
Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile
9
Crystal growth DOI: 10.1002/smll.200701163 Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile Ferdinand Gonzaga, Sherdeep Singh, and Michael A. Brook* The first synthesis of a chelating and reactive surfactant derived from citric acid and a short silicone as hydrophobic tail is described. Aqueous solutions of this reactive amphiphile spontaneously induce gold ion reduction, particle nucleation, and further direct crystal growth. The process, both pH and light dependent, occurs through lipid-directed assembly of metal ions, their reduction and subsequent lipid-directed growth to yield ultrathin (approximately 7 nm thick) quasi two-dimensional gold nanocrystals. 1. Introduction In living organisms, complex interactions between ions and biopolymers acting as templates are used to regulate nucleation and growth of inorganic architectures. Extension of this concept to metallic structures would require the development of suitable precursors, able to interact both with metal ions and the crystalline metal surface. Molecules such as marine siderophores [1,2] –high-affinity iron(III) ligands pro- duced by bacteria–are particularly attractive models, as they can regulate iron acquisition through complexation, controlled self-assembly, [3] and photoinduced reduction processes. [4] The ability of such molecules to bind, pre-organize, and control redox processes, if extended to other metals, would permit materials chemists to synthesize new metallic colloidal architectures and explore their interesting size- and shape- dependent properties. [5] Previous synthetic approaches to create shape-controlled metallic structures involve chemical [6] or photochemical [7] solution-phase methods, or reductions inside soft- colloidal, [8–10] biomimetic, [11,12] or biological [13–18] templates. However, the development of suitable amphiphiles [19] that can dynamically organize metal precursors in solution, initiate reduction processes, and effectively control crystal growth through specific metal–head-group interactions has not been reported. Here we describe the synthesis of such a chelating and reactive lipid, based on citric acid, a molecule broadly involved in biological processes [20] and in siderophore biochemistry. In the presence of gold cations, the lipid induces gold ion reduction, particle nucleation, and further directs gold crystal growth. The process, both pH and light dependent, occurs through lipid-directed assembly and reduction of gold cations into quasi two-dimensional (2D) gold nanocrystals. A single molecule, an ‘‘aurophore,’’ which acts successively as a chelating, reducing, capping, and structure-directing agent drives the entire process, and allows an unambiguous under- standing of how such anisotropic gold crystals are formed. These synthetic siderophore-like reactive amphiphiles, and their organized assemblies, represent a new biomimetic strategy toward nanoengineered metallic architectures. 2. Results and Discussion 2.1. Synthesis of Citric Acid based Reactive Surfactant Citrate reduction is the most popular method to prepare aqueous spherical gold nanoparticles. [21] We reasoned that surfactants chemically derived from citric acid could be used both to induce structure direction and to reductively lead to anisotropic gold crystal growth. A versatile synthetic method permits the preparation of lipid 5 (Scheme 1) through covalent addition of a short and highly hydrophobic silicone tag, without affecting the carboxylic acid units. Citric acid esters are well known but citrate ethers are extremely rare: the methyl ether was incompletely described in the early 1900s and a ring-opening polymerization of propylene or butylene oxide by triethyl citrate, to ultimately give citric acid poly(oxyalk- ene)ethers, has been reported. [22] Otherwise, little is to be found in the literature. Extensive attempts using Williamson etherification to prepare the key intermediate, citric acid allyl ether 3, failed in our hands. The detailed synthetic pathway that was successfully used is illustrated in Scheme 1. The first step involves a classical full papers [ ] Dr. F. Gonzaga, S. Singh, Dr. M. A. Brook Department of Chemistry, McMaster University 1280 Main Street West Hamilton, ON L8S4M1 (Canada) E-mail: [email protected] : Supporting information for this article is available on the WWW under http://www.small-journal.org or from the author. Keywords: biomimetic synthesis crystal growth gold self-assembly surfactants 1390 ß 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim small 2008, 4, No. 9, 1390–1398
-
Upload
ferdinand-gonzaga -
Category
Documents
-
view
216 -
download
0
Transcript of Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile

full papers
1390
Crystal growth
DOI: 10.1002/smll.200701163
Biomimetic Synthesis of Gold Nanocrystals Using aReducing AmphiphileFerdinand Gonzaga, Sherdeep Singh, and Michael A. Brook*
Keywords:� biomimetic synthesis
� crystal growth
� gold
� self-assembly
� surfactants
The first synthesis of a chelating and reactive surfactant derived from citric
acid and a short silicone as hydrophobic tail is described. Aqueous solutions
of this reactive amphiphile spontaneously induce gold ion reduction,
particle nucleation, and further direct crystal growth. The process, both pH
and light dependent, occurs through lipid-directed assembly of metal ions,
their reduction and subsequent lipid-directed growth to yield ultrathin
(approximately 7 nm thick) quasi two-dimensional gold nanocrystals.
1. Introduction
In living organisms, complex interactions between ions and
biopolymers acting as templates are used to regulate
nucleation and growth of inorganic architectures. Extension
of this concept to metallic structures would require the
development of suitable precursors, able to interact both with
metal ions and the crystalline metal surface. Molecules such as
marine siderophores[1,2]–high-affinity iron(III) ligands pro-
duced by bacteria–are particularly attractive models, as they
can regulate iron acquisition through complexation, controlled
self-assembly,[3] and photoinduced reduction processes.[4] The
ability of such molecules to bind, pre-organize, and control
redox processes, if extended to other metals, would permit
materials chemists to synthesize new metallic colloidal
architectures and explore their interesting size- and shape-
dependent properties.[5]
Previous synthetic approaches to create shape-controlled
metallic structures involve chemical[6] or photochemical[7]
solution-phase methods, or reductions inside soft-
colloidal,[8–10] biomimetic,[11,12] or biological [13–18] templates.
However, the development of suitable amphiphiles[19] that can
dynamically organize metal precursors in solution, initiate
reduction processes, and effectively control crystal growth
through specific metal–head-group interactions has not been
reported. Here we describe the synthesis of such a chelating
and reactive lipid, based on citric acid, a molecule broadly
involved in biological processes[20] and in siderophore
[�] Dr. F. Gonzaga, S. Singh, Dr. M. A. Brook
Department of Chemistry, McMaster University 1280 Main Street
West Hamilton, ON L8S4M1 (Canada)
E-mail: [email protected]
: Supporting information for this article is available on the WWWunder http://www.small-journal.org or from the author.
