Basic&Local&Alignment&Search&Tool&(BLAST)&...
39
Joyce Njoki Nzioki BecAILRI Hub, Nairobi, Kenya h;p://hub.africabiosciences.org/ h;p://www.Ilri.org/ [email protected] Basic Local Alignment Search Tool (BLAST) IMBB 19, May 2015
Transcript of Basic&Local&Alignment&Search&Tool&(BLAST)&...

Joyce Njoki Nzioki BecA-‐ILRI Hub, Nairobi, Kenya h;p://hub.africabiosciences.org/ h;p://www.Ilri.org/ [email protected]
Basic Local Alignment Search Tool (BLAST) IMBB
19, May 2015

Sequence comparison ü Newly sequenced DNA data is compared to that already available in biological databases.

Sequence comparison ü Newly sequenced DNA data is compared to that already available in biological databases.
ü Sequence comparison (of DNA / Protein data) is achieved through alignment, the process by which regions of similarity is searched between sequences.

Sequence comparison ü Newly sequenced DNA data is compared to that already available in biological databases.
ü Sequence comparison (of DNA / Protein data) is achieved through alignment, the process by which regions of similarity is searched between sequences.
ü This eases annotaCon of new sequences as biological knowledge from well characterized homologs can be conferred

Some terminology 1. Sequence similarity; this is when two sequences are very alike in
base pair or amino acid sequence ü StaCsCcal measures like E-‐value. P-‐Value and bit score ü Percentage idenCty (% of idenCcal residues between sequences) ü The length of sequence stretch that is similar

Some terminology 1. Sequence similarity; this is when two sequences are very alike in
base pair or amino acid sequence ü StaCsCcal measures like E-‐value. P-‐Value and bit score ü Percentage idenCty (% of idenCcal residues between sequences) ü The length of sequence stretch that is similar
2. Homology; homologs diverse from a common ancestor and homology is inferred by sequence, structural and funcConal similarity ü Orthologs – arise due to a speciaCon event ü Paralogs – arise due to gene duplicaCon within the sequence

Some terminology 1. Sequence similarity; this is when two sequences are very alike in
base pair or amino acid sequence ü StaCsCcal measures like E-‐value. P-‐Value and bit score ü Percentage idenCty (% of idenCcal residues between sequences) ü The length of sequence stretch that is similar
2. Homology; homologs diverse from a common ancestor and homology is inferred by sequence, structural and funcConal similarity ü Orthologs – arise due to a speciaCon event ü Paralogs – arise due to gene duplicaCon within the sequence
If sequence similarity in high enough it can infer homology and consequently structural and funcConal similarity but not in all cases.

Sequence Divergence Sequence diverge over Cme and evoluCon is due to mutaCons 1. SubsCtuCons
GATTCGTACG GATGCGTACG
2. InserCon and deleCons GATTCGT _ _ _ ACG GATTCGTGGTACG
3. DuplicaCon ü Complete gene duplicaCon ü Partly gene duplicaCon ü ParCal polysomy (part of a chromosome duplicates) ü Poliploidy (whole genome duplicaCon)

Sequence Alignment
Pairwise sequence alignment; comparison of two sequences to establish regions of residue similarity Why align sequence; ü IdenCfy sequences with significant similarity / homology ü IdenCfy the domains with sequence, structure & funcConal similarity.
ü Database similarity searching ü Confer funcCon from the known to the unknown

Sequence Alignment There two types of alignment strategies 1. Global alignment; find opCmal alignment over the
enCre length of the two compared sequences
ü Best for highly similar sequence ü May miss important biological relaConships in low similarity

Sequence Alignment 2. Local alignment; aligns short regions of similarity
between sequences.
ü Useful when looking for domains in proteins and gene finding in DNA
ü Best for sequences of low similarity and different length

Searching sequence database ü A query sequence is searched against a database to look for homologs.
ü The algorithm used aligns your query to those in the database and returns highly similar sequences.
ü A scoring procedure is implemented on searches to measure the degree of similarity.
ü Judgment needs to be made on whether the similar sequences are homologous to your query based on scienCfic knowledge
ü There 2 programs for this: 1. BLAST (Altschul et al. 1990) 2. FastA (Pearson and Lipman 1988)

Basic Local Alignment Search Tool
ü Blast is a heurisCc approach used to calculate similarity for biological sequences
ü It finds best local alignment ü Blast displays results as a list of sequence matches ordered by staCsCcal significance (hits)
ü BLAST is useful in idenCfying unknown sequences as well as gene or protein funcCon predicCon.

