
MALAYSIAN GUIDELINES FOR BIOAVAILABILITY AND BIOEQUIVALENCE STUDIES
Upload
sagar-joshiCategory
view
164download
2
Basic PK considerations

Q1)At what point does absorption and elimination begin?
Q2) Why is the graph skewed to the left? Why is it not symmetrical or skewed to the left?
Q3)Why does the graph have a long tail on the right side?

Rate of absorption is very critical!
The graph shows absorption and elimination of same drug but with different doses taken orally

Q4) Why don’ t the absorption phase of the three lines match since the same drug has same absorption mechanism ?
Q5) After about 12 hrs in the graph, why does the one with slowest absorption rate(blue line) also have slowest elimination rate whereas one with the highest absorption rate(red line) also have highest elimination rate (ie blue is above red on the left side)?

Important parameters in bioavailability studies
• Time to Peak (Tmax): the time after admin that it takes for the drug to reach Cmax. This is an indication of the rate of absorption.
• Max. conc. Attained (Cmax): assesses whether conc. is in therapeutic range
• Area Under the Curve (AUC): Total drug abs. - reflects changes in distr. metabolism & excret
• Onset of Action: The time required to reach the MEC. following drug
admin.
• Duration of Action: The time period in which the plasma conc. Exceeds MEC.
• Intensity: the difference in the MEC and Cmax.

Important parameters in bioavailability studies(cont’d)
• Half-life(t1/2): Time taken by body to reduce current Cmax by 50%
(same as time taken to achieve 50 % increase . At 5 half live period 97% of ANY drug will be eliminated regardless of the initial conc.
• Minimum Effective Concentration (MEC): minimum
plasma concerntration of drug required to produce therapeutic effect.
• Minimum Toxic Concentration (MTC)/Maximum Safe Concentration (MSC): The concentration of drug in plasma
above which toxic effects are seen
• Therapeutic Range: Plasma conc. range bet. MEC and MTC.
• Therapeutic index : The ratio of MSC to MEC

Therapeutic range of some drugs
Drug Disease Therapeutic range
Digoxin Congestive Heart failure 0.0005 – 0.002 mcg/ml(very narrow)
Gentamicin Infections 1-10 mcg/ml
Lidocaine Arrhythmias 1-6 mcg/ml
Phenytoin Epillepsy 10-20 mcg/ml
Propanolol Angina 0.02-0.2 mcg/ml
Salicyclic acid Aches and pain 20-100 mcg/ml
Thepphylline Asthma 6-20 mcg/ml
Bramhankar book

Half life of some drug
Drug(use) t1/2 Drug t1/2
Adenosine <10 secondsBuprenorphine(drug addiction)
16–72 hours
Norepinephrine 2 minutes Clonazepam 18–50 hours
Oxaliplatin(chemotherapy)
14 minutes Diazepam 20–100 hours
Salbutamol 1.6 hours Flurazepam 0.8–4.2 days
Zaleplon(sedative hypnotic)
1–2 hoursDonepezil(Alzheimer's)
70 hours (approx.)
Morphine(analgesic)
2–3 hoursFluoxetine(antidepressant)
4–6 days
Methadone(analgesic)
15 hours to 3 days, in rare cases up to 8 days
Dutasteride(enlarged prostate) 5 weeks
Phenytoin( anticonvulsant)
12–42 hoursBedaquiline (2012 MDRT) 5.5 months
wikipedia

• If half life is 1 hr, it does not mean that in 2 hrs ie 2 half life period 100% drug is eliminated but that 75% has been reduced• Similarly 3 half live means 87.5 % of drug has been removed• Steady state will be achieved after 5 half-lives when 96.875% drug iseliminated
• 7th t1/2 99.218%, 10th t1/299.90, 14th t1/2 99.994

Area Under the Curve
AUC2-3 = Cp2 + Cp3 x (t3 - t2)2
AUC 0-∞ = AUC 0-t + Clast /k Clast is the smallest measurable plasmaconc. And k is elimination rate constant


ParacetamolGeneric name – Paracetamol
Brand name – Tylenol, Panadol, Herron, Parasel (Indian), Niko (Nepali)Original innovation as a 500mg tablet branded Tylenol
• All generic brand names have the same 500mg Paracetamol as in the original brand Tylenol.
• The rest of the excipient can be different. However Tylenol is an effort involving 15or more years of research
and hundreds of million dollars invested in R&D involving thousands of people and patients while generic products don’t even undergo any human trials?
• However they are very expensive for the same reason and generic version are cheap
• 20 tab Tylenol = 491 Rs , 20 tab Niko = 50 Rs!!!!!!!!!!!!!!!!!!!!!• The question then comes should we continue to pay high cost for original
drugs or are the cheap generic drugs as good as the original?• Another question is although the clinical trials occurred with the tablet
there is also suspension and solution of paracetamol that haven’t gone through the clinical trials. So on what basis have they been allowed to be in market?

The cost of original drug is very high becausecompany invests lots of time and money

Time vs compounds in a general drug discovery process

Generic drug• A generic drug is a pharmaceutical product, usually
intended to be interchangeable with an innovator product, that is marketed only after patent(exclusive rights to sell) expiry of innovator drug thus not requiring a license from the innovator company and. The brand name of generic name can’t be same as innovator ie Paracetamol made in nepal can’t be named tylenol .
• Generic drug can have differences in color, shape, excipient but not in therapeutic efficacy
• in October 2010 in the UK,generic simvastatin (a cholesterol-lowering medicine) cost £1.12 for a pack of 28 (20mg) compared with approximately £30 for a pack of28 (20mg) of the originator product

A generic Paracetamol formulation
Ingredients 2-7 can be switched with other similar functioning chemicalswhich can influence therapeutic efficacy of the dose

There are many choices for various excipient classes from which many formulation of same drug in same
strength can be madeExcipient
Fillers/Diluent(Allows making a sizable tab if drugcontent is very less)
Lactose,lactose anhydrous, lactose spray dried, directly compressible starch, hydrolyzed starch, MCC, other cellulose derivatives, dibasic calcium phosphate dihydrate, mannitol, sorbitol, sucrose, calcium sulfate dehydrate, dextrose.
Binders(allow drug power to be compressed into a solid tablet)
cellulose, methyl cellulose, polyvinyl pyrrolidine, PEG, gelatin, PVP, HPMC, PEG, sucrose, starch
Disintegrating agent(breaks hard solid tab to free the power drug from tablet)
starch derivatives, clay, cellulose, alginates, PVP(povidone), cross linked Na CMCVeegum HV, Betonite PVP, CMC ,crospovidone, Sodium bicarbonate
Lubricant Stearic acid, stearic acid salt, stearic acid derivatives, talc, PEG, surfactants,waxes, Calcium stearate and magnesium stearatepoly ethylene glycol
Preservative Potassium sorbate,

Change in formulation can modify therapeutic effect
• Digoxin: Doctors in Israel noticed 15 cases of digoxin toxicity between Oct-Dec 1975. It was found that the local manufacturer had changed the formulation to improve dissolution to cause two-fold increase in drug absroption of the new formulation. Digoxin has such a narrow therapeutic range that this increase put the drug in toxic region.
• Phenytoin: In 1969, the tablet diluents of phenytoin was changed from calcium sulfate to lactose which increased drug absorption that caused many cases of toxicity

Bioavailability and Bioequivalence

Bioavailability (BA) is the rate and extent of absorption of unchanged drug from its dosage form into the systemic circulation
Bioequivalence (BE) is the condition wherein the bioavailability of two drug products, containing same amount of active drug and same route, is statistically similar. Two bioequivalent products are therapeutically identical
Bioavailable fraction (F), refers to the fraction of administered dose that enters the systemic circulation
F = Bioavailable dose
Administered dose
F is always expressed in %
Bioavailability & Bioequivalence focus on release of drug substance from its dosage form & subsequent absorption in circulation

Absolute Bioavailability of Nimodipine for different routes:
Q6) So is there bioquivalence between the three results?

This is the end result of a single dose BE study that shows individual BA of both drug products. Q7) Is the generic drug and innovator drug bioequivalent?

Objectives of BA Studies
• Development of suitable dosage form for a New Drug Entity
•Comparison of availability of a drug substance from different form or same dosage form produced by different manufacturers
• Determination of influence of excipients, patient related factors & possible interactions with other drugs
• Development of new drug formulations of existing drugs
• Control of quality of drug products, influence of → processing factors, storage & stability
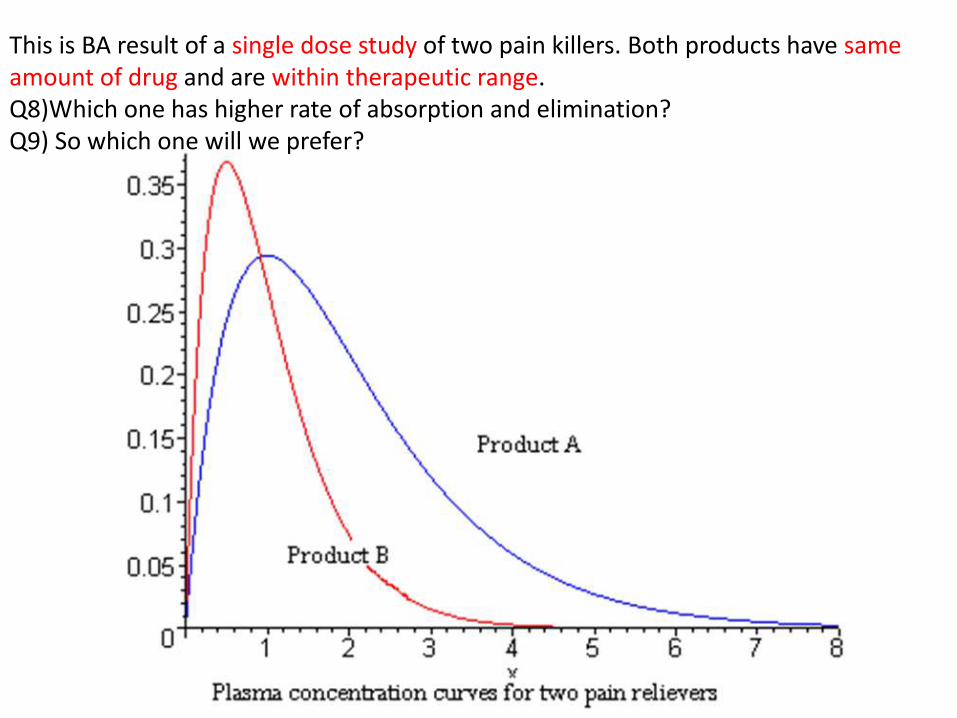
This is BA result of a single dose study of two pain killers. Both products have sameamount of drug and are within therapeutic range.Q8)Which one has higher rate of absorption and elimination?Q9) So which one will we prefer?

Considerations inBA study designIn-vivo Study
• In-vivo means done inside a full living being as opposed to an isolated cell or tissue or machines. Things to consider are
• Bioavailability-Absolute vs Relative
• Healthy people vs Patient
• Single dose vs multiple dose

Bioavailability-Absolute vs Relative
Absolute Bioavailability The fraction of the oral administered dose which is
absorbed intact into the systemic circulation relative to equivalent intravenous dose
F = (AUCtab/AUCIV) x 100%For drugs administered intravenously, bioavailability is
100%
Relative Bioavailability A measure of the fraction of a given drug that is absorbed
intact into the systemic circulation from a dosage form relative to a recognized standard dosage form of that drug.
F = (AUCtest/AUCstd,) x 100%

Why we use IV as standard/ don’t use oral solution as standard?
• Using IV solution allows comparison of BA from oral and parental routes and decide if oral route is appropriate but oral solution doesn’t
• Oral solution limits pharmacokinetic model to one compartment model; cannot apply two compartment model as with IV injection which is more realistic (one compartment means drug going to blood only, two compartment means drug going to blood and then to organs, muscles and fat too)---------------why does this happen Q10)?
• IV route bypasses Absorption and 1st pass metabolism while oral don’t. Thus comparing oral and IV allows to quantify these effects but two oral dosage doesn’t
• However comparison of oral tab and oral solution does help estimate effects of disintegration and dissolution of absorption process. Oral tab has to disintegrate and only then can it be effectively dissolved (since disintegration results in less particle size) whereas oral solution is already in dissolved state and only needs to be absorbed

Single dose vs Multiple dose studies
Single dose BA study • Only a single dose is given to each volunteer. It is preferred
by FDA in most cases• Advantage
– Easy, offer less exposure to drugs to volunteers
• Disadvantage – Cannot account for steady state characteristic of drug– (even with the best efforts to avoid this variability) There may
be excessive variability between volunteers – Sufficiently long sampling periods are needed in order to get
reliable estimate of terminal half life which is needed for correct calculation of total AUC (it is talking about the drug conc. in the tail portion of the graph which takes long time to come to zero)

Single dose (red) multiple dose (blue)

Multiple dose study
• Same dose is given repeatedly to same volunteer for a fixed time interval until a steady state condition is reached. Single dose study is sufficient in most cases. But multiple dose study is required if – There is a difference in the rate of absorption but not
in the extent of absorption.
– There is excessive variability in bioavailability from subject to subject which can be reduced at steady state
– The concentration of the active drug in the blood resulting from a single dose is too low for accurate determination by the analytical method.
– The drug product is an extended-release dosage form.

• Advantages– More accurately reflects the manner in which drug
will be used clinically– Allows blood levels to be measured at same
concerntration encountered therapeutically– Requires collection of fewer blood samples– Higher drug blood concerntration is observed which
makes its determination possible even by less analytical methods
– If fewer subjects are taken, then inter-subject variability is small
– Better evaluation of controlled release formulation– Nonliner pharmacokinetics can be easily detected– Eliminates needs for long wash out period between
doses

• Nonlinear pharmacokinetics means situation when increasing dose does not cause proportional change in plasma concerntration of drug.
• One of the reason this happens is because of carrier mediated absorption process. The carrier proteins are limited and once they are saturated the rate of drug absorption does not change with increase in dose

Relationship between drug concentration and absorption rateFor a passive process (Curve A) and for a carrier-mediated
Process (Curve B).