� 2008 Wiley-VCH Ver
biochemistry. In the presence of gold cations, the lipid induces
gold ion reduction, particle nucleation, and further directs gold
crystal growth. The process, both pH and light dependent,
occurs through lipid-directed assembly and reduction of gold
cations into quasi two-dimensional (2D) gold nanocrystals. A
single molecule, an ‘‘aurophore,’’ which acts successively as a
chelating, reducing, capping, and structure-directing agent
drives the entire process, and allows an unambiguous under-
standing of how such anisotropic gold crystals are formed.
These synthetic siderophore-like reactive amphiphiles, and
their organized assemblies, represent a new biomimetic
strategy toward nanoengineered metallic architectures.
2. Results and Discussion
2.1. Synthesis of Citric Acid based Reactive Surfactant
Citrate reduction is the most popular method to prepare
aqueous spherical gold nanoparticles.[21] We reasoned that
surfactants chemically derived from citric acid could be used
both to induce structure direction and to reductively lead to
anisotropic gold crystal growth. A versatile synthetic method
permits the preparation of lipid 5 (Scheme 1) through covalent
addition of a short and highly hydrophobic silicone tag,
without affecting the carboxylic acid units. Citric acid esters
are well known but citrate ethers are extremely rare: the
methyl ether was incompletely described in the early 1900s and
a ring-opening polymerization of propylene or butylene oxide
by triethyl citrate, to ultimately give citric acid poly(oxyalk-
ene)ethers, has been reported.[22] Otherwise, little is to be
found in the literature. Extensive attempts using Williamson
etherification to prepare the key intermediate, citric acid allyl
ether 3, failed in our hands.
The detailed synthetic pathway that was successfully used
is illustrated in Scheme 1. The first step involves a classical
lag GmbH & Co. KGaA, Weinheim small 2008, 4, No. 9, 1390–1398

Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile
Scheme 1. Synthetic pathway of citric acid-based surfactant 5.
Figure 1. UV/Vis signature of colloidal metallic gold resulting onmixing
lipid 5 and a gold salt, as prepared and after separation of the two
constituents: green product (gold nanoleaves), supernatant (nano-
particles), and their corresponding appearance (inset).
acid-catalyzed benzylation of citric acid. In the crucial step of
the synthesis, palladium-catalyzed allyl transfer from allyl-tert-
butylcarbonate to tribenzyl citrate yielded the desired O-
allyloxy tribenzyl citrate 3 in high yield. Hydrosilylation of 3
with a hydride-terminated trisiloxane led to the benzyl
protected derivative 4 which, after hydrogenolysis over Pd/
C, gave the corresponding surfactant 5 without affecting the
siloxane moiety[23] (yield from citric acid: 78%). All synthetic
steps occur in high to quantitative yields and this methodology
is readily scaled to multigram synthesis (up to 10 g) of citrate
surfactants. It is noteworthy that the flexibility and broad
reactivity of allyl derivative 3 permits the preparation of a wide
range of surfactants, including mono-or bi-catenar surfactants,
with alkyl or silicone hydrophobic chains, as will be described
elsewhere.
2.2. Biomimetic Synthesis of Gold Nanocrystals
Gold crystals were prepared by mixing an aqueous solution
of lipid 5 with a gold salt solution (HAuCl4; tetrachloroauric
acid) under ambient conditions. The initially pale-yellow
solution undergoes a series of color changes (from pale yellow
to colorless, then light pink, and finally dark purple), indicating
the formation of colloidal metallic gold. After 48 h, centrifu-
gation reveals a deep green precipitate that, unusually, has a
continuous absorption from 512 to 900 nm in the UV/Vis and
near-infrared region (NIR) spectroscopy (Figure 1 and inset).
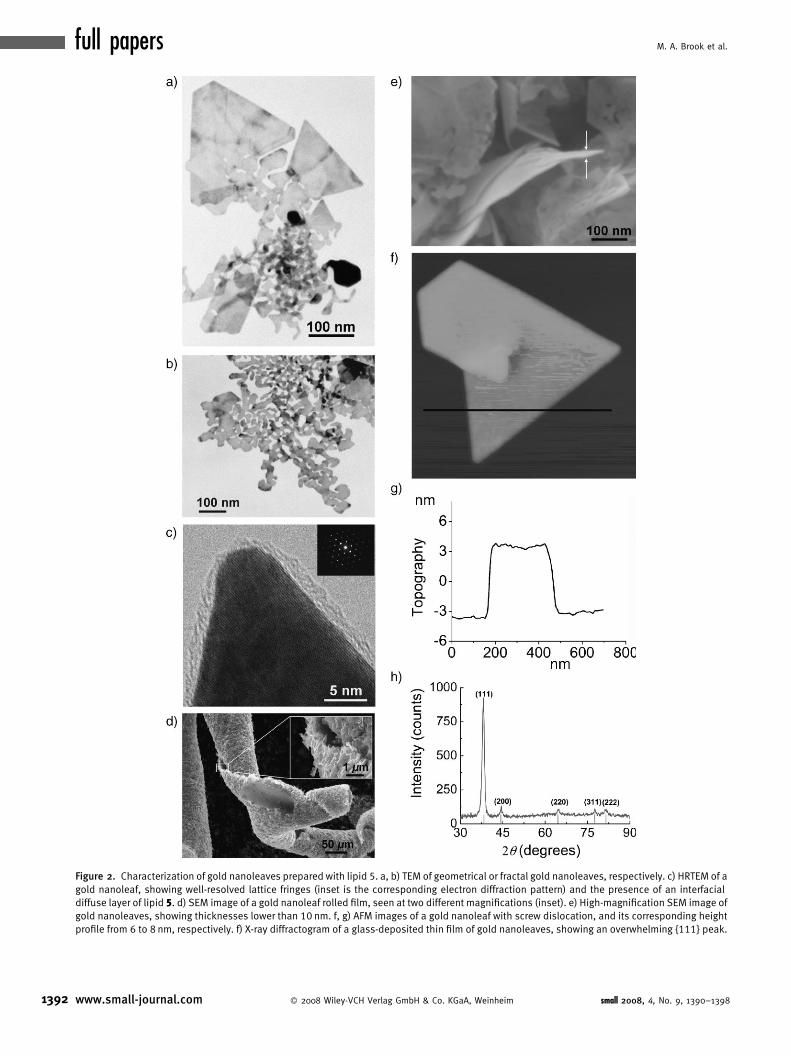
A representative high-magnification transmission electron
microscopy (HRTEM) image of the green product shows the
presence of fluid, round, and interconnected fractal shapes
linked to more geometric platelets (triangles, truncated
triangles, hexagons), with overall sizes of a few hundreds of
nanometers, referred to as gold nanoleaves (Figure 2a and b and
Supporting Information). The nanoleaves and fractal structures
were shown to be crystalline gold using high resolution
transmission electronic microscopy (HRTEM): Figure 2c
small 2008, 4, No. 9, 1390–1398 � 2008 Wiley-VCH Verlag
shows continuous well-resolved crystal lattice fringes, even at
the crystal contours, and the presence of a diffuse layer of lipid
along the gold interface. Energy dispersive X-ray spectroscopy
(EDS) shows the presence of strong peaks for elemental silicon
and gold, confirming the chemical nature of this interfacial layer
(Supporting Information).
