
BindingofImidazoletotheHemeofCytochrome and ... · 1 reduced rapidly (t 1⁄2 1 s). Because the cyt...
10
Binding of Imidazole to the Heme of Cytochrome c 1 and Inhibition of the bc 1 Complex from Rhodobacter sphaeroides I. EQUILIBRIUM AND MODELING STUDIES * □ S Received for publication, March 28, 2010, and in revised form, April 30, 2010 Published, JBC Papers in Press, May 6, 2010, DOI 10.1074/jbc.M110.128058 Oleksandr Kokhan ‡ , Vladimir P. Shinkarev § , and Colin A. Wraight ‡§1 From the ‡ Center for Biophysics and Computational Biology and the § Department of Biochemistry, University of Illinois, Urbana, Illinois 61801 We have used imidazole (Im) and N-methylimidazole (MeIm) as probes of the heme-binding cavity of membrane-bound cyto- chrome (cyt) c 1 in detergent-solubilized bc 1 complex from Rhodobacter sphaeroides. Imidazole binding to cyt c 1 substan- tially lowers the midpoint potential of the heme and fully inhib- its bc 1 complex activity. Temperature dependences showed that binding of Im (K d ≈ 330 M, 25 °C, pH 8) is enthalpically driven (H 0 56 kJ/mol, S 0 121 J/mol/K), whereas binding of MeIm is 30 times weaker (K d ≈ 9.3 mM) and is entropically driven (H 0 47 kJ/mol, S 0 ° 197 J/mol/K). The large enthalpic and entropic contributions suggest significant structural and solva- tion changes in cyt c 1 triggered by ligand binding. Comparison of these results with those obtained previously for soluble cyts c and c 2 suggested that Im binding to cyt c 1 is assisted by forma- tion of hydrogen bonds within the heme cleft. This was strongly supported by molecular dynamics simulations of Im adducts of cyts c, c 2 , and c 1 , which showed hydrogen bonds formed between the N H of Im and the cyt c 1 protein, or with a water molecule sequestered with the ligand in the heme cleft. Studies of the binding of small molecules to the heme in soluble type (or class) I cytochromes c have provided substan- tial insight into the equilibrium and kinetic properties of the heme domain, with longstanding application to understanding protein dynamics, stability, and folding (1–5). In addition, exogenous ligand binding is now proving very relevant to new- found functions of cytochrome (cyt) 2 c in apoptosis and peroxi- dative chemistry (reviewed in Refs. 6 and 7). For soluble cytochromes c and c 2 , exogenous ligands have been shown to bind to the heme with displacement of the native methionine ligand. In most cases, binding occurs preferentially to the ferric iron and causes substantial changes in the redox potential (8, 9) and spectral properties of the cytochrome (10 – 12). These include cyanide, azide, fluoride, imidazole, pyridine, and nitric oxide (8, 9, 13–15). For example, binding of imidazole (Im) to oxidized mitochondrial cyt c or bacterial cyt c 2 produces a blue shift in the Soret region as well as changes in a number of weak absorbance bands between 450 and 600 nm (10 –12). Binding also quenches a characteristic absorption band cen- tered at 695 nm (11, 16 –18), which is attributed to a charge transfer interaction between the oxidized heme iron (Fe 3 ) and the sulfur of the ligating methionine residue (5, 19). For ferrous cyt c, the dissociation constant for Im is more than an order of magnitude weaker (ferrous K d 1 M, ferric K d 30 mM) (9, 10, 12, 20, 21). In contrast to the extensive work on cyt c and c 2 , very little is known about ligand interactions with the heme of cyt c 1 , which is a key component of the cyt bc 1 complex, a ubiquitous and ancient membrane-bound, energy-transducing, electron trans- port protein (22, 23). Cytochrome c 1 is an integral membrane protein with a single hydrophobic transmembrane helix and a globular heme-binding domain with a typical c-type heme covalently attached to the protein by thioether linkages to two cysteine residues. A histidine and a methionine provide the fifth and sixth heme iron ligands, respectively. Although the globu- lar domain shares many of the properties and folds of type I cytochromes c, the sequence has little homology with that of cyt c or c 2 (22, 24, 25), and cyt c 1 is assigned to a separate class. The unusual structural features of cytochrome c 1 have been most convincingly accounted for by Baymann et al. (24). By compar- ing sequences of cyt c 1 from different species, they suggested that the expanded regions of cyt c 1 arose by evolution from a diheme cytochrome c 4 that lost the second heme. This hypoth- esis nicely explains why the globular heme-binding domain of cytochromes c 1 is up to 2 times larger than cyt c, with the major expansion in the C-terminal (or distal) half of the protein. Mutational studies on cyt c 1 have shown that changing the endogenous methionine ligand significantly modifies the mid- point potential of cyt c 1 (26 –29). However, very little has been reported for exogenous ligand interactions with the c 1 heme. Schejter and Berke (30) found cyanide to bind to isolated bovine cyt c 1 , with K d 4mM, but Potter (31) found no effect of 10 mM cyanide on mitochondrial cyt bc 1 , in situ. Osyzcka et al. (32) reported that cyanide binds tightly to ferric cyt c 1 from Rhodobacter capsulatus, with K d 25 M, but only partially inhibits cyt bc 1 . Guergova-Kuras et al. (33) found that His- tagged bc 1 complex from Rhodobacter sphaeroides was inhib- * This work was supported, in whole or in part, by National Institutes of Health Grant R01GM053508. □ S The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3. 1 To whom correspondence should be addressed: Center for Biophysics and Computational Biology, University of Illinois, 156 Davenport Hall, 607 S. Mathews Ave., Urbana, IL 61801. Tel.: 217-333-3245; Fax: 217-244-6615; E-mail: [email protected]. 2 The abbreviations used are: cyt, cytochrome; DAD, 2,3,5,6-tetramethyl-p- phenylenediamine (diaminodurene); DM, dodecyl maltoside; MOPS, 3-(N- morpholino)propanesulfonic acid; Im, imidazole; MeIm, N-methylimida- zole; bc 1 , cyt bc 1 complex or ubiquinol:cyt c oxidoreductase; c 1 -Im, cyt c 1 with bound Im; DQH 2 , 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzohydro- quinone (decyl-ubiquinol); ISP, Rieske iron-sulfur protein; E m7 , midpoint potential at pH 7. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 285, NO. 29, pp. 22513–22521, July 16, 2010 © 2010 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. JULY 16, 2010 • VOLUME 285 • NUMBER 29 JOURNAL OF BIOLOGICAL CHEMISTRY 22513 by guest on May 27, 2018 http://www.jbc.org/ Downloaded from
-
Upload
vuongkhanh -
Category
Documents
-
view
216 -
download
2
Transcript of BindingofImidazoletotheHemeofCytochrome and ... · 1 reduced rapidly (t 1⁄2 1 s). Because the cyt...

Binding of Imidazole to the Heme of Cytochrome c1 andInhibition of the bc1 Complex from Rhodobacter sphaeroidesI. EQUILIBRIUM AND MODELING STUDIES*□S
Received for publication, March 28, 2010, and in revised form, April 30, 2010 Published, JBC Papers in Press, May 6, 2010, DOI 10.1074/jbc.M110.128058
Oleksandr Kokhan‡, Vladimir P. Shinkarev§, and Colin A. Wraight‡§1
From the ‡Center for Biophysics and Computational Biology and the §Department of Biochemistry, University of Illinois,Urbana, Illinois 61801
Wehave used imidazole (Im) andN-methylimidazole (MeIm)as probes of the heme-binding cavity of membrane-bound cyto-chrome (cyt) c1 in detergent-solubilized bc1 complex fromRhodobacter sphaeroides. Imidazole binding to cyt c1 substan-tially lowers themidpoint potential of the heme and fully inhib-its bc1 complex activity. Temperature dependences showed thatbinding of Im (Kd ≈ 330 �M, 25 °C, pH 8) is enthalpically driven(�H0 � �56 kJ/mol, �S0 � �121 J/mol/K), whereas binding ofMeIm is 30 timesweaker (Kd≈9.3mM) and is entropically driven(�H0 � 47 kJ/mol,�S0°� 197 J/mol/K). The large enthalpic andentropic contributions suggest significant structural and solva-tion changes in cyt c1 triggered by ligand binding. Comparisonof these results with those obtained previously for soluble cyts cand c2 suggested that Im binding to cyt c1 is assisted by forma-tion of hydrogen bonds within the heme cleft. This was stronglysupported by molecular dynamics simulations of Im adducts ofcyts c, c2, and c1, which showedhydrogen bonds formedbetweenthe N�H of Im and the cyt c1 protein, or with a water moleculesequestered with the ligand in the heme cleft.
