I. New Journal of Chemistry 2015, 39, 3792.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
44
Poster Communications (PC_5)
Machine learning and essential oils: PDIA3 as a case study
Lorenzo Antonini,a Manuela Sabatino,a Filippo Sapienza,a Eleonora Proia,a Giuliano Paglia,b Margherita Eufemi,b Fabio Altieri,b Rino Ragnoa
a Department of Chemistry and Technology of Drugs, Sapienza University of Rome, Piazzale Aldo moro, 5, 00185, Rome (RM), Italy.
b Department of Biochemical Sciences A. Rossi Fanelli Sapienza University of Rome, Piazzale Aldo moro, 5, 00185, Rome (RM), Italy.
E-mail: [email protected]
In the last ten years, essential oils (EOs) have been widely investigated as antibacterial agents
and, more recently, also evaluated for their potential antiviral and anticancer activity [1, 2, 3].
Although EOs chemical composition is almost always known, a rigorous analysis on the
relationship between composition and assay response is often missing. Machine learning
techniques represent a powerful tool to elucidate the assay outcome, highlight patterns in the data,
and assign “importance” to EOs chemical components in relation to the observed biological activity
[2, 3]. In this work, 30 EOs extracted through steam distillation from Calamintha Glandulosa,
Foeniculum Vulgare, Melissa Altissima, and Ridolfia Segetum were tested to assess their inhibitory
effect on protein disulfide isomerase A3 (PDIA3) using a FITC-insulin assay. PDIA3 is an enzyme
that catalyzes disulfide bond formation through its thiol-oxidoreductase and protein disulfide
isomerase activities. The IC50 returned by the assays and EOs chemical compositions were used
to train machine learning (ML) binary classification (active/inactive) models. Robust statistical
models were obtained through extensive Bayesian hyperparameter optimization and using
dimensionality reduction methods. Algorithms used spanned from classical logistic regression,
support vector machines to decision trees, and gradient boosting. Model’s performances were
assessed using the Matthews correlation coefficient (MCC) metric in fitting and cross-validation
(20% out). The best model was used to perform a feature importance study by means of a model
agnostic method (Permutation Feature Importance) joint with a partial dependence analysis. This
approach allowed to identify the components having the highest impact on the experimentally
observed PDIA3 inhibition. Most promising compounds were selected and sent to biochemical
assay to confirm the predicted importance. This approach is a first attempt to exploit information
from EOs biochemical assays making them available for drug design purposes. It is also a first
attempt to democratize this fast-growing research field, proposing a workflow for results analysis.
[1] Di Martile, M., et al., Cancers 2020, 12, 2650. https://doi.org/10.3390/cancers12092650
[2] Sabatino, M.; et al.; Molecules 2020, 25, 2452. https://doi.org/10.3390/molecules25102452
[3] Papa, R.; et al, Int. J. Mol. Sci. 2020, 21, 9258. https://doi.org/10.3390/ijms21239258

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
45
Poster Communications (PC_6)
From Trabectedin to simplified hit-compounds: design and synthesis using multicomponent reactions
Andrea Bacci,a Samuele Masoni,a Marco De Martino,b Alfredo Fusco,b Eugenio Gaudio,c Antonio Lupia,d Stefano Alcaro,d Simona Rapposelli a
a Department of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy b Department of Molecular Medicine and Medical Biotechnology (DMMBM), University of Naples "Federico
II", Naples, Italy c DTI-Tech, Via Mesolcina 3A, 6500 Bellinzona, Switzerland
d Net4Science Academic Spin-Off, Università "Magna Græcia" di Catanzaro, Campus "S. Venuta", Viale Europa, 88100 Catanzaro, Italy
E-mail: [email protected]
Trabectedin (ET-743), a natural drug approved in 2007 by EMA as potent anticancer agent,
represents a good starting point for the creation of simplified hit-compounds library [1]. With the
aim to search new chemical scaffolds of ET-743, a shape-based screening has been carried out
and let us to identify three different series of hit compounds (series I-III). Multicomponent reactions
(MCRs) are powerful transformations that merge portions of three or more starting materials into a
new compound in a one-pot procedure. In particular, isocyanide-based multicomponent reactions
(IMCRs) are easily performed using available starting materials and tolerate a variety range of
functional groups [2]. In addition, the combination of IMCRs with microwave-assisted reactions
(MAOS) represents a sustainable synthetic approach to the discovery of new molecules with a high
grade of structural diversity [3]. On this basis, we performed the synthesis of different prototypes of
original scaffolds identified through computational studies, by using IMCRs and MAOS. (Fig.1).
Herein we present the design and synthesis of small libraries of hit-compounds carrying the main
structural features of trabectedin. Moreover, preliminary results from in vitro anticancer activity of
synthesized compounds will be discussed.
Figure 1: The representation of the hit compounds designed.
[1] Assi, T., et al., Cancer Treat. Rev., 2019, 72: 37-44. [2] Dömling, A., Ugi I., Angew. Chem. Int. Ed. Engl. 2000, 39(18): 3168-3210.
[3] Fairoosa, J., et al., ChemistrySelect, 2020, 5(17): 5180-5197.
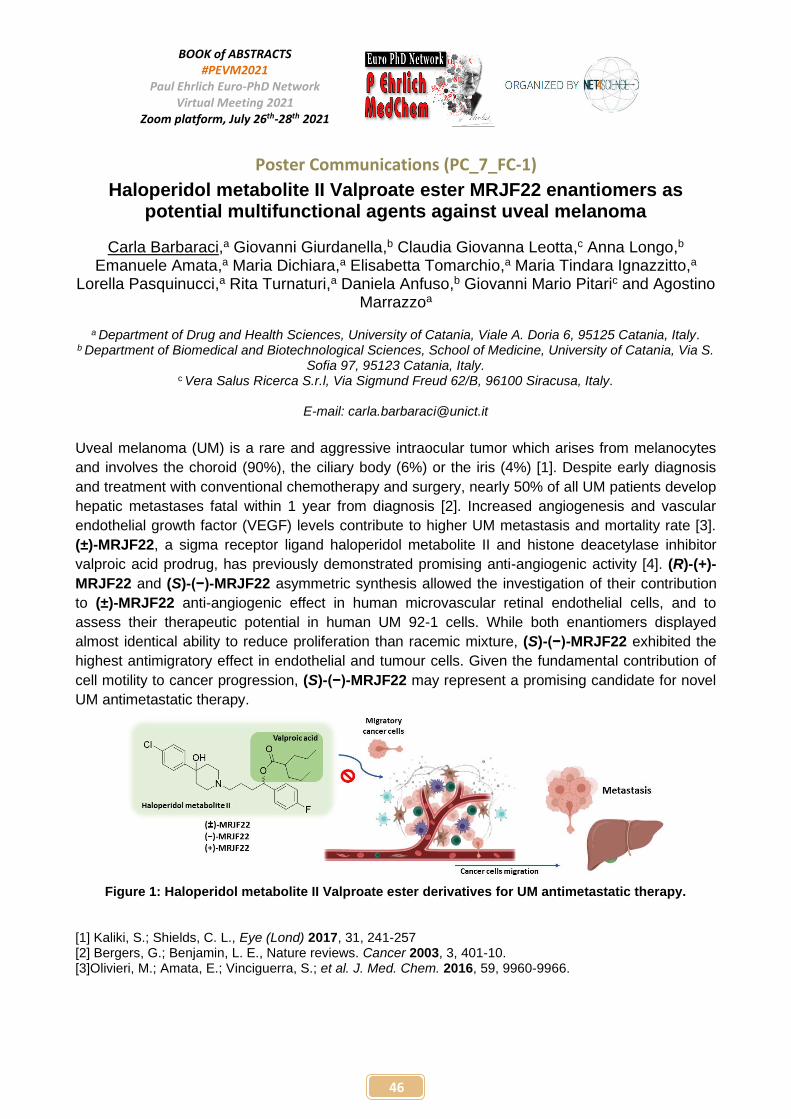
BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
46
Poster Communications (PC_7_FC-1)
Haloperidol metabolite II Valproate ester MRJF22 enantiomers as potential multifunctional agents against uveal melanoma
Carla Barbaraci,a Giovanni Giurdanella,b Claudia Giovanna Leotta,c Anna Longo,b Emanuele Amata,a Maria Dichiara,a Elisabetta Tomarchio,a Maria Tindara Ignazzitto,a
Lorella Pasquinucci,a Rita Turnaturi,a Daniela Anfuso,b Giovanni Mario Pitaric and Agostino Marrazzoa
a Department of Drug and Health Sciences, University of Catania, Viale A. Doria 6, 95125 Catania, Italy. b Department of Biomedical and Biotechnological Sciences, School of Medicine, University of Catania, Via S.
Sofia 97, 95123 Catania, Italy. c Vera Salus Ricerca S.r.l, Via Sigmund Freud 62/B, 96100 Siracusa, Italy.
E-mail: [email protected]
Uveal melanoma (UM) is a rare and aggressive intraocular tumor which arises from melanocytes
and involves the choroid (90%), the ciliary body (6%) or the iris (4%) [1]. Despite early diagnosis
and treatment with conventional chemotherapy and surgery, nearly 50% of all UM patients develop
hepatic metastases fatal within 1 year from diagnosis [2]. Increased angiogenesis and vascular
endothelial growth factor (VEGF) levels contribute to higher UM metastasis and mortality rate [3].
(±)-MRJF22, a sigma receptor ligand haloperidol metabolite II and histone deacetylase inhibitor
valproic acid prodrug, has previously demonstrated promising anti-angiogenic activity [4]. (R)-(+)-
MRJF22 and (S)-(−)-MRJF22 asymmetric synthesis allowed the investigation of their contribution
to (±)-MRJF22 anti-angiogenic effect in human microvascular retinal endothelial cells, and to
assess their therapeutic potential in human UM 92-1 cells. While both enantiomers displayed
almost identical ability to reduce proliferation than racemic mixture, (S)-(−)-MRJF22 exhibited the
highest antimigratory effect in endothelial and tumour cells. Given the fundamental contribution of
cell motility to cancer progression, (S)-(−)-MRJF22 may represent a promising candidate for novel
UM antimetastatic therapy.
Figure 1: Haloperidol metabolite II Valproate ester derivatives for UM antimetastatic therapy.
[1] Kaliki, S.; Shields, C. L., Eye (Lond) 2017, 31, 241-257 [2] Bergers, G.; Benjamin, L. E., Nature reviews. Cancer 2003, 3, 401-10. [3]Olivieri, M.; Amata, E.; Vinciguerra, S.; et al. J. Med. Chem. 2016, 59, 9960-9966.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
47
Poster Communications (PC_8)
Phenylpropiolic acid derivatives as promising anticancer agents
Alexia Barbarossa,a Alessia Carocci,a Carlo Franchini,a Mariangela Marrelli,b Valentina Amodeo,b Filomena Conforti,b Jessica Ceramellab, Domenico Iacopettab, Maria Stefania
Sinicropib
a Department of Pharmacy-Drug Sciences, University of Bari “Aldo Moro” Via Edoardo Orabona, 4, 70126, Bari (BA), Italy.
b Department of Pharmacy Health and Nutritional Sciences, via Pietro Bucci, 87036 Cosenza, Italy.
E-mail: [email protected]
The indole scaffold has been recognized, over the years, as a model for the synthesis of
compounds with anticancer activity by dint of its substantiated ability to act via multiple
mechanisms, which also involve the inhibition of enzymes engaged in DNA replication [1]. In this
regard, in our previous work [2], a series of indole derivatives (I) showed a good antitumor activity.
The lead of the series proved to be a phenylpropiolic ester bearing an indole moiety (1), that
exhibited anticancer activity toward HeLa and the MCF-7 cell lines, with no effects on the viability
of normal cells. This compound was able to induce apoptosis by targeting tubulin and to reduce the
menadione-induced ROS production in 3T3-L1 cells, as confirmed by in vitro and in silico studies.
Basing on these results, to investigate the importance of the indole core, we designed and
synthesized a new series of compounds by substituting the indole nucleus with bioisosteric cores
(I). Furthermore, we explored the possibility to enlarge the biological assays by testing these
compounds on a broader panel of cell lines, including melanoma. As photochemotherapy is one of
the most interesting therapeutic approaches for the treatment of melanoma [3], the photocytotoxic
potential of the synthesized molecules on UVA-irradiated A2058 melanoma cells have been
investigated. Tested compounds were able to affect cell viability in a concentration-dependent
manner, after irradiation at 365 nm for 1h at a dose of 1.08 J/cm2. Herein the results of this study
will be reported.
XOAr
O
ONH
1 I
Figure 1: Phenylpropiolic acid derivatives.
[1] Dadashpour, S., & Emami, S., European journal of medicinal chemistry 2018, 150, 9-29. [2] Iacopetta, D., Catalano, A., Ceramella, J., Barbarossa, A., Carocci, A., Fazio, A., La Torre, C., Caruso, A., Ponassi, M., Rosano, C., Franchini, C., Sinicropi, M. S. Bioorganic Chemistry 2020, 105, 104440. [3] Marrelli, M., Perri, M. R., Amodeo, V., Giordano, F., Statti, G. A., Panno, M. L., Conforti, F. Plants, 2021,10, 123.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
48
Poster Communications (PC_9)
Catechol derivatives as inhibitors of Aβ aggregation: investigating the role of vicinal hydroxy substituents
Filippo Basagni,a Marina Naldi,a Tiziana Ginex,b F. Javier Luque,b Matteo Iurlo,c Massimo Marcaccio,c Anna Minarini,a Manuela Bartolinia and Michela Rosinia
a Department of Pharmacy and Biotechnology, University of Bologna, via Belmeloro 6, 40126-Bologna (IT). b Department of Nutrition, Food Science, and Gastronomy, University of Barcelona, 08921-Santa Coloma de
Gramenet (ES). c Department of Chemistry “Giacomo Ciamician”, University of Bologna, via Selmi 2, 40126-Bologna (IT).
E-mail: [email protected]
The amyloidogenic pathway plays a leading role in the etiopathogenesis of Alzheimer’s disease (AD). Amyloid oligomerization and aggregation represent the triggering step of β-amyloid (Aβ) neurotoxicity, by leading to neurotoxic aggregates formation. Recent findings have highlighted that Aβ soluble oligomers (Aβos) instead of large aggregates are the major culprits of synaptotoxicity, thus underlining the relevance of early inhibiting the aggregation process [1]. Natural polyphenols have been shown to interfere with the protein misfolding cascade at multiple levels [2]. On this basis, we had previously synthesized a series of hydroxycinnamic derivatives, in which the catechol fragment offered a peculiar “on−off” pattern of control of the antiaggregating properties [3]. Herein, to clarify the pivotal role of the catechol feature, we selected the most active antiaggregating compound 1 and synthesized a small set of isomers. Mass spectrometry analysis and thioflavin T-based assay were carried out to check the impact of these structural modifications on the antiaggregating profile of the new analogues. Then, electrochemical analyses were performed to verify whether a correlation exists between oxidative and antiaggregating properties, whereas mass spectrometry studies and quantum mechanical calculations were devoted to the investigation of a putative covalent interaction with Aβ42 as the triggering antiaggregating effect. Synthesized polyphenols demonstrated to exert antiaggregating activities with different mechanisms of action depending on their proelectrophilic character, which arose as essential requirement for activity.
Figure 1. Compound 1 as starting point to investigate antiaggregating mechanisms of catechol
derivatives.
[1] Laurén, J.; Gimbel, D. A.; Nygaard, H. B.; Gilbert, J. W.; Strittmatter, S. M., Nature 2009, 457 (7233), 1128-32. [2] Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.; Masuda, Y.; Takegoshi, K.; Irie, K., Journal of Biological Chemistry 2013, 288 (32), 23212-24. [3] Simoni, E.; Serafini, M.M; Caporaso, R.; Marchetti, C.; Racchi, M.; Minarini, A.; Bartolini, M.; Lanni, C.; Rosini, M., ACS Chemical Neuroscience 2017, 8 (7), 1618-1627.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
49
Poster Communications (PC_10)
Enhancement of ancient Pollino grains with promising nutraceutical properties
Giovanna Basile,a Andrea Castagnello a, Jessica Ceramella a, Domenico Iacopetta a, Francesco Patitucci a, Francesco Puoci a, Maria Stefania Sinicropi a
a Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Arcavacata di Rende (CS).
E-mail: [email protected]
Wheat is the main staple in many diets. Several lines of research have attributed to cereals, in the
context of a balanced diet, a protective function for human health. The phenolic compounds and
others numerous bioactive compounds contained in cereals, indeed, providing health benefits,
and are associated with a reduced risk of chronic diseases and are known for their antiallergic,
antiatherogenic, anti-inflammatory, antimicrobial, antioxidant, antithrombotic, cardioprotective and
vasodilator effects [1,2]. The purpose of this study was the evaluation of the nutraceutical
properties of three varieties of ancient wheat (Maiorca, Carosella and Senatore Cappelli) cultived
close to the “Piana di Sibari”: Cerchiara of Calabria. Their antioxidant activity was evaluated by
comparing it with that of two other commercial varieties. The obtained results are supported the
importances of this ancient wheats as functional foods and encouraged to carry out further
researches regarding the evaluation of their potential anti-inflammatory activity.
[1] Dinu, Whittaker, Pagliai, Benedettelli, & Sofia, Journal of Nutritional Biochemistry 2018 [2] Lilei Yu , Anne-Laure Nanguet and Trust Beta Antioxidants 2013

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
50
Poster Communications (PC_11)
EIF2A as protein target of cannabidiolic acid in glioblastoma cancer
Maria Laura Bellone,a,d Giovanni Appendino,c Nunziatina De Tommasi,a Federica Pollastro,c Fabrizio Dal Piaz b
a Department of Pharmacy, University of Salerno, Via Giovanni Paolo II, 132, 84084, Fisciano, Italy b Department of Medicine and Surgery, University of Salerno, Via Giovanni Paolo II, 132, 84084, Fisciano,
Italy c Department of Pharmaceutical Sciences, University of Eastern Piedmont, Largo Donegani 2/3, 28100,
Novara, Italy d Phd program in Drug Discovery and Development, Department of Pharmacy, University of Salerno, Via
Giovanni Paolo II, 132, 84084, Fisciano, Italy.
E-mail: [email protected]
Phytocannabinoids, the major secondary metabolites of cannabis plants [1], have been shown to
exert a wide diversity of biological activities by targeting different macromolecules. The most
common phytocannabinoids are delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD). Anti-
inflammatory and anti-cancer activities were demonstrated for these both compounds. Based on
these evidences, the present study was focused on the characterization of the biological activity
and the identification of the putative target(s) of cannabidiolic acid (CBDA), cannabigerolic acid
(CBGA) and cannabidivarinic acid (CBDVA) in U87MG, glioblastoma cell lines. Therefore, the
cytotoxicity revealed that CBDA, CBGA and CBDVA were able to prevent cell proliferation only
when low concentrations of fetal bovine serum (FBS) were used in the cell cultures medium. These
data suggested a high affinity of cannabinoids for FBS proteins, but also demonstrated the
potential toxicity of these compounds. To identify the possible target(s) of CBDA, CBGA and
CBDVA in U87MG, a mass spectrometry-based chemical-proteomic approach was carried out.
This study allowed to indicate the eukaryotic initiation factor 2 (EIF2A) [2] as a putative target for
CBDA and CBDVA, whereas CBGA showed a treasurable affinity for it. Since proteomic data
suggested CBDA as the most active molecule, it was regarded as the lead compound for the
subsequent analyses. Therefore, to verify that the bioactive compound was actually able to interact
with the target into the cell, CETSA experiments were performed. They revealed that EIF2A
binding to CBDA induces its thermal stabilization, as inferred by an increase in the denaturation
temperature of EIF2A following incubation of cancer cells with CBDA. The biological activity of
three cannabinoids was studied also in 3D-cultured cells, showing a different cytotoxicity
depending on the concentration of FBS in the upper- and lower-gel, thus confirming the critical role
played by the molecule-FBS interaction. As future perspective both cell-based and cell-free
techniques will be performed in 3D cell culture in order to confirm protein target of our molecules of
interest.
[1] Citti, C.; Linciano, P.; Russo, F.; Luongo, L.; Iannotta, M.; Maione, S.; Laganà, A.; Capriotti, A.L.; Forni, F.; Vandelli, M.A., Scientific reports 2019, 9, 1-13. [2] Komar, A.A.; Merrick, W.C., International journal of molecular sciences 2020, 21, 2054.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
51
Poster Communications (PC_12)
A systematic approach to small molecules in the treatment of viral infections
Roberta Bivacqua,a Marilia Barreca,a Virginia Spanò,a Graciela Andrei,b Stefano Alcaro,c,d Paola Barraja,a and Alessandra Montalbanoa
aDepartment of Biological, Chemical and Pharmaceutical Sciences and Technologies (STEBICEF), University of Palermo, Via Archirafi 32, 90123, Palermo, Italy; b Laboratory of Virology and Chemotherapy, Rega Institute for Medical Research, KU Leuven, Leuven, 3000, Belgium; c Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro, Italy; d Net4Science srl, Academic Spinoff, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro, Italy.
E-mail: [email protected]
Benzotriazole and triazole[4,5-c]pyridine systems have been recently identified as potential anti-
CoVs agents, thanks to their ability to inhibit 3CLpro, the main viral protease, which plays an
essential role in transcription and replication processes [1- 3].
Moreover, 1,2,3-triazole based heterocycles have been widely investigated as promising scaffolds
exhibiting antiviral activities [4]. Thus we decided to explore the chemical space and the antiviral
profile of the class of [1,2,3]triazolo[4,5-h][1,6]naphthyridines (Figure 1).
Figure 1: General structure of [1,2,3]triazolo[4,5-h][1,6]naphthyridines.
In silico screening of a small library of synthetic triazolo-naphthyridines will be performed in order
to identify hit scaffolds able of inhibiting specific viral proteins such as:
• DNA polymerase, helicase (HSV-1, HSV-2, VZV and HCMV);
• 3CLpro and TMPRSS2 serine protease (CoVs). Therefore, a pharmacophore model will be generated in order to rationalize the synthesis of new
derivatives that will be screened for their antiviral properties on HSVs or CoVs infected cell
cultures.
[1] Wu, C.-Y.et al. Chemistry & Biology, 2006, 13, 261–268. [2] Turlington, M. et al. Bioorganic & Medicinal Chemistry Letters., 2013, 23, 6172–6177. [3] Karypidou, K. et al. Bioorganic & Medicinal Chemistry Letters, 2018, 28, 3472-3476. [4] Bozorov, K. et al. Bioorganic & Medicinal Chemistry 2019, 27, 3511–3531.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
52
Poster Communications (PC_13)
Design of dual cyclooxygenase-2 and 5-lipoxygenase inhibitors with iron-chelating properties – molecular docking
Jelena Bošković, Dušan Ružić, Olivera Čudina, Katarina Nikolić, and Vladimir Dobričić
Department of Pharmaceutical Chemistry, University of Belgrade – Faculty of Pharmacy, Vojvode Stepe 450, Belgrade, Serbia.
E-mail: [email protected]
Inflammation has an important function in progression of some diseases, such as cancer [1]. Dual
inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) pathways provides a rational
strategy for development of more effective and safer anti-inflammatory drugs [2]. It is assumed that
compounds bearing sulfohydroxamic acid chelate catalytic iron inside 5-LOX enzyme and there are
only few publications about performed molecular docking studies on these compounds. The main
aim of our study was to establish valid and accurate molecular docking platform for future in silico
screening and design of promising dual COX-2 and 5-LOX inhibitors with terminal sulfohydroxamic
group. GOLD (Genetic Optimisation for Ligand Docking) software v.5.7 was used in order to
predict the binding modes and docking poses of previously published dual COX-2 and 5-LOX
inhibitors [3] compounds in the active sites of COX-1, COX-2 and 5-LOX enzymes. Crystal
structures were downloaded from the PDB website: 5WBE (for COX-1), 1CX2 (for COX-2) and
3O8Y (for 5-LOX) [4]. The first step was to investigate binding modes and docking poses of
sulfohydroxamic analogues taken from literature, which expressed dual COX-2 and 5-LOX
inhibitory activities and to establish correlation between experimentally obtained inhibitory activities
and calculated scoring functions (ChemPLP for COX-1 and COX-2 enzymes, ASPFF for 5-LOX
enzyme). Nine newly designed sulfohydroxamic analogues were docked into COX-1, COX-2 and
5-LOX enzymes. In the case of COX-2 enzyme, all designed compounds showed the same binding
pattern as the co-crystalized ligand, SC-558, while no significant interactions were observed in the
COX-1 enzyme. The compounds had lower ChemPLP scores when docked into COX-1 enzyme
comparing to COX-2, which indicated good in silico predicted COX-2 selectivity. Obtained ASPFF
and docking poses indicate that these compounds are potential 5-LOX chelating inhibitors. In this
study, we developed valid molecular docking models to accelerate in silico identification and
design of dual COX-2 and 5-LOX inhibitors bearing sulfohydroxamic acids.
[1] Ricciotti, E.; Fitzgerald, G. A., Arteriosclerosis, Thrombosis, and Vascular Biology, 2011, 31 (5), 986–
1000.
[2] P, J. J.; Manju, S. L.; Ethiraj, K. R.; Elias, G., European Journal of Pharmaceutical Sciences 2018, 121, 356–381. [3] Kaur, J.; Bhardwaj, A.; Huang, Z.; Knaus, E. E., ChemMedChem 2012, 7 (1), 144–150. [4] www.rcsb.org