Scanning electron microscopy (SEM) at a lower magni-
fication shows that the green precipitate is composed of stacks
of about 200 nm in thickness (Figure 2d and Supporting
Information) of gold nanoleaves. The highest magnifications
(Figure 2e and Supporting Information) confirm that the
individual nanoleaves in the flat or rolled film are extremely
thin, typically less than 8 nm (thicknesses in the range from 6 to
12 nm were measured) as also confirmed by atomic force
microscopy (AFM; Figure 2f and g). The morphology of the
stacks, the curled ribbons shown in Figure 2d, is a consequence
of drying and delamination from the steel SEM stub.
An X-ray diffraction pattern of a thin film of the crystals
deposited on a glass side shows an overwhelming diffraction
peak at �38.2 8 assigned to the {111} facets of a face-centered
cubic (fcc) metal gold structure (Figure 2h), while diffraction
peaks of other facets are much weaker. An average thickness
of 7.7 nm is calculated from the Scherrer equation, which also
demonstrates the extremely low thickness of the nanoleaves.
Under the same conditions, citric acid only yielded roughly
spherical particles (see Supporting Information), indicating
that observed anisotropic growth is related to the surfactancy
of lipid 5 (as citric acid and the lipid only differ by the
hydrophobic silicone moiety, see below). In addition to
their very high aspect ratio, the leaves are even more
remarkable given the fact that crystal growth occurs in
solution, without the use of a preformed template: previous
attempts to produce flat and thin gold crystals (or fractal
crystals) required first the creation of a 2D interface such as a
thermally evaporated film, a Langmuir monolayer, or a
sheared lamellar phase.[23,24,25]
GmbH & Co. KGaA, Weinheim www.small-journal.com 1391
full papers M. A. Brook et al.
Figure 2. Characterization of gold nanoleaves prepared with lipid 5. a, b) TEM of geometrical or fractal gold nanoleaves, respectively. c) HRTEM of a
gold nanoleaf, showing well-resolved lattice fringes (inset is the corresponding electron diffraction pattern) and the presence of an interfacial
diffuse layer of lipid 5. d) SEM image of a gold nanoleaf rolled film, seen at two different magnifications (inset). e) High-magnification SEM image of
gold nanoleaves, showing thicknesses lower than 10 nm. f, g) AFM images of a gold nanoleaf with screw dislocation, and its corresponding height
profile from 6 to 8 nm, respectively. f) X-ray diffractogram of a glass-deposited thin film of gold nanoleaves, showing an overwhelming {111} peak.
1392 www.small-journal.com � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim small 2008, 4, No. 9, 1390–1398

Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile
Having characterized the nature of the crystalline gold
nanoleaves, we wished to optimize the formation of different
structural types. The production of gold nanoleaves induced
by lipid 5 in the presence of white light was found to be
strongly pH dependent. Kinetic studies and TEM analyses
indicate that nanoleaves are already produced in 20 h at pH
6.30. Increasing the pH of the lipid solution by only 1 unit
(from 6.30 to 7.30) considerably decreases the rate of the
process (it requires 48 h at pH 7.30 to obtain the same UV/Vis
signature.
These reactions were re-examined in the absence of white
light. The reduction process still occurs at the three pH values
tested (6.30, 7.30, and 8.30), indicating that the reduction of
gold ions by lipid 5 is not exclusively a light-dependent process.
However, light was found to affect both the rate of the process
and the morphology of the colloidal gold produced. At pH
6.30, micrometer-sized gold nanoplates were obtained (Sup-
porting Information) in only 24 h. The plates aggregate and
settle down in the vial, a behavior which strongly contrasts
with the gold nanoleaves. At higher pHs, no significant
morphological differences were observed, but the rate of the
overall process was significantly impeded in the absence of
light (Supporting Information). Light effects have been
previously described in the plasmon-directed synthesis of
silver nanoprisms,[7] and a recent report by Mirkin and Xue[26]
emphasized the interplay between light and pH effects. In our
system, the decrease in the rate of formation of gold
nanoleaves in the dark supports the idea that optical attractive
forces may be involved in the growth process. However, the
fact that crystals obtained in the dark were larger than those in
the presence of visible light at pH 6.30 also indicates a more
efficient nucleation step in the presence of light: fewer
nucleation events, associated with a highly favored pH for
crystal growth, thus lead to a higher average crystal size. At
higher pH values, the process was already considerably slower
than at pH 6.30 and the absence of light reduced even more the
overall rate of crystal nucleation and growth.
A time-dependent study at pH 7.3, the pH at which the
reaction proceeded at an intermediate rate, was performed
using UV/Vis and TEM to better understand the reaction
sequence leading to the evolution of the gold nanoleaves. In
the early stages of the process (after 12 h of reaction) only
small gold clusters were observed. These particles are
embedded in membranar structures (Figure 3a) and the
strong interactions between the gold clusters are responsible
for the high wavelength-absorbance tail in the UV/Vis spectra
(Supporting Information). After 24–30 h, the sample is
dominated by long and ramified fractal dendrites. Their
lengths vary from 200 nm up to several micrometers, while
their widths are typically lower than 10 nm. However, the
fractal structures are broader at their extremities, indicating
that preferential 2D crystal growth occurs on the periphery of
the arborescent structures (Figure 3b). This observation is
even more pronounced after 36–42 h: almost all dendrites are
now terminated by flat structures, which increasingly evolve
into gold geometric shapes (Figure 3c). After 48 h of growth,
the final nanoleaves are observed (Figure 3d). The TEM
micrographs also reveal the presence of membrane-embedded
growing crystals: the membrane structures contain a high
small 2008, 4, No. 9, 1390–1398 � 2008 Wiley-VCH Verlag
density of gold clusters, which provides further guidance for
the mechanism of crystal growth (Figure 3e). Analysis of large
membranar structures (Figure 3f) reveals that they consist of a
regular arrangement of surfactant bilayers, with a remarkably
low value (<1 nm) for the d spacing (Figure 3g).
