
Turkish Neonatal Society guideline on neonatal infections ...
Upload
brendan-oneillCategory
view
222download
0
� 2007 Wiley-Liss, Inc. American Journal of Medical Genetics Part A 143A:1421–1430 (2007)
Body Fat Distribution and Metabolic Variables inPatients With Neonatal Progeroid Syndrome
Brendan O’Neill, Vinaya Simha, Vani Kotha, and Abhimanyu Garg*Division of Nutrition and Metabolic Diseases, Department of Internal Medicine,
University of Texas Southwestern Medical Center, Dallas, Texas
Received 5 March 2007; Accepted 25 March 2007
Neonatal progeroid syndrome (NPS), also known asWiedemann–Rautenstrauch Syndrome, is a rare autosomalrecessive disorder characterized by accelerated aging andlipodystrophy from birth. Affected children have extremeintrauterine growth retardation, poor postnatal weight gain,and characteristic facial dysmorphic features such as atriangular shape, pinched nose, pseudohydrocephalus withwide fontanelles and prominent subcutaneous (sc) veins.Generalized loss of sc fat has been reported as a cardinalfeature; however, the pattern of fat loss and its associationwith insulin resistance and its metabolic complications havenot been systematically studied. The aim of the current studywas to examine body fat distribution and body compositionin two girls with NPS using anthropometric measures, whole-body magnetic resonance imaging (MRI) and dual energy X-ray absorptiometry (DEXA), and to assess metaboliccomplications such as hyperinsulinemia and dyslipidemia.Both the girls (aged 17 years and 10 years, respectively) hadgeneralized paucity of sc fat on physical examination.However, measurements of skin-fold thickness revealed that
sc fat was decreased over the extremities, but preserved overthe chest and abdomen. MRI studies confirmed the presenceof normal amounts of sc truncal fat, and marked loss of fatfrom the face and distal extremities. Striking fat loss was alsonoted in the paravertebral and lateral gluteal regions.Interestingly, body composition analysis with DEXA scanrevealed a marked reduction in both the fat and lean tissuemass. Fasting glucose, lipids and insulin levels were notelevated. We conclude that patients with NPS do not havegeneralized lipodystrophy as previously reported, but fatloss is confined to the face, distal extremities, and possiblythe paravertebral and lateral gluteal regions. Metabolicabnormalities related to insulin resistance are also uncom-mon in this condition. � 2007 Wiley-Liss, Inc.
Key words: neonatal progeroid syndrome; Wiedemann–Rautenstrauch syndrome; progeria; body fat distribution;adipose tissue; lipodystrophy
How to cite this article: O’Neill B, Simha V, Kotha V, Garg A. 2007. Body fat distribution and metabolicvariables in patients with neonatal progeroid syndrome. Am J Med Genet Part A 143A:1421–1430.
INTRODUCTION
Neonatal progeroid syndrome (NPS, also knownasWiedemann–Rautenstrauch syndrome, OMIM264090) was first described by Rautenstrauch andSnigula in 1977 in two sisters who presented at birthwith features of progeria, or premature aging[Rautenstrauch and Snigula, 1977]. In 1979, Wiede-mann reported two unrelated male infants withstrikingly similar features to the patients describedpreviously, and suggested that they represent apreviously unrecognized progeroid disorder withneonatal onset [Wiedemann, 1979]. This was incontrast to the classicalHutchinson–Gilfordprogeriasyndrome, in which affected children are normal atbirth and develop progeroid features during earlychildhood [Brown, 1992; Eriksson et al., 2003;Hennekam, 2006]. Thus, the essential diagnosticcriteria suggested forNPS are: (1) intrauterine growth
retardation, (2) generalized diminution of subcuta-neous (sc) fat, and (3) an aged or old appearing faceat birth [Hagadorn et al., 1990]. The characteristicfacial features include a triangular shape, pseudohy-drocephalus, wide fontanelles, decreased sc fat,prominent veins, and a beak shaped or pinchednose. Thirty patients with NPS have been describedin the medical literature although a few ofthe reported patients had a strikingly different
Grant sponsor: National Institutes of Health; Grant number: R01-DK54387; Grant sponsor: Southwestern Medical Foundation.
*Correspondence to: Abhimanyu Garg, M.D., Chief, Division ofNutrition and Metabolic Diseases, Department of Internal Medicine,University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd,Y3-222, Dallas, TX 75390. E-mail: [email protected]
DOI 10.1002/ajmg.a.31840

phenotype and do not meet the diagnostic criteria forNPS [Petty et al., 1990; Delatycki et al., 1997; Kartesziet al., 2006]. Considerable phenotypic heterogeneityexists in this syndrome and the underlyinggenetic basis remains unknown. Autosomal reces-sive inheritance is suggested by reports of parentalconsanguinity in three pedigrees [Devos et al., 1981;Bitoun et al., 1995; Hou and Wang, 1995], and sixpedigrees with two affected siblings [Rautenstrauchand Snigula, 1977; Castineyra et al., 1992; Pivnicket al., 2000; Hoppen et al., 2004; Arboleda andArboleda, 2005].
All of the reported patients had generalizeddecrease of sc fat, but in some patients, an abnormalaccumulation of sc fat in the caudal region wasnoticed. However, body fat distribution and bodycomposition have not been systematically studied inthese patients. Therefore, we investigated bodycomposition and phenotypic manifestations in twonew patients with NPS by using anthropometry, dualenergy X-ray absorptiometry (DEXA), and whole-body magnetic resonance imaging (MRI). Further,only few investigators have reported on the meta-bolic abnormalities related to insulin resistance inthese patients [Courtens et al., 1997; Stoll et al., 1998;Pivnick et al., 2000; Arboleda and Arboleda, 2005].Patients with other types of generalized lipodystro-phy, such as congenital generalized lipodystrophy(CGL), have extreme insulin resistance and developabnormal glucose and lipid levels early in life [Garg,2004]. Thus we investigated whether loss of sc fat in
our patients with NPS was associated withdyslipidemia, abnormal glucose tolerance, or hyper-insulinemia.
