
Bleeding disorders(Disorders of Platelets and vessel wall)
57
BLEEDING DISORDERS Dr RAJESH S GUIDE Dr GURDEEP KAUR Madam
Transcript of Bleeding disorders(Disorders of Platelets and vessel wall)

BLEEDING DISORDERSDr RAJESH
S
GUIDE Dr GURDEEP KAUR Madam

BLEEDING DISORDERS
A bleeding tendency is a presentation with bleeding in a patient in whom no anatomical cause for the bleeding (i.e., trauma to a vessel for one of many possible reasons) can be discovered.
It is then inferred that the bleeding is due to a functional impairment of the normal hemostatic process.

What are bleeding disorders and Coagulation disorders??
.
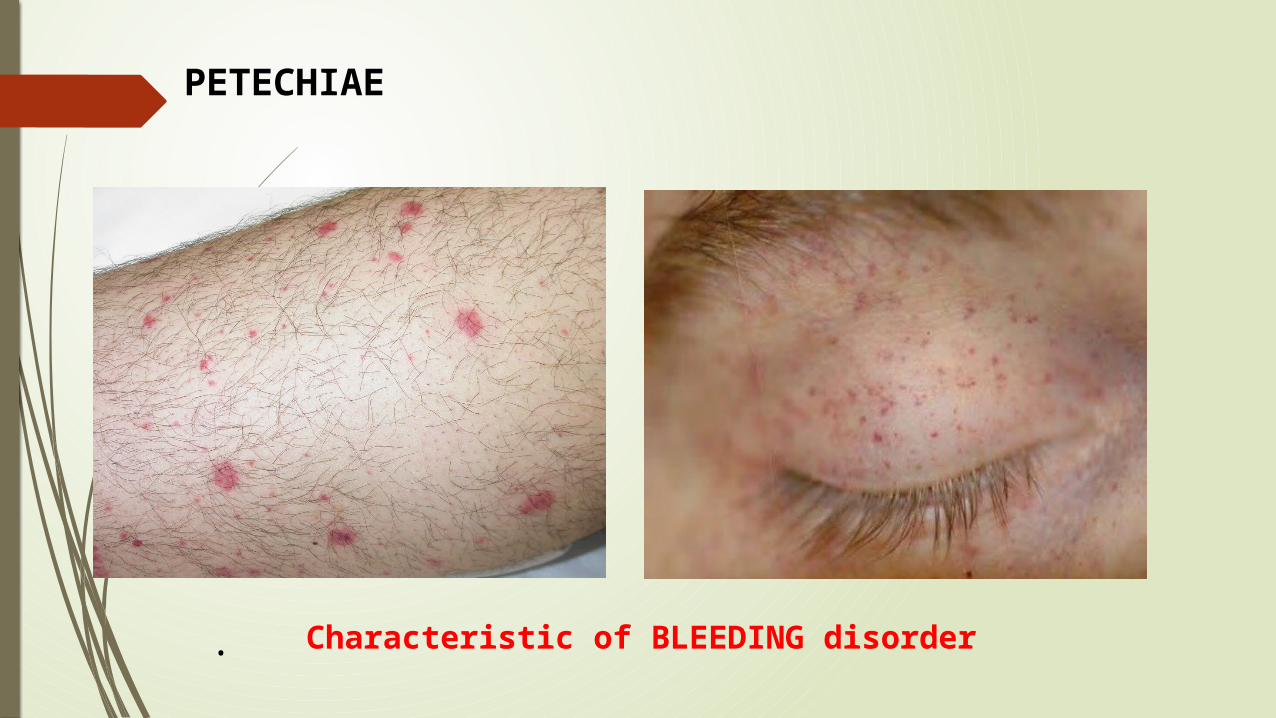
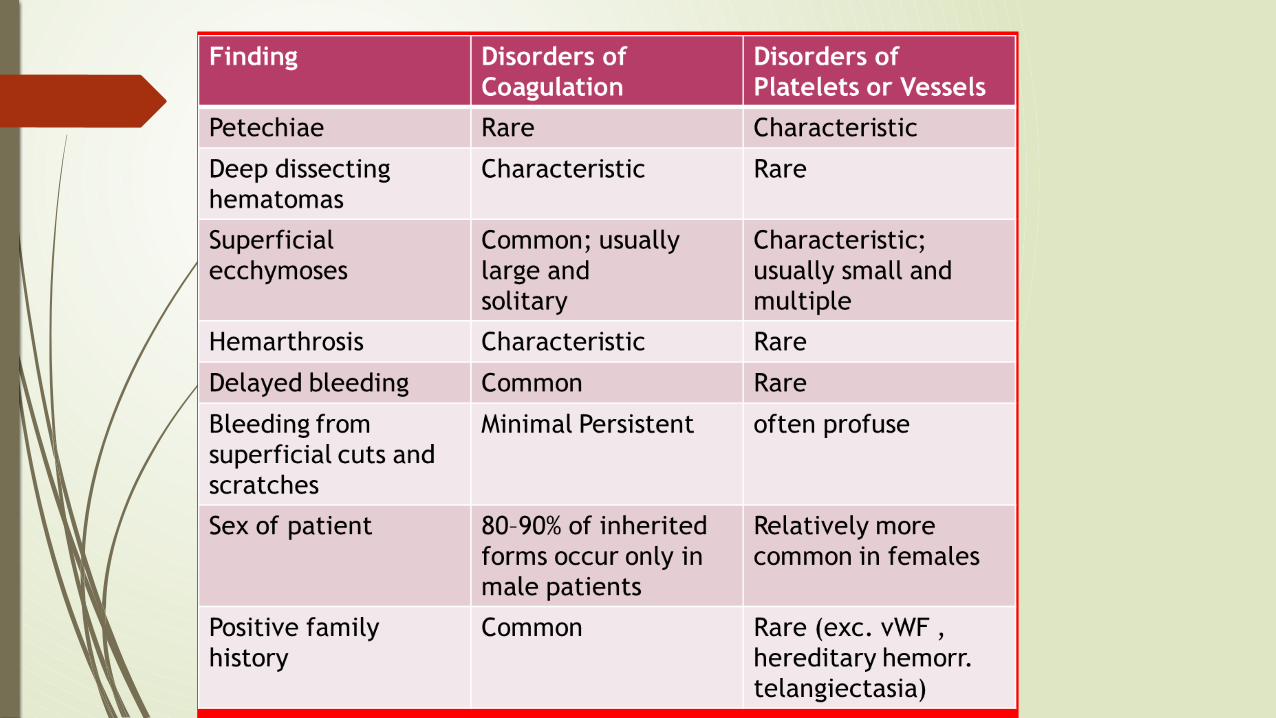
PETECHIAE
Characteristic of BLEEDING disorder
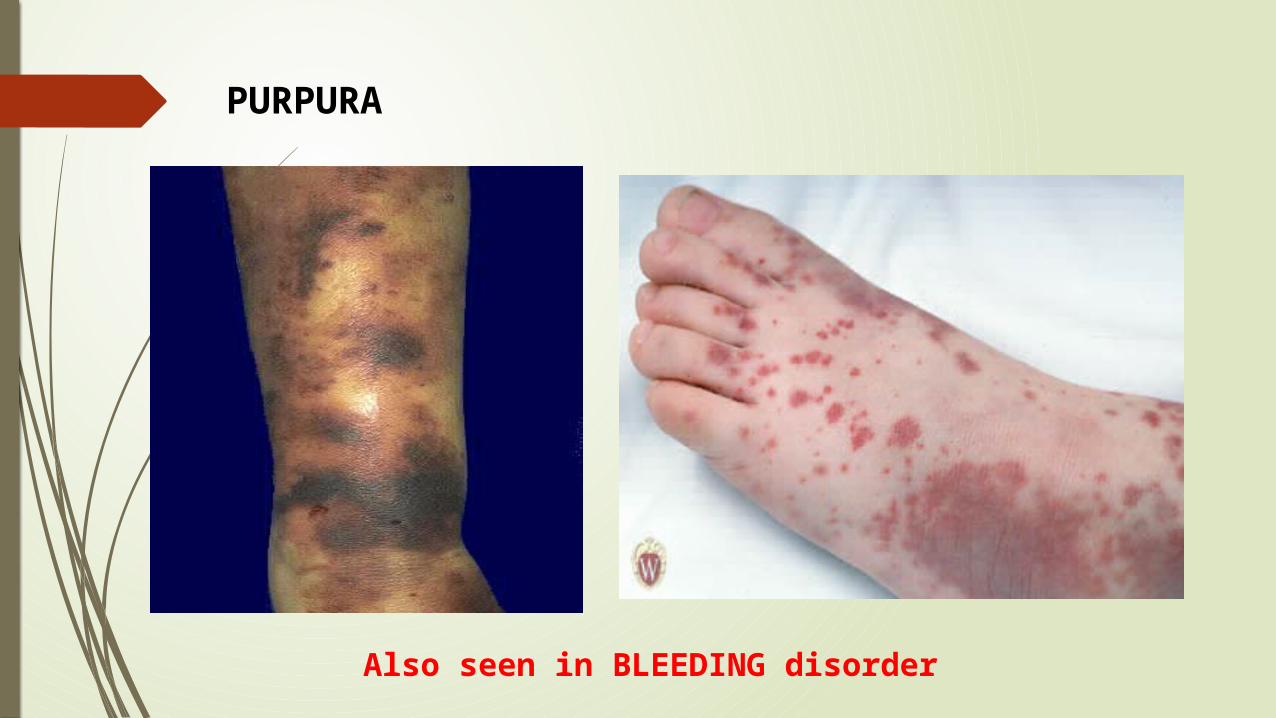
PURPURA
Also seen in BLEEDING disorder

