
Big Question #2 - WordPress.com · Big Question 2 2 3150:153-002/801 . Chapter 1. 6 . Chemical...
18
Big Question 2 3150:153-002/801 1 1. What’s in a solution? How far does a reaction “go”? 2. What factors influence how far a reaction “goes” and how fast it gets there? 3. How do atomic and molecular structure influence observed properties of substances? Big Question #2 What factors influence how far a reaction “goes” and how fast it gets there? Sections 12.1−2 Phase Changes (Part I) A Macroscopic Comparison of Gases, Liquids, and Solids (Review from Ch. 1) State Shape and Volume Compressibility Ability to Flow Gas Conforms to shape and volume of container High High Liquid Conforms to shape of container; Very low Moderate volume limited by surface Solid Maintains its own shape and volume Almost none Almost none Energies of Phase Changes (Some review from Chapter 6, Some New) • heat of fusion (∆H FUS ): enthalpy change for the melting of 1 mole of a substance e.g., H 2 O(s) → H 2 O(l) ∆H = ∆H FUS = +6.02 kJ • heat of vaporization (∆H VAP ): enthalpy change for the vaporization of 1 mole of a substance e.g., H 2 O(l) → H 2 O(g) ∆H = ∆H VAP = +44.0 kJ At a phase change, all added/removed energy goes into the phase change and none goes into changing the temperature. Thus, any measurement of the heat involved when passing through a phase change has to be broken into steps. (Also recall from Chapter 6: Hess’s Law, Specific Heat, q=mcΔT) e.g., Consider converting 1.5 mol of ice at −25 °C to liquid water at 35 °C. What quantity of heat is required by this process in J? The molar heat capacity of ice is 37.6 J·mol –1 ·°C −1 . The molar heat capacity of liquid water is 75.4 J·mol −1 ·°C −1 . The heat of fusion of water is 6.02 kJ·mol −1 . q = ( ) 37.6 J (1.50 mol) 0 C ( 25 C) mol C ° − − ° ° + 6.02 kJ 1000 J (1.50 mol) mol kJ + 75.4 J (1.50 mol) (35 C C) mol C ° − 0 ° ° PROTIP Heat of fusion and heat of vaporization are both ALWAYS endothermic. For a substance, the heat of vaporization is always larger than the heat of fusion.
Transcript of Big Question #2 - WordPress.com · Big Question 2 2 3150:153-002/801 . Chapter 1. 6 . Chemical...

Big Question 2 3150:153-002/801 1
1. What’s in a solution? How far does a reaction “go”? 2. What factors influence how far a reaction “goes” and how fast it gets there? 3. How do atomic and molecular structure influence observed properties of substances?
Big Question #2 What factors influence how far a reaction “goes” and how fast it gets there?
Sections 12.1−2 Phase Changes (Part I) A Macroscopic Comparison of Gases, Liquids, and Solids (Review from Ch. 1) State Shape and Volume Compressibility Ability to Flow Gas Conforms to shape and volume of container High High Liquid Conforms to shape of container; Very low Moderate volume limited by surface Solid Maintains its own shape and volume Almost none Almost none
Energies of Phase Changes (Some review from Chapter 6, Some New) • heat of fusion (∆HFUS): enthalpy change for the melting of 1 mole of a
substance e.g., H2O(s) → H2O(l) ∆H = ∆HFUS = +6.02 kJ
• heat of vaporization (∆HVAP): enthalpy change for the vaporization of 1 mole of a substance
e.g., H2O(l) → H2O(g) ∆H = ∆HVAP = +44.0 kJ
At a phase change, all added/removed energy goes into the phase change and none goes into changing the temperature. Thus, any measurement of the heat involved when passing through a phase change has to be broken into steps. (Also recall from Chapter 6: Hess’s Law, Specific Heat, q=mcΔT)
e.g., Consider converting 1.5 mol of ice at −25 °C to liquid water at 35 °C. What quantity of heat is required by this process in J? The molar heat capacity of ice is 37.6 J·mol–1·°C −1. The molar heat capacity of liquid water is 75.4 J·mol−1·°C −1. The heat of fusion of water is 6.02 kJ·mol−1.
q = ( )37.6 J(1.50 mol) 0 C ( 25 C)mol C
° − − ° °
+ 6.02 kJ 1000 J(1.50 mol)mol kJ
+ 75.4 J(1.50 mol) (35 C C)mol C
° − 0 ° °
PROTIP Heat of fusion and heat of vaporization are both ALWAYS endothermic. For a substance, the heat of vaporization is always larger than the heat of fusion.

