
Bioinformatics - Göteborgs universitetbio.lundberg.gu.se/courses/vt13/sequencing_exome_MD...coding...
60
Bioinformatics Core Facility Next generation sequencing Genomics and Bioinformatics, VT13 Marcela Davila 2013-03-15
Transcript of Bioinformatics - Göteborgs universitetbio.lundberg.gu.se/courses/vt13/sequencing_exome_MD...coding...

Bioinformatics Core Facility
Next generation sequencing Genomics and Bioinformatics, VT13
Marcela Davila
2013-03-15

Bioinformatics Core Facility
Overview
• Next generation sequencing
• Exome sequencing
• Bioinformatics of exome sequencing

Bioinformatics Core Facility
First generation (great cost, intense human effort) 1954 – Sequencing by degradation (Whitfeld PR) 1975 – Chain termination method (Sanger & Coulson) 1977 – Chemical modification (Maxam and Gilbert)
NGS methods
DNA template
Laser beam
Chromatogram
Capillar electrophoresis
Dye-labeled terminator

Bioinformatics Core Facility
Second generation (sincronyzed washing/scanning) SBS – Illumina Pyrosequencing – Roche SBL – AB SOLiD
NGS methods

Bioinformatics Core Facility
1. DNA library preparation (ligation of adapters) 2. Amplification (ePCR, bridge PCR) 3. Sequencing reaction 4. Imaging 5. Decoding
Cyclic array sequencing

Bioinformatics Core Facility
1. DNA library preparation (ligation of adapters) 2. Amplification (ePCR, bridge PCR) 3. Sequencing reaction 4. Imaging 5. Decoding
Cyclic array sequencing

Bioinformatics Core Facility
1. DNA library preparation (ligation of adapters) 2. Amplification (ePCR, bridge PCR) 3. Sequencing reaction 4. Imaging 5. Decoding
Cyclic array sequencing

Bioinformatics Core Facility
SeqBySynthesis - Illumina

Bioinformatics Core Facility
Pyrogram
Pyrosequencing - Roche

Bioinformatics Core Facility
First round
Second round
SeqByLigation - SOLiD

Bioinformatics Core Facility
Color space
Decoded sequence
Base space sequence
SeqByLigation - SOLiD

Bioinformatics Core Facility
Third generation (increase sequencing speed, high throughput, no optics) Semiconductor: Ion Torrent SBS-single molecule: Helicos SBS-single molecule-real time: Pacific Biosciences SBH/SBL- Complete Genomics FRET: VisiGen Protein nanopores: Oxford Nanopore TEM: Halcyon Molecular and ZS Genetics Transistor mediated: IBM STM: Reveo
NGS methods

Bioinformatics Core Facility
IonSensitiveFieldEffectTransistors - Ion Torrent

Bioinformatics Core Facility
Single Molecule Real Time - PacBio
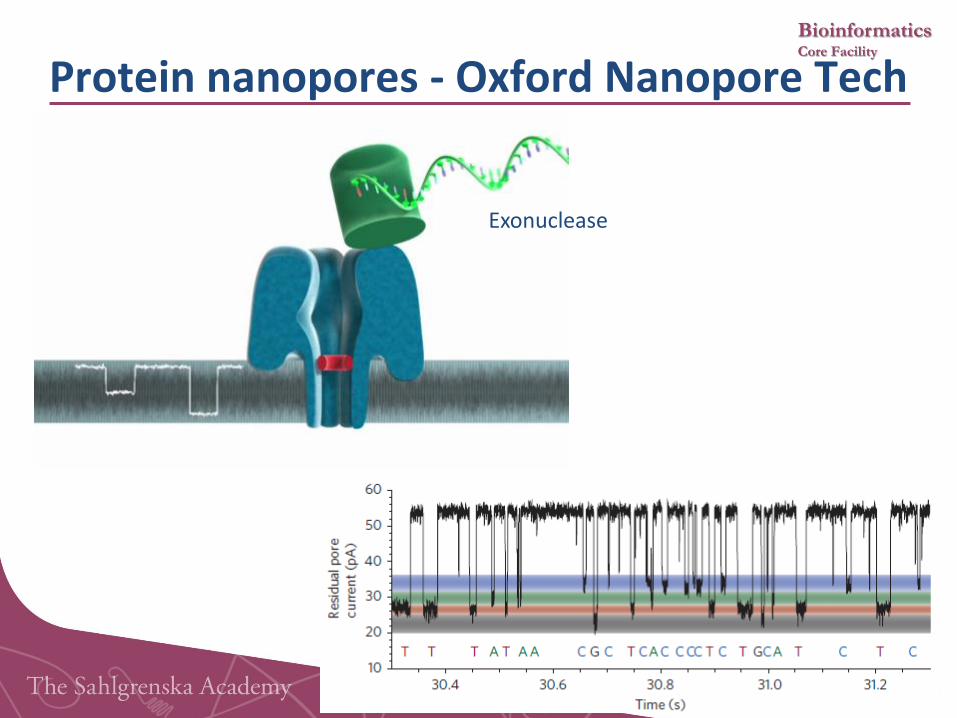
Bioinformatics Core Facility
Exonuclease
Protein nanopores - Oxford Nanopore Tech

