
Biology and Pathogenesis of Thrombosis and Procoagulant ... · gas gangrene are also characterized...
12
CLINICAL MICROBIOLOGY REVIEWS, July 2003, p. 451–462 Vol. 16, No. 3 0893-8512/03/$08.000 DOI: 10.1128/CMR.16.3.451–462.2003 Biology and Pathogenesis of Thrombosis and Procoagulant Activity in Invasive Infections Caused by Group A Streptococci and Clostridium perfringens Amy E. Bryant* Infectious Diseases Section, Veterans Affairs Medical Center, Boise, and Department of Microbiology, University of Idaho, Moscow, Idaho INTRODUCTION .......................................................................................................................................................451 ROLE OF COAGULATION IN INVASIVE GRAM-POSITIVE INFECTIONS ................................................452 THE COAGULATION SYSTEM—A REVIEW ......................................................................................................452 Procoagulants ..........................................................................................................................................................452 Tissue factor ........................................................................................................................................................452 Contact system ....................................................................................................................................................452 Thrombin .............................................................................................................................................................452 Anticoagulants .........................................................................................................................................................452 Antithrombin-heparan system...........................................................................................................................452 Thrombomodulin-protein C-protein S system ................................................................................................453 Tissue factor pathway inhibitor ........................................................................................................................453 Fibrinolytic system..............................................................................................................................................454 The Cellular Players ...............................................................................................................................................454 Endothelial cells ..................................................................................................................................................454 Platelets ................................................................................................................................................................454 Monocytes/macrophages.....................................................................................................................................454 Polymorphonuclear leukocytes ..........................................................................................................................454 Platelet-leukocyte complexes .............................................................................................................................455 Cytokines ..................................................................................................................................................................455 MECHANISMS OF COAGULOPATHY IN INVASIVE S. PYOGENES AND C. PERFRINGENS INFECTIONS ......................................................................................................................................................455 Invasive S. pyogenes Infections ..............................................................................................................................455 C. perfringens Gas Gangrene .................................................................................................................................457 PROMISING ANTITHROMBOTIC AGENTS IN THE TREATMENT OF SEPSIS AND SEPTIC SHOCK.................................................................................................................................................459 Antithrombin ...........................................................................................................................................................459 Tissue Factor Pathway Inhibitor ..........................................................................................................................459 Activated protein C.................................................................................................................................................459 Other Agents............................................................................................................................................................460 CONCLUSIONS .........................................................................................................................................................460 ACKNOWLEDGMENTS ...........................................................................................................................................460 REFERENCES ............................................................................................................................................................460 INTRODUCTION Systemic activation of coagulation and dysregulation of the anticoagulation pathways contribute to the pathogenesis of many diverse disease entities of infectious etiology. For in- stance, the clinical manifestations of thrombotic thrombocyto- penic purpura and hemolytic-uremic syndrome result from oc- clusive microthrombus formation in the arterioles and capillaries of the brain, gastrointestinal tract, kidneys, and other organs (reviewed in reference 56). Similarly, formation of microvascular thrombi contributes to multiple-organ failure in human cases of gram-negative bacteremia (reviewed in ref- erence 101), to purpura fulminans associated with meningo- coccal sepsis (34), and to deep-vein thrombosis associated with Rocky Mountain and Mediterranean spotted fevers. Microvas- cular thrombus formation is also an integral part of the patho- genesis of cardiovascular disease following infection of the endothelium by Chlamydia pneumoniae (35), enterococci (30), or members of the herpesvirus group (103). Similarly, attach- ment of pathogenic organisms such as Staphylococcus aureus, Staphylococcus epidermidis, or Streptococcus sanguis to cardiac valvular endothelium initiates a local inflammatory reaction that triggers activation of the coagulation cascade and results in formation of platelet-fibrin vegetations characteristic of bac- terial endocarditis. Lastly, as we shall see, occlusive microvas- cular thrombosis participates in the rapid destruction of viable tissue in gram-positive necrotizing infections such as invasive streptococcal and clostridial myonecrosis. In each instance, the events that precipitate and sustain the * Mailing address: Infectious Diseases Section, Veterans Affairs Medical Center, 500 West Fort St. (Bldg 45), Boise, ID 83702. Phone: (208) 422-1599. Fax: (208) 422-1365. E-mail: abryant@mindspring .com. 451 on August 9, 2020 by guest http://cmr.asm.org/ Downloaded from
Transcript of Biology and Pathogenesis of Thrombosis and Procoagulant ... · gas gangrene are also characterized...

CLINICAL MICROBIOLOGY REVIEWS, July 2003, p. 451–462 Vol. 16, No. 30893-8512/03/$08.00�0 DOI: 10.1128/CMR.16.3.451–462.2003
Biology and Pathogenesis of Thrombosis and Procoagulant Activity inInvasive Infections Caused by Group A Streptococci and
Clostridium perfringensAmy E. Bryant*
Infectious Diseases Section, Veterans Affairs Medical Center, Boise, and Department of Microbiology,University of Idaho, Moscow, Idaho
INTRODUCTION .......................................................................................................................................................451ROLE OF COAGULATION IN INVASIVE GRAM-POSITIVE INFECTIONS ................................................452THE COAGULATION SYSTEM—A REVIEW ......................................................................................................452
Procoagulants ..........................................................................................................................................................452Tissue factor ........................................................................................................................................................452Contact system ....................................................................................................................................................452Thrombin .............................................................................................................................................................452
Anticoagulants.........................................................................................................................................................452Antithrombin-heparan system...........................................................................................................................452Thrombomodulin-protein C-protein S system ................................................................................................453Tissue factor pathway inhibitor ........................................................................................................................453Fibrinolytic system..............................................................................................................................................454
The Cellular Players...............................................................................................................................................454Endothelial cells..................................................................................................................................................454Platelets ................................................................................................................................................................454Monocytes/macrophages.....................................................................................................................................454Polymorphonuclear leukocytes..........................................................................................................................454Platelet-leukocyte complexes .............................................................................................................................455
Cytokines..................................................................................................................................................................455MECHANISMS OF COAGULOPATHY IN INVASIVE S. PYOGENES AND C. PERFRINGENS
INFECTIONS ......................................................................................................................................................455Invasive S. pyogenes Infections ..............................................................................................................................455C. perfringens Gas Gangrene .................................................................................................................................457
PROMISING ANTITHROMBOTIC AGENTS IN THE TREATMENT OF SEPSIS ANDSEPTIC SHOCK.................................................................................................................................................459
Antithrombin ...........................................................................................................................................................459Tissue Factor Pathway Inhibitor ..........................................................................................................................459Activated protein C.................................................................................................................................................459Other Agents............................................................................................................................................................460
CONCLUSIONS .........................................................................................................................................................460ACKNOWLEDGMENTS ...........................................................................................................................................460REFERENCES ............................................................................................................................................................460
INTRODUCTION
Systemic activation of coagulation and dysregulation of theanticoagulation pathways contribute to the pathogenesis ofmany diverse disease entities of infectious etiology. For in-stance, the clinical manifestations of thrombotic thrombocyto-penic purpura and hemolytic-uremic syndrome result from oc-clusive microthrombus formation in the arterioles andcapillaries of the brain, gastrointestinal tract, kidneys, andother organs (reviewed in reference 56). Similarly, formationof microvascular thrombi contributes to multiple-organ failurein human cases of gram-negative bacteremia (reviewed in ref-
erence 101), to purpura fulminans associated with meningo-coccal sepsis (34), and to deep-vein thrombosis associated withRocky Mountain and Mediterranean spotted fevers. Microvas-cular thrombus formation is also an integral part of the patho-genesis of cardiovascular disease following infection of theendothelium by Chlamydia pneumoniae (35), enterococci (30),or members of the herpesvirus group (103). Similarly, attach-ment of pathogenic organisms such as Staphylococcus aureus,Staphylococcus epidermidis, or Streptococcus sanguis to cardiacvalvular endothelium initiates a local inflammatory reactionthat triggers activation of the coagulation cascade and resultsin formation of platelet-fibrin vegetations characteristic of bac-terial endocarditis. Lastly, as we shall see, occlusive microvas-cular thrombosis participates in the rapid destruction of viabletissue in gram-positive necrotizing infections such as invasivestreptococcal and clostridial myonecrosis.
In each instance, the events that precipitate and sustain the
* Mailing address: Infectious Diseases Section, Veterans AffairsMedical Center, 500 West Fort St. (Bldg 45), Boise, ID 83702. Phone:(208) 422-1599. Fax: (208) 422-1365. E-mail: [email protected].
451
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

coagulopathy (e.g., activation, injury or infection of endothelialcells, leukocytes, or platelets; induction of cytokine synthesis;and direct activation or inhibition of coagulation or anticoag-ulation factors) are as unique as the host responses they evoke,with outcomes ranging from minor local activation to fulmi-nant disseminated intravascular coagulation (DIC) with mul-tiorgan failure. A thorough understanding of these mecha-nisms may suggest novel therapeutic targets for patients withthese devastating infections.
ROLE OF COAGULATION IN INVASIVEGRAM-POSITIVE INFECTIONS
Group A streptococcal necrotizing fasciitis/myonecrosis andClostridium perfringens gas gangrene are two of the most ful-minant gram-positive infections in humans. Tissue destructionassociated with streptococcal toxic shock syndrome (StrepTSS)progresses rapidly to involve an entire extremity (13, 88), andsuch patients require emergent amputation or extensive surgi-cal debridement and prolonged hospitalization (13, 88). Infact, a recent article in the American Journal of Surgery recom-mended radical debridement to maximize limb salvage andsurvival in cases of severe soft tissue infection due to group Astreptococcus (GAS) (79). Rapid destruction of viable, healthytissue is also characteristic of gas gangrene due to C. perfrin-gens. In this infection, the margin between healthy and necrotictissue often advances several inches per hour despite appro-priate antibiotic therapy (53, 55), and radical amputation re-mains the single best life-saving treatment.