Studies of the binding of small molecules to the heme insoluble type (or class) I cytochromes c have provided substan-tial insight into the equilibrium and kinetic properties of theheme domain, with longstanding application to understandingprotein dynamics, stability, and folding (1–5). In addition,exogenous ligand binding is now proving very relevant to new-found functions of cytochrome (cyt)2 c in apoptosis and peroxi-dative chemistry (reviewed in Refs. 6 and 7).For soluble cytochromes c and c2, exogenous ligands have
been shown to bind to the hemewith displacement of the nativemethionine ligand. In most cases, binding occurs preferentiallyto the ferric iron and causes substantial changes in the redox
potential (8, 9) and spectral properties of the cytochrome (10–12). These include cyanide, azide, fluoride, imidazole, pyridine,andnitric oxide (8, 9, 13–15). For example, binding of imidazole(Im) to oxidizedmitochondrial cyt c or bacterial cyt c2 producesa blue shift in the Soret region as well as changes in a number ofweak absorbance bands between 450 and 600 nm (10–12).Binding also quenches a characteristic absorption band cen-tered at 695 nm (11, 16–18), which is attributed to a chargetransfer interaction between the oxidized heme iron (Fe3�) andthe sulfur of the ligating methionine residue (5, 19). For ferrouscyt c, the dissociation constant for Im is more than an order ofmagnitude weaker (ferrous Kd �1 M, ferric Kd � 30 mM) (9, 10,12, 20, 21).In contrast to the extensive work on cyt c and c2, very little is
known about ligand interactions with the heme of cyt c1, whichis a key component of the cyt bc1 complex, a ubiquitous andancientmembrane-bound, energy-transducing, electron trans-port protein (22, 23). Cytochrome c1 is an integral membraneprotein with a single hydrophobic transmembrane helix and aglobular heme-binding domain with a typical c-type hemecovalently attached to the protein by thioether linkages to twocysteine residues. Ahistidine and amethionine provide the fifthand sixth heme iron ligands, respectively. Although the globu-lar domain shares many of the properties and folds of type Icytochromes c, the sequence has little homologywith that of cytc or c2 (22, 24, 25), and cyt c1 is assigned to a separate class. Theunusual structural features of cytochrome c1 have been mostconvincingly accounted for by Baymann et al. (24). By compar-ing sequences of cyt c1 from different species, they suggestedthat the expanded regions of cyt c1 arose by evolution from adiheme cytochrome c4 that lost the second heme. This hypoth-esis nicely explains why the globular heme-binding domain ofcytochromes c1 is up to 2 times larger than cyt c, with themajorexpansion in the C-terminal (or distal) half of the protein.Mutational studies on cyt c1 have shown that changing the
endogenous methionine ligand significantly modifies the mid-point potential of cyt c1 (26–29). However, very little has beenreported for exogenous ligand interactions with the c1 heme.Schejter andBerke (30) found cyanide to bind to isolated bovinecyt c1, withKd � 4mM, but Potter (31) found no effect of 10mM
cyanide on mitochondrial cyt bc1, in situ. Osyzcka et al. (32)reported that cyanide binds tightly to ferric cyt c1 fromRhodobacter capsulatus, with Kd � 25 �M, but only partiallyinhibits cyt bc1. Guergova-Kuras et al. (33) found that His-tagged bc1 complex from Rhodobacter sphaeroides was inhib-
* This work was supported, in whole or in part, by National Institutes of HealthGrant R01GM053508.
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Figs. S1–S3.
1 To whom correspondence should be addressed: Center for Biophysics andComputational Biology, University of Illinois, 156 Davenport Hall, 607 S.Mathews Ave., Urbana, IL 61801. Tel.: 217-333-3245; Fax: 217-244-6615;E-mail: [email protected].
2 The abbreviations used are: cyt, cytochrome; DAD, 2,3,5,6-tetramethyl-p-phenylenediamine (diaminodurene); DM, dodecyl maltoside; MOPS, 3-(N-morpholino)propanesulfonic acid; Im, imidazole; MeIm, N-methylimida-zole; bc1, cyt bc1 complex or ubiquinol:cyt c oxidoreductase; c1-Im, cyt c1
with bound Im; DQH2, 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzohydro-quinone (decyl-ubiquinol); ISP, Rieske iron-sulfur protein; Em7, midpointpotential at pH 7.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 285, NO. 29, pp. 22513–22521, July 16, 2010© 2010 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
JULY 16, 2010 • VOLUME 285 • NUMBER 29 JOURNAL OF BIOLOGICAL CHEMISTRY 22513
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ited when isolated with imidazole. This was attributed to low-ering of Em7 of cyt c1 induced by imidazole binding on the basisof its known interaction with cyt c.In this work, we describe and characterize the reversible
binding of Im to cyt c1 in isolated cyt bc1 complex from R. spha-eroides, causing a large drop inmidpoint potential and full inhi-bition of cyt bc1 activity. We discuss the implications of thethermodynamics of Im binding in terms of the dynamics of theheme domain and the peptide sequence that contains the sixthaxial ligand of the c1 heme iron and use molecular dynamics toillustrate the probable mode of interaction of Im in the hemepocket. The kinetics of Im binding are described in the accom-panying article (70) and reveal novel, transient interactions ofthe ligand with the protein, leading to facilitated entry into theheme pocket.
EXPERIMENTAL PROCEDURES
The strain ofR. sphaeroideswith a hexahistidine tag attachedto the C terminus of cyt bwas generously provided by Dr. A. R.Crofts (strain BC17 carrying the pGB11BH6 plasmid) (33).Cells were grown photosynthetically at 30 °C on Sistrom’smediumwith 2 �g/ml tetracycline and 10 �g/ml of kanamycin.R. sphaeroides cellswere collected by a 10-min centrifugation at8,000 � g and resuspended with 50mMMOPS, pH 7.0, 100 mM
KCl (buffer B). DNase I was added to a concentration of 10�g/ml. The cells were disrupted with a French press at 17,000p.s.i., and debris was removed by a 20-min centrifugation at48,000 � g. The supernatant was centrifuged for 90 min at140,000 � g, and the pellet was resuspended in buffer B.