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
53
Poster Communications (PC_14)
Exploring the MUC1/CIN85 interaction to investigate a small library of peptide inhibitors as potential anti-metastatic agents
Federica Bucolo,a Maria Rita Gulotta,b Ugo Perricone,b and Rosaria Gitto a
a CHIBIOFARAM Department, Università di Messina, Viale Palatucci I,98168-Messina, Italy
b Molecular Informatics Unit, Fondazione Ri.MED, via Filippo Marini 14, 90133-Palermo, Italy.
E-mail: [email protected]
MUC1 is a transmembrane glycoprotein with a physiologically protective role for the cells and a
signalling function. MUC1 extracellular domain is characterized by a variable number of tandem
repeat regions (VNTRs) that are rich in proline residues and highly glycosylated under
physiological conditions. In tumour cells, VNTRs are hypo glycosylated, thus favouring new
protein-protein interactions (PPIs) as found for CIN85 molecule, which is a multifunctional adaptor
protein including SH3 domains, that generally binds proline-rich motifs (PxxP). The association of
CIN85 with MUC1 VNTRs has been correlated to the promotion of cancer progression and
invasiveness [1]. In this work, the MUC1/CIN85 interaction has been investigated by using different
in silico approaches. The aim of this study was to select potential peptide-based inhibitors able to
bind CIN85 SH3 domain to prevent the access to MUC1. Firstly, molecular docking study of the
MUC1 VNTR peptide (PDB: 6KX1) [2] on CIN85 dimer (PDB: 2BZ8) [3] was conducted to explore
MUC1 VNTR binding mode (Figure 1). The resulting docked complex was used to run a molecular
dynamic (MD) simulation to explore frequency and stability of the established interactions. Then, a
structure-based pharmacophore model of CIN85/MUC1 complex was created. Based on the PxxP
motif of MUC1, a similarity search was carried out through SciFinder platform and a database of
peptide analogues containing 4-5 amino acids was created. These peptides were docked on
CIN85 dimer and screened on the pharmacophore model. Finally, the compounds showing the
most promising results were selected and will be assayed in cancer cells.
Financial support of PON FSE-FESR Ricerca e Innovazione 2014-2020 “Dottorati innovativi con
caratterizzazione industriale” (cod. DOT1314952).
Figure 1: On the left, CIN85 dimer (PDB ID: 2BZ8, grey structure) in complex with MUC1 VNTR (PDB
ID: 6KX1, pink peptide); on the right, CIN85/MUC1 interaction diagram.
[1] Cascio S., Farkas A.M., Hughey R.P. and Finn O.J.; Oncotarget 2013, 4, 1686- 1697.
[2] Wakui, H., Tanaka, Y., Ose, T., et al. Chemical Science 2020, 11(19), 4999-5006.
[3] Jozic, D., Cárdenes, N., Deribe, Y. L., et al. Nature structural & molecular biology 2005, 12(11), 972-979.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
54
Poster Communications (PC_15)
Curcumin-based neuroprotective multitarget compounds
Noelia Carmona-Zafra,1 Ángel Cores,1 Juan Domingo Sánchez,1 Sagrario Martín-Aragón,2 Paloma Bermejo,2 Rafael León,3,4 Mercedes Villacampa,1
J. Carlos Menéndez 1
1 Department of Chemistry in Pharmaceutical Sciences (Organic and Medicinal Chemistry Unit), Faculty of Pharmacy, Universidad Complutense, Madrid.
2 Department of Pharmacology, Pharmacognosy and Botanics, Faculty of Pharmacy, Universidad Complutense, Madrid.
3 Instituto Teófilo Hernando y Departamento de Farmacología y Terapéutica, Facultad de Medicina, Universidad Autónoma de Madrid, 28029 Madrid, Spain.
4 Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM-CSIC), Madrid, Spain.
E-mail: [email protected]
Age-related pathologies, including neurodegenerative diseases, have increased due to the
improvements in life expectancy. These disorders are characterized by their multifactorial etiology
and involve several common hallmarks that promote oxidative stress [1, 2]. Curcumin is promising
for the treatment of these diseases because of its good neuroprotective profile that combines
antioxidant, amyloid antiaggregating and metal chelation activities [3]. However, it is metabolically
and chemically unstable.
We propose a family of diversely substituted curcumin analogues with the dicarbonyl structure
rigidified into a 2-pyrrolin-5-one moiety to increase their stability. They have been synthesized
(Figure 1) carrying out a multicomponent procedure to obtain the 2-pyrrolin-5-one core from
primary amines, glyoxal and β-ketoesters [4]. Then, after an acylation step and phosphonate
generation, a final Horner-Wadsworth-Emmons reaction was performed. The library was initially
characterized by studying their antioxidant properties. After cell viability studies, the compounds
were examined in neuroprotection models against hyperphosphorylation and oxidative stress
insults.
Figure 1: General synthetic scheme.
[1] Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Mol Cell Biochem. 2010, 345, 91-104. [2] Johnson, J. A.; Johnson, D. A.; Kraft, A. D.; Calkins, M. J.; Jakel, R. J.; Vargas, M. R.; Chen, P. C. Ann N Y Acad Sci. 2008, 1147, 61–69. [3] Aggarwal, B. B.; Kumar, A.; Aggarwal, M. S.; Shishodia, S. Phytochem. Cancer Chemoprev. 2005, 349-387. [4] Cores, Á.; Estévez, V.; Villacampa, M.; Menéndez, J. C. RSC Adv. 2016, 6, 39433-39443.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
55
Poster Communications (PC_16)
Phytocompounds and their metabolites profiling, as promising multi-target agents for the treatment of Alzheimer's disease
Raffaella Catalano,a,c,d Annalisa Maruca,a,c,d Roberta Rocca,b,c,d Francesco Mesiti,a,c,d Fernanda Borges,e Sofia Benfeito,e Daniel Chavarria,e Stefano Alcaro,a,c,d Francesco
Ortuso a,c,d
a Dipartimento di Scienze della Salute, Università “Magna Grӕcia” di Catanzaro, Viale Europa, 88100 Catanzaro, Italy.
b Dipartimento di Medicina Clinica e Sperimentale, Università "Magna Græcia" di Catanzaro, Viale Europa, 88100 Catanzaro, Italy.
c Net4Science srl, Università “Magna Grӕcia” di Catanzaro, Viale Europa, 88100 Catanzaro, Italy d Associazione CRISEA - Centro di Ricerca e Servizi Avanzati per l’Innovazione Rurale, località Condoleo di
Belcastro (CZ), Italy. e CIQUP/Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, Rua do
Campo Alegre s/n, 4169-007, Porto, Portugal.
E-mail: [email protected]
The innovative paradigm of multi-target drug design has been proposed as a strategy to treat
disorders with complex pathological mechanisms, such as multifactorial neurodegenerative
diseases. Alzheimer's disease (AD) is the most frequent neurodegenerative disorder. Current
treatments rely on the use of single-target drugs. Although they address the disease
symptomatology, they fail to modify the disease progression [1]. Therefore, identifying compounds
able to act on multiple pathways is a very attractive therapeutic strategy for AD [2]. Among several
targets, cholinesterase (ChE), β-secretase1 (BACE-1) and monoamine oxidases (MAOs) play a
pivotal role in the AD network. Based on these hypotheses, in this work we performed the in silico
design of multitarget compounds capable of acting simultaneously on the abovementioned targets.
A library of 1654 phytochemicals (https://phytohub.eu) was screened towards the crystal structures
of the enzymes. Docking results, revealed 9 natural compounds as dual inhibitors of the enzymes.
Furthermore, we computed the structures of phase I metabolites of the best multi-target hits to
compare their pharmacological activity respect to the precursors, towards the same targets. Based
on our theoretical results, we are experimentally investigating their affinity, selectivity and enzyme
inhibition mechanism. The results obtained so far will be presented in this communication.
Figure 1: Major targets involved in the AD pathophysiology and the related multi-target design
strategy. [1] Lane, C. A.; Hardy, J.; Schott, J. M.; Eur J Neurol 2018, 25 (1), 59-70. [2] Zhang, P.; Xu, S.; Zhu, Z.; Xu, J.; Eur J med chem 2019, 176, 228-247. This work was supported by the EU project “ERDF, PON Research and Innovation 2014-2020”.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
56
Poster Communications (PC_17)
An in house library of secondary metabolites as a useful tool for the identification of proteasome activators
Valeria Cavalloro,a Sharon Bryant,b Emanuela Martino,a and Simona Collinac
a Department of earth and Environmental Science, University of Pavia, Via S.Agostino 14 Pavia, 27100, Pavia.
b Inte: Ligand GmbH, Mariahilferstrasse, Vienna, 1070, Austria. c Department of drug science, University of Pavia, Viale Taramelli 12 pavia, 27100, Italy.
E-mail: [email protected]
Proteasome is a multicatalytic target responsable for the degradation of 80-90% of proteins during
cell life. Its main domain is the 20S catalytic core, whose can be associated with regulatory
domains like 11S or 19S, which allows its activation. The 20S core in composed by several
subunits arranged as α1-7β1-7β1-7α1-7. Although proteasome inhibitors are largely studied and three
of them are actually in commerce as anticancer drugs, proteasome activators are still almost
unknown [1]. Evidences seems to suggest that they could be potential agents to contrast
neurodegenerative diseases like Alzheimer, Parkinson and others. Until now, only few small
organic molecules able to activate proteasome are known, mostly acting under an unknown
mechanism of action [2]. A recent work identifies a putative binding site of Chlopromazine, a known
proteasome activator, and its analogues [3].
Starting from the information available, during the period I spent at Inte:Ligand, under the
supervision of Dr. Sharon Bryant, we built a pharmacophoric model suitable for designing novel
proteasome activators. This model was then exploited to screen the in-house library of secondary
metabolites produced by both terrestrial and marine sources, suggesting that a class of alkaloids
produced by a particular genus of seaweed may be able to activate proteasome. To validate our
model, the natural matrix containing the metabolites of interest was collected and extracted by
maceration at room temperature using ethyl acetate as solvent (3 cycles, 2h each). The so
obtained crude extract underwent biological investigation and resulted able to activate proteasome
at non-cytotoxic concentration. The bioguided fractionation of the seaweeds is ongoing.
[1] Manasanch, E.E.; Orlowski, R.Z.. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [2] Leestemaker, Y.; de Jong,A.; Witting,K.F.; Penning,R.; Schuurman,K.; Rodenko,B.; Zaal,E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; Borst,J.; Scheper, W.; Berkers, C.R.; Ovaa, H. Cell Chemical Biology 2017, 24, 725–736. [3] Jones,C.L.; Njomen, E.; Sjogren, B.; Dexheimer, T.S.; Tepe, J.J. ACS Chem. Biol. 2017, 12, 2240−2247.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
57
Poster Communications (PC_18)
Nanoemulsions development as strategy to improve the antitumor properties of Cisplatin and Quercetin
Jessica Ceramella,a Anne-Claire Groo,b Domenico Iacopetta,a Line Séguy,b Annaluisa Mariconda,c Francesco Puoci,a Carmela Saturnino,c Fanny Leroy,d Marc Since,b Pasquale
Longo,e Aurélie Malzert-Fréon,b Maria Stefania Sinicropi a
a Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, 87036 Rende (CS), Italy b Normandie Univ., UniCaen, CERMN, 14000 Caen, France
c Department of Science, University of Basilicata, Viale dell’Ateneo Lucano 10, 85100 Potenza, Italy d Normandie Univ, UniCaen, PLATIN', 14000 Caen, France
e Department of Biology and Chemistry, University of Salerno, Via Giovanni Paolo II, 132, 84084 Fisciano, Italy.
E-mail: [email protected]
The nanoemulsions (NE) development is in continuous progress and represents an exciting
promise in the nanomedicine field for drug delivery in order to improve molecules solubility,
bioavailability and pharmacokinetics [1]. NE have been extensively considered efficient for the
targeted delivery of several anticancer agents [2]. Amongst the anticancer compounds, Cisplatin
and Quercetin are widely used to treat different types of tumor (breast, testicular, ovarian, etc).
However, their scarce solubility and bioavailability, as well as the onset of drug resistance and
dramatic side effects, still represent important hindrances [3,4].
In our approach, to overcome these limitations, three nanoemulsions containing Cisplatin,
Quercetin or both drugs have been formulated, characterized and tested against two human cell
lines, namely human triple negative breast cancer MDA-MB-231 and normal HEK-293 renal cells.
The three types of NE have been obtaining by using a simple and spontaneous process, improving
the antitumor activity of both the molecules and the synergistic effect of the Cisplatin/Quercetin
against the MDA-MB-231. Moreover, the use of NE containing both the drugs reduced the severe
cytotoxic effects of Cisplatin against the human renal HEK-293.
Figure 1: Formulations of Cisplatin and Quercetin NE.
[1] Ashaolu, T.J. Environmental Chemistry Letters (2021): 1-15. [2] Dang, Y., Jianjun, G., 2020. Smart Materials in Medicine 1, 10-19. [3] Aldemir, M., Okulu, E., Kosemehmetoglu, K., Ener, K., Topal, F., Evirgen, O., Gurleyik, E., Avci, A., 2014. Andrologia 46, 1089-1097 [4] Srinivas, K., King, J.W., Howard, L.R., Monrad, J.K., 2010. J Food Eng 100, 208-218.
Improved anticancer properties of NE-CQ
against triple negative MDA-MB-231 breast
cancer cells
NE-CQ protective effect against the Cisplatin
damage induced on HEK-293 cells NE-CQ

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
58
Poster Communications (PC_19)
Investigation of novel N1-substituted quinoxalino-2,3-diones as potent glutamate kainate receptors ligands
Paulina Chałupnik, Alina Vialko and Ewa Szymańska
a Department of Technology and Biotechnology of Drugs, Collegium Medicum, Jagiellonian University, PL 30-688 Kraków, Poland
E-mail: [email protected]
Among ionotropic glutamate receptors (iGluRs), the physiological role of kainate receptors (KARs)
in the central nervous system (CNS) remains the least understood. Kainate receptors, which are
divided into 5 subtypes (GluK1-5) can be found both pre- and postsynaptically and are implicated
in many neurological disorders, including Alzheimer's disease and multiple sclerosis. In this regard,
there is a need for new pharmacological tool compounds, especially subtype-selective compounds,
to explain the role and function of specific KAR subtypes.
The aim of the project was to design and synthesize new potential kainate receptor antagonists,
quinoxalino-2,3-dione derivatives, with structural modifications at the N1 and R7 positions of the
bicyclic core. During the design of the compounds, the main objective was the improvement of
affinity and selectivity at GluK3/GluK1 receptors, based on literature data [1,2]. Furthermore,
additional structure modifications were planned at the N1- position in order to improve solubility.
The project involved molecular docking of the designed structures to the crystal structure of GluK1-
LBD (PDB ID: 6SBT), as well as to a previously constructed model of GluK3-LBD.
Figure 1: General structure of designed compounds.
The target quinoxalino-2,3-diones selected from the molecular modeling results were synthesized
and characterized by receptor binding studies on recombinant iGluRs (GluK1, GluK2, GluK3, and
GluA2). In the present work the obtained results are reported.
[1] Pallesen, J. et al, ACS Chemical Neuroscience 2019, 3, 1841-1853. [2] Møllerud, S. et al, ACS Chemical Neuroscience 2019, 11, 4685-4695.
This work was supported by Jagiellonian University Medical College (grant to. N42/DBS/000042).

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
59
Poster Communications (PC_20)
Synthesis of a new series of optimized Pyrazolo[3,4‐d]pyrimidines as Src Inhibitors
Annarita Cianciusi,a Anna Carbone,a Francesca Musumeci, a and Silvia Schenonea
aDepartment of Pharmacy, University of Genoa, Viale Benedetto XV,3, I 16132 Genoa, Italy.
E-mail: [email protected]
Src is a non-receptor tyrosine kinase (TK) belonging to the Src-family kinases and plays a critical
role in the progression of the cell cycle. Hyperactivation or overexpression of Src are commonly
observed in different pathologies, including cancer [1]. Therefore, Src-family represents a key
target for the design of new therapeutic agents and many small molecule Src inhibitors, belonging
to different chemical classes, have been reported [2]. In this context, my research group
synthesized a wide library of pyrazolo[3,4-d]pyrimidines active as dual Src/Bcr-Abl inhibitors. A
wide-ranging structure−activity (SAR) relationships evaluation of these compounds allowed a deep
understanding of the chemical features needed to increase the activity on Src. In detail, SAR study
suggested that the presence of the aniline moiety at C4 increases the binding affinity towards Src,
regardless of C6 substitution [3]. The introduction of a polar group on the solvent-exposed C6
position afforded compounds endowed with a good balance of activity and pharmacokinetic
properties [4]. In particular, the best results have been obtained with the thioethyl-morpholine
chain. Finally, compounds possessing a p-bromophenyl-2-chloroethyl chain at N-1 showed a better
inhibitory profile than their no brominated analogues. Based on these results we carried out a lead
optimization study, by synthesizing a new series of derivatives (Figure 1) that combine the best
chemical moieties at C4 and C6 with the promising N-1 p-bromo-substituted side chain, to obtain
more active Src inhibitors.
Figure 1: General structure of the novel series of pyrazolo[3,4-d]pyrimidines.
[1] Irby, R. B.; Yeatman, T. J., Oncogene 2000, 19, 5636-5642. [2] Schenone, S.; Manetti, F.; Botta, M., Anti-Cancer Agents in Medicinal Chemistry, 2007, 7, 660-680. [3] Molinari, A.; Fallacara, A.L. et al., Bioorganic & Medicinal Chemistry Letters, 2018, 28, 3454–3457. [4] Tintori, C.; Fallacara, A.L. et al., Journal Medicinal Chemistry, 2015, 58(1), 347–361.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
60
Poster Communications (PC_21)
Design, Synthesis and biological evaluation of hybrid MOR agonist/ HDACi molecules as potential therapeutic agents for chronic pain
treatment
Giuliana Costanzoa, Rita Turnaturib, Carla Barbaracib, Agostino Marrazzob and Lorella Pasquinuccib
a Department of Biomedical and Biotechnological Sciences, University of Catania, Via Santa Sofia, 97,
95123, Catania (Italy) b Department of Drug and Health Science, University of Catania, Viale Andrea Doria, 6, 95125, Catania
(Italy)
E-mail: [email protected]
In pain transmission process new targets have been identified such us the Hystone Deacetylase
(HDAC) enzyme. Recent studies show that epigenetic regulation are involved in development and
maintenance of chronic pain[1]. In fact, HDACi attenuate both neuropathic and inflammatory pain.
Moreover, the histones H3 and H4 hypoacetylation contributes to the decreased expression of mu
opioid receptor (MOR) in the dorsal root ganglion[2]. Then, the development of hybrid MOR
agonist/HDACi molecules could be a good strategy to increase analgesic efficacy of opioids.
Hybrid compounds were designed recurring to the “merging” approach[3]. In vitro their affinity profile
versus opioid receptors are performed through competition binding assays. Moreover, the inhibition
properties in HDAC fluorimetric assays further allow to define in vitro profile of our compounds.
New hybrid ligands have been synthesized, purified and structurally characterized with 1H NMR
,13C NMR and MS spectra. The preclinical evaluation of new hybrid compounds is currently
ongoing.
Figure 1: Rational design of MOR agonist /HDACi hybrid compounds.
[1] Liang, L.; Lutz, BM.; Bekker, A.; Tao, Y.X.; Epigenomics. Epigenetic regulation of chronic pain. 2015, 235,
45.
[2] Xiao-Tao He; Kai-Xiang Zhou; Wen-Jun Zhao; Chen Zhan; Jian-Ping Den; Fa-Ming Chen; Ze-Xu Gu;
Yun-Qing Li and Yu-Lin Dong; Front. Pharmacol. Inhibition of Histone Deacetylases Attenuates Morphine
Tolerance and Restores MOR Expression in the DRG of BCP Rats. 2018, 9, 509.
[3] R. Morphy; Z. Rankovic; Curr.Pharm.Des. Designing multiple ligands - medicinal chemistry strategies and
challenges. 2009, 587, 600.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
61
Poster Communications (PC_22)
Luminescent highly hindered Pt(II) and tryptophan conjugated Ir(III) complexes: the deal-breaker for bioimaging?
Giulia De Soricellis,a Alessia Colombo,a Claudia Dragonetti,a Dominique Roberto,a Daniele Marinotto,b Massimiliano Massi,c Bertrand Carboni d and Veronique Guerchais d
a Dip. di Chimica, Università degli Studi di Milano and INSTM UdR Milano, Via Camillo Golgi 19, 20133-Milan, Italy.
b CNR-SCITEC, Via Alfonso Corti 12, 20133-Milan, Italy c Curtin Institute of Functional Molecules and Interfaces, Curtin University, Kent St, Bentley WA, Perth-6102,
Australia. d,Institut des Sciences Chimiques de Rennes ISCR, University of Rennes, 1263 Avenue Général Leclerc,
35700-Rennes, France.
E-mail: [email protected]
Photoluminescent transition metal complexes play a crucial role in life science, due to their wide-
range versatility. Their optimal photophysical properties such as the long life-time of the excited
states, makes them ideal in the field of bioimaging and photodynamic therapy. Heavy metal
organometallic complexes thus gained increasing interest and extensive studies have been
conducted [1,2]. Furthermore, the organic backbone is suitable for the introduction of biologically
relevant molecules that could serve as targeting vectors. Bioconjugation provides an outstanding
way to localize the photon emission into specific organelles, allowing to both obtain bright images
[3] and to trigger the formation of cytotoxic species that can be exploited to kill cancer cells [4].
For these purposes, a new series of highly hindered Pt(II) complexes were synthesized, aiming to
enhance phosphorescence through inhibition of aggregation processes. Furthermore, a tryptophan
conjugated Ir(III) complex was synthesized and investigated as a luminescent label into brain cells.
Figure 1: Molecular representation of the synthesized Pt(II) and Ir(III) complexes.
[1] Farley, S. J.; Rochester, D. L.; Thompson, A. L.; Howard, J.A. K.; Williams, J. A. G., Inorg. Chem., 2005, 44, 9690-9703. [2] Caporale, C.; Massi, M.; Coord. Chem. Rev., 2018, 363, 71–91. [3] Colombo, A.; Fiorini, F.; Septiadi, D.; Dragonetti, C.; Nisic, F.; Valore, A.; Roberto, D.; Mauro M.; De Cola, L.; Dalton Trans., 2015, 44, 8478-8787. [4] McKenzie, L. K.; Sazanovich, I. V.; Baggaley, E.; Bonneau, M.; Guerchais, V.; Williams, J. A. G.; Weinstein, J. A.; Bryant, H. E.; Chem. Eur. J., 2017, 23, 234 –238.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
62
Poster Communications (PC_23)
Exploring New Scaffolds for the Dual Inhibition of HIV-1 RT Polymerase and Ribonuclease Associated Functions
Serenella Deplano,a Rita Meleddu,a Angela Corona,a Simona Distinto,a Filippo Cottiglia,a Lisa Sequeira,a Daniela Secci,a Alessia Onali,a Erica Sanna,a Francesca Esposito,a Italo
Cirone,a Francesco Ortuso,b Stefano Alcaro,b Enzo Tramontano,a Peter Matyus c and Elias Maccioni a
a Department of Life and Environmental Sciences, University of Cagliari, Cittadella Universitaria di Monserrato, S.P. 8 km 0.700, 09042 Monserrato Cagliari (CA), Italy.
b Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Campus ‘S. Venuta’, Viale Europa, 88100 Catanzaro, Italy.
c Institute of Digital Health Sciences, Faculty of Health and Public Services, Semmelweis University, Ferenc tér 15, Budapest, 1094, Hungary.
E-mail: [email protected]
HIV is the agent responsible for the AIDS (acquired immunodeficiency syndrome), the current
therapeutic protocol for this infection (HAART) consist of the combination of two or more antiviral
agents, targeting different phases of the HIV replication cycle [1]. RT (Reverse Transcriptase) is a
multifunctional enzyme involved in the replication cycle of the virus and is always included as a
target in HAART. This enzyme has two distinct associate functions: RNA/DNA dependent DNA
polymerase (RDDP/DDDP) and Ribonuclease H (RNaseH) [2]. According to HAART basic
principles, the identification of a single agent capable to target two or more phases of the HIV
replication cycle could be an interesting approach to reduce the daily number of pills and the
additive toxicity of the therapy based on the association of different agents. thus, we have
designed and synthetized a new small library of biphenylhydrazo 4-arylthiazoles derivatives
EMAC2056-2071 and evaluated the potential of these derivatives to simultaneously inhibit both
associated functions of HIV RT. All compounds are capable to inhibit both RDDP and RNaseH
functions, indicating that they might represent the starting point for the design of new RT full
activity inhibitors. In particular the compound EMAC2063 was the globally most potent derivative
toward both RNaseH with IC50 4.5 M, and RDDP with IC50 8.0 M.
[1] Gallo, R.C.; Montagnier, L. The discovery of HIV as the cause of AIDS. N. Engl. J. Med. 2003, 349, 2283–2285, doi:10.1056/nejmp038194 [2] Telesnitsky, A.; Goff, S.P. Reverse Transcriptase and the Generation of Retroviral DNA; Cold Spring Harbour Laboratory Press: Cold Spring Harbour, NY, USA, 1997