Hemarthrosis
Characteristic of COAGULATION disorders

Ecchymoses
Typical of coagulation factor disorders
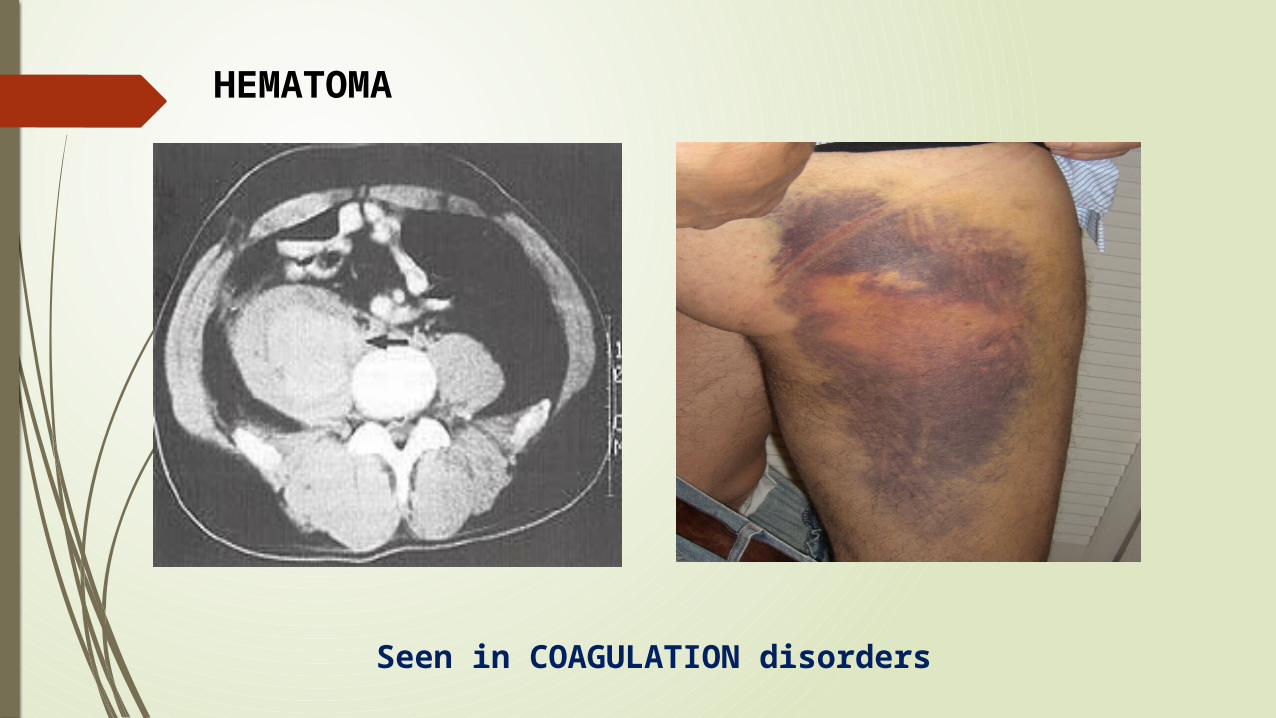
Seen in COAGULATION disorders
HEMATOMA

