
Bioinformatics in next generation sequencing...
6
Bioinformatics in next generation sequencing projects Rickard Sandberg Assistant Professor Department of Cell and Molecular Biology Karolinska Institutet March 2012 Thursday, March 15, 12 Once sequenced the problem becomes computational Computational analyses is the bottleneck • Rapid improvement in sequencing • Still need for customized analysis for most projects Thursday, March 15, 12 Overview of computational analyses Image analysis Base calling Primary Analyses: Mapping (Assembly) Data type specic analyses (e.g. peak calling, calculate expression) Custom project specic analyses ChIP-Seq peak calling RNA-Seq expression levels genome sequence assembled contig Thursday, March 15, 12 Preliminary Analyses Raw Image (TB) Platform-specific analysis using the vendors programs Sequences and Quality scores Text File (GB) Real Time Analysis Thursday, March 15, 12 Sequenced reads >EAS54_6_R1_2_1_413_324 CCCTTCTTGTCTTCAGCGTTTCTCC Fasta file: Fastq file: SOLiD csfasta file >1_39_146_F3 T22100200202311030112002022222002021 >1_39_194_F3 T11022322003020303320012223122202221 SOLiD, QV file >1_39_146_F3 14 6 21 27 5 18 6 15 22 27 18 17 14 18 26 15 24 19 18 18 8 20 17 12 20 6 14 13 23 6 11 12 7 13 4 >1_39_194_F3 26 27 16 27 23 22 23 25 22 10 5 21 4 17 20 26 26 17 25 27 23 25 14 24 26 4 4 4 4 4 4 4 4 4 14 G A A C T C T G C C T T T T T C A G T G A T G A G G A A A G G A G T T C T C T C T G G T C C C C A G a a a b ^ _ U _ a a [ U [ _ Z ] a ` W U _ ^ X ` G T ^ _ \ T M ^ ^ \ _ _ _ \ Z \ Y Q V V X U B B B B Read identifier Quality scores @HWI - EAS269:1:120:1786:18#0/1 +HWI - EAS269:1:120:1786:18#0/1 Thursday, March 15, 12 Phred Quality Score, Q Each base call has an estimate of the probability of being wrong (error probability, p) Q = -10 * log 10 (p) Phred Quality Score Probability of incorrect base call Base call accuracy 10 1 in 10 90 % 20 1 in 100 99 % 30 1 in 1000 99.9 % 40 1 in 10000 99.99 % 50 1 in 100000 99.999 % Thursday, March 15, 12
Transcript of Bioinformatics in next generation sequencing...

Bioinformatics in next generation sequencing projects
Rickard SandbergAssistant ProfessorDepartment of Cell and Molecular BiologyKarolinska Institutet
March 2012
Thursday, March 15, 12
Once sequenced the problembecomes computational
Computational analyses is the bottleneck• Rapid improvement in sequencing• Still need for customized analysis for most projects
Thursday, March 15, 12
Overview of computational analyses
Image analysisBase calling
Primary Analyses: Mapping(Assembly)
Data typespeci!c analyses(e.g. peak calling,
calculate expression)
Custom projectspeci!c analyses
ChIP-Seq peak calling