Cytochrome bc1 complex was isolated from R. sphaeroideschromatophores and purified in a single step by following theprotocol described by Guergova-Kuras et al. (33) with modifi-cations. Unless otherwise specified, bc1 complex was isolated asfollows. 10 ml of chromatophores (A375 � 50 OD units) weremixedwith 10ml ofNi2�-nitrilotriacetic acid resin equilibratedin buffer B (1:2, v/v). Dodecyl maltoside (DM) was added to afinal concentration of 1% (w/v) and stirred at 4 °C for 40 min.The mixture was transferred to a column, and the resin waspacked while eluting. The column (packed volume �3 ml) waswashed with buffer B plus 20 mM cholate, 1 mM ascorbate at 1ml/min for 90–120 min. bc1 complex was eluted with buffer Bplus 20 mM cholate, 1 mM ascorbate, 10 mM EDTA). The bc1complex was washed by repeated dilution with buffer B plus 20mM cholate and concentration using Centriplus (Amicon Inc.,Beverly,MA) concentratorswith 10 kDa cut-off and finally con-centrated to �50 �M, mixed with glycerol (to 20%, v/v), andstored at �80 °C.Ni2�-nitrilotriacetic acid resin was obtained from Qiagen
(Hilden, Germany). Decyl-ubiquinone was either from BioMol(Plymouth Meeting, PA) or from Sigma. Horse heart cyto-chrome c (C2506) and all other chemicals were purchased fromSigma. DM was purchased from Anatrace (Maumee, OH).Decyl-ubiquinol (DQH2) was prepared from decyl-ubiquinoneas described by Trumpower and Edwards (34).The rate of electron transfer from decyl-ubiquinol to cyt c
catalyzed by bc1 was measured from the rate of reduction of cytc using the extinction coefficient ��551–540 � 18.5 mM�1 cm�1
(35), in a buffer containing 50 mM Tris-HCl, pH 8.0, 100 mM
NaCl, 25 �M cyt c, 20 �M DQH2, 0.01% DM, and 10–20 nM bc1complex, at room temperature, using an Agilent 8453 spectro-photometer, unless otherwise stated. Non-enzymatic electrontransport wasmeasured in the same buffer in the absence of theenzyme, and the rate was subtracted from the enzymatic one.Imidazole binding was monitored either by following spec-
tral changes at �A416–400 or by adding ascorbate and diamino-durene (DAD; 2,3,5,6-tetramethyl-p-phenylenediamine) tofinal concentrations of 1mM and 10�M, respectively, at varioustimes after incubation with imidazole and measuring the frac-tion of cyt c1 reduced rapidly (t1⁄2 � 1 s). Because the cyt c1-im-idazole complex has amuch lowermidpoint potential than freecyt c1, only the latter is promptly reduced after mixing withascorbate/DAD. Ferricyanide-oxidized bc1 complex (100 �M
ferricyanide and bc1 complex, containing 0.5–2 �M cyt c1(��552–541 � 20 mM�1 cm�1 (36)) in 50 mM Tris, pH 8.0, 100mMNaCl, 20 mM cholate) was incubated with various amountsof imidazole at room temperature or on ice.Imidazole binding to heme (free or in protein) is well estab-
lished to occur through the deprotonated nitrogen (11, 37).Literature values for the pKa of both imidazole and N-methy-limidazole are 6.95–7.05, and they are generally considered thesame (38, 39). At pH 8, used here, both are 90% deprotonated,and concentrations and concentration-dependent parameters(e.g. Kd) are given in terms of total concentration of Im orMeIm, without correction.Kinetic measurements of reactions with rate constants
slower than 0.5 s�1 were typically performed on a diode-arrayAgilent 8453 spectrophotometer (integration time 100 ms)equipped with a UV blocking filter (Corning CS 0–52, cut-offwavelength 320 nm) to prevent a light-induced reduction ofcyt c1. All reactions faster than 1 s�1 were studied using anApplied Photophysics SX-17MV stopped-flow spectropho-tometer, with mixing time less than 2 ms and integration time2.5 ms. Reactions with rate constants in the 0.5–1 s�1 rangewere typically studied using both instruments to provide over-lap of the measurement ranges.Molecular dynamics simulations of imidazole bound to cyts
c, c2, and c1 were done on the Turing cluster operated by theComputational Science and Engineering program at the Uni-versity of Illinois, using the NAMD molecular dynamics pro-gram (40). All simulations were performed at constant particlenumber, pressure (1 atm) and temperature (300 K), followingthe procedure of Autenrieth et al. (41), with the CHARMM 27force field (42, 43) and standard NAMD parameters.Atom coordinates for simulation of horse heart cyt c with Im
were taken from the Protein Data Bank 1FI7 NMR structure(14). For cyt c2, the coordinates were from the Protein DataBank 1CXA x-ray structure (13). All simulations on ligandinteraction with cyt c1 of R. sphaeroideswere performed on theglobular domain from Protein Data Bank entry 2FYN (44). Allcrystallographic water molecules were removed, and eachprotein was placed in a water-filled box (see supplementalmaterial). Preparation of the imidazole-bound state of cyt c1was initiated by “opening” the heme cleft by pulling on themethionine sulfur atom in a short (1–2-ns) simulation until theforce dropped due to reorientation of the Met-185 side chain(at about 600 piconewtons).
Imidazole Inhibition of the bc1 Complex
22514 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 29 • JULY 16, 2010
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Four simulations (10–25 ns) were performed for each pro-tein: onewith the imidazole ring oriented at 0° (for cyts c and c2,this corresponds to the known structures) and three simula-tions where the imidazole plane was rotated 90, 180, and 270°around the axis perpendicular to the heme plane. For cyt c1, the0° positionwas based on the 1CXA structure for cyt c2, by align-ing the hemes.
RESULTS
Spectral Properties of the Imidazole-Cyt c1 Complex—Incu-bation of fully oxidized R. sphaeroides bc1 complex with Imresulted in significant spectral changes, similar to thosereported for cytochromes c and c2 (11, 12, 45). Themost prom-inent features of the difference spectrumwere a peak at 400 nmand trough at 416 nm, with an isosbestic point at 406 nm (Fig.1), indicating that Im interacts directly with cyt c1, causing ablue shift in the Soret peak at �414 nm. There was also adecrease in absorption around 700 nm (Fig. 1, inset). The wave-length and approximate extinction coefficient (0.2–0.5 mM�1
cm�1) of this feature are consistent with a characteristicabsorbance attributed to the methionine S-heme Fe3� bond,suggesting that Im displaces the methionine ligand from theheme of cyt c1, as previously observed for ferricytochrome c (11,18) and c2 (10, 45, 46). No imidazole-induced spectral changeswere seen for the ascorbate-reduced bc1 complex (cyt c1 andiron-sulfur center reduced, both b-hemes oxidized), indicatingthe sole involvement of ferric cytochrome c1 in the absorbancechanges under oxidizing conditions (Fig. 1, dotted line).Redox Behavior of the Cytochrome c1-Im Complex—In the
presence of imidazole, the cyt c1 heme becomes non-reducibleby ascorbate/DAD (Fig. 2) except on a time scale of severalmin,
which we ascribe to reduction after the release of Im. The mid-point potential of ascorbate is not very well defined, due toirreversibility (47), but Elberry et al. (48) found that isolated bc1complexeswithmutant cyt c1withEm� �50mVwere 25–40%reducible by ascorbate. Thus, the midpoint potential of thec1-Im complex is significantly less than �50 mV. However, it isvery rapidly (�1 s) reduced by sodium dithionite (Eh � �300mV), indicating that at sufficiently low potential, the c1-Imcomplex can be reduced directly rather than after release of Im.This is confirmed by the more distinct peak at 550 nm (Fig. 2B,black). This feature decayed slowly to the native spectrumwitha lifetime of about 90 s, reflecting the slowunbinding of Im fromthe ferrous heme.The implied large drop in midpoint potential of cyt c1 with
bound Im provides a convenient assay of the fraction of cyt c1with bound ligand; only uncomplexed cyt c1 (Em7 � 260 mV(49)) can be quickly reduced by ascorbate/DAD. A similar assaywas used byOsyzcka et al. (32) tomonitor binding of cyanide tocyt bc1.Binding of Imidazole to Cytochrome c1—Binding of imidazole
by cyt c1 is quite tight but slow. At 25 °C, Im binding to cyt c1was sufficiently fast that the kinetics and end point values couldbe readily monitored by the spectral changes in the Soret bandat 416–400 nm. Fig. 3A shows kinetic traces obtained at 25 °Cwith Im concentrations ranging from 0.1 to 5 mM. From theamplitudes of the final absorbance change, we estimated Kd �0.33 0.07 mM (Fig. 3B).At lower temperatures, the rate of binding decreased dra-
matically. At 15 °C, binding could still be assayed by the spectralchange in the Soret band and gave Kd � 0.12 0.015 mM. At0 °C, however, binding was extremely slow, especially at thelowest Im concentrations, and was followed over a period of 10days (Fig. 4B). The extent of binding at each time point wasmeasured by the reducibility of cyt c1 by ascorbate/DAD (Fig.4A). This yielded a dissociation constant, Kd � 40 7 �M (Fig.4C). Combining the dissociation constants at temperatures0, 15, and 25 °C leads to �H0 � �56 6 kJ/mol and �S0 �
FIGURE 1. Spectral changes induced by imidazole binding to ferricya-nide-oxidized (solid lines) and ascorbate-reduced (dotted line) bc1 com-plex from R. sphaeroides at 25 °C. Inset, absorption changes around � � 700nm. Conditions were as follows. 1.5 �M bc1 in 50 mM Tris, pH 8, 100 mM NaCl, 20mM cholate, 100 �M ferricyanide, was mixed with 1 mM imidazole (final con-centration), and spectra were taken at 15, 30, 60, and 300 s after mixing.Interaction of Im with the ascorbate-reduced bc1 complex was measured sim-ilarly, except the buffer contained 1 mM ascorbate and 10 �M DAD in place of100 �M ferricyanide. All difference spectra obtained in the presence of ascor-bate were virtually identical, and only the last one is shown, after 5 min ofincubation with imidazole.
FIGURE 2. The effect of imidazole on the reducibility of cyt c1 in bc1 com-plex. Cyt bc1 stock (�20 �M) in 50 mM Tris, pH 8.0, 100 mM NaCl, 0.01% DM plus100 �M ferricyanide to fully oxidize the complex were incubated on ice for 5 hwithout (A) or with (B) 50 mM imidazole. Samples were then diluted 20-fold, andoxidized spectra were recorded. 1 mM ascorbate, 10 �M DAD was then added,and spectra were taken for up to 1 min to ensure that equilibrium was reached(solid lines). The samples were then reduced with a few crystals of sodium dithio-nite, and fully reduced spectra were obtained after 20–30 s (dashed lines).