Big Question 2 3150:153-002/801 2
Chapter 16 Chemical Kinetics the study of rates of chemical reactions. How fast is a reaction? The kinetics of a reaction must be determined experimentally.
in general, reaction rate = change in concentrationchange in time
rate = Δ(A)Δt
= (A) t
dd
The differential form of the rate shows that the rate = the slope of the tangent line to the curve in a graph of concentration vs. time. (Rates are always described as positive numbers.)
e.g., for 2H2O2 → 2H2O + O2, rate of appearance of H2O = 2Δ(H O)Δt
rate of appearance of O2 = 2Δ(O )Δt
rate of disappearance of H2O2 = − 2 2Δ(H O )Δt
In general, for aA + bB → cC + dD,
− 1 Δ(A)Δta
= − 1 Δ(B)Δtb
= 1 Δ(C)Δtc
= 1 Δ(D)Δtd
e.g., hydrogen peroxide disappears at twice the rate at which oxygen appears: − 2 2Δ(H O )Δt
= 2Δ(O )2Δt
Differential Rate Laws A general relationship between species concentration and reaction rate is called a rate law. Assumptions: 1. only forward reactions (i.e., write rate laws in terms of reactants) 2. equilibrium avoided The rate of reaction depends on concentration, but how exactly? rate ∝ (A)n n = order of reaction
rate = k (A)n k = rate constant
rate = − Δ(A)Δt
= k (A)n ◄a differential rate law: rate as a function of concentration
¡CAUTIONS! 1. n not necessarily equal to stoichiometric coefficient. 2. n and k are experimentally determined. 3. rates vary for each product/reactant. Must specify.
e.g., Consider this reaction: 2ClO2(aq) + 2OH−(aq) → ClO3
−(aq) + ClO2−(aq) + H2O(l)
and the following initial rate data: (ClO2)o/mol·L−1 (OH–)o/mol·L−1 initial rate/mol·L−1·s−1
0.0500 0.100 5.77 × 10−2 0.100 0.100 2.32 × 10−1 0.100 0.050 1.15 × 10−1
Determine the rate law for the reaction and the value of k (with units).
◄ relationships between/among rates in terms of different reactants and products

Big Question 2 3150:153-002/801 3
Integrated Rate Laws Differential rate laws give information about rate as a function of concentration, but what about concentrations as a function of time? We need to integrate the differential rate law. The calculus involved for orders 0, 1, and 2 is below…it’s only necessary to use the final results and not worry about the derivation. Zero-Order Rate = − (A)
t∆
∆= k(A)0 = k
or Rate = − (A)t
dd
= k
− d(A) = k dt
d(A) = − k dt
o
(A)
(A)
(A)d∫ = − k t
0
d∫ t
(A) – (A)o = – k(t – 0)
(A) – (A)o = – kt
Half-Life
let (A) = o(A)2
at t = t½
o(A)2
= – kt½ + (A)o
– o(A)2
= –kt½
o(A)2k
= t½
(A) = −kt + (A)o ◄an integrated rate law: concentration as a function of time
First-Order Rate = – (A)
t∆
∆= k(A)1 or
Rate = – (A)t
dd
= k(A)
– (A)
(A)d
= k dt
(A)(A)
d = – k dt
o
(A)
(A)
(A)(A)
d∫ = – k
t
0
d∫ t
ln(A) – ln(A)o = – k(t – 0) ln(A) – ln(A)o = – kt ln(A) = −kt + ln(A)o
Half-Life
let (A) = o(A)2
at t = t½
ln o(A)2
= – kt½ + ln(A)o
ln(A)o – ln 2 = – kt½+ln(A)o
– ln 2 = –kt½ ln 2 = kt½ ln 2k
= t½
Second-Order Rate = − (A)
t∆
∆= k(A)2 or
Rate = − (A)t
dd
= k(A) 2
– 2
(A)(A)d
= k dt
2
(A)(A)d
= – k dt
o
(A)
2(A)
(A)(A)d
∫ = – k t
0
d∫ t
− 1
(A) −
o
1(A)
−
= – k(t – 0)
– 1
(A)+
o
1(A)
= – kt
1
(A)–
o
1(A)
= kt
1(A)
= kt + o
1(A)
Half-Life
let (A) = o(A)2
at t = t½
o
1(A)
2
= kt½ + o
1(A)
o
2(A)
= kt½ + o
1(A)
o
1(A)
= kt½
o
1(A)k
= t½