RNA-Seq expression levelsgenome sequence
assembled contig
Thursday, March 15, 12
Preliminary Analyses
Raw Image (TB)
Platform-specific analysis using the vendors programs
Sequences and Quality scoresText File (GB)
Real Time Analysis
Thursday, March 15, 12
Sequenced reads
>EAS54_6_R1_2_1_413_324CCCTTCTTGTCTTCAGCGTTTCTCC
Fasta file:
Fastq file:
SOLiD
csfasta file>1_39_146_F3T22100200202311030112002022222002021>1_39_194_F3T11022322003020303320012223122202221
SOLiD, QV file>1_39_146_F314 6 21 27 5 18 6 15 22 27 18 17 14 18 26 15 24 19 18 18 8 20 17 12 20 6 14 13 23 6 11 12 7 13 4 >1_39_194_F326 27 16 27 23 22 23 25 22 10 5 21 4 17 20 26 26 17 25 27 23 25 14 24 26 4 4 4 4 4 4 4 4 4 14
GAACTCTGCCTTTTTCAGTGATGAGGAAAGGAGTTCTCTCTGGTCCCCAG
aaab^_U_aa [U [ _Z ] a `WU_^X`GT^_ \ TM^ ^ \ ___ \ Z \ YQVVXUBBBB
Read identifier
Quality scores
@HWI - EAS269:1:120:1786:18#0/1
+HWI - EAS269:1:120:1786:18#0/1
Thursday, March 15, 12
Phred Quality Score, Q
Each base call has an estimate of the probability of being wrong (error probability, p)
Q = -10 * log10(p)
Phred Quality Score Probability of incorrect base call Base call accuracy10 1 in 10 90 %20 1 in 100 99 %30 1 in 1000 99.9 %40 1 in 10000 99.99 %50 1 in 100000 99.999 %
Thursday, March 15, 12
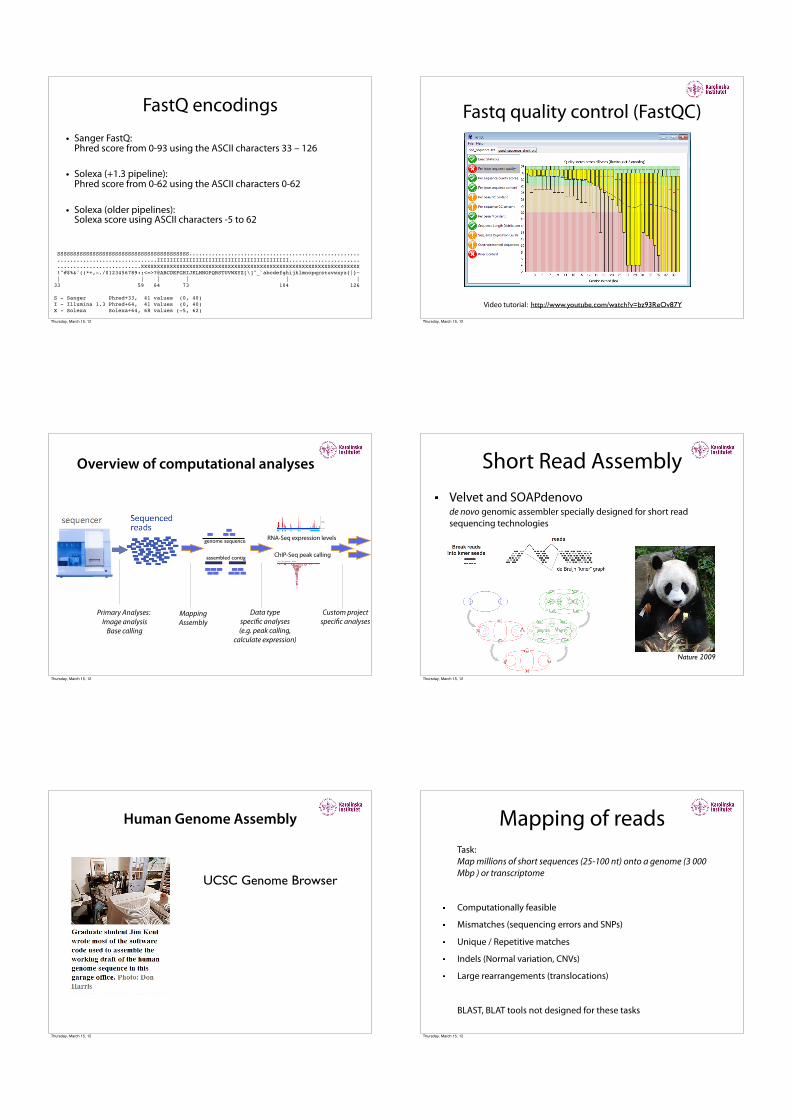
FastQ encodings
• Sanger FastQ: Phred score from 0-93 using the ASCII characters 33 – 126
• Solexa (+1.3 pipeline): Phred score from 0-62 using the ASCII characters 0-62
• Solexa (older pipelines): Solexa score using ASCII characters -5 to 62
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS..................................................... ...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII...................... ..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX !"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~ | | | | | | 33 59 64 73 104 126
S - Sanger Phred+33, 41 values (0, 40) I - Illumina 1.3 Phred+64, 41 values (0, 40) X - Solexa Solexa+64, 68 values (-5, 62)
Thursday, March 15, 12
Fastq quality control (FastQC)
http://www.youtube.com/watch?v=bz93ReOv87YVideo tutorial:
Thursday, March 15, 12
Overview of computational analyses
Image analysisBase calling
Primary Analyses: MappingAssembly
Data typespeci!c analyses(e.g. peak calling,
calculate expression)
Custom projectspeci!c analyses
ChIP-Seq peak calling
RNA-Seq expression levelsgenome sequence
assembled contig
Thursday, March 15, 12
Short Read AssemblyVelvet and SOAPdenovode novo genomic assembler specially designed for short read sequencing technologies
Nature 2009
Thursday, March 15, 12
Human Genome Assembly
UCSC Genome Browser
Thursday, March 15, 12