Both GAS necrotizing fasciitis/myonecrosis and clostridialgas gangrene are also characterized by excruciating pain at theinfection site (13, 53, 88). In StrepTSS, the onset of this painoccurs well before shock, renal impairment, or acute respira-tory distress syndrome are manifest (88). Similarly, the onset ofsevere pain in gas gangrene is “sometimes so sudden as tosuggest a vascular catastrophe” (53).
The mechanisms responsible for the early onset of severepain and the rapid regional destruction of tissues in theseinfections have not been completely elucidated. It has been ourhypothesis that these features are due to microvascular throm-bosis leading to reduced tissue perfusion and to hypoxia andsubsequent regional tissue necrosis. Clinical and experimentalobservations support this concept. First, the speed with whichskin, subcutaneous tissue, fascia, and muscle are destroyed inthese infections is similar to the rate of tissue death followingacute arterial thrombosis. Second, intense pain is a prominentfeature in clinical conditions that involve occlusion of the ar-terial blood supply, such as myocardial infarction. Third, tis-sues which are being rapidly destroyed in the progression ofgas gangrene do not bleed. This age-old observation has be-come dictum for surgeons, who routinely remove necrotic tis-sue until bleeding is encountered. Indeed, histologic examina-tion of necrotic tissues obtained from patients with StrepTSSat biopsy or amputation (13, 88) or experimental animals chal-lenged with group A streptococcus (7, 92) reveals plateletthrombi and fibrin clots in capillaries, postcapillary venules,and arterioles of the affected musculature and soft tissues.Similarly, tissues from experimental animals in the early stagesof gas gangrene demonstrate numerous occlusive thrombithroughout adjacent musculature (19, 20).
Taken together, these observations suggest that severe painand rapid tissue destruction associated with both GAS myone-crosis and clostridial gas gangrene result from vascular occlu-sion that begins as a local ischemic process and expands re-gionally until an entire limb is destroyed. The followingdiscussion reviews the coagulation-anticoagulation systemsand examines the molecular mechanisms contributing to mi-crovascular thrombosis and tissue destruction in these twofulminant infections.
THE COAGULATION SYSTEM—A REVIEW
Procoagulants
Tissue factor. Tissue factor (TF) is the principal activator ofcoagulation in vivo (reviewed in reference 64) (Fig. 1). Whenexpressed by monocytes or endothelial cells following injury,viral infection, or exposure to lipopolysaccharide or cytokines,this membrane glycoprotein forms a proteolytically active com-plex with circulating factor VIIa. This complex initiates thedownstream clotting events of both the intrinsic (via activationof factor IX) and extrinsic (via activation of factor X) coagu-lation pathways, culminating in the conversion of prothrombinto thrombin. Thrombin proteolytically cleaves fibrinogen,yielding fibrin molecules that rapidly polymerize into a stableclot. In the absence of factor VIIa, TF-initiated clotting doesnot occur. TF activity is controlled in vivo by several uniquemechanisms. First, extravascular TF is physically separatedfrom clotting factors in the blood until it is exposed followingvessel injury. Second, TF-mediated procoagulant activity isinhibited by four main anticoagulant systems (see below).Lastly, TF expressed on cell surfaces is encrypted (8) andrequires an activational step to manifest its procoagulant ac-tivity. Such activation in vitro is accomplished by mechanicaldisruption of cells or by calcium ionophore treatment (8), butthe in vivo signal that releases encrypted TF from its dimerizedstate has not been elucidated.
Contact system. Interaction of cell wall components ofgram-negative and gram-positive bacteria can activate the con-tact system, resulting in the generation of activated factor XII(Fig. 1) and the potent vasoactive moiety bradykinin. Bradyki-nin may contribute to the capillary leak, decreased vascularresistance, and hypotension characteristic of septic shock (re-viewed in reference 48).
Thrombin. Thrombin generation can act as a feedback am-plification pathway for coagulation through its ability to acti-vate cofactors V and VIII.
Anticoagulants
The procoagulant response is counterbalanced by four mainanticoagulant systems (Fig. 1): the antithrombin (AT)-heparansystem, the thrombomodulin (TM)-protein C-protein S com-plex, tissue factor pathway inhibitor (TFPI), and the fibrino-lytic system. Genetic or acquired defects in these systems con-tribute to the widespread coagulopathy and microvascularthrombosis associated with sepsis.
Antithrombin-heparan system. AT is a broad-spectrumserine protease inhibitor found in plasma and on the surface ofmicrovascular endothelial cells (reviewed in reference 60). It is
452 BRYANT CLIN. MICROBIOL. REV.
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

best known as an inactivator of thrombin, but it also inactivatesother serine proteases of the coagulation cascade (e.g., factorsXa, IXa, XIa, and VIIa). AT contains both a heparin-bindingdomain and a serine-binding domain at its active site. Thebinding of heparin (either from endogenous heparan sulfatesor from exogenously administered heparin) enhances the in-hibitory effects of AT 1,000- to 10,000-fold. On simultaneousbinding of heparin and the target serine protease, AT under-goes a conformational change that facilitates covalent bondingbetween an arginine residue in AT and the critical active-siteserine residue of the protease. In this complexed form, theprotease is inactive. Specifically in the case of thrombin, onlyhigh-molecular-weight heparin supports the thrombin-ATcomplex formation; low-molecular-weight heparin cannot fa-cilitate AT-mediated inactivation of thrombin.
In the absence of heparin, AT also has potent anti-inflam-matory properties. At high concentrations, AT stimulates in-creased endothelial cell production of prostaglandin I2, re-duces proinflammatory cytokine production by monocytes andendothelial cells (82), and dampens neutrophil activationalresponses.
Thrombomodulin-protein C-protein S system. TM is an in-tegral membrane protein of endothelial cells. When thrombinbinds to TM, it loses its capacity to generate fibrin but gains the
ability to activate protein C. Activated protein C, together withcofactor protein S, inactivates factor Va on platelets, therebyshutting down thrombin synthesis. This complex also increasesthe activity of the fibrinolytic molecule tissue plasminogen ac-tivator (tPA). The procoagulant state during gram-negativebacterial sepsis is enhanced by the downregulation of the an-ticoagulant TM-protein C-protein S system (reviewed in ref-erence 101). This downregulation is mediated by tumor necro-sis factor alpha (TNF-�) (see “Cytokines” below) and occursconcomitantly with increased expression of TF and endothelialcell adherence molecules, such as E-selectin.
Tissue factor pathway inhibitor. TFPI is a multivalentplasma proteinase inhibitor with three tandem Kunitz-typedomains (reviewed in reference 16). In the presence of factorXa, TFPI forms a quaternary complex with TF and factor VIIaon the endothelial cell or platelet surface. In this state, TF isinactivated. The requirement for activated factor X suggeststhat TFPI is effective only after factor Xa has been generated,i.e., after initiation of coagulation. TFPI itself can be inacti-vated by elastase released from activated neutrophils (39),providing one link between inflammation and coagulation.Platelets and endothelial cells are the primary sources of TFPI.Platelets release TFPI on activation by thrombin or other ago-nists (reviewed in reference 16), and heparin administration
FIG. 1. Schematic of the coagulation-anticoagulation system with known interactions of GAS. Solid lines indicate pathways that contribute tocoagulation; and jagged lines indicate pathways that inhibit coagulation or contribute to fibrinolysis. M type 1 and 3 GAS stimulate tissue factorproduction from endothelial cells (ECs) and monocytes (MOs) (21); in addition, direct or indirect injury to ECs by SLO or streptococcal pyrogenicexotoxin B may expose extravascular TF (interaction 1). In experimental GAS necrotizing fasciitis induced by an M type 1 strain of GAS (85) andin humans with StrepTSS (84), the level of factor XII is decreased and the APTT is prolonged; however, the PT is normal, and experimentalanimals display a hypercoagulable state (85) (interaction 2). Streptokinase binds plasminogen, and GAS M protein binds fibrinogen; thisquaternary complex has potent plasminogen activator activity (51, 104) (tPA, tissue plasminogen activator) (interaction 3). In a nonhuman primatemodel of M type 3 GAS necrotizing fasciitis and myonecrosis, animals had increased circulating levels of fibrin degradation products (FDP[interaction 4a]) and thrombin-AT III complexes (interaction 4b), indicating systemic activation of the coagulation system (92) (interaction 4).Some strains of GAS, but notably not M type 1 or 3, bind protein S in plasma (96) (interaction 5). Administration of TFPI to experimental animalswith gram-negative bacteremia or in humans with sepsis has proven benefits; however, the efficacy of TFPI in StrepTSS remains to be determined(interaction 6). (Reprinted from reference 21.)
VOL. 16, 2003 THROMBOSIS IN INVASIVE INFECTIONS 453
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

increases circulating TFPI levels two- to fourfold, presumablyby stimulating the release of TFPI from endothelial cells.
Fibrinolytic system. The enzymatic cleavage of polymerizedfibrin is induced by tissue plasminogen activator (tPA) andurokinase (uPA) released from endothelial cells. These factorsstimulate the conversion of plasminogen to plasmin, whichhydrolyzes fibrin into soluble fragments. Activation of this sys-tem occurs transiently secondary to thrombin generation,which in turn induces the release of tPA from the vascularendothelium. The generation of fibrin accelerates tPA activa-tion of plasminogen by serving as a binding surface for bothtPA and plasminogen. This system can be rapidly curtailed byincreased production of plasminogen activator inhibitor type 1(PAI-1). Several studies have shown that this system is stronglyactivated in humans with sepsis and may be exhausted in pa-tients with septic shock. Recently, novel posttranscriptionalpathways regulating the expression of uPA, tPA, and plasmin-ogen activator inhibitor-1 have been identified (reviewed inreference 40).
The Cellular Players
Endothelial cells. Under normal conditions, the vascularendothelium maintains a profoundly anticoagulant state. Socritical is this function to hemostasis that this status is main-tained by multiple, functionally discrete mechanisms. Theseinclude (i) production of prostaglandin I2, which inhibits acti-vation of platelets and promotes vasodilation; (ii) the presenceof large amounts of endogenous heparan sulfates, which actsynergistically with AT to inactivate thrombin; (iii) the expres-sion of thrombomodulin, TFPI, tPA, and nitric oxide (NO);and (iv) the limited expression of cellular adherence mole-cules. However, when a rapid and focal procoagulant responseis required, the endothelium responds with immediate synthe-sis of platelet-activating factor and expression of the adherencemolecule P-selectin; exposure of subendothelial matrix pro-teins also provides binding sites for platelets. Coagulation issustained by synthesis of von Willebrand factor, PAI, and TF,by upregulation of cellular adherence molecules (e.g., E-selec-tins, intracellular cell adherence molecule type 1 [ICAM-1]),and by exposure of binding sites for coagulation factorcomplexes.