CLINICAL REPORTS
Patient 1
Patient NPS 100.3 is a 17-year-old Hispanic female(Fig. 1), the first child of a nonconsanguineous24-year-old Latin American mother and a 31-year-oldLatin American father (Fig. 2). She has two youngerunaffected siblings. She was born by caesarian at32 weeks of gestation because of marked oligohy-dramnios and breech presentation. Marked intrau-terine growth retardation was noted. Her birthweight was 1,190 g (10th centile), length was 40 cm(39th centile), andoccipitofrontal circumferencewas29 cm (30th centile). At birth, she had a very wideanterior fontanelle and her skin was dry, and deeplywrinkled. She was described as having elfin facies, abeak-shapednose, low-set ears, andupward slantingpalpebral fissures. No dentition was present, and sheappeared tohave a generalizeddecrease in sc fat. Shehad joint contractures in the fingers, knees and toes,and medial deviations of both the hands and feetwere noted. She had normal thyroid function test,echocardiogram, cranial ultrasound, and genitour-inary ultrasound.
During infancy and childhood, she continuedto grow poorly but had normal mental and
FIG. 1. Anterior (A) and posterior (B) views of Patient 1 at 8 years of age, showing decreased sc fat, diminished muscle bulk, a progeroid appearance, anddisproportionately long extremities. Closer viewof the face (C) demonstrateswrinkled skin around themouth, decreased sc fat in the face, upslanting palpebral fissures,and corneal opacity of the right eye. Side view of the face (D) shows pinching of the nose, pseudoexophthalmos, frontal bossing, anddecreased sc fat in the neck. Closerviews of the feet (E) and hands (F) show marked decrease in sc fat in the distal extremities with resultant prominence of subcutaneous veins. C,D,E,F images are at 17years of age. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
1422 O’NEILL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

neuromuscular development. She was noted to havesevere myopia of the left eye at age 4 years, and wasblind in her right eye secondary to severe cornealscarring. There was marked xerophthalmia due todecreased tear production in both the eyes. Nocataracts were identified.
The patient was evaluated at 8 years of age andthen again at 17 years of age. She demonstrated poorweight gain, but her height remained within thenormal range. Her weight was 16 kg (<5th centile)and her height was 132.5 cm (�75th centile) at theageof 8 years. At 17 years of age, her bodymass index(BMI) was very low at 10.9 kg/m2 with a weight of27.6 kg (<5th centile) and height of 158.8 cm (25thcentile.) Muscle bulk and sc fat appeared markedlydiminished. She had prominent sc veins all over herbody, but most dramatically in the hands and feet.Further, she appeared to have long hands and feetalong with long fingers and toes.
Characteristic facial features included prominentmaxillary teeth and a hypoplastic mandible (Fig. 2).Even though her eyeballs were small, she appearedto have exophthalmos, likely due to hypoplasticorbital cavities. The nose was beak shaped, and shehad markedly decreased fat in the cheeks andtemples with prominent sc veins over the forehead.She had abundant dark hair with no graying. Herskull was dolichocephalic.
At 8 years of age, she continued to suffer fromdecreased tear production, requiring the use oflubricating eye drops hourly. At 17 years of age,she was receiving periodic eye care to treat calcifica-tions of her scarred right cornea and she haddeveloped glaucoma and subluxation of the lens inthe left eye.
The patient had normal intelligence and psycho-motor development. She had irregular menstrualcycles since her menarche at the age of 11. She alsosuffered from anemia, which was managed with oraliron supplements and monthly injections of vitaminB12.
Complete blood count, serum chemistries,and thyroid function tests were all within normalrange. Roentgenogram of the skull revealed hypo-telorism, small orbital cavities, dolichocephaly, and
an impacted molar. Karyotype was normal female(46, XX).
Patient 2
Patient NPS 700.7, a 10-year-old Caucasian female,was born at 32 weeks of gestation to healthy,nonconsanguineous parents. The pregnancy wascomplicated by intrauterine growth retardation andgestational diabetes. The birth weight was 1,172 g(10th centile), lengthwas 40.75 cm(39th centile), andher occipitofrontal circumference was 28.5 cm (30thcentile). The Apgar scores were 5 and 9, at 1 and5 min after birth, respectively. She had mild transientneonatal jaundice requiring phototherapy. She hadwide anterior (þ5 SD for term) and posteriorfontanelles with open metopic and sagittal sutures.She had senile facies at birth, with shallow orbits thatmade the globes appear prominent, upwardlysloped palpebral fissures, a small beak-shaped nose,fine hair, and abnormal wrinkling of the skin aroundthe lips. She had markedly diminished sc fat andprominent veins. Normal sc fat was present in thecaudal region. The hands, fingers, and feet weredisproportionately long. At birth, the total handlength was 90th centile for gestational age and themiddle finger length was at the 95th centile. She hada pseudohydrocephalic appearance secondary tohypoplasia of the facial bones. No dentition waspresent. Serum lipids, thyroid function test, andblood amino acid analysis were normal. Skeletalsurvey showed anterior bowing of the tibia and mildbowing of both the ulna and radius bilaterally.
Since birth, her height has remained within normallimits andherweight has remained consistently at the5th centile. She has developed an abnormal accu-mulation of fat in the region of the mons pubis. At 5months of age, iridodonesis, phakodonesis, and acortical haze in the lenses were noted bilaterally. Inaddition, lack of pupillary response to atropine wasnoted.
At age 10 years, her BMI was 11.4 kg/m2 with aheight of 151 cm (90th centile) and a weight of 26 kg(5th centile). She had striking facial features, includ-ing prominent frontal bossing, fat loss from thetemples and visible sc veins in the frontal region(Fig. 3). Her lips were thin and she had a thin, beak-shaped nose. She had thick, curly hair with nograying. Her eyeballs appeared small, yet werebulging forward secondary to small orbital cavities.She had extreme myopia (�26 diopters) andastigmatism requiring contact lenses. The pinnaewere anteverted. Sc fat appeared to be markedlydecreased, particularly in the extremities (Fig. 3), butan accumulation of fat was present in the caudal andmons pubis regions. She appeared to have adiminished muscle mass throughout the body. Herextremities, fingers, and toes were disproportio-nately long and thin. She had mild joint contractures
FIG. 2. NPSPedigrees. Affected individuals are shown asfilledblack symbolsand unaffected subjects as unfilled symbols.
BODY FAT DISTRIBUTION IN NEONATAL PROGEROID SYNDROME 1423
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

in the ankle, knee, and elbow joints. Her psycho-motor development was normal and she performedwell in school. Complete blood count, serumchemistries, and thyroid function testswere allwithinnormal range. Karyotype was normal female (46,XX).
