BioSci D145 lecture 1 page 1 © copyright Bruce Blumberg 2010. All rights reserved BioSci D145...
43
BioSci D145 lecture 1 page 1 © copyright Bruce Blumberg 2010. All rights reserved BioSci D145 Lecture #8 • Bruce Blumberg ([email protected]) – 4103 Nat Sci 2 - office hours Tu, Th 3:30-5:00 (or by appointment) – phone 824-8573 • TA – Bassem Shoucri ([email protected]) – 4351 Nat Sci 2, 824-6873, 3116 – office hours M 2-4 • lectures will be posted on web pages after lecture – http://blumberg.bio.uci.edu/biod145-w2015 – http://blumberg-lab.bio.uci.edu/biod145-w2015 Term papers due Friday, March 6 by 12 midnight (23:59.59) (-1 point for each day late)
-
Upload
nickolas-weaver -
Category
Documents
-
view
223 -
download
0
Transcript of BioSci D145 lecture 1 page 1 © copyright Bruce Blumberg 2010. All rights reserved BioSci D145...

BioSci D145 lecture 1 page 1 ©copyright Bruce Blumberg 2010. All rights reserved
BioSci D145 Lecture #8
• Bruce Blumberg ([email protected])– 4103 Nat Sci 2 - office hours Tu, Th 3:30-5:00 (or by appointment)– phone 824-8573
• TA – Bassem Shoucri ([email protected])– 4351 Nat Sci 2, 824-6873, 3116 – office hours M 2-4
• lectures will be posted on web pages after lecture – http://blumberg.bio.uci.edu/biod145-w2015– http://blumberg-lab.bio.uci.edu/biod145-w2015
Term papers due Friday, March 6 by 12 midnight (23:59.59) (-1 point for each day late)

BioSci D145 lecture 1 page 2 ©copyright Bruce Blumberg 2010. All rights reserved
Term paper requirements and scoring
• Outline - 1 point• Actual paper – 5 pages single spaced 1” margins (references not
included). • Specific aims – 2 points (this should be about 3/4 to one page)
– Write a paragraph introducing the topic, state why it is important and what are the gaps in knowledge that you will address.
– State a hypothesis to be tested– Enumerate 2-3 specific aims in the form of questions that test
your hypothesis. After each one, use a sentence or two to state what you will do to answer these questions
– Finish with a paragraph stating what will be the significance of the research assuming that you successfully execute the proposed experiments.
– It is very important to state the human health relevance of your research (if you are doing something biomedical) or the broader impacts on advancing the frontiers of knowledge (for something that is not relevant to human health).
– This is among the most important parts of any grant application. You have to convince the reviewer here that your work is important and worth funding.

BioSci D145 lecture 1 page 3 ©copyright Bruce Blumberg 2010. All rights reserved
Term paper requirements and scoring
• Background and Significance – 3 points (about 1.5-2.5 pages)– Briefly summarize what is known about the problem.
• Not a comprehensive review, just a summary of the important points.
– Succinctly state what is not known and why it is important that this research be done
• Address knowledge gaps• Are you addressing something controversial?
– talk about the controversy and why your work will address it directly.
– In about one paragraph, state what is important about your proposed research and why will accomplishing it benefit the research community and world at large.
• i.e., what is the potential impact if you are successful• Don’t repeat what was said in specific aims exactly but
obviously they should be related.
• http://blumberg-lab.bio.uci.edu/biod145-w2015/example_grant.pdf

BioSci D145 lecture 1 page 4 ©copyright Bruce Blumberg 2010. All rights reserved
Term paper requirements and scoring
• Research plan – 4 points (about 2.5-3 pages)– In a short paragraph, state what you will do and why it is
important. (I know it seems repetitive by now, but reviewers are busy and will be skimming your grant. You need to hit them over the head a few times before they will get your point).
– Restate each specific aim from the Specific aims section (one by one)
– describe what you will do to address the aim• Break into subaims as appropriate• State the hypothesis to be tested in each• Explain the rationale• Describe briefly what approach you will take• Discuss what you expect to find• Point out any possible problems and alternative approaches
– I am mostly concerned with your hypothesis and rationale here.– Not an all-encompassing proposal – 4-5 years by a small team
(e.g., your PhD thesis research)• http://blumberg-lab.bio.uci.edu/bioD145-w2015/example_grant.pdf