VASCULAR PHASE
PLATELET PHASE
COAGULATION PHASE
HEMOSTASIS

VASCULAR CAUSES OF BLEEDING
HERIDITARY Hereditary hemorrhagic telangiectasia
Ehlers-Danlos syndrome
Marfan’s syndrome
METABOLIC AND INFLAMMATORY Henoch-Schonlein syndrome
Scurvy - Amyloid
Steroid purpura
senile purpura
Rickettsial diseases
Polyclonal gammopathies

Heriditary hemorrhagic telengiectesia(HHT)
Heriditary hemorrhagic telengiectesia(Osler-Weber-Rendu disease) is a disorder where abnormal telengiectatic capillaries result in frequent bleeding episodes,primarily from the nose and gastrointestinal tract.
AVM of lung,brain,and liver may be seen.
pulmonary AVM in 40% of cases(in endoglin gene mutation type).
Expistaxis begins, on average, at the age of 12 and occurs in >95% of affected individuals by middle age.

Rickettsiae causing Rocky Mountain spotted fever, replicate in endothelial cells and damage them and hence causes petechiae and purpura.
Patients with scurvy (vitamin C deficiency) develop painful episodes of perifollicular skin bleeding as well as more systemic bleeding symptoms. Vitamin C is needed to synthesize hydroxyproline, an essential constituent of collagen.
Patients with Cushing's syndrome or on chronic glucocorticoid therapy develop skin bleeding and easy bruising due to atrophy of supporting connective tissue.
ROCKY MOUNTAIN SPOTTED FEVER

Henoch-Schönlein purpura: A distinct, self-limited type of vasculitis that occurs in children and
young adults.
An acute inflammatory reaction with IgA and complement components in capillaries, mesangial tissues, and small arterioles leading to increased vascular permeability and localized hemorrhage.
The syndrome is often preceded by an upper respiratory infection, commonly with streptococcal pharyngitis.
Patients develop a purpuric rash on the extensor surfaces of the arms and legs, usually accompanied by polyarthralgias or arthritis, abdominal pain, and hematuria from focal glomerulonephritis.
Glucocorticoids can provide symptomatic relief but do not alter the course of the illness.

1.THROMBOCYTOPENIA1.INHERITED DISORDERS
2.THROMBOCYTOSIS2.ACQUIRED DISORDERS
PLATELET DISORDERS
QUANTITATIVE DISORDERS
QUALITATIVE DISORDERS

THROMBOCYTOPENIA Thrombocytopenia is defined as a platelet count less than
150,000
2.5 percent of the normal population will have a platelet count lower than this.
Thrombocytopenia results from one or more of three processes:
(1) decreased bone marrow production;
(2) sequestration, usually in an enlarged spleen; and/or
(3) increased platelet destruction.

In evaluating a patient with thrombocytopenia, a key step is to review the peripheral blood smear and to first rule out "pseudothrombocytopenia"
It is an in vitro artifact resulting from platelet agglutination via antibodies ,when the calcium content is decreased by blood collection in EDTA tube.
If a low platelet count is obtained in EDTA-anticoagulated blood,Repeat platelet count determined in blood collected into sodium citrate (blue top tube) or heparin (green top tube).

INFECTION-INDUCED THROMBOCYTOPENIA Most common non-iatrogenic cause of thrombocytopenia.
Can affect both platelet production and platelet survival.
Immune mechanisms are proposed to be the cause, as in infectious mononucleosis,Hiv and other viral Infections.
“A study evaluating the role of bone marrow examination in fever of unknown origin in HIV-infected patients found that for 86% of patients, the same diagnosis was established by less-invasive techniques, notably blood culture.”
Thus, a bone marrow examination is not routinely recommended in evaluation of thrombocytopenia in infections and it is recommended when the diagnosis is needed urgently or when other, less invasive methods have been unsuccessful.

Drug-Induced Thrombocytopenia A predictable decrease in platelet count occurs after treatment with
many chemotherapeutic drugs due to bone marrow suppression.
All drugs should be suspect in a patient with thrombocytopenia without an apparent cause and should be stopped, or substituted, if possible.
Although not as well studied, herbal and over-the-counter preparations may also result in thrombocytopenia.
They are very commonly seen in Quinine and Sulfa drugs. Other examples include Rifampin, Amiodarone, Ampicillin,
Digoxin,Diclofenac,Ibuprofen,Linezolid,Furosemide.

Drug-Induced Thrombocytopenia Classic drug-dependent antibodies are antibodies that react with specific
platelet surface antigens, and result in thrombocytopenia only when the drug is present.
The thrombocytopenia typically occurs after a period of initial exposure (median length 21 days), or upon reexposure, and usually resolves in 7–10 days after drug withdrawal.
The thrombocytopenia caused by the platelet GpIIbIIIa inhibitory drugs, such as abciximab, differs in that it may occur within 24 h of initial exposure. This appears to be due to the presence of naturally occurring antibodies that cross-react with the drug bound to the platelet.