The precise physiologic mechanisms invoked following en-dothelial cell perturbation differ from one vascular bed andone organ to another (reviewed in reference 71). For instance,TM is a more important anticoagulant in vessels of the lungsand heart than in the liver (71). Plasminogen activators alsocontribute to the maintenance of blood fluidity in these organs,but neither TM or tPA plays a significant role in the brain (71).Thus, a delicate and tissue- or organ-specific balance betweenprocoagulant and anticoagulant activities is maintained by acomplex series of mechanisms designed to limit the formationof a hemostatic plug precisely to the site of injury. A shift inthis balance to a prothrombotic, hypercoagulable state hasdevastating consequences as described for the above infections.
Platelets. Under normal conditions, platelets circulate inclose proximity to the vascular endothelium but do not adhereto it. Activation of endothelial cells, exposure of subendothe-lial matrix proteins, or an increase in shear stress (74) stimu-lates the rapid adhesion of platelets at the site. The process ofadhesion triggers a signal transduction pathway that culmi-
nates in activation of platelet receptors (e.g., the glycoproteinheterodimer gpIIbIIIa) and in exposure of phosphatidylserineon the outer plasma membrane surface. Once activated,gpIIbIIIa promotes fibrinogen-dependent platelet-platelet ag-gregation such that platelets from the circulating pool arerecruited to the growing hemostatic plug. Exposure of phos-phatidyl serine accelerates the binding and assembly of acti-vated Factors VIII, V, and X into a functional prothrombinaseenzyme complex for the generation of thrombin. Lastly, adhe-sion and aggregation cause platelets to release intracellulargranule contents, including serotonin from the dense granulesand fibrinogen, P-selectin, clotting factors, �-thromboglobulin,and platelet factor 4 from the alpha granules. The last twosubstances are used clinically as circulating markers of plateletactivation. Thromboxane A2, also released from activated plate-lets, amplifies platelet aggregation and secretion responses,smooth muscle contraction, and vasoconstriction and thus fa-cilitates hemostasis.
Monocytes/macrophages. Originally, TF activity was thoughtto be limited to brain, lungs, and placenta and to sites in otherorgans that do not contact flowing blood. Exposure of thisextravascular pool of TF following vessel injury, or its induc-tion on fixed tissue macrophages or endothelial cells followingviral infection, or exposure to endotoxin or cytokines wasthought to be solely responsible for both the initiation and thepropagation of thrombus formation (63). However, recentstudies have demonstrated detectable TF procoagulant activityin whole-blood samples from healthy individuals (37, 45). Thisactivity was associated principally with the mononuclear cellfraction (45) and was elevated in patients with sickle cell dis-ease (45), severe meningococcal infection (65), or peritonitis(2). Current opinion suggests that this intravascular pool of TFmay contribute significantly to the pathologic propagation ofthrombus formation (46), as is observed in septic states asso-ciated with DIC.
In addition to the expression of surface-bound TF, mono-cytes bind factor X via CD11b/CD18 (5). This binding stimu-lates monocyte release of cathepsin G, which proteolyticallyactivates factor X. Thus, monocytes possess an alternativemeans of initiating procoagulant responses to inflammatorystimuli (69). Interestingly, this binding, as well as the binding ofother ligands, such as fibrinogen, ICAM-1, or iC3b, to the Idomain of the CD11b molecule can be inhibited by both un-fractionated and low-molecular-weight heparin at therapeuti-cally relevant concentrations (67). Thus, this feature is distinctfrom the anticoagulation effects of heparin and may provideadditional clinical benefits (e.g., moderation of leukocyte func-tion) (67).
Polymorphonuclear leukocytes. Polymorphonuclear leuko-cytes (PMNL) also contribute to the procoagulant state. Likemonocytes, stimulated PMNL bind factor X and convert it toits activated form. The generation of thrombin then resultsfrom the interaction of bound factor Xa with the factor V-likeeffector cell protease receptor type 1 expressed on PMNL andendothelial cells (3). In addition, simultaneous engagement ofPMNL ligands with the endothelial cell leukocyte adherencemolecules E-selectin and ICAM-1 results in increased TF anddecreased TM synthesis by the endothelium (78)—a processthat can be inhibited by antibody to TNF-� or by the platelet-activating factor receptor antagonist WEB2086 (78). Further-
454 BRYANT CLIN. MICROBIOL. REV.
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

more, activated and adherent PMNL also release cathepsin G(52) and elastase, which contribute to the procoagulant stateby increasing TF synthesis and cleaving TFPI, AT, and C1inhibitor, respectively. Lastly, enzymes and oxygen radicals re-leased prematurely by activated PMNL damage the endothe-lium and expose matrix proteins that promote platelet aggre-gation and activation.
Platelet-leukocyte complexes. Recent studies have demon-strated both the physiologic and pathologic significance of theinteraction of activated platelets and leukocytes. As part of thenormal inflammatory response, this interaction facilitates therecruitment of PMNL to sites of vascular activation or injury,primes neutrophils for enhanced phagocytosis and killing offoreign microbes (68), enhances monocyte production of cyto-kines (57), and permits chemical cross talk and signaling be-tween these cell types (33, 54). In contrast, these heterotypiccellular complexes also contribute to infectious (19, 20) andnoninfectious (10, 66; P. A. Ward, G. O. Till, and K. J. JohnsonAbstract, Fed. Proc 1312a, 1986) ischemic-tissue injury. Thebalance between controlled hemostasis (physiologic) and wide-spread thrombosis (pathologic) is a fine one, since in bothsettings the events are mediated by increased cell adhesion,PMNL and platelet granule release, superoxide anion genera-tion, and thromboxane and leukotriene synthesis (reviewed inreference 27). In a recent study of platelet function in septicpatients, the numbers of circulating platelet-PMNL complexeswere significantly reduced in individuals with evidence of mul-tiorgan dysfunction compared to those with sepsis alone (36),suggesting that sequestration of these aggregates in the micro-vasculature contributes to organ failure.
Platelet-leukocyte interactions are mediated by several dis-tinct contact mechanisms. Although platelet P-selectin bindingof PMNL glycoproteins has been the paradigm for platelet-PMNL interactions, other investigators have demonstratedthat gpIIbIIIa (CD41/CD61) also participates in the adhesionof thrombin-activated platelets to PMNL in vitro (Fig. 2) (73,83, 107). These studies have shown that this interaction isfibrinogen dependent (73, 107), that CD11b/CD18 serves asthe PMNL ligand for fibrinogen (4, 109), and that this inter-action is further enhanced when the functionally active con-
formation of CD11b/CD18 is expressed (33). These findingshave been assimilated into a multistep adhesion cascade modelin which a platelet P-selectin-dependent recognition step isfollowed by a gpIIbIIIa/CD18-dependent stabilization step (re-viewed in reference 27).
Cytokines
The role of cytokines in the regulation of procoagulant andanticoagulant activities is well established (reviewed in refer-ence 98). TNF-�, when injected into human volunteers, exertsa procoagulant effect by increasing the synthesis of TF (9, 97)and the procoagulant cytokines interleukin-6 (IL-6) and IL-8(100). Neutralization of IL-6 decreases the procoagulant re-sponses induced by TNF-� (99). Interestingly, TNF-inducedactivation of the coagulation cascade is preceded by a transientactivation of the fibinolytic system (reviewed in reference 101);activation of coagulation then triggers a secondary activationof fibrinolysis, which is short-lived due to TNF-induced pro-duction of PAI (77). The net result is a shift to a procoagulantstate. This hypercoagulant state is then sustained by TNF-�-induced suppression of TM synthesis (24, 76; E. Conway andD. Rosenburg, Abstract, Circulation 78: 0462 1998) and bydecreased formation of the activated protein C-protein S com-plex on endothelial cells (24). TNF-� mediates the reduction inTM synthesis by a posttranslational mechanism (76)—a pro-cess that can be counteracted with IL-4 (43). In addition,TNF-� increases production of the acute-phase reactant, C4b-binding protein (C4BP), a molecule that complexes with andsequesters free protein S such that it is no longer available forTM complex formation. Infusion of C4BP with sublethal dosesof Escherichia coli into experimental animals results in a con-sumptive coagulopathy, thrombocytopenia, and microvascularthrombosis (93) similar to that seen in hemolytic-uremic syn-drome, suggesting that inactivation of free protein S may con-tribute to microvascular thrombosis in other diseases. Admin-istration of anti-TNF-� in models of endotoxemia blocks theactivation of fibinolysis but not coagulation (101). In contrast,treatment with anti-IL-6 blocks coagulation but not fibrinolysis.
IL-1 induces TF activity in endothelial cells (12) and mono-cytes (62). Injection of IL-1 into experimental animals (ba-boons) also elicits an initial but transient activation of thefibrinolytic system, followed by a TNF-like shift to a procoagu-lant state (41, 101). Treatment with an IL-1 receptor antago-nist inhibits the activation of both the fibrinolytic and coagu-lation systems in humans with gram-negative bacterial sepsis(15) and in animals with experimental endotoxemia (41).
The anti-inflammatory cytokines IL-4 and IL-10 inhibit IL-1-and lipopolysaccharide-induced TF activity in monocytes (62),and pretreatment of animals with IL-10 limits the size ofthrombi formed in response to stasis-induced venous throm-bosis (29).
MECHANISMS OF COAGULOPATHY IN INVASIVES. PYOGENES AND C. PERFRINGENS INFECTIONS
Invasive S. pyogenes Infections
In 1989, we reported a series of patients from the RockyMountain West with GAS infections associated with the sud-den onset of shock, adult respiratory distress syndrome, renal
FIG. 2. Principal receptor-ligand pairs promoting platelet-leuko-cyte complex formation. PSGL-1; P-selectin glycoprotein 1.