The time-dependent studies (TEM, UV/Vis) described
above permit us to propose a three-step mechanism to explain
the surfactant-induced synthesis of quasi 2D gold nanoplates:
complexation and self-assembly, nucleation, and membrane-
directed crystal growth.
2.3. Complexation and Self-Assembly
There is strong evidence that citrate binds to gold prior to
reduction, both in a historical context, and from our own
results. While binding constants of various metals ions to
citrate are well known, no data are currently available for the
binding constant with gold cations because, in addition to
the high affinity of citrate for gold, redox reactions between
the two species lead to metallic gold and oxidation products of
citric acid (a process used for the Turkevich synthesis of gold
nanoparticles). The two processes, chelation and reduction,
have not previously been independently observed.
On mixing the two precursor solutions, the chelating head
group strongly interacts with gold ions and a resulting citrate–
Au3þ complex is formed. Although the exact nature (e.g.,
monomeric or polymeric) of this complex is not known, UV/
Vis spectroscopy shows the presence of two new absorption
bands at 419 and 448 nm in the early stage of the process that
can only be attributed to such intermediate gold/citrate species
(Supporting Information).
Co-assemblies of membranar aggregates and gold were
also observed during the time-resolved studies of formation of
the nanocrystals. Figure 3f, taken after 12 h of the reduction
process (in the early stage of the reduction) shows such an
assembly, and Figure 3g, a higher magnification of Figure 3f,
shows that those assemblies are made of stacked bilayers of the
amphiphile. The corresponding electron-diffraction pattern,
diffuse and of low intensity, indicates the presence of
polycrystalline gold from small gold clusters. The fact that
such aggregates can be observed by TEM and HRTEM,
without the use of any staining agent is by itself proof of the
presence of metallic centers within the self-assembled
structures. Indeed, noble metals (Ag, Au) as well as other
metallic ions (such as osmium or uranium) are often used as
staining agents in order to visualize surfactant self-assembled
aggregates. For all these reasons, it seems highly probable that
gold ions (which are stable in the same media, under the same
conditions, but in the absence of citrate) must complex with
the citrate-based amphiphile prior to their reduction.
The self-assembly step, which may occur in concert with
the initial complexation, involves the aggregation of these
citrate/gold species into the previously described membrane
structures. The observed bilayer units observed (Figure 3g) are
conceivably the consequence of highly favored hydrophobic
interactions between the siloxane units (the trisiloxane
backbone of 5 is estimated to be comparable, in terms of
hydrophobicity, to a C12 alkyl chain[27]) and reduced
electrostatic repulsion between the citrate polar head groups
GmbH & Co. KGaA, Weinheim www.small-journal.com 1393

full papers M. A. Brook et al.
Figure 3. Directed growth of gold nanoleaves. a–d) Representative TEM images showing the size and shape evolution of colloidal gold prepared on
mixing lipid 5 with a gold salt, at 12 (a), 24 (b), 36 (c), and 48 h (d). The structure evolves from small (<5nm) clusters to ramified dendrites to the
final gold nanoleaves. e) TEM image of growing gold dendrites, showing membrane-embedded gold clusters. f, g) TEM and HRTEM of gold–lipid 5membrane structures, respectively. Well-ordered stacked bilayers can be distinguished. Inset: corresponding electron-diffraction pattern that
appears at pH 6.30 after only 20 h, that is, a 2.4 factor decrease (Supporting Information). A further increase to pH 8.30 drastically slows down the
process, which now only yields irregular nanoparticles, and occasional ultrathin wires (Supporting Information).
1394 www.small-journal.com � 2008 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim small 2008, 4, No. 9, 1390–1398

Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile
following gold ion complexation, which facilitates their
assembly. Existing studies on chelating surfactants support
this proposal: self-assembled columns of stacked lipid bilayers
are obtained when copper is added to liposomes made of a
chelating surfactant.[28] Moreover, natural siderophores such
as Marinobactin E undergo a micelle to vesicle to stacked
bilayer transition through iron coordination.[3,29]
2.4. Nucleation and Membrane-Directed CrystalGrowth
The chemical process behind gold nanoparticle nucleation
is the oxidative-decarboxylation process that ultimately yields
oxidation products of citric acid and a reduced metal center
that subsequently leads to particle nucleation. The formation
of citrate membranar assemblies facilitates the gold crystal
nucleation step, due to excellent interfacial control both
before and after reduction, and local high-concentration
effects that are required for nucleation. In addition, this well-
ordered 2D reactive template can lead to oriented nuclea-
tion,[30] which directs the subsequent growth of the gold
structures into fractal structures and prismatic plates.
Two different mechanisms can be postulated for crystal
growth: an autocatalytic growth of the least-stabilized crystal-
lographic faces (i.e., along the (110) faces), or the sintering of
primary particles into larger crystals. In the first mechanism,
slow diffusion of gold cations within the organic bilayer and
their reduction at the surface of existing nuclei can explain
crystal growth at the most energetic crystal face. In the second
mechanism, aggregation-driven crystal growth occurs through
the sintering of primary gold particles in the organic bilayer
matrix, consistent with the diffusion-limited colloid aggrega-
tion model (DLCA[31]). As gold particles are formed within
the organic matrix they diffuse and adhere to the growing
crystal 2D structure.
Recently, Brust and co-workers[32] showed that 1D
sintering of small hydrophobic gold nanoparticles could be
induced in a matrix of DPPC at the air–water interface, by
compression in a Langmuir–Blodgett film. The key role of the
surfactant DPPC was highlighted, as it acts as a molecular
template, allowing sintering only in certain directions.
Oriented sintering has also been demonstrated for mono-
disperse 2-nm gold nanoparticles, yielding 2-nm diameter
nanowires.[33] Moreover, a very recent report shows how large
triangular silver single crystals can be obtained by mild
annealing of surfactant-capped 5-nm silver nanocrystal mono-
or multilayers.[34] These examples reveal that 1- or 2D
organization of nanoparticles, followed by oriented sintering,
is a powerful tool for the synthesis of new metallic
architectures.
In our case, a preferential 2D sintering could occur in a
lipid bilayer. The interaction between growing crystals and
isolated, surfactant stabilized nanoparticles in this case is
highly favored, as both the capping layer of the particles and
the lipid bilayer membranes are made of the same molecule.
