
Bone Marrow Failure 2008-09
15
BONE MARROW FAILURE & OTHER HAEMATOLOGICAL CHANGES IN SYSTEMIC DISEASE Aims and Objectives Lecture Content • The causes of bone marrow failure • Clinical consequences and management of cytopenias • Pathological features and management of aplastic anaemia • Common causes of white cell abnormalities • Blood changes caused by renal and liver disease, chronic illness and malignancy Learning Objectives At the end of this session you should be able to: • Give an account of the causes of bone marrow failure • Describe the clinical consequences and Management of cytopenias • Describe the causes of leucocytosis and leucopenia • Describe the haematological changes in renal and liver failure • Session content Marrow Failure Many haematological diseases, and especially their treatment with chemotherapy and radiotherapy, cause marrow failure. Marrow failure may be associated with an empty (hypocellular) marrow or a marrow that is infiltrated with disease. This disease squeezes out the normal marrow and competes with normal haemopoetic cells. Some causes of marrow failure are shown in the table below: With a hypocellular marrow With an infiltrated marrow Aplastic anaemia (see below) Secondary cancers (lung, prostate, breast, thyroid, kidney) Myelodysplasia (may be cellular) Chemotherapy Acute leukaemias Radiotherapy Lymphomas Chemotherapy Myeloma The main function of the bone marrow is to produce blood cells so marrow failure results in anaemia, leucopenia and thrombocytopenia. These are collectively called pancytopenia.
-
Upload
karan-bhatt -
Category
Documents
-
view
94 -
download
3
Transcript of Bone Marrow Failure 2008-09

BONE MARROW FAILURE & OTHER HAEMATOLOGICAL CHANGES IN SYSTEMIC
DISEASE
Aims and Objectives Lecture Content • The causes of bone marrow failure • Clinical consequences and management of cytopenias • Pathological features and management of aplastic anaemia • Common causes of white cell abnormalities • Blood changes caused by renal and liver disease, chronic illness and
malignancy Learning Objectives At the end of this session you should be able to: • Give an account of the causes of bone marrow failure • Describe the clinical consequences and Management of cytopenias • Describe the causes of leucocytosis and leucopenia • Describe the haematological changes in renal and liver failure •
Session content
Marrow Failure Many haematological diseases, and especially their treatment with chemotherapy and radiotherapy, cause marrow failure. Marrow failure may be associated with an empty (hypocellular) marrow or a marrow that is infiltrated with disease. This disease squeezes out the normal marrow and competes with normal haemopoetic cells. Some causes of marrow failure are shown in the table below:
With a hypocellular marrow With an infiltrated marrow Aplastic anaemia (see below) Secondary cancers (lung, prostate, breast,
thyroid, kidney) Myelodysplasia (may be cellular) Chemotherapy Acute leukaemias Radiotherapy Lymphomas Chemotherapy Myeloma The main function of the bone marrow is to produce blood cells so marrow failure results in anaemia, leucopenia and thrombocytopenia. These are collectively called pancytopenia.