VOL. 16, 2003 THROMBOSIS IN INVASIVE INFECTIONS 455
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

failure, bacteremia, and death (88). Similar cases emergedworldwide (reviewed in reference 86), prompting an officialcase definition of StrepTSS by the Centers for Disease ControlWorking Group on Streptococcal Infections (108a). Despitebetter clinical recognition of StrepTSS and intense research onstreptococcal virulence factors, morbidity is high and mortalityremains between 30 and 70% (13).
Clinical reports and epidemiologic studies have repeatedlyshown an association between invasive infections (i.e., bacte-remia, necrotizing fasciitis, myonecrosis, and StrepTSS) andGAS strains of M protein types 1, 3, 12, and 28. Among pa-tients having a defined portal of entry, M type 1 and 3 GASaccount for over 40% of all isolates (reviewed in reference 13).Further, among the 50% of all StrepTSS patients having nodefined portal of entry, virtually all strains of GAS isolated areof M type 1 or 3. Thus, invasive infections due to M type 1 and3 GAS are characterized by coagulopathy and rapid tissuedestruction.
Numerous studies have investigated the interactions of GASwith the coagulation system (Fig. 1). In vitro work has dem-onstrated that M protein binds fibrinogen and that streptoki-nase forms a high-affinity complex with plasminogen. Whenthis latter complex interacts with M-protein-bound fibrinogenon the surface of GAS, it acquires potent plasminogen activa-tor activity that is not inhibitable by antiproteases such as�2-antiplasmin (Fig. 1) (51, 104). Further, in a murine model ofM type 1 GAS necrotizing fasciitis, Sriskandan et al. haverecently shown that the activated partial thromboplastin time(APTT) was prolonged and was associated with reduced levelsof factor XII and prekallikrein (Fig. 1) (85). Similarly, in asmall study of humans with StrepTSS, seven of seven patientshad a significantly elevated APTT compared to that of patientswith GAS infections not associated with shock and organ fail-ure (84). From these findings, it might be predicted that pa-tients with severe GAS infections would develop a largelyfibrinolytic clinical picture and perhaps a bleeding diathesis.
However, in the murine model mentioned above, the pro-thrombin time (PT) was normal and mice actually demon-strated a hypercoagulable state despite prolongation of theAPTT (85). Further, our recent studies of the responses ofexperimental animals (baboons) to GAS infection have dem-onstrated an intense, systemic activation of the coagulationsystem. Specifically, we have shown that bacteremia due to Mtype 3 GAS was associated with a profound drop in the plasmafibrinogen concentration (to 1% of baseline control values), a50% reduction in platelet count, and marked increases in thelevels of fibrin degradation products (�640 �g/dl) and throm-bin-antithrombin and plasmin-antiplasmin complexes (87).Lastly, the marked DIC observed in the lungs (60%), adrenals(100%), and kidneys (80%) of primates with streptococcalbacteremia (87) and in tissues from humans with StrepTSS alsosuggests that the hemostatic balance is shifted toward a patho-logic procoagulant state in severe GAS infections. Indeed,coagulopathy is part of the case definition of StrepTSS (108a).
In the experimental nonhuman primate model of GAS bac-teremia, pretreatment of animals with neutralizing monoclonalantibody against TNF improved survival but did not reversethe observed coagulopathy (87). This suggested that (i) thecoagulopathy associated with GAS bacteremia was not strictlyan epiphenomenon of cytokine generation in the septic state
and (ii) specific bacterium-host cell interactions drive the co-agulation response to GAS.
Our recent investigations of this latter hypothesis demon-strated that killed, washed M type 1 and 3 strains of GASisolated from patients with invasive infection stimulated the invitro production of TF (21). Interestingly, GAS appeared tohave an M-type-specific predilection for the TF-producing cellsthey stimulated. Specifically, M type 3 GAS elicited high levelsof TF from endothelial cells but not monocytes, whereas withM type 1 strains, the converse was observed (21). This disparityexisted although both M types elicited high levels of TNF-�and IL-8 from cultured monocytes (21) and enhanced leuko-cyte adherence molecule expression in endothelial cells (myunpublished observations). Such tissue-specific tropism amongdifferent GAS strains is not unique to this setting. For instance,it has long been recognized that some M types of GAS areprimarily skin associated whereas others are throat strains.Recently, Kalia et al. have demonstrated that this type of tissuetropism and niche separation is associated with specific pat-terns found in the gene locus for M protein (42). Together,these findings support the concept that specific bacterium-hostcell interactions contribute to the coagulopathy associated withStrepTSS.
That these responses are unique to GAS is suggested by thefact that other gram-positive cocci do not stimulate these crit-ical endothelial cells responses controlling inflammation andhemostasis. Specifically, Noel et al. (59) have shown that S.aureus, Enterococcus faecalis, and Streptococcus pneumoniaecould not induce E-selectin expression in cultured endothelialcells. Similarly, Veltrop et al. demonstrated that S. sanguis andS. epidermidis could not elicit TF from human vascular endo-thelial cells (102). In addition, clinical isolates of S. aureus didnot elicit the expression of endothelial cell E-selectin orICAM-1 (90) or induce TF synthesis (102).
TF-mediated coagulation by GAS could also be initiated bydirect injury of the endothelium by soluble streptococcal viru-lence factors such as streptolysin O (SLO) or the cysteineprotease. For instance, Kappur et al. have shown that the GAScysteine protease induces the detachment of endothelial cellsin culture (44). Shanley et al. have shown that this protease,when instilled directly into the lungs of rats, augments lungendothelial cell injury induced by instillation of either strepto-coccal cell wall antigen or SLO (80). Thus, in some settings,SLO or the streptococcal cysteine protease could augmentcoagulation by damaging the endothelium and exposing sub-endothelial TF and matrix proteins. However, it should benoted that only 50 to 70% of isolates of GAS from StrepTSSpatients produce the cysteine protease precursor streptococcalpyrogenic exotoxin B (23, 38, 88).
Sublytic concentrations of SLO and a related toxin, perfrin-golysin O, from C. perfringens each stimulate the functionalupregulation of PMNL CD11b/CD18 (18, 22) and prime neu-trophils for enhanced respiratory burst activity (18). Thus, it islikely that these thiol-activated toxins may indirectly contributeto coagulation by stimulating CD11b/CD18-dependent activa-tion of factor X (69) (see above section on platelet-leukocytecomplexes) and by stimulating premature degranulation of hy-peradherent PMNL. In addition, toxin-induced functional up-regulation of CD11b/CD18 may serve to stabilize gpIIbIIIa-mediated intravascular aggregates of platelets and PMNL.
456 BRYANT CLIN. MICROBIOL. REV.
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from
Such aggregates appear to occlude vessels at the site of GASinfection in experimental animals (92) and may contribute tothe local intense pain and rapid destruction of adjacent healthytissue characteristic of human cases of streptococcal myositisand necrotizing fasciitis.
For a hypercoagulable state to progress to clinical DIC ininvasive S. pyogenes infections, the opposing anticoagulant sys-tems (e.g., TFPI, TM-protein C-protein S) must also be func-tionally downregulated (Fig. 1). Interestingly, certain GASstrains bind protein S in its plasma and endothelial cell-boundforms (96). In addition, some S. pyogenes strains bind C4BP(95), a serum protein that complexes with free protein S. Interms of the coagulation system, we suggest that streptococcalbinding of protein S would effectively remove this moleculefrom participation in the anticoagulant system and therebypromote coagulation. However, the role of protein S binding inthe pathogenesis of invasive streptococcal disease remains un-clear since GAS serotypes that are commonly associated withinvasive infections (i.e., M type 1 or 3) do not bind protein S(49) or C4BP (95). Lastly, novel anticoagulant strategies suchas those involving activated protein C and TFPI may holdpromise for the treatment of invasive streptococcal infectionsby limiting both the local and systemic manifestations of co-agulopathy. However, these modalities remain to be evaluatedin animal models of GAS bacteremia and soft tissue infection.
In summary, GAS possess unique cell-associated compo-nents and soluble factors that elicit important functional re-sponses in cells of the coagulation-hemostatic system. In vivo,these responses probably contribute to intravascular thrombo-sis and leukostasis, multiple-organ failure, and rapid tissuedestruction characteristic of StrepTSS.
C. perfringens Gas Gangrene
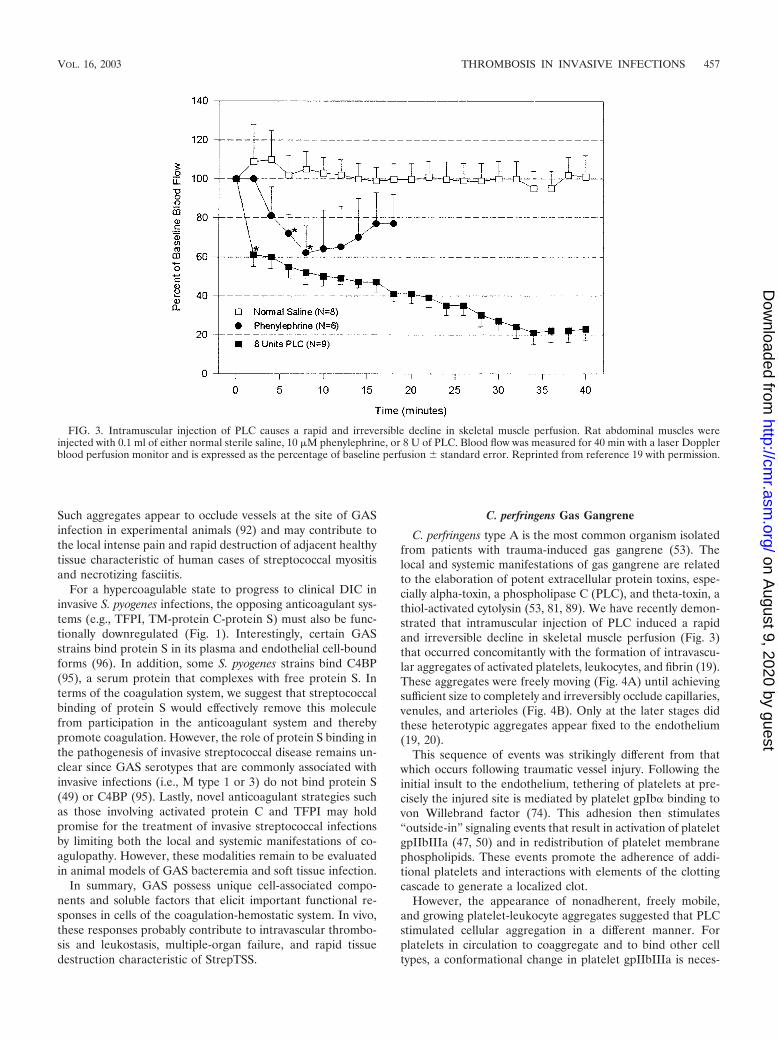
C. perfringens type A is the most common organism isolatedfrom patients with trauma-induced gas gangrene (53). Thelocal and systemic manifestations of gas gangrene are relatedto the elaboration of potent extracellular protein toxins, espe-cially alpha-toxin, a phospholipase C (PLC), and theta-toxin, athiol-activated cytolysin (53, 81, 89). We have recently demon-strated that intramuscular injection of PLC induced a rapidand irreversible decline in skeletal muscle perfusion (Fig. 3)that occurred concomitantly with the formation of intravascu-lar aggregates of activated platelets, leukocytes, and fibrin (19).These aggregates were freely moving (Fig. 4A) until achievingsufficient size to completely and irreversibly occlude capillaries,venules, and arterioles (Fig. 4B). Only at the later stages didthese heterotypic aggregates appear fixed to the endothelium(19, 20).