• Disadvantage
– Tedious, requires more time to complete
– More costly to conduct (prolonged dosing to subjects)
– Poor compliance by subjects
– Greater exposure of subjects to test drug which increases chances for adverse reactions
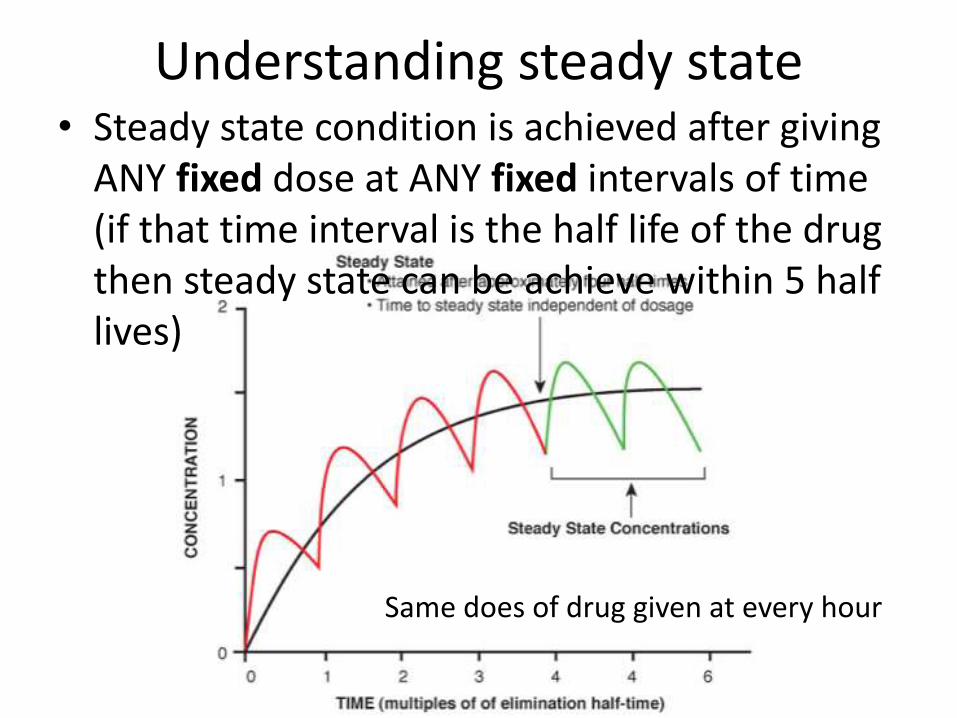
Understanding steady state• Steady state condition is achieved after giving
ANY fixed dose at ANY fixed intervals of time (if that time interval is the half life of the drug then steady state can be achieve within 5 half lives)
Same does of drug given at every hour

Human volunteerHealthy vs Patients, Male vs Female
• Our goal is not clinical trials• Aim is to only focus on effects of drug formulation on BA or
BE and to do this we need to keep all other variable, involving people such as age, sex, state of health, race etc as a constant
• To do so, we need a group of people who are very similar to each other in terms of physiology. Since absorption and thus BA can be influenced by gender and disease and we don’t want that, female and patients are biased because including them will create unnecessary variance in data which may be over or under analyzed. After all, if a new formulation has influenced drug release and absorption in such a way that BA has gone to toxic or sub-therapeutic levels it will be reflected in both male, females , children, old or patients. The degree of change may vary between age and gender and state of health but none the less change will or won’t be seen which is the only data we want.

• Criteria of volunteers– Healthy, male, 20-40 years– Female only for oral contraceptives– Minimum number of subject to avoid inter-subject
variability about 24-36– Obtain permission from volunteers– Pre-medical checkup to exclude people with any disease
that can interfere with BA– Volunteer must not take any other medicine for at least
a week and fast overnight and 4 hr after dosing– The volume and type of fluid and food must be specified– If same subject if studied twice, there must be gap
period of minimum 10 biological half lives (to allow drugs to be COMPLETELY washed out since in the tail portion of graph drug is still in body but its conc may be too low to detect)

Reports on PK difference between the SexesIn 2003, Schwartz published a paper on the influence of sex on PK.He noted:• Absorption was not significantly affected by sex, but
that rates may be slightly slower in women.• Bioavailability, for CYP3A substrates in particular,
may be somewhat higher in women compared to men, resulting in greater exposure due to lower clearance.
• The role of sex on pharmacokinetics, when considered in conjunction with genetics, age, disease, and social habits is not yet known in the clinical setting and needs more study
Schwartz JB. The influence of sex on pharmacokinetics. Clin Parmacokinet 2003;42(2):107-21.

Measurement of Bioavailability
• We need such measurement that can be related to no effect, therapeutic effect or toxic effect of drug.
• Pharmacokinetic Methods– Plasma level-time studies
– Urinary excretion studies
• Pharmacodynamic Methods– Acute pharmacological response
– Therapeutic response

Pharmacokinetic Methods
• Pharmacokinetic deals with studying drug concerntration in plasma as a effect of absorption, distribution, metabolism and elimination ie What body does to drug?
• The assumption is that unchanged drug concerntration in plasma or sometime in urine correlates to drug concerntration at site of action inside body which then correlates to the response produced.
• This assumption is not valid for prodrugs!

Plasma Level-Time Studies• Principle: Drug’s response can be measured as a function of
drug concerntration in blood or in some cases urine. • Drug dosing can be single or multipleA) Single dose study• Process involves Collecting blood samples periodically and
make plot of drug conc. Versus corresponding time. • Period and Frequency of blood sampling:Blood sample is drawn for period of about 3 drug half-lives to capture at least 80% of AUC
– Sampling frequency during absorption phase • At least 3 sampling
– Sampling frequency during elimination phase• if one compartment model 3 sampling • and if two compartment model 5-6 sampling• For IV dosage sampling must start from 5 mins after dosing and then
for each 15 min interval for 3 half live period

• Parameters of interest are Cmax, Tmax and AUC0-t,AUC∞
• The extent of Bioavailability for single dose can be measured as
B) Multiple dose study
• Process involves steady state condition which can be achieved by a fixed drug dose administration for at least 5 biological t1/2
with dosing interval equal to or greater than t1/2
• Period and Frequency of blood sampling:
• Blood sample should be taken at the end of previous dosing interval and
• 8-10 times after the administration of the next dose
Absolute bioavailability Relative bioavailability

• Parameters of interest are Css,max , τ , AUCss
where ss means steady state, τ is the dosing interval
• The extent of bioavailability is given as

Pilot study• There is no defined time interval to draw blood.
• The ideal time intervals is found by first doing small scale trial study, called pilot study, with few volunteers to know the optimum times at which to draw blood so that the graph comes out very smooth
• The pilot study can also be used to validate analytical methodology, assess PK variability, determine volunteer size and determine the length of the washout period needed between treatments.

Urinary Excretion Studies• This method is based on the principle that urinary
excretion of drug is directly proportional to the plasma concerntration of drug.
• To use this method a minimum of 20% of administered drug has to be excreted unchanged from kidney
• Plasma study is preferred, however, it is useful for – Drugs that are extensively unchanged in the urine eg
thiazide diuretics and sulphonamides
– Drugs that have urine as site of action eg urinary antiseptics such as nitrofurantoin and hexamine

Plasma-time curve vs Urine Excretion rate curve

The process involves • Water-loading 400ml after fasting overnight and 200 ml with drug
and 200ml hourly for next 4 hours• collection of urine at regular intervals, not specific time (ideally
less than one drug t1/2) for a time-span equal to 7 biological half-lives which ensure 99% drug collection
• analysis of unchanged drug in the collected samples• Determination of the amount of drug excreted in each interval and
cumulative amount excretedFor valid results, following criteria must be met too• At each sample collection , total emptying of the bladder is needed
to avoid addition of residual amount to the next urine sample• Frequent sampling of urine is also essential in the beginning in
order to compute correctly the rate of absorption• The fraction excreted unchanged in the urine must remain constant

• Advantages– Urine has a small volume and can show high
concerntration for same amount of drug as compared to plasma that has very high volume. Thus this method requires less sensitive analytical method while plasma requires more sensitive analytical techniques which may not present
– Convenient and Non-invasive and thus better subject compliance
– When coupled with plasma level-time study, it can estimate renal clearance
– Direct measurement of BA, both absolute and relative without need for fitting data to a mathematical model
– Derive other information such as 1st order elimination and metabolism, excretion and absorption rate constant

Limitations
• Can’t compute Vd (volume of distribution) and Clt (systemic clearence at time t)
• Difficult to apply in case where drug is slowly released or has long half-life cause concerntration in urine will be very low
• Parameter of interest is
(dXu/dt)maxMax urinary excretion rate like Cmax
(tu)max time for max excretion rate like Tmax
Xu∞cumulative amount of drug excreted in the urine

• The extent of bioavailability is given by
• For single dose
• For multiple dose

51
The following plasma and urinary data are obtained following intravenous administration of 250 mg tetracycline to a 70 kg subject:
t(h) C,plasma
(ug/ml)
t(h) X,urine
(mg)
1.5 2.40 3 29.4
4 1.71 5 16.6
6 1.50 7 13.0
8 1.2 9 12.0
10 1.05 11 10.0
12 0.90 13 8.0
73 62.0

Acute Pharmacological Response Method
• Acute Pharmacological Response means any physiologic changes that occur shortly after administration of drug. It is not necessarily a curative response eg changes in ECG, blood pressure, pupil diameter etc
• A dose-response graph or pharmacological effect-time curve is made
• This method is used when PK methods is difficult, inaccurate or non-reproducible
• Measurement of response for at least three half lives is done
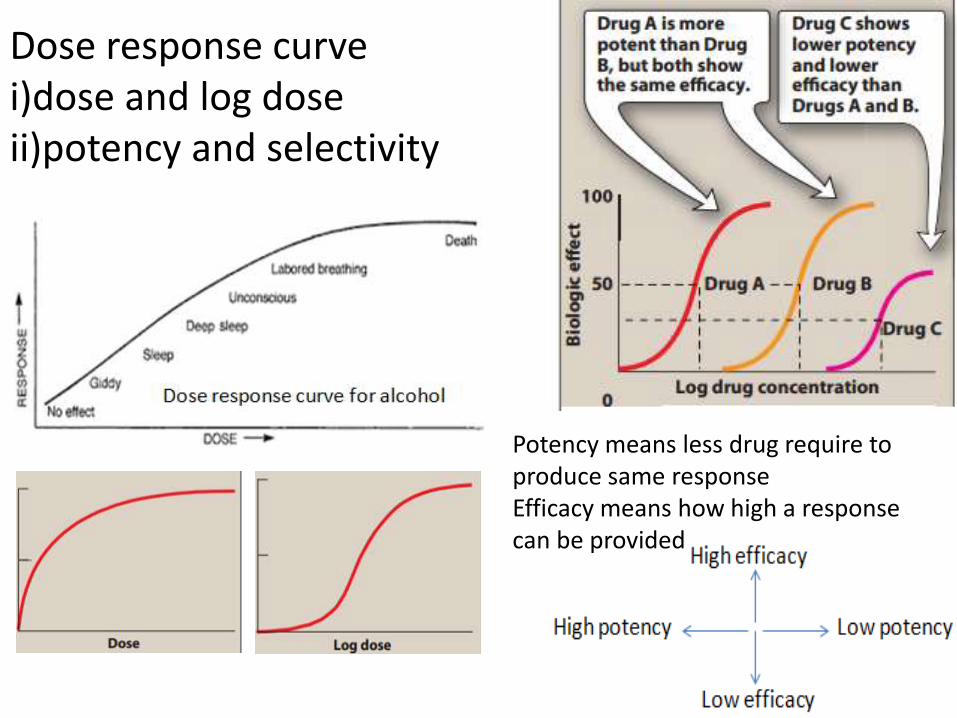
Dose response curve i)dose and log doseii)potency and selectivity
Potency means less drug require to produce same response Efficacy means how high a response can be provided

•When separate dose response graph is plotted for therapeutic and adverse effect in log dose style, then it becomes easy to define drug’s therapeutic window. •A therapeutic window describes the range of drug dose which is compromise between wanting to attain highest therapeutic response with the highest of dose but being careful not to cross region of minimum unwanted effect

Disadvantage
• Pharmacological response is naturally more variable among persons (ie some are more resistant to alcohol while other pass out with a few glass), which prevents proper correlation between measured response and drug available from formulation
• The observed effect may be due to an active metabolite

Pharmacokinetics and pharmacodynamics of a single bolus of propofol 2% in healthy volunteers.
• This study was undertaken to assess the bioequivalence (normally injectable don’t need to be checked for BS/BE cause they are 100% available since the beginning , but propolol is not a solution but an emulsion, drugs within emulsion aren’t 100% bioavailable since the start but is released slowly) between a new formulation of propofol 2% and the commercially available product Diprivan. Secondary objectives were to compare the times to onset of and emergence from hypnosis, the hemodynamic effects, and the safety profiles. Twelve healthy male volunteers were included in a randomized crossover study. Subjects were administered a 2-mg/kg single bolus injection of each formulation separated by a 7- to 10-day washout period. Plasma propofol was determined by reversed-phase liquid chromatography with fluorescence detection. Eleven subjects completed the study, and both formulations were considered bioequivalent. There were no serious or severe adverse events. The concentration-time profiles of all the subjects could adequately be described using a three-compartment model. The mean times to cessation of counting out loud (17 vs. 18 s) and to eye opening (245 vs. 244 s) were not statistically different between treatment groups. Moreover, they seem to show some degree of pharmacodynamic bioequivalence, although a higher number of subjects are necessary to unequivocally demonstrate it.
http://onlinelibrary.wiley.com/doi/10.1177/0091270003251391/abstract


Therapeutic Response Method• This method is based on observing clinical
response to a drug formulation given to patients who are suffering from the associated disease the drug was meant to be used. Here also dose response curve is used.
• Application is limited to drugs that only act at site of administration and are not meant to be absorbed into the blood such as topical antifungal creams and sucralfate used in ulcer therapy or drugs that can harm healthy people such as anticancer drug etoposide and anti-schizophrenic clozapine

It has many drawbacks• In the second period of crossover BE studies, in which each
of the two group of patients receive both standard and test drug products, the two groups might be more healthy than at beginning of study which defeats the purpose of selecting patients
• Unless multiple dose study is done, a patient won’t be able to take his medicine multiple times a day because single dose study requires a washout period between two doses which can about 5 or more half lives. (During this time patients can’t take another dose ie he will be in a period where drug is lower than therapeutic range. This might worsen the patient's health)
• Many patients are on more than one drug which can interact which the study drug and make BA study difficult to understand
• Measurement of observed response is too improper between two dosage form of the same drug to allow a good BA study


Up to now summaryWe are studying BA because:• to evaluate effect of formulation on BA to avoid cases like phenytoin
toxicity where we came to understand that change in formulation can place BA outside the therapeutic range
• BA data is needed to do BE, ie to compare generic paracetamol (Niko) and proprietary paracetamol (tylenol)
4 methods to do In-vivo (within living being) BA study. • Plasma – plasma drug conc vs time graph, method of choice• Urine – plasma drug conc vs time graph, drugs that act on bladder• Acute –dose/log[dose] vs response graph, • Therapeutic - dose/log[dose] vs response graph, done on patients for
locally acting drugs such as antifungal cream, BE study problem• For every method, three things to consider
– Volunteer type and size (healthy 20-40 aged male, same race, around 20-30 people)
– Single or multiple dose ( single mostly preferred by FDA)– Absolute or relative (absolute gives data about effect of 1st pass metabolism
and rate of absorption)

In-Vitro BA study• In-vivo study is expensive and difficult to perform• Could there be a simpler approach to evaluate BA
that doesn’t involve data from blood?• There is. It is the in-vitro dissolution study. It
involves studying rate and extent of drug dissolution/release from the dosage form as compared to rate and extent of drug absorption from stomach.
• Fortunately these two correlate well in some cases(Thank god for Statistics!) and hence dissolution study is well established to even act as a quality parameter for EVERY SINGLE BATCH of the same drug produced by ALL companies

Process involves putting drug product inside one of the vessel and allowing it to dissolve by rotating the paddle/basket. A few ml volume is drawn regularly and drug concerntration is known by UV spectrophotometer method. A graph between time and respective conc is plotted.