HEPARIN INDUCED THROMBOCYOPENIA Heparin has been used as an anticoagulant medication since the late
1930s and is one of the most prescribed drugs.
However, clinicians recognized that some patients developed a syndrome of immune-mediated thrombocytopenia and thrombosis, which came to be called heparin-induced thrombocytopenia (HIT).
Drug-induced thrombocytopenia due to heparin differs from that seen with other drugs in two major ways.
(1) The thrombocytopenia is not usually severe, with nadir counts rarely <20,000/L.
(2) Heparin-induced thrombocytopenia (HIT) is not associated with bleeding and, in fact, markedly increases the risk of thrombosis.

HIT occurs when IgG antibodies develop against neoantigens created by multimolecular heparin/platelet factor 4 (PF4) complexes.
The antiheparin/PF4 antibody can activate platelets through the FcRIIa receptor and also activate monocytes and endothelial cells.
Surgical patients develop the antibodies more often than medical or obstetric patients because surgery results in platelet activation and PF4 release from platelet alpha granules.
In general, antibodies can be identified in up to 15% of orthopedic surgical patients and up to 50% of cardiopulmonary bypass patients.
Therapeutic heparin doses are not required to stimulate antibody production; even small amounts of heparin can result in antibody formation and clinical HIT.

Patients baseline platelet counts should be obtained before initiating heparin therapy. A falling platelet count during therapy raises the suspicion of HIT, and in the usual presentation
The platelet count begins to drop after 5 to 10 days of heparin therapy. Patients with recent heparin exposure (within 100 days) may have platelet counts that fall before day 5, often within the first 24 hours. Delayed-onset cases also occur but are uncommon.


WHEN TO GO FOR DIAGNOSTIC TESTING? The 4 T's have been recommended to be used in a diagnostic
algorithm for HIT: (Pre test Probability)
1.Thrombocytopenia
2.Timing of platelet count drop
3.Thrombosis and other sequelae such as localized skin reactions
4.oTher causes of thrombocytopenia not evident
Each feature is given a score and patients are classified as high, intermediate, or low probability.
Such clinical scoring systems have good Negative predictive value for patients with low scores;
patients with moderate or high clinical probability usually require lab testing

LABORATORY TESTING
1.ELISA (with PF4/polyanion as the antigen)
• This tests for presence of antibodies in serum against heparin and PF4 complexes.
• Since many patients develop antibodies but do not develop clinical HIT, the test has a low specificity for the diagnosis of HIT.
2.Platelet activation assays (Seratonin Release Assays)
• which measures the ability of the patient's serum to activate platelets in the presence of heparin in a concentration-dependent manner.
• This test has lower sensitivity
but higher specificity than the ELISA.
• This remains to be GOLD STANDARD test in HIT diagnosis.
Neverthless, HIT remains to be a CLINICAL DIAGNOSIS

Go for Imaging Studies( Atleast lower extremity duplex Dopplers)
The direct thrombin inhibitors (DTIs) argatroban and lepirudin are to be started
If thrombosis present – treat for 3 to 6 monthsIf no evidence – treat for 1 month
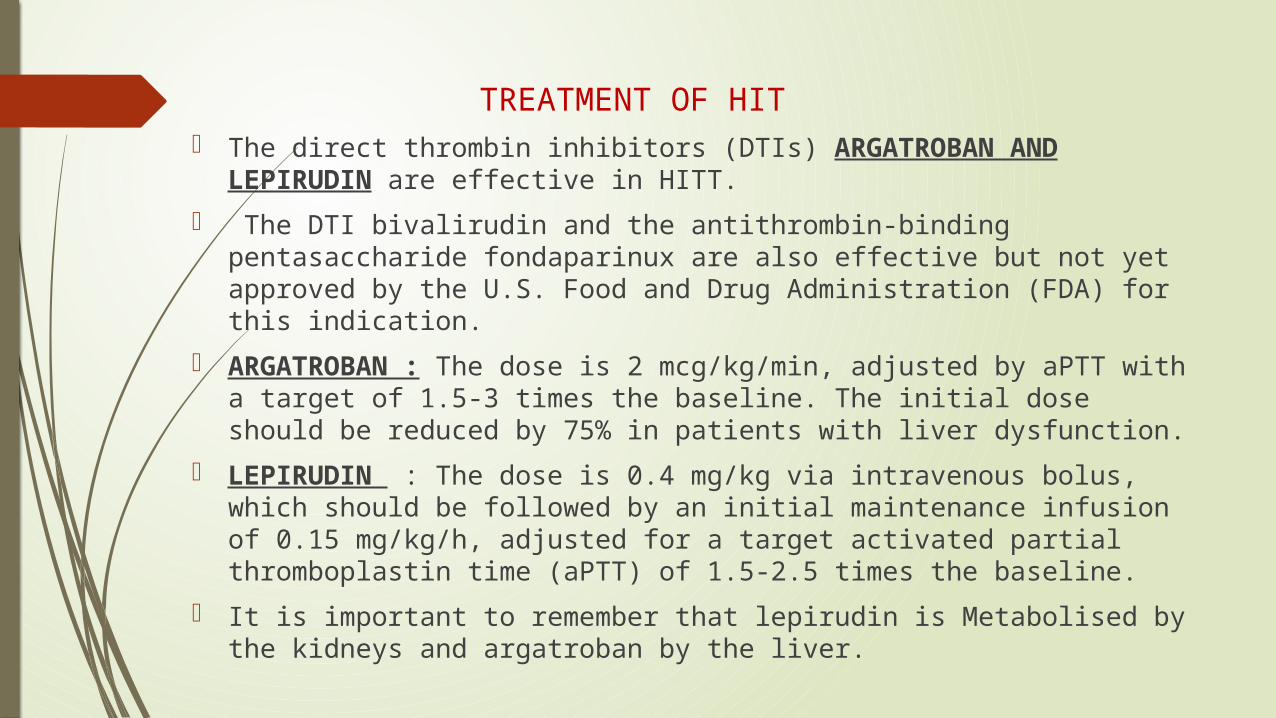
TREATMENT OF HIT The direct thrombin inhibitors (DTIs) ARGATROBAN AND LEPIRUDIN
are effective in HITT.
The DTI bivalirudin and the antithrombin-binding pentasaccharide fondaparinux are also effective but not yet approved by the U.S. Food and Drug Administration (FDA) for this indication.
ARGATROBAN : The dose is 2 mcg/kg/min, adjusted by aPTT with a target of 1.5-3 times the baseline. The initial dose should be reduced by 75% in patients with liver dysfunction.
LEPIRUDIN : The dose is 0.4 mg/kg via intravenous bolus, which should be followed by an initial maintenance infusion of 0.15 mg/kg/h, adjusted for a target activated partial thromboplastin time (aPTT) of 1.5-2.5 times the baseline.
It is important to remember that lepirudin is Metabolised by the kidneys and argatroban by the liver.