This sequence of events was strikingly different from thatwhich occurs following traumatic vessel injury. Following theinitial insult to the endothelium, tethering of platelets at pre-cisely the injured site is mediated by platelet gpIb� binding tovon Willebrand factor (74). This adhesion then stimulates“outside-in” signaling events that result in activation of plateletgpIIbIIIa (47, 50) and in redistribution of platelet membranephospholipids. These events promote the adherence of addi-tional platelets and interactions with elements of the clottingcascade to generate a localized clot.
However, the appearance of nonadherent, freely mobile,and growing platelet-leukocyte aggregates suggested that PLCstimulated cellular aggregation in a different manner. Forplatelets in circulation to coaggregate and to bind other celltypes, a conformational change in platelet gpIIbIIIa is neces-
FIG. 3. Intramuscular injection of PLC causes a rapid and irreversible decline in skeletal muscle perfusion. Rat abdominal muscles wereinjected with 0.1 ml of either normal sterile saline, 10 �M phenylephrine, or 8 U of PLC. Blood flow was measured for 40 min with a laser Dopplerblood perfusion monitor and is expressed as the percentage of baseline perfusion � standard error. Reprinted from reference 19 with permission.
VOL. 16, 2003 THROMBOSIS IN INVASIVE INFECTIONS 457
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from
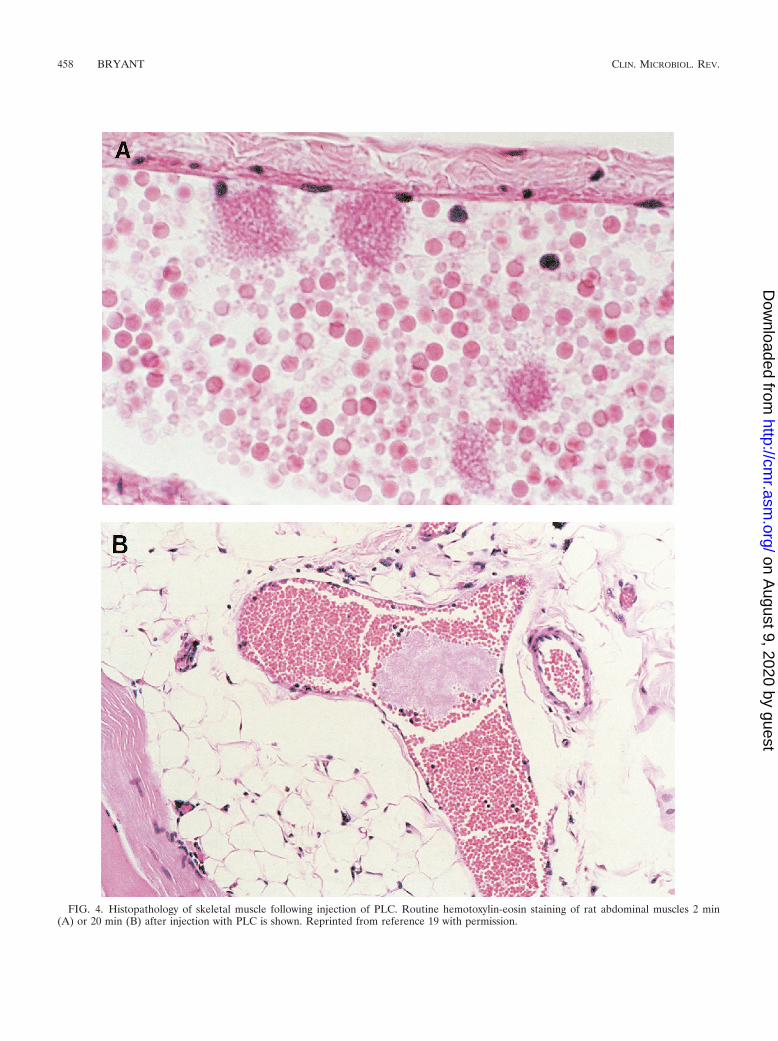
FIG. 4. Histopathology of skeletal muscle following injection of PLC. Routine hemotoxylin-eosin staining of rat abdominal muscles 2 min(A) or 20 min (B) after injection with PLC is shown. Reprinted from reference 19 with permission.
458 BRYANT CLIN. MICROBIOL. REV.
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

sary to permit fibrinogen binding (reviewed in reference 14).Indeed, flow cytometric analyses of PLC-treated whole blooddemonstrated that the formation of both platelet-platelet andplatelet-leukocyte aggregates was mediated by the activation ofplatelet gpIIbIIIa (20).
Because several pathways are known to contribute to theactivation of this adherence molecule, we next investigated theintracellular signaling events leading to functional upregula-tion of gpIIbIIIa induced by PLC. Classically, increases incytosolic calcium levels and activation of protein kinase C(PKC) independently regulate many cellular functions includ-ing activation of platelet gpIIbIIIa. Further, calcium mobiliza-tion and PKC activation act synergistically to elicit the fullphysiological responses of platelets (reviewed in reference 58).Our studies of these signaling pathways demonstrated thatPLC-induced activation of gpIIbIIIa is highly calcium depen-dent but, surprisingly, PKC independent even though PKC isstrongly activated by PLC treatment (17). This finding wasunexpected since most agonists that directly or indirectly acti-vate PKC also activate gpIIbIIIa. However, Tapley and Murrayhave previously demonstrated that treatment of platelets withC. perfringens PLC results in the proteolytic cleavage of solublePKC into a calcium- and phosphospholipid-independent butenzymatically active low-molecular-weight form of the kinase(91). Since PKC-mediated signal transduction requires reloca-tion of the enzyme from subcellular sites to membrane regions(reviewed in reference 28), it is possible that PLC-inducedcleavage of PKC prevents translocation of the truncated kinaseto the plasma membrane, where it would normally contributeto gpIIbIIIa activation. Definitive resolution of this apparentparadox requires further studies.
Increases in intracellular calcium levels in platelets occurprimarily by two mechanisms: receptor-mediated opening ofplasma membrane calcium channels (75) and store-operatedcalcium entry (reviewed in references 32 and 70). In store-operated calcium entry, agonist-induced depletion of calciumfrom intracellular stores triggers the opening of plasma mem-brane calcium channels and the influx of extracellular calcium.Pharmacologic inhibitors of intracellular calcium release andof store-operated plasma membrane calcium channels blockedboth gpIIbIIIa activation and calcium mobilization in PLC-treated platelets (17).
Thus, one can conclude that PLC initiates an “inside-out”signaling cascade that begins with the depletion of internalcalcium stores, is sustained by an influx of calcium throughstore-sensitive channels, and culminates in functional activa-tion in gpIIbIIIa. Further, the lack of involvement of PKCsuggests that the response to PLC does not follow receptor-linked, G-protein-mediated signaling with inositol trisphos-phate generation. This conclusion is further supported by thefact that prostaglandin E1, a known inhibitor of G�i-mediatedsignaling, had no effect on PLC-induced activation ofgpIIbIIIa. Instead, neutralization studies with monoclonal an-tibody against PLC suggest that activation of gpIIbIIIa is adirect consequence of the PLC and/or sphingomyelinase activ-ities of the toxin and not receptor occupancy. This model doesnot, however, exclude a potentiating role of other mediators(e.g., thromboxane production) in the PLC-induced effects.
In summary, these findings suggest that calcium channelblockade and/or therapeutic strategies targeting gpIIbIIIa,
such as those currently used to treat acute myocardial infarc-tion, may prevent vascular occlusion, maintain tissue viability,and provide an alternative to radical amputation for patientswith clostridial gas gangrene. Experimental studies to evaluatethese therapeutic approaches are under way in my laboratory.
PROMISING ANTITHROMBOTIC AGENTS IN THETREATMENT OF SEPSIS AND SEPTIC SHOCK
Antithrombin
A meta-analysis of four small, randomized, placebo-con-trolled studies of AT in severe sepsis demonstrated a trendtoward reduction of 30-day all-cause mortality in the groupreceiving AT compared to placebo. This trend was also wasassociated with shortened stays in the intensive care unit (31).Recently, a larger study (2,314 patients) examined the efficacyof high-dose AT alone and in combination with heparin for thetreatment of severe sepsis (106). The 28-day all-cause mortalitywas not different between the treatment and placebo groups,and increased bleeding was associated with combination AT-heparin therapy. However, the difference in survival betweenthose receiving AT alone versus placebo became significantafter 90 days (106). Over this extended period, patients whoreceived AT without heparin also demonstrated increased im-provements in their quality of life (72).