Big Question 2 3150:153-002/801 4
Summary of Rate Laws Order Differential Rate Law Integrated Rate Law (straight-line form)
0 Rate = − (A)t
dd
= k (A) = −kt + (A)o
1 Rate = − (A)t
dd
= k(A) ln(A) = −kt + ln(A)o
2 Rate = − (A)t
dd
= k(A)2 1
(A)= kt +
o
1(A)
Using the Integrated Rate Laws Example: What are the order and rate constant at 25 °C for the decomposition of N2O5?
2N2O5(g) → 4NO2(g) + O2(g) Experimental method: place 0.1000 mol of N2O5 in a 1.000 L flask and measure (N2O5) as a function of time. Experimental data: time/s (N2O5)/mol·L−1 0 0.1000 50 0.0707 100 0.0500 200 0.0250 300 0.0125 400 0.00625 Approach: Make a guess at the reaction order, n. Plot the experimental data using the corresponding integrated rate law. The integrated rate law that gives a straight line will give the values of n and k.
Integrated rate law plots and reaction orders.
Half-Life, t½ the amount of time required for the reactant concentration to decrease to half its initial value
i.e., the amount of time to reach (A) = 12(A)o
You can derive the formulas for half-life from the integrated rate laws; see above. Only the first-order half-life formula is independent of the initial concentration.
Big Question 2 5 3150:153-002/801
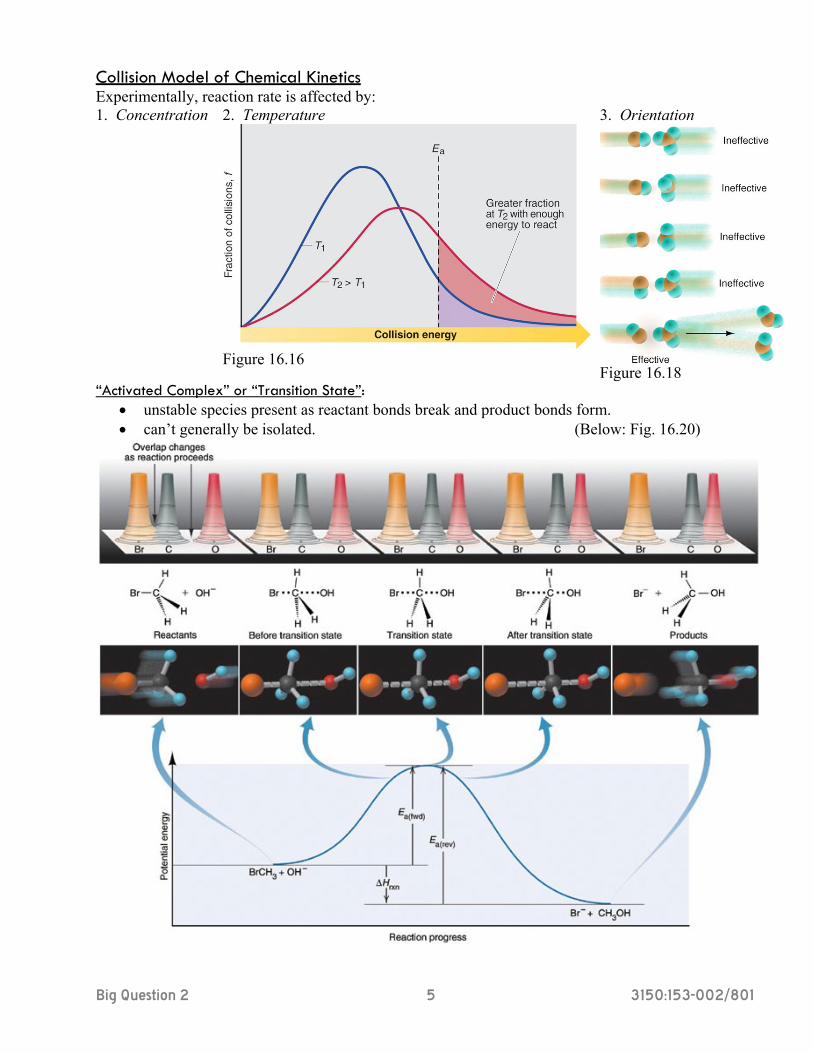
Collision Model of Chemical Kinetics Experimentally, reaction rate is affected by: 1. Concentration 2. Temperature
Figure 16.16
3. Orientation
Figure 16.18
“Activated Complex” or “Transition State”: • unstable species present as reactant bonds break and product bonds form. • can’t generally be isolated. (Below: Fig. 16.20)