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
63
Poster Communications (PC_24)
Polysaccharides in Drug delivery: the role of Calcium chloride in the alginate hydrogel printability
Giovanni Falcone,a,b Tiziana Espositoa, Rita P. Aquino,a Giulia Auriemmaa, Pasquale Del Gaudio, a Paola Russo a
a Department of Pharmacy, University of Salerno, Fisciano, SA, Italy.
b PhD Program in Drug Discovery and Development, University of Salerno, Fisciano, SA, Italy. E-mail: [email protected]
Since 2015, the 3D printing technologies, due to their rapid prototyping, and high manufacturing
flexibility, have been identified from pharmaceutical researchers as the keystone to switch from
traditional to personalized therapeutic approach [1, 2]. Among others, semi solid extrusion (SSE)
3D printing can be considered the most promising technology for the drug delivery systems
production. In the SSE printing process, characterized by mild working conditions and relative ease
in drug loading, the 3D objects are obtained through the extrusion of polymeric paste or gels onto a
building plate via a syringe like system [3]. Predictably, the success of the printing process is
strictly related to the optimization of the printing parameters, according to the gel characteristics
[4]. Considering the physico-chemical properties of feedstocks, natural polymers, widely used in
pharmaceutical compounding could fulfil the SSE 3D printing technological needs [5]. For these
reasons, in this work the attention was focused on the alginate matrices obtained through the
crosslinking with divalent cation, a successful duo already used in other technological processes.
In details, the influence of calcium chloride at different ratio (10; 12; 15; 18; 20; 25 mM), selected
as crosslinking agent, on the hydrogels properties, i.e. rheological and physico-chemical, was
investigated, to deeply know the process that can lead to customizable drug platform using
alginate ink. The rheological data combined with the morphological analysis of printed forms
allowed us to identify the relationship between printability and the share thinning behaviour of gels.
While the alginate pre-crosslinking strongly improved the DDS shape retention after printing, the
same cannot be said for the shape after drying, which was completely lost. Thus, with the aim to
reduce the matrix collapsing after DDS drying, some functional excipients such us lactose,
mannitol, talc, or cellulose were tested. Among them, the mannitol showed the best result,
improving the shape retention after drying, without affecting the gel extrudability.
Figure 2: From alginate hydrogel crosslinking to SSE 3D-printing of DDS, through a deep physico-chemical characterization.
[1] Aquino, R. P.; Barile, S.; Grasso, A.; Saviano, M., Futures 2018, 103, 35-50. [2] Vithani, K.; Goyanes, A.; Jannin, V.; Basit, A. W.; Gaisford, S.; Boyd, B. J, Pharmaceutical research 2019,
36, (1), 1-20. [3] Azad, M. A.; Olawuni, D.; Kimbell, G.; Badruddoza, A. Z. M.; Hossain, M.; Sultana, T., Pharmaceutics
2020, 12, (2), 124. [4] Hazur, J.; Detsch, R.; Karakaya, E.; Kaschta, J.; Teßmar, J.; Schneidereit, D.; Friedrich, O.; Schubert, D.
W.; Boccaccini, A. R., Biofabrication 2020, 12, (4), 045004. [5] Paxton, N.; Smolan, W.; Böck, T.; Melchels, F.; Groll, J.; Jungst, T., Biofabrication 2017, 9, (4), 044107.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
64
Poster Communications (PC_25)
Identification of novel indole-2-carboxamides as heme oxygenase-1 (HO-1) inhibitors
Antonino N. Fallica,a Giuseppe Floresta,b Valeria Sorrenti,a Valeria Pittalà,a Antonio Rescifina a
a Department of Drug and Health Sciences, University of Catania, V.le Andrea Doria 6, 95125 Catania, Italy b Department of Analytics, Environmental & Forensics, King’s College London, Stamford Street, London SE1
9NH, UK
E-mail: [email protected]
Heme oxygenase (HO) is an enzymatic family responsible for the degradation of heme [1]. Among
the two identified isoforms, HO-1 represents the stress-induced isozyme whose over-expression in
tumor cells has been associated to heightened tumor aggressiveness and poor chances of
survival. Therefore, HO-1 inhibition represents a valid strategy for novel anticancer therapies. Our
research group has gained a strong background on the design of HO-1 inhibitors whose chemical
structure can be split up in three main portions: i) a lipophilic moiety responsible for potency and
isozyme selectivity; ii) a central connecting chain; iii) an imidazole ring essential for the inhibitory
activity [2-3]. As a continuation of our previous work in which was evaluated the effectiveness of
different heterocyclic rings serving as lipophilic moieties [4], we virtually evaluated the HO-1
inhibitory properties of a novel class of compounds based on the indole-2-carboxamide chemical
structure (Figure 1). Compounds with the best predicted IC50 values have been synthesized and
their experimental IC50 values have been evaluated with an in vitro HO-1 enzymatic assay. Results
showed that this novel class of compounds could be further explored for the identification of potent
HO-1 inhibitors.
Figure 1: General structure of novel HO-1 inhibitors based on the indole-2-carboxamide chemical structure.
[1] Abraham NG; Kappas A., Pharmacological Reviews 2008, 60 (1), 79-127. [2] Salerno L; Floresta G; Ciaffaglione V; Gentile D; Margani F; Turnaturi R; Rescifina A; Pittalà V., European Journal of Medicinal Chemistry 2019, 167, 439-453. [3] Intagliata S; Salerno L; Ciaffaglione V; Leonardi C; Fallica AN; Carota G; Amata E; Marrazzo A; Pittalà V; Romeo G., European Journal of Medicinal Chemistry 2019, 183, 111703. [4] Salerno L; Pittalà V; Romeo G; Modica MN; Marrazzo A; Siracusa MA; Sorrenti V; Di Giacomo C; Vanella L; Parayath NN; Greish K., European Journal of Medicinal Chemistry 2015, 96, 162-172.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
65
Poster Communications (PC_26)
Molecular modeling analysis of clinically relevant mutations associated
with SARS-CoV-2 nucleoside RNA-dependent RNA polymerase
inhibitors
Carmen Gratteri,a Anna Artese,a,b Francesca Alessandra Ambrosio,a Isabella Romeoa,b
and Stefano Alcaroa,b
a Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Campus “S. Venuta”,
Catanzaro, Italy. b Net4Science Academic Spin-off, Università “Magna Græcia” di Catanzaro, Campus “S. Venuta”, Catanzaro,
Italy.
E-mail: [email protected]
Infection with SARS-CoV-2 is associated with substantial morbidity and mortality that calls for
urgent effective therapeutic measures. To date, a wide variety of antiviral treatments for COVID-19
has been investigated, involving many repurposed drugs. SARS-CoV-2 RNA-dependent RNA
polymerase (RdRp, encoded by nsp12-nsp7-nsp8), which plays an essential role in replication and
transcription of the viral genome, has been targeted by numerous inhibitors. The most promising
broad-spectrum class of viral RdRp inhibitors are nucleoside analogues (NAs). Currently,
Remdesivir (GS-5734) is the only FDA approved drug for the SARS-CoV-2 treatment [1]. However,
numerous mutations in the viral genome were detected and could compromise the effectiveness of
both Remdesivir and other potential inhibitors. In particular, RdRp P323L substitution was found
predominantly in a large number of infected patients [2]. The aim of the present study is to
investigate the molecular recognition of Remdesivir and other NAs towards wild-type (WT) and
mutated RdRp sequences using in silico approaches.
Figure 1. RdRp 3D structure.
[1] Goran, K.; Hauke, S.H.; Dimitry, T.; Christian, D.; Florian, S.; Jana, S.; Lucas F.; Aaron S.; Claudia H., Patrick, C., Mechanism of Sars-Cov-2 polymerase stalling by remdesivir. Nat Commun 2021, 12 (279).
[2] Subrata, K.,B.; Sonchita, R.M., Spike protein D614G and RdRP P323L: the SARS-COV-2 mutations associated with severity of COVID-19. Genomics & Inform 2020, 18 (4).

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
66
Poster Communications (PC_27)
Capsicum annuum as source of bioactive compounds with multi-targeting anti-cancer and anti-obesity profile vs hCAs isoforms
Gianmarco Gualtieri,a Annalisa Maruca,a,b,d Roberta Rocca,a,c,d Giosuè Costa,a,b,d and Stefano Alcaroa,b,d
a Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro, Italy
b Net4Science Srl, Università “Magna Græcia” di Catanzaro, Viale Europa, 88100, Catanzaro Italy c Dipartimento di Medicina Clinica e Sperimentale, Università "Magna Græcia" di Catanzaro, Viale Europa,
88100, Catanzaro, Italy d Associazione CRISEA - Centro di Ricerca e Servizi Avanzati per l’Innovazione Rurale, località Condoleo di
Belcastro, Catanzaro, Italy
E-mail: [email protected]
Increasing evidence suggests that the Mediterranean diet is correlated with a reduced risk of
developing cardiovascular diseases, cancer, and mental disorders [1]. Hot pepper (Capsicum
annuum) represents one of the most common functional foods which takes part in this nutritional
model, with a cardioprotective, anti-cancer, anti-obesity, antioxidant, and anti-diabetic profile [2].
These potential polypharmacological properties are due to its bioactive spicy molecules named
capsaicinoids, including capsaicin (trans-8-methyl-N-vanillyl-6-nonenamide), the most extensively
studied. Scientific data support different capsaicin’s beneficial effects, proposing that many of them
may not be related to the activation of Transient Receptor Potential Vanilloid 1 (TRPV1) [3].
Figure 3. Principal steps for the discovery of multi-targeting bioactivity towards hCAs for compounds
extracted from Capsicum annum.
Starting from the above considerations, here we present application and results of in silico and in
vitro techniques to assess the inhibitory activity of capsaicin and analogues against tumor-
associated hCAs IX, XII and against obesity-related hCA V (figure 1).
[1] Martini, D., Nutrients 2019, 11(8), 1802. [2] Maruca, A.; Catalano, R.; Bagetta, D.; Mesiti, F.; Ambrosio, F. A.; Romeo, I.; Moraca, F.; Rocca, R.; Ortuso, F.; Artese, A.; Costa, G.; Alcaro, S.; Lupia, A., European Journal of Medicinal Chemistry 2019, 181, 1-23. [3] Papoiu, A. D.; Yosipovitch, G., Expert Opinion on Pharmacotherapy 2010, 11, 1359–1371.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
67
Poster Communications (PC_28)
Identification of new inhibitors of protein arginine methyltransferases by a multi-substrate-adduct approach
Giulia Iannelli,a Ciro Milite,a Alessandra Feoli, a Jean Cavarelli,b Sabrina Castellano,a
Gianluca Sbardella.a
a Epigenetic Med Chem Lab, Dipartimento di Farmacia, Università degli Studi di Salerno, Via Giovanni Paolo II 132, I-84084 Fisciano, Salerno, Italy; b Istitut de Génétique et de Biologie Moléculaire et Cellulaire,
Université de Strasbourg, CNRS UMR 7104, INSERM U 964, Illkirch, F-67404, France.
E-mail: [email protected]
The methylation of arginine residues is a common post-translational modification, performed by a
family of methyltransferases known as PRMTs (Protein Arginine Methyltransferases). In humans,
methylation is carried out by nine members of the PRMTs that are ubiquitously expressed [1].
Arginine methylation is known to play a key role in gene regulation due to the ability of the PRMTs
to deposit activating or repressive “histone marks”. This modification correlates PRMTs to several
biological processes, including transcription, DNA repair, protein stability, cell signaling, pre-mRNA
splicing and receptor trafficking. Therefore, aberrant activity of PRMTs is involved in many
pathological conditions like inflammation, neurodegeneration and cancer. According to these
evidences, PRMTs have been identified as promising therapeutic targets [2],[3].
Starting from EML108, an inhibitor of PRMTs previously identified by us [4], we deconstructed our
compound and then performed a medicinal chemistry optimization campaign.
Here we report a multi-substrate-adduct approach to develop new powerful and selective
compounds (Figure 1).
Figure 1: Multi-substrate adduct approach.
[1].Bedford, M. T.; Clarke, S. G., Mol. Cell 2009, 33 (1), 1-13. [2].Yang, Y.; Bedford, M. T., Nat. Rev. Cancer 2013, 13 (1), 37-50. [3].Hashimoto, M.; Murata, K.; Ishida, J.; Kanou, A.; Kasuya, Y.; Fukamizu, A., J. Biol. Chem. 2016, 291 (5), 2237-2245. [4].Ragno, R.; Simeoni, S.; Castellano, S.; Vicidomini, C.; Mai, A.; Caroli, A.; Tramontano, A.; Bonaccini, C.; Trojer, P.; Bauer, I.; Brosch, G.; Sbardella, G., J. Med. Chem. 2007, 50 (6), 1241-1253.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
68
Poster Communications (PC_29_FC-2)
Design, synthesis and biological evaluation of new 4-oxo-1,4-dihydroquinolin-3-adamtilamides derivates to develop CB2R fluorescent
probes Francesca Intranuovo,a Maria Majellaro,b Francesca Serena Abatematteo,a Claudia Gioè,b
Giuseppe Felice Mangiatordi,c Pietro Delre,c Angela Stefanachi,a Joseph Brea,c Maria Isabel Loza,c Nicola Antonio Colabufo,a,e Eddy Sotelo,b Carmen Abate,a Marialessandra Continoa.
aDipartimento di Farmacia-Scienze del Farmaco, Universita degli Studi di Bari ALDO MORO, via Orabona 4, 70125, Bari, Italy; bComBioMed Research Group, Centro de Química Biológica y Materiales Moleculares (CIQUS), Department of Organic Chemistry, Faculty of Pharmacy, University of Santiago de Compostela (CIQUS). Campus Vida, 15782, Santiago de Compostela, Spain; cCNR - Istituto di Cristallografia, Via Amendola, 122/o, 70126, Bari, Italy; dInnopharma Screening Platform, BioFarma Research Group, Center for Research in Molecular Medicine and Chronic Diseases (CIMUS), University of Santiago de Compostela, 15782, Santiago de Compostela, Spain; eBiofordrug s.r.l, Spin-off dell’Università degli Studi di Bari ALDO MORO, via Dante 99, 70019, Triggiano, Bari, Italy
E-mail: [email protected]
Abstract CB2R, formerly known as the "peripheral cannabinoid receptor" because firstly identified
at the peripheral level (in the cells and organs of the immune system), has been found to be
overexpressed in pathological processes related to inflammation (neuroinflammation and cancer).
CB2R fluorescent probes have been developed as “green” and safe diagnostic tools alternative to
the classic radioligands, commonly employed to study this target. Keeping this in mind, we recently
developed as CB2R fluo-ligand, the compound 1 [1,2] (Figure 1), bearing a quinolone core
(responsible for CB2R affinity and selectivity) linked, by an hexamethylene linker, to the 4-N,N-
dimethylaminophthalimide (as green emitting tag). Compound 1 showed a good affinity towards
CB2R (Ki = 130 nM) and a very high selectivity, since it is devoid of affinity towards the CB1R
subtype (14 %@ 1μM). In order to improve the pharmacodynamic properties of compound 1, we
proceeded with its optimization by: i) the insertion of substituents on the quinolone core to improve
CB2R affinity; ii) the modification of the length of the spacer linking the fluorescent tag to the
quinolone scaffold; iii) the introducti on of alternative fluorescent tags with an emission spectrum
shifted towards longer wavelengths (Red and NIR regions) to have greater versatility for other
fluorescence techniques. The compounds resulting from this study were tested for their affinity and
selectivity towards CB2R, as well as for their fluorescent properties. Molecular docking simulations
complemented the experimental findings providing a molecular rationale behind the observed
CB2R affinities, hence paving the way for a rational design of new fluorescent probes.
Figure 1: Lead compound 1.
[1] Spinelli, F. et al. J. Med. Chem. 2020, 188, 112037. [2] Spinelli, F. et al. J. Med. Chem. 2017, 60 (24), 9913–9931.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
69
Poster Communications (PC_30)
Chalcones with anti-HIV-1 protease activity: 3D-QSAR study, design of novel inhibitors, molecular docking and in silico ADMET
characterization
Milan Jovanovic,a Nemanja Turkovic,b Branka Ivkovic,a Zorica Vujic,a Katarina Nikolic,a and Sonja Grubišićc
a Faculty of Pharmacy - Department of Pharmaceutical Chemistry, , University of Belgrade, Vojvode Stepe 450, Belgrade, Republic of Serbia.
b Agency for Medicines and Medical Devices of Montenegro - Import/export authorizations for medicines, certificates and expert opinions department, Bulevar Ivana Crnojevića 64A, Podgorica, Montenegro.
c University of Belgrade – Institute of Chemistry, Technology and Metallurgy, Department of Chemistry, Njegoševa 12, Belgrade, Republic of Serbia.
E-mail: [email protected]
HIV protease inhibition represents one of key components of the AIDS therapy [1]. Chalcone
derivatives have been shown to inhibit HIV-1 protease and represent interesting drug candidates
for research [2]. A 3D-QSAR (3D-Quantitative structure-activity relationship) study has been
performed on a dataset with 20 chalcones obtained from our previous study [3] in order to develop
a model and to define key structural characteristics which enhance anti-HIV-protease activity. The
good prediction capability of the created model has been confirmed with internal and external
validation. All dataset compounds fit into the defined applicability domain. MESPs, HOMO-LUMO
and Mulliken analysis based on functional density theory have also been conducted in order to
further explain the pharmacophoric features of the chalcone derivatives. Based on this information,
new derivatives have been designed and their activities have been predicted with the previously
developed model. Compounds with the highest predicted activities have been subjected to
molecular docking study in order to foresee the interactions they can form with the HIV-1 protease.
In silico ADMET profiling of all compounds has been done in order single out the drug candidates
with optimal pharmacokinetic and toxicity attributes.
[1] Sharp PM, Hahn BH, Cold Spring Harbor Perspectives in Medicine 2011, 1 (1), 1-22. [2] Nowakowska Z Eur J Med Chem 2007, 42 (2), 125-37. [3] Turkovic N, Ivkovic B, Kotur-Stevuljevic J, Tasic M, Marković B, Vujic Z, Current Pharmaceutical Design
2020, 26, 802-814.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
70
Poster Communications (PC_31)
Design and Synthesis of Dual D2/5-HT3Rs Ligands as Potential Drugs in
the Management of Schizophrenia
Radomir Juza,a,c Premysl Vlcek,a Eva Mezeiova,a,b Kamil Musilek,c Ondrej Soukup,b Jan Korabecny,a,b
a National Institute of Mental Health, Topolova 748, 250 67 Klecany, Czech Republic b Biomedical Research Centre, University Hospital Hradec Kralove, Sokolska 581, 500 05 Hradec Kralove,
Czech Republic c Department of Chemistry, University of Hradec Kralove, Hradecka 1285, 500 01 Hradec Kralove, Czech
Republic
E-mail: [email protected]
Schizophrenia is a serious mental disorder with approximately 1% of lifetime prevalence [1].
According to various studies, there is no response to pharmacological intervention even when
choosing different combinations of antipsychotics in 13-50 % of all patients [2]. Current
pharmacotherapy of schizophrenia is based on targeted alternations mainly of serotoninergic and
dopaminergic systems by the second-generation antipsychotics (SGA) [3]. Aripiprazole, is a partial
agonist of D2R that keeps a certain amount of dopamine activity on D2R when it is deficient, and,
conversely, it inhibits dopamine activity when this one is overstimulated [4]. Current antipsychotics
do not show effect on 5-HT3Rs (with the exception of clozapine), whose modulation demonstrably
improves sensorial resistance and reduces negative symptoms of schizophrenia according to the
PANSS scale [5]. Meta-analyses of augmentation of current dopaminergic pharmacotherapy by
setrons (antagonists of 5-HT3R - ondansetron, tropisetron, granisetron) confirmed the mentioned
clinical effect [5].
Based on the the observation with setrons, we present a novel family of D2/5-HT3Rs ligands
combining various arylpiperazine scaffold and aryl(piperazine-1-yl)methanone moiety tethered by
aliphatic linker of varying length. Our combination merges the affinity towards both receptors,
namely D2/5-HT3Rs. Within our contribution, we will report design of the compounds in detail, their
in vitro data (cytotoxicity, prediction of blood-brain barrier penetration) and in vivo behavior
(pharmacokinetic analysis and acute toxicity).
This work was supported by Czech Science Foundation (project nr. 19-11332S) and University of Hradec Kralove (Faculty of Science, no. SV2104-2021, VT2019‐2021)
[1] Messias, E.M.D.; Chen, Ch-Y.; Eaton, W.W., Psychiatric Clinic of North America 2007, 30, 323-338 [2] Bebawy, M.; Chetty, M., BioEssays 2008, 30, 183-188 [3] Holder, SD.; Edmunds, AL.; Morgan, S., FP essentials 2017, 455, 23-29 [4] Burris, KD.; Molski, TF.; Xu, C.; Ryan, E.; Tottori, K.; Kikuchi, T.; Yocca, FD.; Molinoff, PB., Journal of Pharmacology and Experimental Therapeutics 2002, 302, 381-389 [5] Kishi, T.; Mukai, T.; Matsuda, Y.; Iwata, N., NeuroMolecular Medicine 2014, 16, 61-69

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
71
Poster Communications (PC_32)
5-arylideneimidazolones possessing fluorine as potential solution for cancer multidrug resistance
Aneta Kaczor,a Jácint Borzán, b Nikoletta Szemerédi, b Gabriella Spengler, b and Jadwiga Handzlik a
a Department of Technology and Biotechnology of Drugs, Faculty of Pharmacy, Jagiellonian University Medical College, 9 Medyczna Street, 30-688 Kraków, Poland
b Department of Medical Microbiology, Albert Szent-Györgyi Health Center and Faculty of Medicine, University of Szeged, Semmelweis utca 6, 6725 Szeged, Hungary
E-mail: [email protected]
Multidrug resistance (MDR) in chemotherapy is nowadays an increasing global problem. Various
approaches were proposed in order to overcome cancer MDR. Adjuvants of chemotherapeutic
drugs, which will be able to block at least one mechanism of resistance e.g. ABCB1 efflux pump,
are one of potential solutions [1]. Moreover, selective cytotoxicity towards MDR cancer cells is a
desirable activity. This kind of activity was previously observed for imidazolone derivatives with
fluorine atom, while blockage of the ABCB1 efflux pump was described for other imidazolone
derivatives [2]. For this reason, further research studies for adjuvants in cancer, involving design,
synthesis and biological evaluation, were carried out in the group of 5-arylideneimidazolones.
Previous results were the basis for creation of current general structure and modifications.
Selected compounds were obtained in 4-6-step synthesis. Main route consists of 3-4-stages i.e. (i)
Knoevenagel condensation, (ii) S-methylation, (iii) reaction with amine, which in some cases was
followed by (iv) Dimroth rearrangement. Part of amine intermediates were obtained in 2-step
Gabriel synthesis. Final products were tested for cytotoxicity in parental (PAR) and multidrug
resistant (MDR) T-lymphoma cancer cell lines as well as mouse fibroblasts NIH/3T3, furthermore
the selectivity of compounds was determined towards cancer cells. Then, rhodamine 123
accumulation assay was carried out to determine ability to block ABCB1 efflux pump. Moreover,
checkerboard combination assays of 5-arylideneimidazolones and doxorubicin were performed.
New imidazolones with fluorobenzylidene moiety were designed. Eighteen final products were
synthesized and tested in biological assays. Most of them proved higher cytotoxicity towards MDR
than NIH/3T3 cells, furthermore many of them exhibited synergism with the standard
chemotherapeutic drug doxorubicin. In rhodamine 123 accumulation assay imidazolones showed
up to 51 % of potency of the strong ABCB1 inhibitor, tariquidar.
To sum up, results obtained for 5-arylideneimidazolones possessing fluorine seem to be promising
and pointed out adjuvant activity selective towards multidrug resistant cells. This work was
financially supported by grant of Polish Ministry of Science and Higher Education (grant number
0169/DIA/2017/46).
[1] Dong, J.; Qin, Z.; Zhang, W.D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R. Jr; Chen, Z.S.; Cheng, X.D.; Qin, J.J. Drug Resistant Update 2020, 49, 100681. [2] Kaczor, A.; Szemerédi, N.; Kucwaj-Brysz, K.; Dąbrowska, M.; Starek, M.; Latacz, G.; Spengler, G.; Handzlik, J., ChemMedChem 2021, DOI: 10.1002/cmdc.202100252.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
72
Poster Communications (PC_33)
Water-soluble aminofullerenes as effcient delivery agents for siRNA
transfection
Julia Korzuch,a Monika Rak,b Katarzyna Balin,c Maciej Zubko,d,e Olga Głowacka,b Mateusz Dulski,d Robert Musioł,a Zbigniew Madeja,b Maciej Serda a
a Institute of Chemistry, University of Silesia in Katowice, Katowice, 40-006, Poland.
b Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Kraków, Poland. c Institute of Physics and Silesian Center for Education and Interdisciplinary Research, University of Silesia in
Katowice, 75 Pułku Piechoty 1A, 41-500 Chorzów, Poland. d Institute of Materials Science, University of Silesia in Katowice, Chorzow, 75 Pulku Piechoty, 1A, 41-500,
Poland. e University of Hradec Králové, Faculty of Science, Department of Physics, Rokitanského 62, 500-03, Hradec
Králové, Czech Republic.
E-mail: [email protected]
Nanoparticles for cancer treatment have increased their popularity over the last decade and
changed classical medical chemistry. In recent years combination of small molecules and more
complex biologicals entered the pharmaceutical reasearch and development e.g with nucleic acid-
based therapy [1]. A combination of gene therapy with targeted drugs can significantly improve
current treatment options. It can be used to knock out the genes responsible for multidrug
resistance. The gene inhibition can be achieved by the technique called RNA interference. So
called siRNA – small interfering RNA - is a short (21-25 nucleotides), double-stranded RNA
molecule. Although siRNA is highly immunogenic and has a poor pharmacokinetic profile in the
presence of an engineered, efficient transfection agent can be delivered to the cytoplasm and
degrade specific mRNA. Therefore we are presenting water-soluble cationic C60Hexakisamino for
non-viral siRNA transfection. Fullerene derivatives have been previously reported as gene vectors
but never for siRNA transfection [2]. We used a double-step Bingel-Hirsch protocol for introducing
ethylenediamine malonate to the fullerene scaffold. The structure was confirmed by 13C NMR,
ESI-MS, FT-IR and X-ray photoelectron spectroscopy. The efficiency of C60Heksakisamino was
proven by in vitro tests using prostate cancer cell line DU145 expressing GFP protein.
[1] Dammes, N.; Peer, D., Trends in Pharmacological Sciences 2020, Vol. 41, No. 10 [2] Sigwalt, D., Holler, M., Iehl, J., Nierengarten, J.-F., Nothisen, M., Morin, E., & Remy, J.-S., Chemical Communications, 2011,47(16), 4640

