
Biology 644: Bioinformatics - Riley...
26
Biology 644: Bioinformatics
Transcript of Biology 644: Bioinformatics - Riley...

Biology 644: Bioinformatics
Biology 644: Bioinformatics
Biology 644: Bioinformatics
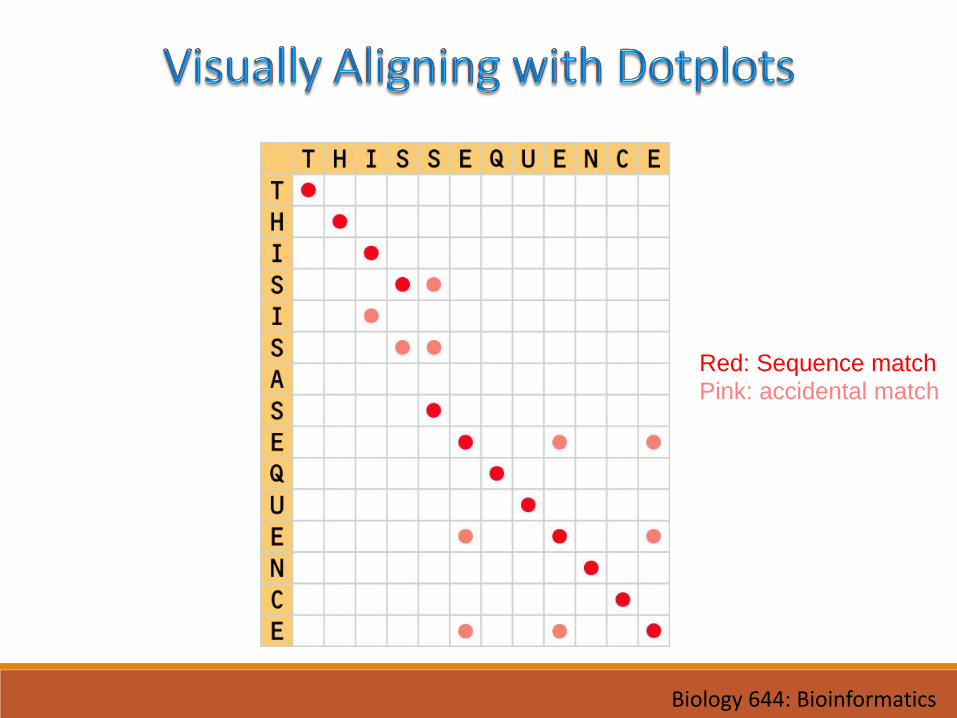
Red: Sequence match
Pink: accidental match
Biology 644: Bioinformatics
Biology 644: Bioinformatics
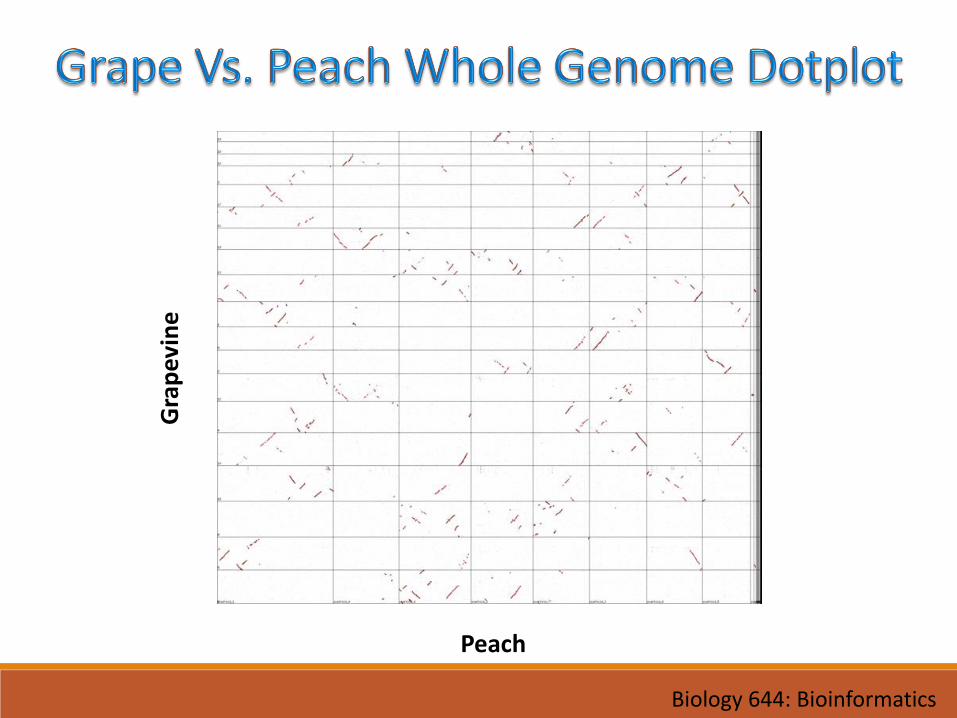
Peach
Gra
pev
ine
Biology 644: Bioinformatics