Big Question 2 6 3150:153-002/801
activation energy, Ea: energy required to overcome the barrier to reaction. Evaluating Ea
1889, Svante Arrhenius k = Zρ RTaE
e−
= A RTaE
e−
ln k = ln RTAaE
e−
= ln A + ln RTaE
e−
= ln A − RT
aE
ln k = –R
aE 1T
+ ln A
Determine k at various temperatures; plot ln k vs. 1T
.
Or measure k at two temperatures (T1 and T2):
ln 2
1
kk
= R
aE
1 2
1 1T T
−
Catalysts • increase rate of reaction • are not consumed overall by reaction (disappear, but later reappear) • provide alternative reaction pathways with lower activation energies homogeneous catalyst: in same phase as reactants (e.g., all aqueous or all gaseous)
heterogeneous catalyst: in different phase from reactants (e.g., solid surface for gases to react on)
¡CAUTIONS! • need R = 8.3145 J·mol–1·K–1 • T must be in K • Ea will be calculated in J, not kJ
Z: frequency factor ρ: steric factor (ratio between the experimental value of the rate constant and the one predicted by collision theory)

Big Question 2 7 3150:153-002/801
Reaction Mechanisms reaction mechanism: sequence of individual steps by which a reaction proceeds.
The slowest step governs the overall rate. Mechanistic steps MUST o add to overall reaction. o be physically reasonable. (For example, termolecular steps are unreasonable.) o correlate with observed rate law.
e.g., NO2 + CO → CO2 + NO observed rate law: rate = k(NO2)2 e.g., 2 NO + H2 → N2O + H2O observed rate law: rate = k(NO)2(H2)
Proposed mechanism: 2 NO N2O2 fast N2O2 + H2 → N2O + H2O slow
The slow second step causes some N2O2 to build up waiting to react. The first step reaches equilibrium.
K = 2 22
(N O )(NO)
, so (N2O2) = K(NO)2
The slow step governs the overall rate of the reaction: rate = k(N2O2)(H2) But N2O2 can not be included in the rate law since it is not a reactant…..

Big Question 2 8 3150:153-002/801
3150:154 Experiment 4 (Chemical Kinetics)
6 I–(aq) + BrO3–(aq) + 6 H3O+ (aq) → 3 I2 (aq) + Br–(aq) + 9 H2O(l)
rate = rate of loss of BrO3– = − 3[BrO ]
t
−∆∆
rate = 13 (rate of formation of I2) = + 1
32Δ[I ]
Δt
rate = k [I–]m [BrO3–]n [H3O+]p
The three pairings can be accomplished in a total of four experiments at room temperature, as covered in the Procedure. Experiments 3 and 4 in Table 2 in the Procedure fulfill the requirements of case a: [I–]3 ≠ [I–]4 but [BrO3
–]3 = [BrO3–]4 and [H3O+]3 = [H3O+]4
Given these concentration values, we can use equation 4 to write the initial rates of reaction in the two experiments: rate3 = k ([I–]3)m ([BrO3
–]4)n ([H3O+]4)p rate4 = k ([I–]4)m ([BrO3
–]4)n ([H3O+]4)p The ratio of the rates is
( )( )
33 3
4 44
[I ]rate [I ]rate [I ][I ]
m m
m
− −
−−
= =
In equation 5, rate3, rate4, [I–]3 and [I–]4 are numbers that you measure experimentally. Only the value of the exponent m is unknown. Solve for m using the laws of logarithms:
3 3
4 4
rate [I ]ln lnrate [I ]
m−
−
=
( )( )
3 4
3 4
ln rate rateln [I ] [I ]
m− −
=
Table 2. Components of Solutions A and B for Room Temperature Experiments
Exp. Components of Solution A (mL) Components of Solution B (mL)
H2O Na2S2O3 KI KBrO3 HCl
1 0 2.00 2.00 4.00 2.00
2 0 2.00 2.00 2.00 4.00
3 0 2.00 4.00 2.00 2.00
4 2.00 2.00 2.00 2.00 2.00
3H 0 2.00 4.00 2.00 2.00
3C 0 2.00 4.00 2.00 2.00