How does blast work ü BLAST is based on smith and waterman algorithm ü It uses a word method to align a query sequence in a database. “it uses words rather than individual residues”

How does blast work ü BLAST is based on smith and waterman algorithm ü It uses a word method to align a query sequence in a database. “it uses words rather than individual residues”
ü NucleoCde query is split into words of defined length GTAAAATCAAGT (word length of 7) GTAAAAT TAAAATC AAAATCA AAATCAA AATCAAG ATCAAGT

How does BLAST work 1. The protein query sequence is split into words of defined length
P D E R T Y H I (word length of three) 123 234 345 456 567 678 PDE DER ERT RTY TYH YHI

How does BLAST work 1. The protein query sequence is split into words of defined length
P D E R T Y H I (word length of three) 123 234 345 456 567 678 PDE DER ERT RTY TYH YHI
2. BLAST finds (The neighborhood) all related words with conservaCve subsCtuCons introduced. PDE DER ERT RTY TYH YHI PDD DDR ERY RTF TYY YHI PDN EDR ERF RFY TTH THV PDR EER DRT KTY FTH THA

How does BLAST work 1. The protein query sequence is split into words of defined length
P D E R T Y H I (word length of three) 123 234 345 456 567 678 PDE DER ERT RTY TYH YHI
2. BLAST finds (The neighborhood) all related words with conservaCve subsCtuCons introduced. PDE DER ERT RTY TYH YHI PDD DDR ERY RTF TYY YHI PDN EDR ERF RFY TTH THV PDR EER DRT KTY FTH THA
3. Define a cut off (T) to select only related words

How does BLAST work 4. The database is searched for all hit to neighborhood words.
GTAAAATCAAGTCCAGTATGACCT
TCAAGTCCA “for nucleoCde one exact match”
P D E R T Y H I S P Y E R S F T Y E R Y A P Y
“Protein two neighboring matches within 40 residues”

How does BLAST work 5. Pairwise alignment by extending hits in both direcCons.
P D E R T Y H I P D D R T _ H L
Score =12
Score 12+8 = 20
Score 20+5 = 25

How does BLAST work 5. Pairwise alignment by extending hits in both direcCons.
P D E R T Y H I P D D R T _ H L
6. Extension conCnues, Cll some point mismatches and gaps drop the score. When the score drops to a given threshold (X) extension is terminated. (PAM and BLOSUM for scoring)
Score =12
Score 12+8 = 20
Score 20+5 = 25

How does BLAST work 5. Pairwise alignment by extending hits in both direcCons.
P D E R T Y H I P D D R T _ H L
6. Extension conCnues, Cll some point mismatches and gaps drop the score. When the score drops to a given threshold (X) extension is terminated. (PAM and BLOSUM for scoring)
7. Any extended segment that scores higher that threshold (S) is called a HSP (High scoring Segment Pair)
Score =12
Score 12+8 = 20
Score 20+5 = 25

How does BLAST work 8. Blast returns sequences with HSP which have staCsCcally
significant scores. • The parameters: W-‐word length and T (cut off for hits)
influence the sensiCvity of the method • The choice of W influence X as larger word lengths result in
higher scores hence X needs be higher to get same sensiCvity.
• The larger the W gives fewer hits, but likely more accurate. • Word size for protein sequences is usually smaller than that
of nucleoCde database searches.

Scoring system NucleoCdes

Scoring systems PROTEINS 1. BLOSUM (BLOcks of amino acid SubsCtuCon MAtrix)
ü BLOSUM matrix is derived from conserved un-‐gapped blocks of protein sequence alignments.
ü BLOSUM matrices are directly calculated across varying evoluConary distance:
ü BLOSUM-‐45 represents sequences with 45% idenCty ü BLOSUM-‐80 represents sequences with 80% idenCty ü The higher the BLOSUM matrix the more closely related the sequences should be

Scoring systems PROTEINS 2. PAM (Point Accepted MutaCons) • Point accepted mutaCon based on the fact that amino acids of the same size, charge or hydrophobicity are likely to be subsCtuted for each other
• PAM-‐1 matrix represents an average change of 1% (1 subsCtuCon in 100 residues).
• PAM-‐1 would be suitable for very closely related sequences
• PAM-‐250 = 250 mutaCons in 100 residues. • Hence higher PAM matrices are used for sequences of greater evoluConary distance