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
73
Poster Communications (PC_34)
Non-acidic bifunctional benzothiazole-based thiazolidinones with antimicrobial and aldose reductase inhibitory activity as a promising
therapeutic strategy for sepsis
Antonios Kousaxidis,a Lucia Kovacikova,b Ioannis Nicolaou,a Milan Stefek,b Athina Geronikaki a
a School of Health, Department of Pharmacy, Aristotle University of Thessaloniki, 54124, Greece.
b Institute of Experimental Pharmacology and Toxicology, CEM, SAS, Dúbravská cesta 9, 84104 Bratislava, Slovakia.
E-mail: [email protected]
Sepsis is a life-threatening disease which affects millions of people worldwide. Microbial infections
which lead to sepsis syndrome are associated with an increased production of inflammatory
molecules [1]. Aldose reductase has recently emerged as a molecular target that is involved in
various inflammatory diseases, including sepsis [1, 2]. A series of previously synthesized
benzothiazole-based thiazolidinones [3] that exhibited strong antibacterial and antifungal activities
has been evaluated for inhibition efficacy against aldose reductase and selectivity towards
aldehyde reductase under in vitro conditions. The most promising inhibitor 5 was characterized
with IC50 value of 3.99 μM and a moderate selectivity. Molecular docking simulations revealed the
binding mode of compounds at the active site of human aldose reductase. Moreover, owning to the
absence of an acidic pharmacophore, good membrane permeation of the novel aldose reductase
inhibitors was predicted. Excellent ‘drug-likeness’ was assessed for most of the compounds by
applying the criteria of Lipinski’s ‘rule of five’.
Figure 1: Compound 5 is a moderate aldose reductase inhibitor with satisfactory selectivity towards aldehyde reductase exhibiting antifungal as well as antimicrobial activity. Docking analysis revealed the binding mode at the active site of human aldose reductase. [1] Ramana K. V., Willis M. S., White M. D., Horton J. W., Dimaio J. M., Srivastava D., Bhatnagar A., Srivastava S. K., Circulation 2006, 114 (17), 1838-46. [2] Kousaxidis A., Petrou A., Lavrentaki V., Fesatidou M., Nicolaou I., Geronikaki A., Eur J Med Chem 2020, 207 (112742), 1-42. [3] Haroun M., Tratrat C., Kositzi K., Tsolaki E., Petrou A., Aldhubiab B., Attimarad M., Harsha S., Geronikaki A., Venugopala K. N., Elsewedy H. S., Sokovic M., Glamoclija J., Ciric A., Curr Top Med Chem 2018, 18, 75-87.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
74
Poster Communications (PC_35)
Derivatives of DL76 as histamine H3 receptor ligands with monoamine oxidase B inhibitory activities
Ewelina Królicka,a Agnieszka Olejarz-Maciej,a Agata Siwek,b Agata Doroz-Płonka,a David Reiner-Link,c Annika Frank,c Holger Stark,c Katarzyna Kieć-Kononowicz,a Dorota
Łażewska a
a Department of Technology and Biotechnology of Drugs, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9, Kraków 30-688, Poland.
b Department of Pharmacobiology, Faculty of Pharmacy, Jagiellonian University Medical College, Medyczna 9, 30-688 Kraków, Poland.
c Institute of Pharmaceutical and Medicinal Chemistry, Heinrich Heine University Düsseldorf, Universitaetsstr. 1, 40225 Duesseldorf, Germany.
E-mail: [email protected]
Parkinson's disease (PD) is a progressive disorder characterized by a severe deficiency of
dopamine (DA) (80-90%) in the striatum. Currently, in the symptomatic treatment of PD, are used
monoamine oxidase (MAO) B inhibitors, such as selegiline [1]. MAO B is an enzyme that breaks
down DA in the brain. Histamine H3 receptors (H3R), widely distributed in the human brain, affect
the release of histamine and other neurotransmitters, including DA. Blocking of H3R enhanced DA
neurotransmission in the brain. Therefore, the combination of MAO B inhibition with H3R receptor
antagonism could be a promising concept for PD treatment.
Continuing our previous work [2], analogs of the compound 1-(3-(4-tert-butylphenoxy)
propyl)piperidine (DL76) were designed and synthesized. Three types of modifications were
introduced as shown in Figure 1. All compounds synthesized were tested for affinity at human H3R
(hH3R) stably expressed in CHO or HEK293 cells. Inhibitory activity toward human MAO B (hMAO
B) was evaluated using a fluorometric MAO assay. Compounds showed different inhibitory activity
for both biological targets. The most promising dual-targeting ligand proved to be 1-(3-(4-(tert-
butyl)phenoxy)propyl)-2-methylpyrrolidine (DL123) (hH3R: Ki = 25 nM; hMAO BIC50 = 4 nM).
Figure 1: Types of modification on the lead structure DL76.
The research was partly funded by Jagiellonian University Medical College grant no. N42/DBS/000124 as well by the EU-COST Action 15135.
[1] Bloem, B.R.; Okun M.S.; Klein, C., The Lancet 2021 – in press.
[2] Łażewska. D.; Olejarz-Maciej, A.; Reiner, D.; et al., Int. J. Mol. Sci 2020, 21 (10), 3411.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
75
Poster Communications (PC_36)
Investigations of the antiproliferative mechanism of furanylvinylquinoline derivatives
Michał Kuczak,a Patryk Małecki,a Anna Mrozek-Wilczkiewicz,b Robert Musioł a
a Institute of Chemistry, University of Silesia, Szkolna 9 Street, 40-006, Katowice b A.Chełkowski Institute of Physics and Silesian Centre for Education and Interdisciplinary, University of
Silesia, 75 Pułku Piechoty 1 Street, 41-500, Chorzów
E-mail: [email protected]
The persistently high incidence of cancer and the consequent deaths of many people is
undoubtedly a social problem. Due to the complex signaling pathways in cancer cells, many
cytostatics are directed towards a specific molecular target. Focusing on this issue, it is crucial to
design and synthesise new compounds with multi-targeted antiproliferative properties against
cancer cells. Notable, an additional significant problem related to the drug resistance of cancer
cells is the numerous mutations that rapidly alter the conformations of the molecular structures
used as targets for anticancer drugs. Therefore, recently, a polypharmacological approach has
become important among the strategies leading to the discovery of new agents used in medicinal
chemistry. The concept of a privileged structure is often used to implement this approach [1]. The
well-described privileged structures are small-molecule heterocyclic compounds. Among them,
quinoline is noticed for its broad spectrum of biological properties [2]. The diverse biological
activities of compounds based on quinoline scaffold are determined by the location of the
substituents. In recent years, our research has focused on investigating the anticancer
mechanisms of action of new styrylquinoline derivatives [3]. Based on our previous results, we
decided to modify the styryl substituent by replacing the benzene ring with a furan. Consequently, a
series of furanylvinylquinoline derivatives were synthesised. The cytotoxicity of the compounds was
studied in vitro on a panel of human cancer cells lines differing in p53 protein status i. a. colon
carcinoma (HCT 116), pancreatic (PANC-1), glioblastoma (U251). Additionally, the selectivity of
the tested analogues was determined by testing their toxicity towards normal human dermal
fibroblasts (NHDF). Among the analysed compounds, one showing the best anticancer properties
was selected. Its antiproliferative activity was evaluated on 3D spheroids. The potential mechanism
of action in cancer cell lines with wild-type, deletion and p53 mutations was then identified. The
effect of the derivative on the cell cycle and the type of cell death were investigated using flow
cytometry. Differences in expression levels of selected genes and proteins indicated a significant
influence of p53 status in the mechanism of action of the derivative. Moreover, the time-dependent
increase of intracellular reactive oxygen species (ROS) as well as changes in expression of
molecular targets associated with oxidative stress were presented. The results indicated a
significant involvement of the tested compound in oxidoreductive disturbances in cancer cells.
The financial support of the National Science Centre grant 2018/31/B/NZ7/02122 is greatly appreciated.
[1] Welsch, M.E.; Snyder, S.A.; Stockwell, B.R., Curr Opin Chem Biol, 2010, 14, 347-361. [2] Musiol, R., Expert Opin Drug Discov, 2017, 12, 583-597. [3] Mrozek-Wilczkiewicz, A.; Kuczak, M.; Malarz, K.; Cieslik, W.; Spaczynska, E.; Musiol, R., Eur J Med Chem, 2019, 177, 338-349.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
76
Poster Communications (PC_37_FC-3)
Novel Proteasome Inhibitors based on γ-lactams for cancer treatment
Roberta Listro,a Rita Stabile,a Daniela Rossi,a Pasquale Linciano,a Alessio Malacrida,b Giosuè Costa,c and Simona Collina a
a Department of Drug Sciences of the University of Pavia, Via Taramelli 12, 27100 Pavia, Italy b School of Medicine and Surgery of the University of Milan-Bicocca, Via Cadore 48, 20900 Monza, Italy c Dipartimento di Scienze della Salute - Net4Science Academic Spin-Off, Università “Magna Græcia” di
Catanzaro, Viale Europa, 88100 Catanzaro, Italy.
E-mail: [email protected]
Cancer is a class of diseases characterized by out-of-control cell growth. Despite the attempts that
have been made both in academic and industry settings to develop new effective drugs, tumors
remain one of the major causes of death in the world. Indeed, about 14.1 million new cases of
cancer occur every year with 15.7% death rate [1]. Within this context, my PhD project was
focused on multiple myeloma (MM), one of the most aggressive cancer characterized by
increasing incidence, difficult diagnosis, and a missing tangible cure. It is well known that
proteasome works as degradation machine of misfolded proteins involved in cancer etiology and
therefore it plays a key role in MM Since the publication of the first phase 1 trials of bortezomib 20
years ago, this proteasome inhibitor (PI) has contributed substantially to the observed
improvement in survival in MM patients. Therefore, proteasome is validated drug targets in
oncology, and numerous structurally diverse inhibitors of natural and nonnatural origin have been
reported so far [2].
Recently, we discovered a secondary metabolite (HibA) from Hibiscus sabdariffa Linn endowed
with an interesting profile against MM [3]. Starting from this compound as template, and following a
medicinal chemistry approach, during this 3-years period structurally related compounds have
been acquired or synthesized and a focused library based on a γ-lactam synthetic scaffold has
been designed. To validate our strategy, two members of the library have been synthesized in
homochiral form and their activity on RPMI8226 cell lines evaluated. Eutomers of both compounds
show good IC50 value against MM, and significantly inhibit proteasome with IC50 values in the
micromolar range. Basing on the encouraging results obtained, considering the synthetic feasibility
and the compound stability, other members of the compound library have been synthesized. Their
anticancer properties are under investigation.
The results herein presented will constitute the starting point for the development of potential
anticancer molecules based on γ-lactams useful as proteasome inhibitors.
[1] Behrooz, A.; Masoumeh R. and Meisam, O. Orient. J. Chem. 2018, 34, 2002 [2] Lee, J. H., Jung, K., Quach, C. H. T., Park, J. W., Moon, S. H., Cho, Y. S., Lee, K. Scientific Reports 2018, 8, 1. [3] Malacrida, A., Cavalloro, V., Martino, E., Cassetti, A., Nicolini, G., Rigolio, R., Cavaletti, G., Mannucci, B., Vasile, F., Di Giacomo, M., Collina, S., Miloso, M. Molecules 2019, 24, 13

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
77
Poster Communications (PC_38)
Computational Study of CK1-CRBN and CK2-CRBN Complexes
Laura Márquez-Cantudo, Claire Coderch, Ana Ramos, Beatriz de Pascual-Teresa
Departamento de Química y Bioquímica, Facultad de Farmacia, Universidad San Pablo-CEU, CEU
Universities, Urbanización Montepríncipe, 28925, Alcorcón, Madrid, Spain.
E-mail: [email protected]
PROTACs (PROteolysis TArgeting Chimaeras) are heterobifunctional molecules that mediate
selective degradation of a protein of interest (POI) by recruiting a given E3 ligase [1]. One of the
POIs of our research group is Casein Kinase II (CK2), which is a validated target for the treatment
of cancer [2]. Until recently, we have focused our efforts on the design and synthesis of dual-
targeted inhibitors against CK2 and one of its phosphorylation targets, HDAC [3, 4]. Now our aim is
to achieve the selective degradation of this protein by recruiting CRL4CRBN as E3 ligase. The E3
ligase CRL4CRBN linked to Immunomodulatory Drugs (IMiDs) such as thalidomide, lenalidomide or
pomalidomide that are capable of ubiquitinating CK1 [5]. However, despite the high structural
homology with CK2, surprisingly the latter is not a substrate of this E3 ligase.
In the recent years the formation of a stable ternary complex (E3: PROTAC: POI) has acquired
special importance [1]. Thus, we want to perform a conformational study, by means of
computational methods, of CK1-CRBN and CK1-Lenalidomide-CRBN complexes and compare
them to that of the corresponding CK2-CRBN and CK2-Lenalidomide-CRBN complexes. Our final
aim is to understand the structural differences and dynamic behaviors in both sets of complexes
that will be exploited for the design of CK2 selective PROTACs.
Acknowledgements: Financial support from RTI2018-093539-B-I00 (MICIU/FEDER, UE) is kindly
acknowledged. Laura Marquez-Cantudo thanks Universidad San Pablo CEU and Banco Santander for a
Young Researcher contract.
[1]. Smith, B. E.; Wang, S. L.; Jaime-Figueroa, S.; Crews, C.M.; Harbin, A.; Wang, J.; Hamman, B. D. Nature Communications 2019, 10, 131. [2]. Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O'Brien, S. E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; Proffitt, C.; Schwaebe, M. K.; Ryckman, D. M.; Rice, W. G.; Anderes, K. Cancer Research 2010, 70, 10288-10298. [3]. Swider, R.; Maslyk, M.; Zapico, JM.; Coderch, C.; Panchuk, R.; Skorokhyd, N.; Schnitzler, A.; Niefind, K.; de Pascual-Teresa, B.; Ramos, A. RSC Advances, 2015, 5 (89), 72482-94. [4]. Rangasamy, L.; Ortín, I.; Zapico, J. M.; Coderch, C.; Ramos, A.; de Pascual-Teresa, B. ACS Medicinal Chemistry Letters 2020, 11, 713-719. [5]. Chamberlain, P. P.; Cathers, B. E. Drug Discovery Today: Technologies 2019, 31, 29-34.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
78
Poster Communications (PC_39)
Novel benzenesulfonate scaffolds with a high anticancer activity and G2/M cell cycle arrest
Jacek Mularski,a Michał Kuczak,a,b Anna Mrozek-Wilczkiewicz,b Robert Musioła and
Katarzyna Malarzb
a Institute of Chemistry, University of Silesia in Katowice, 75 Pułku Piechoty 1a, 41-500 Chorzów, Poland
b Chełkowski Institute of Physics and Silesian Centre for Education and Interdisciplinary Research, University of Silesia in Katowice, 75 Pułku Piechoty 1a, 41-500 Chorzów, Poland
E-mail: [email protected]
Sulfonates, unlike their derivatives, sulphonamides, have rarely been investigated for their anti-
cancer activity. Unlike the well-known sulphonamides, esters are mainly used as convenient
intermediates in chemical synthesis. We present the first in-depth investigation of quinazoline
sulfonates. A small series of derivatives were synthesized and tested for their anticancer activity.
Based on their structural similarity, these compounds resemble tyrosine kinase inhibitors and the
p53 reactivator CP-31398. Their biological activity profile, however, was more related to
sulphonamides because there was a strong cell cycle arrest in the G2/M phase. Our results
became increasingly interesting as tosylate was not only active at the submicromolar level but also
had a good selectivity profile and a specific mechanism of action.
N
N
O
OCH3
S
OO
OO
N
N
O
OCH3
S
OO
CH3
Cl
N
N
O
OCH3
S
OO
CH3
N
N
OS
OO
CH3
OCH3
BS1 BS3 BS4 BS5
No. Activity IC50 Value [μM]
K562 HCT 116 p53+/+ HCT 116 p53−/− U-251 PANC-1 NHDF
BS1 0.172 ± 0.034 0.880 ± 0.086 0.563 ± 0.121 1.897 ± 0.649 3.981 ± 0.597 12.540 ± 0.855
BS3 0.078 ± 0.027 0.363 ± 0.028 0.239 ± 0.030 1.757 ± 0.388 0.097 ± 0.030 9.415 ± 1.652
BS4 0.173 ± 0.031 1.567 ± 0.357 3.724 ± 0.487 1.907 ± 0.214 0.235 ± 0.042 >25
BS5 10.190 ± 0.819 >25 >25 >25 >25 >25
CP-31398 3.087 ± 0.360 18.63 ± 0.92 26.28 ± 1.41 18.77 ± 1.65 >25 12.26 ± 0.54
Imatinib 0.133 ± 0.030 44.55 ± 2.41 51.21 ± 4.09 >25 >25 >25
Figure 1: Structures and anticancer activity of tested sulfonates.
[1] Malarz, K.; Mularski, J.; Kuczak, M.; Mrozek-Wilczkiewicz, A.; Musiol, R., Cancers 2021, 13, 1790.
The reported studies are financial supported by the Polish National Center for Science (NCN, grant no 2019/35/B/NZ5/04208).

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
79
Poster Communications (PC_40)
Ebselen-inspired multitarget compounds as potential neuroprotective agents.
Miguel Muñoz,a Giorgio Giorgi,a J. Carlos Menéndez a.
a Unidad de Química Orgánica y Farmacéutica, Departamento de Química en Ciencias Farmacéuticas, Facultad de Farmacia, Universidad Complutense, 28040, Madrid, Spain
E-mail: [email protected]
Ebselen (EBS) is a non-toxic organo-selenium compound that has attracted considerable interest
in medicinal chemistry. It is understood as a benchmark compound in view of its promising
antioxidant function. [1] Moreover, EBS exhibits a broad spectrum of biological activities (anti-
inflammatory, anti-tumoral, antibacterial, neuroprotective, cytoprotective etc.), having been
effective in the treatment of a range of maladies including diabetes, neurodegenerative diseases,
cancer.
Neurodegeneration of the central nervous system is characterized by a progressive loss of
neuronal structure and function resulting in physical and mental impairments. Historically, “one
molecule – one target – one disease” paradigm has been the basis of drug discovery. This
approach has not been successful for neurodegenerative disease, due to the multifactorial nature
of their pathological mechanisms. In spite of that, many of these diseases share similar cellular
hallmarks: redox homeostasis, mitochondrial dysfunction, inflammation, among others. Thus, the
interest in modern multi-target drug proposal has been relatively improved over the past two
decades. [2]
In particular, the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS)
and, therefore, oxidative damage may not be the primary cause of etiology of this kind of diseases
but play an important role in the progression of the diseases.
Having all into account, the design, synthesis and characterization of a library of hybrid compounds
that combine the core of ebselen with antioxidant and anti-inflammatory elements was the main
objective. In this case, all derivatives were obtained using ultrasound activation for the synthesis of
Na2Se2 as a source of selenium. Moreover, using a conventionally applied nuclear magnetic
resonance spectroscopy (NMR) assay, all derivates were tested as antioxidants by GPx-like
catalyst model. [3]
[1]. Ruberte AC, Sanmartin C, Aydillo C, Sharma AK, Plano D. Development and Therapeutic Potential of
Selenazo Compounds. J Med Chem. 2020 Feb 27; 63(4):1473-1489.
[2]. Zhou, J., Jiang, X., He, S., Jiang, H., Feng, F., Liu, W., et al. (2019). Rational design of multitarget-
directed ligands: strategies and emerging paradigms. J. Med. Chem. 2019, 62, 20, 8881–8914
[3]. Kumakura, F., Mishra, B., Priyadarsini, K.I. and Iwaoka, M. (2010), A Water-Soluble Cyclic Selenide with
Enhanced Glutathione Peroxidase-Like Catalytic Activities. Eur. J. Org. Chem., 2010: 440-445.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
80
Poster Communications (PC_41)
Naturally occurring homoisoflavonoids as novel tools against breast cancer
Maria Antonietta Occhiuzzi,a Bruno Rizzuti,b Giuseppina Ioele,a Michele De Luca, a Gaetano Ragno,a Giancarlo Statti,a Antonio Garofaloa and Fedora Grandea
a Department of Pharmacy, Health and Nutritional Sciences, University of Calabria, Ampl. Polifunzionale, Via
P. Bucci, 87036 Rende (CS), Italy b CNR-NANOTEC, Licryl-UOS Cosenza and CEMIF.CAL, Department of Physics, University of Calabria, Via
P. Bucci, 87036 Rende (CS), Italy
E-mail: [email protected]
Estrogens exert a panel of biological activities mainly through the estrogen receptors (ER) α and β,
which belong to the nuclear receptor superfamily. Diverse studies have shown that several
chemical compounds known as phytoestrogens, derived from plants, demonstrate the ability to
bind to these receptors, producing estrogenic or anti-estrogenic activity. Therefore, their biological
effects could be potentially useful for the formulation of specific nutraceuticals or pharmaceutics. In
particular, homoisoflavonoids, phenolic compounds found abundant in bulbs of Scilla scilloide,
showed antiproliferative properties in different cancer models. Herein we investigate the capability
of two isomeric homoisoflavonoids, I and II (Figure 1) to bind to the active site of both ERα and
ERβ [1,2]. Molecular docking studies on the ERs crystallographic structure have been accordingly
carried out and the results obtained indicate that the two isomers are able to interact with the key
residues of the active site of both receptors, also showing a great conformational adaptability in
their binding geometry. These results suggest that the studied homoisoflavonoids represent
promising starting points for the development of selective agents useful in hormone replacement
therapies or as complements in the treatment of hormone-dependent breast cancers.
Figure 1. Chemical structure of studied homoisoflavonoids.
[1] Traboulsi, T.; El Ezzy, M.; Gleason, J.L.; Mader, S. Journal of Molecular Endocrinology 2017, 58, R15–
R31.
[2] Grande, F.; Rizzuti, B.; Occhiuzzi, M. A.; Ioele, G.; et al. Molecules 2018, 23, 894-908.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
81
Poster Communications (PC_42_FC-4)
New cytisine-based multitarget compounds for neurodegenerative diseases
Emmanuel Orocio,a José Clerigué,a M. Teresa Ramos,a Rafael Leónb and J. Carlos Menéndeza
a Departamento de Química en Ciencias Farmacéuticas, Unidad de Química Orgánica y Farmacéutica, Universidad Complutense de Madrid, Pza. de Ramón y Cajal s/n, 28040, Madrid, Spain.
b Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM-CSIC), Calle Juan de la Cierva 3, 28006, Madrid, Spain.
E-mail: [email protected]
Neurodegenerative diseases (NDDs) are one of the major concerns in medical sciences, as they
are connected to the aging of world population. Due to their complex etiology, early diagnosis and
effective treatments are not available in clinical practice, so new strategies are in development in
order to overcome these drawbacks. The design of multitarget directed ligands (MTDLs) seems to
be one of the most promising emerging approaches in this area [1]. MTLDs are molecules that
contain several moieties that display pharmacological activities on different targets.
Considering the key role of oxidative stress in the pathogenesis of NDDs [2], our group has been
interested in the design and synthesis of new MTLDs by combining two moieties with antioxidant
properties. Our design is based on cytisine, an alkaloid with activity on α4β2 and α7 nicotinic
receptors, which is involved in the regulation of the Nrf2 transcription factor [3], one of the most
important antioxidant mechanisms in animal cells, to which we added the neuroprotective and
antioxidant properties of melatonin or phenolic acids. New MTLDs are currently in pharmacological
study to assess their neuroprotective activity and mechanism of action. Moreover, we are working
in the design of new MTDLs based on cytisine analogues and other frameworks with well-known
antioxidant and/or neuroprotective properties.
Figure 1: General design of a new family of multitarget directed ligands.
[1] Singh M., Kaur M., Chadha N., Silakari O. Molecular Diversity 2016, 20 (1), 271-297. [2] Andersen J.K. Nature Medicine 2004, 10, 18-25. [3] Bencherif M. Acta Pharmacologica Sinica 2009, 30 (6), 702-714.
NH
NH
ONH
O NH
NH
OHN
NH
NN
R1
R2
HN
NH
NN
O
R
R3
R4
Melatonin-like moietyNrf2 inducer, antioxidant,
antiinflamatory, radical scavenger, neurogenic.
Cytisine deaza analogueNrf2 inducer, antioxidant,
neuroprotective.
Michael aceptor linkerNrf2 inducer.
Amide bondAt this position, it is esencial for antioxidant activity of melatonin.
Cytisine diazabicyclic analogueNrf2 inducer, antioxidant,
neuroprotective.
Cinnamic amideNrf2 inducer, antioxidant,
radical scavenger.
A B C