Warfarin therapy in HIT Warfarin should be postponed until substantial platelet recovery. If
warfarin has already been started, vitamin K should be given.
Introduction of warfarin alone in the setting of HIT or HITT may precipitate thrombosis, particularly venous gangrene, presumably due to clotting activation and severely reduced levels of proteins C and S.
HIT patients typically present with an international normalized ratio (INR) greater than 4, which corresponds to severe protein C depletion. Preferably, warfarin should not started before the thrombocyte count is greater than 150 x 109/L.
Platelet transfusions should be avoided in heparin-induced thrombocytopenia (HIT), as they may increase the thrombogenic effect.

Immune Thrombocytopenic Purpura (ITP)
an acquired disorder in which there is immune-mediated destruction of platelets and possibly inhibition of platelet release from the megakaryocyte.
Childhood form (most < 10 yrs old)
May follow viral infection, vaccination
Peak incidence in fall & winter
~50% receive some treatment
≥75% in remission within 6 mo
Adult form
No prodrome
Chronic, recurrences common
Spontaneous remission rate about 5%
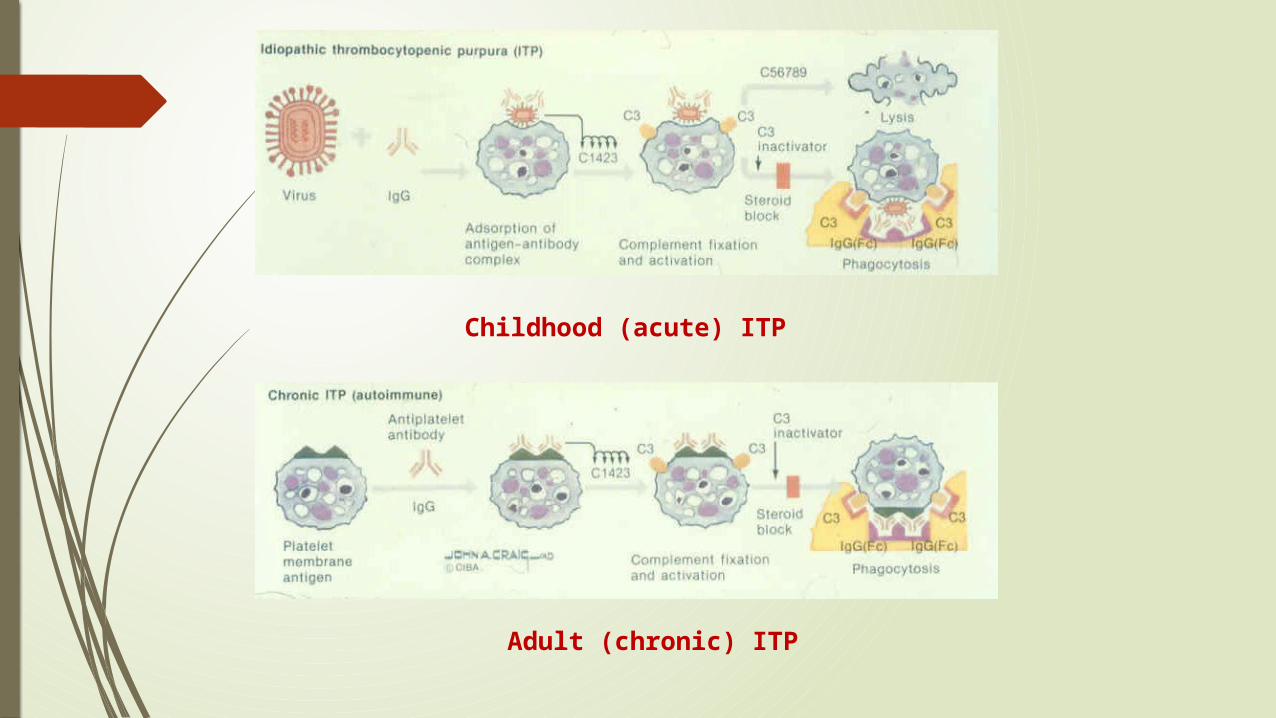
Childhood (acute) ITP
Adult (chronic) ITP

CLINICAL FEATURES ITP is characterized by mucocutaneous bleeding and a low, often very low,
platelet count, with an otherwise normal peripheral blood cells and smear.
Patients usually present either with ecchymoses and petechiae, or with thrombocytopenia incidentally found on a routine CBC.
Mucocutaneous bleeding, such as oral mucosa, gastrointestinal, or heavy menstrual bleeding, may be present. Rarely, life-threatening, including central nervous system, bleeding can occur. Wet purpura (blood blisters in the mouth) and retinal hemorrhages may herald life-threatening bleeding.

LABORATORY TESTINGLaboratory testing for antibodies (serologic testing) is usually not helpful due to the low sensitivity and specificity of the current tests.
Bone marrow examination :can be reserved for older adults (usually >60 years)
in patients who do not respond to initial therapy.
Peripheral blood smear : May show large platelets
Iron deficiency anemia may be present.
Testing for HIV infection and hepatitis C (to rule out secondary causes)
Serologic testing for SLE, serum protein electrophoresis, and immunoglobulin levels to potentially detect hypogammaglobulinemia .
Direct antiglobulin testing (Coombs test) to rule out combined autoimmune hemolytic anemia with ITP (Evans syndrome).