Mapping of readsTask: Map millions of short sequences (25-100 nt) onto a genome (3 000 Mbp ) or transcriptome
Computationally feasible
Mismatches (sequencing errors and SNPs)
Unique / Repetitive matches
Indels (Normal variation, CNVs)
Large rearrangements (translocations)
BLAST, BLAT tools not designed for these tasks
Thursday, March 15, 12

MAQ bowtie
Thursday, March 15, 12
Commonly used programs
Program Approach Comments
BowtieBurrow-Wheeler
Transformation (BWT) Illumina, (SOLiD), fast
MAQ Spaced Seed Indexing Illumina, (SOLiD), SNPs
BWA Burrow-Wheeler Transformation (BWT)
Illumina, (SOLiD), indels
NovoalignNeedleman-Wunch
AlignmentIllumina, indels, slower, free (single proc mode)
ZOOM Designed spaced seeds Illumina, fast, indels, not free
Mappers from Illumina (ELAND) and SOLiD (bioscope/mapreads)
http://en.wikipedia.org/wiki/List_of_sequence_alignment_software#Short-Read_Sequence_Alignment
Thursday, March 15, 12
Storing mapped Alignments
Formats for storing alignments should include:
genomic coordinates
mismatches, insertion, deletions etc.
quality information
Thursday, March 15, 12
Samtools
Sequence Alignment Map (SAM)
Generic Alignment format
Supports long and short reads
Human readable, !exible and compact
Emerging standard
h"p://samtools.sourceforge.net/
Li6H.*,6Handsaker6B.*,6Wysoker6A.,6Fennell6T.,6Ruan6J.,6Homer6N.,6Marth6G.,6Abecasis6G.,6Durbin6R.6and610006Genome6Project6Data6Processing6Subgroup6(2009)6The6Sequence6alignment/map6(SAM)6format6and6SAMtools.6BioinformaScs,625,62078W9.6[PMID:619505943]
Thursday, March 15, 12
SAM Example
16 chr Y 616000 255 22M731N28M* 0 0 ATTTCGACCATGATCATCGAACCTTCCCCTGGATCCACTTCCACGATCAC#9 ; -7 +2@4 : 2=20 - 14= : ><?< ; : BB? : 4<BB?ABBBBABCBBBBC=BB NM: i : 0XS: A:-
Bit field, where 16 means reverse strand
Start position
Alignment structure. Here: 22 aligned bases,then 731 bases intron, then 28 aligned bases
HWI - EAS269:1:114:1242:1582#0
Thursday, March 15, 12
CIGAR Format
M, match/mismatch
I, insertion
D, deletion
S, softclip
...
Ref: GCATTCAGATGCAGTACGC
Read: ccTCAG--GCAGTAgtg
Pos: 5
CIGAR: 2S4M3D6M3S
Thursday, March 15, 12

Samtools for SAM/BAM !les
Library and software package (C, Java)
Creating, sorting, indexing SAM & BAM
Visualizing alignments in command
SNP calling
Short indel detection
BAM (Binary representation of SAM) ~25% "le size reduction
Thursday, March 15, 12
Overview of computational analyses
Image analysisBase calling
Primary Analyses: MappingAssembly
Data typespeci!c analyses(e.g. peak calling,
calculate expression)
Custom projectspeci!c analyses
ChIP-Seq peak calling
RNA-Seq expression levelsgenome sequence
assembled contig
Thursday, March 15, 12
Visualization
Integrated Genome Viewer (Broad Inst.)