METHODS
The study was approved by the InstitutionalReview Board of the University of Texas South-western Medical Center and written informed con-sent was taken from both the patients or their legalguardians. Informed consent was also obtained froma 10-year-old non-Hispanic girl and her legalguardians to participate in the study. MRI studieson this normal volunteer were used as a control.
Anthropometric Measurements
Height and body weight were measured bystandard procedures. Skin-fold thickness was mea-sured using a Lange skin-fold caliper (CambridgeScientific Industries, Cambridge, MD) thrice on eachsite, and the mean value was calculated. The skin-foldthickness measurements were obtained at the follow-ing 12 sites; chin, on the right side of body at thechest, midaxillary, abdominal, subscapular, suprai-liac, biceps, triceps, forearm, hip, thigh, and calf.
DEXA
Wholebodyand regional fat in thehead, trunk, andupper and lower extremities were determined using
a DEXA scan with a multiple detector fan-beamHologic QDR-2000 densitometer (Hologic, Inc.,Waltham, MA). The proportion of fat in individualregions as well as whole-body fat was calculated aspercentage of body mass. Data were also obtainedfor measurement of lean tissue mass and bonemineral density. Appendicular skeletal muscle masswas calculated as fat-free soft tissue in the upper andlower extremities, and total body skeletal musclemass was estimated by dividing the appendicularskeletal muscle mass by 0.75 [Proctor et al., 1999].
MRI
Whole-body MRI studies were performed using a1.5 T imaging device (Phillips Medical Systems, Best,The Netherlands) and 5.2-2 software. The patientswere evaluated using 10-mm-thick T1 imagingtechnique with repetition time of 580 msec and anecho time of 8 msec and a 384� 512 matrix combinedwith a 45-cm field of view. Adipose tissue distributionwas assessed by visual inspection of the films.
Blood Samples
Blood was collected after a 12 hr overnight fastfor analysis of serum lipoproteins, insulin, leptin,glucose, and a chemistry profile as previouslydescribed [Garg, 2000].
Biochemical Analyses
Serum chemistries, along with glucose andlipids, were measured as part of the systematic
FIG. 3. Anterior (A) andposterior (B) view of Patient 2 demonstrating decreased sc fat, diminished muscle bulk, and disproportionately long extremities. Closer viewof the face (C) shows decreased sc fat in the cheeks, temples, and anteverted pinnae. Side view of the face (D) shows pinched nose, pseudoexophthalmos, frontalbossing anddecreased sc fat in the neck. Closer viewsof the feet (E) andhands (F) showmarkedlydecreased fat in thedistal extremities. All images are at 10 years of age.[Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
1424 O’NEILL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a
multichannel analysis using automated equipment(Beckman, Fullerton, CA). Plasma insulin and leptinlevels were determined by radio-immunoassay usingcommercial kits (Linco Research, Inc., St. Charles,MO).
Genetic Analysis
GenomicDNAwas isolated from theblood sample,and the exons and splice-site junctions of LMNA genewere amplified. The PCR product was purifiedand sequenced using ABI PRISM 3100 (AppliedBiosystems, Foster City, CA). Sequences werecompared using Vector NTi Suite 6 software (Infor-Max, Inc., Bethesda, MD) and by visual inspection.The details of the procedure have been describedbefore [Chen et al., 2003].
RESULTS
Skin-Fold Measurements
Even though both the patients had a ‘‘generalizedskinny appearance,’’ measurement of skin-foldthickness revealed that fat loss was confined to theextremities (Fig. 4). They had normal skin-foldthickness in the abdominal and suprailiac regions,while at the thigh, calf, and triceps; it was near orbelow the 10th centile of matched controls (datafrom MI Goran, Los Angeles, CA). Patient 2, who hadmore sc fat than Patient 1, had skin-fold thickness
above the median in the subscapular area, chest, andaxilla.
Magnetic Resonance Imaging
Whole-body MRI images revealed a similar patternof body fat distribution in both patients. In Patient 1,sc fat was markedly decreased in the face, particu-larly over the temples (Fig. 5). Retro-orbital fat waspresent, but the absolute amount was less thannormal secondary to the small size of the orbitalcavities. In addition, sagittal views of the headrevealed thickening of the calvarium and decreasedsc fat overlying the spinous processes. The facial andretro-orbital fat of Patient 2 was obscured byinterference from metallic dental braces present atthe timeof evaluation, and thereforeher imageswerenot included in Figure 5. In Patient 1, there wasmarkedly decreased sc fat over the paravertebralregion of the trunk and neck (Fig. 6). Patient 2 alsohad decreased fat over this area, but to a lesser extentthan Patient 1. Both patients had qualitatively normalamounts of sc fat over the remaining areas of thetrunk, consistent with the normal skin-fold thicknessmeasuredover the abdominal and suprailiac regions.Intra-abdominal fat (intraperitoneal and retroperito-neal fat) was markedly reduced in both patients. Thelateral gluteal area had markedly reduced sc fat, incontrast with a normal amount over the medialgluteal area, in both patients. An apparent increase insc fat in the area of the mons pubis was evident in
FIG. 4. Skin-fold thickness at various anatomical sites in patients with neonatal progeroid syndrome. Skin-fold measurements were obtained at age 8 and 10 years,respectively, in Patients 1 and 2. The shaded bars represent the median, 10th and 90th centile values of skin-fold thickness for normal girls aged 4–10 years with averagebody mass index of 19.1� 4.7 kg/M2 (mean� SD, data supplied by MI Goran, Los Angeles, CA).
BODY FAT DISTRIBUTION IN NEONATAL PROGEROID SYNDROME 1425
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a
Patient 2. Both patients had sc fat in the proximalextremities that was proportional to muscle mass.However, the distal extremities had markedlydecreased sc fat, most evident in the palms andaround the ankles (Fig. 7). Intermuscular fat wasdecreased to absent, and there was a qualitativereduction in muscle mass in all muscle groupscompared to controls.