Rh0(D) immune globulin therapy:
Only in Rh + Patients
at 50–75 g/kg
The mechanism of action of anti-D is not fully understood however, after administration the anti-D coated red blood cell complexes saturate Fcγ receptors sites on macrophages ,resulting in preferential destruction of RBCs, therefore sparing antibody-coated platelets.

STEROIDS:
Prednisone to be given at 1 mg/kg.
Until platelet counts comes back to normal levels
It takes around two to four weeks.
Intravenous Immunoglobulin(Ivig): Intravenous gamma globulin (IVIgG), which is pooled, primarily IgG
antibodies, also blocks the Fc receptor system, but appears to work primarily through different mechanism(s).
IVIgG is dosed at 2 g/kg total, given in divided doses over 2–5 days.
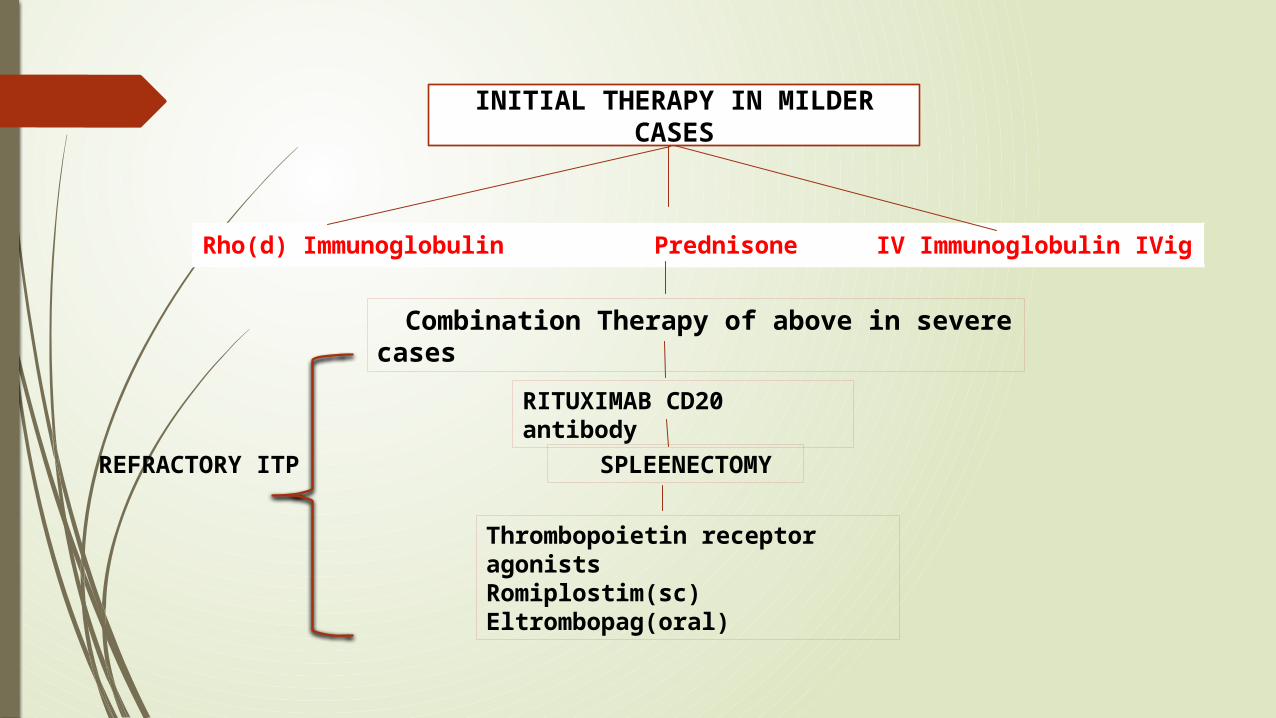
Rho(d) Immunoglobulin
Prednisone IV Immunoglobulin IVig
INITIAL THERAPY IN MILDER CASES
Combination Therapy of above in severe cases
RITUXIMAB CD20 antibody
SPLEENECTOMY
Thrombopoietin receptor agonists Romiplostim(sc)Eltrombopag(oral)
REFRACTORY ITP

INHERITED THROMBOCYTOPENIA
AUTOSOMAL DOMINANT • May-Hegglin
anomaly • Sebastian• Epstein's• Fechtner
syndromes,
• A common feature of these disorders is large platelets
AUTOSOMAL RECESSIVE • congenital
amegakaryocytic thrombocytopenia
• Thrombocytopenia with absent radii
• Bernard Soulier syndrome
X-linked disorders
• Wiskott-Aldrich syndrome