Imidazole Inhibition of the bc1 Complex
JULY 16, 2010 • VOLUME 285 • NUMBER 29 JOURNAL OF BIOLOGICAL CHEMISTRY 22515
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
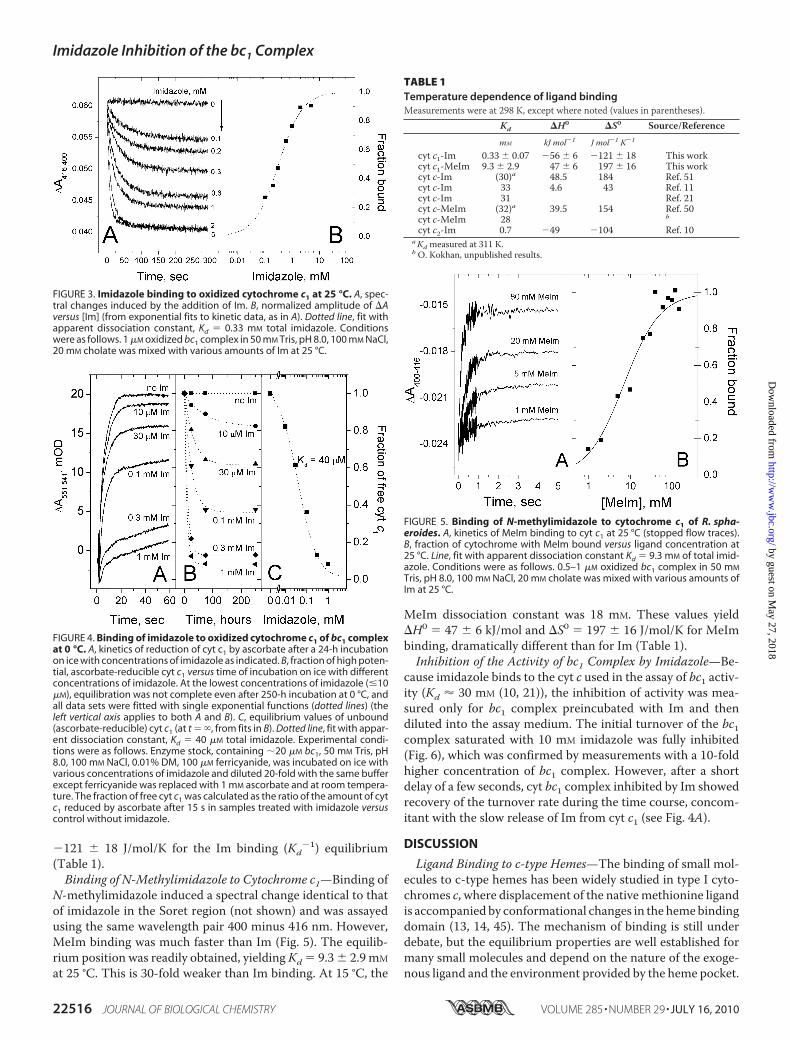
�121 18 J/mol/K for the Im binding (Kd�1) equilibrium
(Table 1).Binding of N-Methylimidazole to Cytochrome c1—Binding of
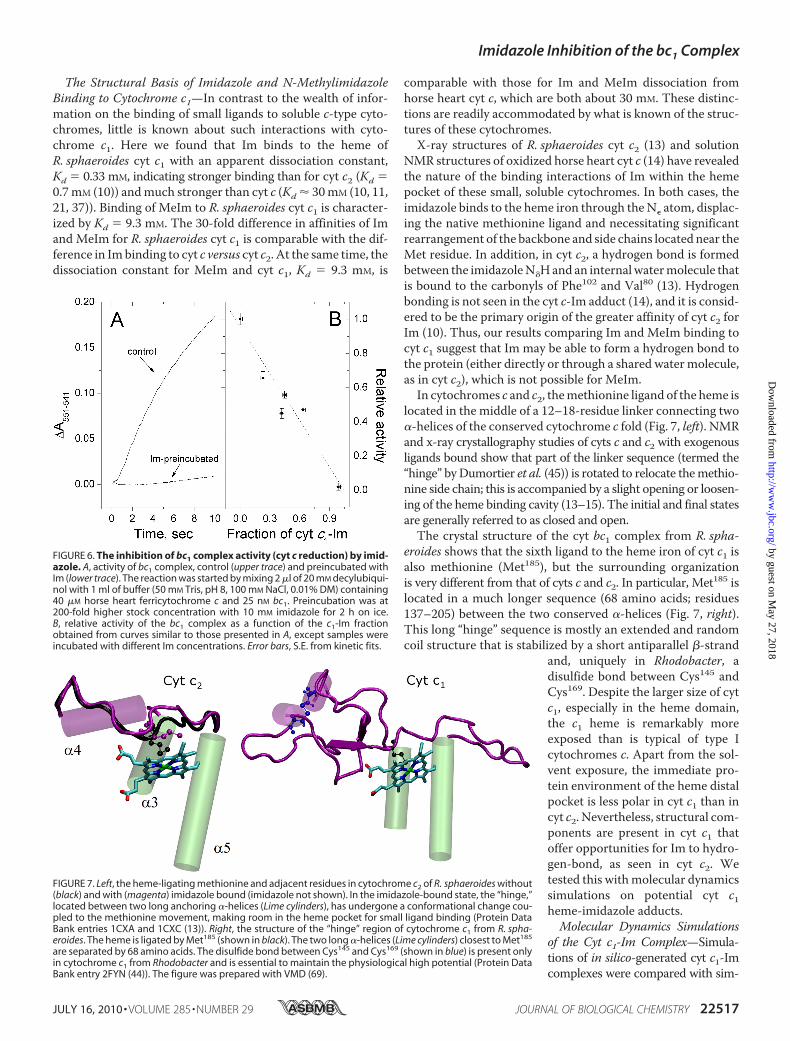
N-methylimidazole induced a spectral change identical to thatof imidazole in the Soret region (not shown) and was assayedusing the same wavelength pair 400 minus 416 nm. However,MeIm binding was much faster than Im (Fig. 5). The equilib-rium position was readily obtained, yieldingKd � 9.3 2.9 mM
at 25 °C. This is 30-fold weaker than Im binding. At 15 °C, the
MeIm dissociation constant was 18 mM. These values yield�H0 � 47 6 kJ/mol and �S0 � 197 16 J/mol/K for MeImbinding, dramatically different than for Im (Table 1).Inhibition of the Activity of bc1 Complex by Imidazole—Be-
cause imidazole binds to the cyt c used in the assay of bc1 activ-ity (Kd � 30 mM (10, 21)), the inhibition of activity was mea-sured only for bc1 complex preincubated with Im and thendiluted into the assay medium. The initial turnover of the bc1complex saturated with 10 mM imidazole was fully inhibited(Fig. 6), which was confirmed by measurements with a 10-foldhigher concentration of bc1 complex. However, after a shortdelay of a few seconds, cyt bc1 complex inhibited by Im showedrecovery of the turnover rate during the time course, concom-itant with the slow release of Im from cyt c1 (see Fig. 4A).
DISCUSSION
Ligand Binding to c-type Hemes—The binding of small mol-ecules to c-type hemes has been widely studied in type I cyto-chromes c, where displacement of the nativemethionine ligandis accompanied by conformational changes in the hemebindingdomain (13, 14, 45). The mechanism of binding is still underdebate, but the equilibrium properties are well established formany small molecules and depend on the nature of the exoge-nous ligand and the environment provided by the heme pocket.
FIGURE 3. Imidazole binding to oxidized cytochrome c1 at 25 °C. A, spec-tral changes induced by the addition of Im. B, normalized amplitude of �Aversus [Im] (from exponential fits to kinetic data, as in A). Dotted line, fit withapparent dissociation constant, Kd � 0.33 mM total imidazole. Conditionswere as follows. 1 �M oxidized bc1 complex in 50 mM Tris, pH 8.0, 100 mM NaCl,20 mM cholate was mixed with various amounts of Im at 25 °C.