Tissue Factor Pathway Inhibitor
In patients with disease conditions associated with DIC, amassive production and continuous exposure of excess TF isthought to exhaust the available TFPI (reviewed in reference64). This concept formed the basis for several efficacy studiesof TFPI in both humans and experimental animals with gram-negative bacterial sepsis. Administration of TFPI limited thedevelopment of acute lung and renal injury (108), DIC, andmortality (26) in baboons with E. coli bacteremia and reducedmortality in mice with polymicrobial intra-abdominal infection(61). A safety study of TFPI in humans with sepsis has recentlybeen completed (1). Although not geared to demonstrate ef-ficacy, this study noted improvements in pulmonary, cardiovas-cular, and coagulation scores, a reduction of the circulatinglevels of IL-6 and thrombin-AT complexes, and a trend towardincreased survival in patients receiving recombinant TFPI (1).
Activated protein C
A recent large clinical trial (1,690 patients) investigated theefficacy and safety of activated protein C (APC) replacementtherapy in patients with severe sepsis (11). Causes of infectionin patients with severe sepsis included S. aureus (�14%) andother staphylococci (�6%), S. pneumoniae (�11%) and otherstreptococcal (�9%) and enterococcal (�7%) species, multi-ple gram-negative organisms (�48%), and a few fungal spe-cies. The incidence of infections due to gram-positive versusgram-negative organisms was similar between the placebo andtreatment groups. Administration of APC resulted in a 6.1%reduction in the risk of death compared to the placebo group(11) but was associated with an increased risk of severe bleed-ing.
VOL. 16, 2003 THROMBOSIS IN INVASIVE INFECTIONS 459
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

Other Agents
Low-molecular-weight thrombin inhibitors, complementregulators, PAF antagonists, nitric oxide inhibitors, andcaspase inhibitors are all undergoing investigation for possibletherapeutic benefit in patients with sepsis. For a review, seereference 6.
CONCLUSIONS
The severe pain and rapid tissue destruction characteristic ofinvasive streptococcal and clostridial infections probably resultfrom hypercoagulation and vascular occlusion mediated byunique interactions of the organisms and their toxins with thehuman coagulation system. Understanding the specific mech-anisms by which bacterial virulence factors interact with hu-man cells may offer new insights into the pathogenesis of theseinfections while providing novel therapeutic targets to limit theseverity of these invasive diseases.
ACKNOWLEDGMENTS
This work was supported by grants from the U.S. Department ofVeterans Affairs and the National Institutes of Health (COBRENCRR P20RR15587).
REFERENCES
1. Abraham, E., K. Reinhart, P. Svoboda, A. Seibert, D. Olthoff, A. DalNogare, R. Postier, G. Hempelmann, T. Butler, E. Martin, C. Zwingelstein,S. Percell, V. Shu, A. Leighton, and A. A. Creasey. 2001. Assessment of thesafety of recombinant tissue factor pathway inhibitor in patients with severesepsis: a multicenter, randomized, placebo-controlled single-blind, doseescalation study. Crit. Care Med. 29:2081–2089.
2. Almdahl, S. M., J. H. Brox, and B. Osterud. 1987. Mononuclear phagocytethromboplastin and endotoxin in patients with secondary bacterial perito-nitis. Scand. J. Gastroenterol. 22:914–918.
3. Altieri, D. C. 1995. Xa receptor EPR-1. FASEB J. 9:860–865.4. Altieri, D. C., R. Bader, P. M. Mannucci, and T. S. Edgington. 1988.
Oligospecificity of the cellular adhesion receptor Mac-1 encompasses aninducible recognition specificity for fibrinogen. J. Cell Biol. 107:1893–1990.
5. Altieri, D. C., and T. S. Edgington. 1988. The saturable high affinity asso-ciation of factor X to ADP-stimulated monocytes defines a novel functionof the Mac-1 receptor. J. Biol. Chem. 25:7007–7019.
6. Anel, R. L., and A. Kumar. 2001. Experimental and emerging therapies forsepsis and septic shock. Expert Opin. Investig. Drugs 10:1471–1485.
7. Ashbaugh, C. D., H. B. Warren, V. J. Carey, and M. R. Wessels. 1998.Molecular analysis of the role of the group A streptococcal cysteine pro-tease, hyaluronic acid capsule, and M protein in a murine model of humaninvasive soft-tissue infection. J. Clin. Investig. 102:550–560.
8. Bach, R. R. 1998. Mechanism of tissue factor activation on cells. BloodCoagul. Fibrinolysis 9:S37–S45.
9. Bauer, K. A., H. ten Cate, S. Barzeger, D. R. Spriggs, and M. L. Sherman.1989. Tumor necrosis factor infusions have a procoagulant effect on thehemostatic mechanism of humans. Blood 74:165–175.
10. Bednar, M., B. Smith, A. Pinto, and K. M. Mullane. 1985. Neutrophildepletion suppresses111In-labeled platelet accumulation in infarcted myo-cardium. J. Cardiovasc. Pharmacol. 7:906–912.
11. Bernard, G. R., J. L. Vincent, P. F. Laterre, S. P. LaRosa, J. F. Dhainaut,A. Lopez-Rodriguez, J. S. Steingrub, G. E. Garber, J. D. Helterbrand, E. W.Ely, and C. J. Fisher, Jr. 2001. Efficacy and safety of recombinant humanactivated protein C for severe sepsis. N. Eng. J. Med. 344:699–709.
12. Bevilacqua, M. P., J. S. Pober, G. R. Majeau, R. S. Cotran, and M. A.Gimbrone, Jr. 1984. Interleukin 1 (IL-1) induces biosynthesis and cellsurface expression of procoagulant activity in human vascular endothelialcells. J. Exp. Med. 160:618–623.
13. Bisno, A. L., and D. L. Stevens. 1996. Streptococcal infections in skin andsoft tissues. N. Engl. J. Med. 334:240–245.
14. Blockmans, D., H. Deckmyn, and J. Vermylen. 1995. Platelet activation.Blood Rev. 9:143–156.
15. Boermeester, M. A., P. Van Leeuwen, S. M. Coyle, et al. 1995. Interleukin-1receptor blockade in patients with sepsis syndrome: evidence that interleu-kin-1 contributes to the release of interleukin-6, elastase and phospholipaseA2, and to the activation of the complement, coagulation and fibrnolyticsystem. Arch. Surg. 130:739–748.
16. Broze, G. J., Jr. 1995. Tissue factor pathway inhibitor and the revised theoryof coagulation. Annu. Rev. Med. 46:103–112.
17. Bryant, A. E., C. R. Bayer, S. M. Hayes-Schroer, and D. L. Stevens. 2003.Activation of platelet gpIIbIIIa by phospholipase C from Clostridium per-fringens involves store-operated calcium entry. J. Infect. Dis. 187:408–417.
18. Bryant, A. E., R. Bergstrom, G. A. Zimmerman, J. L. Salyer, H. R. Hill,R. K. Tweten, H. Sato, and D. L. Stevens. 1993. Clostridium perfringensinvasiveness is enhanced by effects of theta toxin upon PMNL structure andfunction: the roles of leukocytotoxicity and expression of CD11/CD18 ad-herence glycoprotein. FEMS Immunol. Med. Microbiol. 7:321–336.
19. Bryant, A. E., R. Y. Z. Chen, Y. Nagata, Y. Wang, C. H. Lee, S. Finegold,P. H. Guth, and D. L. Stevens. 2000. Clostridial gas gangrene I: cellular andmolecular mechanisms of microvascular dysfunction induced by exotoxinsof C. perfringens. J. Infect. Dis. 182:799–807.
20. Bryant, A. E., R. Y. Z. Chen, Y. Nagata, Y. Wang, C. H. Lee, S. Finegold,P. H. Guth, and D. L. Stevens. 2000. Clostridial gas gangrene II: phospho-lipase C-induced activation of platelet gpIIb/IIIa mediates vascular occlu-sion and myonecrosis in C. perfringens gas gangrene. J. Infect. Dis. 182:808–815.
21. Bryant, A. E., S. M. Hayes-Schroer, and D. L. Stevens. 2003. M type 1 and3 group A streptococci stimulate tissue factor-mediated procoagulant ac-tivity in human monocytes and endothelial cells. Infect. Immun. 71:1903–1910.
22. Bryant, A. E., M. A. Kehoe, and D. L. Stevens. 1992. Streptococcal pyro-genic exotoxin A and streptolysin O enhance PMNL binding to proteinmatrixes. J. Infect. Dis. 166:165–169.
23. Chaussee, M. S., J. Liu, D. L. Stevens, and J. J. Ferretti. 1996. Genetic andphenotypic diversity among isolates of Streptococcus pyogenes from invasiveinfections. J. Infect. Dis. 173:901–908.
24. Clauss, M., J. Ryan, and D. Stern. 1992. Modulation of endothelial cellhemostatic properties by TNF: insights into the role of endothelium in thehost response to inflammatory stimuli, p. 49–63. In B. Beutler (ed.), Tumornecrosis factors: the molecules and their emerging role in medicine RavenPress, New York, N.Y.
25. Reference deleted.26. Creasey, A. A., A. C. K. Chang, L. Feigen, T. C. Wun, F. B. Taylor, and L. B.
Hinshaw. 1993. Tissue factor pathway inhibitor reduces mortality fromEscherichia coli septic shock. J. Clin. Investig. 91:2850–2860.
27. De Gaetano, G., C. Cerletti, and V. Evangelista. 1999. Recent advances inplatelet-polymorphonuclear leukocyte interaction. Haemostasis 29:41–49.
28. Dorn, G. W., and D. Mochly-Rosen. 2002. Intracellular transport mecha-nisms of signal transducers. Annu. Rev. Physiol. 64:407–429.
29. Downing, L. J., R. M. Strieter, A. M. Kadell, C. A. Wilke, J. C. Austin, B. D.Hare, M. D. Burdick, L. J. Greenfield, and T. W. Wakefield. 1998. IL-10regulates thrombus-induced vein wall inflammation and thrombosis. Immu-nology 161:1471–1476.
