
Basic Approach to Dissolution Method Development – Challenges and Regulatory Issues
133
Dr. Harshal Ashok Pawar Assistant Professor & HOD (Quality Assurance), Dr. L. H. Hiranandani College of Pharmacy, Ulhasnagar Email : [email protected] , [email protected]
-
Upload
dr-harshal-pawar -
Category
Health & Medicine
-
view
242 -
download
1
Transcript of Basic Approach to Dissolution Method Development – Challenges and Regulatory Issues

Dr. Harshal Ashok PawarAssistant Professor & HOD (Quality Assurance),
Dr. L. H. Hiranandani College of Pharmacy, UlhasnagarEmail : [email protected] , [email protected]

CONTENTS
Theoretical aspect
Instrumentation
Pre-requisite
Selection of Dissolution medium
Optimization of dissolution parameters Optimization of dissolution parameters
Specifications /Acceptance criteria
Comparison of Dissolution Profiling
Biovaivers

Theoretical AspectTheoretical Aspect

DISSOLUTION
In the body, a pharmaceuticalactive ingredient must be “insolution” before it can beabsorbed by the blood andabsorbed by the blood andultimately carried to thereceptor site to render atherapeutic effect (in vivo).
Dissolution is the processby which that activeingredient enters intosolvent to yield a solution.

DISSOLUTION SOLUBILITY
Dissolution rate is defined asthe amount of solid substancethat goes into solution perunit time under standard
Absolute solubility is definedas the maximum amount ofsolute dissolved in a givensolvent under standardunit time under standard
conditions of temperature,pH, solvent composition andconstant solid surface area.
solvent under standardconditions of temperature ,pressure and pH.
It is a dynamic process. It is a static process.

MECHAMISM OF DISSOLUTION
Initial mechanical lag
Wetting of dosage form
Penetration of dissolution medium
Disintegration Disintegration
Deaggregation
Dissolution
Occlusion of some particles

Dissolution process of solid dosage Forms :
DISINTEGRATION DISSOLUTION
DISSOLUTION ABSORPTION
IN-VIVO
TABLETS OR CAPSULES
GRANULES OR AGGREGATES
DRUG IN SOLUTION
(IN-VITRO OR IN-
DRUG IN BLOOD,OTHER
FLUIDS,AND IN-VIVO
DISAGGREGATION
DISSOLUTION
AGGREGATES
FINE PARTICLES
(IN-VITRO OR IN-VIVO)
FLUIDS,AND TISSUES

Dissolution TestingDissolution is one the three primary tests used to
release a finished drug product:
Assay – determines the overall potency of the batch and ensuresthe accuracy of the finished drug product.
Dose Uniformity – determines the consistency among the Dose Uniformity – determines the consistency among theindividual dosage units and ensures the precision of themanufacturing process.
Dissolution – ensures that the performance of the finished drugproduct is consistent with the release rates of the API as determinedin bioavailability studies during the clinical trials.

Which product to be tested?USP <1088>:
“No product, including suspensions and chewable tablets,should be developed without dissolution or drug releasecharacterization where a solid phase exists.”
and
“Dissolution testing is required for all solid oral Pharmacopeial“Dissolution testing is required for all solid oral Pharmacopeialdosage forms in which absorption of the drug is necessaryfor the product to exert the desired therapeutic effect.
Exceptions are for tablets meeting a requirement forcompleteness of solution or for rapid (10 to 15 minutes)disintegration for soluble or radiolabeled drugs.”

NEED FOR DISSOLUTION TESTING
Development and Optimization of dosage forms.
Batch to batch drug release uniformity.
Ensures quality, safety, efficacy and stability of the product.
Evaluation of IVIV Correlation / bioavailability .
To support waiver for bioequivalence requirement.
For assessing Pre and Post approval changes in manufacturing
process, or formulation

Dissolution and Drug Development
Drug
Clinical trial (IVIV)
Manufacturing
(Quality control)
Discovery ( Little to no dissolution)
Characterization (Intrinsic dissolution)
Drug development (DMD)
control)

InstrumentationInstrumentation

I.P U.S.P B.P E.P
TYPE 1 Paddleapparatus
Basket apparatus
Basket apparatus
Basket apparatus
TYPE 2 Basket apparatus
Paddleapparatus
Paddleapparatus
Paddleapparatus
TYPE 3 Reciprocating cylinder
Flow through cell
Flow through cell
TYPE 4 Flow through cell
TYPE 5 Paddle over disk
TYPE 6 Rotatingcylinder
TYPE 7 Reciprocating disk

APPARATUS-1(ROTATING BASKET)
DESIGN:
Vessel: -Made of borosilicate glass.
-Semi hemispherical bottom
-Capacity 1000ml
Shaft : -Stainless steel type 316
-Rotates smoothly without
significance wobble(100 rpm)
-Speed regulator
Water bath:-Maintained at 37±0.5ºC
USE: Tablets, capsules, delayed release
suppositories, floating dosage forms.