Thrombotic Thrombocytopenic Purpura Classic pentad:
Microangiopathic hemolytic anemia Thrombocytopenia Renal involvement Neurologic signs Fever
Most cases in adults are caused by acquired autoantibodies that inhibit ADAMTS13(a metalloprotease that cleaves vWF within platelet-rich thrombi)
Congenital form (Upshaw-Schulman syndrome) is the result of a deficiency of ADAMTS13
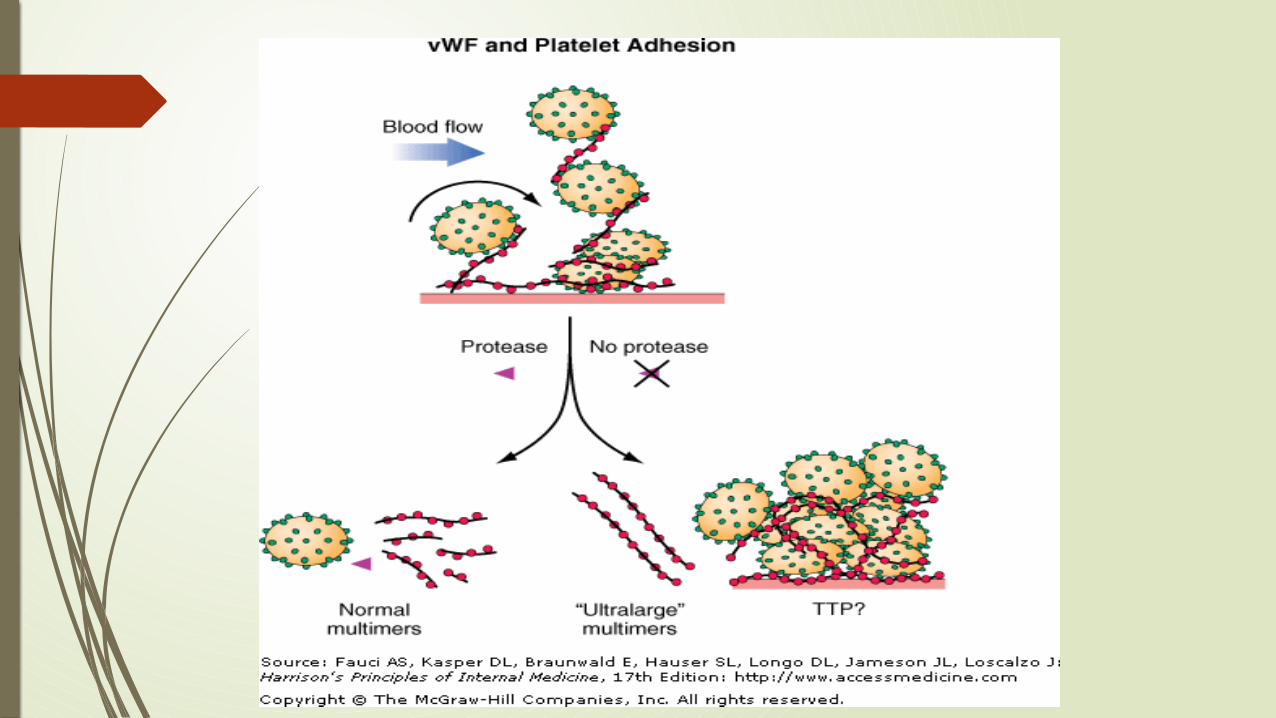

DIAGNOSISTTP is a devastating disease if not diagnosed and treated promptly.
Findings to support the TTP diagnosis include an
Increased lactate dehydrogenase
Increased indirect bilirubin
Decreased haptoglobin
Increased reticulocyte count
Negative direct antiglobulin test.
The peripheral smear may show evidence of schistocytes .

TREATMENT Plasma exchange remains the mainstay of treatment of TTP.
Plasma exchange is continued until the platelet count is normal and signs of hemolysis are resolved for at least 2 days.
The use of glucocorticoids seems a reasonable approach, but should only be used as an adjunct to plasma exchange.
Immunomodulatory therapies have been reported to be successful in refractory or relapsing TTP, including rituximab, vincristine, cyclophosphamide, and splenectomy.
A significant relapse rate is noted, 25–45% within 30 days of initial "remission," and 12–40% with late relapses

THROMBOCYTOSIS
Thrombocytosis is almost always due either to
Iron deficiency
Inflammation, cancer, or infection (reactive thrombocytosis)
An underlying myeloproliferative process [essential thrombocythemia or polycythemia vera)

QUALITATIVE DISORDERS OF PLATELETS
INHERITED Glanzmann's thrombasthenia
(absence of the platelet GpIIbIIIa receptor)
Bernard Soulier syndrome (absence of the platelet GpIb-IX-V receptor).
ACQUIRED Antiplatelet therapy
Uremia.
Cardiopulmonary bypass
Myeloproliferative and myelodysplastic syndromes


GLANZMANN THROMBASTHENIA AR; Mutation in IIb-IIIa, the most abundant platelet surface
receptor
Fundamental defect of thrombasthenic patients is the inability of the platelets to aggregate.
Clinical features include bleeding in skin, mucous membrane (petichiae, echymoses), recurrent epistaxis, GI hemorrhage, menorrhagia.
Bleeding time prolonged. The hallmark of the disease is severely reduced or absent platelet
aggregation in response to multiple agonists ie ADP, thrombin, or collagen (except Ristocetin)

BERNARD-SOULIER SYNDROME
AR; characterized by moderate to severe thrombocytopenia, giant platelets, and profuse/spontaneous bleeding.
Basis for the disease is deficiency or dysfunction of the GP Ib-V-IX complex.
Prolonged bleeding time, thrombocytopenia (plt<20 K), peripheral smear shows large platelets
Aggregation studies show normal aggregation in response to all agonists except Ristocetin (opposite pattern than thrombasthenia)

VON WILLEBRAND DISEASE
von Willebrand factor Synthesis in endothelium and megakaryocytes Forms large multimer Carrier of factor VIII Anchors platelets to subendothelium Bridge between platelets
Inheritance - autosomal dominant
Incidence - 1/5-10,000
Clinical features - mucocutaneous bleeding

TYPES
1. Type I(70-80%) :a mild-to-moderate quantitative deficiency in vWF
2. Type II(10-15%) : is due to qualitative abnormalities of vWF and is subdivided into type 2A VWD,2B,2M are due to various mutations causing functional defects.
Type 2N - Mutations in VWF that preclude binding of FVIII. As FVIII is stabilized by binding to VWF, the FVIII in patients with type 2N VWD has a very short half-life, and the FVIII level is markedly decreased. This is sometimes termed autosomal hemophilia.
3. Type III : a severe quantitative deficiency associated with very little or no detectable plasma or platelet vWF, have a profound bleeding disorder.

Screening tests typically include
Prothrombin time (PT)
Activated partial thromboplastin time (aPTT),
FVIII level
Ristocetin cofactor (RCoF) activity
vWF antigen (vWF:Ag).
vonWillebrand typeAssay 1 2 3
vWF antigen ß Normal ßßvWF activity ß ß ßßMultimer analysis Normal Normal Absent

DDAVP (deamino-8-arginine vasopressin) plasma VWF levels by stimulating secretion from endothelium Duration of response is variable Not generally used in type 2 disease Dosage 0.3 µg/kg q 12 hr IV Used in Type 1 Vwf disease
Cryoprecipitate Source of fibrinogen, factor VIII and VWF Only plasma fraction that consistently contains VWF multimers Used in Type 2 and 3 Vwf disease.