30. Drake, T. A., G. M. Rodgers, and M. A. Sande. 1984. Tissue factor is a majorstimulus for vegetation formation in enterococcal endocarditis in rabbits.J. Clin. Investig. 73:1750–1753.
31. Eisele, B., M. Lamy, L. G. Thijs, H. O. Keinecke, H. P. Schuster, F. R.Matthias, F. Fourrier, H. Heinrichs, and U. Delvos. 1998. Antithrombin IIIin patients with severe sepsis. A randomized, placebo-controlled, double-blind multicenter trial plus a meta analysis on all randomized, placebo-controlled, double-blind trials with antithrombin III in severe sepsis. Inten-sive Care Med. 24:663–672.
32. Elliott, A. C. 2001. Recent developments in non-excitable cell calcium entry.Cell Calcium 30:73–93.
33. Evangelista, V., S. Manarini, S. Rotondo, N. Martelli, R. Polischuk, J. L.McGregor, G. De Gaetano, and C. Cerletti. 1996. Platelet/polymorphonu-clear leukocyte interaction in dynamic conditions: evidence of adhesioncascade and cross talk between P-selectin and the �2 integrin CD11b/CD18.Blood 88:4183–4194.
34. Faust, S. N., M. Levin, O. Harrison, R. D. Goldin, M. S. Lockhart, S.Kondaveeti, Z. Laszik, C. T. Esmon, and R. S. Heydermann. 2001. Dys-function of the endothelial protein C activation pathway in severe menin-gococcal sepsis. N. Engl. J. Med. 345:408–416.
35. Fryer, R. H., E. P. Schwobe, M. L. Woods, and G. M. Rodgers. 1997.Chlamydia species infect human vascular endothelial cells and induce pro-coagulant activity. J. Investig. Med. 45:168–174.
36. Gawaz, M., T. Dickfeld, C. Bogner, S. Fateh-Moghadam, and F. J. Neu-mann. 1997. Platelet function in septic multiple organ dysfunction syn-drome. Intensive Care Med. 23:379–385.
37. Giesen, P. L. A., U. Rauch, B. Bohrmann, D. Kling, M. Roque, J. T. Fallon,J. J. Badimon, J. Himber, M. A. Riederer, and Y. Nemerson. 1999. Blood-borne tissue factor: another view of thrombosis. Proc. Natl. Acad. Sci. USA96:2311–2315.
38. Hauser, A. R., D. L. Stevens, E. L. Kaplan, and P. M. Schlievert. 1991.Molecular analysis of pyrogenic exotoxins from Streptococcus pyogenes iso-lates associated with toxic shock-like syndrome. J. Clin. Microbiol. 29:1562–1567.
39. Higuchi, D., T.-C. Wun, K. M. Likert, and G. J. Broze, Jr. 1992. The effectof leukocyte elastase on tissue factor pathway inhibitor. Blood 79:1712–1719.
460 BRYANT CLIN. MICROBIOL. REV.
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

40. Idell, S. 2002. Endothelium and disordered fibrin turnover in the injuredlung: newly recognized pathways. Crit Care Med. 30:S274–S280.
41. Jansen, P. M., M. A. Boermeester, E. Fischer, et al. 1995. Contribution ofinterleukin 1 to activation of coagulation and fibrinolysis, to neutrophildegranulation and the release of sPLA2 in sepsis. Studies in non-humanprimates following interleukin 1� administration and during lethal bacte-remia. Blood 86:1027–1034.
42. Kalia, A., B. G. Spratt, M. C. Enright, and D. E. Bessen. 2002. Influence ofrecombination and niche separation on the population genetic structure ofthe pathogen Streptococcus pyogenes. Infect. Immun. 70:1971–1983.
43. Kapiotis, S., J. Besemer, D. Bevec, P. Valent, P. Bettelheim, K. Lechner,and W. Speiser. 1991. Interleukin-4 counteracts pyrogen-induced down-regulation of thrombomodulin in cultured human vascular endothelial cells.Blood 78:410–415.
44. Kappur, V., M. W. Majesky, L. L. Li, R. A. Black, and J. M. Musser. 1993.Cleavage of interleukin 1� (IL-1�) precursor to produce active IL-1� by aconserved extracellular cysteine protease from Streptococcus pyogenes. Proc.Natl. Acad. Sci. USA 90:7676–7680.
45. Key, N. S., A. Slungaard, L. Dandelet, S. C. Nelson, C. Moertel, L. A. Styles,F. A. Kuypers, and R. R. Bach. 1998. Whole blood tissue factor procoagu-lant activity is elevated in patients with sickle cell disease. Blood 91:4216–4223.
46. Konigsberg, W., D. Kirchhofer, M. A. Riederer, and Y. Nemerson. 2001.The TF:VIIa complex: clinical significance, structure-function relationshipsand its role in signaling and metastasis. Thromb. Haemostasis 86:757–771.
47. Law, D. A., L. Nannizzi-Alaimo, and D. R. Phillips. 1996. Outside-in inte-grin signal transduction: �IIb�3-(gpIIbIIIa) tyrosine phosphorylation in-duced by platelet aggregation. J. Biol. Chem. 271:10811–10815.
48. Levi, M. 2000. Keep in contact: the role of the contact system in infectionand sepsis. Crit. Care Med. 28:3765–3766.
49. Liang, O. D., K. T. Preissner, and G. S. Chhatwal. 1997. The hemopexin-type repeats of human vitronectin are recognized by Streptococcus pyogenes.Biochem. Biophys. Res Commun. 234:445–449.
50. Lipfert, L., B. Haimovich, M. D. Schaller, B. S. Cobb, J. T. Parsons, andJ. S. Brugge. 1992. Integrin-dependent phosphorylation and activation ofthe protein tyrosine kinase pp125FAK in platelets. J. Cell Biol. 119:905–912.
51. Lottenberg, R., C. C. Broder, and M. D. P. Boyle. 1987. Identification of aspecific receptor for plasmin on a group A Streptococcus. Infect. Immun.55:1914–1928.
52. Macgregor, I. R., A. M. Perrie, S. C. Donnelly, and C. Haslett. 1997.Modulation of human endothelial thrombomodulin by neutrophils andtheir release products. Am. J. Respir. Crit. Care Med. 155:47–52.
53. MacLennan, J. D. 1962. The histotoxic clostridial infections of man. Bac-teriol. Rev. 26:177–276.
54. Maugeri, N., V. Evangelista, P. Piccaroni, G. Dell’Elba, A. Celardo, G. DeGaetano, and C. Cerletti. 1992. Transcellular metabolism of arachidonicacid: increased platelet thromboxane generation in the presence of acti-vated polymorphonuclear leukocytes. Blood 80:447–451.
55. McNee, J. W., and J. S. Dunn. 1917. The method of spread of gas gangreneinto living muscle. Br. Med. J. 1:727–729.
56. Moake, J. L. 2002. Thrombotic microangiopathies. N. Engl. J. Med. 347:589–600.
57. Neumann, F. J., N. Marx, M. Gawaz, K. Brand, I. Ott, C. Rokitta, C.Sticherling, C. Meinl, A. May, and A. Schomig. 1997. Induction of cytokineexpression in leukocytes by binding of thrombin-stimulated platelets. Cir-culation 95:2387–2394.
58. Nishizuka, Y. 1984. The role of protein kinase C in cell surface signaltransduction and tumour promotion. Nature 308:693–698.
59. Noel, R. F., T. T. Sato, C. Mendez, M. C. Johnson, and T. H. Pohlman.1995. Activation of human endothelial cells by viable or heat-killed gram-negative bacteria requires soluble CD14. Infect. Immun. 63:4046–4053.
60. Opal, S. M., C. M. Kessler, J. Roemisch, and S. Knaub. 2002. Antithrom-bin, heparin, and heparan sulfate. Crit. Care Med. 30:S325–S331.
61. Opal, S. M., J. E. Palardy, N. A. Parejo, and A. A. Creasey. 2001. Theactivity of tissue factor pathway inhibitor in experimental models of supe-rantigen-induced shock and polymicrobial intra-abdominal sepsis. Crit.Care Med. 29:205–207.
62. Osnes, L. T., A. B. Westvik, G. B. Joo, C. Okkenhaug, and P. Kierulf. 1996.Inhibition of IL-1 induced tissue factor (TF) synthesis and procoagulantactivity (PCA) in purified human monocytes by IL-4, IL-10, and IL-13.Cytokine 8:822–827.
63. Osterud, B., M. S. Bajaj, and S. P. Bajaj. 1995. Sites of tissue factorpathway inhibitor (TFPI) and tissue factor expression under physiologicand pathologic conditions. Thromb. Haemostasis 73:873–875.
64. Osterud, B., and E. Bjorklid. 2001. The tissue factor pathway in dissemi-nated intravascular coagulation. Semin. Thromb. Hemostasis 27:605–617.
65. Osterud, B., and T. Flaegstad. 1983. Increased thromboplastin activity inmonocytes of patients with meningococcal infection: Related to an un-favourable prognosis. Thromb. Haemostasis 49:5–7.
66. Palabrica, T., R. Lobb, B. C. Furie, M. Aronovitz, C. H. Benjamin, Y.-M.Hsu, S. A. Sajer, and B. Furie. 1992. Leukocyte accumulation promoting
fibrin deposition is mediated in vivo by P-selectin on adherent platelets.Nature 359:848–852.
67. Peter, K., M. Schwarz, C. Conradt, T. Nordt, M. Moser, W. Kubler, and C.Bode. 1999. Heparin inhibits ligand binding to the leukocyte integrin Mac-1(CD11b/CD18). Circulation 100:1533–1539.
68. Peters, M. J., G. Dixon, K. T. Kotowicz, D. J. Hatch, R. S. Heyderman, andN. J. Klein. 1999. Circulating platelet-neutrophil complexes represent asubpopulation of activated neutrophils primed for adhesion, phagocytosisand intracellular killing. Br. J. Haematol. 106:391–399.
69. Plescia, J., and D. C. Altieri. 1996. Activation of Mac-1 (CD11b.CD18)-bound factor X by released cathepsin G defines an alternative pathway ofleucocyte initiation of coagulation. Biochem. J. 319:873–879.