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
82
Poster Communications (PC_43)
Development of machine learning models to predict bioactivity of inhibitors across three kinases representative for the kinome.
Deborah Palazzotti,a Maria-Anna Trapotsi,b Layla Hosseini-Gerami,b Andrea Astolfi,a Maria Letizia Barrecaa and Andreas Benderb
a Department of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123-Perugia, Italy. b Centre for Molecular Informatics, Department of Chemistry, Cambridge University, Lensfield Road, CB2
1EW-Cambridge, United Kingdom.
E-mail: [email protected]
Understanding the selectivity of kinase inhibitors is a challenging task in drug discovery [1].
Recently, using a collection of inhibitors with known multi-kinase activity has led to the conclusion
that testing new candidate compounds against three specific kinases may be sufficient for a first-
pass evaluation of the bioactivity profile of inhibitors [2].
Therefore, the aim of our work is to provide a computational tool able to assess the potential
compound activity against a set of three kinases, namely ABL1, KDR and GSK3β. To this end, we
used 4587 bioactivity data retrieved from ChEMBL and 13162 from ExCAPE database.
Classification models were trained with Random Forest (RF) and extended connectivity fingerprints
(ECFPs) were used as compounds’ descriptors. RF models produced an overall balanced
accuracy (BA) with values of 0.74 (ABL1), 0.82 (KDR) and 0.83 (GSK3β). Subsequently, due to the
class imbalance of the dataset, oversampling and undersampling techniques were applied to
improve the model performance. We found that the oversampled RF model performed similarly to
the model generated with real data (ABL1 and KDR). In contrast, the undersampling approach,
applied to GSK3β, resulted in an improvement of the model accuracy with a BA value which
increased from 0.83 to 0.94.
Currently, these models are being experimentally and prospectively validated using an external
test set of FDA-approved kinase inhibitors.
[1] Davis, M.I.; Hunt, J.; Herrgard, S.; Ciceri, P.; Wodicka, M.L.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P., Nature biotechnology 2011, 29 (11), 1046-1051. [2] Laufkötter, O.; Laufer, S.; Bajorath, J., European Journal of Medicinal Chemistry 2020, 204, 112641.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
83
Poster Communications (PC_44)
Identification of novel dual inhibitors towards SET/EED domains of PRC2 through a structure-based drug design approach
Giulia Panzarella,a Roberta Rocca,b,c Annalisa Maruca,a,c Raffaella Catalano,a,c Francesco Ortuso,a,c and Stefano Alcaroa,c
a Dipartimento di Scienze della Salute, Università “Magna Grӕcia” di Catanzaro, Viale Europa, 88100 Catanzaro, Italy.
b Dipartimento di Medicina Clinica e Sperimentale, Università "Magna Græcia" di Catanzaro, Viale Europa, 88100 Catanzaro, Italy.
c Net4Science srl, Università “Magna Grӕcia” di Catanzaro, Viale Europa, 88100 Catanzaro, Italy.
E-mail: [email protected]
PRC2 belongs to Polycomb group (PcG) proteins, which are epigenetic regulators of transcription
through post-translational modification (PTM) of histones. It is highly expressed in proliferating
cells, where it induces gene silencing by its methyltransferase activity on the lysine 27 of the
histone H3 [1]. PRC2 has a catalytic subunit, known as EZH2, containing the SET enzymatic
domain and requires the EED subunit for promoting its activity [2]. In this regard, PRC2 inhibition is
a promising approach for treating complicated PRC2-dependent diseases, such as cancer. By
drawing inspiration from the polypharmacology paradigm [3], we developed a Structure-Based
Virtual Screening (SBVS) for the design of small molecules with potential dual activity towards
SET/EED PRC2 domains, in order to prevent cell proliferation and stages of cancer advancement.
Thus, the Mu.Ta.Lig. Chemotheca [4] entities were screened through pharmacophore and docking
approaches, and shared hits have been chosen. These efforts led to select some interesting
compounds with a potential dual SET/EED binding profile.
This work is supported by EU PON fundings.
Figure 1: Workflow study and PRC2 inhibition implication.
[1] Justin, N.; Zhang, Y.; Tarricone, C.; et al. Nat Commun. 2016, 7, 11316.
[2] Chittock, E.; Latwiel, S.; et al. Biochemical Society Transactions. 2017 45, 193-205. [3] Alcaro, S.; Ortuso, F., Eur J Med Chem. 2020, 192, 112188.
[4] Ortuso, F.; Bagetta, D.; Maruca, A.; et al. Front Chem. 2018, 19, 6-130.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
84
Poster Communications (PC_45)
Natural compounds could become an answer to the antimicrobial resistance.
Maria Rosaria Paravati,a Giosuè Costa,a, b, c and Stefano Alcaroa, c
a Dipartimento di “Science della Salute”, Università “Magna Græcia”, Campus Salvatore Venuta, 88100, Catanzaro, Italy.
b Net4Science srl, c/o Università “Magna Græcia” di Catanzaro, 88100, Catanzaro, Italy. c Associazione CRISEA – Centro di Ricerca e Servizi Avanzati per l’innovazione Rurale,
località Condoleo di Belcastro (CZ), Italy.
E-mail: [email protected]
Antimicrobial resistance is currently a serious problem for the treatment of bacterial and fungal
infections. Many microorganisms develop and transmit new resistance mechanisms that cancel the
action of the antimicrobial drugs in clinical use. Many of the numerous natural compounds possess
antimicrobial activity, therefore they could represent a valid alternative to the antimicrobial drugs
currently in use, which can be ineffective. In this study, considering three natural compounds, was
performed a conformation analysis using crystallographic models obtained from the database
Protein Data Bank [1]. In particular, the natural compound selected for the study are:
epigallocatechin gallate, a phenolic antioxidant found in a number of plants such as green and
black tea [2], complexed with D80A-Fructofuranosidase from Xanthophyllomyces Dendrorhous;
curcumin, complexed with streptococcal dehydrogenase, this compound is a phytopolylphenol
pigment isolated from the plant Curcuma longa [2]; and phloretin, a natural dihydrochalcone found
in apples and many other fruits [3] that is complexed with a Multidrug Binding Protein Ttgr from
Pseudomonas putida.
Figure 1: 2D Structures of respectively: A) epigallocatechin gallate, B) curcumin and C) phloretin.
[1] RCSB PDB: www.rcsb.org
[2] National Cancer Institute (NCI): www.ncit.nci.nih.gov/ncitbrowser/
[3] DrugBank: https://www.drugbank.ca/drugs/DB07810

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
85
Poster Communications (PC_46)
Sonchus asper (L.) Hill from Campania region: chemical composition and bioactivity
Valentina Parisi,a Valentina Santoro,a Luigi Milella, b Fabrizio Dal Piaz c and Nunziatina De Tommasia
a Department of Pharmacy, Università degli Studi di Salerno, Via Giovanni Paolo II, 84084- Fisciano, Italia. b Department of Science, Università degli Studi della Basilicata, Viale dell’Ateneo Lucano n° 10, 85100-
Potenza, Italia. c Department of Medicine and Surgery, Università degli studi di Salerno, Via S. Allende n° 43, 84081-
Baronissi, Italia.
E-mail: [email protected]
Spontaneous edible plants, which have been an essential component of rural diet since ancient
times, have now been almost replaced by limited varieties cultivated during the era of globalization.
In fact, the selection of more productive cultivars and the industrial cultivation has led to an
impoverishment of the diet, both in terms of choice and nutrition. In contrast to this event and with
the aim to promote the use of forgotten species used in Campanian tradition, a chemical-biological
study on Sonchus asper (L.) Hill was carried out. Sonchus asper (Asteraceae) is a spontaneous
plant that, especially in the past, was harvested and consumed as vegetables by the population of
the inland areas of the Campania region. Previous investigations reported the presences of
polyphenols, terpenes, carotenoids as main specialized metabolites [1, 2]. The plant is also used
for the treatment of several human disorder such as gastrointestinal infection, wounds and burns
cough, diabetes, and inflammation disease [3]. Raw and cooked plant, harvest in Benevento, was
subjected to extraction through green technologies such as Microwave Assisted Extraction (MAE),
and Ultrasound Assisted Extraction (UAE) using hydroalcoholic solvents. The extracts were
subjected to LC-HRESIMS/MS analyses to perform a quali-quantitative profile of bioactive
molecules and were tested to evaluate their antioxidant activity in vitro. Flavonoids, fatty acids, and
phenolic acids classes were identified as major components, while lactone sesquiterpenes,
glycosides and coumarins were found in traces. In particular, the presence of polyunsaturated fatty
acids, and polyphenols, both in the cooked and raw matrix confirms the interesting nutritional value
of the species [4], whose usual reintroduction in the diet would represent a healthy and sustainable
choice.
[1] Giambanelli, E.; Filippo D'Antuono, L.; Romero‐González, R.; Garrido Frenich, A., Journal of the science of food and agriculture 2018, 98 (3), 945-954. [2] Xu, Y.-J.; Sun, S.-B.; Sun, L.-M.; Qiu, D.-F.; Liu, X.-J.; Jiang, Z.-B.; Yuan, C.-S., Food chemistry 2008, 111 (1), 92-97. [3] Khan, I. U.; Khan, F. U.; Hussain, J.; Badshah, S.; Muhammad, N.; Khan, R. A.; Kait, C. F.; Ali, M. A.; Khan, H.; Aslam, M. W., Asian Journal of Chemistry 2014, 26 (9), 2699. [4] Guil‐Guerrero, J.L.; Giménez‐Giménez, A.; Rodríguez‐García, I.; Torija‐Isasa, M.E., Journal of the Science of Food and Agriculture 1998, 76, 628-632.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
86
Poster Communications (PC_47)
Inhibition of ZIKA virus replication by novel inhibitors of NS2B/NS3 complex
Desirée Pecora,a Giuseppe La Regina,a Antonio Coluccia,a Marianna Nalli,a Jin-Ching Lee,b and Romano Silvestri.a
a Laboratory Affiliated to Institute Pasteur Italy − Cenci Bolognetti Foundation, Department of Drug Chemistry and Technologies, Sapienza University of Rome, I00185 Rome, Italy
b Department of Biotechnology, College of Life Science, College of Medicine, and Drug Development and Value Creation Research Center, Kaohsiung Medical University, Kaohsiung 807, Taiwan.
E-mail: [email protected]
Zika virus (ZIKV) is an RNA virus of the Flaviviridae family, which is responsible for a condition
known as Zika fever. While initially, the infection was endemic only in Africa and Asia, now it is
spread all over the world. ZIKV is a viral ailment transmitted by Aedes mosquitoes, mainly located
in the equatorial zone. The symptoms are generally mild. Nevertheless, during pregnancy this
infection can cause the birth of infants with microcephaly or other inborn malformation. The NS2B /
NS3 viral protease complex is implicated in virus replication and immune system escape, so that
there is growing interest in the design of new ZIKV inhibitors with this target. Preliminary studies
with molecular models provided insights into the molecular determinants responsible for its high
affinity toward the target enzyme. On these bases, we have designed and synthesized 14 new
potential allosteric inhibitors of the NS2B / NS3 complex, characterized by an indole with a benzyl
or benzoyl group in position 3 [1].
Two of these new compounds, showed strong activity in both enzymatic and cellular assays. As a
proof of concept, the most promising compound was evaluated in a mouse animal model. (Figure
1). Not only the compound has significantly reduced NS2B / NS3 synthesis and viral replication,
but also it prevented the mice from a life-threatening infection showing a powerful reduction of the
brain damages produced by the viral infection. These results pave the way to new ZIKV drug
candidates able to cross the blood-brain barrier to reach the neural cells.
Figure 1: Proposed binding mode and biological evaluation of the derivative.
[1] A. Coluccia et al. ACS Med. Chem. Lett. 2020, 11, 10, 1869–1874.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
87
Poster Communications (PC_48)
Synthesis of enantiopure benzothiazepines and benzodiazepines for
their study as calcium channel blockers and neuroprotectors.
J.M. Pérez,a Á. Cores,a M. Villacamapa,a J.C. Menéndez.a
a Unidad de Química Orgánica y Farmacéutica, Departamento de Química en Ciencias Farmacéuticas, Facultad de Farmacia, Universidad Complutense, 28040, Madrid, España.
E-mail: [email protected]
The improvement in lifespan promotes an increase in age-related diseases, such as
neurodegenerative diseases. Neurodegenerative diseases (NND) are characterized by the loss of
neurons in the brain and / or spinal cord and have become one of the most important health
problems worldwide. These diseases are multifactorial pathologies with different etiologies sharing
many mechanistic pathways: protein misfolding, neuroinflammation, impaired mitochondrial
function, increased oxidative stress, mitochondrial dysfunction, alterations in calcium homeostasis,
among others [1].
Dysregulation of intracellular calcium is one of the most striking hallmarks of NDD. Furthermore,
lower control of Ca2 + concentrations is connected by a complex biochemical network with other
dysfunctions, including oxidative stress, energy impairment, and inadequate proteostasis.
The mNCX transporter is a mitochondrial sodium-calcium exchanger and has an important role in
the control of intraneuronal Ca+2 homeostasis [2]. The NND are characterized by an excess of
calcium in the cell cytoplasm. For this reason, it´s important to tackle the problem of the lack of
mNCX agonists.
The benzothiazepine derivative known as CGP-37157 is become in a milestone because was the
first compound reported as a selective antagonist of mNCX. CGP-37157 has been shown to be
neuroprotective in various cell models. However, one of the main limitations is the fact that it has
always been tested as a racemic mixture due to pure enantiomers have not been obtained yet [3].
Attaining an enantiopure CGP-37157 may well improve selectivity and potency as an antagonist of
mNCX.
The CGP-37157 enantiopures were obtained after racemic mixture resolution through respective
diastereoisomers by the formation of an intermediate linked to a molecule of known chirality.
[1] Pei, Y. Lilly, M. J. Owen, D. J. D’Souza, L. J. Tang, X. Q. Yu, J. Nazarbaghi, R. Hunter, A. Anderson, C. M. Glasco, S. Ede, N. J. James, I. W. Maitra, U. Chandrasekaran, S. Moos, W. H. Ghosh, S. S. J. Org. Chem. 2003, 68, 92-103. [2] Vaghy, P. L.; Johnson, J. D.; Matlib, M. A.; Wang, T.; Schwartz, A. J. Biol. Chem. 1982, 257, 6000-6002. [3] Chiesi, M.; Schwaller, R.; Eichenberger, K. Biochem. Pharmacol. 1988, 37, 4399-4403.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
88
Poster Communications (PC_49)
Design and synthesis of novel NNRTIS inhibitors for the treatment of AIDS
Anthi Petrou,1 Athina Geronikaki,1 Phaedra Eleftheriou,2 Melpomeni G. Akrivou,3 Ioannis
Vizirianakis,3 Maria Fesatidou1
E-mail: [email protected]
1 Department of Biomedical Sciences, School of Health, International Hellenic University, 57400
Thessaloniki, Greece. 2 School of Pharmacy, Aristotle University of Thessaloniki, Thessaloniki, 54124, Greece.
3 School of Pharmacy Department of Pharmacology and Pharmacognosy, Aristotle University of Thessaloniki, 54124, Greece.
HIV is the causative agent of acquired immunodeficiency syndrome (AIDS), an infectious disease with increasing incidence worldwide. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) play an important role in the treatment of AIDS. Although, many compounds are already being used as anti-HIV drugs, research for the development of new inhibitors continues as the virus develops resistant strains. Taking into account the best features of available NNRTIs we design a novel series of inhibitors. PASS (Prediction of activity spectra for substances) prediction program and molecular docking studies were used for the selection of the designed compounds for the synthesis. Selected compounds were synthesized using conventional and microwave irradiation methods and HIV RT inhibitory action was evaluated by colorimetric photometric immunoassay. The evaluation of HIV-1 RT inhibitory activity revealed that seven compounds out of 24 have significantly lower ΙC50 values than nevirapine (0.3 μΜ). In particularly, three of the compounds exhibited IC50 values lower than 5 nM and two compounds 9 and 10 exhibited very good inhibitory activity with IC50 1 nM. It was observed that the activity of compounds depends not only on the nature of substituent and it position in benzothiazole ring but also on the nature and position of substituents in benzene ring.
Figure 1: Docking of TMC125 (yellow) and compounds 1 (green), 9(blue), 14 (light blue) and 27 (purple) to the allosteric site of HIV-1 RT.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
89
Poster Communications (PC_50)
In Silico Evaluation of the Effectivity of Approved Protease Inhibitors against the Main Protease of the Novel SARS-CoV-2 Virus
Phaedra Eleftheriou,1 Dionysia Amanatidou,1 Anthi Petrou,2 Athina Geronikaki 2
1 Department of Biomedical Sciences, School of Health, International Hellenic University, 57400
Thessaloniki, Greece. 2 School of Pharmacy, Aristotle University of Thessaloniki, Thessaloniki, 54124, Greece.
E-mail: [email protected]
Novel coronavirus SARS-CoV-2, is responsible for the pandemic, which first appeared in Wuhan, China, causing the death of approximately 10000 people daily. Coronavirus, has 79.5% sequence identity with SARS-CoV-1 and the same strategy for host cell invasion through the ACE-2 surface protein. Since the development of novel drugs is a long-lasting process, researchers look for effective substances among drugs already approved or developed for other purposes. In this work, the 3D structure of the SARS-CoV-2 main protease was compared with the 3D structures of seven proteases, which are drug targets, and docking studies to the SARS-CoV-2 protease structure of thirty-four approved and on-trial protease inhibitors was performed. Increased 3D structural similarity between the SARS-CoV-2 main protease, the HCV protease and α-thrombin was found. According to docking analysis the most promising results were found for HCV protease, DPP-4, α-thrombin and coagulation Factor Xa known inhibitors, with several of them exhibiting estimated free binding energy lower than −8.00 kcal/mol and better prediction results than reference compounds. Since some of the compounds are well-tolerated drugs, the promising in silico results may warrant further evaluation for viral anticipation. DPP-4 inhibitors with anti-viral action may be more useful for infected patients with diabetes, while anti-coagulant treatment is proposed in severe SARS-CoV-2 induced pneumonia.
Figure 1: The 3D structural alignment between the SARS-CoV-2 main protease and the HIV-1 protease (A), and the HCV protease (B).

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
90
Poster Communications (PC_51)
Detection of structural features influencing compound physicochemical properties with the use of SHAP values
Agnieszka Pocha,a Rafał Jankowski,a and Sabina Podlewskab,c
a Faculty of Mathematics and Computer Science, Jagiellonian University, 6 S. Łojasiewicza Street, 30-348 Kraków, Poland
b Maj Institute of Pharmacology, Polish Academy of Sciences, 12 Smętna Street, 31-343 Kraków, Poland c Department of Technology and Biotechnology of Drugs, Faculty of Pharmacy, Jagiellonian University
Medical College, 9 Medyczna Street, 30-688 Kraków, Poland
E-mail: [email protected]
Provision of desired affinity profile of compounds towards defined set of protein targets is just the
first step in the long and expensive drug design pipelines. Another important factor, which can
determine whether particular compound can become a future drug are its physicochemical and
pharmacokinetic properties [1].
There exists a wide range of computational approaches enabling evaluation of selected
physicochemical and ADMET properties. Most of them works in such a way that they provide
information about the predicted value of particular parameter. The estimates are most often made
by machine learning (ML) models of various type [2].
In the study, we developed a methodology based on the SHapley Additive exPlanations (SHAP)
values [3] to explain ML models predictions. It uses key-based compound representations
(MACCS keys and Klekota&Roth Fingerprint were used in our study) and indicates the contribution
of particular structural feature to the ML model output. We focused on the assessment of metabolic
stability, but the methodology can be applied to the whole range of properties. In future work, we
plan to extend it also to the evaluation of solubility, biological membranes permeability, binding to
plasma proteins, hERG channels blocking and hepatotoxicity.
The indication of structural features important for compound physicochemistry can be of great help
in the design of structures with improved physicochemical and ADMET profile.
Acknowledgments
The study was supported by the grant OPUS 2018/31/B/NZ2/00165 financed by the National
Science Centre, Poland (www.ncn.gov.pl)
[1] Rao, V. S.; Srinivas, K. Modern drug discovery process: An in silico approach. J. Bioinf. Seq. Anal. 2011, 5, 89–94. [2] Varnek, A.; Baskin, I. Machine learning methods for property prediction in chemoinformatics: quo vadis? J. Chem. Inf. Model. 2012, 6, 1413–1437. [3] Lundberg, S.M.; Lee, S. An Unified Approach to Interpreting Model Predictions. In: Advances in Neural Information Processing Systems 30, Guyon, I.; Luxburg, U.V.; Bengio, S.; Wallach, H.; Fergus, R.; Vishwanathan, S.; Garnett, R. (eds) 2017, Curran Associates Inc, 4765-4774.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
91
Poster Communications (PC_52_FC-5)
A novel scaffold for potent and selective inhibition of tumor-related carbonic anhydrase isoforms IX and XII
Virginia Pontecorvi,a Paolo Guglielmi,a Emanuela Berrino,a Secci Daniela,a
a Department of “Chimica e Tecnologie del Farmaco”, Sapienza University of Rome, P.le A. Moro 5, 00185 Rome, Italy
E-mail: [email protected]
Human carbonic anhydrases (hCAs) are known as useful targets for antitumoral therapy.
Particularly, the selective inhibition of tumor-related isoforms IX and XII has been recognized as a
promising therapy tool for the treatment of solid tumors [1]. Different inhibitors have been devised
based on the structure and the mechanism of these enzymes [2]. The ability of the coumarin
structure-based compounds to inhibit hCAs prompted us to design a series of derivatives
originated from a molecular simplification of the coumarin nucleus (Scheme 1a) [3]. The novel
pyran-2-one scaffold has been endowed with carboxamidic linker to achieve a final structure
bearing different substituents. The latter allow the designed compounds to mime the coumarin
structure but also to interact with the active site by modulating their physic-chemical properties. In
order to achieve the amides, the synthetic strategy based on the one-pot coupling reaction of the
coumalic acid with different amines has been performed as shown in Scheme 1b.
Scheme 1. a) Design of the novel hCAs inhibitors; b) one-pot synthesis of the cumalic acids amides.
The activity and selectivity of these inhibitors towards different isoforms of human carbonic
anhydrase (hCA I, II, IX and XII) were evaluated showing to be inactive against the two cytosolic
off-target hCA I and II (hCA I, II > 100 µM). Conversely, these compounds inhibited hCA IX and XII
in the low nanomolar range (0.072 µM < hCA IX < 3.69 µM; 0.068 µM < hCA XII < 0.80 µM).
[1] Kumar, S.; Rulhania, S.; Jaswal, S.; Monga, V. Eur. J. Med. Chem. 2021, 209, 112923.
[2] Supuran, C.T. J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360.
[3] Cornelio, B.; Laronze-Cochard, M.; Miambo, R.; De Grandis, M.; Riccioni, R.; Borisova, B.; Dontchev, D.;
Machado, C.; Ceruso, M.; Fontana, A.; et al. Eur. J. Med. Chem. 2019, 175, 40–48.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
92
Poster Communications (PC_53)
SARS-CoV2 spike emerging mutants effects on the ACE-2 molecular recognition: A theoretical study
Francesca Procopio,a Stefano Alcaro,a,b and Francesco Ortusoa,b
a Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, Campus “S. Venuta”,
Catanzaro, Italy. b Net4Science Academic Spin-off, Università “Magna Græcia” di Catanzaro, Campus “S. Venuta”, Catanzaro,
Italy.
E-mail: [email protected]
Spike glycoprotein mediates the viral cell entry of novel Severe Acute Respiratory Syndrome
Coronavirus 2 (SARS-CoV2) by binding the human receptor Angiotensin Converting Enzyme 2
(ACE2) [1]. Through the virus genome sequencing, several Spike Receptor Binding Domain (RBD)
mutations have been identified. These Spike polymorphisms could modify the ACE2 recognition
enhancing virus diffusion and immune escape capability, with repercussion on vaccines
effectiveness [2]. In the present study, GISAID database (https://www.gisaid.org/) has been
searched for the higher frequency Spike RBD mutations. With the aim to explore the effects of the
most frequent (> 1%) Spike RDB mutations on ACE2 recognition, Molecular Dynamics simulations
on wilde-type and selected mutated Spike RBD models have been performed, either in unbounded
or in ACE2 bounded state. Furthermore, Grid-Based Pharmacophore Models (GBPM) analysis has
been performed to highlight most relevant residues involved in the Spike-ACE2 complex
stabilization [3] [4].
Figure 1. Spike-ACE2 complex 3D structure (PDB: 6LZG). In red the most frequent mutations on the
RBD analyzed in the present study.
N439K
N501Y
S477N
T478K
K417N/T
E484K
[1] M. Letko, A. Marzi e V. Munster, Nat. Microbiol, n. 5, pp. 562-569, 2020.
[2] R. Chen, X. Zhang e J. e. a. Case, Nat Med, vol. 27, pp. 717-726, 2021.
[3] F. Ortuso, T. Langer e S. Alcaro, Bioinformatics, vol. 22, n. 12, pp. 1449-55, 2006.
[4] F. Ortuso, P. H. G. Daniele Mercatelli e F. M. Giorgi, J Biomol Struct Dyn, vol. 13, pp. 1-11, 2021.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
93
Poster Communications (PC_54)
3-D QSAR and COMBINE models to predict the bioactivity of potential inhibitors of SARS-CoV-2 main protease
Eleonora Proia,a Manuela Sabatino,a Lorenzo Antonini,a Filippo Sapienzaa and Rino Ragnoa
a Department of Chemistry and Technology of Drugs, Sapienza University of Rome, Piazzale Aldo Moro, 5, 00185, Rome (RM), Italy
E-mail: [email protected]
The main protease (Mpro) of SARS-Cov-2 is an essential enzyme for maturation of functional
proteins implicated in viral replication and transcription. The peculiarity of its specific cleavage site
joint with its high degree of conservation among all coronaviruses promote it as an attractive target
to develop broad-spectrum inhibitors, with high selectivity and tolerable safety profile [1]. The
present study provides a combined usage of three-dimensional quantitative structure–activity
relationships (3-D QSAR) and comparative molecular binding energy (COMBINE) analysis to build
robust and predictive statistical models through the well-established web portal www.3d-qsar.com
[2]. Models were trained on experimental binding poses of co-crystallized Mpro-inhibitors and
validated on available literature data. After an exhaustive search of optimal parameters to enhance
both robustness and predictiveness of the models, we obtained final statistical values of r2, q2 and
SDEP with reliable performances. Despite the different nature (ligand-based and structure-based)
of the employed methods, their outcome converged. The obtained results will guide future rational
design and/or virtual screening campaigns with the aim of discovering new potential anti-
coronavirus candidates, minimizing both time and financial resources.
Figure 1: Graphical output of 3-D QSAR and COMBINE techniques in Mpro binding site.
[1] Cannalire, R.; Cerchia, C.; Beccari, A.R. ; Di Leva, F.S.; Summa V., Journal of Medicinal Chemistry 2020. [2] Ragno, R., Journal of Computer-Aided Molecular Design 2019, 33 (9), 855-864