Dual Energy X-Ray Absorptiometry (DEXA)
The results of body composition analysis by DEXAare summarized in Table I. The total and regionalbody fat percentages in both the patients werereduced compared to age-, race-, and sex-matchednormals [Mazess et al., 1990; Wong et al., 2002].However, the values were within 2 SD of the meanexcept for the arms in Patient 1. The data furthershow a decreased fat free mass and skeletal musclemass at or greater than 2 SD below the mean [Wonget al., 2002; Poortmans et al., 2005]. In Patient 1, thetotal skeletalmusclemasswas approximately half thenormal value. In Patient 2, the total skeletal musclemass was 3.12 kg less than age-matched controls.However, when expressed as a percentage of totalbody mass, the overall skeletal muscle mass was
found tobe innormal proportion. This is likely due toa concurrent decrease in adipose tissue and totalbody mass. Both patients did appear to have a lowerproportion of skeletal muscle in the legs comparedto normal controls. Total body fat percentage wasdecreased in both, but to a lesser extent than the fatfree mass. Whole-body bone mineral density wasmore than 1 SD below the mean for control subjectsin both Patients 1 and 2 [Horlick et al., 2004].
Metabolic Variables
The results of plasma glucose, insulin, and lipidmeasurements are shown in Table II. No significantabnormalities were noted. Consistent with thedecreased adiposity, serum leptin levels were low(<7th centile).
Genetic Analysis
Sequencing of the exons and splice sites of theLMNA gene revealed no disease causing variants inboth the patients.
DISCUSSION
We present the first systematic evaluation of bodyfat distribution in NPS. Based on visual inspectionalone, earlier reports had indicated a generalized lossof sc fat in these patients similar to CGL. Our patientsalso appeared to have generalized loss of sc fat onphysical examination, but detailed analysis revealedthat fat loss is confined to certain regions such asthe face and distal extremities. Further, unlike CGLwhich is associated with muscle hypertrophy [Garget al., 2000], patients with NPS also have concomitantloss of lean mass which may contribute to their thinstature and previous descriptions of generalizedlipodystrophy.
A specific pattern of severely diminished sc fat wasfound in Patient 1 and a similar, although lessstriking, pattern was found in Patient 2. The patternconsisted of decreased sc fat in the paravertebralregion, lateral gluteal area, face, and distal extremi-ties. In addition, both the patients had markedlydecreased intra-abdominal and intermuscular fat.Previously, autopsy of one patient with NPS whodied at day 104of life due to cardiorespiratory failure,showed a complete absence of adipose tissue in themesentery [Hagadorn et al., 1990]. The diminished fatin the distal extremities by MRI was corroborated bythe presence of prominent sc veins in the hands andfeet (Figs. 1 and 3). However, prominent veins werealso visualized in less affected body regions, possiblydue to hypoplasia of the dermis. A thin, hypoplasticdermis was found on skin histology in one patient[Rudin et al., 1988]. The finding of prominent scvessels in all regions may have lead many pastauthors to report a generalized lack of sc fat in
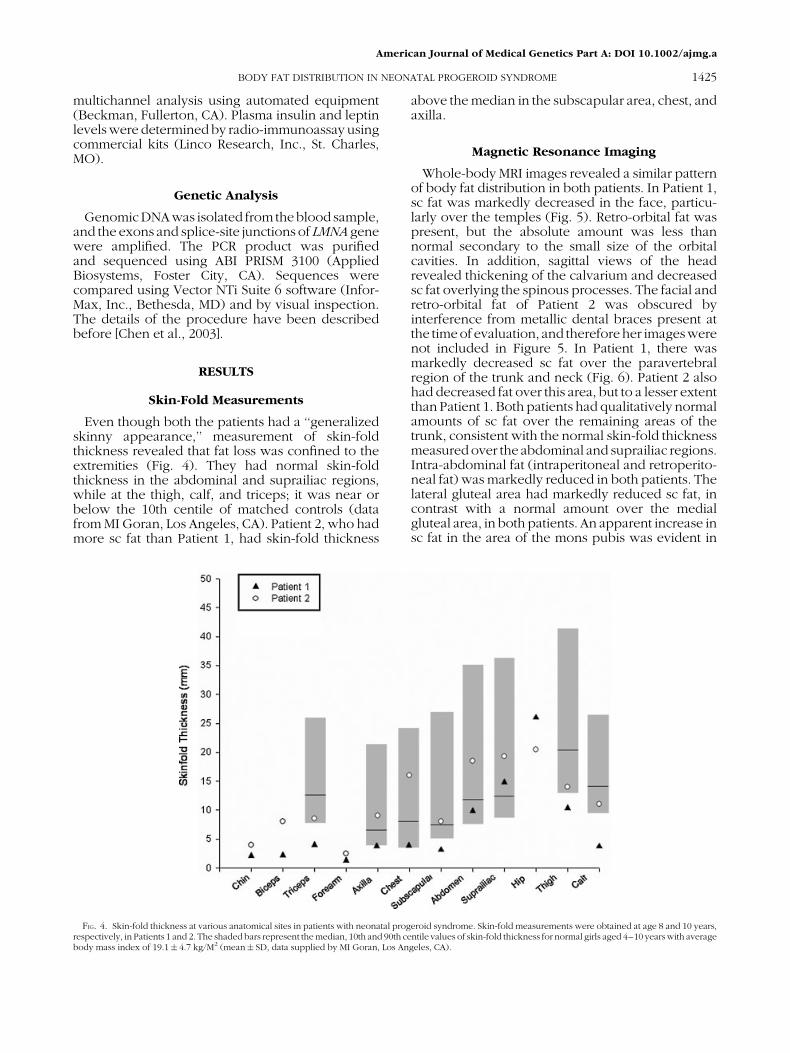
FIG. 5. Transaxial T1MRIs of the head at the level of the orbits of Patient 1 (A)and a normal 10-year-old female subject (B) show decreased fat in the temples.Retro-orbital fat is present, but theorbital cavities arehypoplastic in Patient 1 (A)compared to normal (B). T1-weighted MRI of the midline sagittal section of thehead and upper thorax of Patient 1 (C) shows decreased fat around the chin,neck, and skull compared to a normal female subject (D). The spinousprocesses are accentuated due to diminished paraspinal fat in Patient 1 (C). Thecalvarium of Patient 1 (C) is thick compared to normal (D).