BioSci 145B lecture 6 page 5 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting• Transgenesis is mostly a gain-of-function technique
– Loss-of-function preferred for identifying gene function
• Targeted gene disruption is very desirable– to understand function of newly identified genes
• e.g., from genome projects• Or gene by gene
– produce a mutation and evaluate the requirements for your gene of interest
– good to create mouse models for human diseases• knockout the same gene disrupted in a human and may
be able to understand disease better and develop efficacious treatments
• excellent review is Müller (1999) Mechanisms of Development 82, 3-21.

BioSci 145B lecture 6 page 6 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting (contd)• enabling technology is embryonic
stem (ES) cells (or iPS cells)– these can be cultured but
retain the ability to colonize the germ line
– essential for transmission of engineered mutations
– derived from inner cell mass of blastula stage embryos
– grown on lethally irradiated “feeder” cells which help to mimic the in vivo condition
• essential for maintaining stem cell phenotype• Also problematic for human ES lines
• ES cells are very touchy in culture– lose ability to colonize germ line with time– easily infected by “mysterious microorganisms” that inhibit ability
to colonize germ line• ko labs maintain separate hoods and incubators for ES cell
work– ES cells depend critically on the culture conditions maintain an
uncommitted, undifferentiated state that allows germ line transmission.

BioSci 145B lecture 6 page 7 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting (contd)
• isolate genomic clones from ES cell library
• Restriction map – Especially exons/introns
• Make targeting construct– Want ~5kb genomic regions
flanking targeted region– Must disrupt essential exon– Want no functional protein– Verify in cell culture– often useful to fuse reporter gene to the coding region of the
protein• gene expression can be readily monitored
– Insert dominant selectable marker within replacement region– negative selection marker is located outside the region targeted
to be replaced• Electroporate DNA into ES cells, select colonies resistant to positive
selection• Integration positive cells then subjected to negative selection
– homologous recombinants lose this marker

BioSci 145B lecture 6 page 8 ©copyright Bruce Blumberg 2009. All rights reserved
Gene Targeting (contd)
• Targeting vector
• Electroporate into ES cells
• Recombination
• Selection
• identification