FIGURE 4. Binding of imidazole to oxidized cytochrome c1 of bc1 complexat 0 °C. A, kinetics of reduction of cyt c1 by ascorbate after a 24-h incubationon ice with concentrations of imidazole as indicated. B, fraction of high poten-tial, ascorbate-reducible cyt c1 versus time of incubation on ice with differentconcentrations of imidazole. At the lowest concentrations of imidazole (�10�M), equilibration was not complete even after 250-h incubation at 0 °C, andall data sets were fitted with single exponential functions (dotted lines) (theleft vertical axis applies to both A and B). C, equilibrium values of unbound(ascorbate-reducible) cyt c1 (at t � , from fits in B). Dotted line, fit with appar-ent dissociation constant, Kd � 40 �M total imidazole. Experimental condi-tions were as follows. Enzyme stock, containing �20 �M bc1, 50 mM Tris, pH8.0, 100 mM NaCl, 0.01% DM, 100 �M ferricyanide, was incubated on ice withvarious concentrations of imidazole and diluted 20-fold with the same bufferexcept ferricyanide was replaced with 1 mM ascorbate and at room tempera-ture. The fraction of free cyt c1 was calculated as the ratio of the amount of cytc1 reduced by ascorbate after 15 s in samples treated with imidazole versuscontrol without imidazole.
FIGURE 5. Binding of N-methylimidazole to cytochrome c1 of R. spha-eroides. A, kinetics of MeIm binding to cyt c1 at 25 °C (stopped flow traces).B, fraction of cytochrome with MeIm bound versus ligand concentration at25 °C. Line, fit with apparent dissociation constant Kd � 9.3 mM of total imid-azole. Conditions were as follows. 0.5–1 �M oxidized bc1 complex in 50 mM
Tris, pH 8.0, 100 mM NaCl, 20 mM cholate was mixed with various amounts ofIm at 25 °C.
TABLE 1Temperature dependence of ligand bindingMeasurements were at 298 K, except where noted (values in parentheses).
Kd �H0 �S0 Source/Reference
mM kJ mol�1 J mol�1 K�1
cyt c1-Im 0.33 0.07 �56 6 �121 18 This workcyt c1-MeIm 9.3 2.9 47 6 197 16 This workcyt c-Im (30)a 48.5 184 Ref. 51cyt c-Im 33 4.6 43 Ref. 11cyt c-Im 31 Ref. 21cyt c-MeIm (32)a 39.5 154 Ref. 50cyt c-MeIm 28 b
cyt c2-Im 0.7 �49 �104 Ref. 10aKd measured at 311 K.b O. Kokhan, unpublished results.
Imidazole Inhibition of the bc1 Complex
22516 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 29 • JULY 16, 2010
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from
The Structural Basis of Imidazole and N-MethylimidazoleBinding to Cytochrome c1—In contrast to the wealth of infor-mation on the binding of small ligands to soluble c-type cyto-chromes, little is known about such interactions with cyto-chrome c1. Here we found that Im binds to the heme ofR. sphaeroides cyt c1 with an apparent dissociation constant,Kd � 0.33 mM, indicating stronger binding than for cyt c2 (Kd �0.7mM (10)) andmuch stronger than cyt c (Kd � 30mM (10, 11,21, 37)). Binding of MeIm to R. sphaeroides cyt c1 is character-ized by Kd � 9.3 mM. The 30-fold difference in affinities of Imand MeIm for R. sphaeroides cyt c1 is comparable with the dif-ference in Imbinding to cyt c versus cyt c2. At the same time, thedissociation constant for MeIm and cyt c1, Kd � 9.3 mM, is
comparable with those for Im and MeIm dissociation fromhorse heart cyt c, which are both about 30 mM. These distinc-tions are readily accommodated by what is known of the struc-tures of these cytochromes.X-ray structures of R. sphaeroides cyt c2 (13) and solution
NMR structures of oxidized horse heart cyt c (14) have revealedthe nature of the binding interactions of Im within the hemepocket of these small, soluble cytochromes. In both cases, theimidazole binds to the heme iron through theN� atom, displac-ing the native methionine ligand and necessitating significantrearrangement of the backbone and side chains located near theMet residue. In addition, in cyt c2, a hydrogen bond is formedbetween the imidazoleN�Hand an internalwatermolecule thatis bound to the carbonyls of Phe102 and Val80 (13). Hydrogenbonding is not seen in the cyt c-Im adduct (14), and it is consid-ered to be the primary origin of the greater affinity of cyt c2 forIm (10). Thus, our results comparing Im and MeIm binding tocyt c1 suggest that Im may be able to form a hydrogen bond tothe protein (either directly or through a shared water molecule,as in cyt c2), which is not possible for MeIm.
In cytochromes c and c2, themethionine ligand of the heme islocated in the middle of a 12–18-residue linker connecting two�-helices of the conserved cytochrome c fold (Fig. 7, left). NMRand x-ray crystallography studies of cyts c and c2 with exogenousligands bound show that part of the linker sequence (termed the“hinge” byDumortier et al. (45)) is rotated to relocate themethio-nine side chain; this is accompanied by a slight opening or loosen-ing of the heme binding cavity (13–15). The initial and final statesare generally referred to as closed and open.The crystal structure of the cyt bc1 complex from R. spha-
eroides shows that the sixth ligand to the heme iron of cyt c1 isalso methionine (Met185), but the surrounding organizationis very different from that of cyts c and c2. In particular, Met185 islocated in a much longer sequence (68 amino acids; residues137–205) between the two conserved �-helices (Fig. 7, right).This long “hinge” sequence is mostly an extended and randomcoil structure that is stabilized by a short antiparallel �-strand
and, uniquely in Rhodobacter, adisulfide bond between Cys145 andCys169. Despite the larger size of cytc1, especially in the heme domain,the c1 heme is remarkably moreexposed than is typical of type Icytochromes c. Apart from the sol-vent exposure, the immediate pro-tein environment of the heme distalpocket is less polar in cyt c1 than incyt c2. Nevertheless, structural com-ponents are present in cyt c1 thatoffer opportunities for Im to hydro-gen-bond, as seen in cyt c2. Wetested this withmolecular dynamicssimulations on potential cyt c1heme-imidazole adducts.Molecular Dynamics Simulations
of the Cyt c1-Im Complex—Simula-tions of in silico-generated cyt c1-Imcomplexes were compared with sim-
FIGURE 6. The inhibition of bc1 complex activity (cyt c reduction) by imid-azole. A, activity of bc1 complex, control (upper trace) and preincubated withIm (lower trace). The reaction was started by mixing 2 �l of 20 mM decylubiqui-nol with 1 ml of buffer (50 mM Tris, pH 8, 100 mM NaCl, 0.01% DM) containing40 �M horse heart ferricytochrome c and 25 nM bc1. Preincubation was at200-fold higher stock concentration with 10 mM imidazole for 2 h on ice.B, relative activity of the bc1 complex as a function of the c1-Im fractionobtained from curves similar to those presented in A, except samples wereincubated with different Im concentrations. Error bars, S.E. from kinetic fits.
FIGURE 7. Left, the heme-ligating methionine and adjacent residues in cytochrome c2 of R. sphaeroides without(black) and with (magenta) imidazole bound (imidazole not shown). In the imidazole-bound state, the “hinge,”located between two long anchoring �-helices (Lime cylinders), has undergone a conformational change cou-pled to the methionine movement, making room in the heme pocket for small ligand binding (Protein DataBank entries 1CXA and 1CXC (13)). Right, the structure of the “hinge” region of cytochrome c1 from R. spha-eroides. The heme is ligated by Met185 (shown in black). The two long �-helices (Lime cylinders) closest to Met185
are separated by 68 amino acids. The disulfide bond between Cys145 and Cys169 (shown in blue) is present onlyin cytochrome c1 from Rhodobacter and is essential to maintain the physiological high potential (Protein DataBank entry 2FYN (44)). The figure was prepared with VMD (69).
Imidazole Inhibition of the bc1 Complex
JULY 16, 2010 • VOLUME 285 • NUMBER 29 JOURNAL OF BIOLOGICAL CHEMISTRY 22517
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

ulations of the known structures of Im adducts of cyts c and c2(imidazole orientations denoted “0°”) and with starting structureswhere the Im was placed in three other orientations, rotated 90,180, and 270° about the Fe-N axis. In 10–20 ns trajectories, therotation of the imidazole plane and water contact with the imid-azole N� were followed (see supplemental material).