The Apparatus• Aims to simulate the environment a solid drug product
encounters in the stomach or intestine and obtain in-vitro dissolution as an alternative to in-vivo dissolution. It consists of
• Rotating paddle (to simulate how stomach stirs up food)
• Water bath for temperature control (mostly set to 37 degree)
• A transparent U-shaped vessel holding an aqueous fluid in which drug will dissolve. Various such fluids are– Pure water, 0.1N HCl, Phosphate buffer at 4.5 or 6.8, SDF
or SIF
• Maintain sink condition (only some do)• Use of enzymes not needed unless drug is very
sensitive to GI enzymes

USP APP. DESCRIPTOIN DOSAGE FORM must be solid forms)
Type 1 Basket apparatus Immediate release(IR), Chewable tablets, controlled released tablets
Type 2 Paddle apparatus IR, mouth dissolving tablets, suspensions, Chewable tablets, controlled released tablets
Type 3 Reciprocating cylinder Chewable tablets, controlled released
Type 4 Flow through cell apparatus
Formulation of poorly soluble drugs, implants, powders and granules
Type 5 Paddle over disk Transdermal
Type 6 cylinder Transdermal
Type 7 Reciprocating holder Transdermal, controlled released
Various USP Dissolution Apparatus

Concept of Controlled release formulation
Transdermal patch
http://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=bfdc4a52-a3a8-4f1e-8ab3-5c37d7a7d0a2&type=display
4 dose of 50mg immediate release vs single 200mg dose of controlled release

APPARATUS 1- BASKET APPARATUS
• Dosage form contained within basket• Dissolution occurs within Basket
• Drug product– floating capsules/tablets
• Rotating speed at about 50 to 100 rpm
• Operating temp is 37 deg
• Standard volume: 900/1000 ml
• Disadvantage– Formulation may clog to 40 mesh screen
68


USP Apparatus 2 – Paddle
• It has a paddle for rotation• Dosage form sinks to the bottom
and might require sinkers if they float
• Useful for Tablets, Capsules• Rotating speed at about 25 to 50
rpm, operating temp is 37 deg• Standard volume: 900/1000 ml
• Disadvantages– Floating dosage forms
require sinker– Positioning of tablet can not
be fixed which leads to uneven exposure of water flow around the tablet which creates uneven dissolution
70


Apparatus 3 – Reciprocating cylinder

• The dosage unit is placed in reciprocating cylinder that contains the solvent . The cylinder is allowed to move in upward and downward direction constantly which provides the stirring action. Release of drug into solvent within the cylinder measured.
• Useful for: Tablets, Beads, controlled release formulations• Standard volume: 200-250 ml/station• Advantages: Easy to change the pH-profiles• Disadvantages: Small volume (max. 250 ml)

Apparatus 4 – Flow-Through Cell

• The assembly consists of a reservoir and a pump for the Dissolution Medium which is injected into a flow-through cell containing the dosage form
• Only one to Maintains perfect sink conditions• Useful for: Tablets, Beads, controlled release formulations• Standard volume: 200-250 ml/station• Water flow rate : 240-960 ml/h• Operating temp 37 degree• Useful for: Low solubility drugs, Micro particulates, Implants,
Suppositories, Controlled release formulations• Advantages:• 1. Easy to change media pH• 2. low solubility drugs• 3. Sink conditions• Disadvantages:• 1. Deaeration necessary• 2. High volumes of media

USP Apparatus 5 - Paddle Over Disk
76

• It the same as Apparatus 2 but with the addition of a stainless steel disk at the bottom for holding the transdermal patch at the bottom of the vessel.
• Useful for: Transdermal patches• It operates at 32 deg C (taking consideration that skin is relatively cooler
than inside ob body)
• Rotating speed at about 25 to 50 rpm
• Standard volume: 900/1000 ml

USP Apparatus 6 - Cylinder
78

• It is same as Apparatus 1 except the basket has been replaced with a stainless steel cylinder
• On it’s surface, a transdermal patch is stuck
• The temperature is maintained at 32°C ± 0.5°C
• Drug product– mainly transdermal
• Rotating speed is specific for each product
• Standard volume: 900/1000 ml

USP Apparatus 7 – Reciprocating Holder
80

The assembly consists of a centrally fixed sample holder which is inside a chamber containing 50-200ml dissolution medium. The dosage unit is placed inside the holder which has pores from which drugs can release out. The holder moves up and down within the chamber (earlier there was no sample holder and the whole cylinder moved up and down). The temperature, inside the containers is 32 ± 0.5 °C. For Coated tablet drug delivery system attach each system to be tested to a suitable Sample holder

Various use of Dissolution data
• Day to day dissolution checking for QC purpose
• Avoid doing in-vivo BE study
• IVIVC

Dissolution Acceptance criteria(for daily QC purposes between batches)• We want to establish if the new formulation or
batch passes an existing dissolution standard or not
• We have only one time-drug release graph • Based on Q values• Q is the % of drug content dissolved in a given
time period eg 90% in 30 mins in 0.1N HClmedium
• This value is specified in the monograph for each existing drug

Dissolution Acceptance criteria

Dissolution Acceptance criteria
• In first stage six dosage units are tested. If their Q value is greater than or equal to Q+5 %, then dissolution is passed.
• If not then more of six dosage units is taken and now the average Q of 12 (6+6) dosage units must be equal or greater than Q, while no dosage unit can be less than Q-15%
• In final stage, twelve more units are evaluated. Now the average Q of total 24 units must be greater than or equal to Q

Comparison of dissolution profile(to avoid doing in-vivo BE study)
• Here we two time-drug release graph
• One for test formulation, another for a standard drug product
• We must not select this standard on our own, we have use official one
• Process involves a model-independent method based on determination of difference factor f1 and similarity factor f2

• Where
n = number of dissolution time point
Rt = dissolution value of the reference drug product at time t
Tt = dissolution value of the test drug product at time t

• Following conditions must be met• Minimum of three dissolution time points must
be are measured• 12 dosage units must be used for both standard
and test• The dissolution value to be considered must not
be more than 85% in 15 min or less(f2 test is not required for very rapid dissolving products)
• Standard deviation of mean dissolution for each test and standard unit should not be more than 10%

Comparison of Dissolution profile
Difference factor f1
Similarity factor f2
Inference
0 100 Dissolution profiles are identical
≤15 ≥50 Similarity or equivalence of two profile

Requirement for f2 test during biowaiver
Note- we are still checking BE, but instead of comparing generic and innovator plasma-conc time profiles in human we are comparing dissolution data of the two products obtained by dissolution apparatus
‘Very rapidly’ dissolving products
– At least 85% of the labeled amount is released within 15 minutes or less from the test and comparator product
– In this case, profile comparison is not needed
‘Rapidly’ dissolving products
– At least 85% of the labeled amount is released within 30 minutes or less from the test and comparator product
– Profile comparison (e.g., f2 testing) required

Uses/Objectives of Dissolution profile Comparison
• It is an alternate (but least desired) BE study method which can be used for biowaiver of– lower dose strength in proportion to higher drug
product containing same drug and same excipient (this drug may not be BCS class I)
– BCS class I drug
• To check product quality (for day to day use) if switch to – new formulation composition, new equipment, new site
of manufacture (it is because new site may not have implemented same standard in building and equipment)
– Different source/grade of raw material
– Scale up process

Dissolution test Use summary(which test when to use)
1) To do day to day testing for quality consistency
of each test batch of a certain formulation, check
dissolution profile of single batch and compare
with Q value provided in Pharmacopeia. Use S1,S2,S3
table to decide pass or fail
2) For one time Biowaiver ie avoid doing BE study
create and compare dissolution profile between test
and official ref product given in orange book using f1&f2. Use table to decide
Once the dissolution of test is found to be similar for your own formulation, then for every batch of the test we don’t need to repeat this again and again. We then go with above procedure by simply providing a good Q value and compare with it
3) A higher level application is IVIVC. It’s use is to claim dissolution as a quality determining factor which then allows for rationality of above two tests. Process is complex

Orange BookAn FDA published list that tells which is the ref product to do BE study withAlso has patent expiration dates
PharmacopeiaAn official book that contains quality standardsfor starting materials used in drug formulation and on the finished product (does not have formulation, Different nations have their own but International isPublished by WHO)


IVIVC
IVIVC has been defined by the FDA as “a
predictive mathematical model describing the
relationship between an in-vitro property of a
dosage form and an in-vivo response”.
Generally; the in-vitro property is the rate or
extent of drug dissolution or release while the
in-vivo response is the plasma drug
concentration or amount of drug absorbed.(can
also be amount of drug excreted)

Categories of IVIVCLevel A • Level A correlation is a point to point correlation
between in-vivo dissolution to in-vivo absorption• In-vivo correlation of this type is desired as it can be
used as an alternate to in-vivo data and can support biowaiver to do BE/BA study
Level B • Level B uses the principles of statistical moment
analysis (fancy word for mean, variance, skewnessand Kurtosis). The mean in vitro dissolution time (MDT) is compared either to the mean residence time (MRT) or to the mean in vivo dissolution time
• Not to be relied for biowaiver

Level C
• Level C establishes a single point relationship between a dissolution parameter, eg time to disintegrate drug or time to have 10% drug dissolved and a pharmacokinetic parameter, such as AUC, CMax and Tmax
• It can be used while making pilot formulation but cannot support biowaiver
Multiple Level C
• A multiple Level C correlation relates one or several pharmacokinetic parameters of interest to the amount of drug dissolved at several time points of the dissolution profile. If this relation exist over the entire dissolution curve then, it can support for biowaiver


BCS classification

BCS theoretical origination
Stomach fluidAqueous
PlasmaAqueous
stomach wall
Lipophillic
Drug dissolves here
Drug penetrate here
Drug absorb here

BCS originationAbsorption from stomach to blood requires two processes• Drug must dissolve into the aqueous stomach fluid • It must penetrate the lipophillic stomach membrane• Drugs with high hydrophillicity will easily dissolve in
aqueous stomach fluid but diffusion across the lipophillic membrane will be very difficult which results less absorption
• Drugs with high lipophillicity drug will not dissolve into the aqueous stomach fluid and even if it was somehow made to dissolve by use of surfactants or other types of excipients in the formulation, then it will get stuck on the lipophillic membrane and not diffuse into blood
• Thus , drugs need a balance of both hydrophillicity and lipophillicity. These two factors are very important to consider for formulation design.

• (Also true for during drug design where similar case happen when drug needs to go from blood into its receptor located inside the cells and demands crossing that cell’s membrane. This is the reason that Lipinski rule of five restricts LogPwithin −0.4 to +5.6 range and Polar surface area to be no greater than 140 Ǻ2)
• Also note that BCS does not dictate the therapeutic range of drug since solubility, permeability are PK parameters while therapeutic range is PD parameter ie it is not stated that BCS class I has wide therapeutic index or class IV has narrow therapeutic index

BCS was first proposed in 1995 by : Amidon GJ, Lennernäs H, Shap VP, Crison JR. A theoretical basis for a bio-pharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20.
BCS Class Examples
I Diltiazem, Propanolol, Metoprolol
II Nifedipine, Carbamazepine,Naproen
III Insulin,Metformin,Cimetidine,Paracetamol
IV Taxol, Chlorthiazide,Furosemide

www.Tsrlinc.net/search.cfmBSC database (contain about 400 drugs as of 2014 end)


Application of BCS
1) Biowaiver for BCS Class I BE study (doesn't apply to BA study to determine effect of food)
2) Judge scope of IVIVC and hence justify use of dissolution data as an alternative to plasma conc-time data
3) Single or multiple point dissolution
(single point is typically for QC purpose multiple point is typically for R&D purpose)


Criteria for Biowaiver on BCS system:• Product must be oral immediate-release products that are absorbed
throughout the intestinal tract(and not through a specific part such as from intestine only, this is termed as drug having absorption window)
• should contain a BCS class 1 compound• should be rapidly dissolving in USP apparatus 1 or 2(≥ 85 % within
30 minutes) in pH 1.2, 4.5 and 6.8 and • it’s highly soluble (highest dose must be soluble in 250ml of water in
wide pH range 1-7) and • have high permeability (more than 90% in an in-vitro setting which
mimic drug passage through intestine such as Caco 2 cell permeability test)
• should not contain excipient which could influence the absorption of the compound such as surfactants, absorption enhancers, prolong or shorten GI transit time
• should not contain an compound with a narrow therapeutic index; • and should not be designed to be absorbed from the oral cavity ie
buccal or sublingual
ACTA MEDICA (Hradec Králové) 2011; 54(1):3–8

Need for varied pH(1.2, 4.5 and 6.8)
• The pH in the GIT is not same. Stomach is acidic wheras parts of intestine are more basic. Thus three pH settings are used to mimic absorption environment encountered by drug as it passes through stomach, and small intestine(jejunum, ileum) in fasting condition.(both BA/BE study only consider fasting state to avoid variability in results due to food)

BCS and IVIVC
Q12) Why do think BCS Class 3 and 4 have little or no IVIVC expectation ?