BioSci 145B lecture 6 page 9 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting (contd)
• Technique (contd)– homologous recombination is
verified by Southern blotting– factors affecting targeting
frequency (success)• length of homologous
regions, more is better.– 0.5 kb is minimum
length for shortest arm
• isogenic DNA (ie, from the ES cells) used for targeting construct is best
• locus targeted. This may result from differences in chromatin structure and accessibility
– Expand ES cell colonies
BioSci 145B lecture 6 page 10 ©copyright Bruce Blumberg 2009. All rights reserved
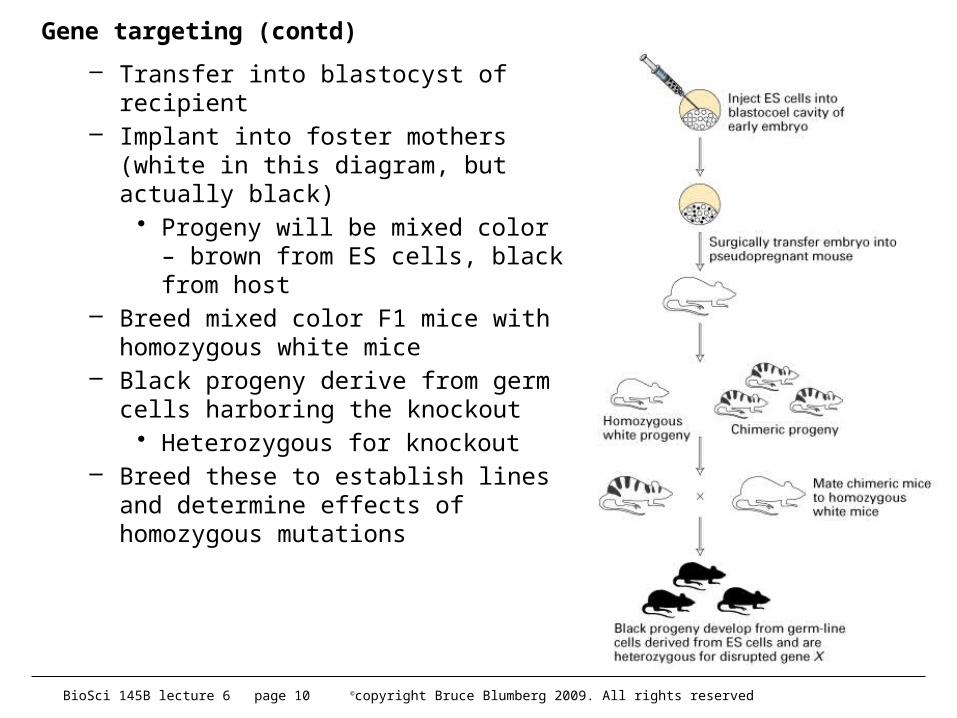
Gene targeting (contd)
– Transfer into blastocyst of recipient– Implant into foster mothers (white in
this diagram, but actually black)• Progeny will be mixed color –
brown from ES cells, black from host
– Breed mixed color F1 mice with homozygous white mice
– Black progeny derive from germ cells harboring the knockout
• Heterozygous for knockout– Breed these to establish lines and
determine effects of homozygous mutations

BioSci 145B lecture 6 page 11 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting (contd)
• problems and pitfalls– incomplete knockouts, ie, protein function is not lost
• but such weak alleles may be informative– alteration of expression of adjacent genes
• region removed may contain regulatory elements• may remove unintended genes (e.g. on opposite strand)
– interference from selection cassette• strong promoters driving these may cause phenotypes

BioSci 145B lecture 6 page 12 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting (contd)
• Applications– creating loss-of-function alleles– introducing subtle mutations– chromosome engineering– marking gene with reporter, enabling whole mount detection of
expression pattern (knock-in)

Example of a “knock-in” model
BioSci D145 lecture 8 page 13 ©copyright Bruce Blumberg 2009 All rights reserved
Long arm7kbp
Kpn
I
Kpn
I
Pst
I
Eco
RI
XhoI
Bam
HI
Bam
HI
Eco
RI
Bam
HI
Eco
RI
loxP
loxP
E2 E3 E4 E5 E6E7 E8 E9
Short arm1.3kbp
E3Neo I8 PGK-DTA
BGH-3’UTR
Human PXR LBD to C terminus
E2 E3
Bam
HI
Bam
HI
NeoNeoAL2 SXR RC RV5
Southern Probe
E9
E9E3E2
Wild type allele
Targeting vector
Targeted allele
Cre-recombined allele
Long arm7kbp
Kpn
I
Kpn
I
Pst
I
Eco
RI
XhoI
Bam
HI
Bam
HI
Eco
RI
Bam
HI
Eco
RI
loxP
loxP
E2 E3 E4 E5 E6E7 E8 E9
Short arm1.3kbp
E3Neo I8 PGK-DTA
BGH-3’UTR
Human PXR LBD to C terminus
E2 E3
Bam
HI
Bam
HI
NeoNeoAL2 SXR RC RV5
Southern Probe
E9
E9E3E2
Wild type allele
Targeting vector
Targeted allele
Cre-recombined allele

BioSci 145B lecture 6 page 14 ©copyright Bruce Blumberg 2009. All rights reserved
Gene targeting (contd)
• Applications– creating loss-of-function alleles– introducing subtle mutations– chromosome engineering– marking gene with reporter, enabling whole mount detection of
expression pattern (knock-in)
• advantages– can generate a true loss-of-function alleles– precise control over integration sites– prescreening of ES cells for phenotypes possible– can also “knock in” genes
• disadvantages– not trivial to set up– may not be possible to study dominant lethal phenotypes– non-specific embryonic lethality is common (~30%)– difficulties related to selection cassette