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
94
Poster Communications (PC_55)
A- And C-Ring Steroidal Olefins Versus Epoxides as Aromatase Inhibitors: Design, Synthesis, Biochemical Activity Evaluation, and
Structure-Activity Relationships
Fernanda M.F. Roleira,1 Saul C. Costa,2 Ana R. Gomes,1 Carla L. Varela,1 Cristina Amaral,3 Tiago Augusto,3 Georgina Correia-da-Silva,3 Natércia Teixeira,3 Elisiário J.
Tavares-da-Silva1
1Univ Coimbra, CIEPQPF, FFUC, Laboratory of Pharmaceutical Chemistry, Azinhaga de Santa Comba, Pólo III - Pólo das Ciências da Saúde, 3000-548 Coimbra, Portugal.
2Univ Coimbra, FFUC, Laboratory of Pharmaceutical Chemistry, Azinhaga de Santa Comba, Pólo III - Pólo das Ciências da Saúde, 3000-548 Coimbra, Portugal.
3UCIBIO.REQUIMTE, Laboratory of Biochemistry, Department of Biological Sciences, Faculty of Pharmacy, University of Porto, 4050-313 Porto, Portugal.
E-mail: [email protected]
Breast cancer is the most frequently diagnosed cancer in women worldwide [1] and about 80% are
estrogen dependent. Thus, one of the therapeutic strategies resides in using drugs that reduce the
circulating estrogen levels, such as aromatase inhibitors (AIs). AIs undoubtedly have a major role
in the treatment of breast cancer, in all stages. However, there are only three AIs in clinical use
with several associated side effects. Therefore, research to discover new AIs is very relevant. Our
group investigates in the medicinal chemistry and cell biology fields related with the steroidal AIs
[2]. Steroidal olefins and epoxides have shown strong activity as AIs. In this work, we intend to go
further in this research by designing, synthesizing, and studying new olefins and epoxides as
steroidal AIs. Based on this, we have studied some olefins and the corresponding epoxides at C-1,
C-2 and C-9, C-11 positions (Figure1). We found that A-ring epoxides were less potent than A-ring
olefins. On the contrary, C-ring epoxides were more potent than C-ring olefins, leading to the
discovery of a very strong aromatase inhibitor, with an IC50 of 0.011 μM (the steroidal AI in clinical
use, Exemestane, presents an IC50 of 0.050 μM). Overall, this work can contribute to the discovery
of new lead compounds which can be translated to new drugs for the treatment of hormone-
dependent breast cancer.
Figure 1: Steroidal olefins and epoxides as AIs.
[1] Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics
2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA
Cancer J Clin. 2020;0(0):394–424.
[2] Roleira FMF, Varela C, Amaral C, Costa SC, Correia-Da-Silva G, Moraca F, et al. C-6α- vs C-7α-
Substituted Steroidal Aromatase Inhibitors: Which Is Better? Synthesis, Biochemical Evaluation, Docking
Studies, and Structure-Activity Relationships. J Med Chem. 2019;62(7):3636–57.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
95
Poster Communications (PC_56_FC-6)
Hybrid design, synthesis and in vitro biological evaluation of 1H-indazoles as MAO B inhibitors: effect of 1,2,4-oxadiazole bioisosteric
replacement of the amide linker
Mariagrazia Rullo,a Marco Catto,a Anna Linusson,b Cosimo Damiano Altomare,a and Leonardo Pisania
aDepartment of Pharmacy-Drug Sciences, University of Bari “Aldo Moro”, via Orabona 4, 70125, Bari, Italy b Department of Chemistry, Umeå University, 90187, Umeå, Sweden
E-mail: [email protected]
Monoamine oxidases (MAOs) A and B are flavin-containing oxidoreductases responsible for the
oxidative deamination of different amines including neurotransmitters and xenobiotics. Being
involved in the onset of oxidative stress conditions, these enzymes are potential targets for the
treatment of multifactorial neurodegenerative diseases such as Alzheimer's Disease (AD) [1]. In
this context, 1H-indazole [2] nucleus was chosen as a privileged scaffold for blocking MAO B
activity, crucial for AD neurotoxic cascade, and as a structural motif recurrent in many other
inhibitors targeting disease-related enzymes [3]. The rational design started from the hybridization
of already-known MAO inhibitors and envisaged amide-to-1,2,4-oxadiazoles bioisosteric
replacement as a key structural modification. Some novel indazole-containing molecules displayed
outstanding inhibitory potency in the nanomolar range along with high selectivity toward MAO B
(e.g., #1-4). The early drug-like profiling assessed kinetic solubility at physiological pH along with
stability in buffered solutions (pH 7.4 and pH 2) and human serum for the most promising indazole-
bearing MAO B inhibitors. Modeling studies allowed to retrieve reliable binding poses for the best
hits (Figure 1) and shed light on key binding interactions.
Figure 1: Design of single-targeting MAO B inhibitors bearing-indazole scaffold.
[1] Pisani, L.; Iacobazzi, R. M.; Catto, M.; Rullo, M.; Farina, R.; Denora,N.; Cellamare, S.; Altomare, C. D. European Journal of Medicinal Chemistry 2019, 161, 292-309. [2] Tzvetkov, N. T.; Hinz, S.; Küppers, P.; Gastreich, M. and Müller, C. E. Journal of Medicinal Chemistry 2014, 57, 15, 6679-6703. [3] Kamenecka, T.; Habel, J.; Duckett, D.; Chen, W.; Ling, Y. Y.; Frackowiak, B.; Jiang, R.; Shin, Y.; Song, X. and LoGrasso, P. V. The Journal of Biological Chemistry 2009, 284, 19, 12853-12861.
LINKER
NH
N
R3
R2
R1
Nitrogenheterocycle
B
Lipophilic Motif A
O
NH
HN
O
SNH
O O NO
N
O
Linker modifications:
IC50 mM or % inhibition at 10 mM
hMAO A hMAO B
1 0.07215 ± 3 %
14 ± 5 % 0.052
28 ± 1 % 0.025
2
3
4 3.16 0.015
SI
> 140
> 192
210
> 400
#
ON
N

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
96
Poster Communications (PC_57)
Design, synthesis, and biological profiling of new H2S-releasing hybrid
molecules for treatment of neurodegenerative diseases
Massimiliano Runfola,a Simona Sestito,b Luca Cerri,a Veronica La Rocca,c,d Michele Lai,c
Sheraz Gul,e Ylenia Zambito,a and Simona Rapposellia
a Department of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy
b Dept. of Chemistry and Pharmacy, University of Sassari, Via Vienna 2, 071006 Sassari, Italy
c Retrovirus Center and Virology Section, Department of Translational Research and New Technologies in
Medicine and Surgery, University of Pisa, 56100 Pisa, Italy. d Sant'Anna School of Advanced Studies, 56100 Pisa, Italy.
e Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Schnackenburgallee 114, 22525
Hamburg, Germany.
E-mail: [email protected]
Neurodegenerative diseases (NDDs) represent one of the main challenges for the 21st century
medical research. To date, no disease-modifying agent able to halt or slow down NDDs’
progression has been yet developed. However, the identification and profiling of new molecular
pathways contributing to neurodegeneration along with recent developments in MedChem
strategies are starting to make room for new hopes in the drug discovery process for NDDs. As a
matter of fact, the discovery and development of polypharmacological drugs are proving to be a
valuable approach to these complex pathologies with promising results both in preclinical and
clinical studies [1]. In this context, we recently developed a new class of hybrid small molecules
bearing pharmacophoric moieties of approved anti-NDDs drugs and capable of releasing H2S, a
gasotransmitter with a key role in pathophysiological mechanisms involved in neurodegeneration
[2]. Among this class, H2S-releasing derivatives of Rivastigmine, a cholinesterase inhibitor
currently prescribed for treatment of Alzheimer’s and Parkinson’s dementias, have demonstrated
interesting results. Particularly, compound MP30 showed exciting effects in providing protection
against several models of neuroinflammation and in reactivating the autophagy-lysosomal pathway
in an mTORC-dependent manner. Prompted by these results, we decided to further advance in the
drug development of this compound investigating its pharmacokinetics properties, representing
one of the major critical point in the development of multi-target drugs [3]. Here, we report the
chemical strategy and the pharmacological investigation that led us to the discovery of MP30 along
with the biological studies over its BBB permeation, tissue distribution, off-target liability, and
metabolic stability.
[1] Benek, O; Korabecny, J; Soukup, O. Trends in Pharmacological Sciences. 2020, 41 (7), 434-345.
[2] Sestito, S.; Daniele, S., Pietrobono, D.; Citi, V.; Bellusci, L.; Chiellini, G.; Calderone, V.; Martini, C.;
Rapposelli, S. Scientific reports 2019, 9(1), 1-11.
[3] Maramai, S.; Benchekroun, M.; Gabr, M. T.; Yahiaoui, S. BioMed Research International, 2020, 20
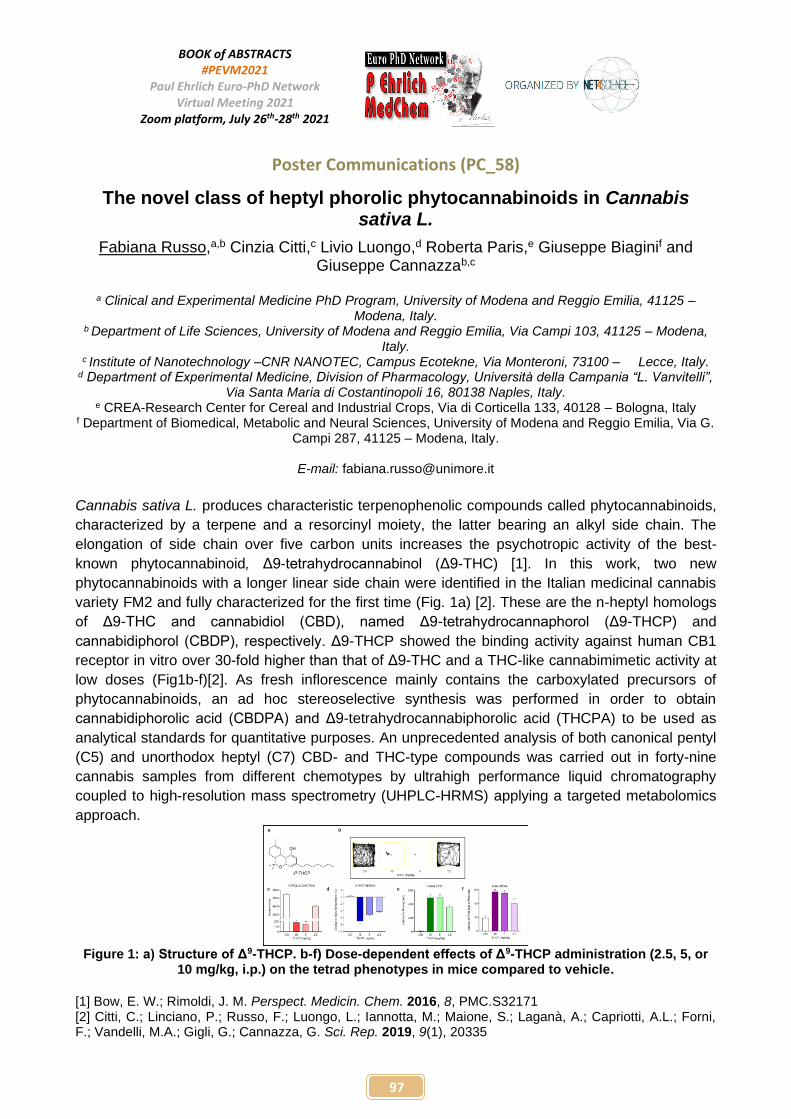
BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
97
Poster Communications (PC_58)
The novel class of heptyl phorolic phytocannabinoids in Cannabis sativa L.
Fabiana Russo,a,b Cinzia Citti,c Livio Luongo,d Roberta Paris,e Giuseppe Biaginif and Giuseppe Cannazzab,c
a Clinical and Experimental Medicine PhD Program, University of Modena and Reggio Emilia, 41125 – Modena, Italy.
b Department of Life Sciences, University of Modena and Reggio Emilia, Via Campi 103, 41125 – Modena, Italy.
c Institute of Nanotechnology –CNR NANOTEC, Campus Ecotekne, Via Monteroni, 73100 – Lecce, Italy. d Department of Experimental Medicine, Division of Pharmacology, Università della Campania “L. Vanvitelli”,
Via Santa Maria di Costantinopoli 16, 80138 Naples, Italy. e CREA-Research Center for Cereal and Industrial Crops, Via di Corticella 133, 40128 – Bologna, Italy
f Department of Biomedical, Metabolic and Neural Sciences, University of Modena and Reggio Emilia, Via G. Campi 287, 41125 – Modena, Italy.
E-mail: [email protected]
Cannabis sativa L. produces characteristic terpenophenolic compounds called phytocannabinoids,
characterized by a terpene and a resorcinyl moiety, the latter bearing an alkyl side chain. The
elongation of side chain over five carbon units increases the psychotropic activity of the best-
known phytocannabinoid, Δ9-tetrahydrocannabinol (Δ9-THC) [1]. In this work, two new
phytocannabinoids with a longer linear side chain were identified in the Italian medicinal cannabis
variety FM2 and fully characterized for the first time (Fig. 1a) [2]. These are the n-heptyl homologs
of Δ9-THC and cannabidiol (CBD), named Δ9-tetrahydrocannaphorol (Δ9-THCP) and
cannabidiphorol (CBDP), respectively. Δ9-THCP showed the binding activity against human CB1
receptor in vitro over 30-fold higher than that of Δ9-THC and a THC-like cannabimimetic activity at
low doses (Fig1b-f)[2]. As fresh inflorescence mainly contains the carboxylated precursors of
phytocannabinoids, an ad hoc stereoselective synthesis was performed in order to obtain
cannabidiphorolic acid (CBDPA) and Δ9-tetrahydrocannabiphorolic acid (THCPA) to be used as
analytical standards for quantitative purposes. An unprecedented analysis of both canonical pentyl
(C5) and unorthodox heptyl (C7) CBD- and THC-type compounds was carried out in forty-nine
cannabis samples from different chemotypes by ultrahigh performance liquid chromatography
coupled to high-resolution mass spectrometry (UHPLC-HRMS) applying a targeted metabolomics
approach.
Figure 1: a) Structure of Δ9-THCP. b-f) Dose-dependent effects of Δ9-THCP administration (2.5, 5, or
10 mg/kg, i.p.) on the tetrad phenotypes in mice compared to vehicle.
[1] Bow, E. W.; Rimoldi, J. M. Perspect. Medicin. Chem. 2016, 8, PMC.S32171 [2] Citti, C.; Linciano, P.; Russo, F.; Luongo, L.; Iannotta, M.; Maione, S.; Laganà, A.; Capriotti, A.L.; Forni, F.; Vandelli, M.A.; Gigli, G.; Cannazza, G. Sci. Rep. 2019, 9(1), 20335

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
98
Poster Communications (PC_59)
Push and pull strategy in styrylquinoline derivatives against neurodegenerative diseases
Álvaro Sarabia Vallejo,a Marta Piquero Martí,a Pilar López-Alvarado Gutiérrez,a José Carlos Menéndez Ramosa
a Unit of Organic and Pharmaceutical Chemistry, Department of Chemistry in Pharmaceutical Sciences, School of Pharmacy, Complutense University of Madrid, 28003 Madrid, Spain.
E-mail: [email protected]
At present, neurodegenerative diseases represent one of the main health issues in our society
since the remarkable increase in life expectancy has caused them to be an appreciable burden.
Protein misfolding is commonly found is this type of maladies, which leads to alterations in protein
structure and activity failures, giving rise to dysfunctions and anomalies. Among these diseases,
Alzheimer´s Disease (AD) is of special relevance, affecting nowadays around 50 million people [1].
The aetiology of AD is known to involve several alterations, but is not completely understood.
These processes are deposition of amyloid beta-peptide and hyperphosphorylated tau
proteinoxidative stress, neuroinflammation, mitochondrial dysfunction and an imbalance in
glutamatergic and cholinergic tone. Some commercially available drugs achieve a temporary relief
of the symptoms of AD, but none of them addresses the cause nor cures the disease. Though, a
treatment for AD is still to be found. Multitarget ligands, binding to different targets and regulating
several pathways at the same time, are of particular interest for multifactorial diseases like AD.
Additionally, theranostic compounds provide therapy and diagnostic information simultaneously.
In this context, and on the basis of a common scaffold of styrylquinoline, our research group
disclosed a novel family of molecules with promising profiles [2] [3]. These compounds were found
to exhibit remarkable properties, including inhibition of tau protein aggregation, neuroprotective and
antioxidant activity [3]. Furthermore, beta amyloid detection was possible due to their fluorescence
emission in the near-infrared range. These styrylquinoline derivatives beared in their structure an
electronic-density donor group in one side of the molecule, like amine groups, and an electronic-
density acceptor group in the other side, like cyano groups or BODIPY´s. This push and pull
strategy allows these compounds to shift their native fluorescent emission towards the near
infrared (NIR), making them appropriate NIR probes for diagnostic purposes. In addition, new
derivatives with boron complexes as electron withdrawing groups are being synthesized to
evaluate their emission behaviour and pharmacological properties.
[1] Lane CA, Hardy J, Schott JM. Eur J Neurol. 2018;25(1):59-70. doi:10.1111/ene.13439 [2] Staderini M, Aulić, SA, Bartolini M, et al 2012. doi:10.1021/ml3003605 [3] Piquero Martí M. PhD Thesis, Universidad Complutense de Madrid, 2020. Supervisors: P. López-Alvarado, J. C. Menéndez

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
99
Poster Communications (PC_60)
Development and application of a biophysical platform for the identification of fragments for the development of SETD8 ligands
Giuliana Sarno,a Ciro Milite,a Alessandra Feoli, a Sabrina Castellano,a Sergio Valente,b Antonello Mai,b Gianluca Sbardella,.a
a Department of Pharmacy, University of Salerno, Via Giovanni Paolo II 132, Fisciano, I-84084 Salerno, Italy;
b Department of Chemistry and Technologies of Drugs, Sapienza University of Rome, P.le A. Moro 5, 00185, Rome, Italy.
E-mail: [email protected]
SETD8/SET8/Pr-SET7/KMT5A is the only known protein lysine methyltransferase (PKMT) that
catalyses the monomethylation of histone H4 Lys20 (H4K20me1). Besides H4K20, SETD8
methylates lysine residues of many other proteins, such as the proliferating cell nuclear antigen
(PCNA) and the tumour suppressor p53 [1]. For this reason, a dysregulation of SETD8 is related to
different pathological conditions, including cancer [2,3]. Despite, the steadily growing interest in
physiological and pathological roles of SETD8, to date only few selective inhibitors of this protein
have been reported. In this project, the development and the optimization of a method for the
screening of a fragment library has been proposed to identify new chemical scaffolds that can be
used to develop potent and selective inhibitors of SETD8. Fragment-based drug discovery (FBDD)
approach (Figure 1), that was applied, consists of initial hits identification by Differential Scanning
Fluorimetry (DSF), a high-throughput method to detect fragments that alter the thermal stability of
the target protein, and then hits validation by Surface Plasmon Resonance (SPR), a biophysical,
label-free method for measuring molecular interactions. The low chemical complexity of the
identified fragments will allow a more efficient exploration of chemical space. Thus, their binding
properties to the target and biophysical information will be used to guide the iterations of evolution
and optimization of the fragments using medicinal chemistry strategies.
Figure 1: Fragment-based drug discovery (FBDD) approach.
[1] Milite, C.; Feoli, A.; Viviano, M.; et al. Clin Epigenetics 2016, 8, 102. [2] Lazarus, K. A.; Hadi, F.; Zambon, E.; et al. Nat Commun 2018, 9 (1), 3327. [3] Veschi, V.; Liu, Z.; Voss, T. C.; et al. J. Cancer Cell 2017, 31 (1), 50-63.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
100
Poster Communications (PC_61)
Investigating the Anticancer Activity of Isatin/Dihydropyrazole
Hybrids
Daniela Secci,a Rita Meleddu,a Vilma Petrikaite,b,c Simona Distinto,a Antonella Arridu,a Rossella Angius,d Lorenzo Serusi,a Laura Skarnulyte,b Ugne Endriulaityte,b Migle
Paškevičiute,b Filippo Cottiglia,a Marco Gaspari,e Domenico Taverna,e Serenella Deplano,a Benedetta Fois,a and Elias Maccionia
a Department of Life and Environmental Sciences, University of Cagliari, Via Ospedale 72, 09124 Cagliari,
Italy. b Department of Drug Chemistry, Faculty of Pharmacy, Lithuanian University of Health Sciences, 50162
Kaunas, Lithuania. c Institute of Biotechnology, Vilnius University, LT-10257 Vilnius, Lithuania.
d Laboratorio NMR e Tecnologie Bioanalitiche, Sardegna Ricerche, Pula, 09010 Cagliari, Italy. e Dipartimento di Medicina Sperimentale e Clinica, Università “Magna Græcia”, Viale Europa, 88100
Catanzaro, Italy.
E-mail: [email protected]
Pursuing on the research for novel and potentially multi-target anticancer agents we have
investigated structural modification on differently substituted indolinones[1][2]. Nowadays, both
academia and industry are focusing towards the development of molecules with multi-target profile,
able to inhibit or modify different targets involved in cell growth, apoptosis, signal transduction,
angiogenesis and metastasis. Isatin has already been introduced in different kinase inhibitor
anticancer drugs, such as Sunitinib and Nintedanib[3]. On these basis, different series of
molecules were synthesized and their structure activity relationships investigated. Within
synthesised compounds EMAC 4001, EMAC 4007 and EMAC 4008, resulted as promising being
substituted in the position 5 of isatin nucleus with -Cl, -CH3 and -OCH3 respectively. These
compounds exhibited an interesting activity profile not only due to their antiproliferative activity and
EC50 values in the low micromolar range, but also due to their specific cell death mechanism.
Indeed, studies to investigate their mechanisms of cell death, in the cell lines A549, IGR39 and
U87, revealed that, EMAC 4001 and EMAC 4008 induced necrosis in less than 1% of tested cells,
suggesting that cell deaths occurred due to a specific mechanism and not due to generalised
toxicity. Prompted by these information, we are actively proceeding with the optimization of this
scaffold in order to improve the activity and to further investigate their mechanism of actions.
Therefore, modifications of the thiazoline ring, by adding small substituents on the nitrogen atom
are in progress as well as other modifications that consider the dihydro-pyrazole ring by either
removing or partially modify the nucleus. Meanwhile we are focusing toward the optimisation of the
drug like properties in order to improve water solubility by adding more polar and/or salifiable
groups. The project is progress and the most recent results will be presented.
[1] G. Bianco et al. ACS Med. Chem. Lett., Aug. 2017, 8, 792–796, [2] C. Melis et al. ACS Med. Chem. Lett., Jul. 2018, 9, 725–729, [3] C. Melis et al.J. Enzyme Inhib. Med. Chem., Dec. 2017, 32, 68–73,