Bioinformatics Core Facility
Modifications of a single gene over 10,000 of human diseases (½ have a gene associated)
http://www.who.int/
DISEASE GENE MUTATION
Thalassaemia HBB ∆ frameshift
Sickle cell anemia HBB G6V
Cystic Fibrosis CFTR G542X …
Fragile X syndrome FMR1 CGG expansion
Huntington’s HTT CAG +36 repeats
Tay sachs HEXA 65 single base mutations
14 splice site lesions
10 deletions
2 insertions
Monogenic diseases

Bioinformatics Core Facility
AAGCCTA
AAGCTTA
Human genome 3 billion bps
3 million differences
(0.1%)
AAGCCTA
AAG--TA
AAG--TA
AAGCTTA
Ability to influence: Disease risk Drug efficacy and side-effects Heritable phenotypes
SNPs
Lactose intolerance rs4988235 triggers obesity rs9939609 coronary heart disease rs1333049

Bioinformatics Core Facility
85% of the disease-causing mutations are located in protein coding regions
UAG GGU ACU
* G T
Splice sites/branch site UTRs Coding regions
Genome (3GB) vs exome (50 MB)
Exome sequencing

Bioinformatics Core Facility
Illumina sequencing workflow
Library prep Cluster generation
Sequencing, imaging, data generation

Bioinformatics Core Facility
Genomic DNA (2-3ug)
Library prep
Biotin probes
Streptavidin beads
150-200 bp 250-275 bp
300-400 bp
Shearing
A overhang
End repair
Adapter ligation
PCR amp. Denaturation Hybridization Capture Index ligation

Bioinformatics Core Facility
Amplification
Template hybridization and extension
Primer hybridization
Cluster generation - cBOT

Bioinformatics Core Facility
Sequencing by Synthesis

Bioinformatics Core Facility
Different recepies
Single end (SE)
Paired-end (PE)
Mate-pair (MP)
200-500 bp
2-5 Kb
R2
R1
R1
R1
R2