Biology 644: Bioinformatics
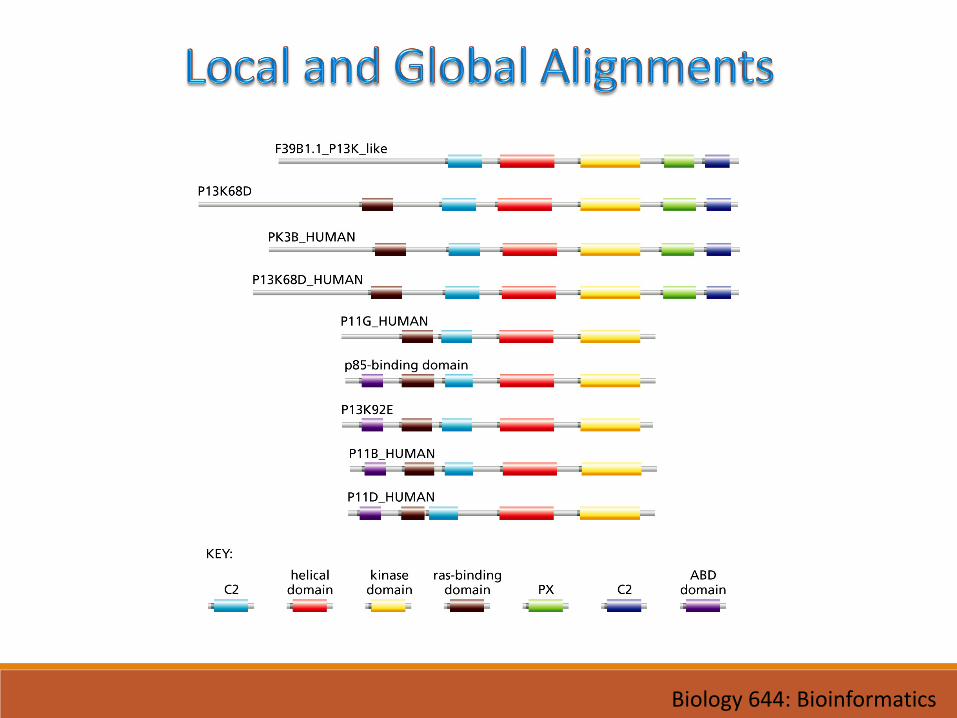
• Use local alignment (BLAST, Smith-Waterman) when:
• Searching databases for biologically related sequences
• Analyzing multidomain proteins
• Use global alignment (CLUSTALW, Needleman-Wunsch, etc.) when:
• Aligning several close members of a protein family
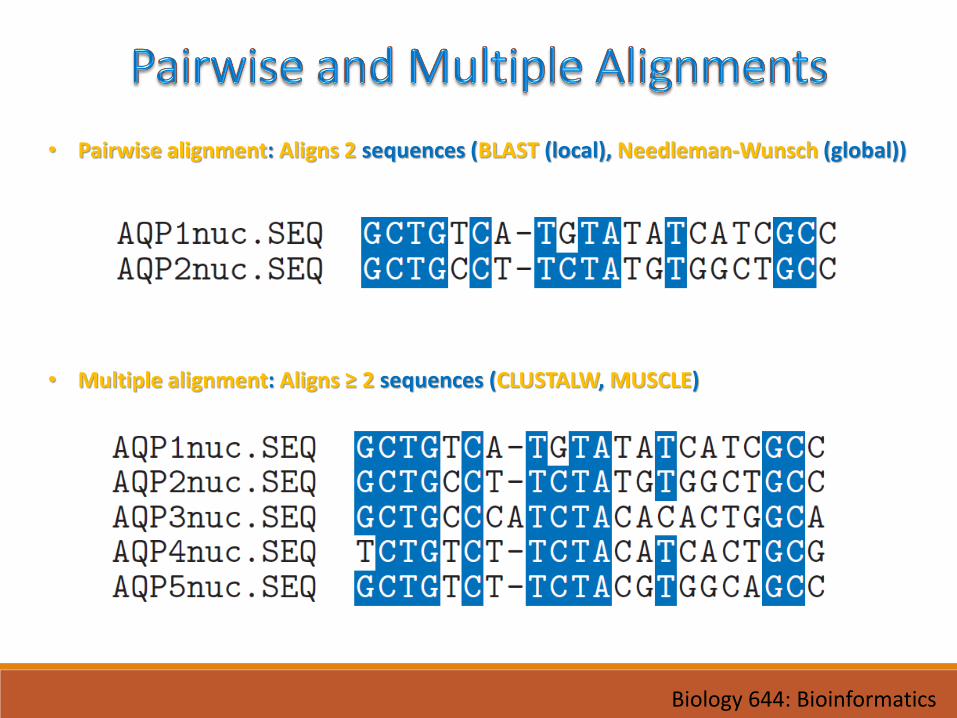
• Pairwise alignment: Aligns 2 sequences (BLAST (local), Needleman-Wunsch (global))
• Multiple alignment: Aligns ≥ 2 sequences (CLUSTALW, MUSCLE)
Biology 644: Bioinformatics