BioSci D145 lecture 8 page 15 ©copyright Bruce Blumberg 2009 All rights reserved
Conditional gene targeting
• Many gene knockouts are embryonic lethal– some of these are appropriate and expected
• gene activity is required early– others result from failure to form and/or maintain the placenta
• ~30% of all knockouts• Clearly a big obstacle for gene analysis
• How can this be overcome?– Generate conditional knockouts either in particular tissues or
after critical developmental windows pass– Sauer (1998) Methods 14, 381-392.

BioSci D145 lecture 8 page 16 ©copyright Bruce Blumberg 2009 All rights reserved
Conditional gene targeting - contd
• Approach– recombinases perform
site-specific excision between recognition sites
– FLP system from yeast• doesn’t work well
– Cre/lox system from bacteriophage P1
• P1 is a temperate phage that hops into and out of the bacterial genome
• recombination requires – 34 bp recognition sites
locus of crossover x in P1(loxP)
– Cre recombinase• if loxP sites are directly repeated then deletions• if inverted repeats then inversions result
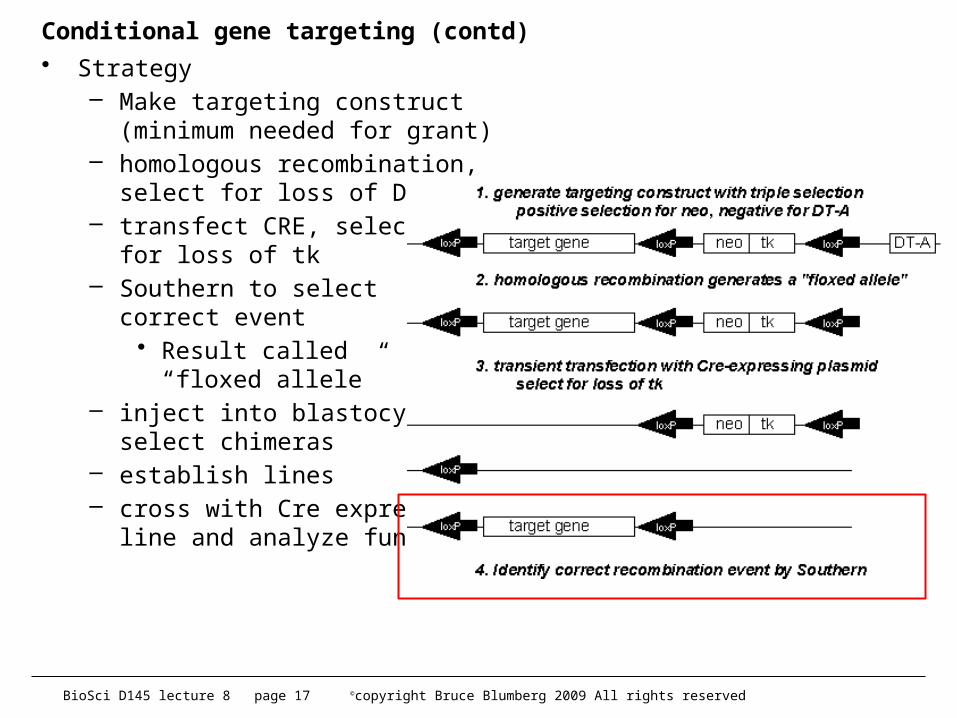
BioSci D145 lecture 8 page 17 ©copyright Bruce Blumberg 2009 All rights reserved
Conditional gene targeting (contd)• Strategy
– Make targeting construct (minimum needed for grant)
– homologous recombination,select for loss of DT-A
– transfect CRE, select for loss of tk
– Southern to select correct event
• Result called“floxed allele”
– inject into blastocysts,select chimeras
– establish lines – cross with Cre expressing
line and analyze function