Big Question 2 9 3150:153-002/801
Chapter 20 Chemical Thermodynamics First Law of Thermodynamics Energy can be converted from one form to another, but can not be created or destroyed.
ΔEUNIV = ΔESYS + ΔESURR = 0 Reaction Spontaneity “spontaneous”: occurring without outside intervention Entropy, S Entropy is a measure of the tendency of energy to spread out, to
“diffuse”, to become “less concentrated” a measure of the number of ways energy can be
distributed among the motions of particles. a measure of a driving force. not necessarily conserved. Entropy is NOT a driving force itself disorder or a measure of disorder applicable to macroscopic objects Ludwig Boltzmann (late 1800s): S = k ln W
Second Law of Thermodynamics In any spontaneous process, the entropy of the universe increases.
Experimentally, for a reversible process, ΔSSYS = SYS
Tq and ΔSSURR = SURR
Tq at constant T
Example: An iron skillet at 500 K is allowed to cool in the kitchen (300 K). The skillet transfers 500 J of heat to the kitchen. Why does this occur spontaneously?

Big Question 2 10 3150:153-002/801
Alternate form of Second Law: Energy always flows as heat from hot objects to cold ones.
The Second Law allows predictions about what should occur, but says nothing about when. Spontaneous processes can be very, very slow. Something holds back some spontaneous processes; otherwise all spontaneous processes would be instant.
Obstructions to the Second Law make life possible! ΔS and Spontaneity Recap
ΔSUNIV + : spontaneous process
ΔSUNIV − : nonspontaneous process; reverse process spontaneous
ΔSUNIV = 0 : process at equilibrium (no net tendency in either direction)
Gibbs Free Energy, ΔG We would like to be able to predict spontaneity based on a system variable rather than a universe one.
ΔSUNIV = ΔSSYS + ΔSSURR (constant T)
and ΔH = qSYS (constant P)
Heat is exchanged between the system and surroundings:
ΔHSYS = qSYS = −qSURR = −ΔHSURR
And ΔSSURR = SURR
Tq = SURRΔ
TH = − SYSΔ
TH ◄ΔSSURR in terms of system
Substituting:
ΔSUNIV = ΔSSYS − SYSΔT
H ◄ΔSUNIV in terms of system
Multiplying by −T:
−TΔSUNIV = −TΔSSYS + ΔHSYS = ΔHSYS − TΔSSYS ◄ TΔS: energy units
ΔGSYS = ΔHSYS − TΔSSYS ◄ ΔG ≡ Gibbs Free Energy
ΔG = ΔH –TΔS
ΔG and Spontaneity
ΔSUNIV = − SYSΔT
G
ΔG − : spontaneous process ΔG + : nonspontaneous process; reverse process spontaneous ΔG = 0 : process at equilibrium (no net tendency in either direction)
Gibbs Free Energy is the amount of energy that is free to do useful work in a spontaneous process.
ΔG = wMAX (in theory—you never get that much work in real processes)
• If ΔG −, work that can be done on surroundings by system
• If ΔG +, work that must be done on system by surroundings to force the nonspontaneous process to occur.