1426 O’NEILL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a
patients with a similar appearance to Patients 1 and 2.Some have described the presence of caudal fataccumulation in patients with NPS. We found a sharpdecrease in fat in the lateral gluteal region, with
sparing of the medial gluteal region, which createsthe impression of caudal fat accumulation. In fact,this is not a true accumulation. However, a trueaccumulation was found in Patient 2 in the region of
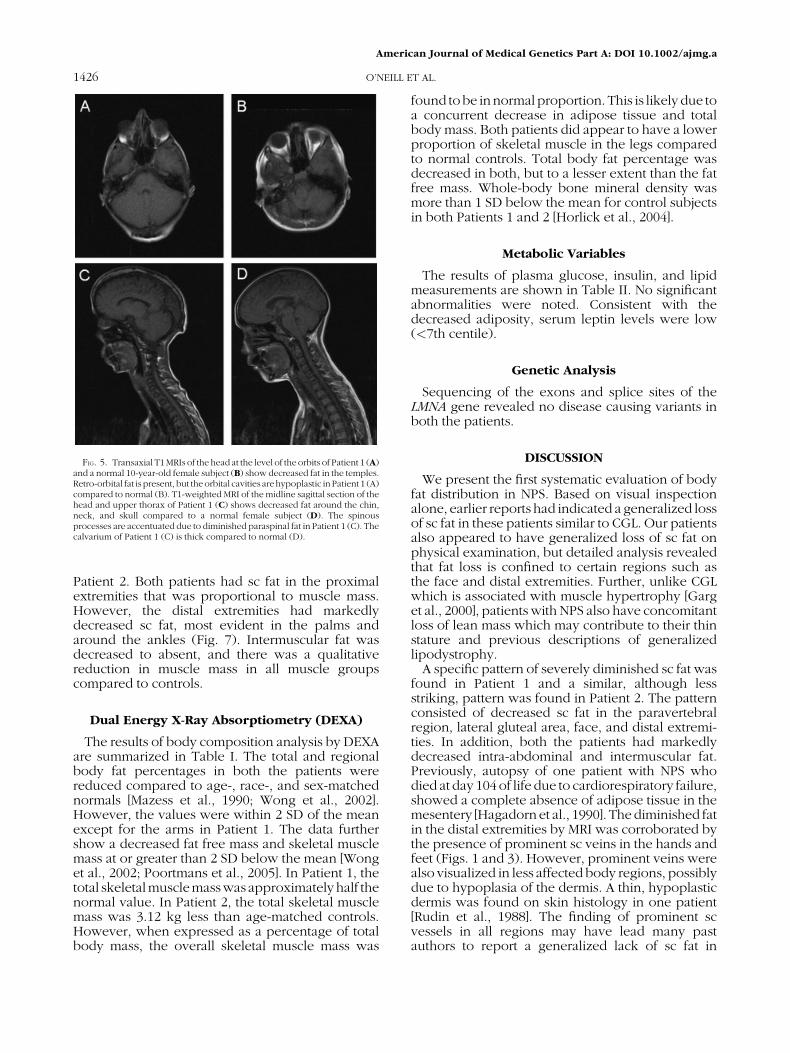
FIG. 6. Transaxial T1MRIs at the level of the kidney in Patient 1 (A), Patient 2 (B), and a normal 10-year-old female (C) show decreased intra-abdominal fat in Patients1 (A) and2 (B), andmarkedlydecreasedparavertebral fat in Patient 1 (A). At the level of the lumbar spine, decreased paravertebral fat andmusculature is noted inPatient1 (D) and Patient 2 (E) relative to control (F). In the gluteal region, a transaxial slice 3.5 cm below the hip joint, Patient 1 (G) and Patient 2 (H) show decreased fat in theposterolateral gluteal area compared to control (I).
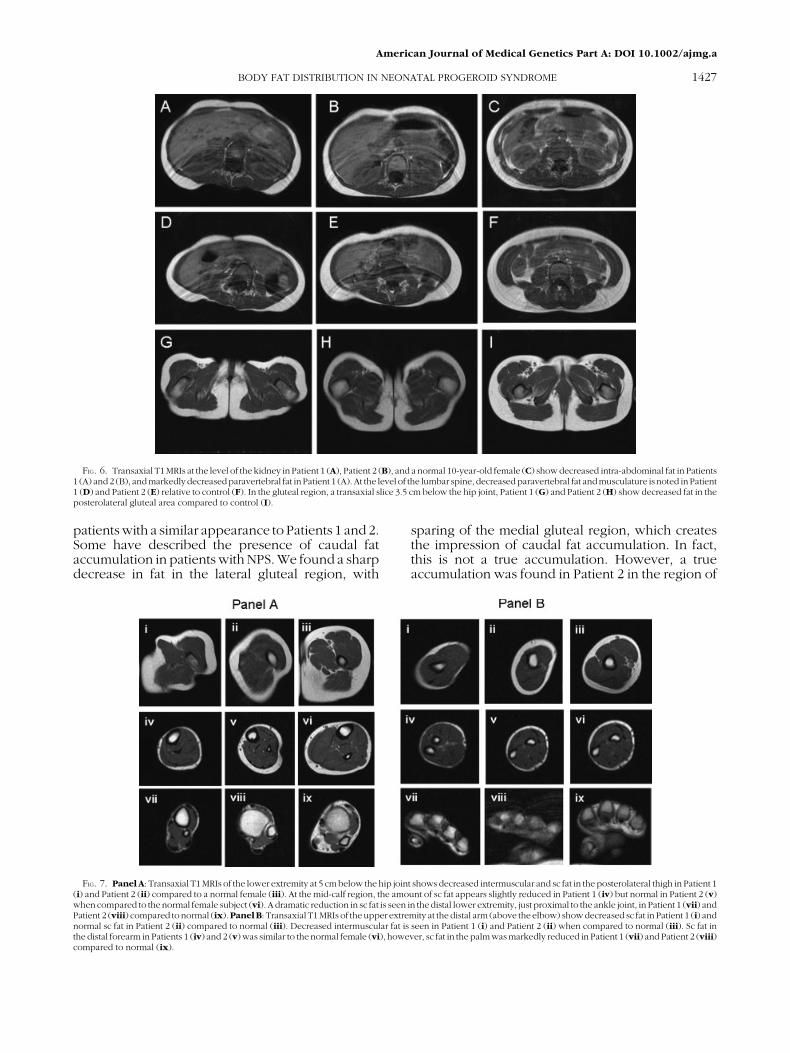
FIG. 7. Panel A: Transaxial T1 MRIs of the lower extremity at 5 cm below the hip joint shows decreased intermuscular and sc fat in the posterolateral thigh in Patient 1(i) and Patient 2 (ii) compared to a normal female (iii). At the mid-calf region, the amount of sc fat appears slightly reduced in Patient 1 (iv) but normal in Patient 2 (v)when compared to the normal female subject (vi). A dramatic reduction in sc fat is seen in the distal lower extremity, just proximal to the ankle joint, in Patient 1 (vii) andPatient 2 (viii) compared to normal (ix).Panel B: Transaxial T1MRIs of the upper extremity at the distal arm (above the elbow) show decreased sc fat in Patient 1 (i) andnormal sc fat in Patient 2 (ii) compared to normal (iii). Decreased intermuscular fat is seen in Patient 1 (i) and Patient 2 (ii) when compared to normal (iii). Sc fat inthe distal forearm in Patients 1 (iv) and 2 (v) was similar to the normal female (vi), however, sc fat in the palm was markedly reduced in Patient 1 (vii) and Patient 2 (viii)compared to normal (ix).