The slow kinetics of this process would highly favor growth
into prismatic or triangular flat nanocrystals: indeed, it has
previously been demonstrated that in the citric acid catalyzed
synthesis of gold nanocrystals the formation of gold nano-
small 2008, 4, No. 9, 1390–1398 � 2008 Wiley-VCH Verlag
triangles was kinetically controlled and highly favored at low
temperature, while spherical particles were obtained in boiling
water.[35]
The product crystals, fractal, or prismatic (Figure 3)
present a flat surface, indicating anisotropic growth, even from
the very first stages of the process. This can result from an
oriented nucleation step (which will thus induce anisotropic
growth), and a specific stabilization of the (111) facets of the
crystals by the citrate head groups such that growth occurs at
the edges. Several studies support the latter observation:
citrate adlayers have been experimentally observed at the Au
(111) surface by scanning probe microscopy. Citrate anions
form very well ordered (4� 2H3) adlayers and these ordered
adlayers were found to be extremely stable, which is in
accordance with the fact that the citrate anion is one of the best
stabilizers for metallic nanomaterials.[36] Furthermore, the
presence of chloride ions from the precursor chloroaurate
derivative is also known to promote the growth of (111)-
oriented triangular/truncated triangular particles.[35] Thus, the
driving force of the process can be seen as the formation of
highly stabilized (111) facets, along with the creation of stable
bi- or multilayers of citrate amphiphiles on those facets.
Several reports have described the synthesis of gold
nanostructures using citric acid and surfactants, in particular
CTAB: extensive work by Murphy et al. has shown that gold
nanorods could be obtained in high yield using a seeded
growth approach.[37] Following this approach, Mirkin and co-
workers reported the first chemical and aqueous procedure to
prepare triangular nanoplates in high yield.[38] Recently, a
thermal-reduction approach described the synthesis of gold
plates having three different particle size range.[39] In all these
studies, preferential growth was attributed to selective
adsorption of CTAB molecules on specific facets of the crystal.
The concentration of surfactant and the ratio of surfactant/
metal salt used in our procedures are considerably lower than
in the seeded growth procedure (less than 5 mM versus 0.1 M,
respectively), which could indicate a stronger binding of the
citrate head group compared to the alkylammonium head
group of CTAB. Moreover, the kinetics of the two processes,
CTAB versus 5, are also very different.
We believe that, as with citric acid, our amphiphile is able
to form stable adlayers at the (111) facets of the growing
crystals, and this even at the very early stage of their growth.
The presence of hydrophobic tails on the amphiphile would
certainly increase the stability of such adlayers. This has
previously been demonstrated with the alkyl chains of CTAB-
coated gold nanorods (where a bilayer of CTAB was
evidenced at the surface of the gold crystals).[36]
The presence of such a stable silicone-based lipid bilayer at
the (111) facets of the gold crystals would then favor the
growth in the direction normal to the (111) basal plane, as was
observed experimentally: the fractal crystals grow preferen-
tially from their extremities, where the amphiphile stabiliza-
tion is the weakest.
2.5. Fractal Versus Prismatic Crystalline Plates
The high-temperature synthesis of gold nanoplates
catalyzed by citric acid and CTAB, leads to fractal structures
GmbH & Co. KGaA, Weinheim www.small-journal.com 1395

full papers M. A. Brook et al.
1396
with a central core, an overall flat appearance and external
sheetlike structures in the very initial stages of the process (10 s
for the reaction performed at 85 8C).[40] However, unlike the
citrate stabilized crystals reported here, those structures are
not stable and rapidly evolve to more thermodynamically
stable triangular or hexagonal plates.
At the outset, in our case, growth of the 2D crystal normal
to the (111) plane occurs giving fractal structures. Initially,
gold clusters are rapidly formed and randomly sinter in a
kinetically controlled process that leads to formation of 2D
fractal ‘‘leaves’’ (Figure 2a and b), which continue to grow at
the extremities. Eventually, as formation of new particles, and
their migration to the growing crystal surface, becomes more
measured the more stable prismatic plates are nucleated and
then effectively grow: in all cases, we have observed a fractal
core followed by nucleation and growth of prismatic plates
(triangles or hexagons) at the peripheral ‘‘tips’’ of the leaves
where curvature is high. Depending on the specific reaction
conditions, it is possible to isolate purely fractal crystals,
particularly at low conversion. However, at longer reaction
times (it takes 48 h to complete the process) nucleation and
growth of prismatic structures occurs from the tips of the
fractal ‘‘leaves,’’ leading subsequently to large thermodyna-
mically favored prismatic structures at the periphery of a
fractal core: in all cases, very high aspect-ratio gold crystals are
observed.
This three-step model is consistent with the previously
postulated mechanism in which crystal nucleation and initial
growth occur within an interfacial layer such as a thin gel-like
film.[30] It further highlights that the sintering of fluid-like
metallic particles, also observed in the biological synthesis of
gold nanoprisms[18] (but for which the identity of structure-
directing agent(s) remains unknown), could also be induced by
a single molecule with self-assembly properties. The essential
roles of self-assembled lipid membrane and hydrophobic
interactions were explicitly demonstrated by adding a
destructuring agent such as THF (tetrahydrofuran) to the
reaction medium: reduction still occurs but leads now to gold
particles of ill-defined shape. Thus, the morphological control
of crystal growth due to the lipidic scaffold is lost
simultaneously with the ability of the surfactant to aggregate
in the binary solvent system (see Figure S4 in Supporting
Information). In such a situation, the system is returned to a
state similar to that with citric acid alone.
The sensitivity of this model to pH should be addressed.
Metal chelation, and also carboxylate-based surfactant self-
assembly, are known to be strongly pH dependent. The
protonation state of the citric acid head group of 5 will thus
be sensitive to pH, which is reflected in the variations
observed in the rate of formation and morphologies of the
gold structures found at different pH values. Similar effects
were previously reported in the photoreduction of iron(III)–
citrate complexes to yield oxo- or hydroxy-iron(II) spe-
cies,[41] and also in the photochemistry of natural iron–
siderophore complexes.[4] However, while siderophores
manipulate the redox-chemistry of ionic iron in order to
facilitate its uptake, the gold–citrate system leads to further
reduction giving metallic structures, due to the instability of
intermediate gold oxidation states.
www.small-journal.com � 2008 Wiley-VCH Verlag Gm
3. Conclusions
The biomimetic assembly and reduction of metal ions by
aurophore 5 is a highly controlled route to exceptionally thin
gold nanoleaves. Their formation depends on a delicate
balance between nucleation rate and crystal growth in a
membrane-assisted process. The relatively slow reduction
rates of gold in the presence of silicone citrate surfactant 5,
when compared to the thermal reduction of gold by citrate
anions, allows the template to fully exert control over the
nucleation, diffusion, and growth of gold crystals. More
important, however, is the exploitation of biomimetic
hydrophobic and co-operative effects of reactive lipids that
mimic natural siderophores and direct metal reactivity and
assembly at the organic–metal interface. The flexibility in the
synthesis of citric acid based silicones, their ability to complex
a wide variety of cations, and the rich lyotropic behavior of
surfactants, including hexagonal and other packing arrange-
ments, suggests that this method may be exploited for the
synthesis of a wide range of structured materials based on gold
and other metals.