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
101
Poster Communications (PC_62)
Esters of chlorogenic acid as -glucosidase inhibitors with a potential antiviral activity. Synthesis, molecular docking and in vitro assays
Riccardo Semproli,a Daniela Ubiali,a Teodora Bavaro,a Maria Cristina De Rosa,b Davide Pirolli,b Benedetta Righino,b Federica Zaccheria,c and Nicoletta Ravasioc
a Department of Drug Sciences, University of Pavia, Viale Taramelli 12, 27100 Pavia, Italy. b Istituto di Scienze e Tecnologie Chimiche “G. Natta” (SCITEC), CNR, Largo Francesco Vito 1, 00168
Roma, Italy. c Istituto di Scienze e Tecnologie Chimiche “G. Natta” (SCITEC), CNR, Via Golgi 19, 20133 Milano, Italy.
E-mail: [email protected]
Biosynthesis and processing of the oligosaccharide chains of N-linked glycoproteins, which are
components of the viral envelope proteins, take place in the endoplasmic reticulum and are finely
tuned by host glycosidases. These oligosaccharide "trimming" reactions enable glycoproteins to
fold correctly and to interact with chaperone proteins for transport through the Golgi apparatus [1].
Inhibition of glycosidases, especially -glucosidases I and II (GluI/II), prevents the assembly of
functional glycoproteins. As a result, non-native conformers and incompletely assembled oligomers
are not further processed and, if misfolded persistently, they are degraded, thus impairing the
construction of viral glycoproteins and inhibiting viral replication.
Many polyhydroxylated alkaloids are active as glycosidase inhibitors. For example, iminocyclitol
2,5-dideoxy-2,5-imino-D-mannitol and D-1-deoxynojirimycin are inhibitors of both - and -
glucosidases [2].
Chlorogenic acid is a natural polyphenol widespread in plants, particularly in green coffee, which
has shown antiviral activity [3]. CGA has been easily functionalized through a solvent- and metal-
free direct esterification with fatty alcohols, in the presence of a heterogeneous catalyst, to give a
small library of CGA derivatives with a tunable hydrophilic/hydrophobic balance.
Yeast and human GluI share similar substrate specificity, pH optimum, and inhibitor sensitivity.
Thus, the yeast enzyme has been described as a good experimental model of human enzyme [4].
Preliminary computational chemistry studies indicated that CGA esters have a binding affinity for
the yeast enzyme higher than CGA, thus allowing a deeper insight into the molecular mechanism
of this interaction.
In vitro inhibition assays of CGA and CGA esters against yeast GluI are currently in progress.
Microbiological assays will be also performed to assess a correlation between the protective effect
of CGA esters towards viral infections and improper protein unfolding depending on GluI.
[1] Fukushi, M.; Yoshinaka, Y.; Matsuoka Y. et al. J. Virol. 2012, 86, 11745-11753 [2] Kiappes, J. L.; Hill, M.L.; Alonzi, D.S.; Miller, J.L.; Iwaki, R.; Sayce, A.C.; Caputo, A.T.; Kato, A.; Zitzmann, N. ACS Chem. Biol. 2018, 13, 60−65 [3] Ding, Y.; Cao, Z.; Cao, L.; Ding, G.; Wang, Z.; Xiao, W. Scient. Rep. 2017, 7, 45723 [4] Barker, M.K.; Rose D.R. J. Biol. Chem. 2013, 288, 13563-13574

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
102
Poster Communications (PC_63)
Structure-activity relationship in the group of 1,3,5-triazine derivatives with various halogen substitution
Sylwia Sudoł,1 Katarzyna Kucwaj-Brysz,1 Rafał Kurczab,2 Grzegorz Satała,2 Jadwiga Handzlik1
1 Department of Technology and Biotechnology of Drugs, Faculty of Pharmacy, Jagiellonian University, Medical College, Medyczna 9, PL 30-688 Kraków, Poland.
2 Department of Medicinal Chemistry, Maj Institute of Pharmacology, Polish Academy of Sciences, Smętna 12, PL 31-343 Kraków, Poland.
E-mail: [email protected]
Serotonin 5-HT6 receptors have been a popular protein target for over 20 years in the
search for new therapeutic agents for the treatment of diseases and dysfunctions of the central
nervous system, including depression, Alzheimer's disease, schizophrenia or obesity [1]. However,
none from the 5-HT6R agents have reached pharmaceutical market yet, thus searching for the
structurally novel, highly active 5-HT6R ligands with desired pharmacokinetic profile is both
challenging and necessary field of drug development research.
The subject of the presented research is a series of 16 novel triazine-based derivatives (1-
16): substituted with two fluorine atoms (1-4), fluorine and chlorine atoms (5-12) and CF3
containing (13-16) in the aromatic ring (R1) and varying in branching (R2) of the linker (Fig. 1).
Figure 1. General structure for investigated compounds.
All the investigated derivatives showed high affinity with Ki < 100 nM. The most active compound
(Ki = 5 nM) is even more active than already reported di-chloro-substituted derivatives [2].
The structures of the triazine-based derivatives match quite well to triangle topology
characteristic for 5-HT6R antagonists [3], despite the fact that this class of compounds does not
contain sulfone nor indole moiety, as majority of 5-HT6R ligands [4]. Pharmacophore-based
analysis, performed within this study led to the highlighting the differences of presented derivatives
from other reported 5-HT6R ligands.
The project is financed by the National Science Center grant No. NCN 2018/31 / B / NZ7 / 02160.
[1] Yun and Rhim, Exp Neurobiol. 2011, 4, 159; [2] Sudoł et al. Eur. J. Med. Chem. 2020, 203, 11252916 [3] López-
Rodríguez et al. J. Med. Chem. 2005, 48, 4216–4219. [4] Ivanenkov et al. Rev. Neurosci. 2014, 25 451–467.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
103
Poster Communications (PC_64)
A Multi-Level Computational Approach in the Discovery of New Quorum Sensing Inhibitors for P. aeruginosa
Tatiana F. Vieira,a Cristina Sousa,a and Sérgio F. Sousaa
aUCIBIO/REQUIMTE, BioSIM Departamento de Biomedicina, Faculdade de Medicina, Universidade do Porto, Alameda Professor Hernâni Monteiro, 4200-319 Porto, Portugal
E-mail: [email protected]
Pseudomonas aeruginosa is an opportunistic Gram-negative pathogen. It causes acute and
chronic infections especially in immune-compromised and hospitalized patients [1]. Infections
caused by P. aeruginosa are very difficult to eradicate because the bacteria can be organized in
structured microbial communities forming a biofilm.
Biofilms are structured microbial communities of surface-attached cells embedded in a self-
produced matrix of extracellular polymeric substances (EPS) that can be formed in a variety of
biological and industrial surfaces [2]. Controlling biofilm formation and development
A biofilms structural database was created to quickly assess all the structural information on
different protein structures involved in biofilm formation, development, and virulence available [3].
Here we report the optimization of a methodology using docking and virtual screening to identify
new drugs against a specific quorum-sensing system in P. aeruginosa responsible for biofilm
development, the PQS system.
Large databases of compounds (such as IBS InterbioScreen, Mu.Ta.Lig Chemotheca,
Chimiothèque Nationale and ZINC), were screened after careful validation of the Virtual Screening
protocol. Subsequently, molecular dynamics and free energy calculation methods were performed
in the top 5 results of each database to further validate the results, calculate the binding free
energy, and have a better understanding of the protein-ligand interactions.
[1] Bodey, G. P., Bolivar, R., Fainstein, V. & Jadeja, L., Clin. Infect. Dis. 1983, 5, 279–313 [2] Welch, M., Hodgkinson, J. T., Gross, J., Spring, D. R. & Sams, T., Biochemistry 2013, 52, 4433–4438 [3] Vieira, T., Magalhães, R. P., Fernandes, H. S., Simões, M. & Sousa, S. F., Trends Biotechnol. 2020, 38, 937-940
This work was supported by national funds from Fundacao para a Ciencia e a Tecnologia [grant numbers UIDP/04378/2020 and UIDB/04378/2020, SFRH/BD/137844/2018, 2020.01423.CEECIND]. Some of the calculations were produced with the support of INCD funded by FCT and FEDER under project 01/SAICT/2016 number 022153 and projects CPCA/A00/7140/2020 and CPCA/A00/7145/2020.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
104
Poster Communications (PC_65)
Physico-chemical characterization and in vitro cytotoxicity of Brij O2-stabilized gliadin-based nanoparticles
Silvia Voci,a Agnese Gagliardi,a Maria Cristina Salvatici,b Massimo Fresta,a Donato Coscoa
a Dipartimento di Scienze della Salute, Università “Magna Græcia”, Campus Universitario “S. Venuta”, I-88100, Catanzaro, Italy
b Institute of Chemistry of Organometallic Compounds (ICCOM)-Electron Microscopy Centre (Ce.M.E.), National Research Council (CNR), via Madonna del Piano n. 10, 50019 Sesto Fiorentino, Firenze, Italy.
E-mail: [email protected]
Natural proteins are emerging as promising raw materials to be employed in the development of
nanoparticles for drug delivery. In this investigation, gliadin, the prolamin-rich protein contained in
wheat grains, was used as biocompatible material to obtain a novel nanoformulation. In detail,
gliadin-based nanoparticles were prepared by the nanoprecipitation of different amounts of the
biomaterial (0.2-1.6 mg/ml), previously dissolved in an ethanol/water mixture. The organic phase
was mixed to the aqueous one by means of a homogenizer, and the suspension placed on a
magnetic stirrer to promote the evaporation of the organic solvent. The physico-chemical features
of the nanoparticles were evaluated by dynamic and static multiple light scattering. Among the
various surfactants used, the addition of 0.1% w/v of the Super Refined grade Brij O2 (SR BO2)
promoted the formation of negatively charged nanostructures, characterized by a mean diameter of
⁓150 nm. The emulsifier prevented the destabilization of the colloidal architecture up to 50 °C.
Mannitol demonstrated to be a suitable cryoprotectant to obtain a freeze-dried formulation. The
gliadin matrix promoted the entrapment and a controlled release of both hydrophilic and lipophilic
model compounds. SR BO2-stabilized gliadin-based nanoparticles showed a safety profile on
normal and tumor cell lines, up to 25 µg/ml of biomaterial. The obtained results demonstrate the
potential application of gliadin nanostructures as drug carriers [1].
Figure 1: Development of SR BO2-stabilized gliadin nanoparticles.
[1] Voci, S.; Gagliardi, A.; Salvatici, M.C.; Fresta, M.; Cosco, D. Development of polyoxyethylene (2) oleyl ether-gliadin nanoparticles: Characterization and in vitro cytotoxicity. European Journal of Pharmaceutical Sciences 2021,162, 105849.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
105
Poster Communications (PC_66)
Synthesis of new thiopyrano[2,3-d]thiazole derivatives bearing pyrazoline moiety as potential biological active compounds
Ihor Yushyn,a Roman Lesyka
a Department of Pharmaceutical, Organic and Bioorganic Chemistry, Danylo Halytsky Lviv National Medical University, Pekarska 69, 79010, Lviv, Ukraine
E-mail: [email protected]
Thiopyrano[2,3-d]thiazole derivatives exhibit various pharmacological activities due to similar steric effects to 5-ene-4-thiazolidinones [1] and may be considered as possible drug-candidates. Pharmacological profiles of thiopyrano[2,3-d]thiazole derivatives have been studied successfully as potential anticancer, antiviral [2], and antibacterial [3] agents and in combination with pharmacologically attractive pyrazoline fragment [4] could prevent to potentiation of biological activity. The efficient method for target pyrazoline-thiopyrano[2,3-d]thiazole derivatives 4 synthesis starting from 5-(4-methoxyphenyl)-3-naphthalen-2-yl-4,5-dihydro-1H-pyrazole 1 via acylation reaction with crotonic anhydride was accomplished in 1-[5-(4-methoxyphenyl)-3-naphthalen-2-yl-4,5-dihydropyrazol-1-yl]-but-2-en-1-one 2. The target synthesis of 6-[5-(4-methoxyphenyl)-3-naphthalen-2-yl-4,5-dihydropyrazole-1-carbonyl]-5-methyl-3,5,6,7-tetrahydro-thiopyrano[2,3-d]thiazol-2-one 4 was accomplished by reaction of hetero-Diels-Alder using 1-[5-(4-methoxyphenyl)-3-naphthalen-2-yl-4,5-dihydropyrazol-1-yl]-but-2-en-1-one 2 as the dienophile with appropriate 5-substituted aryl/heteryl isorhodanine (4-thioxo-2-thiazolidinone) 3 (Fig.1). The structures of compounds have been determined by 1H NMR and LCMS. Anticancer activity studies of target compound are in progress.
2
3
41
Toluene, reflux,2h
Hydroquinone
AcOH, reflux,2h
Figure 1: Synthesis of 6-[5-(4-methoxyphenyl)-3-naphthalen-2-yl-4,5-dihydropyrazole-1-carbonyl]-5-methyl-3,5,6,7-tetrahydro-thiopyrano[2,3-d]thiazol-2-ones.
[1] Kaminskyy, D., Kryshchyshyn, A., & Lesyk, R. 5-Ene-4-thiazolidinones–An efficient tool in medicinal chemistry. European journal of medicinal chemistry, 2017, 140, 542-594. [2] Lozynskyi, A., Golota, S., Zimenkovsky, B., Atamanyuk, D., Gzella, A., & Lesyk, R.. Synthesis, anticancer and antiviral activities of novel thiopyrano [2,3-d] thiazole-6-carbaldehydes. Phosphorus, Sulfur, and Silicon and the Related Elements, 2016, 191 (9), 1245-1249. [3] Metwally, N. H., Badawy, M. A., & Okpy, D. S.. Green synthesis of some new thiopyrano [2,3-d][1,3] thiazoles using lemon juice and their antibacterial activity. Synthetic Communications, 2018, 48(19), 2496-2509. [4] Karabacak, M., Altıntop, M. D., İbrahim Çiftci, H., Koga, R., Otsuka, M., Fujita, M., & Özdemir, A.. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules, 2015, 20(10), 19066-19084.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
106
Poster Communications (PC_67)
1,3,4-oxadiazol-2-one derivatives submitted into the 3D-QSAR study
Agata Zięba,a Tuomo Laitinen,b Jayendra Z. Patelc, Antti Posob,d, Dariusz Matosiuka, Agnieszka A. Kaczora,b
a Department of Synthesis and Chemical Technology of Pharmaceutical Substances with Computer Modeling Laboratory, Faculty of Pharmacy, 4A Chodzki St, PL-20059 Lublin, Poland
b School of Pharmacy, University of Eastern Finland, Yliopistonranta 1, P.O. Box 1627, FI-70211 Kuopio, Finland
c Division of Pharmaceutical Chemistry and Technology, University of Helsinki, Viikinkaari 5, Biocenter 2, FI-00790 Helsinki, Finland
d Department. of Internal Medicine VIII, University Hospital Tübingen, Otfried-Müller-Strasse 14, DE-72076 Tübingen, Germany
E-mail: [email protected]
The Endocannabinoid System and its activity recently became a hot topic that is widely examined
by medicinal chemists. Several studies suggest that medication affecting endocannabinoid
signaling can be used in the treatment of several complex, currently incurable diseases e.g.
chronic pain or schizophrenia[1], [2]. One of the enzymes responsible for the degradation of
endogenous cannabinoids is termed fatty acid-amide hydrolase. It is suggested that inhibition of
this enzyme enhances the signaling within the endocannabinoid system and comprises a potential
therapeutic strategy in treating mentioned above diseases [3]. One way to design selective and
potent FAAH inhibitors is to thoughtfully examine the structure-activity relationship among a series
of compounds with experimentally determined inhibitory activity. Quantitative-structure activity
relationship techniques (QSAR) can be easily implemented for such process. Therefore, we
decided to take advantage of the currently available X-ray structure of fatty acid-amide hydrolase
and construct 3D-QSAR (the CoMFa and CoMSIA) models for a series of previously published
compounds (1,3,4-oxadiazol-2-one derivatives)[4], [5]. These models were characterized by a high
statistical significance which was evaluated with the use of multiple techniques. Therefore, we
believe that promissing results obtained in this study will contribute to a better understanding of the
structure-activity relationship among FAAH inhibitors and assist in designing novel, more potent
compounds.
[1] J. Desfossés, E. Stip, L. A. Bentaleb, i S. Potvin, „Endocannabinoids and Schizophrenia”, Pharmaceuticals, t. 3, nr 10, s. 3101–3126, paź. 2010, doi: 10.3390/ph3103101. [2] N. Barrie i N. Manolios, „The endocannabinoid system in pain and inflammation: Its relevance to rheumatic disease”, Eur. J. Rheumatol., t. 4, nr 3, s. 210–218, wrz. 2017, doi: 10.5152/eurjrheum.2017.17025. [3] K. Ahn, D. S. Johnson, i B. F. Cravatt, „Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders”, Expert Opin. Drug Discov., t. 4, nr 7, s. 763–784, lip. 2009, doi: 10.1517/17460440903018857. [4] J. Z. Patel i in., „Revisiting 1,3,4-Oxadiazol-2-ones: Utilization in the Development of ABHD6 Inhibitors”, Bioorg. Med. Chem., t. 23, nr 19, s. 6335–6345, paź. 2015, doi: 10.1016/j.bmc.2015.08.030. [5] J. Z. Patel i in., „Chiral 1,3,4-Oxadiazol-2-ones as Highly Selective FAAH Inhibitors”, J. Med. Chem., t. 56, nr 21, s. 8484–8496, lis. 2013, doi: 10.1021/jm400923s.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
107
Author index
Legend Prefix_# progressive number PENP Paul Ehrlich Network Projects
PEEPA Paul Ehrlich Euro-PhD Award
FC Flash PENP Communication
PC Poster Communication
Abate C. PC_29_FC-2 ................................................. 68
Abatematteo F.S. PC_29_FC-2 ................................................. 68
Acosta L. PC_1 .............................................................. 40
Akrivou M.G. PC_49 ............................................................ 88
Alajarín R. PEEPA-6 ....................................................... 26
Alaviuhkola J. FC-3 ............................................................... 34
Alcaro S. PC_12 ............................................................ 51 PC_16 ............................................................ 55 PC_23 ............................................................ 62 PC_26 ............................................................ 65 PC_27 ............................................................ 66 PC_44 ............................................................ 83 PC_45 ............................................................ 84 PC_53 ............................................................ 92 PC_6 .............................................................. 45 PEEPA-1 ....................................................... 21 PEEPA-8 ....................................................... 28 PENP-1 .......................................................... 10 PENP-2 .......................................................... 11
Altieri F. PC_5 .............................................................. 44
Altomare C.D. PC_56_FC-6 ................................................. 95
Amanatidou D. PC_50 ............................................................ 89
Amaral C. PC_55 ............................................................ 94
Amata E. PC_7_FC-1 ................................................... 46
Ambrosio F.A. PC_26 ............................................................ 65 PEEPA-1 ....................................................... 21
Amodeo V. PC_8 .............................................................. 47
Anceschi L.
PC_3 ............................................................. 42 Andrei G.
PC_12 ........................................................... 51 Anfuso D.
PC_7_FC-1 .................................................. 46 Angeli A.
PEEPA-7 ...................................................... 27 Angius R.
PC_61 ......................................................... 100 Annunziata F.
PC_4 ............................................................. 43 Antonini L.
PC_5 ............................................................. 44 PC_54 ........................................................... 93
Appendino G. PC_11 ........................................................... 50
Aquino R.P. PC_24 ........................................................... 63
Arridu A. PC_61 ......................................................... 100
Artese A. PC_26 ........................................................... 65 PEEPA-1 ...................................................... 21
Astolfi A. PC_43 ........................................................... 82
Augusto T. PC_55 ........................................................... 94
Auriemma G. PC_24 ........................................................... 63
Bacci A. PC_6 ............................................................. 45
Bai R. PENP-1 ......................................................... 10
Balin K. PC_33 ........................................................... 72
Barbaraci C. PC_21 ........................................................... 60 PC_7_FC-1 .................................................. 46
Barbarossa A. PC_8 ............................................................. 47
Barraja P. PC_12 ........................................................... 51

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
108
PENP-1 .......................................................... 10 Barreca M.
PC_12 ............................................................ 51 PENP-1 .......................................................... 10
Barreca M.L. PC_43 ............................................................ 82
Barreiro S. PEEPA-8 ....................................................... 28
Bartolini M. PC_9 .............................................................. 48
Basagni F. PC_9 .............................................................. 48
Basile G. PC_10 ............................................................ 49
Bavaro T. PC_62 .......................................................... 101
Bellone M.L. PC_11 ............................................................ 50
Bender A. PC_43 ............................................................ 82
Benfeito S. PC_16 ............................................................ 55 PEEPA-4 ....................................................... 24
Bermejo P. PC_15 ............................................................ 54 PENP-5 .......................................................... 14
Berrino E. PC_52_FC-5 ................................................. 91
Bertoni F. PENP-1 .......................................................... 10
Biagini G. PC_58 ............................................................ 97
Bivacqua R. PC_12 ............................................................ 51
Bolognesi M.L. PENP-10 ....................................................... 19
Borges F. PC_16 ............................................................ 55 PEEPA-4 ....................................................... 24 PEEPA-8 ....................................................... 28 PENP-3 .......................................................... 12
Bortolozzi R. PENP-1 .......................................................... 10
Borzán J. PC_32 ............................................................ 71
Bošković J. PC_13 ............................................................ 52
Bouton J. PENP-9 .......................................................... 18
Bouz G. PEEPA-2 ....................................................... 22
Brazzolotto X. PEEPA-4 ...................................................... 24
Brea J. PC_29_FC-2 ................................................ 68
Brighenti V. PC_3 ............................................................. 42
Bryant S. PC_17 ........................................................... 56
Bucolo F. PC_14 ........................................................... 53
Bufano M. FC-1 .............................................................. 32
Cagide F. PEEPA-4 ...................................................... 24
Caljon G. PENP-9 ......................................................... 18
Cannazza G. PC_58 ........................................................... 97
Capasso C. PEEPA-7 ...................................................... 27
Carbone A. PC_20 ........................................................... 59
Carboni B. PC_22 ........................................................... 61
Cardoso Santos C. PENP-9 ......................................................... 18
Carmona-Zafra N. PC_15 ........................................................... 54
Carocci A. PC_8 ............................................................. 47
Castagnello A. PC_10 ........................................................... 49
Castellano S. PC_28 ........................................................... 67 PC_60 ........................................................... 99
Catalano R. PC_16 ........................................................... 55 PC_44 ........................................................... 83
Catalanotti B. PENP-4 ......................................................... 13
Catto M. PC_56_FC-6 ................................................ 95
Cavalloro V. PC_17 ........................................................... 56
Cavarelli J. PC_28 ........................................................... 67
Ceramella J. PC_10 ........................................................... 49 PC_18 ........................................................... 57 PC_8 ............................................................. 47 PEEPA-3 ...................................................... 23