As a control, the 0° (crystallographic) orientation in cyt c2maintained a stable position about�17° from the starting posi-tion, with r.m.s fluctuations of12°. The “270°” simulation alsoquickly found the same stable orientation. In contrast, the “90°”and “180°” orientations in cyt c2 both rotated to about 200° (i.e.�180° from the orientation adopted by the 0° starting struc-ture), with similar r.m.s fluctuations. This presumably reflectssome barriers to rotation that are not readily surmounted inshort trajectories.For cytochrome c1, the 90, 180, and 270° imidazole posi-
tions all reoriented quickly (�50 ps) to about 110°, with r.m.sfluctuations of 15°, whereas the 0° orientation rotated toabout 20° but was less stable. Cyt c exhibited significantlylarger fluctuations, especially for the 180 and 270° startingpositions. The 0 and 90° orientations settled into apparentlystable positions between 115 and 135°. In all cases, majorfluctuations were between orientations related by �90 or180° flips.Water contact with the imidazole N� was categorized
according to number of waters, distance, and stability (seesupplemental material). Cyt c was by far the “wettest,” withup to seven water molecules within 4 Å of N�, but with rapidexchange between many different water molecule identitiesfor all starting Im positions. The “0°” (NMR structure) ori-entation exhibited the most stable water associations butshowed exchange of dozens of water molecules. For cyt c2,N� was contacted by 0–2 water molecules (very rarely three).For the 0° (crystallographic orientation) and 270° startingorientations, only 1 or 2 waters were present at all times, andthe same water molecule maintained the closest contact formore than 98% of the time, consistent with its function as ahydrogen bonding bridge between the imidazole and thepeptide backbone.In the case of cyt c1, 1–3 water molecules were generally
present (within 4 Å of N�) and very rarely as many as 4. In threeorientations, all waters entered and left the cavity quite rapidly.However, in the 180° starting orientation of the Im, one partic-ular molecule was closest to the imidazole N� more than 93% ofthe time, reflecting the simultaneous formation of three stablehydrogen bonds withN� of imidazole and the carbonyl oxygensof Met185 and Pro186 (Fig. 8).Two other molecular dynamics simulations of cyt c1 (90 and
270°) converged to the same �110° angle but without a stablybound water molecule. In both of these simulations, waters inthe heme binding cavity were exchanged much more quicklythan seen for the “180°” trajectory. Instead, direct hydrogenbonds were formed between the N� atom of imidazole and thecarbonyl oxygens of either Ala184 orMet185, respectively, in the90 and 270° simulations (see supplemental material). Thus, arole for hydrogen bonding of imidazole in the cyt c1 heme isstrongly implicated by the molecular dynamics simulations,although the precise configuration is uncertain.
Thermodynamics of Ligand Binding—The equilibrium bind-ing of Im to cyt c1 is driven by a large enthalpy change (�H0 ��56 kJ/mol), and the reaction exhibits a significant decrease inentropy (�S0 � �121 J/mol/K). These values for cyt c1 are sim-ilar to those found for Im binding to cyt c2: �H0 � �49 kJ/mol,and �S0 � �104 J/mol/K (10).3 The thermodynamic parame-ters for cyts c1 and c2 are consistent with a significant confor-mational change and bond rearrangement accompanying theenthalpically favorable insertion of Im (see Table 1).In contrast, MeIm binding to cyt c1 is entropically driven,
whereas the enthalpy change is positive (�H0 � 46 kJ/mol and�S0 � 197 J/mol/K). These values are very similar to thosereported by Tang and co-workers (50, 51) for both Im andMeIm binding to cyt c. They are quantitatively different fromthe earlier work of Schejter and Aviram (11), but all reports onthe binding of Im by cyt c and bymetmyoglobin andmethemo-globin are qualitatively similar in showing binding to beentropically driven (11, 52, 53).The quantitative differences between the enthalpic and
entropic contributions to Im and MeIm binding to cyt c1 areextreme but are well supported by the activation energies forbinding and release rates of the two ligands (see accompanyingarticle (70)). Furthermore, the qualitative differences reflect thechemical attributes of the two ligands. The magnitudes of thethermodynamic contributions for both ligands strongly impli-cate the protein and significant solvation responses, and thequalitative differences between Im and MeIm reveal the
3 Dumortier et al. (10) appear to misstate their own findings for the tem-perature dependence of their equilibrium and kinetic data, both by afactor of 2.6. The values we quote were obtained by plotting the data intheir Table III.
FIGURE 8. Snapshot from a molecular dynamics simulation of cyt c1 withimidazole initially in the 180° orientation. Here, a water molecule wastrapped near imidazole and remained in approximately the same positionover the entire simulation. In most frames, it simultaneously formed hydro-gen bonds with the N� atom of imidazole and the carbonyl oxygens of Met185
and Pro186. This binding mode is very similar to the binding configuration ofimidazole to cyt c2 seen in the 1CXA structure and maintained in the 0° sim-ulation for cyt c2. The figure was prepared in VMD (69).
Imidazole Inhibition of the bc1 Complex
22518 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 29 • JULY 16, 2010
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

strongly correlated nature of the enthalpy and entropy changes,associated with hydrogen bond formation (54, 55).The enthalpic drive of Imbinding is consistent with its ability
to form one or more hydrogen bonds through N�H, eitherdirectly to the protein (supplemental Fig. S3) or through asequestered water molecule (Fig. 8). The smaller size of Imcompared with MeIm may also allow a somewhat strongerFe–N bond, providing additional enthalpic drive. However, thefavorable enthalpy is substantially countered by strong negativeentropy contributions. These will include degrees of freedomthat are lost upon sequestration of the ligand, which should besimilar for both Im andMeIm, and additional entropy losses forany bound water(s) that are likely to be unique to Im. Entropiccontributions may also arise from structural reorganization ofthe protein, and the ability of Im to engage in hydrogen bondswithin the reconfigured heme cleft will limit the flexibility ofthe protein and contribute a further loss of entropy.In the case ofMeIm, binding is enthalpically unfavorable but
is driven by a large positive entropy. The inability to form ahydrogen bond in the final heme-ligated state and steric clashesbetween themethyl group and the heme and protein contributeto the positive enthalpy. Steric conflicts may also weaken theFe–N bond. The lack of a hydrogen bond within the heme cleftwill also contribute to the substantial entropy increase by allow-ing greater conformational mobility of the protein. An addi-tional entropy increase is expected for transfer of the hydro-phobicmethyl group from the aqueous phase to the heme cleft.Electrochemical Properties of the Cyt c1-Im Complex—Bind-
ing of Im to horse heart cyt c results in a �400-mV potentialdrop, to �135 mV (8, 9), consistent with replacement of thenative methionine ligand, yielding an imidazole-histidineligated hemewith properties similar to those of a typical bis-hisheme. In both R. capsulatus and R. sphaeroides cyt c1, substitu-tion of histidine for methionine as the sixth ligand led to pho-tosynthetic incompetence, which was attributed to the lowerredox potential of themutant cyt c1 (26, 29, 56).However, redoxtitrations of the Met 3 His mutation in R. capsulatus cyt c1showed amajor titration component with Em7 � �140mV (26,28, 56), whereas the samemutation in cyt c1 fromR. sphaeroidesyielded a significantly higher Em7 � �74 mV (29). The unreli-ability of the growth criterion is evident in comparing the func-tionality of R. sphaeroides mutant bc1 complexes with theMet3 His substitution and those with the structurally stabi-lizing disulfide link (Cyt145-Cys169) removed, which haveEm7�48–64mV, depending on the specific mutation (57). The latterare able to grow photosynthetically despite the lower midpointpotential.Guergova-Kuras et al. (33) reported that the R. sphaeroides