Anaemia This component of marrow failure is usually the simplest to treat, with red cell transfusions. For in-patients it is usual to keep the haemoglobin around 9-10 g/dl. In an adult one unit of red cells raises the haemoglobin by approximately 1 g/dl. Packed cells (or plasma reduced red cells) are the preferred product as plasma proteins are not usually required by the patient, (but are needed for manufacture of albumin and coagulation factors) and the less volume transfused the less risk of volume overload. Severe anaemia (haematocrit less than .30) prolongs the bleeding time and may worsen thrombocytopenic bleeding. Patients who have had many previous transfusions may have been immunised to produce antibodies against red cell, white cell and platelet antigens. The latter are usually directed against HLA (human leucocyte) antigens present on both white cells and platelets. Red cell antibodies may result in incompatible cross-matches and difficulty selecting compatible units of blood. HLA antibodies may react with dead white cells and platelets in transfused red cell units causing non-haemolytic febrile transfusion reactions (NHFTR's). Testing blood for infectious disease that may be transmitted by blood has reduced the number of immunocompromised patients infected by blood transfusion. Patients whose treatment may include bone marrow transplantation should ideally be transfused with blood not containing the cytomegalovirus (CMV) which is carried in dormant form in the lymphocytes of previously infected donors. When transfused to immunocompromised recipients severe CMV infection may result. Only 40% of donors in the UK are CMV negative. In practice the CMV antibody status of the patient is established as soon as possible after admission, and if they are CMV antibody positive (previously infected) there is no point in continuing to transfuse CMV negative blood products. Leucopenia Whilst lymphopenia contributes to the immune deficit of patients with marrow failure it is the neutrophil count which most determines short term prognosis. A neutrophil count of less than 1.0 X 109/l is associated with risk of pyogenic (bacterial) infection. Below 0.5 X 109/l this risk is severe. In the most severely immunocompromised patients e.g. those having marrow transplants it is usual to employ a regimen of barrier isolation in filtered air rooms with a diet of sterile food. Conventional isolation is of doubtful value in the prevention of infection in neutropenic patients. A compromise used in this district is to ban visitors with active infections, remove food with a high bacterial count (unpeelable fruit, cheese, pepper) and carers in physical contact with the patient to wear gloves and plastic pinafores. The high incidence of bacterial pathogens and antibiotic resistance found in hospitals may make the patient safer at home. All haematology units have protocols for the management of febrile neutropenic episodes (FNE's). One temperature reading of 38.5oC or above (or two of 38oC separated by less than an hour) should prompt clinical examination and taking of blood cultures, throat swab and mid-stream urine. Broad spectrum antibiotics are then started. These may be modified when the results of antibiotic sensitivity testing become available or if the FNE does not respond to treatment. Continuing pyrexia after changing antibiotics usually results in the administration of intravenous anti-fungal agents such as amphotericin. All neutropenic patients should be treated with prophylactic oral antifungal agents such as nystatin or amphotericin, which are not absorbed, but help prevent oral thrush. Prophylactic antibiotics are not usually employed because of problems with

acquisition of bacterial resistance. One exception is the use of low doses of cotrimoxazole to prevent pneumocystis pneumonia in post-transplant and AIDS patients. Leucocyte transfusions may be obtained from normal donors treated with G-CSF or patients with chronic granulocytic leukaemia (who have a great increase in circulating neutrophils) by using the cell separator. However they are very rarely employed because of lack good clinical evidence of their effectiveness. Lymphopenia results in increased susceptibility to viral infections and pneumocystis. Prophylactic aciclovir is given to patients with severe lymphopenia to prevent herpes virus infections until their helper T-lymphocyte count is over 0.2 X 109/l. Marrow transplant procedures, particularly when associated with GVHD cause prolonged suppression of cell-mediated immunity, as do chemotherapies containing nucleoside analogues such as fludarabine. Thrombocytopenia Patients with a platelet count of less than 10 X 109/l due to marrow failure usually have daily transfusions of platelet concentrate extracted from normal blood donations less than 72 hours old. Platelets have a short lifespan so unlike red cell transfusions, there may not be a sustained increase in the platelet count 24 hours after the transfusion. The effectiveness of platelet transfusions is judged by 1) cessation of bleeding and 2) an increase of platelet count measured one hour after transfusion of >20 X 109/l over the baseline level. Platelet transfusions may be ineffective due to HLA antibodies in the recipient or increased consumption at sites of bleeding. Relatives, or HLA matched donors may be platelet-pheresed on the cell separator. Aplastic Anaemia The main features of this disorder are a pancytopenia in the blood (reduction in white cells, red cells and platelets) with a hypoplastic (empty) marrow. The reticulocyte count is low. The clinical manifestations are infection, bleeding and anaemia. Causes of Aplastic Anaemia Congenital: • Fanconi's congenital aplastic anaemia - rare
Acquired: • Idiopathic Many of these cases are in fact autoimmune in nature, the target of
the immune reaction being the bone marrow stem cells. Serum or cellular factors in blood or bone marrow (particularly T suppressor lymphocytes) can be shown to suppress the growth of bone marrow cells in in-vitro culture.
• Radiation. Radiotherapy, nuclear accidents and explosions, radioisotopes. • Chemicals. Benzene TNT, DDT • Drugs. Bone marrow hypoplasia is a predictable effect of many cytotoxic
agents, and frequent blood counts are necessary to ensure a safe dose is not exceeded. Other drugs cause aplastic anaemia by an unpredictable idiosyncratic reaction e.g. chloramphenicol, phenylbutazone, gold,