Apparatus 1 - Basket

Advantages
Full pH change during the test
Can be easily automated which is important for routine investigations.
Disadvantages
Basket screen is clogged with
gummy particles.
Hydrodynamic dead zoneunder the basket
Degassing is particularly important
Mesh gets corroded by HCl solution.

APPARATUS-2 (PADDLE)
DESIGN:
Vessel: -Same as basket apparatus
Shaft: -The blade passes through the shaft so that the bottom of the blade fuses with bottom of the shaft.
Stirring elements: -Made of tefflon
For laboratory purposeFor laboratory purpose
-Stainless steel 316
Water-bath: -Maintains at 37±0.5°C
Sinkers : -Platinum wire used to prevent
tablet/capsule from floating

Advantages
Easy to use
Robust
Can be easily automated which is important for
routine investigations
Disadvantages Disadvantages
pH/media change is often difficult
Sinkers for floating dosage forms

Paddle apparatus:

APPARATUS-3(RECIPROCATING CYLINDER)Tester was designed to test the dissolution rates of extended releaseproducts or any dosage form requiring release profiling at multiple pHlevels.
DESIGN:
Vessel: -Set of cylindrical flat bottom glass vessels
-Set of reciprocating cylinders
-stainless steel fittings(type 316) and
screens made of nonsorbing or screens made of nonsorbing or
non-reactive materials.
Agitation type: -Reciprocating at 5-35 rpm
Volume of dissolution medium:-200-250ml
Water bath:- Maintain at 37±0.5°C
USE: Chewable Tablets, beads, controlled and
Extended release formulations, Soft gel cap,
Non Disintegrating type products

METHOD(Reciprocating cylinder):
Place the stated volume of dissolution medium in each vessel of the apparatus, assemble the apparatus, equilibrate the dissolution medium to 37±0.5 and remove the thermometer
Place one dosage form unit in each of the cylinders taking care to exclude the air bubbles from the surface of each dosage unit and immediately operate the apparatus as specified in the monograph.
During the upward and downward stroke, the reciprocating cylinder During the upward and downward stroke, the reciprocating cylinder moves through a total distance of 9.9 to 10.1cm.
Within the time interval specified, raise the cylinders and withdraw a portion of the solution under test from a zone midway between the surface of the dissolution medium and bottom of each vessel.

Advantages Easy to change the pH
pH-profiles
Hydrodynamics can be directly influenced by varying the dip rate
Disadvantages Small volume (max. 250 ml)
Little experience
Limited data

APPARATUS-4 (FLOW THROUGH CELL)
DESIGN:
Reservoir : -For dissolution medium
Pump : -Forces dissolution medium through cell
-Holding a sample
-Flow rate 10-100ml/min
-Laminar flow is maintained
-Peristaltic/centrifugal pumps are not recommended
Water bath:- Maintain at 37±0.5°C
USE:
Modified-release dosage forms and immediate-release dosage forms
Soft gelatin capsules, beaded products, suppositories, or injectable-depot dosage forms
Suspension-type extended-release dosage forms for oral or parenteral use, or ocular application.