His-tagged bc1 complex isolated with imidazole in the elutionbuffer was less than 5% active and had two fractions of cyt c1;70% showed a midpoint potential of �146 mV, a decrease ofonly �120 mV, whereas the remaining fraction had the nativehigh potential of �270 mV. However, we found that the cytc1-Im complex was not reducible by ascorbate and concludedthat the c1-Im midpoint potential is significantly lower than�50 mV, which would be more consistent with the establishedresponse of soluble cyt c to Im binding. It seems very likely thatthe previous redox titration of cyt c1 does not represent a true
equilibrium condition, which would require maintaining a veryhigh concentration of imidazole, sufficient to saturate the lowaffinity of the reduced heme. For titration of cyt c, Liu et al. (9)used concentrations of 4–6 M imidazole and derivatives. Atnon-saturating levels, the release of Im upon heme reductionand the slow kinetics of the binding equilibrium can beexpected to lead to non-equilibrium titration that would over-estimate the midpoint potential and give rise to heterogeneousbehavior. Furthermore, in our hands, oxidized cyt c1 in isolatedbc1 complex is extremely susceptible to inactivation whendilute, and this has so far precluded us performing any redoxtitrations that are reversible in terms of enzyme activity.Imidazole Inhibition of bc1 ComplexActivity—We found that
activity assays of the imidazole-inhibited bc1 complex fromR. sphaeroides display two distinct phases. During the initialphase, the catalytic turnover rate was indistinguishable fromthe non-catalytic rate of reduction of cyt c by DQH2 and cer-tainly nomore than 1% of control values. However, after a clearlag (�5 s), the bc1 complex-catalyzed rate of cyt c reductionbegan to accelerate (Fig. 7A). This behavior was qualitativelyconsistent and to be expected from unbinding of the ligandupon dilution. However, it was quantitatively very variable,with the time and extent of recovery dependent on detergent,lipid, and bc1 preparation, and is currently under furtherinvestigation.In normal turnover of the bc1 complex, or ubiquinol:cyto-
chrome c oxidoreductase, quinol is oxidized in a bifurcatedprocess that directs the two electrons along separate pathways,in a process known as theQ-cycle. One electron passes througha sequence of high potential components, via the 2Fe2S clusterof the iron-sulfur protein (ISP) followed by cytochrome c1 andthen to a soluble c-type cytochrome, whereas the other electrongoes through a low potential sequence, via two b-type hemes toa quinone (reviewed in Refs. 58–61).In its position between ISP and cyt c, cyt c1 participates in two
electron transfer reactions, and lowering of the Em of cyt c1 byIm is a plausible mechanism of inhibition of bc1 activity. How-ever, it is unlikely to be a direct effect on the rate of electrontransfer, becauseMarcus theory does not give a good account ofthe absolute values of the electron transfer rates between ISP,c1, and c or c2. Nevertheless, it is instructive in estimating thepossible response to changes in driving force (�Em). The elec-tron transfer rate between ISP and cyt c1 in the R. sphaeroidesbc1 complex is 8.104 s�1 at pH 8, where �G0 � 0 mV (62), andthe rate between cyt c1 and bound cyt c in yeast bc1 complex isabout 104 s�1, also with �G0 � 0 mV (63). For a reorganizationenergy of 1 eV, Marcus theory predicts that a 300-mV decreasein themidpoint potential of cyt c1 wouldmake the ISP3 c1 rate3 orders of magnitude slower (i.e. �102 s�1). At the same time,the rate of electron transfer from c1 to c should be increased byabout 100-fold (i.e. �106 s�1). Thus, ISP3 c1 electron transferwould become rate-limiting in this segment of the high poten-tial chain. However, this is much faster than the observed rateandwould not limit turnover. A 400-mVdecrease inEm, as seenfor soluble cyt c, would cause roughly 10-fold larger changes butwould not substantially alter this conclusion even if Marcustheory were applicable. In fact, neither of these individual elec-tron transfer rates is very dependent on driving force (62–64).
Imidazole Inhibition of the bc1 Complex
JULY 16, 2010 • VOLUME 285 • NUMBER 29 JOURNAL OF BIOLOGICAL CHEMISTRY 22519
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Instead, we note that if the Em of cyt c1 is lowered by 300mV,the population of reduced c1 formed in the ISP3 c1 step will bedecreased by a factor of 10�5 (a 400-mV decrease in Em wouldlower the population by almost 10�7-fold). This would have adramatic effect on the overall rate of electron transfer from ISPto cyt c, especially if the intrinsic electron transfer rate from c1to c is not substantially accelerated by the increased potentialdrop. Thus, the large decrease in equilibrium population ofreduced c1 will be the major inhibitory influence on the turn-over time of cyt c reduction.Ligand Binding and Function—The affinities of imidazole
and N-methylimidazole binding to cyt c1 differ by a factor 30and exhibit very different temperature dependences. Compar-ison with the binding of imidazole to cyts c and c2 suggests thatimidazole may form a hydrogen bond within the heme pocketof cyt c1, and molecular dynamics simulations gave strong sup-port to this. The binding of Im to cyt c1 was found to be enthal-pically driven, whereas MeIm was entropically driven. Thismarked qualitative difference was accounted for in terms oftheir hydrogen bonding abilities and hydrophobicity. The pro-pensity for cyt c1 to bind small molecules that displace thenative methionine ligand may render it capable of performingnon-standard reactions, such as peroxidation, and the genera-tion of signaling molecules, as is now well recognized for cyt c(65–68). The kinetics and mechanistic implications of imidaz-ole binding to cyt c1 are described in the following article (70).
REFERENCES1. Scott, R. A., and Mauk, A. G. (eds) (1996) Cytochrome c: A Multidisci-
plinary Approach, University Science Books, Sausalito, CA2. Battistuzzi, G., Borsari, M., and Sola, M. (2001) Antioxid. Redox Signal. 3,
279–2913. Englander, S. W., Mayne, L., and Krishna, M. M. (2007) Q. Rev. Biophys.
40, 287–3264. Maity, H., Maity, M., and Englander, S. W. (2004) J. Mol. Biol. 343,
223–2335. Moore, G. R., and Pettigrew, G. W. (1990) Cytochromes c: Evolutionary,
Structural and Physiochemical Aspects, Springer, Berlin6. Caroppi, P., Sinibaldi, F., Fiorucci, L., and Santucci, R. (2009) Curr. Med.
Chem. 16, 4058–40657. Kagan, V. E., Bayir, H. A., Belikova, N. A., Kapralov, O., Tyurina, Y. Y.,
Tyurin, V. A., Jiang, J., Stoyanovsky, D. A., Wipf, P., Kochanek, P. M.,Greenberger, J. S., Pitt, B., Shvedova, A. A., and Borisenko, G. (2009) FreeRadic. Biol. Med. 46, 1439–1453
8. Battistuzzi, G., Borsari, M., Cowan, J. A., Ranieri, A., and Sola, M. (2002)J. Am. Chem. Soc. 124, 5315–5324
9. Liu, G., Shao, W., Zhu, S., and Tang, W. (1995) J. Inorg. Biochem. 60,123–131
10. Dumortier, C., Meyer, T. E., and Cusanovich,M. A. (1999)Arch. Biochem.Biophys. 371, 142–148
11. Schejter, A., and Aviram, I. (1969) Biochemistry 8, 149–15312. Viola, F., Aime, S., Coletta, M., Desideri, A., Fasano, M., Paoletti, S., Tar-
ricone, C., and Ascenzi, P. (1996) J. Inorg. Biochem. 62, 213–22213. Axelrod, H. L., Feher, G., Allen, J. P., Chirino, A. J., Day, M.W., Hsu, B. T.,
and Rees, D. C. (1994) Acta Crystallogr. D 50, 596–60214. Banci, L., Bertini, I., Liu, G., Lu, J., Reddig, T., Tang, W., Wu, Y., Yao, Y.,
and Zhu, D. (2001) J. Biol. Inorg. Chem. 6, 628–63715. Yao, Y., Qian, C., Ye, K.,Wang, J., Bai, Z., andTang,W. (2002) J. Biol. Inorg.
Chem. 7, 539–54716. Greenwood, C., and Palmer, G. (1965) J. Biol. Chem. 240, 3660–366317. Pettigrew, G. W., Aviram, I., and Schejter, A. (1975) Biochem. J. 149,
155–16718. Schejter, A., and George, P. (1964) Biochemistry 3, 1045–1049
19. Taler, G., Schejter, A., Navon, G., Vig, I., and Margoliash, E. (1995) Bio-chemistry 34, 14209–14212
20. Ikeda-Saito, M., and Iizuka, T. (1975) Biochim. Biophys. Acta 393,335–342
21. Sutin, N., and Yandell, J. K. (1972) J. Biol. Chem. 247, 6932–693622. Berry, E. A., Guergova-Kuras, M., Huang, L. S., and Crofts, A. R. (2000)
Annu. Rev. Biochem. 69, 1005–107523. Crofts, A. R. (2004) Annu. Rev. Physiol. 66, 689–73324. Baymann, F., Lebrun, E., and Nitschke, W. (2004) Proc. Natl. Acad. Sci.