Blast Flavors Some Flavors of BLAST
ucleotide rotein N
NN
N
N
N
P
P
blastx
tblastn
tblastx
P P P P P P
P P P P P P P P P P P P
P P P P P P
Query Database Program
blastp
blastn
P P N N
PP




Graphical BLAST results
ü This is a graphical view of the distribuCon of BLAST hits on the query sequence
ü The length of the hits shows the query coverage and regions of similarity
Query Sequence
Blast Hits, A mouse over gives you the details. Click to view alignment

Hit List BLAST result Hit list gives the iden-fy of sequences similar to your query sequence ranked by similarity
Bit score values < 50 unreliable
% Query coverage
E-‐value
Sequence definiCon, click to view the pairwise alignment
Accession number, Link to the record in GenBank

Pairwise alignment (Protein)

Pairwise alignment (nucleoCde)

BLAST result interpretaCon • How do you make your conclusion on homology: • E-‐value = Expected value. (this indicates the probability that the blast hit may have occurred by random chance).
• The lower the E-‐value (or the closer it is to 0) the more significant the hit. To be certain of homology your E-‐value must be below 10-‐4 or 0.001.
• % idenCty the higher the idenCty the increasing likelihood of homology.
• Query coverage – if a hit has high query coverage and similarity in increases the chances of homology.

Summary for nucleoCde sequence
Length Database Purpose BLAST Program
20 bp or longer NucleoCde IdenCfy the query sequence
blastn megablast
Find similar nucleoCde sequence.
blastn
Find similar proteins to translated query in a translated nucleoCde database
tblastx
Protein Find proteins coded in my query DNA sequence
blastx
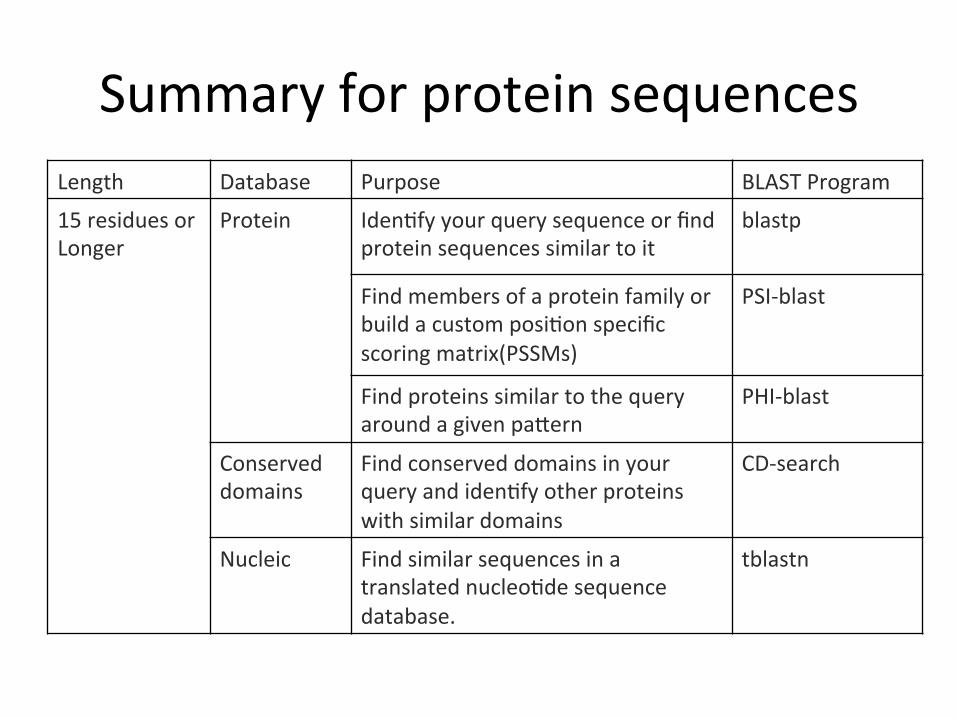
Summary for protein sequences Length Database Purpose BLAST Program
15 residues or Longer
Protein IdenCfy your query sequence or find protein sequences similar to it
blastp
Find members of a protein family or build a custom posiCon specific scoring matrix(PSSMs)
PSI-‐blast
Find proteins similar to the query around a given parern
PHI-‐blast
Conserved domains
Find conserved domains in your query and idenCfy other proteins with similar domains
CD-‐search
Nucleic Find similar sequences in a translated nucleoCde sequence database.
tblastn

The End