Bioinformatics Core Facility
Fastq format
R1 R2
@HWI-H200:53:D08U2ACXX:5:1101:1231:2012 1:N:0:
GCATTTTAGTAGAACCAGNCATTTCCCCCNACNTCNNTNCGNNANNNNTAA
+
@CCFFFFFHFFHHJJJJJ#3<FGIJJJJJ#1?###################
@HWI-H200:53:D08U2ACXX:5:1101:1184:2013 1:N:0:
TATATTTAATGTACTTTCNTATTTTATATNCANTATNTNATANANNNNTTG
+
CC@FFFFFHHHFFFFHIG#3AFGIIIHIJ#2A#1:C###############
@HWI-H200:53:D08U2ACXX:5:1101:1151:2035 1:N:0:
TTTTGCCTTGTTGCCCAGGTTGGTCTCGAACTCCTGGGCTCAAGGGATATG
+
@CCFFFFFHHHHHJJJGIGJJIIBHHHHIIGBGHGCHIIIHHGIGIJGHIF
@HWI-H200:53:D08U2ACXX:5:1101:1248:2055 1:N:0:
CAGGAACAGAATGAATGAGCGAAACAAATTCCCCTTGAGCTTCACTTGTTG
+
CCCFFFFFHHHHH######################################
@HWI-H200:53:D08U2ACXX:5:1101:1235:2080 1:N:0:
ATGGTCTATTAAGTATGCAATAGTATTTTGTCTAAAACAATAATGTACATA
+
@@@FADDFHHHGHFHHGEIHIJGAIFHIIIIJIHIIJHIJIJJJHFHDHII
@HWI-H200:53:D08U2ACXX:5:1101:1165:2081 1:N:0:
ATAACAATGACAATAGAATTTGGGGACTCAGGAGGAAAGGGAGGGAAGCGG
+
CCCFFFFFGHHHHJGHIIIJJJJJJJJIIIJJIGGIJJJJGIIGIIIIIJJ
@HWI-H200:53:D08U2ACXX:5:1101:1231:2012 2:N:0:
TACTNNTANNTNCAGANCAGTTTAAATAAATAAAACATNCACCAGTATGTA
+
@BCF##22##2#2<CG#2AEFGIHJIIJJJFIJJJJJJ#0?GGGBFHIJGH
@HWI-H200:53:D08U2ACXX:5:1101:1184:2013 2:N:0:
ACATCAAAGNTNAAAGNTCACAAACTATATATTATATANTGTACATAAAAT
+
B@@FFF22GG2j3<CG#3AFHIJJJGJJJJJJJJIJJJ#0?FGHJJJJGJG
@HWI-H200:53:D08U2ACXX:5:1101:1151:2035 2:N:0:
CAAACTAACCANGCGGACTTCATTGCTTTTAGAGGACACAATTAATTCTCT
+
CCCFFFFFHHH#2<CGIJBHJJIJJGIGJIIFGGIJJJIIJHIJIGIJIJI
@HWI-H200:53:D08U2ACXX:5:1101:1248:2055 2:N:0:
TATACAATCAANGCACAATCTATTAGAATGGGAAGAGACCCTGGAGATAAT
+
CCCFFFFFHHH#2AFHIJIHHHJJJJJJIJJJJJJJJJJJJIJJHEGHGG<
@HWI-H200:53:D08U2ACXX:5:1101:1235:2080 2:N:0:
AATCCCAACACTTTGGGAGGCTGAGGTGGGTGGATCACTTGGGGTCAGGAG
+
B@?DFBFFHHHHHIJJIJIJJJJIGI:DGI?F@GBFGIIGAGIIBF>HGIH
@HWI-H200:53:D08U2ACXX:5:1101:1165:2081 2:N:0:
GCTGTGTTAGCTTCTTTGTCCTATTGAAATGCAAAGATAGGCTGACTAACT
+
CC@FFFFFHHHHHJJJJI?CHFHGJJJJJIIJJJJIIJJGFHIJJJJJJJE
Single end (SE) Paired-end (PE)