• Viruses. The viruses of hepatitis are toxic for the bone marrow stem cells as well as the hepatocytes, and a severe type of aplastic anaemia may follow all types of viral hepatitis.
Management of Aplastic Anaemia • Remove the cause, if known • Supportive treatment - red cell transfusion, antibiotics, platelet transfusions.. • Stimulate the residual bone marrow stem cells - anabolic steroids. For severe cases of aplastic anaemia defined as having neutrophil counts less than 0.5 X 109/l, platelets less than 20 X 109/l, and a transfusion dependent haemoglobin, the following treatments may be used: • Bone marrow transplantation if aged <45 years with an HLA matched sibling
donor available. • Anti-thymocyte-globulin (ALG). This may work by removing inhibitory T-cells
from the bone marrow but causes temporary thrombocytopenia and serum sickness.
• Other immunosuppressants e.g. cyclosporine, high dose steroids.
Myelodysplastic syndromes/myelodysplasia (MDS) This is a group of acquired clonal disorders arising in haemopoietic stem cells. Patients are usually over the age of 70 years and the disease is characterised by cytopenias in the blood, most commonly anaemia, with morphological evidence of abnormalities (dysplasia) in the blood and bone marrow cells. The disease is usually idiopathic in origin but can also arise as a consequence of previous cytotoxic chemotherapy or radiotherapy given for the treatment of another tumour. Despite the low blood counts, the bone marrow is usually cellular or hypercellular (distinguishing it from aplastic anaemia) and there is a tendency for MDS to progress to acute myeloid leukaemia (AML). Diagnosis is made on the basis of typical dysplastic changes in the blood and bone marrow and associated clonal cytogenetic abnormalities present on chromosomal examination of bone marrow cells. Prognosis This is largely related to three variables – the percentage of immature ‘blast’ cells in the bone marrow (low blasts=best); cytogenetic abnormalities; and the number of cytopenias in the blood (anaemia, thrombocytopenia, neutropenia. More cytopenias=worse).
Treatment Supportive e.g. with red cell transfusions.Patients on long-term transfusions may need iron chelation therapy. Some patients may respond to recombinant erythropoietin therapy. Cytotoxic chemotherapy this can be used when the disease transforms to AML but has a very poor response rate. Differentiation therapy

there has been some success with the agent 5-azacytidine which is thought to work by accelerating differentiation of the MDS clone and allowing normal stem cells to regain dominance. Stem cell transplantation this is the only definitive cure for MDS but is only applicable to a small fraction of younger patients.

Haematological Changes in Systemic Disease
White cell changes in disease Neutrophilia neutrophil count above 7.5x109/l • Bacterial infection localized eg abscess or disseminated eg.
septicaemia • Inflammation/necrosis eg MI, vasculitis • Malignant disease • Myeloproliferative disease eg chronic myeloid leukaemia • Metabolic disease eg uraemia, gout • Corticosteroid therapy Leukaemoid reaction WBC > 50x109/l not due to leukaemia Lymphocytes or neutrophils ± immature forms • severe infections bacterial eg pneumonia • viral eg infectious mononucleosis • severe haemorrhage or haemolysis • malignant disease • intoxications burns, eclampsia Leukoerythroblastic change presence in blood of nucleated RBC and primitive WBC • Marrow invasion metastatic tumour, haematological
malignancy (eg myeloma, lymphoma), fibrosis • Severe illness trauma, septicaemia, massive
haemolysis
Neutropenia neutrophils < 2x109/l Can be isolated neutropenia or part of reduction of all blood cells (pancytopenia) • Isolated • Drugs eg phenylbutazone, co-trimoxazole,
carbimazole, anti-psychotics • Racial • Congenital including Kostmann’s syndrome, cyclic • Infections eg hepatitis, typhoid, TB, malaria • Auto-immune e.g Felty’s, SLE, idiopathic
Pancytopenia • Marrow failure from any cause (aplastic anaemia, megaloblastic anaemia,
irradiation, malignant infiltration etc)