BCS and IVIVCDrug absorption requires a drug to first dissolve in the
stomach fluid and then permeate through the GI epithelium into the plasma. The dissolution apparatus can only account for solubility factor in that it is trying to mimic drug dissolution in the stomach but not the permeability factor. Thus this situation creates a limitation when trying to use in-vitro dissolution data to account in-vivo concerntration since solubility (ie in-vivo dissolution) but not permeability can be accounted. Thus only in drugs belonging to BCS class I where there is high solubility and high permeability can a confident IVIVC be established to grant a biowaiver ie if a drug shows sufficient dissolution throughout the GI tract, then the high permeability property of such drugs allows us to be confident about a good absorption.

• For the same reason, BCS class III and IV have limited or no IVIVC ie test that factor only solubility are not suited in situation where permeability is the limiting factor for absorption.
• In case of BCS Class II, IVIVC is expected which seems contradictory but is corrected if the condition “…if in-vitro dissolution rate is similar to in-vivo dissolution rate” is met. It means if drug release in dissolution apparatus is happening at the same rate as drug dissolution in stomach, then since permeability is high enough we can expect IVIVC.
• Although class I drugs are quickly absorbed than class II under same condition due to difference in solubility, our goal is not to take any decision based on how fast or how slow absorption occurs but to be able to correlate data from the dissolution machine and in-vivo absorption so that dissolution data can be used in place of absorption data. This makes things cheap, easy and less time consuming.
• Even at that, the condition “if dissolution rate is slower than the gastric emptying rate” has to be met which means that drug cannot be completely absorbed only in stomach but throughout the GI tract which requires it to happen slower than rate of drug passing through the gut

BCS and Dissolution Methodology(single point or multiple point)
BSC class Dissolution methodology
I Single point if 85% drug release in 15 min(this is termed very rapid dissolution)Multiple point if less than 85 % drug release in 15 mins
II Multiple point
III Same as class I
IV Multiple point

Single point and multiple point dissolution

Case of paracetamol• Paracetamol is BCS class 3. Thus its has little or no IVIVC .There can
be no BCS biowaiver too• Technically BE/BA study is important however, generic paracetamol
are not constrained by BA/BE study• The reason seems to be that it is an old drug and borderlines BCS
class I property (permeability higher than 80 vs needed 90% to be in class I). Before concerns of BA/BE came forward it was already globally used by all under different brand names (ie different excipient) and they were efficacious too.
• Deu to this observation, regulatory body has granted biowaiver to paracetamol formulation if – it is a immediate release solid dosage form, – has in-vitro dissolution profile similarity with a ref product and – uses a recommended set of excipient.
• Even if there is differences in absoprtion, it is not given serious thought due to paracetamol’s wide therapeutic index and high elimination, any extra drug in body is not a serious concern.
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 95, NO. 1, JANUARY 200

• Equivalence is relative term that compares drug products with respect to a specific characteristic or function or to a defined set of standards
• Chemical Equivalence indicates that two drug products have the same active drug
Eg paracetamol tablet 500mg and paracetamolpediatric suspension 125 mg/ml
• RLD (Reference Listed Drug) A Reference Listed Drug (RLD) is an approved drug product to which new generic versions are compared to show that they are bioequivalent.
• Over-the-Counter Drugs (OTC) FDA defines OTC drugs as safe and effective for use by the general public without a doctor's prescription.

Pharmaceutical Equivalents FDA considers drug products
to be pharmaceutical equivalents if they meet these three criteria: 1. they contain the same active ingredient(s)2. they are of the same dosage form and route of
administration 3. they are identical in strength or concentration• Pharmaceutically equivalent drug products may
differ in characteristics such as• shape• release mechanism• labeling (to some extent)• excipient (including colors, flavors, preservatives)

Note• Observed in vivo differences in BA from two
pharmaceutically equivalent solid oral products may be due to differences in drug dissolution in vivo (which could be effect of formulation). This why we created an in-vitro test that expects to mimic in-vivo dissolution and judge BA based on this info.
• Dissolution test can only account solubility parameter but not permeability parameter. This limitation prevents use of dissolution data to be correlated to in-vivo in cases where permeability problems is the cause of low BA

Therapeutic Equivalence (TE)Drug products classified as therapeutically equivalent can be substituted with the full expectation that the substituted product will produce the same clinical effect and safety profile as the prescribed product. Drug products are considered to be therapeutically equivalent only if they meet these criteria: • they are pharmaceutical equivalents (contain the
same active ingredient(s); dosage form and route of administration; and strength.)
• they are assigned by FDA the same therapeutic equivalence codes (TEC) starting with the letter "A”.
• assigns therapeutic equivalence codes (TEC) based on data that a drug sponsor submits in an ANDA to scientifically demonstrate that its product is bioequivalent

Therapeutic Equivalence (TE) Codes The coding
system for therapeutic equivalence evaluations allows
users to determine whether FDA has evaluated a
particular approved product as therapeutically
equivalent to other pharmaceutically equivalent
products.
• A drug product is deemed to be therapeutically equivalent is rated "A"
• A drug product that is not deemed to be therapeutically equivalent is rated “B“
Over-the-counter drugs are not assigned TE codes.

Chemically equivalent but not pharmaceutically equivalent
• Tabets
• Suspension
• Injection
• Emulsion

Bioequivalence denotes that the drug substance
in two or more dosage form, reaches the systemic
circulation at the same relative rate and to the same
relative extent ie their plasma conc-time profile will
not be statistically significant differences.
Bioequivalence is only studied in pharmaceutically
equivalent products. Two bioequivalent products
are therapeutically equivalent.

BE study• BE study is needed in cases
– To gain confidence in therapeutic efficacy of generic product, especially if they have a narrow therapeutic range
– Extensions of innovator products ie salt or prodrugs– variations that require bioequivalence testing, such as Scale Up and Post
Approval Changes (SUPAC)– between early clinical trial products and to-be-marketed products
• When to do BE study?– BE study is an important report to be presented by the generic company
before seeking marketing permission– Also need by innovator company to extend their patent life in which
case they make salts or produrg of original drug
• Q12) The makers of “tylenol” (innovator of 500mg tablet paracetamol) tablet have decided to introduce 400mg tablets which first of such dose strength in market (ie there is either 500mg or 325 mg paracetamol in market). Comment on need of BA/BE study for this reduced dosage form.

BE study design
• BE requires BA from both generic and innovator product
• So same consideration for Volunteer, single vsmultiple, PK or PD measurement apply
• (minimum of 12 to usually 24-36 volunteers are used generally)
• Two new factors involves1) Ways of grouping volunteers, one for test and
other for standard2) There is a defined criteria for equivalence or not

BE study design(self note form book)
• Parallel or random design– Assign test or standard randomly to 2 equal groups– Used in half life is very long– In a parallel design the inter-subject variability being very high,
the sensitivity of the test is considerably reduced. Thus requiring a larger number of subjects compared to a crossover design, to attain the same sensitivity.
• Crossover design (FDA preferred method)– Assign both test and standard to all with a wash out period of 5
half lives to avoid period/carryover effect– Technically called – 2 treatment(means test and ref drug product)– 2 period (means before and after washout period)– 2 sequence (ie first ‘Test’ and then ‘ref’ or first ‘Ref’ and ‘Test’)
crossover design– Advantage is that inter-subject variation is negated

A is reference drug productB is generic drug product

This is for a cross over design

Statistical criteria• We do not simply overlap the plasma time profile
of both generic and reference drug and visually to decide on bioequivalence. We need to do statistical treatment.
• Idea is to check not the equality of products butthe difference between them. The rationale is that the two drug product are never the same , there is always bound to be some difference. Even two batches are rarely identical.
• The FDA guidelines for bioavailability studies state that “Generic Products whose rate and extent of absorption differ by 20% or less as compared to proprietary are generally bioequivalent”

Manufacturer’s risk vs Consumers risk• Manufacturer’s risk means concluding nonbioequivalence when
actually there is bioequivalence (good product is seen bad)• Consumers risk means concluding bioequivalence when actually
there is nonbioequivalence (bad product seen as good)• Regulatory authorities are more interested in consumer safety.
Thus they have set the consumer’s risk to 20% and let the pharmaceutical company decide how much manufacturer’s risk they are willing to accept.
• Statistical methods (by taking into account mean Cmax or mean AUC from both generic and reference) determine whether the risk of accepting BE when actually they aren’t equivalent t is within ±20%
• Manufacturer’s risk can be lowered by selecting proper sample size of volunteers (there is a statistical formula for this)

Various statistical tests are• ANOVA (hypothesis approach - is considered inferior)
– If F > F crit at α = 0.05 or p < 0.05 then not bioequivalence
– Not good way of concluding bioequivalence
• TOST (2 one-tailed t- test)(hypothesis approach -improvement over weakness of ANOVA)
– To conclude bioequivalence t > t crit at a= 0.05 or p ≤ 0.05
• 90% Confidence interval approach (superior approach)
– To conclude bioequivalence, the confidence interval of the difference of mean Cmax or AUC of generic and innovator should be contained within the limits of 0.80–1.25 of reference
Indian J Pharmacol |August 2004 |Vol 36 |Issue 4 |209-216
Except in ANOVA, the Cmax and AUC data must be log transformed first in case of TOST and confidence intervals to convert into normal distribution

Basic statisticA) Descriptive Statistics
• Descriptive statistics aims only to gather data data and check its mean , median, mode standard deviation, range, histogram etc. It only helps to describe, show or summarize data in a meaningful way.
• Eg in a census data Descriptive statistics lets us know the distribution of population by age, sex, location, religion, income level etc in a graphical charts

Basic statisticB) Inferential Statistics
• Inferential statistics aims to relate information taken from a sample data and apply it to the entire population
• This is where statistics starts becoming cool and have real world application
• Eg A new drug is tested on a few thousand people and if found to be safe and effective, the sameresults are thought to be safe and effective for 7 billion human in earth
• BE study is inferential analysis

Statistical tests1)Hypothesis testing
– ANOVA (conventional 2 sided method), TOST (interval based)
2)Confidence interval approachHypothesis testing involves the construction of two statements:
– the null hypothesis and – alternative hypothesis.
The null hypothesis reflects that there will be no observed effect for our experiment (two drug products will show same Cmax or AUC in same population ie products are bioequivalent)
The alternative hypothesis reflects that there will be an observed effect for our experiment (two drug products will show different Cmax or AUC in same population ie products are not bioequivalent). Conventionally, the hypothesis one desires to prove must be stated in alternative hypothesis. So things are kind of opposite for BE study by this conventional hypothesis testing.

• one rejects the null hypothesis in favor of the alternative hypothesis if the evidence is sufficiently strong against the null hypothesis.
• Fisher, R.A. The Design of Experiments, Oliver and Boyd, London, 1935
• “The null hypothesis is never proved or established, but is possibly disproved in the course of experimentation. Every experiment may be said to exist only in order to give the facts a chance of disproving the null hypothesis”
• It’s like a court trial where every person is assumed innocent to begin with and if there is sufficient evidence to reject his innocent, then only can the person be said guilty.

• ANOVA (analysis of variance) is a hypothesis testing method. The hypothesis are
• H0 :µT=µR(i.e. products are bioequivalent),• H1 :µT≠µR (i.e. products are not bioinequivalent) • where µT and µR represent the expected mean
bioavailability of the test and reference formulations(Cmaxor AUC), respectively.
• This type of hypothesis statement which checks for ‘equality or not’ is criticized and considered the weakest approach for BE study.
• The fundamental idea behind the criticism, as I have to understand is that, “no two different things can be equal-eg two drugs from different batch, two orange of same species but different tree”. Thus is it impractical to check similarity or not when we clearly know the two drug products are bound to be different and hence null hypothesis will always be rejected. (Refer below paper)
1) ANOVA test to evaluate BE

• Decision Rule
By F values
– if F (calculated value) > FCV(df1,df2)we reject the null hypothesis
(null hypothesis being that products are bioequivalent ieproducts are not bioequivalent) (FCV is obtained from a F table with α= 0.05 for degree of freedom . There are F table with α= 0.1,0.05,0.01 etc. we use 0.05. It means saying we are 95% sure the mean Cmax from generic is not equal to mean Cmax from ref )
By P values– If P<0.05 we can reject null hypothesis, statistical significant
difference exists between two means(not bioequivalent)
– If P≥0.05 we cannot reject null hypothesis, statistical significant difference doesn’t exist between two means
(Note- P value is stronger than F value and going by its decision is better)


Since F is not > F crit we cannot say that products are not non-bioequivalent. However, non-rejection of null hypothesis is not taken as ‘proof’ of its validity ie we can’t say that since we can’t prove that there is difference in the two means of Cmax, hence they MUST BE bioequivalent. Stated differently, absence of evidence does not imply evidence of absence because small samples could easily be used to show non-significant difference when there is actually significance difference.

• Moral of ANOVA study: ANOVA can say that the two drug products are not bioequivalent but it cannot really say if products are bioequivalent which is of our prime interest since null hypothesis, which claims for BE, will always be rejected owning to some difference between the mean Cmax or mean AUC of two drug products.
• Frequent mistake: The absence of statistical significance has been interpreted incorrectly as absence of clinically relevant differences.
• There is another test called one-tailed or two tailed student’s t test where we find T value and generate P value from that T. But this is used for normally or symmetrically distributed data which is not compatible with our BA data. Thus Cmax ou AUC must be log transformed which turns it into normal distribution.