Bioinformatics Core Facility
1) @SEQ_ID instrument:run:flowcell:lane:tile:x:y pair:fail:control:index 2) sequence 3) marker 4) quality
1) @HWI-H200:53:D08U2ACXX:5:1101:1231:2012 1:N:0:
2) GCATTTTAGTAGAACCAGNCATTTCCCCCNACNTCNNTNCGNNANNNNTAA
3) +
4) @CCFFFFFHFFHHJJJJJ#3<FGIJJJJJ#1?###################
Fastq format
31 37 39 18 16 2
Bioinformatics Core Facility
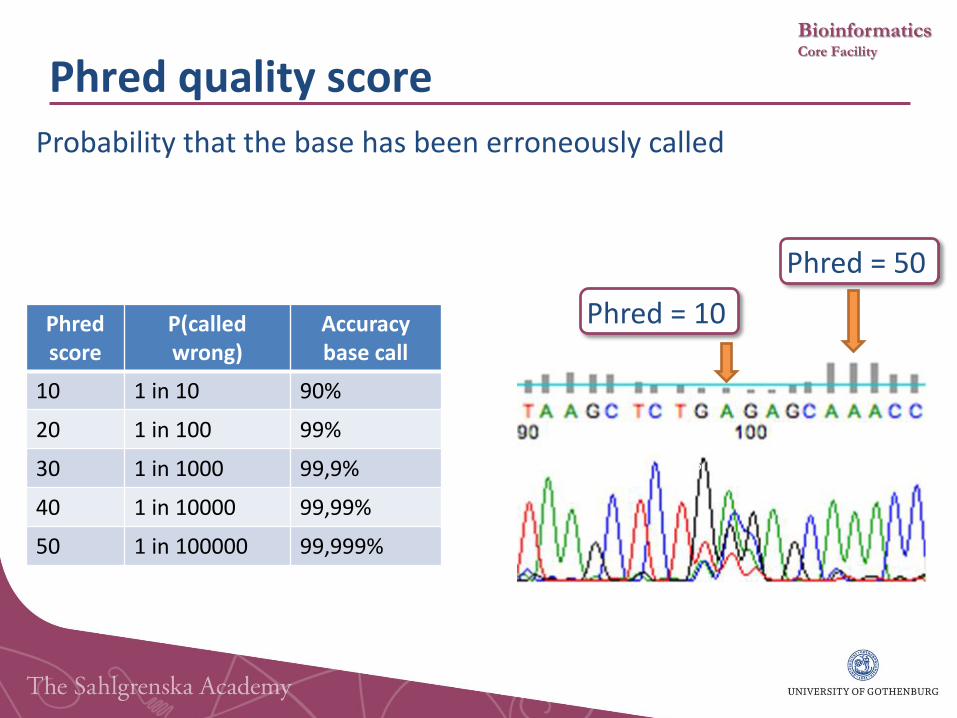
Phred quality score
LIMS
Phred = 50
Probability that the base has been erroneously called
Phred score
P(called wrong)
Accuracy base call
10 1 in 10 90%
20 1 in 100 99%
30 1 in 1000 99,9%
40 1 in 10000 99,99%
50 1 in 100000 99,999%
Phred = 10

Bioinformatics Core Facility
First stage: Quality and Mapping
Quality Check Quality Filter
Mapping to reference genome Realignment and recalibration
SNV detection Peak detection Transcript abundance estimation
Exome-seq RNA-seq ChIP-Seq

Bioinformatics Core Facility
Quality check
Application which reads raw sequence data from high throughput sequencers and runs a set of quality checks to produce a report which allows you to quickly assess the overall quality of your run
FastQC, PrinSeq

Bioinformatics Core Facility
Per base sequence quality

Bioinformatics Core Facility
Per base sequence content
RNA-seq
bad adapter clipping
Bioinformatics Core Facility
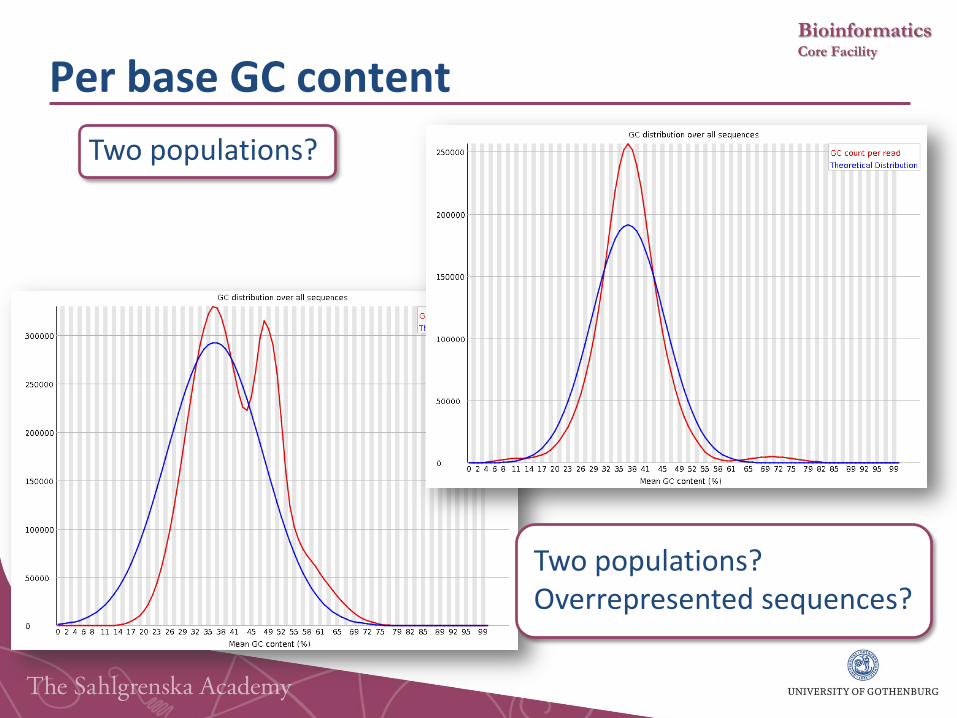
Per base GC content
Two populations?
Two populations? Overrepresented sequences?