Biology 644: Bioinformatics
Two kinds of computational problems:
• Tractable: Runs in a “reasonable" amount of time
• Can Find best solution• Example: Pairwise Local and Pairwise Global Sequence Alignment
• Intractable: Requires huge amount of computer time to find best solution
• Would take longer than the age of the universe• Famous examples:
• Traveling salesman problem• Multiple sequence alignment
• Heuristic solution: finds a good approximate answer in a reasonableamount of time

Biology 644: Bioinformatics
Local Alignment Global Alignment
2 SequencesBLAST Family (Heuristic)
Smith-Waterman (Optimal)Needleman-Wunsch
(Optimal)
>2 SequencesRun many Local Heuristic
Alignments
CLUSTAL FamilyMUSCLET-Coffee
(All Heuristic)

Biology 644: Bioinformatics
+ Identical or similar amino acid substitutions
- Penalties for dissimilar amino acid substitutions
- Penalties for gap openings
- Penalties for gap extensions
final alignment score
+

Biology 644: Bioinformatics
• Used to determine the probability of homology between two or more sequences.
• Substitutions that are favored through evolutionary time are given a positive score
• Substitutions that are disfavored through evolutionary time are given a negative score
• Substitutions that are more likely should get a higher score
• Substitutions that are less likely should get a lower score
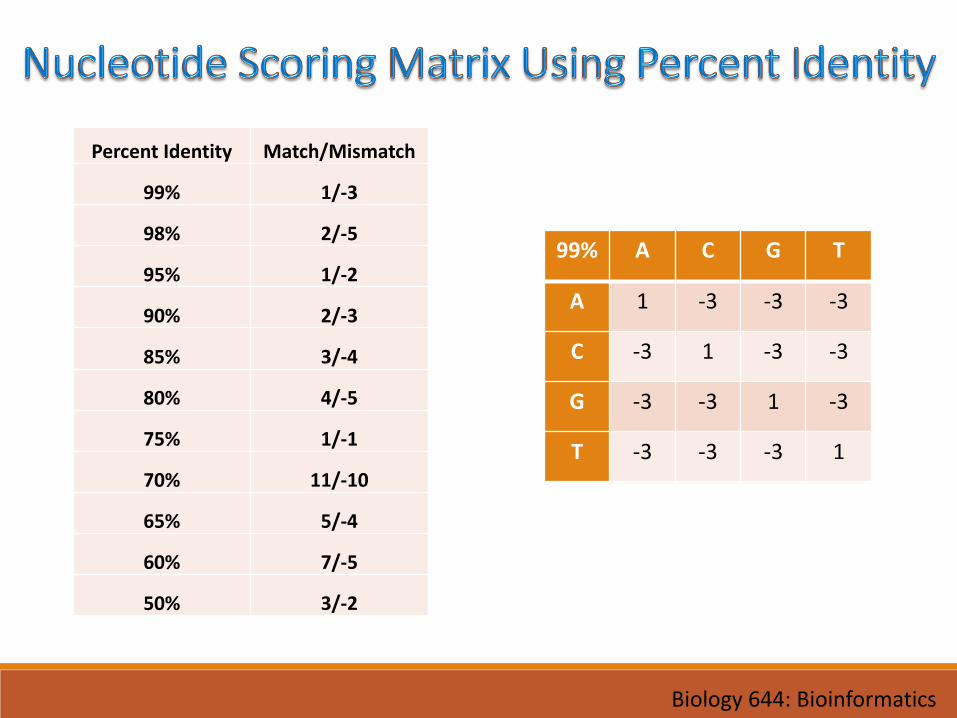
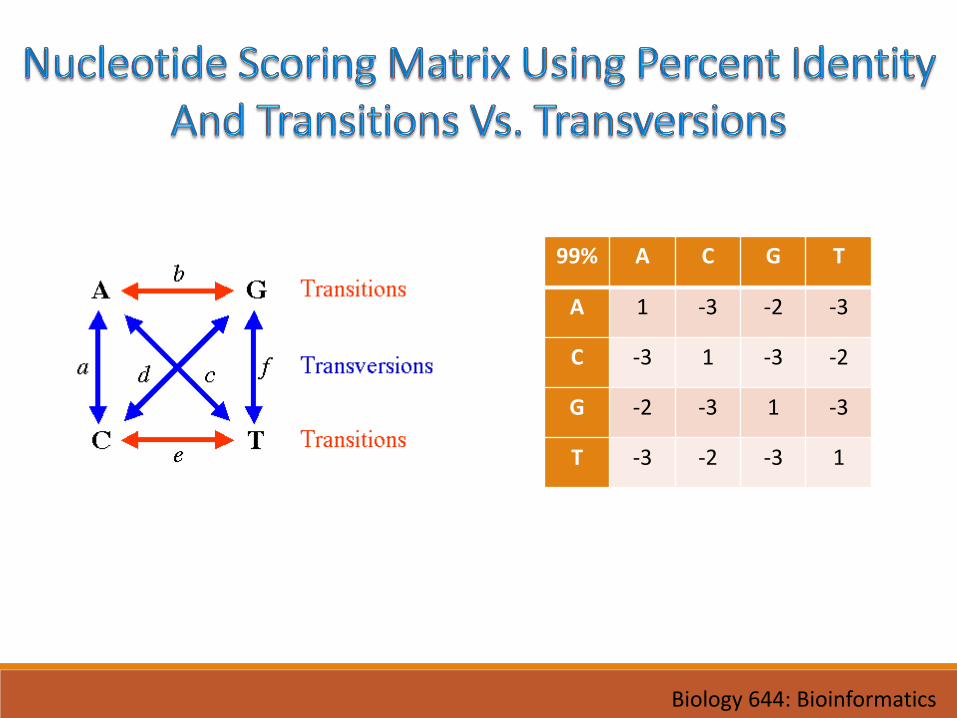
• For DNA• Percent Identity• Transitions Vs. Transversions
• For Proteins• Log-odds Substitution Matrices
• PAM• BLOSUM
Biology 644: Bioinformatics
Percent Identity Match/Mismatch
99% 1/-3
98% 2/-5
95% 1/-2
90% 2/-3
85% 3/-4
80% 4/-5
75% 1/-1
70% 11/-10
65% 5/-4
60% 7/-5
50% 3/-2
99% A C G T
A 1 -3 -3 -3
C -3 1 -3 -3
G -3 -3 1 -3
T -3 -3 -3 1
Biology 644: Bioinformatics
99% A C G T
A 1 -3 -2 -3
C -3 1 -3 -2
G -2 -3 1 -3
T -3 -2 -3 1