• Hypersplenism

Eosinophilia more than 0.5x109/l Causes: • Allergies asthma, drugs, hayfever • Parasites ankylostoma, ascaris, filaria • Skin diseases eczema, psoriasis, dermatitis
herpetiformis • Malignancy Hodgkin’s disease • Inflammatory disease Sarcoidosis, polyarteritis nodosa • Hypereosinophilic syndrome • Eosinophilic leukaemia Lymphocytosis more than 3.5x109/l Causes: Acute infection usually viral e.g. rubella, mumps, infection.
mononucleosis • Chronic infection B, brucellosis, hepatitis • Thyrotoxicosis • Chronic lymphocytic leukaemia • other leukaemias and lymphomas
Acute Phase Reactants General indicators of systemic disease: inflammatory response to tissue damage. Examples include fibrinogen, complement, C-reactive protein, haptoglobin, ferritin, serum amyloid protein Measures of acute phase reaction is useful to indicate presence and extent of inflammation and to follow response over time eg to therapy. The commonly used indicators are ESR, plasma viscosity and CRP: Erythrocyte sedimentation rate (ESR) Degree of red cell sedimentation through plasma over a one hour period. • Cheap test • Dependent on plasma concentration of large proteins eg immunoglobulins,
fibrinogen • Normal range men <5mm/hr; women <15mm/hr but both increase with age • Raised especially in chronic infections, myeloma, disseminated malignancy,
autoimmune disease • Red cell concentration influences ESR – low in polycythaemia, high in anaemia Plasma viscosity • Normal range 1.5-1.7mPa/s • Test takes 15 minutes

• Increases only slightly with age • Unaffected by red cell changes C-reactive protein • Rapid increase in response to tissue injury (4-6 hours) • Highly sensitive • Not affected by red cell changes*
Liver Disease
Bleeding tendency • Deficiency of: vitamin-K dependent factors (II, VII, IX, X); factor V; fibrinogen • Functional abnormalities of fibrinogen • Increased fibrinolytic activity • Decreased platelets (hypersplenism, direct effect of alcohol) • Portal hypertension leading to varices
Anaemia Due to: • Bleeding and iron deficiency • Alcohol – direct toxic effect • Folate deficiency – leading to megaloblastic anaemia • Hypersplenism • Haemolysis – caused by alcohol, copper (Wilson’s disease), autoimmune (a/w
hepatitis) • Changes in red cell morphology - macrocytosis, target cells, spur cells
Chronic Renal Failure
Anaemia Due to
• decreased erythropoietin production • iron deficiency (blood loss during dialysis, venesection, poor platelet function) • aluminium toxicity • folate deficiency (chronic dialysis) • haemolysis (haemolytic uraemic syndrome - HUS) • anaemia of chronic disease
Changes in red cell morphology burr cells, fragmented cells in HUS
Bleeding tendency
• Abnormal platelet function • Thrombocytopenia - autoimmune, HUS
Anaemia Potential causes • Anaemia of chronic disease • Iron deficiency due to chronic bleeding esp. GI tract tumours • Immune haemolytic anaemia esp NHL • Pure red cell aplasia a/w thymoma

• Bone marrow infiltration (if extensive) • Folate deficiency due to anorexia, drugs
Polycythaemia Erythropoietin producing tumours eg kidney, cerebellum, liver
Platelets and coagulation
• Thrombocytosis associated with GI bleeding,
reactive • DIC mucin-secreting carcinoma • Coagulation factor antibody eg to factor VIII
Hypersplenism and Hyposplenism
Hypersplenism Splenomegaly from any cause can cause anaemia, leucopenia and thrombocytopenia. Due to increased pooling, sequestration and destruction of blood cells by the enlarged spleen.
Hyposplenism Causes: • Splenectomy • Sickle cell disease • Essential thrombocythaemia • Adult coeliac disease Splenic function can also be impaired by corticosteroids, radiation.
Haematological changes:
Red cells Target cells, Howell-Jolly bodies (DNA remnants), siderotic granules (iron containing), nucleated RBC.
White cells Early after splenectomy there is neutrophilia. Later lymphocytosis, monocytosis.
Platelets Early post-splenectomy there is marked thrombocytosis Long-term platelets are slightly elevated