BioSci D145 lecture 8 page 18 ©copyright Bruce Blumberg 2009 All rights reserved
Conditional gene targeting (contd)
– Tissue- or stage-specific knockouts from crossingfloxed mouse with specific Cre-expressing line
– requirement for Cre lines
• must be well characterized
– promoters can’t be leaky
• Andras Nagy’s database of Cre lines and other knockout resources http://nagy.mshri.on.ca/cre_new/index.php

Figure 5.25 Floxing mice

BioSci D145 lecture 8 page 20 ©copyright Bruce Blumberg 2009 All rights reserved
Conditional gene targeting (contd)
• advantages– can target recombination to specific tissues and times– can study genes that are embryonic lethal when disrupted– can use for marker eviction– can study the role of a single gene in many different tissues with
a single mouse line– can use for engineering translocations and inversions on
chromosomes
• disadvantages– not trivial to set up, more difficult than std ko but more
information possible– requirement for Cre lines
• must be well characterized regarding site and time of expression
• promoters can’t be leaky (expressed when/where not intended)

BioSci D145 lecture 8 page 21 ©copyright Bruce Blumberg 2009 All rights reserved
Other approaches to genome manipulation
• Transgenic and knockout technology is species dependent (doesn’t work in all species – need ES cell equivalent). How else can we accomplish gene disruption in a targeted way ?
– RNAi approaches (Boutros, Luo papers this week)– Nuclease based methods - introduce double-stranded breaks
• ZFN – zinc finger nucleases• TALEN nucleases• Meganuclease
– CRISPR/Cas – RNA guided nuclease (paper this week)
• Meganucleases– Based on naturally occurring restriction enzymes with extended
DNA binding specificity (relatively limited application)• TALEN and ZFN nucleases
– Artificial fusion proteins combining an engineered DNA binding domain fused to a nonspecific nuclease domain from FokI restriction enzyme
– ZNF – zinc finger repeats– TALE – repeats – Main limitation is the sequence specificity that can be engineered
in.

BioSci D145 lecture 8 page 22 ©copyright Bruce Blumberg 2009 All rights reserved
CRISPR-Cas gene editing
• CRISPR = clustered, regularly interspaced, short palindromic repeatSander & Joung (2014) CRIRISPR-Cas systems for editing, regulating
and targeting genomes, Nat Biotech 32: 347-355– Cas9 – RNA-guided nuclease– Functions as a bacterial immune system.
Foreign DNA is incorporated between CRISPR repeat sequences, transcription generates crRNA.
– Hybridizes to tracrRNA (also encoded by CRISPR system), guides Cas9 nuclease to the target
– Cas9 nuclease introduces double strandedbreaks into target
– Repair of this break can introduce deletionsframeshifts, etc.

BioSci D145 lecture 8 page 23 ©copyright Bruce Blumberg 2009 All rights reserved
CRISPR-Cas gene editing
• CRISPR/Cas9 (contd)– For gene targeting, we would fuse
the target RNA to the tracrRNA andintroduce this into the target cell,embryo, etc together with Cas9 nuclease
– Introduces ds breaks at target sequencewhich may introduce desirable mutations.
– Potential issues• Can’t specify what happens after
ds break (deletion, frameshift,insertion, etc
• Some question about specificityof the crRNA introduced
• Some sequence preferences of Cas9nuclease may limit utility

BioSci D145 lecture 8 page 24 ©copyright Bruce Blumberg 2009 All rights reserved
CRISPR-Cas gene editing - applications

BioSci D145 lecture 8 page 25 ©copyright Bruce Blumberg 2009 All rights reserved
Generating phenocopies of mutant alleles
• How to inactivate endogenous genes in a targeted but general way?– Important new development is RNAi – RNA
interference– Observation is that introduction of double-
stranded RNAs into cells lead to destruction of corresponding mRNA (if there is one)
– Principle is siRNA – small interfering RNAs– These generate small single stranded RNAs
that target mRNAs for destruction by– RISC – RNA interference silencing complex– First applied in C. elegans where it works
extremely well• Can introduce siRNA into cells even by
feeding to the worms!• Works very well in Drosophila• variably in mammalian cells• Poorly in Xenopus – Why?
Xenopus has an endogenous helicase