TOST (2 one-sided t test)• TOST also requires hypothesis but it put the statement of
bioequivalence in the alternate hypothesis and uses 2 one sided hypothesis for each null and alternative. (One sided means a < x and a > y as opposed to y < a < x being two sided)
• It aims to test the joint null hypothesis that our mean difference between test and reference products is not larger than + 0.20 µR nor below - 0.20 µR which is the recommended range of equivalence
– H01 :µT- µR ≤ -δ and H02 :µT- µR≥ +δ (not bioequivalent)
– H11 :µT- µR> -δ and H12 :µT- µR< +δ (bioequivalence)
where µT and µR = mean Cmax or mean AUC of test and
reference and ±δ = ± 0.20 µR,
By rejecting both null hypotheses, we can conclude that the
difference of mean Cmax or AUC falls within the range
specified ie ± 0.20 µR

Decision rule
• Reject both H0 if t ≤ -tCV or t ≥ tCV in α = 0.05
• s is the root mean square error (RMSE) from the analysis of variance and n is the sample size ie number of patients or volunteers and v = volunteers - 2
• To get tCV look in one tailed t-table at ‘t(1-α)’ iet0.95 column and V row ie (volunter-2) row (if 12 volunteer then look in row 12-2=10)
• (This formula is valid only for crossover design)

• TOST for parallel design
Degree of freedom = N -2
n1 + n2 = N (total volunteers)


Confidence interval (for the difference of two mean)
• Most used method
• Make sure to use log transformed data
• Is actually a two tailed t test each tail having α=0.05
• No hypothesis statement required

Given the following data of a 2X2 crossover study
Period 2Period 12x2 BE trial
N=12
(T)
70, 90, 95, 70, 60,
70
(R)
75, 95, 90, 80, 70,
85
Sequence 1
(R)
40, 50, 70, 80, 70,
95
(T)
75, 85, 80, 90, 50,
65
Sequence 2

First, log-transform the data
Period 2Period 12x2 BE trial
N=12
4.2485, 4.4998,
4.5539, 4.2485,
4.0943, 4.2485
4.3175, 4.5539,
4.4998, 4.3820,
4.2485, 4.4427
Sequence 1
3.6889, 3,9120,
4,2485, 4.3820,
4.2485, 4.5539
4.3175, 4.4427,
4.3820, 4,4998,
3,9120, 4.1744
Sequence 2

Second, calculate the arithmetic mean of each period and sequence
Period 2Period 12x2 BE trial
N=12
T = 4.316R = 4.407Sequence 1 (BA)
R = 4.172T = 4.288Sequence 2 (AB)

Note the difference between Arithmetic Mean and Least Square
Mean• The arithmetic mean (AM) of T (or R) is the
mean of all observations with T (or R) irrespective of its group or sequence– All observations have the same weight
• The LSM of T (or R) is the mean of the two sequence by period means– In case of balanced studies AM = LSM – In case of unbalanced studies observations in
sequences with less subjects have more weight– In case of a large unbalance between sequences due
to drop-outs or withdrawals the bias of the AM is notable

Third, calculate the LSM of T and R
Period 2Period 12x2 BE trial
N=12
T= 4.316R = 4.407Sequence 1 (BA)
R = 4,172T= 4.288Sequence 2 (AB)
R = 4.2898 T = 4.3018

Fourth, calculate the point estimate
• F = LSM Test (A) – LSM Reference (B)
• F = 4.30183 – 4.28985 = 0.01198
• Fifth step! Back-transform to the original scale
• Point estimate = eF = e0.01198 = 1.01205
• Five very simple steps to calculate the point estimate!!!

Now we need to calculate the variability!
• Step 1: Calculate the difference between periods for each subject and divide it by 2: (P2-P1)/2
• Step 2: Calculate the mean of these differences within each sequence to obtain 2 means: d1 and d2
• Step 3:Calculate the difference between “the difference in each subject” and “its corresponding sequence mean”. And square it.
• Step 4: Sum these squared differences
• Step 5: Divide it by (n1+n2-2), where n1 and n2 is the number of subjects in each sequence. In this example 6+6-2 = 10– This value multiplied by 2 is the MSE
– CV (%) = 100 x √eMSE-1

This can be done easily in a spreadsheet!
I II Step 1 Step 1 Step 3 Step 3 Step 4
R T P2-P1 (P2-P1)/2 d - mean d squared Sum = 0,23114064
4,31748811 4,24849524 -0,06899287 -0,03449644 0,01140614 0,0001301
4,55387689 4,49980967 -0,05406722 -0,02703361 0,01886897 0,00035604 Step 5
4,49980967 4,55387689 0,05406722 0,02703361 0,07293619 0,00531969 Sigma2(d) = 0,02311406
4,38202663 4,24849524 -0,13353139 -0,0667657 -0,02086312 0,00043527 MSE= 0,04622813
4,24849524 4,09434456 -0,15415068 -0,07707534 -0,03117276 0,00097174 CV = 21,7516218
4,44265126 4,24849524 -0,19415601 -0,09707801 -0,05117543 0,00261892
Step 2 Mean d1 = -0,09180516 -0,04590258
n1 = 6
T R
4,3175 3,6889 -0,62860866 -0,31430433 -0,25642187 0,06575218
4,4427 3,9120 -0,53062825 -0,26531413 -0,20743167 0,0430279
4,3820 4,2485 -0,13353139 -0,0667657 -0,00888324 7,8912E-05
4,4998 4,3820 -0,11778304 -0,05889152 -0,00100906 1,0182E-06
3,9120 4,2485 0,33647224 0,16823612 0,22611858 0,05112961
4,1744 4,5539 0,37948962 0,18974481 0,24762727 0,06131926
Step 2 Mean d2 = -0,11576491 -0,05788246
n2 = 6
PERIOD

Step 1: Calculate the difference between periods for each subject and divide it by 2: (P2-P1)/2
I II Step 1 Step 1
R T P2-P1 (P2-P1)/2
4,31748811 4,24849524 -0,06899287 -0,03449644
4,55387689 4,49980967 -0,05406722 -0,02703361
4,49980967 4,55387689 0,05406722 0,02703361
4,38202663 4,24849524 -0,13353139 -0,0667657
4,24849524 4,09434456 -0,15415068 -0,07707534
4,44265126 4,24849524 -0,19415601 -0,09707801
Step 2 Mean d1 = -0,09180516 -0,04590258
n1 = 6
T R
4,3175 3,6889 -0,62860866 -0,31430433
4,4427 3,9120 -0,53062825 -0,26531413
4,3820 4,2485 -0,13353139 -0,0667657
4,4998 4,3820 -0,11778304 -0,05889152
3,9120 4,2485 0,33647224 0,16823612
4,1744 4,5539 0,37948962 0,18974481
Step 2 Mean d2 = -0,11576491 -0,05788246
n2 = 6
PERIOD

Step 2: Calculate the mean of these differences within each sequence to obtain 2 means: d1 & d2
I II Step 1 Step 1
R T P2-P1 (P2-P1)/2
4,31748811 4,24849524 -0,06899287 -0,03449644
4,55387689 4,49980967 -0,05406722 -0,02703361
4,49980967 4,55387689 0,05406722 0,02703361
4,38202663 4,24849524 -0,13353139 -0,0667657
4,24849524 4,09434456 -0,15415068 -0,07707534
4,44265126 4,24849524 -0,19415601 -0,09707801
Step 2 Mean d1 = -0,09180516 -0,04590258
n1 = 6
T R
4,3175 3,6889 -0,62860866 -0,31430433
4,4427 3,9120 -0,53062825 -0,26531413
4,3820 4,2485 -0,13353139 -0,0667657
4,4998 4,3820 -0,11778304 -0,05889152
3,9120 4,2485 0,33647224 0,16823612
4,1744 4,5539 0,37948962 0,18974481
Step 2 Mean d2 = -0,11576491 -0,05788246
n2 = 6
PERIOD

I II Step 1 Step 1 Step 3 Step 3
R T P2-P1 (P2-P1)/2 d - mean d squared
4,31748811 4,24849524 -0,06899287 -0,03449644 0,01140614 0,0001301
4,55387689 4,49980967 -0,05406722 -0,02703361 0,01886897 0,00035604
4,49980967 4,55387689 0,05406722 0,02703361 0,07293619 0,00531969
4,38202663 4,24849524 -0,13353139 -0,0667657 -0,02086312 0,00043527
4,24849524 4,09434456 -0,15415068 -0,07707534 -0,03117276 0,00097174
4,44265126 4,24849524 -0,19415601 -0,09707801 -0,05117543 0,00261892
Step 2 Mean d1 = -0,09180516 -0,04590258
n1 = 6
T R
4,3175 3,6889 -0,62860866 -0,31430433 -0,25642187 0,06575218
4,4427 3,9120 -0,53062825 -0,26531413 -0,20743167 0,0430279
4,3820 4,2485 -0,13353139 -0,0667657 -0,00888324 7,8912E-05
4,4998 4,3820 -0,11778304 -0,05889152 -0,00100906 1,0182E-06
3,9120 4,2485 0,33647224 0,16823612 0,22611858 0,05112961
4,1744 4,5539 0,37948962 0,18974481 0,24762727 0,06131926
Step 2 Mean d2 = -0,11576491 -0,05788246
n2 = 6
PERIOD
Step 3: Squared differences

Step 3 Step 4
squared Sum = 0,23114064
0,0001301
0,00035604 Step 5
0,00531969 Sigma2(d) = 0,02311406
0,00043527 MSE= 0,04622813
0,00097174 CV = 21,7516218
0,00261892
0,06575218
0,0430279
7,8912E-05
1,0182E-06
0,05112961
0,06131926
Step 4: Sum these squared differences

Step 3 Step 4
squared Sum = 0,23114064
0,0001301
0,00035604 Step 5
0,00531969 Sigma2(d) = 0,02311406
0,00043527 MSE= 0,04622813
0,00097174 CV = 21,7516218
0,00261892
0,06575218
0,0430279
7,8912E-05
1,0182E-06
0,05112961
0,06131926
Step 5: Divide the sum by n1+n2-2

Calculate the confidence interval withpoint estimate and variability
• Step 11: In log-scale
• 90% CI: F ± t(0.1, n1+n2-2)-√((Sigma2(d) x (1/n1+1/n2))
• F has been calculated before
• The t value is obtained in t-Studient tables with 0.1 alpha and n1+n2-2 degrees of freedom
– Or in MS Excel with the formula =DISTR.T.INV(0.1; n1+n2-2)
• Sigma2(d) has been calculated before.

Final calculation: the 90% CI
• Log-scale 90% CI: F±t(0.1, n1+n2-2)-√((Sigma2(d)·(1/n1+1/n2))
• F = 0.01198
• t(0.1, n1+n2-2) = 1.8124611
• Sigma2(d) = 0.02311406
• 90% CI: LL = -0.14711 to UL= 0,17107
• Step 12: Back transform the limits with eLL and eUL
• eLL = e-0.14711 = 0.8632 and eUL = e0.17107 = 1.1866


• Step 2) Find ratio of mean Cmax of test to mean Cmax of ref and if this ratio falls under the before interval, then bioequivalence is inferred
Interpretation of confidence intervals• For example, in a study the observed ratio for Cmax
between generic and innovator is 0.95 (representing a 5% difference between products). If the 90% confidence interval was 0.85 to 1.01, this means that we can be confident that if the same study was conducted 100 times, then 90 of those times the observed result for the ratio of Cmax would lie somewhere in the range 0.85 to 1.01.
• In 127 generic drugs applications to the US Food and Drug Administration in 1997 the mean difference was 3.3% for AUC and 4.3% for Cmax (20% is the cut off value. Thus we see generic products are very close to innovator products)
Henney J. JAMA 1999;21:1995.

Summary of test for 2X2 crossover study
Test Table Dergree of freedom α
ANOVA F (1,volunteer-2)1is row
0.05
TOST t (one tailed) Total Voulnteer-2 0.05
Confidence interval Confidence interval(which is actually a two tailed t test at α= 0.05)
Total volunteers 0.05, but since it is two tailed, 0.05+0.05 = 0.1 (two tailed)

BiowaiversIt means situation where in-vitro dissolution study can be accepted as an alternative to in-vivo study to determine BE. It does not mean no need to do BE or BA
The term “biowaiver” is de-fined by the WHO as “approval of generic solid oral formulation of an active pharmaceutical ingredient based on strictly defined dissolution criteria as a surrogate for an in vivo bioequivalence test”.
A) BCS based (already studied)B) Non-BCS based
– Dosage from– Dose strength and formulation proportionality – Route of administration– Local efficacy– Non-therapeutic but just diagnostic use

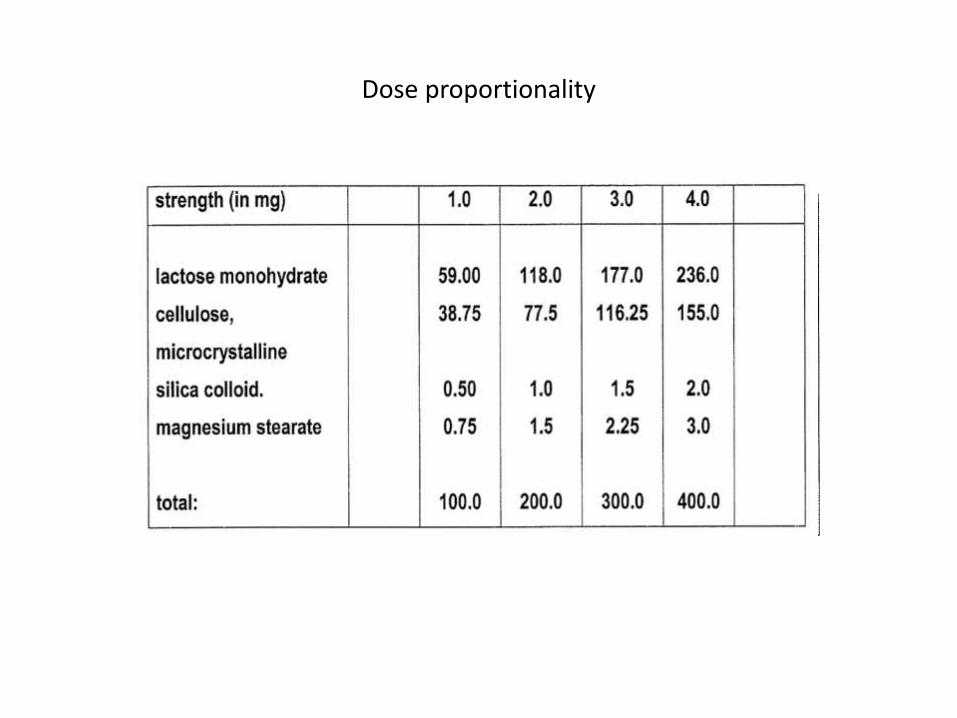
Non-BCS based cases1) The drug differs only in strength of active
compounds, provided it satisfies following conditions
• Linear PK (change in dose causes change in • The formulations have dose proportionality
(Same ratio between active substance and excipient)
• Both strengths must be produced by same manufacturer at the same production site
• BA/BE study exist for the highest strength • Under same test conditions, the in vitro
dissolution rate is the same