Antifibrinolytic therapy using either E-aminocaproic acid or tranexamic acid is an important therapy, either alone or in an adjunctive capacity, particularly for the prevention or treatment of mucosal bleeding.
particularly useful in prophylaxis for dental procedures, with DDAVP for dental extractions and tonsillectomy, menorrhagia, and prostate procedures.
It is contraindicated in the setting of upper urinary tract bleeding, due to the risk of ureteral obstruction.

WHY IS THROMBOCYTOPENIA IN MALARIA?
The Plasmodium species have no direct effect on platelets. Instead Plasmodium attacks red blood cells (RBCs) and when the are damaged they are trapped by the spleen which filters the blood.
Because there are so many RBCs damaged the spleen takes up an unusually high number of RBCs and becomes enlarged (splenomegaly).
The platelets get trapped in the now clogged spleen and this causes the thrombocytopenia.
Source:
Cohen & Powderly: Infectious Diseases, 2nd ed., Copyright © 2004 Mosby

IMMUNE-MEDIATED THROMBOCYTOPENIA OF MALARIA(J G Kelton, J Keystone, J Moore, G Denomme, E Tozman, M Glynn, P B Neame, J Gauldie, and J Jensen)
We studied 28 patients with malarial infections and noted that 16 of 17 thrombocytopenic patients had elevated levels of platelet-associated IgG (PAIgG).
In all thrombocytopenic patients studied, the level of PAIgG returned to normal as the platelet count rose to normal levels.
The thrombocytopenia that complicates at least some malarial infections is caused by immune mechanisms; specific IgG binds to platelet-bound malaria antigen through the Fab portion of the immunoglobulin molecule.

THROMBOCYTOPENIA IN DENGUE
Three possible triggers to induce thrombocytopenia in dengue virus infection. (Funahara Y, Ogawa K, Fujita N, Okuno Y.)
1) DV antigen attached to human platelets without immune-mediated reaction.(direct lysis)
2) A decrease in platelet count was more markedly demonstrated by the binding of anti-DV antibody on the DV antigen associated with platelets than by the binding of the antigen-antibody complex on platelets.(immune mediated)
3) a modulation of endothelial cell by the infection of DV to the cell was suggested as one of the causes of the thrombocytopenia.(endothelium-ADAMTS13 related).

CAPILLARY LEAKAGE IN DENGUE Angiopoietin-1 (Ang-1) is stored in platelets and activates the
endothelial cell-specific tyrosine kinase receptor Tie-2, which in turn leads to enhanced endothelial cell survival and stabilization and maintains vascular integrity.
On the other hand, the angiopoietin-2 (Ang-2), is derived from endothelium and stored in Weibel-Palade bodies (WPBs).
There was an inverse correlation between angiopoietin-1 and markers of plasma leakage and a positive correlation between angiopoietin-2 and markers of plasma leakage
The VWF activation factor was also higher in children with DHF/DSS: it was highest in children who died.
In all probability, high circulating levels of VWF in an active conformation, together with low ADAMTS-13 levels, contribute to the thrombocytopenia and complications of dengue
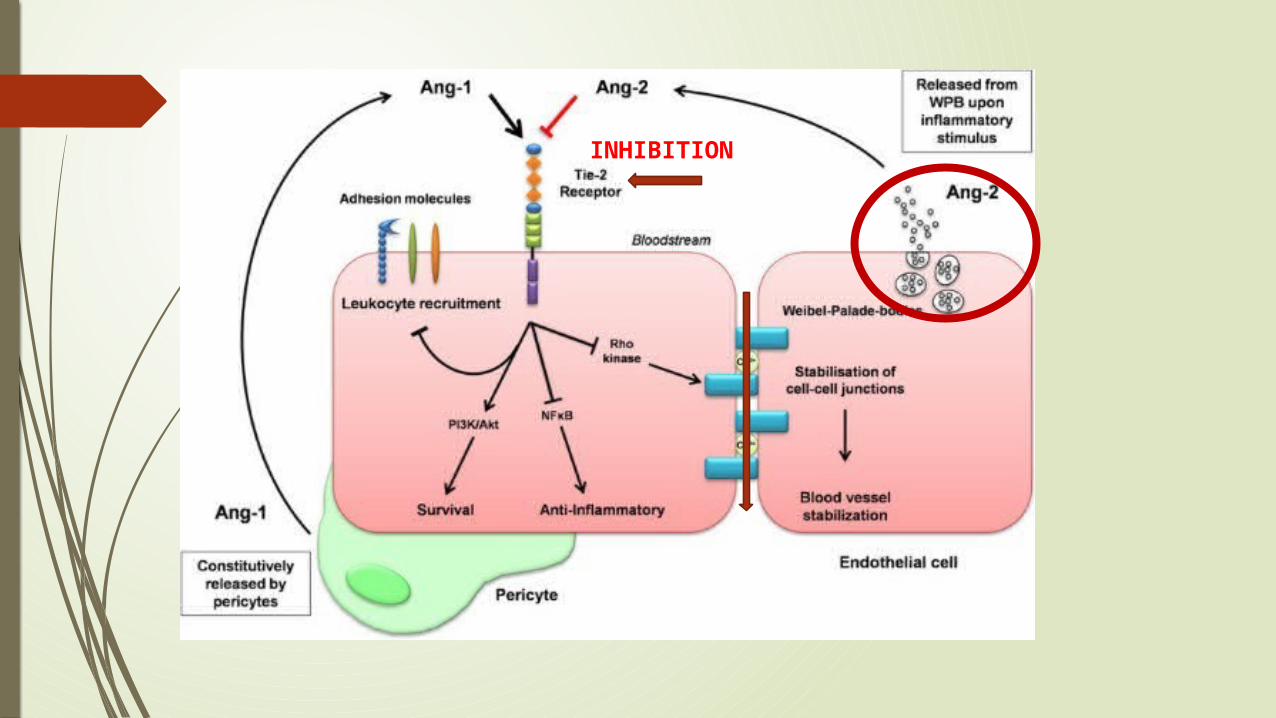
INHIBITION

THANK YOU


