BioSci D145 lecture 9 page 26 ©copyright Bruce Blumberg 2010. All rights reserved
RNAi (contd)
• Dicer complex generates short duplexes from dsRNA in the cell– Important to have 2-nt
overhangs• siRNAs are generated from these
fragments– Antisense strand binds to
mRNA and this recruits the RISC - RNAi silencing complex
– Complex leads to mRNA cleavage and destruction
• Two important reviews to read– McManus and Sharp (2002)
Nature Reviews Genetics 3, 737-747
– Dykxhoorn et al. (2003) Nature Reviews, Molecular Cellular Biology 4, 457-467

BioSci D145 lecture 9 page 27 ©copyright Bruce Blumberg 2010. All rights reserved
RNAi (contd)
• Micro RNAs are small cellular RNAs that previously lacked any known function– Always form a hairpin structure
with mismatches in stem• Micro RNAs direct gene silencing via
translational repression– (miRNAs) are mismatched
duplexes that dicer processes into stRNAs (small temporal RNAs)
– Use same cellular complex as siRNAs
– Perfect matches -> target cleavage
– Imperfect matches -> translational repression of target
• Two important papers– Giraldez et al (2005) Science
308, 833-838 (microRNAs regulate brain morphogenesis)
– Lecellier et al (2005) Science 308, 557-560 (microRNA mediates antiviral defenses in human cells)

BioSci D145 lecture 9 page 28 ©copyright Bruce Blumberg 2010. All rights reserved
RNAi (contd)
• Parallels between siRNA and miRNA-directed RNAi
BioSci D145 lecture 9 page 29 ©copyright Bruce Blumberg 2010. All rights reserved
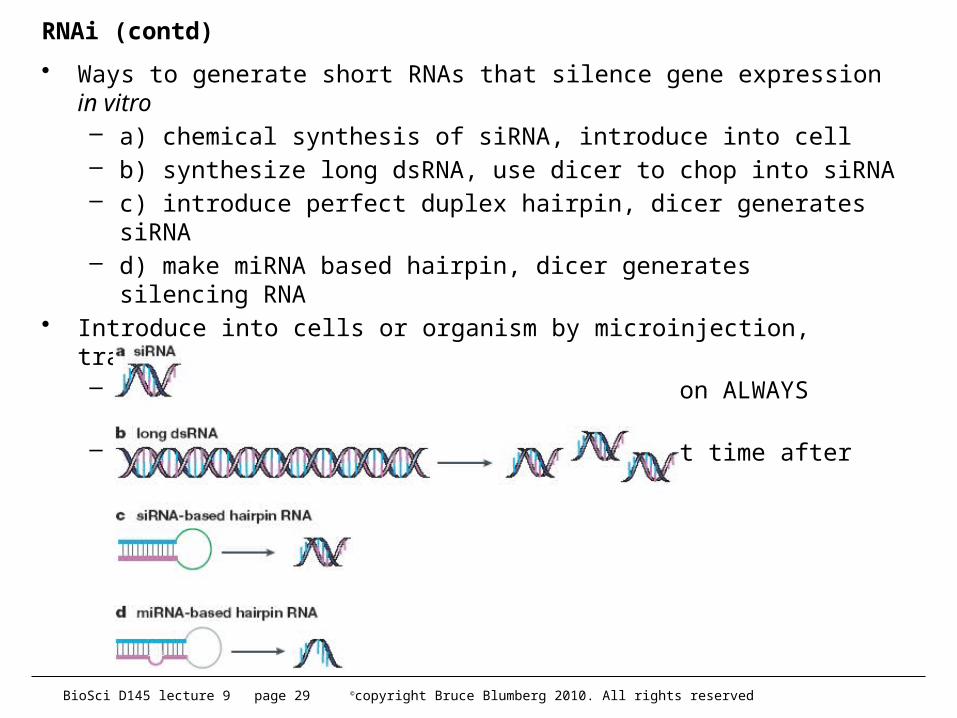
RNAi (contd)
• Ways to generate short RNAs that silence gene expression in vitro– a) chemical synthesis of siRNA, introduce into cell– b) synthesize long dsRNA, use dicer to chop into siRNA– c) introduce perfect duplex hairpin, dicer generates siRNA– d) make miRNA based hairpin, dicer generates silencing RNA
• Introduce into cells or organism by microinjection, transfection, etc.– Expression is transient, loss of function ALWAYS partial– can only generate phenotypes for a short time after introduction