BODY FAT DISTRIBUTION IN NEONATAL PROGEROID SYNDROME 1427
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a
the mons pubis. This is only the second report ofsuch an accumulation in NPS [Pivnick et al., 2000].Besides lipodystrophic changes, the total skeletalmuscle mass was markedly reduced in both ourpatients.
Patients with generalized lipodystrophy oftenpresent with hypertriglyceridemia and insulin resis-tance. However, we did not notice any significantmetabolic abnormalities in our patients, and onlyfew previous reports have described mild to mode-rate hypertriglyceridemia which sometimes maybe transient [Courtens et al., 1997; Stoll et al., 1998;Pivnick et al., 2000; Arboleda and Arboleda, 2005].One of the patients described by Pivnick et al. [2000]had a serum triglyceride concentration of 352 mg/dlas a neonatewhichnormalizedby7weeksof age andremained in the normal range through 21 months ofage. Another patient, a 16-month-old boy, reported
by the same authors had a serum triglycerideconcentration of 412 mg/dl. This patient alsohad hyperinsulinemia with normal fasting plasmaglucose and a normal hemoglobin A1c. The patientof Stoll et al. [1998], at 31/2 years of age, had elevatedserum triglycerides and mild hepatomegaly, and wasalso reported to have insulin resistance based onhyperinsulinemic euglycemic clamp study. Thepatient described by Arboleda and Arboleda [2005]had a serum triglyceride level of 345mg/dl at 12 yearsof age, but this normalized when evaluated 1 yearlater. Thus, hypertriglyceridemia appears to beinfrequent in this syndrome and mild when it doesoccur. Diabetes or impaired glucose tolerance hasnot been reported. It is not clear why lipodystrophyin NPS is not commonly associated with insulinresistance like in other generalized and partiallipodystrophy syndromes. Frequent comorbidities
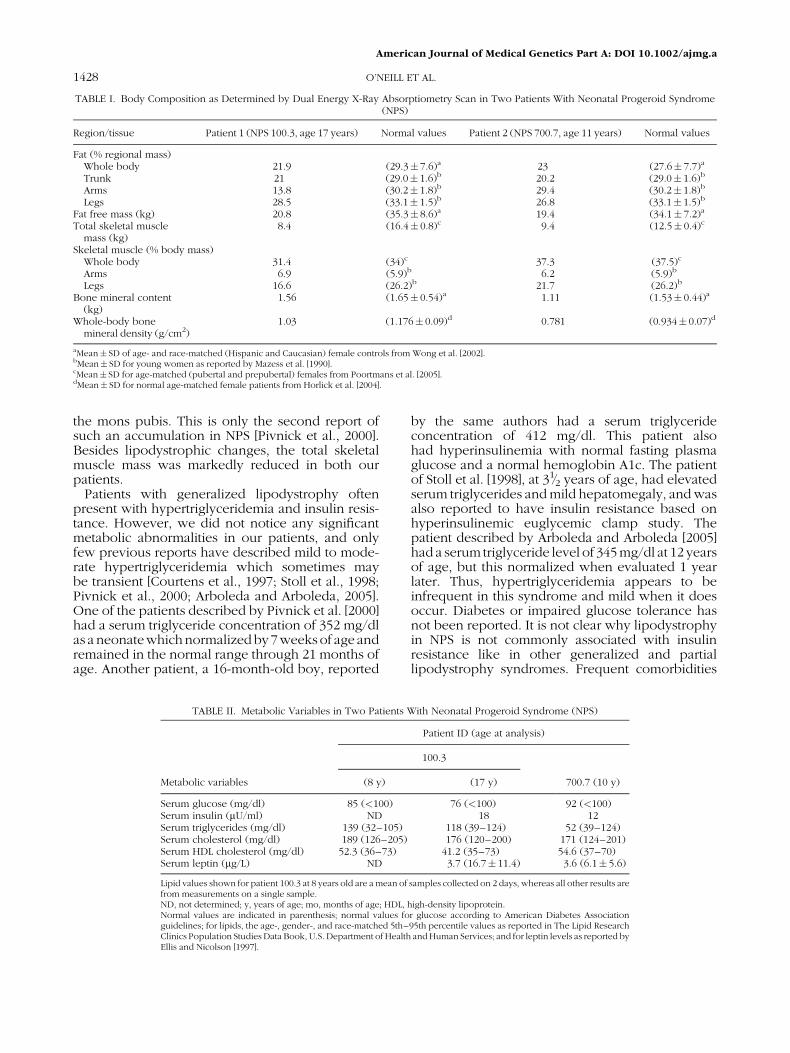
TABLE I. Body Composition as Determined by Dual Energy X-Ray Absorptiometry Scan in Two Patients With Neonatal Progeroid Syndrome(NPS)
Region/tissue Patient 1 (NPS 100.3, age 17 years) Normal values Patient 2 (NPS 700.7, age 11 years) Normal values
Fat (% regional mass)Whole body 21.9 (29.3� 7.6)a 23 (27.6� 7.7)a
Trunk 21 (29.0� 1.6)b 20.2 (29.0� 1.6)b
Arms 13.8 (30.2� 1.8)b 29.4 (30.2� 1.8)b
Legs 28.5 (33.1� 1.5)b 26.8 (33.1� 1.5)b
Fat free mass (kg) 20.8 (35.3� 8.6)a 19.4 (34.1� 7.2)a
Total skeletal musclemass (kg)
8.4 (16.4� 0.8)c 9.4 (12.5� 0.4)c
Skeletal muscle (% body mass)Whole body 31.4 (34)c 37.3 (37.5)c
Arms 6.9 (5.9)b 6.2 (5.9)b
Legs 16.6 (26.2)b 21.7 (26.2)b
Bone mineral content(kg)
1.56 (1.65� 0.54)a 1.11 (1.53� 0.44)a
Whole-body bonemineral density (g/cm2)
1.03 (1.176� 0.09)d 0.781 (0.934� 0.07)d
aMean� SD of age- and race-matched (Hispanic and Caucasian) female controls from Wong et al. [2002].bMean� SD for young women as reported by Mazess et al. [1990].cMean� SD for age-matched (pubertal and prepubertal) females from Poortmans et al. [2005].dMean� SD for normal age-matched female patients from Horlick et al. [2004].