2) The drug product has slightly reformulated or manufacturing method has been slightly modified by the original manufacturer in ways that can convincingly be argued to be irrelevant for the BE
3) An acceptable IVIVC Bioavailability and bioequivalence are considered self evident for these non-
solid products and thus biowaivered, 4)Product is in form of Solutions (elixir, syrup, tincture), with same
concerntration of active ingredient and has no excipient known to significantly alter absorption. Emulsion and suspension are not biowaivedcause unlike solution drugs from suspension or emulsion are not 100% available even when given in injection form
5) Topical products meant for local effect (cream, ointment, gel)6) Orally administered drugs not intended to be absorbed (antacid or radio-
opaque medium)7) Product administered by inhalation as a gas or vapour or parentral products
including lyophilized product or sterile power which are intended to be used as parental soln
(lyophilized product appear solid but in contact with water, they immediately turn into solution, like putting cotton candy in mouth)
(Remember that biowaiver is only valid for solid, oral doses and not to liquid oral or liquid injectables)
Dose proportionality

Biowaivers accepted by FDA Cefadroxil
Galantamine HBr
Labetalol
Levetiracetam
Levofloxin
Memantine HCl
Metoprolol
Ofloxacin
Pramipexoledihydrochloride
Pregabalin
Propanolol
Ramelteon
Rivastigmine HCl
Sotalol HCl
Tiagabine HCl
Timolol
Venafaxine HCl

1) WHO and EMCA allows biowaiver for class III drugs which are very close to being class I (which is what III/I means)
2)Even drugs for sensitive diseases such as AIDS and TB Can be biowaived
Only drugs solubility and permeability (and metabolism), therapeutic range etc matter to biopharmaceutics. No need to be extra cautious just because the disease is very big.
WHO biowaivers
HIV/AIDS and related diseases
Anti-TB medicines
Lamivudine (Class I)Stavudine (Class I)Zidovudine (Class I)
Ethambutol (Class III/I)Isoniazid (Class III/I)Levofloxacin (Class I)Ofloxacin (Class I)Pyrazinamide (Class III/I)

• (IN 2005) WHO EXTENSIONS TO THE SCOPE OF APPLICATION OF THE BIOWAIVER
• In the "Multisource document" WHO has broadened the scope of application of the Biowaiver in three directions:
• 1) The criteria for classification as a Class I API have been relaxed with respect to both the dose: solubility ratio and permeability requirements.
• 2) The new version of the document allows pharmaceutical products containing Class III APIs to be considered for a Biowaiver, with application of more stringent dissolution criteria.
• 3) The new version of the document further allows pharmaceutical products containing BCS class II APIs that are weak acids which can meet a dose: solubility ratio of 250 ml or less at pH 6.8 to be eligible for a Biowaiver, with the requirement that they dissolve rapidly at pH 6.8 and similarly to the comparator product at pH 1.2 and 4.5 too.
Working document QAS/04.109/Rev.1

For pharmaceutical products containing BCS class III(highly soluble, low permeability) API a biowaiver can be only considered if both the multisource (generic) and the comparator (original) product • are very rapidly dissolving; 85% or more
dissolution of the labelled amount of the API should be achieved within 15min in standard media at pH 1.2, 4.5 and 6.8 using the paddle apparatus at 75rpm or alternatively the basket apparatus at 100 rpm.
• Generally the risks of an inappropriate biowaiver decision should be more critically reviewed (side specific absorption, induction/competition at the absorption side, excipient composition and therapeutic risks, etc.) than for BCS class I drugs.

For pharmaceutical products containing APIs with high solubility at pH 6.8 but not at pH 1.2 or 4.5 and with high permeability (by definition, BCS class II compounds with weak acidic properties), these are eligible for a Biowaiver provided that the multisource product:
• is rapidly dissolving i.e. 85% or more dissolution of the labelled amount of the API should be achieved within 30 min in standard media at pH 6.8 using the paddle apparatus at 75 rpm or alternatively the basket apparatus at 100rpm, and
• The multisource product exhibits similar dissolution profiles, as determined with the f2value or equivalent statistical evaluation, to those of the comparator product in buffers at all three pH values (pH 1.2, 4.5 and 6.8).

BE study- to do or not to do vs how to do?1) Is the drug product, (not drug itself) in solid form or liquid form?
– Solid form needs BE (all kinds of tablet, suppository, transdermal patch)– Liquid form (solution, syrup, tincture, elixir, injections) needs no any kind of in
vivo or in vitro BE– However emulsion and suspension either oral or suspension do need BE
• BE is mandatory for all solid, oral products. It is not mandatory for solution state oral or injectable products
2) If for solid products then, is the solid form in oral tablet or not? 2.1) If solid and oral tablet and then first consider biowaiver cases if2.11) the drug fall under BCS class12.12) Doesn’t have a narrow Therapeutic range2.13) Doesn’t have absorption window (ie we want drugs that are
absorbed through the GIT and not from a specific region called absorption window such as sublingual, buccal, exclusively at small intestine etc)
2.14) shows immediate release profileIf these criteria are met, then do BE study using in-vitro dissolution study
profile

2.2) Solid but non-oral tablets or not to be swallowed(suppository, sublingual, buccal tablets) and liquid but not solution (suspension and emulsion) are not eligible for any kind of biowaivers and hence must do in-vivo study
3) Whether in-vivo or in-vitro, there is need of an official drug product to compare with. That is found from FDA’s ‘orange book’.
4) in-vitro dissolution profile
• check dissolution in 3 pH of 1.2, 4.6 and 6.8 for both ref and test products
• If ≥ 85% drug released in 15 mins in all pH, no need to do profile comparison ( best case scenario)
• If not then do f2 similarity test which must be above 50

5) For in-vivo method need following things
– Min 12 Healthy, fasting male volunteers
– Single dose study is preferred by FDA
– 2X2 crossover study is preferred
– Drug plasma conc is known by HPLC equipment
– The proper time to draw blood samples is known by a pilot study involving few volunteers
– There should be washout period of about 5 half lives to avoid carry over effect
– Need to do statistical clauclation (go for TOST or confidence intervals over ANOVA)

BA study BE study
The objective is to simply Study BA of singledrug product
The objective is to Compare BA between Two product;usually generic and innovator
Any IV, oral or suspension form of the drug can be used to judge relative BA
The innovator product must be chosenofficially using FDA’s orange book
It has no any criteria to judge pass or fail. However, by comparing two BA study various differences can be pointed out such as duration of action, onset of action, Cmax, tmax etc
Since it is a comparative test, it has a strict criteria for pass or fail as defined by bioequivalence if there is max of ±20% deviation from standard product
Study effect of formulation parameter such as change in dosage (such as tablet to suspension or solution),change of excipient, dose change has made product toxic or sub therapeutic to avoid cases like Digoxin and Phenytoin toxicity
To compare original product that comes with confidence of around 15 years of development time, cost of more than 800 million $ and tested on thousands of people vs a generic product which do not do clinical studies but have simply changed the excipient or their proportion in the formulation.

Over coming BA problems• PK approach
– Develop new drug with favorable PK• Oral penicillin's that don’t degrade in stomach
– Prodrug design
• Biological approach– Change route of administration
• Pharmaceutical approach– Enhancing drug solubility (BCS class II)
– Enhancing drug permeability (BCS class III)
– Enhancing of drug stability (BCS class V)
– Enhancing of GI retention (BCS class II,III or V)

Penicillin G is hydrolyzed in stomach while penicillin V is not. Thus it can be givenorally. Since the structure has altered, penicillin V is a different drug and needs to go through
it’s separate clinical trials which is costly and time consuming. Thus Pharmaceutical methods are in demand.
Another perspective is a successful drug development needs to think about PK considerations very early in the program and if possible during the design phase itself
Improving PK by developing a new drug entirely
Cannot be given orally Can be given Orally

Class V consists of drugs that have variable solubility or permeability. Although these two property is generally fixed for a any chemical, it is variable in these cases because their chemical structure contains functional groups that is subjected to variation by effect of pH ie they are acidic or basic drugs. They can exist can a neutral form or salt form. Salt form has more solubility but reduced permeability

Basic concepts
• Enhanced solubility means same volume of solvent can dissolve
more solute
• Rapid dissolution means, drug dissolves very fast , although solubility
in the constant volume of solvent is not increasing
Rapid dissolution is even better than high solubility because drug will start to permeate immediately showing quicker onset of action. Body does not wait for the complete drug in dose to be dissolved.
• Solubility vs Dissolution• Solubility is “how much solute goes into a fixed vol”, only considers amount
• Dissolution is “how fast a solute goes into a fixed vol”, considers rate not amount

All lines indicate a drug with same dose. The difference in the graphs is due to difference inSolubility. Q13)If the solubility or dissolution were further enhanced, is there chance to cross MTC?

Enhancing drug solubility1. Micronization: It involves size reduction of drug
particles to 1-10 µm. Spray drying process are used or air attrition process (fluid energy or jet mill) or colloid mills Eg griseofulvin
2. Nanonization: It involves size reduction of drug particles to 200-600 nm. The power is formulated into a suspension which fine enough to even be given by injection eg amphotericin. Two of the technology currently in use is Supercritical Fluid recrystallization and Homogenization in water or other solvents or colloid mills


http://www.google.com/patents/EP2488160A1?cl=en

Enhanced dissolution of formulation that use nanoparticles
Advances in Nanoparticles, 2013, 2, 51-59

A) Ordinary marketted suspension of 500mg curmurin having microparticles
B) Novel 500mg Nano suspension of curmurin

Percentage of dissolved Curcumin from nanocrystal-loaded capsules (formulation A) compared to micro- crystal-loaded capsules (formulation B) in water ((a): Top); buffer at pH 1.2 ((b): Middle) and pH 6.8 ((c): Bottom).
In water
In pH 1.2
In pH 6.8

3)Jet millIn an jet mill, the drug to be micronized is made to circulate in a chamber where compressed air is released. Size reduction occurs as a result of the drug particles colliding with each other at very high speed as they are carried by the strong wind. No mechanical grinding is involved. Such process can result in product in the size range of 0.25 to 15 microns
http://www.jetpulverizer.com/how-jet-mills-work.php

4) Spray Freezing into Liquid (SFL)
• A drug solution/emulsion/suspension is sprayed into cryogenic liquids (made by cooling gases such as nitrogen, argon until they are in liquid phase) which creates very small frozen particles. Then they are lyophilized which results in dry and free- flowing micronized powers. Not only do they have very high surface area due to nano size but lyophillzationchanges their crystal state into a more soluble amorphous form which have high rate of solubility.
5) Supercritical Fluid (SCF) Recrystallization
• A drug when dissolved in a SCF, it can be recrystallized as nano-sized particles. Supercritical fluids are fluids whose temperature and pressure are greater than its critical temperature and critical pressure, which allows it to have property of both liquid and a gas eg CO2.

6) Colloid mill : (Colloid refer to particles size smaller than 10-3 m and greater than 10-9. They are an intermediate between true solution and suspension when only considering particle size. Depending on the machine size reduction to micro and nano size is possible.)
A colloid mill is a machine that is used to reduce the particle size of a solid in suspension in a liquid, or to reduce the droplet size of an emulsion. Colloid mills has a motor that turns at high speeds (2000 - 18000 RPM) which creates high levels of hydraulic shear. They are similar to jet mill in that, jet mill uses strong wind currents while colloid mill generates strong liquid currents to smash drug particle against one another. Colloid mill are innately in suspension or emulsion from because the shearing force is created by liquid

Hydraulic shear
• Motion in fluids such as air and water behave like a stack of cards tumbling
• When the top layer moves, it’s motion is opposed by the friction with lower layers. Not all of the cards tumble.
• That is why if we stir a pond at the centre, the motion dies as it move away from Centre
• Or when we throw stone in water, the wave created propagates a certain distance and then dies down
• The friction between the subsequent layers opposes motion• If more stirring is provided, more friction between adjacent layers
occur• This friction can be used to grind drug particle in between the layers

Understanding Super critical fluid This is phase diagram of water


1)what will happen if you continuously pressurize boiling water? (it will turn solid -Follow the vertical red line at 100 C towards up)2)What will happen if you subject ice into vacuum? (ice will directly convert into vapor – follow the vertical red line at 0 C downwardsQ) What will happen if we cross the critical point? (we get super critical fluids)

Ans) Beyond Critical point water has both property of liquid and vapor/gas) and is termed super critical fluid
This term is valid for any other solvents or gas
Precipitation done in such solventsResults in particle size of nano sizes


• Till now, students generally know of a single way of causing crystallization- supersaturation followed by cooling
• Now a new concept called, phase separation is introduced
• Eg Dissolve water insoluble, non-polar compound such as cinnamic ester into alcohol. Into it add enough water, the ester will ppt out. It is because alcohol will mix with water and the solution becomes more polar and the non-polar ester is forced to phase out ie turn solid from dissolved state.