4. Experimental Section
Chemicals: Citric acid (99.5R%, Aldrich), benzyl alcohol
(Certified, Fisher Scientific), p-toluenesulfonic acid monohydrate
(pTSA, Aldrich), palladium acetate (99.98%, Aldrich), triphenyl-
phosphine (99%, Aldrich), 1,1,1,3,3,5,5-heptamethyltrisiloxane
(95%, Gelest), platinum–divinyltetramethyldisiloxane complex
(Karstedt’s catalyst) in xylene (Gelest), palladium on activated
charcoal (Degussa type E101NE/W, wet/Pd 10% dry-weight
basis, water 50%, Aldrich), Celite (Aldrich), were used as received.
Allyl-tert-butyl carbonate was prepared as previously
described.[42] Solvents were dried over activated alumina. NMR
solvents (CDCl3, CD3COCD3, and CD3OD) were obtained from
Cambridge Isotope Laboratories. All syntheses were carried out in
dry apparatus under a dry nitrogen atmosphere utilizing conven-
tional bench-top techniques. Water was purified by a Millipore
purification system (resistance¼18.1MO cm).
Spectroscopic characterization of organic compounds: 1H NMR
Fourier spectra were recorded on a Bruker AC-200 (200MHz)
spectrometer. Chemical shifts for 1H NMR spectra are reported
with respect to the following standards: residual chloroform set at
7.24 ppm, CD2HOD set at 3.30 ppm, and tetramethylsilane set at
0 ppm. J-modulated 13C NMR were performed on a Bruker AC-200
(at 50.3MHz for carbon). 13C NMR spectra are reported with
respect to the following standards: chloroform set at 77 ppm and
tetramethylsilane set at 0 ppm. Coupling constants (J) are
recorded in Hertz (Hz). The abbreviations s¼ singlet, d¼doublet,
ublet, t¼ triplet, dd¼doublet of doublets, m¼multiplet, are used
in reporting the spectra. Pneumatically assisted electrospray
ionization mass spectrometry (ESMS) was performed on a
Micromass Quattro-LC triple quadrupole mass spectrometer with
dichloromethane, dichloromethane/methanol (50:50) or metha-
nol as the mobile phase at a flow rate of 15mL minS1, with use of
a Brownlee Microgradient syringe pump. Samples were dissolved
bH & Co. KGaA, Weinheim small 2008, 4, No. 9, 1390–1398

Biomimetic Synthesis of Gold Nanocrystals Using a Reducing Amphiphile
in dichloromethane/methanol (50:50) or pure methanol. Ammo-
nia or NH4OAc was added for analysis in the negative mode; for
analysis in the positive mode, formic acid was added. Mass
spectra were reported as percent intensity (%) versus mass/
charge (m/z) ratio.
Synthesis of lipid 5: 3-Hydroxy-pentanedioic acid dibenzyl 3-
benzyloxycarbonyl ester (tribenzyl citrate) (2): Citric acid (1)
(9.00 g, 46.8mmol), benzyl alcohol (29.1mL, 280.8mmol), and
toluene (250mL) were placed in a 500mL round-bottomed flask. A
catalytic amount of pTSA (0.20 g) was added and the reaction
mixture was then stirred under reflux with azeotropic removal of
the water produced during the ester formation (Dean-Stark). After
18 h, the mixture was allowed to cool to room temperature. The
solvent and unreacted benzyl alcohol were removed in vacuo. The
residue was dissolved in EtOAc, and washed twice with saturated
aqueous NaHCO3, twice with water, and finally twice with brine.
The organic phase was dried over Na2SO4, and the solvent was
removed under reduced pressure, leaving an oily substance that
solidifies slowly at room temperature. Recrystallization in hexanes/
EtOAc (95:5) afforded 21.6 g (quantitative) of the crystalline title
compound (white needles).1H NMR (acetone-d6, 200MHz): d 2.83 (br s, 1H); 2.90, 3.04
(2d, 4H, J¼15.5 Hz); 5.07 (s, 6H); 7.30–7.35 (m, 15H). 13C NMR
(acetone-d6, 200MHz): d 43.89; 66.70; 67.76; 74.16; 128.83;
129.11; 136.53; 136.88; 169.96; 173.52. IR (cm�1): 3493, 3066,
3034, 2955, 1738, 1498, 1456, 1382, 1341, 1214, 1186, 1174,
1079, 972, 750, 697. MS: ES-positive mode: (m/z): 463.3 (MþHþ)
(calculated: M¼ 462.50). 480.4 (MþNH4þ), 485.3 (MþNaþ).
3-Allyloxy-3-benzyloxycarbonyl-pentanedioic acid dibenzyl es-
ter (3): Under a nitrogen atmosphere, compound (2) (10.32 g, 22.3
mmol) was introduced in a round-bottomed flask, followed by allyl-
tert-butylcarbonate (5.29g, 33.5 mmol) and 110mL of dry toluene.
Palladium acetate (30mg, 0.13 mmol) and triphenylphosphine
(0.31 g, 1.18 mmol) were then added, and this mixture was refluxed
overnight. After being cooled to room temperature, the reaction
medium was washed with dilute aqueous NaHCO3, water, then
brine. The organic phase was dried over Na2SO4, and the volatiles
removed in vacuo. The residue was purified by chromatography over
a silica gel column, eluting with increasing amounts of EtOAc in
hexanes (9:1 to 4:1), to afford 8.97 g of the title compound (93%) as
a clear oil.1H NMR (acetone-d6, 200MHz): d 3.109, 3.27 (2d, 4H,
J¼15.7Hz); 4.03 (d, 2H, J¼5.2 Hz); 4.90–5.20 (m, 8H); 5.60–
5.90 (m, 1H); 7.34 (br.s, 15H). 13C NMR (acetone-d6, 200MHz): d
39.86; 66.20; 66.73; 67.58; 79.21; 116.36; 128.78; 128.83;
129.15; 135.24; 136.55; 136.89; 169.91; 170.64. IR (cm�1):
3454, 3091, 3067, 3035, 2954, 2892, 1957, 1878, 1739, 1499,
1456, 1383, 1346, 1280, 1213, 1170, 1062, 996, 924, 751, 698,
583. MS: ES-positive mode: (m/z) 503.4 (MþHþ), calculated:
M¼502.57; 520.4 (MþNH4þ), 525.3 (MþNaþ).