METHOD(Flow through cell):
The flow through cell is transparent & inert mounted vertically with filters.
Standard cell diameters are 12 & 22.6 mm.
The bottom cone usually filled with glass beads of 1 mm diameter.
Tablet holder used for positioning special dosage form e.g. inlay tablets.
Place the glass beads into the cell as specified in the monograph. Place the glass beads into the cell as specified in the monograph.
Place one dosage unit on top of the beads or on a wire carrier.
Assemble the filter head and fix the parts together by means of a suitable clamping device.
Introduce by the pump of the dissolution medium warmed to 37±0.5 through the bottom of the cell to obtain the flow rate specified and measured with an accuracy of 5%.
Collect the eluate by fractions at each of the times stated.
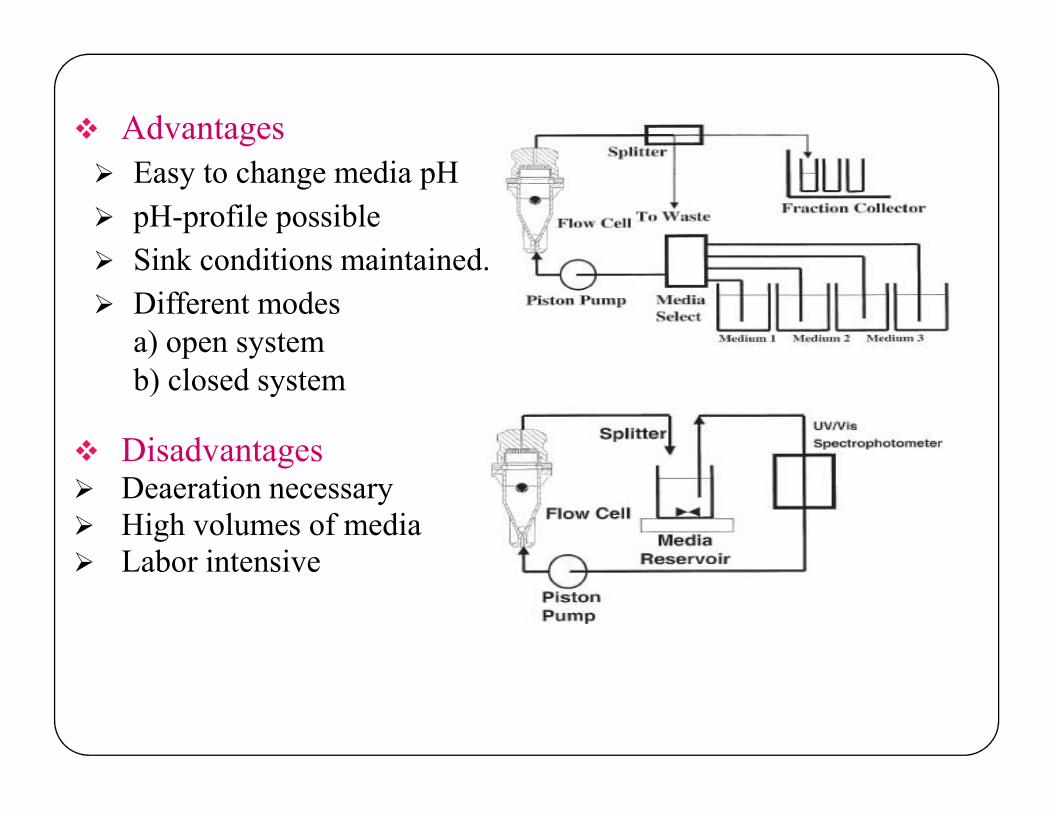
Advantages
Easy to change media pH
pH-profile possible
Sink conditions maintained.
Different modesa) open systemb) closed system
Disadvantages Deaeration necessary High volumes of media Labor intensive
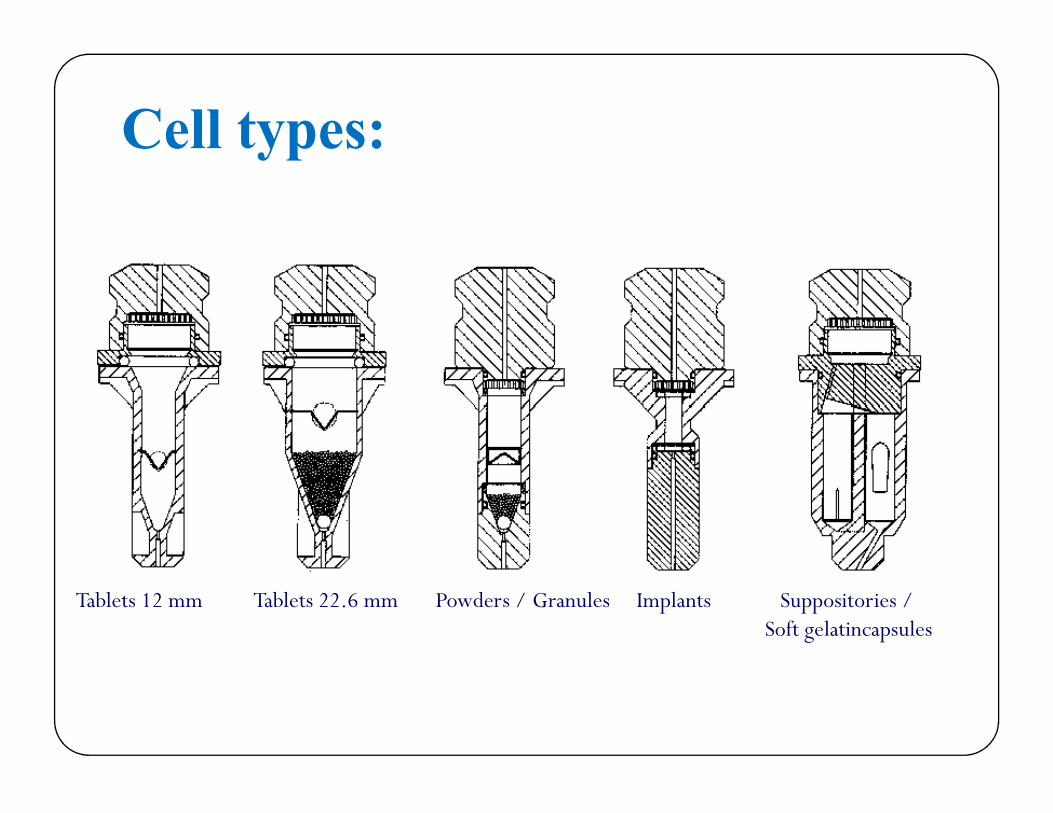
Cell types:
Tablets 12 mm Tablets 22.6 mm Powders / Granules Implants Suppositories / Soft gelatincapsules