7) Evaporative precipitation into Aqueous solution(EPAS)• EPAS uses rapid phase separation to nucleate and grow
nanoparticles and micro particles of lipophilic drugs. The drug is first dissolved in a low boiling point organic solvent. Then it is heated to a temperature above it’s BP but under high pressure. At this high pressure, it won’t evaporate (BP increases with increased pressure). Then the drug + solvent solution is spayed through fine atomizing nozzle into a heated aqueous solution at normal pressure. The normal atmosphere pressure, plus hot aqueous solution causes the organic solvent to instantly vaporize and such rapid removal of solvent causes a very fine dispersion of drug particle because the drug particles don’t get enough time to merge into bigger crystals. Surfactants are added to the mixture of organic and aqueous solution to optimize and stabilize particle formation
• Comparatively dissolution rates are faster for the SFL particles than EPAS.* *Eur J Pharm Biopharm. 2005 May;60(1):81-
9.(http://www.ncbi.nlm.nih.gov/pubmed/15848060)

8)Use of salt form• Salt are more soluble than original neutral forms eg
acid salt of atropine ie atropine HCl and basic salt of penicillin ie Sodium penicillin
• The selection of the counter-ion (the non-drug other part of the salt)must be selected on basis on its safety, therapeutic indication and route of administration
• Limitation– Drug needs to have acidic (eg COOH)or basic Functional
groups (eg NH2) or else not possible– Weakly acidic or weakly basic drugs don’t completely
ionize and thus are difficult to completely turn into salt– The salt may be hygroscopic (readily absorbs water) ,
exhibit polymorphism or has poor processing characteristics
– Reconversion of salt into original less soluble acid or base form on the surface of solid dosage form or in the stomach

Why salt have higher solubility?• Solubility appears to be a physical event but is actually a
chemical event. Salts can form ions which can form electrostatic bonds with polar solvents like water. The energy provided by this bonding is sufficient to break the intermolecular force between the ions in their solid crystalline state hence causing the salt to dissolve.

Why steroids won’t dissolve in water?
• First thing, stop saying drugs don’t dissolve in water. Technically they dissolve, it’s just a matter of how much they dissolve.
• In contrast to salt, steroids are very hydrophobic (just look how many rings they have!). It provides no scope for any electrostatic bonding. Thus they have very limited solubility in water
• However, they have scope of hydrophobic bonding ( not ionic or covalent, it is simple van der wall bond exclusive to carbons). Thus they easily dissolve in non-polar solvents such as hexane and benzene. Also hexane and benzene are not micible in water for the same reason.
• Conclusion : Like dissolves like (ionic in polar, hydrophobic in non-polar)


9) Use of surfactants:
• Surfactants are amphoteric compounds that can lower surface tension. They improve solubility by promoting wettability. (ie provide more contact between water and drug particles so more electrostatic interactions can occur). They are also used to stabilize drug suspensions.
• They need to be used below their critical micelle concerntration (CMC) or else they form micelles which entrap drug within them which prevents its dissolution
• Eg steroids such as spironolactone which is very lipophilic drug


Anionic
Cationic
Zwitterionic
Neutral


Imagine the surface of leaf to be the surface of a hydrophobic drug. If water doesn’t come in “molecular” contact with drug, there is no chance of solubility. Adding
surfactant can lower the surface tension such that water now spreads on the surface like that of wooden table. More contact of drug with water means more solubility
Water can’t wet the hydrophobic surface of taro
Water easily wets table wood

Watch on Youtube7.2 surfactants and Surface tension

Physically a solid can be inside water but weather water wets it or not depends on both the hydrophobicity of
solid and surface tension of water molecularlyVideo below shows magic sand

Conclusion of both videos (surfactant +solubility concept)
• If the magic sand was placed inside soap water instead of pure water, the decreased surface tension of water would have promoted “wettability” ie water will spread over the solid and it won’t be as dry as with pure water.
• What does this have to do with enhancing a Drug solubility?
• While we can’t change drugs hydrophobicity, we can certainly alter water’s surface tension. Lowering surface tension promotes more wettability ie more spreading of water which provides more physical contact between water and drug molecule which provides opportunity for chemical interaction ie form electrostatic bonds with cation and anions of salt or from dipole bonds or H bonds non-ionic but polar drug (sugar is not salt but has high solubility due to H bonds) with which can break the lattice of the solid crystal.

Hexane vs Hexanol vs Hexanol and surfactant
• Hexane – Practically insoluble cause no scope for any electrostatic bonding
• Hexanol – Hydroxyl group provides scope for electrostatic interaction but it can’t overcome the hydrophobic interaction between cyclohexane groups, thus not soluble
• Hexanol and surfactant – Surfactant increase wettability, allow more water-compound electrostatic interaction which relatively increases solubility than with no surfactant (*whether this increase allows hexanol to dissolve is to be tested practically)

10) Use of precipitation inhibitors• Drug can precipitate in the stomach if it encounters
super-saturation condition. This is unwanted and can be blocked by use of inert water soluble polymers such as HPMC,PVP,PVA,PEG. They work because– Increases viscosity thereby reducing rate of crystallization– Provides steric barrier to crystal growth– Absorb (stick) onto surface of host crystals, reduce the
crystal growth rate of the host and produce smaller crystals
• Such expcipients can maintain a supersaturated solution for low solubility drug. Such unnaturally highly concentrated solution can be absorbed in both higher rate and higher amount thus improving BA
• (just think a 5% sugar solution and 25 % sugar solution, which will show greater glucose level increase in blood and when?

11) Alteration of pH of the drug microenvironment• Excipients that alter pH can support salt formation
of drug in-situ (compound is not in salt form in the dosage form but converts to salt once inside GI tract) in case of tablets.
• For drug salt solution which are mostly used by IV, buffers are used to maintain the pH of solution and keep the drug in solution in salt form egphenytoin injection
• Generally used with salt form of drugs• (consider what happens in Na acetate solution
encounters HCl. The result is acetic acid and NaCl. Neutral acetic acid is less soluble than it’s sodium salt. If that soln is buffered , then it won’t happen )

12) Use of Amorphs, Anhydrates, Solvates form of drug
These refer to various polymorphic form of the drug iechemically same but different in physical amkeup
• Amorphous form is drug is more soluble than crystalline form because solubility mean breaking the lattice structure which is less ordered in amorphous form than in crystal form
• Anhydrate is more soluble than hydrate because hydrate is already chemically associated with solvent molecules Eg anhydrous CuSO4 vs CuSO4.5H20
(Remember – solubility is a chemical process)
• Solvate more soluble than non-solvate
• Solvate means the crystal state of drug contains a solvent. If the solvent is water then termed hydrate.

In molecular level, amorphous state is less disordered than crystalline state. Less energy is required to break apart the disordered amorphous state. It means amorphous form willdissolve more quickly (and also that amorphous form are less stable than crystalline form)
Crystalline vs amorphous form

13) Precipitation
• In this method poorly aqueous soluble drug is dissolved in a suitable organic solvent followed by its rapid mixing with a non-solvent which causes precipitation of drug in nanosized particles. Such product are termed hydrosol.

14) Hydrotrophy
• Hydrotrophy is the process where increase in solubility of drug in water beyond the normal saturation point is due to the presence of large amount of additives called hydrotropic agents. They are organic salts of alkali metal such as sodium benzoate, urea, sodium salicylate, nicotinamide, sodium acetate and sodium citrate. eg sodium salt of Ibuprofen in Ibuprofen, sodium benzoate in Carbamazepine, and sodium salicylate in Paracetamol

• Up to now: In all previous methods only the drug was considered. • It’s size was reduced
– By physical process (Jet mill, colloid mill)– By recrystallizing in a setting that promotes crystals of small size
• recrystallization in a super critical fluid (SCF)• Crystallization due to phase separation (EPAS)more complex• Crystallization due to phase separation (precipitation) simple process
• it was turned into salt, • It salt state was maintained by altering pH of solvent• Wettability, water’s contact with it, was increased (surfactant)• it’s precipitation from super saturated system was inhibited • It solubility was increased beyond its saturation point (hydrotrophy)• (note - the above three methods enhance solubility while other cause rapid
dissolution)• Carrier based methods: Now below methods consider drug which will be in a
mixed state with another molecule called carrier. The carrier is inert and either water soluble or not. The process involved results a solid mixture of drug and carrier which can be put into a tablet or suspension accordingly. The presence of carrier improves solubility by creating a very fine dispersion of drug when in contact with liquid.
• Water soluble carriers: MCC,starch• Water insoluble carriers: Bentonite

15) Solvent depositionIn this method a poorly aqueous soluble drug is dissolved in an organic solvent like alcohol and deposited on an inert, hydrophilic, solid matrix carriers such as starch or MCC by evaporation of solvent (filtration won’t work cause drug will also flow with solvent). The result is a solid mass which is pulverized, sieved and used for tablet making. In the stomach, the carrier is dissolved leaving behind very fine dispersion of drug.
16) Selective Adsorption on Insoluble carriersProcess similar to above but instead of hydrophilic carriers, here the carrier is inert, inorganic clays like bentonite. Although the carrier are insoluble, the drug absorbed on it’s surface is held by weak bonding and is quickly released in contact with solvent in form of very fine dispersion.

17) Solid Solutions(Solid solution can be easily understood if we think about alloys such as steel which is carbon dissolved in iron. Such combination improves strength and prevents rusting. In pharmaceutical context, a drug and solvent solid “solution” can increase solubility)
It is a physical mixture of a drug and a carrier which exist as a single solid such that he two components do not react with each other but the drug is being dispersed in the carrier in molecular level, thus names solid solution. Thy are prepared by fusion method whereby a physical mixture of solute and solvent are melted together followed by rapid solidification. Such system are called melts. eg griseofulvin-succinic acid melts dissolved 6-7 times faster than pure griseofulvin. Enhanced solubility results when the soluble carrier dissolves rapidly leaving behind the insoluble drug in a state of very fine microcrystalline dispersion which dissolves rapidly

Types of solid solution
A) based on miscibility of drug and carrier– Continuous solid solution is one in which the drug and
carrier are miscible in all proportions. This is an ideal but unrealistic situation
– Discontinuous solid solution is the one where solubility of each of the component in the other is limited
B) Based on distribution– Substitution crystalline solid solution is where drug
molecule replaces carrier molecule in it’s crystal lattice
– Interstitial crystalline solid solution is where drug molecule occupy the interstitial spaces (in between space)in the crystal lattice of carrier molecules
When the resultant solid solution is a homogenous transparent and brittle system, it is called as glass solution

Red is drug and black is carrier

18) Eutectic mixtureA eutectic mixture is defined as a mixture of two or more components which usually do not interact to form a new chemical compound but, which at certain ratios, inhibit the crystallization process of one another resulting in a system having a lower melting point than either of the component.These are simply a intimate blend of two solid cyrstals, one is drug and other is carrier however neither one is uniformly mixed in the other as in solid solution just that very small crystal of one is together with very small crystal of another. They are also prepared by fusion process. The difference from solid solution is that miscibility is not occurring in molecular level like sugar dissolved in water but on physical level only.Eg paractamol-urea, griseofulvin-urea, griseofulvin-succinic acidBoth solid solution and eutectic mixture are easy to prepare, simply melt the mixture of two compounds and cool rapidly, economical and requires no solventsThis method cannot be applied to:• Drugs which fail to crystalize from the mixed melt• Drugs or carriers which decompose below or at their melting point

Point M is melting point of A while point N is Melting Point of BObserve that at eutectic point, E, the Melting point of both
A and B is lowered ie it is below Point M and H

19) Solid dispersion/co-precipitation/co-evaporate
In this technique the drug and carrier are dissolved in a common volatile organic solvent such as alcohol and the liquid is removed under reduced pressure (note-boiling point is reduced at low pressure) or by freeze-drying which results in amorphous precipitation of drug in the crystalline carrier. Thus it can be applied to thermolabilesubstance. Solubility increase is a result of both amorphous form and formation of very fine dispersion after the carrier dissolves. (It is different than solid solution and eutectic mixture in that the process uses drying of volatile solvent instead of melting mixtures and is different than solvent deposition in that both drug and carrier are in dissolved state instead of only the drug being in solution)

Solid dispersion Eutectic, Solid solution
Dissolve drug and carrier in common volatile organic solution
Mix two solid drug
Remove solvent in low pressure
This causes rapid precipitation of both components
Very small sized drug in amorphous form is dispersed in carrier
Heat them until both melts and fuses
Cool them to obtain a single mass of (drug and carrier) mixture
Drug is dispersed crystal form in carrier in very small size to molecular level
Carrier releases drug as a very fine dispersion
Pulverize to get powder form
Carrier dissolves and releases drug in a very fine dispersion

The carrier is same as in solid solution and eutectic mixture.
Limitation:
• Since carrier is hydrophillic and drug is hydrophobic, it is difficult to find a common solvent to dissolve both
• The product is often soft, waxy and possesses poor compressibility and flowability (tablet making would be problematic)
• Difficult to reproduce
• The dispersion are physically unstable

20) Molecular encapsulation with cyclodextrins(inclusion)
Cyclodextrin are polymeric oligoscahharidescontaining 6,7 and 8 glucopyranose. They form a structure which as hydrophillic exterior and hydrophibic cavity. The cavity is of suitable size to host hydrophobic molecules and this form of drug and cyclodentrin is called molecular inclusion complex. The complex has good solubility and releases drug when in contact with water to produce a very fine dispersion which will have enhanced solubility due to small size. Cyclodextrinitself is not absorbed in stomach or intestine but degraded in the colon.


• The cavity of cyclodextrin can host drug inside it• It is not a hole where drug just drops into!• The cavity provides hydrophobic interaction with the
drug of low water solubility
At molecular level everything is chemistry!! Because nothing can exits without bonds

Enhancement of Permeability across the bio membrane
• Good Solubility alone is not a determining factor for good absorption. Since the drug needs to cross the biological membrane , permeability is also important especially for drugs with good water solubility. Techniques that improve permeability focus on lipid based carriers.
• Lipids are a group of hydrophobic naturally or synthetic molecules that include fats, waxes, sterols, fat-soluble vitamins (such as vitamins A, D, E, and K), monoglycerides, diglycerides, triglycerides, phospholipids, and others.


Lipid based technology
• Lipids based formulations can be manufactured as solutions, suspensions, emulsions, self-emulsifying systems and micro-emulsions and even tablets and apart from increasing permeability of hydrophilic drugs, they present opportunity to bypass aqueous solubility enhancing methods for lipophilic drugs (it means just make oily solution of lipophilic drug)

Various advantage of lipid based formulationsPharmaceutical advantage• Opportunity to formulate sustained release product• Flexibility to make various dosage from can be made Physiochemical advantage• Improved permeation of drugs • Improved solubility of drugs • Stabilization of drugs against hydrolysis or oxidationPharmacokinetic advantages• Reduced plasma profile variability• Potential for drug targeting applicationPharmacodynamic advantage• Reduced toxicity• Consistency in drug response

Lipid solutions and suspension
• Triglycerides can be used as a solvent to dissolve lipophilic drugs . The solution is encapsulated in soft gels eg Vit K soft gels. In the stomach the drug diffuses from the oily phase in the aqueous gut in a steady manner.
• This does not directly improve permeability and is actually a clever bypass of water solubility limitation. Remember that drugs are not 100% water insoluble. They are just not good at it like sugar or glucose. If the usual dose is introduce in water they start aggregating and won’t permeate easily. A clever way around this problem is to continuously release drug in small amount, such that they won’t aggregate but be dissolved and now they can easily permeate too since they are not a big aggregate.