U.S.A. 101, 17737–1774025. Bertini, I., Cavallaro, G., and Rosato, A. (2006) Chem. Rev. 106, 90–11526. Darrouzet, E., Mandaci, S., Li, J., Qin, H., Knaff, D. B., andDaldal, F. (1999)
Biochemistry 38, 7908–791727. Gray, K. A., Davidson, E., and Daldal, F. (1992) Biochemistry 31,
11864–1187328. Li, J., Darrouzet, E., Dhawan, I. K., Johnson, M. K., Osyczka, A., Daldal, F.,
and Knaff, D. B. (2002) Biochim. Biophys. Acta 1556, 175–18629. Zhang, H., Osyczka, A., Moser, C. C., and Dutton, P. L. (2006) Biochemis-
try 45, 14247–1425530. Schejter, A., and Berke, G. (1968) Biochim. Biophys. Acta 162, 459–46131. Potter, V. R. (1941) J. Biol. Chem. 137, 13–2032. Osyczka, A., Moser, C. C., and Dutton, P. L. (2004) Biochim. Biophys. Acta
1655, 71–7633. Guergova-Kuras, M., Salcedo-Hernandez, R., Bechmann, G., Kuras, R.,
Gennis, R. B., and Crofts, A. R. (1999) Protein Expr. Purif. 15, 370–38034. Trumpower, B. L., and Edwards, C. A. (1979) J. Biol. Chem. 254,
8697–870635. Margoliash, E., and Frohwirt, N. (1959) Biochem. J. 71, 570–57236. Wood, P. M. (1980) Biochem. J. 192, 761–76437. Saleem,M.M.M., andWilson,M.T. (1988) InorganicaChimicaActa153,
93–9838. Perrin, D. D. (1965, supplement 1972) Dissociation Constants of Organic
Bases in Aqueous Solution, pp. 190–193, Butterworths, London39. Tanokura, M. (1983) Biochim. Biophys. Acta 742, 576–58540. Phillips, J. C., Braun, R., Wang, W., Gumbart, J. C., Tajkhorshid, E., Villa,
E., Chipot, C., Skeel, R. D., Kale, L., and Schulten, K. (2005) J. Comp. Chem.26, 1781–1802
41. Autenrieth, F., Tajkhorshid, E., Baudry, J., and Luthey-Schulten, Z. (2004)J. Comput. Chem. 25, 1613–1622
42. Brooks, B. R., Brooks, C. L., 3rd, Mackerell, A. D., Jr., Nilsson, L., Petrella,R. J., Roux, B., Won, Y., Archontis, G., Bartels, C., Boresch, S., Caflisch, A.,Caves, L., Cui,Q., Dinner, A. R., Feig,M., Fischer, S., Gao, J., Hodoscek,M.,Im,W., Kuczera, K., Lazaridis, T., Ma, J., Ovchinnikov, V., Paci, E., Pastor,R. W., Post, C. B., Pu, J. Z., Schaefer, M., Tidor, B., Venable, R. M., Wood-cock, H. L.,Wu, X., Yang,W., York, D.M., and Karplus, M. (2009) J. Com-put. Chem. 30, 1545–1614
43. MacKerell, A. D., Bashford, D., Bellot, M., Dunbrack, R. L., Jr., Evansec,J. D., Field, M. J., Fisher, S., Gao, J., Guo, H., Ha, S., Joseph, D., Kuchnir, L.,Kuczera, K., Lau, F. T. K., Mattos, C., Michnick, S., Ngo, T., Nguyen, D. T.,Prodhom, B., Reiher, I.W. E., Roux, B., Schlenkrich,M., Smith, J., Stote, R.,Straub, J., Watanabe, M., Wiorkiewicz-Kuczera, J., Yin, D., and Karplus,M. (1998) J. Phys. Chem. B 102, 3586–3616
44. Esser, L., Gong, X., Yang, S., Yu, L., Yu, C. A., and Xia, D. (2006) Proc. Natl.Acad. Sci. U.S.A. 103, 13045–13050
45. Dumortier, C., Holt, J. M., Meyer, T. E., and Cusanovich, M. A. (1998)J. Biol. Chem. 273, 25647–25653
46. Dumortier, C., Fitch, J., Van Petegem, F., Vermeulen, W., Meyer, T. E.,Van Beeumen, J. J., and Cusanovich, M. A. (2004) Biochemistry 43,7717–7724
47. Clark, W. M. (1972) Oxidation-Reduction Potentials of Organic Systems,pp. 469–471, Robert E. Krieger Publishing, Huntington, NY
48. Elberry,M., Xiao, K., Esser, L., Xia, D., Yu, L., andYu, C. A. (2006)Biochim.Biophys. Acta 1757, 835–840
49. Margalit, R., and Schejter, A. (1973) Eur. J. Biochem. 32, 492–49950. Shao, W., Liu, G., and Tang, W. (1995) J. Inorg. Biochem. 57, 103–11351. Shao, W., Wei, J., and Tang, W. (1992) Acta Chim. Sin. 50, 1129–113352. Stephanos, J. J., Farina, S. A., and Addison, A.W. (1996) Biochim. Biophys.
Acta 1295, 209–221
Imidazole Inhibition of the bc1 Complex
22520 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 29 • JULY 16, 2010
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

53. Zakariassen, H., and Sørlie, M. (2007) Thermochimica Acta 464, 24–2854. Cooper, A. (2005) Biophys. Chem. 115, 89–9755. Dunitz, J. D. (1995) Chem. Biol. 2, 709–71256. Gray, K. A., Dutton, P. L., and Daldal, F. (1994) Biochemistry 33,
723–73357. Elberry, M., Yu, L., and Yu, C. A. (2006) Biochemistry 45, 4991–499758. Cape, J. L., Bowman,M.K., andKramer,D.M. (2006)Trends Plant. Sci.11,
46–5559. Covian, R., and Trumpower, B. L. (2008) Biochim. Biophys. Acta 1777,
1079–109160. Crofts, A. R., Holland, J. T., Victoria, D., Kolling, D. R., Dikanov, S. A.,
Gilbreth, R., Lhee, S., Kuras, R., and Kuras, M. G. (2008) Biochim. Biophys.Acta 1777, 1001–1019
61. Osyczka, A., Moser, C. C., and Dutton, P. L. (2005) Trends Biochem. Sci.30, 176–182
62. Engstrom, G., Xiao, K., Yu, C. A., Yu, L., Durham, B., andMillett, F. (2002)
J. Biol. Chem. 277, 31072–3107863. Engstrom, G., Rajagukguk, R., Saunders, A. J., Patel, C. N., Rajagukguk, S.,
Merbitz-Zahradnik, T., Xiao, K., Pielak, G. J., Trumpower, B., Yu, C. A.,Yu, L., Durham, B., and Millett, F. (2003) Biochemistry 42, 2816–2824
64. Millett, F., and Durham, B. (2004) Photosynth. Res. 82, 1–1665. Driscoll,W. J., Chaturvedi, S., andMueller, G. P. (2007) J. Biol. Chem. 282,
22353–2236366. Kalanxhi, E., and Wallace, C. J. (2007) Biochem. J. 407, 179–18767. Radi, R., Thomson, L., Rubbo, H., and Prodanov, E. (1991) Arch. Biochem.
Biophys. 288, 112–11768. Stewart, J. M., Blakely, J. A., and Johnson, M. D. (2000) Biochem. Cell Biol.
78, 675–68169. Humphrey, W., Dalke, A., and Schulten, K. (1996) J. Mol. Graph. 14,
33–3870. Kokhan, O., Shinkarev, V. P., andWraight, C. A. (2010) J. Biol. Chem. 285,
22522–22531
Imidazole Inhibition of the bc1 Complex
JULY 16, 2010 • VOLUME 285 • NUMBER 29 JOURNAL OF BIOLOGICAL CHEMISTRY 22521
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Oleksandr Kokhan, Vladimir P. Shinkarev and Colin A. WraightSTUDIES
: I. EQUILIBRIUM AND MODELINGRhodobacter sphaeroidesComplex from 1bc and Inhibition of the 1cBinding of Imidazole to the Heme of Cytochrome
doi: 10.1074/jbc.M110.128058 originally published online May 6, 20102010, 285:22513-22521.J. Biol. Chem.
10.1074/jbc.M110.128058Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2010/05/06/M110.128058.DC1
http://www.jbc.org/content/285/29/22513.full.html#ref-list-1
This article cites 66 references, 14 of which can be accessed free at
by guest on May 27, 2018
http://ww
w.jbc.org/
Dow
nloaded from


