Biology 644: Bioinformatics
• Dayhoff - 1978 - Estimates the rate at which each possible residue in a sequence changes to each other residue over time
• Uses an evolutionary model to find mutation rates in closely related sequences and then extrapolate to greater evolutionary distances
• Aligned closely-related proteins, counted amino acid substitutions at each position
• PAM: Point (or percent) accepted mutations
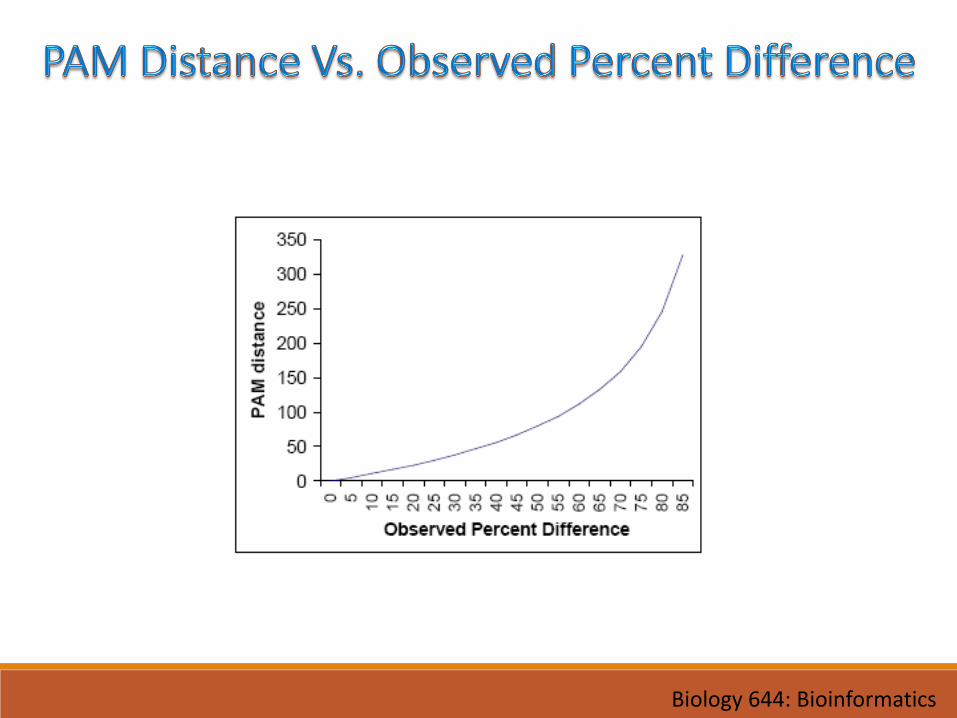
• One PAM unit: 1 change per 100 residues (~10M years)• 1 PAM = PAM1 = 1% average change of all amino acid positions
• After 100 PAMs of evolution, not every residue will have changed• some residues may have mutated several times• some residues may have returned to their original state• some residues may not changed at all

Biology 644: Bioinformatics
• The act of an amino acid substitution is considered as a Markov model characterized as independent steps of evolutions
• A Markov model is characterized by a series of changes of state in a system such that a change from one state to another does not depend on the previous history of the state• Assumes independence between positions over time where• P(A → B) = P(B → A)
• Use of the Markov model makes it possible to extrapolate from amino acid substitutions observed over a relatively short period of evolutionary time to longer periods of evolutionary time by taking powers of the PAM1 matrix
• PAMX = Scoring matrix for X evolutionary steps
• PAM2 = PAM1 × PAM1 = (PAM1)2
• PAM3 = PAM2 × PAM1 = (PAM1)3
.• PAMX = PAM(X-1) × PAM1 = (PAM1)X
.• PAM250 = PAM249 × PAM1 = (PAM1)250

Biology 644: Bioinformatics
• Find set of very similar sequences (99% identity)
• Make global alignment (71 groups)
• Count the substitutions (1572 changes)
• Calculate mutation frequencies
• Normalize by mutability of respective amino acid• Makes comparisons possible
• Multiply to wanted evolutionary distance x
• Calculate log-odds score for [P(A→B) + P(B→A)]/2 and round• S > 0: More often than per chance• S = 0: Number expected by chance• S < 0: Less often than per chance ji
ji
jipp
qS
,
, log

Biology 644: Bioinformatics
• Closely related sequences → 1 evolutionary step
• PAM1 describes likelihood of changes for one small evolutionary step
• Longer evolutionary spans are modeled through successive small steps
• PAM2 = PAM1 × PAM1 = (PAM1)2
• PAM3 = PAM2 × PAM1 = (PAM1)3
.• PAMX = PAM(X-1) × PAM1 = (PAM1)X
.• PAM250 = PAM249 × PAM1 = (PAM1)250
• Markov chain property (independence)
• Greater divergence achieved through matrix multiplication
• Larger PAM number: more similar values in the matrix
Biology 644: Bioinformatics