Custom tracks at UCSC Genome Browser
Thursday, March 15, 12
Visualization
Thursday, March 15, 12
Integrated Genome Viewer
Imports many mentioned formats (SAM, BAM, BED etc)
Excellent for visualization of RNA-Sequencing or ChIP-sequencing data
Can also download/visualize data from public or private servers
Thursday, March 15, 12
UCSC Genome Browser
Recently introduced new formats for e#cient viewing of large data sets:
- BedGraph
- BigWig
Add as custom tracks (slower)
Thursday, March 15, 12

Peak characteristics di"er with signal
Thursday, March 15, 12
Peak characteristics di"er with signal
H3K4me3: Sharp promoter peaksH3K36me3: Broad transcription elongation signal
Thursday, March 15, 12
Important !le formats
Sequences: FastQ
Aligned reads: SAM/BAM
Genome annotations: Bed, G$
Coverage: Wig, (Tdf )
http://genome.ucsc.edu/FAQ/FAQformat.html
Thursday, March 15, 12
BED format
chrom6W6The6name6of6the6chromosome6(e.g.6chr3,6chrY,6chr2_random)6or6scaffold6(e.g.6scaffold10671).
chromStart6W6The6starSng6posiSon6of6the6feature6in6the6chromosome6or6scaffold.6The6first6base6in6a6chromosome6is6numbered60.
chromEnd6W6The6ending6posiSon6of6the6feature6in6the6chromosome6or6scaffold.6The6chromEnd6base6is6not6included6in6the6display6of6the6feature.6
For6example,6the6first61006bases6of6a6chromosome6are6defined6as6chromStart=0,6chromEnd=100,6and6span6the6bases6numbered60W99.
http://genome.ucsc.edu/FAQ/FAQformat.html
track name=pairedReads description="Clone Paired Reads" useScore=1chr22 1000 5000
Thursday, March 15, 12
BED continued
strand - Defines the strand - either '+' or '-'.thickStart - The starting position at which the feature is drawn thickly (for example, the start codon in gene displays).thickEnd - The ending position at which the feature is drawn thickly (for example, the stop codon in gene displays).itemRgb - An RGB value of the form R,G,B (e.g. 255,0,0). If the track line itemRgb attribute is set to "On", this RBG value will determine the display color of the data contained in this BED line. NOTE: It is recommended that a simple color scheme (eight colors or less) be used with this attribute to avoid overwhelming the color resources of the Genome Browser and your Internet browser.blockCount - The number of blocks (exons) in the BED line.blockSizes - A comma-separated list of the block sizes. The number of items in this list should correspond to blockCount.blockStarts - A comma-separated list of block starts. All of the blockStart positions should be calculated relative to chromStart. The number of items in this list should correspond to blockCount.
track name=pairedReads description="Clone Paired Reads" useScore=1chr22 2000 6000 cloneB 900 - 2000 6000 0 2 433,399, 0,3601
Thursday, March 15, 12
Variable step Fixed step
variableStep chrom=chr2300701 12.5300702 12.5300703 12.5300704 12.5300705 12.5is equivalent to:variableStep chrom=chr2 span=5300701 12.5
fixedStep chrom=chr3 start=400601 step=100112233
WIG format
Wiggle format (WIG) allows the display of continuous-valued data in a track format
Thursday, March 15, 12

Data Repositories
Short Read Archive (fastq) [discontinued!]http://www.ncbi.nlm.nih.gov/sraEuropean Nucleotide Archive
Gene Expression Omnibus (bed, wig, fastq)http://www.ncbi.nlm.nih.gov/geo/
Thursday, March 15, 12
SEQAnswers, an active forum for discussions on next-generation sequencing methods and bioinformatics
http://seqanswers.com/Thursday, March 15, 12
Thursday, March 15, 12


