Bioinformatics Core Facility
Quality Check – more stats
Per base N content Sequence duplication levels
Overrepresented sequences

Bioinformatics Core Facility
Contamination
Good Bad
Screen a library of sequences in FastQ format against a set of sequence databases so you can see if the composition of the library matches with what you expect.
%m
app
ed
FastQScreen, BLAST

Bioinformatics Core Facility
Quality filter
@HWI-H200:53:D08U2ACXX:5:1101:1231:2012 1:N:0:
GCATTTTAGTAGAACCAGNCATTTCCCCCNACNTCNNTNCGNNANNNNTAA
+
@CCFFFFFHFFHHJJJJJ#3<FGIJJJJJ#1?###################
X nts
Low quality
Ambiguous bases
A collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing.

Bioinformatics Core Facility
Per base sequence quality
Before trimming After trimming

Bioinformatics Core Facility
Mapping
CTACTACATCGATCTACGCAGCTACTACACGTGCTGGGACGC REF
TCGATCGACG CACGTGCTGG CTACT CGACGCAGATACT ATCGAGCGAC TGCTGGAACGC TACATCGATC CACGTGCTGGAAC CTACTACA TCGACGC CTACTACA GGAACGC
READS
WHERE to place the reads? a) Unique reads b) Everywhere possible c) Choose one randomly d) Use pair-end data
HOW to place the reads? a) Ungapped b) Gapped
Bfast, BioScope, Bowtie, BWA CLC bio, CloudBurst, Eland/Eland2, GenomeMapper, GnuMap, Karma, MAQ, MOM, Mosaik …

Bioinformatics Core Facility
Recalibration and realignment
Differenciate between polymorphisms and sequencing errors Correct alignments due to the presence of indels

Bioinformatics Core Facility
SAM/BAM format
SAM (Sequence Alignment/Map) http://samtools.sourceforge.net/SAM1.pdf BAM (Binary Alignment/Map) compression, allows random access
HWI-H200:53:D08U2ACXX:6:1108:18555:16623 99 chr1 10001 60 45M6S = 10174 224
TAACCCTAACCCTAACCCTAACCCTAACCCTAACCCT
AACCCTAAAGATCG @?@DDDBDAH??FHDGFFFHIIIGDGEHHI<ABHICHIEHCDD3BDEDGEC MD:Z:45 RG:Z:1 XG:i:0 AM:i:0 NM:i:0
SM:i:0 XM:i:0 XO:i:0 XT:A:M
HWI-H200:53:D08U2ACXX:6:1101:9568:123823 99 chr1 10003 11 1S46M1S = 10204 252
GACCCTGACCCTGACCCTAACCCTAACCCTAACCCTA
ACCCCAAACCC @@CFBDFFDFHHFGIIEHGGGD@GGHDGGFHGGEHEGCGHGGHGEHGC MD:Z:5A5A28T2C2 RG:Z:1 XG:i:0
AM:i:11 NM:i:4 SM:i:11 XM:i:4 XO:i:0 XT:A:M
HWI-H200:53:D08U2ACXX:6:1302:17187:33007 97 chr1 10003 0 51M chrM 430 0
ACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAA
CCCTAACCCTAACC CCCFFFFFHHHHHJJJJJJIIIIJJJJJIIJJJJJJJJJJJJJJJJJIIGI X0:i:513 MD:Z:51 RG:Z:1 XG:i:0
AM:i:0 NM:i:0 SM:i:0 XM:i:0 XO:i:0 XT:A:R
HWI-H200:53:D08U2ACXX:6:1104:2930:78353 177 chr1 10004 0 51M chr22 38431286 0
CCCTAACCCTAACCCTAACCCTAACCCTAACCCTAAC
CCTAACCCTAACCC IIGAF?JJIGADJIGGD?GHGEEEIHGCCGIIHIHHIHFDHDHDDDDB@@B X0:i:515 MD:Z:51 RG:Z:1 XG:i:0
AM:i:0 NM:i:0 SM:i:0 XM:i:0 XO:i:0 XT:A:R
HWI-H200:53:D08U2ACXX:6:1205:3665:10423 99 chr1 10054 0 51M = 10366 363
CTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTA
ACCCTA CCCFFFFFDBHHHIGEGEHEHIJJIHIFGIIGEHIGH9FGHHIIJJGGI=C X0:i:502 MD:Z:51 RG:Z:1 XG:i:0 AM:i:0
NM:i:0 SM:i:0 XM:i:0 XO:i:0 XT:A:R
HWI-H200:53:D08U2ACXX:6:1101:4778:107011 163 chr1 10056 0 51M = 10355 350
AACCCTAACCCTAACCCTAACCCTAACCCTAACCCTA
ACCCTAACCCTAAC CCCFFFFFHHHHGJJJJJJJJJJIJJIJJIIHGIJECEHIJ;FGEIIEHCA X0:i:508 MD:Z:51 RG:Z:1 XG:i:0
AM:i:0 NM:i:0 SM:i:0 XM:i:0 XO:i:0 XT:A:R
Query name HWI-H200:53:D08U2ACXX:6:1108:18555:16623
Flag 99
Reference name chr1
Leftmost position 10001
Mapping quality 60
CIGAR string 45M6S
Mate reference =
Mate position 10174
Insert size 224
Query sequence TAACCCTAACCCTAACCCTAACCCTAACCCTAACCCT..
Quality @?@DDDBDAH??FHDGFFFHIIIGDGEHHI<ABHICH..
Optional fields MD:Z:45 RG:Z:1 XG:i:0 AM:i:0 NM:i:0 SM:i:0
XM:i:0 XO:i:0 XT:A:M