Complications: Increased risk of fulminant infections especially with encapsulated bacteria, malaria • S. pneumoniae • N. meningitidis • H. influenzae
Require life-long prophylactic penicillin (or erythromycin if penicillin-allergic) Immunisation against pneumococcus, Hib, meningococcus (prior to splenectomy if elective)
Cases for self directed learning Case 1 History Mrs M.O. was a 22-year-old housewife. She gave a six week history of easy bruising and repeated infections, including otitis externa, vaginal thrush, URTI and gum infections. She had had heavy periods, 6-7/28 for a few months. There was no significant previous medical history. Recent drug treatment had been with oxytetracycline, amoxycillin and nystatin. She had one brother and one half sister, both well. On examination Pale. There were 0.5 cm lymph nodes in the anterior triangle of the neck. The gums around the lower left wisdom teeth were oedematous and inflamed. Investigations: Blood Count WBC X109/l
RBC X1012/l
Hb g/dl
Hct ratio
MCV Fl
MCH pg
MCHC g/dl
RDW Plt X109/l
4-11 M 4.7-6.1 F 4.2-5.4
M 13-17.5 F 12-16
M .42-.50 F .37-.47
80-99 27-31 32-35 11.5-14.5 150-400
1.9 2.20 7.3 .021 95.0 33.2 34.8 12.0 12 Differential: Neuts 9%, Lymphs 88%, Mono 2%, Eos 1% Reticulocyte count: 0.1% (normal 0.2-2%) Blood group: O Rh Positive, no atypical red cell antibodies. Serum B12 706 pg/ml (normal 150-700), Red cell folate 645 pg/ml (normal 150-650) Bone marrow aspirate: no particles, reduced numbers of erythroid and myeloid precursors. The majority of cells are lymphocytes. Bone marrow trephine biopsy: Grossly hypocellular marrow with scattered eosinophils, plasma cells and lymphocytes. No erythroid precursors or megakaryocytes. The appearances are of aplastic anaemia. Patients tissue type: A2 A3 B7 B15(62) Brother's tissue type: A1 A3 B7 B8 Treatment and progress

A transfusion of blood and platelets was given. Her IUCD was removed, and she was started on norethisterone. Metronidazole was administered for her gingivitis. Treatment for her aplastic anaemia was started with anti-lymphocyte globulin cyclosporin and prednisolone. Ten days after her last ALG treatment she developed a pyrexia of 38.5oC, arthralgia and an erythematous rash. This settled with further steroid treatment. Over the next three months her blood counts gradually improved and one year later her blood count showed WBC 3.8 (neuts 2.2), Hb 12.3, platelets 70.

Questions 1. Define pancytopenia. 2. What levels of platelets are associated with bleeding, and what level of
neutrophils is associated with infection. 3. What are the causes of aplastic anaemia? 4. What is serum sickness?
Case 2 A 45 year old man with long-standing rheumatoid arthritis is found to have the following blood count at a clinic visit: Hb 9.1g/dl (13-16.5) MCV 81fl (80-98) WBC and platelets normal His current drugs include sulphasalazine and naproxen. Further investigations show: serum iron 5 mol/l (9-30) total iron binding capacity 40 mol/l (45-70) ferritin 200 g/l (16-323) Questions 1. What is/are the possible cause(s) of the anaemia? 2. What other investigations might help you to reach a diagnosis? 3. What treatments may be of use in improving the anaemia? Case 3 A 54 year old woman is admitted after having vomited blood. On direct questioning she says she has been an alcoholic for several years. On examination she is jaundiced and has a palpable liver and moderately enlarged spleen. Full blood count: Hb 8.1g/dl (11.5-15.5) MCV 106fl (80-98) Platelets 125 x109/l (150-400) Clotting screen: INR 2.8 APTT 54 seconds/control 32 TT 21 sec/con12 Questions 1. What are the possible causes of the haematemesis? 2. What are the possible causes of the anaemia? 3. How would you manage this patient?