70. Rosado, J. A., and S. O. Sage. 2000. The actin cytoskeleton in store-mediated calcium entry. J. Physiol. 526:221–229.
71. Rosenberg, R. D., and W. C. Aird. 1999. Vascular bed-specific hemostasisand hypercoagulable states. N. Engl. J. Med. 340:1555–1564.
72. Rublee, D., S. M. Opal, W. Schramm, H. O. Keinecke, and S. Knaub. 2002.Quality of life effects of antithrombin III in sepsis survivors: results from theKyberSept trial [ISRCTN22931023]. Crit. Care 6:349–356.
73. Ruf, A., R. F. Schlenk, A. Maras, E. Morgenstern, and H. Patsheke. 1992.Contact-induced neutrophil activation by platelets in human cell suspen-sions and whole blood. Blood 80:1238–1246.
74. Ruggeri, Z. M. 1997. Mechanisms initiating platelet thrombus formation.Thromb. Haemostasis 78:611–616.
75. Sage, S. O., J. E. Merritt, T. J. Hallam, and T. J. Rink. 1989. Receptor-mediated calcium entry in fura-2 loaded human platelets stimulated withADP and thrombin. Biochem. J. 258:923–926.
76. Scarpati, E. M., and J. E. Sadler. 1989. Regulation of endothelial cellcoagulant properties. J. Biol. Chem. 264:20705–20713.
77. Schleef, R., M. P. Bevilacqua, M. Sawdey, M. Gimbrone, and D. Loskutoff.1988. Cytokine activation of vascular endothelium. Effects on tissue plas-minogen activator and type 1 plasminogen activator inhibitor. J. Biol.Chem. 263:5797–5803.
78. Schmid, E., T. H. Muller, R. M. Budzinski, K. Binder, and K. Pfizenmaier.1995. Signaling by E-selectin and ICAM-1 induces endothelial tissue factorproduction via autocrine secretion of platelet-activating factor and tumornecrosis factor �. J. Interferon Cytokine Res. 15:819–825.
79. Schurr, M., S. Engelhardt, and R. Helgerson. 1998. Limb salvage for strep-tococcal gangrene of the extremity. Am. J. Surg. 175:213–217.
80. Shanley, T. P., D. Schrier, V. Kapur, M. Kehoe, J. M. Musser, and P. A.Ward. 1996. Streptococcal cysteine protease augments lung injury inducedby products of group A streptococci. Infect. Immun. 64:870–877.
81. Smith, L. D. S. 1975. Clostridium perfringens, p. 115–176. In L. D. S. Smith(ed.), The pathogenic anaerobic bacteria. Charles C Thomas, Springfield,Ill.
82. Souter, P., S. Thomas, A. R. Hubbard, S. Poole, J. Romisch, and E. Gray.2001. Antithrombin inhibits lipopolysaccharide-induced tissue factor andinterleukin-6 production by mononuclear cells, human umbilical vein en-dothelial cells and whole blood. Crit. Care Med. 29:134–139.
83. Spangenberg, R., H. Redlich, I. Bergmann, W. Losche, M. Gotzrath, and B.Kehrel. 1993. The platelet glycoprotein IIb/IIIa complex is involved in theadhesion of activated platelets to leukocytes. Thromb. Haemostasis 70:514–521.
84. Sriskandan, S., and J. Cohen. 2000. Kallikrein-kinin system activation instreptococcal toxic shock syndrome. Clin. Infect. Dis. 30:961–962.
85. Sriskandan, S., G. Kemball-Cook, D. Moyes, J. Canvin, E. Tuddenham,and J. Cohen. 2000. Contact activation in shock caused by invasive group AStreptococcus pyogenes. Crit. Care Med. 28:3684–3691.
86. Stevens, D. L. 1992. Invasive group A Streptococcus infections. Clin. InfectDis. 14:2–13.
87. Stevens, D. L., A. E. Bryant, S. P. Hackett, A. Chang, G. Peer, S. Kosanke,T. Emerson, and L. Hinshaw. 1996. Group A streptococcal bacteremia: therole of tumor necrosis factor in shock and organ failure. J. Infect. Dis.173:619–626.
88. Stevens, D. L., M. H. Tanner, J. Winship, R. Swarts, K. M. Reis, P. M.Schlievert, and E. Kaplan. 1989. Reappearance of scarlet fever toxin Aamong streptococci in the Rocky Mountain West: severe group A strepto-coccal infections associated with a toxic shock-like syndrome. N. Engl.J. Med. 321:1–7.
89. Stevens, D. L., B. E. Troyer, D. T. Merrick, J. E. Mitten, and R. D. Olson.1988. Lethal effects and cardiovascular effects of purified alpha- and theta-toxins from Clostridium perfringens. J. Infect. Dis. 157:272–279.
90. Strindhall, J., P.-E. Lindgren, S. Lofgren, and E. Kihlstrom. 2002. Varia-tions among clinical isolates of Staphylococcus aureus to induce expressionof E-selectin and ICAM-1 in human endothelial cells. FEMS Immunol.Med. Microbiol. 32:227–235.
91. Tapley, P. M., and A. W. Murray. 1984. Platelet Ca2�-activated, phospho-lipid-dependent protein kinase: evidence for proteolytic activation of theenzyme in cells treated with phospholipase C. Biochem. Biophys. Res.Commun. 118:835–841.
92. Taylor, F. B., Jr., A. E. Bryant, K. E. Blick, E. Hack, P. M. Jansen, S. D.Kosanke, and D. L. Stevens. 1999. Staging of the baboon response to group
VOL. 16, 2003 THROMBOSIS IN INVASIVE INFECTIONS 461
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

A Streptococcus administered intramuscularly: a descriptive study of theclinical symptoms and clinical chemical response. Clin. Infect. Dis. 29:167–177.
93. Taylor, F. B., Jr., B. Dahlback, A. C. Chang, M. S. Lockhart, K. Hatanaka,G. Peer, and C. T. Esmon. 1995. Role of free protein S and C4b bindingprotein in regulation of the coagulant response to Escherichia coli. Blood86:2642–2652.
94. Reference deleted.95. Thern, A., L. Stenberg, B. Dahlback, and G. Lindahl. 1995. Ig-binding
surface proteins of Streptococcus pyogenes also bind human C4b-bindingprotein (C4BP), a regulatory component of the complement system. J. Im-munol. 154:375–386.
96. Valentin-Weigand, P., J. Grulich-Henn, G. S. Chhatwal, G. Muller-Berghaus, H. Blobel, and K. T. Preissner. 1988. Mediation of adherence ofstreptococci to human endothelial cells by complement S protein (vitronec-tin). Infect. Immun. 56:2851–2855.
97. van der Poll, T., H. R. Buller, H. ten Cate, et al. 1990. Activation ofcoagulation after administration of tumor necrosis factor to normal sub-jects. N. Engl. J. Med. 322:1622–1627.
98. van der Poll, T., E. De Jonge, and M. Levi. 2001. Regulatory role ofcytokines in disseminated intravascular coagulation. Semin. Thromb. He-mostasis 27:639–651.
99. van der Poll, T., M. Levi, C. E. Hack, et al. 1994. Elimination of interleukin6 attenuates coagulation activation in experimental endotoxemia in chim-panzees. J. Exp. Med. 179:1253–1259.
100. van der Poll, T., M. Levi, and S. J. H. Van Deventer. 1990. TNF and thedysbalance between coagulant and anticoagulant mechanisms in septice-mia. Intensive Care Emerg. Med. 14:269–273.
101. van Gorp, E. C. M., C. Suharti, H. ten Cate, W. M. V. Dolmans, J. W. M.van der Meer, J. W. ten Cate, and D. P. M. Brandjes. 1999. Review:infectious diseases and coagulation disorders. J. Infect. Dis. 180:176–186.
102. Veltrop, M. H. A. M., H. Beehuizen, and J. Thompson. 1999. Bacterialspecies- and strain-dependent induction of tissue factor in human vascularendothelial cells. Infect. Immun. 67:6130–6138.
103. Visser, M. R., and G. M. Versellotti. 1993. Herpes simplex virus and ath-erosclerosis. Eur. Heart J. 14:39–41.
104. Wang, H., R. Lottenberg, and M. Boyle. 1994. Analysis of plasmin(ogen)acquisition by clinical isolates of group A streptococci incubated in humanplasma. J. Infect. Dis. 169:143–149.
105. Reference deleted.106. Warren, B. L., A. Eid, P. Singer, S. S. Pillay, P. Carl, I. Novak, P. Chalupa,
A. Atherstone, I. Penzes, A. Kubler, S. Knaub, H. O. Keinecke, H. Hein-richs, F. Schindel, M. Juers, R. C. Bone, S. M. Opal, and KyberSept TrialStudy Group. 2002. Caring for the critically ill patient. High-dose anti-thrombin III in severe sepsis: a randomized controlled trial. JAMA 286:1869–1878.
107. Weber, C., and T. A. Springer. 1997. Neutrophil accumulation on activated,surface-adherent platelets in flow is mediated by interaction of Mac-1 withfibrinogen bound to �IIb�3 and stimulated by platelet-activating factor.J. Clin. Investig. 100:2085–2093.
108. Welty-Wolf, K. E., M. S. Carraway, D. L. Miller, T. L. Ortel, M. Ezban, A. J.Ghio, S. Idell, and C. A. Piantadosi. 2001. Coagulation blockade preventssepsis-induced respiratory and renal failure in baboons. Am. J. Respir. Crit.Care Med. 164:1988–1996.
108a.Working Group on Severe Streptococcal Infections. 1993. Defining thegroup A streptococcal toxic shock syndrome: rationale and consensus def-inition. JAMA 269:390–391.
109. Wright, S. D., J. I. Weitz, A. D. Huang, S. M. Levin, and S. C. Silverstein.1988. Complement receptor type three (CD11b/CD18) of human polymor-phonuclear leukocytes recognizes fibrinogen. Proc. Natl. Acad. Sci. USA85:7734–7738.
462 BRYANT CLIN. MICROBIOL. REV.
on August 9, 2020 by guest
http://cmr.asm
.org/D
ownloaded from


