TABLE II. Metabolic Variables in Two Patients With Neonatal Progeroid Syndrome (NPS)
Metabolic variables
Patient ID (age at analysis)
100.3
700.7 (10 y)(8 y) (17 y)
Serum glucose (mg/dl) 85 (<100) 76 (<100) 92 (<100)Serum insulin (mU/ml) ND 18 12Serum triglycerides (mg/dl) 139 (32–105) 118 (39–124) 52 (39–124)Serum cholesterol (mg/dl) 189 (126–205) 176 (120–200) 171 (124–201)Serum HDL cholesterol (mg/dl) 52.3 (36–73) 41.2 (35–73) 54.6 (37–70)Serum leptin (mg/L) ND 3.7 (16.7� 11.4) 3.6 (6.1� 5.6)
Lipid values shown for patient 100.3 at 8 years old are a mean of samples collected on 2 days, whereas all other results arefrom measurements on a single sample.ND, not determined; y, years of age; mo, months of age; HDL, high-density lipoprotein.Normal values are indicated in parenthesis; normal values for glucose according to American Diabetes Associationguidelines; for lipids, the age-, gender-, and race-matched 5th–95th percentile values as reported in The Lipid ResearchClinics Population Studies Data Book, U.S. Department of Health and Human Services; and for leptin levels as reported byEllis and Nicolson [1997].
1428 O’NEILL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

leading to poor energy intake, the young age ofmost reported cases, and concomitant loss of bothadipose tissue and lean mass may modulate insulinresistance.
Other endocrine abnormalities have also beenonly rarely reported in this syndrome. Neonatalhypoglycemia and hypothyroidism have beenreported [Rudin et al., 1988; Bitoun et al., 1995], butthe majority of patients demonstrated normal pitui-tary, thyroid, and adrenal functions [Stoll et al., 1998;Pivnick et al., 2000; Korniszewski et al., 2001].
Extensive reviews of the literature document theclinical heterogeneity of NPS [Arboleda et al., 1997;Courtens et al., 1997; Pivnick et al., 2000; Kornis-zewski et al., 2001]. In particular, prominent hetero-geneity exists in patient survival and psychomotordevelopment. Including our patients, 11 of 35 havedied before 6 years of age. Of these, six died ofrecurrent and severe respiratory infections andtwo due to unspecified respiratory failure. Incontrast, seven patients in the literature and bothour patients have survived into the second decade oflife [Wiedemann, 1979; Rautenstrauch et al., 1994;Pivnick et al., 2000; Arboleda and Arboleda, 2005]. Ofthese, the patient of Rautenstrauch et al. [1994]suffered from severe motor and mild mental retarda-tion. Our patients have survived to the ages of17 and 11 years, respectively, and have completelynormal psychomotor development. Both ourpatients demonstrate ophthalmologic findingswhich may be another feature of this syndrome.
The differential diagnosis of NPS includes Hutch-inson–Gilford progeria syndrome and mandibuloa-cral dysplasia, but both of these are distinguishedfrom NPS because affected patients are normal atbirth [Cao and Hegele, 2003]. Hallerman–Streiffsyndrome has progeroid features at birth with shortstature, but can be differentiated by the presence ofnormal adipose tissue [Hagadorn et al., 1990].Patients with congenital cutis laxa syndrome canpresent with a prematurely aged appearance at birth,but this syndrome can be differentiated by itscharacteristic lax skin and dissimilar facial features[Almeida et al., 2005]. CGL is included in thedifferential, but this disorder is distinguished by itscharacteristic extreme lack of fat, muscular appear-ance, acromegaloid features, impaired glucosetolerance, and hypertriglyceridemia [Garg, 2004].The genetic basis of NPS remains to be determined.Since LMNA mutations have been described inHutchinson–Gilford progeria and other atypicalprogeroid disorders, we screened our patients alsofor mutations in this gene. However, no diseasecausing variants in LMNA were identified similar tothe previous report [Cao and Hegele, 2003].
In conclusion, we have found that patients withNPS do not have generalized lipodystrophy aspreviously described, but a distinct pattern of fat lossconfined to the face, distal extremities, paravertebral
area, lateral gluteal area, and intra-abdominal area.Concomitant loss of lean tissue might contribute tothe appearance of generalized lipodystrophy. Unlikeother formsof genetic lipodystrophies, thesepatientsdo not exhibit marked metabolic abnormalitiesrelated to insulin resistance.
ACKNOWLEDGMENTS
We are grateful to Dr. Katherine Jacob forassistance with patient evaluation, to Meredith Millayand Sarah Mayhew for technical assistance, and tothe nursing services of the General Clinical ResearchCenter at the University of Texas SouthwesternMedical Center, Dallas for patient care. The studywas supported in part by the National Institutes ofHealth grant R01-DK54387 and by the SouthwesternMedical Foundation.
REFERENCES
Almeida P, Hernandez J, Marti M, Hernandez B. 2005. Whatsyndrome is this? Wiedemann-Rautenstrauch syndrome.Pediatr Dermatol 22:75–78.
Arboleda H, Arboleda G. 2005. Follow-up study of Wiedemann-Rautenstrauch syndrome: Long-term survival and comparisonwith Rautenstrauch’s patient ‘‘G’’. Birth Defects Res A Clin MolTeratol 73:562–568.
Arboleda H, Quintero L, Yunis E. 1997. Wiedemann-Rauten-strauch neonatal progeroid syndrome: Report of three newpatients. J Med Genet 34:433–437.
Bitoun P, Lachassine E, Sellier N, Sauvion S, Gaudelus J. 1995. TheWiedemann-Rautenstrauch neonatal progeroid syndrome: Acase report and review of the literature. Clin Dysmorphol4:239–245.
Brown WT. 1992. Progeria: A human-disease model of acceler-ated aging. Am J Clin Nutr 55:1222S–1224S.