Case 4 A 46 year old man with known poorly controlled long-standing insulin dependent diabetes mellitus gives a history of increasing lethargy. Physical examination is unremarkable. FBC: Hb 8.7g/dl (13-16.5) MCV 95fl (80-98) WBC and platelets normal Glucose 17 mmol/l Urea 45 mmol/l (3-7.5) Creatinine 496 mol/l (80-133) Na 141 mmol/l (136-145) K 5.9 mmol/l (3.5-5)
Questions 1. Explain what has happened to the renal function and the likely cause. 2. What is the likely reason for the anaemia? How would you investigate and treat this?
Case 5 A 63-year-old man with a known diagnosis of myelodysplasia attends the A&E department with a 48-hour history of painless haematuria. He looks pale but is otherwise well. Blood count shows: WBC X109/l
RBC X1012/l
Hb g/dl
Hct ratio
MCV Fl
MCH pg
MCHC g/dl
RDW Plt X109/l
4-11 M 4.7-6.1 F 4.2-5.4
M 13-17.5 F 12-16
M .42-.50 F .37-.47
80-99 27-31 32-35 11.5-14.5
150-400
1.2 2.29 8.7 .224 98.0 30.8 34.8 11.0 32 Urine analysis reveals frank haematuria with proteinuria ++ Questions 1. How is the diagnosis of myelodysplasia made? 2. How would you investigate the cause of his haematuria? 3. How would you manage his haematuria?
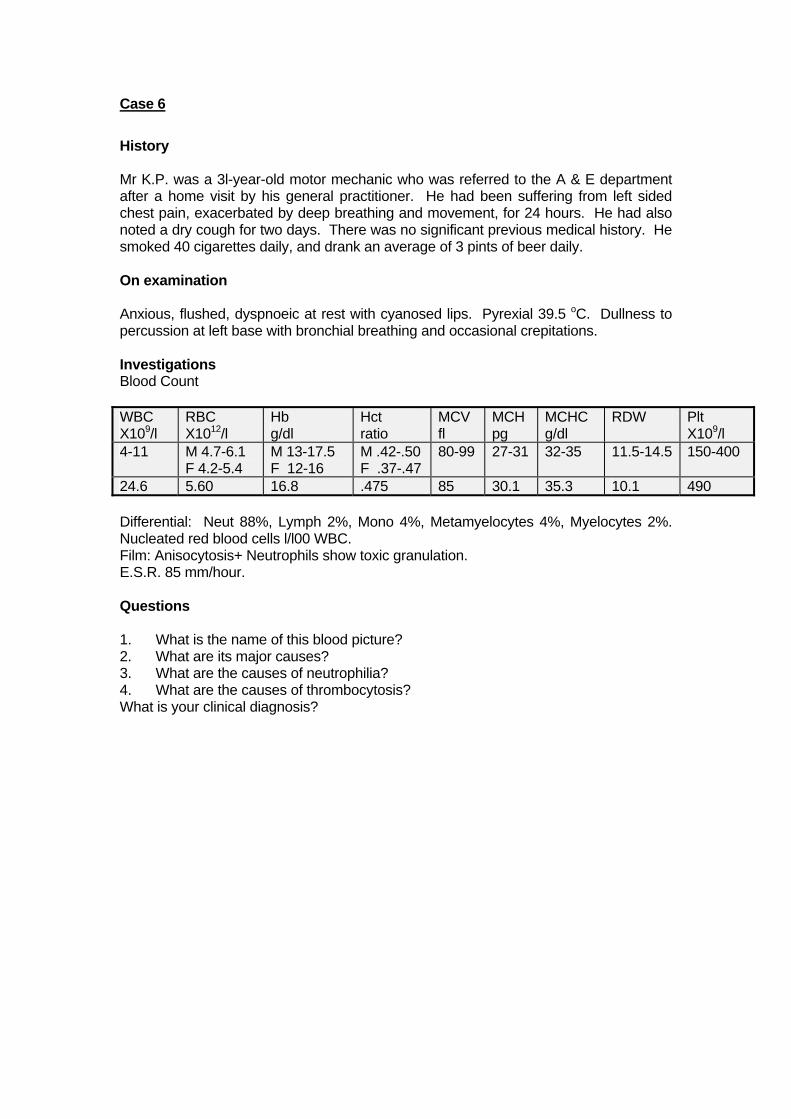
Case 6
History Mr K.P. was a 3l-year-old motor mechanic who was referred to the A & E department after a home visit by his general practitioner. He had been suffering from left sided chest pain, exacerbated by deep breathing and movement, for 24 hours. He had also noted a dry cough for two days. There was no significant previous medical history. He smoked 40 cigarettes daily, and drank an average of 3 pints of beer daily. On examination Anxious, flushed, dyspnoeic at rest with cyanosed lips. Pyrexial 39.5 oC. Dullness to percussion at left base with bronchial breathing and occasional crepitations. Investigations Blood Count WBC X109/l
RBC X1012/l
Hb g/dl
Hct ratio
MCV fl
MCH pg
MCHC g/dl
RDW Plt X109/l
4-11 M 4.7-6.1 F 4.2-5.4
M 13-17.5 F 12-16
M .42-.50 F .37-.47
80-99 27-31 32-35 11.5-14.5 150-400
24.6 5.60 16.8 .475 85 30.1 35.3 10.1 490 Differential: Neut 88%, Lymph 2%, Mono 4%, Metamyelocytes 4%, Myelocytes 2%. Nucleated red blood cells l/l00 WBC. Film: Anisocytosis+ Neutrophils show toxic granulation. E.S.R. 85 mm/hour. Questions 1. What is the name of this blood picture? 2. What are its major causes? 3. What are the causes of neutrophilia? 4. What are the causes of thrombocytosis? What is your clinical diagnosis?


