3-(1,1,1,3,3,5,5-Heptamethyltrisiloxane)-propyloxy-3-benzy-
loxy-carbonyl-pentanedioic acid dibenzyl ester (4): In a round-
bottom flask was introduced (3) (5.03 g, 10 mmol) in 25mL of dry
toluene, followed by 1,1,1,3,3,5,5-heptamethyltrisiloxane (2.90 g,
13 mmol) in of dry toluene (10mL). Karstedt’s platinum
hydrosilylation catalyst (platinum–divinyltetramethyldisiloxane
complex, solution in xylenes: Karstedt’s catalyst, 0.02mL) was
added, and the mixture was stirred at room temperature in a dry
small 2008, 4, No. 9, 1390–1398 � 2008 Wiley-VCH Verlag
atmosphere for 24 h. The volatiles were then removed in vacuo
without heating, and the residue was purified by chromatography
over a silica gel column, eluting with hexanes/ethyl acetate (15:1
to 4:1) to afford 6.08 g (84%) of the title compound as a colorless
oil.1H NMR (CDCl3, 200MHz): d 0.02 (s, 6H); 0.05 (s, 6H); 0.09 (s,
9H); 0.37 (m, 2H); 1.47 (m, 2H); 3.10, 3.27 (2d, 4H, J¼15.7 Hz);
3.34 (t, 2H, J¼6.8 Hz); 5.08 (s, 4H); 5.12 (s, 2H); 7.32 (br.s, 15H).13C NMR (CDCl3, 200MHz): d 0.82; 2.03; 2.57; 14.68; 24.32;
39.82; 67.18; 67.99; 68.16; 77.13; 128.95; 129.11; 129.25;
136.06; 136.35; 170.43; 171.20. IR (cm�1): 3092, 3068, 3036,
2958, 2899, 1742, 1499, 1456, 1383, 1346, 1258, 1214, 1167,
1138, 1047, 842, 796, 751, 697, 582, 491. MS: ES-positive
mode: (m/z): 725.3 (MþHþ) (calculated: M¼725.08).
Surfactant 5: 3-(1,1,1,3,3,5,5-heptamethyltrisiloxane)-propyl-
oxy-3-carboxy-pentanedioic acid: Benzyl-protected silicone-surfac-
tant 4 was dissolved in THF/MeOH (9:1, typically 25mL g�1). Then,
a 5% molar amount of 10% Pd/C was added, and hydrogen was
bubbled into the mixture under stirring at rt. Completion of the
reaction was checked by 1H NMR, following the disappearance of
the benzylic protons. After completion, the catalyst was removed
by filtration over a 0.45mm Teflon filter. Activated carbon was
added to the filtrate, and the mixture was stirred for more 3 h,
before being filtrated again. Volatiles were removed by flushing
nitrogen through the solution, until ca. 95% was removed, then in
vacuo without heating to afford the corresponding silicone-
carboxylic acid surfactant in a quantitative or almost quantitative
yield.1H NMR (CD3OD, 200MHz): d 0.02 (s, 6H); 0.07 (s, 6H); 0.09
(s, 9H); 0.54 (m, 2H); 1.58 (m, 2H); 2.97, 3.13 (2d, 4H,
J¼15.8 Hz); 3.45 (t, 2H, J¼6.8Hz). 13C NMR (CDCl3, 200MHz):
d �1.81; 0.75; 1.26; 14.34; 24.14; 38.98; 67.25; 78.65; 172.88;
173.47. MS: IR (cm�1): 3670–2153 (broad), 2959, 2617, 1721,
1426, 1400, 1293, 1258, 1225, 1204, 1147, 1050, 841, 796,
689, 643, 620. Elemental analysis: C: 42.36% H: 7.53%
(Calculated: C: 42.26% H: 7.54%). MS: ES-positive mode: (m/z):
472.1870 (MþNH4þ) (calculated: MþNH4
þ¼472.1854).
Synthesis of gold nanoleaves: Synthesis of the different metal
nanostructures involves initially the preparation of surfactant 5
aqueous solutions, typically 50mL of 5.0mM stock solutions. The
following procedure was used: in a beaker, compound 5 (0.114 g;
0.25 mmol) was partially dissolved in about 35mL of water; the
initial pH value was about 2.75. The pH was adjusted to the
required value by slowly adding aliquots of a 1 N aqueous NaOH
solution, while monitoring with a color digital pH meter. Complete
dissolution was achieved using brief sonication. The solution was
then carefully transferred to a 50mL volumetric flask, and water
was added to the mark. The pH of the total volume solution was
checked again. Stock solutions were always used immediately
after preparation.
In a typical experiment, 1mL of a freshly prepared tetrachlor-
oauric(III) acid aqueous solution (2.5mM) was added to 2mL of a
surfactant 5 aqueous solution (pH 7.30; 5.0mM), in a glass
scintillation vial. The resulting mixture was briefly hand shaken
and left at room temperature under ambient and static conditions
(no stirring). Light was provided by two classical fluorescent tubes
(white light type) located approximately 2 m above the vial. The
mixture undergoes a series of color changes, from pale yellow
GmbH & Co. KGaA, Weinheim www.small-journal.com 1397

full papers M. A. Brook et al.
1398
initially, to colorless, then light pink, purple, and finally dark
purple due to the formation of the green products, which slowly
starts to settle down after ca. 48 h.
Successive centrifugations (4000 rpm; 15min) followed by
redispersion in water were used in order to separate the gold
nanoleaves from a small amount of gold nanoparticles also
produced. The purification process was improved by allowing the
gold nanoleaves to slowly settle down in the vials (after 5 days):
the pink upper layer of gold nanoparticles was then easily
removed by a Pasteur pipette. Redispersion in water followed by a
single centrifugation cycle led to pure gold nanoleaves.
For the dark experiments, the two solutions were mixed in the
dark, and the sample vials were double-wrapped in aluminum foil
to prevent any exposure to light.
Acknowledgements
We thank the Natural Sciences and Engineering Research
Council of Canada for financial support of this research. We are
grateful to the Canadian Centre for Electron Microscopy
(McMaster University) and to Marcia West and the McMaster
Hospital Electron microscopy facility staff for extensive support
with TEM. W. Gong (Brockhouse Institute for Materials
Research, McMaster University) is acknowledged for X-Ray
analysis and helpful discussions.