Bioinformatics Core Facility
SAM/BAM format
Flag 99
CIGAR string 45M6S
Optional fields MD:Z:45 RG:Z:1 XG:i:0 AM:i:0 NM:i:0 SM:i:0
XM:i:0 XO:i:0 XT:A:M
Flags: http://picard.sourceforge.net/
explain-flags.html
R2 R1

Bioinformatics Core Facility
SAM/BAM format
Flag 99
CIGAR string 45M6S
Optional fields MD:Z:45 RG:Z:1 XG:i:0 AM:i:0 NM:i:0 SM:i:0
XM:i:0 XO:i:0 XT:A:M
6M 2I 4M 1D 2M
3S 11M
6M 14N 8M
14M with NM=1

Bioinformatics Core Facility
SAM/BAM format
Flag 99
CIGAR string 45M6S
Optional fields MD:Z:45 RG:Z:1 XG:i:0 AM:i:0 NM:i:0 SM:i:0
XM:i:0 XO:i:0 XT:A:M

Bioinformatics Core Facility
Second stage: process of the data
Quality Check Quality Filter
Mapping to reference genome Realignment and recalibration
SNV detection Peak detection Transcript abundance estimation
Exome-seq RNA-seq ChIP-Seq

Bioinformatics Core Facility
Different pipelines
RNA-seq
ChIP-Seq Exome-seq
Variant calling Annotation Custom filtering of variants
Transcripts assembly Comparison to annotation Detection of: Diff. Exp. genes/transc. Novel transcripts
Peak calling Enriched regions Diff. Profile analysis Motif discovery Gene set analysis Relation to gene structure
Quality Check Quality Filter
Mapping to reference genome Realignment and recalibration

Bioinformatics Core Facility
Variant calling
CTACTACATCGATCTACGCAGCTACTACACGTGCTGGGACGC REF
TCGATCGACG CACGTGCTGG CTACT CGACGCAGATACT ATCGAGCGAC TGCTGGAACGC TACATCGATC CACGTGCTGGAAC CTACTACA TCGACGC CTACTACA
READS
Is it a variant allele? What is the most likely genotype?
SOAP2, samtools, GATK, Beagle, CRISP, Dindel, FreeBayes, SeqEM, VarScans
P(GG|D) = 0.06 P(GT|D) = 0.94 P(TT|D) = 3 × 10−11

Bioinformatics Core Facility
VCF format Variant call format http://www.1000genomes.org/node/101