Big Question 2 11 3150:153-002/801
Many bodily processes are kept from reaching equilibrium by biochemical coupling. A system at equilibrium can not do any useful work.
Equilibrium: point of minimum free energy and maximum entropy
Third Law of Thermodynamics The entropy of a perfect, crystalline substance at absolute zero is 0. Standard State conditions compounds • gas: pressure of 1 bar (100 kPa, ~1 atm) • pure liquid/solid • solution: 1 M concentration
elements • form in which element exists at 1 bar and T specified ΔHF° and ΔGF° of an element in its standard state ≡ 0/
ΔH, ΔS, T, and Spontaneity 1. ΔH −, ΔS +: always spontaneous (ΔG always −) because both ΔH and ΔS are favorable 2. ΔH +, ΔS −: always nonspontaneous (ΔG always +) because both ΔH and ΔS are unfavorable 3. ΔH −, ΔS −: sign of ΔG depends on T and magnitudes of ΔH and ΔS 4. ΔH +, ΔS +: sign of ΔG depends on T and magnitudes of ΔH and ΔS

Big Question 2 12 3150:153-002/801
For reactions that change in spontaneity depending on T, we can calculate the “crossover temperature”: Example: Hg(l) Hg(g) ΔH° = 60.83 kJ ΔS° = 97.49 J·K−1
ΔG, ΔG°, and K ΔG° = free energy change when reactants in standard states are converted to products in standard states
Why bother with ΔG°?
1. Can compare relative tendencies of reactions to occur
2. Need it to calculate ΔG under nonstandard conditions ΔG = ΔG° + RT ln Q
3. Provides information about equilibrium position
At equilibrium, ΔG = 0 and Q = K
Example: What is ΔG° for the auto-ionization of water?

Big Question 2 13 3150:153-002/801
Chapter 21 Electrochemistry
Review: Reduction-Oxidation Reactions (See Chapter 4) Driving force: transfer/shift of electrons
“LEO the lion says GER” Loss of Electrons is Oxidation; Gain of Electrons is Reduction
“OIL RIG” Oxidation Is Loss; Reduction Is Gain
“ELMO” Electron Loss Means Oxidation
The substance that is oxidized ≡ the reducing agent. The substance that is reduced ≡ the oxidizing agent.
A reaction is a “redox reaction” if the oxidation numbers of one or more atoms changes. e.g.,
2Zn(s) + O2(g) → 2ZnO(s) ON of Zn changes from 0 to +2 ON of O changes from 0 to –2
∴ This is a redox reaction, and
Zn was oxidized and was the reducing agent, and
O2 was reduced and was the oxidizing agent.
Electrochemistry • the study of interchange of chemical and electrical energy • involves redox reactions
Goal: to be able to do useful things (work) by generating an electric current.
Much of the experimental evidence for thermodynamic concepts arose from electrochemical experiments!
Experimentally, this reaction is spontaneous (i.e., ΔG° is negative): Zn(s) + Cu2+(aq) → Cu(s) + Zn2+(aq)
The problem is that the electron transfer occurs at the interface between the substances. To do useful electrical work, we need to force the electrons to travel through a wire; i.e., the reduction and oxidation processes must be separated!

Big Question 2 14 3150:153-002/801
We need to add an apparatus that will allow electrical neutrality to be maintained in both beakers without allowing the contents of the beakers to mix.
• Salt bridge o allows ions to flow, maintaining electrical neutrality o contains a strong electrolyte in a gelatinous matrix or a porous/absorbent substance saturated
with a strong electrolytes solution, with the ends closed with a glass frit. o is an “external source” of ions.
• Porous disk o prevents solutions from mixing significantly but allows ions to cross, maintaining charge balance. o is an “internal source” of ions.
voltaic (galvanic) cell: device in which chemical energy is converted to electrical energy electrodes: metallic conductors that make electrical contact with contents of half-cells. anode: electrode at which oxidation occurs cathode: electrode at which reduction occurs In the salt bridge: anions flow toward/into anode compartment cations flow toward/into cathode compartment