Cao H, Hegele RA. 2003. LMNA is mutated in Hutchinson-Gilfordprogeria (MIM 176670) but not in Wiedemann-Rautenstrauchprogeroid syndrome (MIM264090). JHum Genet 48:271–274.
Castineyra G, Panal M, Lopez Presas H, Goldschmidt E, SanchezJM. 1992. Two sibs with Wiedemann-Rautenstrauch syn-drome: Possibilities of prenatal diagnosis by ultrasound. J MedGenet 29:434–436.
Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, ShafeghatiY, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS,Kennedy BK, Oshima J. 2003. LMNA mutations in atypicalWerner’s syndrome. Lancet 362:440–445.
Courtens W, Nuytinck L, Fricx C, Andre J, Vamos E. 1997. Aprobable case of Wiedemann-Rautenstrauch syndrome orneonatal progeroid syndrome and review of the literature.Clin Dysmorphol 6:219–227.
Delatycki MB, Cleary MA, Bankier A, McDougall PN, AhluwaliaJS, Chow CW, Cooke-Yarborough CM. 1997. A maternallytransmitted lethal neonatal progeroid syndrome with promi-nent genitourinary and gastrointestinal features. J Med Genet34:520–524.
Devos EA, Leroy JG, Frijns JP, Van den Berghe H. 1981. TheWiedemann-Rautenstrauch or neonatal progeroid syndrome.Report of a patient with consanguineous parents. Eur J Pediatr136:245–248.
Ellis KJ, Nicolson M. 1997. Leptin levels and body fatness inchildren: Effects of gender, ethnicity, and sexual develop-ment. Pediatr Res 42:484–488.
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L,Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E,Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. 2003.
BODY FAT DISTRIBUTION IN NEONATAL PROGEROID SYNDROME 1429
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a

Recurrent de novo point mutations in lamin A causeHutchinson-Gilford progeria syndrome. Nature 423:293–298.
Garg A. 2000. Gender differences in the prevalence of metaboliccomplications in familial partial lipodystrophy (Dunniganvariety). J Clin Endocrinol Metab 85:1776–1782.
Garg A. 2004. Acquired and inherited lipodystrophies. N EnglJ Med 350:1220–1234.
Garg A, Stray-Gundersen J, Parsons D, Bertocci LA. 2000. Skeletalmuscle morphology and exercise response in congenitalgeneralized lipodystrophy. Diabetes Care 23:1545–1550.
Hagadorn JI, Wilson WG, Hogge WA, Callicott JH, Beale EF. 1990.Neonatal progeroid syndrome: More than one disease? Am JMed Genet 35:91–94.
Hennekam RC. 2006. Hutchinson-Gilford progeria syndrome:Review of the phenotype. Am J Med Genet Part A 140A:2603–2624.
Hoppen T, Naumann A, Theile U, Rister M. 2004. [Siblings withneonatal progeroid syndrome (Wiedemann-Rautenstrauch)].Klin Padiatr 216:70–71.
Horlick M, Wang J, Pierson RN Jr, Thornton JC. 2004. Predictionmodels for evaluation of total-body bone mass with dual-energy X-ray absorptiometry among children and adoles-cents. Pediatrics 114:e337–e345.
Hou JW, Wang TR. 1995. Clinical variability in neonatal progeroidsyndrome. Am J Med Genet 58:195–196.
Karteszi J, Kosztolanyi G, Czako M, Hadzsiev K, Morava E. 2006.Transient progeroid phenotype and lipodystrophy in mosaicpolyploidy. Clin Dysmorphol 15:29–31.
Korniszewski L, Nowak R, Okninska-Hoffmann E, Skorka A,Gieruszczak-Bialek D, Sawadro-Rochowska M. 2001. Wiede-mann-Rautenstrauch (neonatal progeroid) syndrome: Newcase with normal telomere length in skin fibroblasts. Am J MedGenet 103:144–148.
Mazess RB, Barden HS, Bisek JP, Hanson J. 1990. Dual-energy x-ray absorptiometry for total-body and regional bone-mineraland soft-tissue composition. Am J Clin Nutr 51:1106–1112.
Petty EM, Laxova R, Wiedemann HR. 1990. Previously unrecog-nized congenital progeroid disorder. Am JMedGenet 35:383–387.
Pivnick EK, Angle B, Kaufman RA, Hall BD, Pitukcheewanont P,Hersh JH, Fowlkes JL, Sanders LP, O’Brien JM, Carroll GS,Gunther WM, Morrow HG, Burghen GA, Ward JC. 2000.Neonatal progeroid (Wiedemann-Rautenstrauch) syndrome:Report of five new cases and review. Am J Med Genet 90:131–140.
Poortmans JR, Boisseau N,Moraine JJ, Moreno-Reyes R, GoldmanS. 2005. Estimation of total-body skeletal muscle mass inchildren and adolescents. Med Sci Sports Exerc 37:316–322.
Proctor DN, O’Brien PC, Atkinson EJ, Nair KS. 1999. Comparisonof techniques to estimate total body skeletal muscle massin people of different age groups. Am J Physiol 277:E489–E495.
Rautenstrauch T, Snigula F. 1977. Progeria: A cell culture studyand clinical report of familial incidence. Eur J Pediatr 124:101–111.
Rautenstrauch T, Snigula F, Wiedemann HR. 1994. Neonatalprogeroid syndrome (Wiedemann-Rautenstrauch). A follow-up study. Klin Padiatr 206:440–443.
RudinC, Thommen L, Fliegel C, SteinmannB, BuhlerU. 1988. Theneonatal pseudo-hydrocephalic progeroid syndrome (Wie-demann-Rautenstrauch). Report of a new patient and reviewof the literature. Eur J Pediatr 147:433–438.
Stoll C, Labay F, Geisert J, Alembik Y. 1998. Wiedemann-Rautenstrauch syndrome. A case report and review of theliterature. Genet Couns 9:119–124.
Wiedemann HR. 1979. An unidentified neonatal progeroidsyndrome: Follow-up report. Eur J Pediatr 130:65–70.
Wong WW, Hergenroeder AC, Stuff JE, Butte NF, Smith EO, EllisKJ. 2002. Evaluating body fat in girls and female adolescents:Advantages and disadvantages of dual-energy X-ray absorp-tiometry. Am J Clin Nutr 76:384–389.
1430 O’NEILL ET AL.
American Journal of Medical Genetics Part A: DOI 10.1002/ajmg.a


