BioSci D145 lecture 8 page 30 ©copyright Bruce Blumberg 2009 All rights reserved
RNAi (contd)
• Ways to generate short silencing RNAs in vivo – need continuing expression to generate stable phenotype– a) produce long hairpin
from pol II promoter, letdicer make siRNA
– b) produce two transcriptsfrom pol III promoter, letanneal in cells
– c) produce a short hairpinfrom pol III promoter (orviral vector), let dicer generate siRNAs
– d) produce imperfecthairpin from pol II promoter, let dicergenerate miRNAs that direct gene silencing

BioSci D145 lecture 9 page 31 ©copyright Bruce Blumberg 2010. All rights reserved
RNAi (contd)
• RNAi for whole genome functional analysis– First generate library of constructs that generates siRNA or stRNA
• Or synthesize a complete library of siRNAs– Introduce these into cells, embyos (fly, frog, mouse) or animals
(C. elegans, plants)• For C. elegans, make the library in E. coli and simply feed
bacteria to worms• Must microinject or transfect with other animals
– Evaluate phenotypes
• IMPORTANT!– RNAi is inherently transient unless you are expressing the siRNAs
from a stably integrated plasmid– RNAi is a knock-down (not knock-out) method.

BioSci D145 lecture 8 page 32 ©copyright Bruce Blumberg 2009 All rights reserved
Phenotypic rescue by RNAi – synthetic lethal and related approaches• How can we find other members of pathways we already know
something about?– Or, how can we find drugs that act on a pathway to kill cells (e.g.,
cancer cells?)– Synthetic lethal is one relatively new and promising approach
• 2 mutations are synthetic lethal if either single mutation is viable but the double mutant is lethal

BioSci D145 lecture 8 page 33 ©copyright Bruce Blumberg 2009 All rights reserved
Phenotypic rescue by RNAi – synthetic lethal and related approaches• How can we find other members of pathways we already know
something about?– In cancer screening, what if we combine a mutation and
a drug to find a combination that increases the kill rate, or reveals a phenotype that is similar to the total loss of function?
• Could find novel drug targets (pathways that kill cells in presence of siRNA+drug (usually sublethal amount of drug)
• Can find genes that are targeted by drugs – pathway analysis• Could find new biomarkers that are required for cell viability
following drug treatment

BioSci D145 lecture 8 page 34 ©copyright Bruce Blumberg 2009 All rights reserved
Mohr et al., 2010, Genomic Screening with RNAi: Results and Challenges. Annual Review of Biochemistry, 29: 37-64

BioSci D145 lecture 8 page 35 ©copyright Bruce Blumberg 2009 All rights reserved
Genome wide analysis of gene function
• How to mutate all genes in a given genome?– Easy with microbial genomes – can mutate all yeast genes by
homologous recombination– Recombine in selectable marker– Propagate strain and analyze phenotypes
Target gene
Selectable marker(antibiotic resistance)
Homology region
Unique oligonucleotide“barcodes” for PCR

BioSci D145 lecture 8 page 36 ©copyright Bruce Blumberg 2009 All rights reserved
Genome wide analysis of gene function (contd)
• How about gene targeting in other organisms– With more complex genomes and more genes?– Huge undertaking to specifically target 30K+ genes in
mammalian cells• Difficulty• Expense• Inability to target all possible loci
– Some efforts to make mouse collection • Lexicon Genetics has a collection of ES cells
– Drosophila collection as well– Driving force behind these efforts is
• Genome annotation• Drug target discovery (Lexicon)• Functional analysis