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
109
Cerri L. PC_57 ............................................................ 96
Chałupnik P. PC_19 ............................................................ 58
Chavarria D. PC_16 ............................................................ 55 PEEPA-4 ....................................................... 24 PEEPA-8 ....................................................... 28
Cianciusi A. PC_20 ............................................................ 59
Cirone I. PC_23 ............................................................ 62
Citti C. PC_58 ............................................................ 97
Clerigué J. PC_42_FC-4 ................................................. 81
Codeluppi A. PC_3 .............................................................. 42
Coderch C. PC_1 .............................................................. 40 PC_38 ............................................................ 77 PEEPA-5 ....................................................... 25
Colabufo N.A. PC_29_FC-2 ................................................. 68
Collina S. PC_17 ............................................................ 56 PC_37_FC-3 ................................................. 76
Colombo A. PC_22 ............................................................ 61
Coluccia A. FC-1 ............................................................... 32 FC-4 ............................................................... 35 FC-5 ............................................................... 36 PC_47 ............................................................ 86
Conforti F. PC_8 .............................................................. 47
Conti P. PC_4 .............................................................. 43
Contino M. PC_29_FC-2 ................................................. 68
Cores Á. PC_15 ............................................................ 54 PC_48 ............................................................ 87
Coricello A. PENP-2 .......................................................... 11
Corona A. PC_23 ............................................................ 62
Correia-da-Silva G. PC_55 ............................................................ 94
Corsi L. PC_3 .............................................................. 42
Cosco D. PC_65 ......................................................... 104
Costa G. PC_27 ........................................................... 66 PC_37_FC-3 ................................................ 76 PC_45 ........................................................... 84
Costa S.C. PC_55 ........................................................... 94
Costanzo G. PC_21 ........................................................... 60
Costi M.P. FC-7 .............................................................. 38
Cottiglia F. PC_23 ........................................................... 62 PC_61 ......................................................... 100
Čudina O. PC_13 ........................................................... 52
D’Amore V.M. PENP-7 ......................................................... 16
Da Silva O. PEEPA-4 ...................................................... 24
Dal Piaz F. PC_11 ........................................................... 50 PC_46 ........................................................... 85
Daniela S. PC_52_FC-5 ................................................ 91
de Almeida Fiuza L.F. PENP-9 ......................................................... 18
de Frutos S. PEEPA-6 ...................................................... 26
De Luca L. PEEPA-10 .................................................... 30 PEEPA-7 ...................................................... 27
De Luca M. PC_41 ........................................................... 80
De Martino M. PC_6 ............................................................. 45
de Pascual-Teresa B. PC_1 ............................................................. 40 PC_38 ........................................................... 77 PEEPA-5 ...................................................... 25
De Rosa M.C. PC_62 ......................................................... 101
De Simone G. PEEPA-7 ...................................................... 27
De Soricellis G. PC_22 ........................................................... 61
De Tommasi N. PC_11 ........................................................... 50 PC_46 ........................................................... 85
Del Gaudio P.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
110
PC_24 ............................................................ 63 Del Genio V.
FC-2 ............................................................... 33 del Pino J.
PENP-5 .......................................................... 14 Deleu M.
PENP-8 .......................................................... 17 Delre P.
PC_29_FC-2 ................................................. 68 Deplano S.
PC_23 ............................................................ 62 PC_61 .......................................................... 100
Di Fiore A. PEEPA-7 ....................................................... 27
Di Geronimo B. PEEPA-5 ....................................................... 25
Di Leva F.S. PENP-7 .......................................................... 16
Di Maro S. PENP-7 .......................................................... 16
Dias J. PEEPA-4 ....................................................... 24
Dichiara M. PC_7_FC-1 ................................................... 46
Distinto S. PC_23 ............................................................ 62 PC_61 .......................................................... 100
Dobričić V. PC_13 ............................................................ 52
Doležal M. PEEPA-2 ....................................................... 22
Doroz-Płonka A. PC_35 ............................................................ 74
Dragonetti C. PC_22 ............................................................ 61
Dulski M. PC_33 ............................................................ 72
Eleftheriou P. PC_49 ............................................................ 88 PC_50 ............................................................ 89
Elz S. PEEPA-9 ....................................................... 29
Endriulaityte U. PC_61 .......................................................... 100
Esposito F. PC_23 ............................................................ 62
Esposito T. PC_24 ............................................................ 63
Eufemi M. PC_5 .............................................................. 44
Falanga A.
FC-2 .............................................................. 33 Falcone G.
PC_24 ........................................................... 63 Fallica A.N.
PC_25 ........................................................... 64 Fayeulle A.
PENP-8 ......................................................... 17 Feliciello A.
PENP-4 ......................................................... 13 Feoli A.
PC_28 ........................................................... 67 PC_60 ........................................................... 99
Fernandes C. PEEPA-4 ...................................................... 24 PEEPA-8 ...................................................... 28
Ferreira Nunes D. PENP-9 ......................................................... 18
Fesatidou M. PC_49 ........................................................... 88
Fiorillo B. PENP-4 ......................................................... 13
Fiorini G. FC-7 .............................................................. 38
Floresta G. PC_25 ........................................................... 64
Fois B. PC_61 ......................................................... 100
Franche A. PENP-8 ......................................................... 17
Franchini C. PC_8 ............................................................. 47
Frank A. PC_35 ........................................................... 74
Fresta M. PC_65 ......................................................... 104
Fusco A. PC_6 ............................................................. 45
Gagliardi A. PC_65 ......................................................... 104
Galdiero M. FC-2 .............................................................. 33
Galdiero S. FC-2 .............................................................. 33
Galera-Prat A. FC-3 .............................................................. 34
Gamarro F. FC-7 .............................................................. 38
Gandolfi R. PC_4 ............................................................. 43
García-Hernández R. FC-7 .............................................................. 38

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
111
García-Marín J. PEEPA-6 ....................................................... 26
Garofalo A. PC_41 ............................................................ 80
Garrido J. PEEPA-4 ....................................................... 24
Gaspar A. PEEPA-8 ....................................................... 28
Gaspari M. PC_61 .......................................................... 100
Gaudio E. PC_6 .............................................................. 45 PENP-1 .......................................................... 10
Geronikaki A. PC_34 ............................................................ 73 PC_49 ............................................................ 88 PC_50 ............................................................ 89
Gianni S. FC-5 ............................................................... 36
Gianni T. PENP-7 .......................................................... 16
Gil-Martins E. PEEPA-4 ....................................................... 24
Ginex T. PC_9 .............................................................. 48
Gioè C. PC_29_FC-2 ................................................. 68
Giorgi G. PC_40 ............................................................ 79
Gitto R. PC_14 ............................................................ 53 PEEPA-10 ..................................................... 30 PEEPA-7 ....................................................... 27
Giurdanella G. PC_7_FC-1 ................................................... 46
Głowacka O. PC_33 ............................................................ 72
Gomes A.R. PC_55 ............................................................ 94
Gómez-Carpintero J. PENP-5 .......................................................... 14
González J.F. PENP-5 .......................................................... 14
Grande F. PC_41 ............................................................ 80
Gratteri C. PC_26 ............................................................ 65 PENP-2 .......................................................... 11
Griera M. PEEPA-6 ....................................................... 26
Groo A.C.
PC_18 ........................................................... 57 Grubišić S.
PC_30 ........................................................... 69 Guaglio A.
PC_4 ............................................................. 43 Gualtieri G.
PC_27 ........................................................... 66 Guerchais V.
PC_22 ........................................................... 61 Guglielmi P.
PC_52_FC-5 ................................................ 91 Gul S.
PC_57 ........................................................... 96 PEEPA-8 ...................................................... 28
Gulotta M.R. PC_14 ........................................................... 53
Gutiérrez P.L.A. PC_59 ........................................................... 98
Hamel E. PENP-1 ......................................................... 10
Handzlik J. PC_32 ........................................................... 71 PC_63 ......................................................... 102
Hosseini-Gerami L. PC_43 ........................................................... 82
Hulpia F. PENP-9 ......................................................... 18
Iacopetta D. PC_10 ........................................................... 49 PC_18 ........................................................... 57 PC_8 ............................................................. 47 PEEPA-3 ...................................................... 23
Iannelli G. PC_28 ........................................................... 67
Ignazzitto M.T. PC_7_FC-1 .................................................. 46
Intranuovo F. PC_29_FC-2 ................................................ 68
Ioele G. PC_41 ........................................................... 80
Iurlo M. PC_9 ............................................................. 48
Ivkovic B. PC_30 ........................................................... 69
Jankowski R. PC_50 ........................................................... 90
Jovanovic M. PC_30 ........................................................... 69
Juza R. PC_31 ........................................................... 70
Kaczor A.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
112
PC_32 ............................................................ 71 Kaczor A.A.
PC_67 .......................................................... 106 Karalic I.
PENP-9 .......................................................... 18 Karcz T.
PEEPA-9 ....................................................... 29 Keminer O.
PEEPA-8 ....................................................... 28 Kessler H.
PENP-7 .......................................................... 16 Kieć-Kononowicz K.
PC_35 ............................................................ 74 PEEPA-9 ....................................................... 29
Korabecny J. PC_31 ............................................................ 70
Korzuch J. PC_33 ............................................................ 72
Kotańska M. PEEPA-9 ....................................................... 29
Kousaxidis A. PC_34 ............................................................ 73
Kovacikova L. PC_34 ............................................................ 73
Królicka E. PC_35 ............................................................ 74
Kucwaj-Brysz K. PC_63 .......................................................... 102
Kuczak M. PC_36 ............................................................ 75 PC_39 ............................................................ 78
Kuder K.J. PEEPA-9 ....................................................... 29
Kurczab R. PC_63 .......................................................... 102
La Regina G. FC-4 ............................................................... 35 FC-5 ............................................................... 36 PC_47 ............................................................ 86
La Rocca V. PC_57 ............................................................ 96
Lai M. PC_57 ............................................................ 96
Laitinen T. PC_67 .......................................................... 106
Langer T. PEEPA-10 ..................................................... 30
Łażewska D. PC_35 ............................................................ 74
Lee J.C. FC-4 ............................................................... 35
PC_47 ........................................................... 86 Lehtiö L.
FC-3 .............................................................. 34 León R.
PC_15 ........................................................... 54 PC_42_FC-4 ................................................ 81
Léonard E. PENP-8 ......................................................... 17
Leotta C.G. PC_7_FC-1 .................................................. 46
Leroy F. PC_18 ........................................................... 57
Lesyk R. PC_66 ......................................................... 105
Lezoualc’h F. FC-1 .............................................................. 32
Lin C. PENP-9 ......................................................... 18
Linciano P. PC_37_FC-3 ................................................ 76
Lins L. PENP-8 ......................................................... 17
Linusson A. PC_56_FC-6 ................................................ 95
Listro R. PC_37_FC-3 ................................................ 76
Longo A. PC_7_FC-1 .................................................. 46
Longo P. PC_18 ........................................................... 57 PEEPA-3 ...................................................... 23
Loza M.I. PC_29_FC-2 ................................................ 68
Luongo L. PC_58 ........................................................... 97
Lupia A. PC_6 ............................................................. 45 PENP-2 ......................................................... 11
Luque F.J. PC_9 ............................................................. 48
Maccioni E. PC_23 ........................................................... 62 PC_61 ......................................................... 100 PENP-3 ......................................................... 12
Madeja Z. PC_33 ........................................................... 72
Maes L. PENP-9 ......................................................... 18
Mai A. PC_60 ........................................................... 99
Majellaro M.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
113
PC_29_FC-2 ................................................. 68 Maksimainen M.M.
FC-3 ............................................................... 34 Malacrida A.
PC_37_FC-3 ................................................. 76 Malarz K.
PC_39 ............................................................ 78 Małecki P.
PC_36 ............................................................ 75 Malzert-Fréon A.
PC_18 ............................................................ 57 Mancuso F.
PEEPA-7 ....................................................... 27 Mangiatordi G.F.
PC_29_FC-2 ................................................. 68 Manzano J.I.
FC-7 ............................................................... 38 Marcaccio M.
PC_9 .............................................................. 48 Mariconda A.
PC_18 ............................................................ 57 PEEPA-3 ....................................................... 23
Marinelli L. PENP-7 .......................................................... 16
Marinotto D. PC_22 ............................................................ 61
Marquez L. PC_1 .............................................................. 40
Márquez-Cantudo L. PC_38 ............................................................ 77
Marrazzo A. PC_21 ............................................................ 60 PC_7_FC-1 ................................................... 46
Marrelli M. PC_8 .............................................................. 47
Martín-Aragón S. PC_15 ............................................................ 54 PENP-5 .......................................................... 14
Martino E. PC_17 ............................................................ 56
Martins E.G. PEEPA-8 ....................................................... 28
Maruca A. PC_16 ............................................................ 55 PC_27 ............................................................ 66 PC_44 ............................................................ 83 PEEPA-8 ....................................................... 28
Masoni S. PC_6 .............................................................. 45
Massari S. FC-3 ............................................................... 34
Massi M. PC_22 ........................................................... 61
Matosiuk D. PC_67 ......................................................... 106
Matyus P. PC_23 ........................................................... 62
Meleddu R. PC_23 ........................................................... 62 PC_61 ......................................................... 100
Menéndez J.C. PC_15 ........................................................... 54 PC_40 ........................................................... 79 PC_42_FC-4 ................................................ 81 PC_48 ........................................................... 87 PENP-10 ....................................................... 19 PENP-5 ......................................................... 14
Menéndez Ramos J.C. PC_59 ........................................................... 98
Mesiti F. PC_16 ........................................................... 55 PEEPA-8 ...................................................... 28
Mezeiova E. PC_31 ........................................................... 70
Milella L. PC_46 ........................................................... 85
Milite C. PC_28 ........................................................... 67 PC_60 ........................................................... 99
Minarini A. PC_9 ............................................................. 48
Montalbano A. PC_12 ........................................................... 51 PENP-1 ......................................................... 10
Moraca F. PENP-4 ......................................................... 13
Moreira O.C. PENP-9 ......................................................... 18
Moyano P. PENP-5 ......................................................... 14
Mrozek-Wilczkiewicz A. PC_36 ........................................................... 75 PC_39 ........................................................... 78
Mularski J. PC_39 ........................................................... 78
Muñoz M. PC_40 ........................................................... 79
Murthy S. FC-3 .............................................................. 34
Musco G. PENP-7 ......................................................... 16
Musilek K.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
114
PC_31 ............................................................ 70 Musioł R.
PC_33 ............................................................ 72 PC_36 ............................................................ 75 PC_39 ............................................................ 78
Musumeci F. PC_20 ............................................................ 59
Nachon F. PEEPA-4 ....................................................... 24
Naldi M. PC_9 .............................................................. 48
Nalli M. FC-4 ............................................................... 35 PC_47 ............................................................ 86
Nicolaou I. PC_34 ............................................................ 73
Nikolic K. PC_30 ............................................................ 69
Nikolić K. PC_13 ............................................................ 52
Nizi M.G. FC-3 ............................................................... 34
Occhiuzzi M.A. PC_41 ............................................................ 80
Olejarz-Maciej A. PC_35 ............................................................ 74
Oliveira P.J. PEEPA-4 ....................................................... 24
Onali A. PC_23 ............................................................ 62
Orocio E. PC_42_FC-4 ................................................. 81
Ortín I. PC_1 .............................................................. 40
Ortuso F. PC_16 ............................................................ 55 PC_23 ............................................................ 62 PC_44 ............................................................ 83 PC_53 ............................................................ 92
Paglia G. PC_5 .............................................................. 44
Palazzotti D. PC_43 ............................................................ 82
Panzarella G. PC_44 ............................................................ 83
Paravati M.R. PC_45 ............................................................ 84
Paris R. PC_58 ............................................................ 97
Parisi V. PC_46 ............................................................ 85
Paškevičiute Mi. PC_61 ......................................................... 100
Pasquinucci L. PC_21 ........................................................... 60 PC_7_FC-1 .................................................. 46
Pastor M. PC_1 ............................................................. 40 PEEPA-5 ...................................................... 25
Patel J.Z. PC_67 ......................................................... 106
Patitucci F. PC_10 ........................................................... 49
Pecora D. PC_47 ........................................................... 86
Pedone E. PENP-4 ......................................................... 13
Pellati F. PC_3 ............................................................. 42
Perea-Martínez A. FC-7 .............................................................. 38
Pérez J.M. PC_48 ........................................................... 87
Perricone U. PC_14 ........................................................... 53 PEEPA-10 .................................................... 30
Petrikaite V. PC_61 ......................................................... 100
Petrou A. PC_49 ........................................................... 88 PC_50 ........................................................... 89
Pezron I. PENP-8 ......................................................... 17
Pinzi L. PENP-6 ......................................................... 15
Piquero Martí M. PC_59 ........................................................... 98
Pirolli D. PC_62 ......................................................... 101
Pisani L. PC_56_FC-6 ................................................ 95
Pitari G.M. PC_7_FC-1 .................................................. 46
Pittalà V. PC_25 ........................................................... 64
Pocha A. PC_51 ........................................................... 90
Pockes S. PEEPA-9 ...................................................... 29
Podlewska S. PC_50 ........................................................... 90
Pollastro F.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
115
PC_11 ............................................................ 50 Pontecorvi V.
PC_52_FC-5 ................................................. 91 Poso A.
PC_67 .......................................................... 106 Procopio F.
PC_53 ............................................................ 92 Proia E.
PC_5 .............................................................. 44 PC_54 ............................................................ 93
Puoci F. PC_10 ............................................................ 49 PC_18 ............................................................ 57
Puxeddu M. FC-4 ............................................................... 35
Ragno G. PC_41 ............................................................ 80
Ragno R. PC_5 .............................................................. 44 PC_54 ............................................................ 93
Raimondi M.V. PENP-1 .......................................................... 10
Rak M. PC_33 ............................................................ 72
Ramos A. PC_38 ............................................................ 77
Ramos A.M. PC_1 .............................................................. 40
Ramos M.T. PC_42_FC-4 ................................................. 81
Rapposelli S. PC_57 ............................................................ 96 PC_6 .............................................................. 45
Rastelli G. PENP-6 .......................................................... 15
Ravasio N. PC_62 .......................................................... 101
Reiner-Link D. PC_35 ............................................................ 74
Remião F. PEEPA-4 ....................................................... 24
Rescifina A. PC_25 ............................................................ 64
Richards N. PENP-2 .......................................................... 11
Righino B. PC_62 .......................................................... 101
Rizzuti B. P_41 ............................................................... 80
Roberto D. PC_22 ............................................................ 61
Rocca R. PC_16 ........................................................... 55 PC_27 ........................................................... 66 PC_44 ........................................................... 83 PEEPA-8 ...................................................... 28 PENP-1 ......................................................... 10
Rodriguez-Puyol D. PEEPA-6 ...................................................... 26
Rodriguez-Puyol M. PEEPA-6 ...................................................... 26
Roleira F.M.F. PC_55 ........................................................... 94
Romeo I. PC_26 ........................................................... 65
Rosano C. PEEPA-3 ...................................................... 23
Rosini M. PC_9 ............................................................. 48
Rossi D. PC_37_FC-3 ................................................ 76
Rullo M. PC_56_FC-6 ................................................ 95
Runfola M. PC_57 ........................................................... 96
Russo F. PC_58 ........................................................... 97
Russo P. PC_24 ........................................................... 63
Ružić D. PC_13 ........................................................... 52
Sabatino M. PC_5 ............................................................. 44 PC_54 ........................................................... 93
Sadek B. PEEPA-9 ...................................................... 29
Salvatici M.C. PC_65 ......................................................... 104
Sánchez J.D. PC_15 ........................................................... 54
Sanna E. PC_23 ........................................................... 62
Santoro V. PC_46 ........................................................... 85
Sapienza F. PC_5 ............................................................. 44 PC_54 ........................................................... 93
Sarabia Vallejo Á. PC_59 ........................................................... 98
Sarno G. PC_60 ........................................................... 99
Satała G.

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
116
PC_63 .......................................................... 102 Saturnino C.
PC_18 ............................................................ 57 PEEPA-3 ....................................................... 23
Sbardella G. PC_28 ............................................................ 67 PC_60 ............................................................ 99
Schenone S. PC_20 ............................................................ 59
Sebastiani J. FC-5 ............................................................... 36
Secci D. PC_23 ............................................................ 62 PC_61 .......................................................... 100
Séguy L. PC_18 ............................................................ 57
Semproli R. PC_62 .......................................................... 101
Sequeira L. PC_23 ............................................................ 62 PENP-3 .......................................................... 12
Serda M. PC_33 ............................................................ 72
Serusi L. PC_61 .......................................................... 100
Sestito S. PC_57 ............................................................ 96
Silva R. PEEPA-4 ....................................................... 24 PEEPA-8 ....................................................... 28
Silva V. PEEPA-4 ....................................................... 24
Silvestri R. FC-1 ............................................................... 32 FC-4 ............................................................... 35 FC-5 ............................................................... 36 PC_47 ............................................................ 86
Since M. PC_18 ............................................................ 57
Sinicropi M.S. PC_10 ............................................................ 49 PC_18 ............................................................ 57 PC_8 .............................................................. 47 PEEPA-3 ....................................................... 23
Sirignano M. PEEPA-3 ....................................................... 23
Sisouklath K. FC-6 ............................................................... 37
Siwek A. PC_35 ............................................................ 74
Skarnulyte L. PC_61 ......................................................... 100
Soares C. PEEPA-4 ...................................................... 24
Soares P. PEEPA-4 ...................................................... 24
Soeiro M.N.C. PENP-9 ......................................................... 18
Sorrenti V. PC_25 ........................................................... 64
Sotelo E. PC_29_FC-2 ................................................ 68
Soukup O. PC_31 ........................................................... 70
Sousa C. PC_64 ......................................................... 103
Sousa S.F. PC_64 ......................................................... 103
Sowa S.T. FC-3 .............................................................. 34
Spanò V. PC_12 ........................................................... 51 PENP-1 ......................................................... 10
Spengler G. PC_32 ........................................................... 71
Stabile R. PC_37_FC-3 ................................................ 76
Stark H. PC_35 ........................................................... 74 PEEPA-9 ...................................................... 29
Statti G. PC_41 ........................................................... 80
Stefan E. PENP-4 ......................................................... 13
Stefanachi A. PC_29_FC-2 ................................................ 68
Stefek M. PC_34 ........................................................... 73
Sudoł S. PC_63 ......................................................... 102
Supuran C.T. PEEPA-7 ...................................................... 27 PENP-3 ......................................................... 12
Szczepańska K. PEEPA-9 ...................................................... 29
Szemerédi N. PC_32 ........................................................... 71
Szymańska E. PC_19 ........................................................... 58
Tabarrini O. FC-3 .............................................................. 34

BOOK of ABSTRACTS #PEVM2021
Paul Ehrlich Euro-PhD Network Virtual Meeting 2021
Zoom platform, July 26th-28th 2021
117
Tagliazucchi L. FC-7 ............................................................... 38
Tamborini L. PC_4 .............................................................. 43
Tavares-da-Silva E.J. PC_55 ............................................................ 94
Taverna D. PC_61 .......................................................... 100
Teixeira N. PC_55 ............................................................ 94
Tinivella A. PENP-6 .......................................................... 15
Tomarchio E. PC_7_FC-1 ................................................... 46
Tomassi S. PENP-7 .......................................................... 16
Tramontano E. PC_23 ............................................................ 62
Trapotsi M.A. PC_43 ............................................................ 82
Turkovic N. PC_30 ............................................................ 69
Turnaturi R. PC_21 ............................................................ 60 PC_7_FC-1 ................................................... 46
Ubiali D. PC_62 .......................................................... 101
Uliassi E. PENP-10 ....................................................... 19
Uriarte E. PENP-3 .......................................................... 12
Valente S. PC_60 ............................................................ 99
Van Calenbergh S. PENP-9 .......................................................... 18
Vaquero J.J. PEEPA-6 ....................................................... 26
Varela C.L. PC_55 ............................................................ 94
Ventura S. PEEPA-10 .................................................... 30
Vialko A. PC_19 ........................................................... 58
Vieira T.F. PC_64 ......................................................... 103
Villacamapa M. PC_48 ........................................................... 87
Villacampa M. PC_15 ........................................................... 54
Viola G. PENP-1 ......................................................... 10
Vittorio S. PEEPA-10 .................................................... 30
Vizirianakis I. PC_49 ........................................................... 88
Vlcek P. PC_31 ........................................................... 70
Voci S. PC_65 ......................................................... 104
Vrabel M. PEEPA-7 ...................................................... 27
Vujic Z. PC_30 ........................................................... 69
Yushyn I. PC_66 ......................................................... 105
Zaccheria F. PC_62 ......................................................... 101
Zambito Y. PC_57 ........................................................... 96
Zapico J.M. PC_1 ............................................................. 40
Zięba A. PC_67 ......................................................... 106
Zitko J. PEEPA-2 ...................................................... 22
Zubko M. PC_33 ........................................................... 72


