Bioinformatics Core Facility
VCF format - Header

Bioinformatics Core Facility
VCF format - Body

Bioinformatics Core Facility
Variant annotation
CTACTACATCGATCTACGCAGCTACTACACGTGCTGGGACGC REF
TCGATCGACG CACGTGCTGG CTACT CGACGCAGATACT ATCGAGCGAC TGCTGGAACGC TACATCGATC CACGTGCTGGAAC CTACTACA TCGACGC CTACTACA
READS
Annovar, SIFT, PP2, dbSNP, GO, KEGG, OMIM
In which gene is it located? Name, Description, OMIM, Pathway, GO, Expression profiles . . .
Where in the gene is it located? Intron, exon, UTR, intergenic region, splice site
Is there any AA change? GAA -> GAG = E->E GTT -> CTT = V->L TGG -> TGA = W->X TGA -> CGA = X->R
What impact does the AA change have? Damaging, benign
Is it a known SNP?

Bioinformatics Core Facility
SIFT: aminoacid change impact

Bioinformatics Core Facility
Variant list (TSV format)

Bioinformatics Core Facility
Variant list (TSV format)

Bioinformatics Core Facility
On target coverage
S1 S2 S3
Gene1 100 200 50
Gene2 50 0 50
Gene3 50 0 55
Gene4 10 10 55
Coverage 52.5X 52.5X 52.5X

Bioinformatics Core Facility
IGV – Integrative Genome Viewer
reads
gene
coverage
location genome
My VCF
My BAM

Bioinformatics Core Facility
UCSC browser
Variation track
gene variants
My VCF
My BAM

Bioinformatics Core Facility
Third stage: Making sense of the data
Exome sequencing
cases
Coding variants
Controls Genetic variation DBs
Disease model Disease knowledge
Candidate genes
Family filters
Your real work begins…

Bioinformatics Core Facility
Structural variants Genomic rearrangements that affect >50 bp of sequence, including deletions, novel insertions, inversions, mobile-element transpositions, duplications and translocations
VariationHunter, BreakDancer, MoDil, MoGul, HYDRA, Corona, SPANNER, Genome STRiP, FusionMap, Tophat-fusion

Bioinformatics Core Facility
Structural variants
VariationHunter, BreakDancer, MoDil, MoGul, HYDRA, Corona, SPANNER, Genome STRiP, FusionMap, Tophat-fusion
#Chr1 Pos1 Orientation1 Chr2 Pos2 Orientation2 Type Size Score num_Reads num_Reads
_lib
Allele_
Freq
chr1 155618385 0+15- chr1 155733377 15+0- ITX 114519 99 15 A.bam|15 -2.96
chr2 89104418 0+39- chr2 96517634 0+40- INV 7412990 99 39 A.bam|39 0.38
chr1 143210211 16+14- chr4 49504867 16+0- CTX -220 99 14 A.bam|14 7.81
chr4 84221717 14+0- chr4 84222031 0+15- DEL 385 99 14 A.bam|14 -2.84
chr5 21497327 12+2- chr5 34197428 18+0- INV 12699904 99 12 A.bam|12 0.46
chr2 230045514 19+112- chr5 71146737 1+116- CTX -220 99 112 A.bam|112 3.56

Bioinformatics Core Facility
Copy Number Variants
ExomeCNV, controlFreec, aCGH, CNVnator, CNVseq, mrsfast
CHR START END ECN ALTERATION
7 49300000 49400000 3 gain
7 53950000 54000000 4 gain
7 57950000 58050000 1 loss
7 58050000 58100000 4 gain
7 79000000 79050000 3 gain
8 99700000 122450000 3 gain
8 122450000 122500000 6 gain
8 122500000 129900000 3 gain
8 129900000 129950000 5 gain
8 129950000 146364022 3 gain
Change the number of base pairs in the genome. fluorescent in situ hybridization (FISH), spectral karyotyping array comparative genomic hybridization (ACGH), SNP arrays

Bioinformatics Core Facility
Bioinformatics Core Facility

Bioinformatics Core Facility
NGS analysis Phylogeny
Pipelines and algorithms Gene and protein annotation
DE analysis
GWAS
Expression analysis
Contact us
to include
YOUR analysis!!
Today we have a focus on


