Big Question 2 15 3150:153-002/801
Cell Potential, E (E°) measured in volts 1 volt = 1 joule energy per coulomb charge transferred [1 V = 1 J∙C−1]
E°CELL = E°OVERALL = E°OXIDATION + E°REDUCTION = E°CATHODE – E°ANODE E°OXIDATION = – E°ANODE
E°REDUCTION = E°CATHODE
e.g., the two tabulated reductions Zn2+ + 2 e– → Zn E°RED = −0.76 V
and Cu2+ + 2 e– → Cu E°RED = +0.34 V
Reverse Zn half-reaction: Zn → Zn2+ + 2 e– E°OX = +0.76 V (= − E°ANODE = − (−0.76 V))
Add to Cu half-reaction: Cu2+ + 2 e– → Cu E°RED = +0.34 V (= E°CATHODE)
For Zn + Cu2+ → Cu + Zn2+: E°CELL = +1.10 V
We need a reference to measure half-cell E°s…
“Standard Hydrogen Electrode” (SHE) (really a half-cell) Assigned exactly zero volts
H2(g) → 2H+(aq) + 2e− OR 2H+(aq) + 2e− → H2(g)
Cell Shorthand Notation
anode solid
species oxidized, if different
oxidation product
species reduced
reduction product
cathode solid, if different
e.g., For Zn(s) + Cu2+(aq) → Cu(s) + Zn2+(aq): Zn(s) | Zn2+(aq) || Cu2+(aq) | Cu(s)
e.g., For 2H+(aq) + Fe(s) → H2(g) + Fe2+(aq): Fe(s) | Fe2+(aq) || H+(aq) | H2(g) | Pt(s)
E° and Reaction Spontaneity—or—Predicting Redox Reactions Redox reactions that have a positive overall E° are spontaneous as written.
∆G = –nFE ∆G° = –nFE° n = moles e− transferred n, F always positive F = Faraday constant ∴∆G, ∆G° negative 96,485 C∙mol−1 if E, E° positive

Big Question 2 16 3150:153-002/801
Example: Can acid oxidize iron? Nernst Equation Cell potentials at other-than-standard conditions
ECELL = E°CELL −RTnF ln Q
R = 8.314 J∙mol−1∙K−1 T in Kelvin n = moles e− transferred F = 96,485 C∙mol−1 Q = reaction quotient Application: Concentration Cells Same half-reactions, but different concentrations.
Cell will run so as to
equalize concentrations.

Big Question 2 17 3150:153-002/801
I-70 Indianapolis
Application: Measurement of Equilibrium Constants
ECELL = E°CELL −RTnF ln Q At equilibrium, E = 0 and Q = K, so ln K = nF
RTE°
A Couple Selected Redox/Electrochemistry Applications • Corrosion (ingredients: anode + cathode + electrolyte + conductor)
Some metals form durable oxide coatings that prevent further oxidation. Many do not, however, like iron…..
~Fig. 21.22B Corrosion Prevention
1. Galvanizing—coat steel with a layer of zinc Fe → Fe2+ + 2 e− E°OX = +0.44 V
Zn → Zn2+ + 2 e− E°OX = +0.76 V
2. Alloying—add metals that form a durable oxide coating, such as Cr or Ni
3. Cathodic Protection—connect iron to a metal with a more positive E°OX with a wire.
o used for buried tanks/pipes, piers, lock gates, ship hulls, bridge decks o some cathodic protection uses external current to force the protected steel to be the cathode.
Fe → Fe2+ + 2 e− E°OX = +0.44 V Mg → Mg2+ + 2 e− E°OX = +2.37 V

Big Question 2 18 3150:153-002/801
“The advantage of cathodic protection is that it can halt the progress of corrosion without the removal of chloride-contaminated concrete. Corrosion requires an anode, a point on the reinforcing steel where ions are released. Cathodic protection is the application of direct current such that the steel becomes cathodic to artificial anodes located on the deck. These anodes usually consist of sheets of thin wire mesh. A relatively small DC rectifier operating on “AC” line voltage and a control panel are normally located beneath the bridge. “Cathodic protection systems do not need to operate 24 hours per day to be beneficial. Therefore, they can be powered by solar panels or in line with highway lighting systems. Cathodic protection systems should be considered for locations where traffic maintenance costs are very high and where a few years of additional service between repairs would be advantageous.”
Source: Indiana Department of Transportation • Electrolytic Cells
Using electric current to drive nonspontaneous reaction
e.g., plating Cr(s) on iron
Fe → Fe2+ + 2 e− E°OX = +0.44 V Cr3+ + 3 e− → Cr E°RED = –0.74 V
e.g., How many grams of solid chromium could be deposited by running 10 A of current through a Cr3+ solution for 2 hours?
2 hr ×60 min 60 s 10 C mol e 1 mol Cr 52.00 g Cr 13 g Cr
hr min s 96485 C 3 mol e mol Cr
−
−× × × × × =