Biology 644: Bioinformatics

Biology 644: Bioinformatics
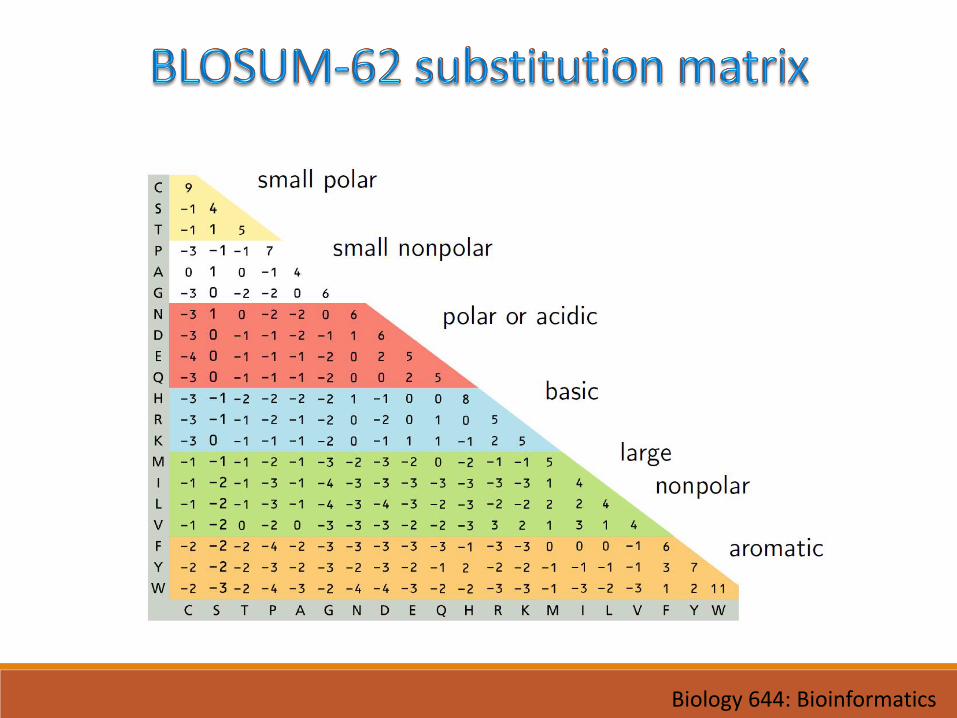
• Heniko & Heniko 1992, 1996, very widely used
• BLOSUMX identifies sequences that are X% similar to the query sequence• BLOSUM62 was created using sequences sharing no more than 62% identity
• Uses different training sets to observe actual mutations rates for sequences with different percent identity• No evolutionary model and extrapolation through matrix identification
• Scan databases for motifs via local alignments• a BLOCK (motif) = ungapped in local alignment• One protein can contain many blocks (motifs)• Make groups of blocks (motifs)• ~2000 blocks, >500 protein families
• Still assumes independence between positions

Biology 644: Bioinformatics
• Larger sample size than PAM
• Different training sets for each % identity
• Looked at closely and distantly related blocks explicitly without PAM assumptions
• Smaller numbers model more distantly related proteins:
• BLOSUM45 captures greater evolutionary distance than BLOSUM90
• Opposite of PAM matrices!

Biology 644: Bioinformatics
• Calculate substitution scores for sequences of X% identity
• Create sets of blocks of no-gap local alignments
• Very similar sequences are grouped and weighted
• Avoids bias due to repetition in the genome
• Count the substitutions
• Calculate log-odds scores for all pairs and round
Biology 644: Bioinformatics

Biology 644: Bioinformatics
• The PAM family• Matrices are based on global alignments of closely related proteins.• The PAM1 is the matrix calculated from comparisons of sequences with no more than
1% divergence• Other PAM matrices are extrapolated from PAM1 via matrix multiplication
• The BLOSUM family• Matrices are based on local alignments (blocks)• All BLOSUM matrices are based on observed alignments• (BLOSUM62 is a matrix calculated from comparisons of sequences with no less than 62%
divergence)
• Higher numbers in the PAM matrix naming scheme model greater evolutionary distance
• Lower numbers in the BLOSUM matrix naming scheme model greater evolutionary distance
• Similar Evolutionary Distances• PAM120 <----> BLOSUM80• PAM160 <----> BLOSUM62• PAM250 <----> BLOSUM45

Biology 644: Bioinformatics
• Two main strategies• Linear gap penalty: G(n) = a·n
• Penalize each gap the same whether contiguous or not
• Affine gap penalty: G(n) = O + E·n• More evolutionary sound - a series of k indels more commonly arise from
a single indel event rather than a series of k individual nucleotide indelevents
• Opening a gap costs more than extending it
• How large O and E should be depends on the Substitution Matrix used
• Gap Penalties are just as important as which Substitution Matrix to use• Too large: no gaps created• Too small: too many gaps created
ATT--ACG
ATTGAACG
ATT-A-CG
ATTGAACGis more likely than


