Partition vs diffusion
• Diffusion of solute happens until both sides of semi-permeable membrane has equal conc
• Partition of solute happens between immiscible liquids where solute diffusion from more soluble to less soluble liquid, however the final conc of solute in the low layers of liquids won’t be same the solubility of solute is not same in both

Emulsions, microemulsions and Self Emulsifying Drug Delivery System (SEDDS) and Self Micro Emulsifying Drug Delivery System (SMEDDS)• Emulsions are tiny stabilized dispersions of oil in water(or water in oil too). This solves solubility problem as well as providing a greater surface area for drug release releasewhich facilitates permeability.
• Self emulsifying systems are mixtures of oils, surfactants and co-solvents /co-surfactants. Once administered in to the GI system, they are diluted with gastrointestinal fluid and the gastric motility provides the agitation for the formation of a fine oil-in-water (o/w) micro emulsion (SMEDDS). The difference between a SEDDS and SMEDDS is that the former when diluted results in a droplet size between 100 & 300 nm and the later results in a droplet size of less than 50 nm.

Lipid Nanoparticles (SLNs, NLCs & lipid drug conjugates)(all have single layer of outermost lipid)Solid Lipid Nanoparticles (SLC)• It has a single lipid bilayer which encloses a solid lipid core and the size of the
whole lipid particle is in nano micron size of about 100-1000uM. They have less drug loading capacity and tend to eject out drug in storage. (Earlier we were making the drug particle very small, now the drug carrier which is lipid is made very small).
Nanostructured lipid carrier (NLC) • Instead of pure solid lipid core, it is a mixture of both solid and liquid lipid which
has been size reduced to nano range. This increased drug loading capacityLipid drug conjugate nanoparticle • It is simply covalent bonding between drug and a lipid (just like cyclodextrin drug
complex) which have been size reduced to nano range. This increased drug loading capacity
Lipid nanoemulsion• It contains a single layer of lipid layer enclosing an oily fluid in which drug is
dissolved. The size of this structure is in nano range. It presents very high surface area from which drugs can slowly be diffused in water


Liposomes• Liposomes are spherical vessel made with
phospholipids bilayer which can hold an aqueous fluid. Thus It can entrap soluble drug in the hydrophilic core while the phospholipid outer part is the same thing that makes cell membrane and it can fuse with GI membrane it releases drug into the membrane and thus aids penetration. It can also be used to improve solubility of hydrophobic substances in which case the drug is entrapped with the bilayer. Liposome formulation are typically liquid preparation.Liposomes are biocompatible and biodegradable. Even though they fuse with biomembrane, they don’t cause toxicity


Surfactant Liposomes
Cyclodextrin

Cyclodextrin vs LiposomeCyclodextrin Liposome
Hydrophobic core Hydrophilic core
Hydrophillic surface, is not absorbed
Hydrophillic surface thatmimics cell membrane
Promotes solubility of hydrophobic drug by releasingthe host drug when in contact with water to create a very fine molecular dispersion of drug which dissolves rapidly
Promotes permeability of soluble drug by fusing with the cell membrane and pinching off the drug inside the membrane
Remember when using Surfactant to increase solubilityWe have to use in small conc. to prevent micelle formation

Ion Pairing
• Drugs that exist in ionized form or can get ionized (because they have acidic or basic functional group)and can’t penetrate the cell membrane. In such conditions their charge can be neutralized by pairing with an endogenous lipophilic ion of opposite charge eg mucin. The complex is neutral and very lipophillic, hence permeates easily. After absorption, the complex dissociates which releases the drug again in ionic state.

Endogenous lipophillic counter-ion• They are charged ligands which occur have a lilophillic portion
too eg mesylate (MES), acetate (AC), pyruvate (PYRU), nicotinate (NIC), hydrogenfumarate (HFUM), hydrogenmaleate (HMAL), p-toluenesulfonate (PTS), caproate(CPR), deoxycholate (DOC) and prostaglandin E1 anion (PGE1)
NICMES
HFUM
CPR
PYRU
DOCHMAL
PGE1


Penetration Enhancers
• These are compounds which facilitate the transport of drugs across the biomembrane. Normally macromolecules such protein or peptides are too big to permeate but with the chemical permeation enhancers even they can be easily absorbed. Eg sodium lauyrl sulphate, Cholic acid, Sodium deoxyglycolate, Urea, methyl pyrollidine, oleic acid etc
• Urea increases permeability of transdermal patches by hydrating the skin and formation of hydrophilic diffusion channels within the barrier
• Oleic acid greatly increased the flux of many drugs such as increasing the flux of salicylic acid 28-fold and 5-flurouracil flux 56-fold through human skin membrane in vitro

BA enhancement through enhancement of drugs stability
Enteric coating: Some drugs are degraded in acidic condition of stomach such as erythromycin, pancreatin, benzimidizoles, omeprazole but not in the relatively basic condition of intestine. Since drugs has to go through stomach first, this might be problematic. To overcome acid liability of drugs, the drug tablets are coated with acid resistant polymers which prevent acidic exposure to drugs. However we do want the drugs to be exposed in the intestines which is relatively basic than stomach and degradation is avoided. Thus the coating has to come off at the intestine.Enteric coating uses acid resistent polymers to coat tablets so as to prevent exposure of acid labile drugs such as erythromycin, pancreatin, benzimidizoles, omeprazole . The coating dissolves under basic condition in the intestine and drug gets absorbed from there.

Omeprazole in acidic condition

Complexation
• Complexation technique involves forming a weak and thus temporary chemical bond with drugs which improves it’s stability. Complexating agent can be caffeine, sodium salicylate, sodium benzoate and nicotinamide
• The complexed drug is not compatible to enzymatic binding site and hence it avoids being metabolized

Use of metabolic inhibitors
• Metabolism is chemical degradation of compounds and co-administeration of metabolic inhibitor thus increases bioavailbility for drugs. Primary site of metabolism is the liver but it can also happen in the intestinal wall known as prehepatic metabolism. These metabolic inhibitors can come from other drugs or diet, egincrease in cyclosporin BA by co-administration of ketoconazole which inhibits P-gp and grapefruit juice inhibits intestinal, not hepatic CYP3A4. Erythromycin is another metabolic inhibitor that can block liver enzymes

BA enhancement through GI retension
• It is a form of controlled drug delivery where drug product is kept in the stomach or intestine until all the dose has been absorbed.
• The excipients achieve this by being bioadhesiveor that swell on hydration which causes it to float in the stomach.
• Doing this– Increases contact with epithelial surfaces
– Prolongs residence time in the stomach
– Delaying intestinal transit

The answer to the little boy’s innocent question is thatAny epithelium(skin or walls of hollow organs) of body whether it be skin or the GI tract, nasal, ear, eye is capableof putting drug into the bloodstream. Once drug enter
the blood stream it can go anywhere within body
A suppository. It is inserted into the rectum

How safe is 1000mg paracetamoltaken every 6 hrs? very safe
• In a study, conducted by McNeil, who are the original innovators of paracetamol, in 1992 on healthy subjects plasma acetaminophen concentrations were determined after repeated doses of 1000 mg every six hours for two days (4 g / day), and they are shown in Figure 3-5. As predicted by acetaminophen’s short t½ and the dosing interval, there is minimal accumulation and acetaminophen is almost completely eliminated from the plasma at 12 hours following the last dose. The mean Cmax,ss at steady state with repeat doses of 1000 mg every six hours is 11.4 ± 3.8 m g/mL
http://www.fda.gov/ohrms/dockets/ac/02/briefing/3882B1_13_McNeil-Acetaminophen.htm#_Toc18717574

• short plasma half-life (t½) that ranges from 2 to 3 hours in healthy young and elderly adults and from 1.5 to 2.9 hours in children
• Because of its rapid clearance, repeated doses do not lead to accumulation of acetaminophen plasma concentrations
• Prospective pharmacokinetic studies show that with repeat doses of 1 and 1.5 g every 6 hours (ie total of 4 and 6 g per day) acetaminophen plasma concentrations reach steady-state levels within 10 to 15 hours and do not accumulate to higher levels with continued dosing. These results are consistent with the short elimination half-life of two to three hours for acetaminophen and the recommended dosing interval of four to six hours.

AnswersQ1) Both start from zero. Body does not wait for all of the drug from the
dose to be absorbed. But absorption rate is very high due to sink conditions, thus effect is increased in drug conc in plasma. Elimination only starts lowering drug concerntration once absorption is complete which happens at the peak of curve
Q2) The skewed graph indicates high rate of absorption and slow rate of elimination. Gastric fluid is about 250ml, blood is 5 lt and urine is about 250 ml. This difference in solvent volume creates a sink condition which makes sure that diffusion based absorption is always happening at a high conc difference which is why absorption phase is very steep. However, for the same reason , sink condition cannot maintained during elimination from blood to urine and thus rate of elimination is slow and gradual. Absorption comes before elimination. These combined effect causes the graph to be skewed to the left.
Q3) The tail reflects very, very slow rate of elimination caused by very , very slow rate of diffusion caused by very, very low conc gradient which naturally occurs as drug is eventually eliminated/cleared from plasma

Answers
Q4) The non-overlap during absorption of different does indicate different rate of diffusion, which is the absorption mechanism. Even though drug is same, it’s dose ie concerntration in stomach will be different. Higher dose puts a higher initial concerntration in stomach which results in greater concerntration difference between stomach and blood and consequently increases rate of diffusion. (This is also true for non-overlap during elimination phase)

Q6) Can’t say by just looking at graph. The time and associated Conc. data needs to be statistically treated to find how just how different the two BA curves are.
Q7) Trick question. Bioequivalence is not compared between different routes (since different routes naturally have different bioavailability)
Q8) Look for steepness or slope of line ie red one
Q9) Look for duration of action and consider that both are within therapeutic range

Q10) Why using oral solution limits one compartment modeling but using IV injection allows two compartment modeling?
It has to do with amount of drug in blood which is 100% in IV but less in oral solution for same starting dose. Drug gets distributed from blood to other compartments ie (organs, fat, muscle) by function of concerntration gradient which is high in IV dosage and thus more probable but not oral dosage (since limited absorption and 1st pass liver metabolism).
Q11) It is because the dissolution apparatus can only account for solubility, not permeability. BSC class 3 and 4 have low permeability issue. Thus the in vitro dissolution study which can only account for solubility in form of rate of drug release can’t be related to drug absorption in-vivo because permeability is low and can’t be factored
Q12) BA study can be done on anything. However, since 400mg paracetamol is the first of it’s kind, it has no pharmaceutical equivalent drug products to compare with. Hence BE study cannot and need not be done.
Q13) There is a chance but it can easily be controlled by the dosing.

More QnA
• Why salts are more soluble in water than neutral form?• Solubility in water is an event characterized by breakdown of
solutes molecules from their solid structure and their dispersionwhich is both caused and stabilized by formation of electrostatic bond between water and solute instead of chemical bonds between solute themselves which causes solute to dissociate from its solid state. Electronegativity difference between Oxygen and hydrogen creates polarity in their bond with oxygen having more share of electron than hydrogen. This polarity is used to make e electrostatic bond with neutral solute termed dipole-dipole interaction. But as salts can exist as ion within their solid form, the electrostatic bond transforms into dipole-ion interaction which is stronger and thus more stable than dipole-dipole. This increased stability cause’s solute to preferentially bond to water instead among themselves ietheir solid structure breaks down and hence it dissolves.

Q) Why amorphous form is more soluble than crystalline form?• Solubility in water is an event characterized by breakdown
of solutes molecules from their solid structure and their subsequent stabilization by solvent. Since amorphous form has less ordered structure than crystalline, its molecules are susceptible to being easily broken down, hence the increased solubility.
Q) Why the BA study is not affected by only considering healthy males and not females or patients? • The BA study aims to simply check formulation effect on
plasma profile of drug product by estimating if drug conc is in toxic, therapeutic or ineffective region as determined by the therapeutic range. It is not checking clinical effect of drug effectiveness in male vs female or healthy vs sick people (that has already been done and passed during clinical trials). Since therapeutic range is same for both sexes and healthy and normal people, technically any group can be used. Only one group need be chosen which conventionally is set as males.

• What problem will come if crossover BE study is applied to pateints?
• During crossover studies it is expected that in each period the volunteers be at same state of health so as to minimize variance in BA due to health condition. But with patients once they take drugs in first period, they get better and in the second phase they are not in the same ‘unhealthy’ condition as during the first period. This can cause unwanted biasness in data.
• Why cross-over method is preferred to parallel method in BE study?
• In cross over study every person is sampled twice, one for test and other for standard drug product. Thus any difference seen is purely due to formulation effect since both formulation were tested in the same person. However in parallel, one group receives test and other receives standard drug product. Thus the difference observed is not purely due to formulation difference but PK variation between the two groups.

Q) Why during BA study volunteers are kept in fasting state before giving drug?• Food can interfere with absorption. They can increase
absorption by blocking metabolic enzymes or decrease absorption by increasing GI peristaltic movement. Thus BA of drug is over or under estimated. Thus fasting condition is followed to avoid such variance.
Q)What problem will come if single dose protocol is applied to patients? Show though illustration• Single dose BA study requires sampling for about at
least 3-5 half lives to get proper estimation of AUC. Until that period is over patient can’t take his next dose of same drug or if he has more than one disease then he will miss other drug dose. Depending of the drug’s half life this period can be a whole day or more and in this time patient health can be compromised.

Q) What parameter determines the length of a BA or BE study period?
Ans. Study needs enough data within 5 half-life ie to process 96% elimination
Q) Why is solubility and dissolution tested over a wide pH range?
Ans. The wide pH range is to account for drug absorption through the Gut ie from acidic stomach to basic ileum and jejunum of small intestines. Also it allows satisfaction of one of the criteria of biowaiver which is that drug must not be absorbed from a specific part in gut but throughout the gut.


