[1] C. G. Trick, Curr. Microbiol. 1989, 18, 375.[2] G. Granger, N. M. Price, Limnol. Oceanogr. 1999, 44, 541.[3] J. S. Martinez, G. P. Zhang, P. D. Holt, H.-T. Jung, C. J. Carrano, M. G.
Haygood, A. Butler, Science 2000, 287, 1245.[4] K. Barbeau, E. L. Rue, K.W. Bruland, A. Butler,Nature 2001,413, 409.[5] C. Burda, X. Chen, R. Naranayan, M. A. El-Sayed, Chem. Rev. 2005,
105, 1025.
[6] Y. Sun, Y. Xia, Science . 2002, 298, 2176.[7] R. Jin, Y. C. Cao, E. Hao, G. S. Metraux, G. C. Schatz, C. A. Mirkin,
Nature 2003, 425, 487.[8] M. P. Pileni, Nat. Mater. 2003, 2, 145.[9] Y. Song, R. M. Garcia, R. M. Dorin, H. Wang, Y. Qiu, J. A. Shelnutt,
Angew. Chem, Int. Ed. 2006, 45, 8126.[10] M. Ambrosi, E. Fratini, V. Alfredsson, B. W. Ninham, R. Giorgi, P.
LoNostro, P. Baglioni, J. Am. Chem. Soc. 2006, 128, 7209.[11] M. Reches, E. Gazit, Science 2003, 300, 625.[12] I. A. Banerjee, L. Yu, H. Matsui, Proc. Natl. Acad. Sci. USA 2003,
100, 14678.
[13] C. Mao, C. E. Flynn, A. Hayhurst, R. Sweeney, J. Qi, B. Iverson, G.
Georgiou, A. M. Belcher, Science 2004, 303, 213.
www.small-journal.com � 2008 Wiley-VCH Verlag Gm
[14] T. Scheibel, R. Parthasarathy, G. Sawicki, X.-M. Lin, H. Jaeger, S. L.
Lindquist, Proc. Natl. Acad. Sci. USA 2003, 100, 4527.[15] P. Mukerjhee, A. Ahmad, D. Mandal, S. Senapati, S. R. Sainkar, M.
I. Khan, R. Ramani, R. Parischa, P. V. Ajayakumar, M. Alam, M.
Sastry, R. Kumar, Angew. Chem, Int. Ed. Eng. 2001, 40, 3585.[16] T. Klaus, R. Joerger, E. Olsson, C. G. Granqvist, Proc. Natl. Acad. Sci.
USA 1999, 96, 13611.[17] L. A. Gugliotti, D. L. Feldheim, B. E. Eaton, Science 2004, 304, 850.[18] S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad, M. Sastry,
Nat. Mater. 2004, 3, 482.[19] M. Li, H. Schnablegger, S. Mann, Nature 1999, 402, 393.[20] J. P. Glusker, Acc. Chem. Res. 1980, 13, 345.[21] M. C. Daniel, D. Astruc, Chem. Rev. 2004, 104, 293.[22] G. P. Touey, H. E. Davis, US Patent No. 3 278 593, 1966.[23] Silicones are susceptible to redistribution/metathesis under
acidic and basic conditions: M. A. Brook, Silicon in Organic,
Organometallic, and Polymer Chemistry, Wiley, New York 2000.[24] M. Sastry, A. Swami, S. Manda, P. R. Selvakannan, J. Mater. Chem.
2005, 15, 3161.[25] M. E. Meyre, O. Lambert, C. Faure, J. Mater. Chem. 2006, 16, 3552.[26] C. Xue, C. A. Mirkin, Angew. Chem. Int. Ed. 2007, 46, 2036.[27] M. Gradzielski, H. Hoffmann, P. W. Robisch, Tenside, Surfactants,
Deterg. 1990, 27, 366.[28] T. A. Waggoner, J. A. Last, P. G. Kotula, D. Y. Sasaki, J. Am. Chem.
Soc. 2001, 123, 496.[29] T. Owen, R. Pynn, J. S.Martinez, A. Butler, Langmuir 2005, 21, 12109.[30] H. Colfen, S. Mann, Angew. Chem. Int. Ed. 2003, 42, 2350.[31] M. Y. Lin, H. M. Lindsay, D. A. Weitz, R. C. Ball, R. Klein, P. Meakin,
Nature 1989, 339, 360.[32] K. Nørgaard, L. Iversen, C. J. Kiely, M. Brust, T. Bjørnholm, Adv.
Mater. 2002, 14, 1126.[33] A. Halder, N. Ravishankar, Adv. Mater. 2007, 19, 1854.[34] A. Courty, A.-I. Henry, N. Goubet, M.-P. Pileni, Nat. Mater. 2007, 6,
900.
[35] S. S. Shankar, S. Bhargava, M. Sastry, J. Nanosci. Nanotech. 2005,5, 1721.
[36] Y. Lin, G.-B. Pan, G.-J. Su, X.-H. Fang, L.-J. Wan, C.-L. Bai, Langmuir
2003, 19, 10000.[37] a) C. J. Murphy, A. M. Gole, S. E. Hunyadi, C. J. Orendorff, Inorg.
Chem. 2006, 45, 7544; b) L. Gou, C. J. Murphy, Chem. Mater. 2005,17, 3668; c) J. Gao, C. M. Bender, C. J. Murphy, Langmuir 2003, 19,9065; d) A. Gole, C. J. Murphy, Chem. Mater. 2004, 16, 3633.
[38] J. E. Millstone, S. Park, K. L. Shuford, L. Qin, G. C. Schatz, C. A.
Mirkin, J. Am. Chem. Soc. 2005, 127, 5312.[39] H. C. Chu, C. H. Kuo, M. H. Huang, Inorg. Chem. 2006, 45, 808.[40] W.-L. Huang, C.-H. Chen, M. H. Huang, J. Phys. Chem. C 2007, 111,
2533.
[41] H. B. Abrahamson, A. B. Rezvani, J. G. Brushmiller, Inorg. Chim.
Acta 1994, 226, 117.[42] F. Houlihan, F. Bouchard, J. M. J. Frechet, C. G. Willson, Can. J.
Chem. 1985, 63, 153.
bH & Co. KGaA, Weinheim
Received: November 24, 2007Revised: February 20, 2008
small 2008, 4, No. 9, 1390–1398


