BioSci D145 lecture 8 page 37 ©copyright Bruce Blumberg 2009 All rights reserved
Figure 5.18 The three major types of mutagen

BioSci D145 lecture 8 page 38 ©copyright Bruce Blumberg 2009 All rights reserved
Genome wide analysis of gene function (contd)
• Main method for gene targeting in more complex organisms is random insertional mutagenesis– Transposon mutagenesis
• Bacteria – Tn transposons• Yeast - Ty transposons• Drosophila - P- elements• Vertebrates - Sleeping Beauty transposons
– Viral infection• Typically retroviruses – host range selectivity is obstacle
– Gene or enhancer trapping – modified viruses or transposons

BioSci D145 lecture 8 page 39 ©copyright Bruce Blumberg 2009 All rights reserved
Insertional mutagenesis - Gene trapping – enhancer trip
• enhancer trap is designed to bring inserted reporter gene under the control of local regulatory sequences– put a reporter gene adjacent to a weak promoter (enhancer-less),
e.g. a retrovirus with enhancers removed from the LTRs– may or may not disrupt expression – Hopkins zebrafish group used unmodified virus
• viruses and transposable elements can deliver DNA to random locations– can disrupt gene function – put inserted gene under the
control of adjacent regulatory sequences
– BOTH

BioSci D145 lecture 8 page 40 ©copyright Bruce Blumberg 2009 All rights reserved
Insertional mutagenesis - Gene trapping –enhancer trap (contd)
Insertional mutagenesis by the Tol2 transposon-mediated enhancer trap approach generated mutations in two developmental genes: tcf7 and synembryn-like. Nagayoshi S, Hayashi E, Abe G, Osato N, Asakawa K, Urasaki A, Horikawa K, Ikeo K, Takeda H, Kawakami K. Development 2008 Jan;135(1):159-69.

BioSci D145 lecture 8 page 41 ©copyright Bruce Blumberg 2009 All rights reserved
Insertional mutagenesis - Gene trapping –enhancer trap (contd) stopped here 2015• enhancer trap (contd)
– expression only when integrate into an active transcription unit• reporter expression duplicates the temporal and spatial
pattern of the endogenous gene– reporters used
• -galalactosidase was the most widely used reporter• GFP is now popular• -lactamase is seeing increasing use
– advantages• relatively simple to perform• active promoters frequently targeted, perhaps due to open
chromatin– Disadvantages
• Inactive promoters probably not targeted • insertional mutagenesis not the goal, and not frequent
– overall frequency is not that high• relies on transposon or retroviruses to get insertion
– may not be available for all systems, requires transgenesis or good viral vectors

BioSci D145 lecture 8 page 42 ©copyright Bruce Blumberg 2009 All rights reserved
Insertional mutagenesis – Gene trapping (contd)
• expressed gene trap (many variations possible)– goal -> ablate expression of endogenous gene, replace with
transgene– Make insertion construct – reporter, selection, polyA sites
• No promoter but has a splice-acceptor sequence 5’ of reporter• Can only be expressed if spliced into an endogenous mRNA
– Transfer into embryonic cells, generate a library of insertional mutagens
• Mouse, Drospophila, zebrafish, frog– reporter expression duplicates the temporal and spatial pattern of
the endogenous gene

BioSci D145 lecture 8 page 43 ©copyright Bruce Blumberg 2009 All rights reserved
Insertional mutagenesis - Gene trapping (contd)
• Expressed gene trapping (contd) – advantages
• insertional mutagen– gives information about expression patterns– can be made homozygous to generate phenotypes
• higher efficiency than original trapping methods• selectable markers allow identification of mutants
– many fewer to screen– dual selection strategies possible
– disadvantages• overall frequency is still not that high• frequency of integration into transcription unit is not high
either• relies on transposon or retroviruses to get insertion
– may not be available in your favorite system.– Uses
• Insertional mutagenesis• Marking genes to identify interesting ones• Gene cloning• http://www.genetrap.org/