Flow-Through Cell:

APPARATUS-5(PADDLE-OVER-DISK)
DESIGN:
Vessel
Shaft
Stirring elements- rotating speed 25-50 rpm
Sample holder:-disk assembly that hold a product in such a way Sample holder:-disk assembly that hold a product in such a way
that release surface is parallel with paddle
-Paddle is directly attached over disk assembly
-Samples are drawn between surface off the medium
and top of the paddle blade
Volume:900ml
Temperature:32°C (Similar to skin)

USE: Transdermal patches, ointments, floaters , emulsions.
Modification: Disk design and volume
Advantages:
Easy to handle
Sink conditions are maintained.
Membrane effect is minimum.
i.e. drug is placed on a disc at the bottom.
Disadvantages:Disadvantages:
Disk assembly restricts the patch size
17 mesh is standard(others available)
Accommodates patches up to 90mm.

METHOD(Paddle over disk)
This method is used for testing the release of drugs from transdermal products.
The apparatus consists of a sample holder or disc assembly that holds the product.
The entire preparation is placed in a dissolution flask filled with specified medium maintained at 32ºC.specified medium maintained at 32ºC.
The paddle is placed directly over the disc assembly.
The disk assembly holds the system flat and is positioned such that release surface is placed parallel with the bottom of the paddle blade. Vessel is covered to minimize evaporation during test.
Samples are drawn midway between the surface of dissolution medium and the top of the paddle blade at specified times.

APPARATUS-6(ROTATING CYLINDER)
DESIGN:
Vessel:- In place of basket, cylinder is used.
Shaft :-Stainless steel 316
Sample :- Mounted to cuprophan (inner porous cellulosic material)
an entire system adheres to cylinder.an entire system adheres to cylinder.
- Dosage unit is placed in cylinder and release from side out.
Water-bath: maintained at 32±0.5°C
USE:
Transdermal patches cannot be cut into small size.
Solid dosage forms, pH profile , small volumes

METHOD( Rotating cylinder):
Use the assembly from apparatus 1 except to replace the basket and shaft with a stainless steel cylinder stirring element.
The temperature is maintained at 32±0.5°C.
The dosage unit is placed on the cylinder with side out .
The dosage unit is placed to the exterior of the cylinder such that long axis of the system fits around the circumference of the cylinder long axis of the system fits around the circumference of the cylinder and removes trapped air bubbles.
Place the cylinder in the apparatus and immediately rotate at the rate specified in the individual monograph.
Samples are drawn midway between the surface of the dissolution medium and the top of the rotating cylinder for analysis.

Carefully apply theadhesive coated side of thesystem to the exterior ofthe cylinder with the longaxis of the system fittingaxis of the system fittingaround the circumferenceof the cylinder.

Rotating cylinder:
Advantages: -Equipment (apparatus 1)available with the manufacturers can be used with modification as apparatus 6.
Disadvantages:-Large volume of medium is required.
-Drug gets diluted & causes difficulties in analysis
-Difficult to clean the cylinder.

APPARATUS-7(RECIPROCATING-DISK)DESIGN:
Vessel:-Flat bottomed cylindrical vessel
-Volume of dissolution medium
Shaft :
Sample : -Placed on disk shaped holders
Agitation :-Reciprocation
shaft
disk
dissolution medium
constant temp water bath Agitation :-Reciprocation
-Reciprocating frequency 30 cycle/sec
Water-bath:-Maintain at 32±0.5°C
USE:
Non-disintegrating, oral modified-release dosage forms, stents, and implants
Transdermal dosage forms.

METHOD(Reciprocating disk):
The assembly consists of a set of volumetrically calibrated solution containers made of glass or suitable inert material, a motor , a drive assembly used to reciprocate the system vertically.
The samples are placed on the disk shaped holders using cuprophan supports
The test is carried out at 32°C.
The reciprocating frequency is 30cycles/min. The reciprocating frequency is 30cycles/min.
Advantages:-Convenient method for selecting the volume of the
medium. Modifications can been made to accommodate
300 mL vessels
-sink conditions can be maintained.
Disadvantages: -Investment is high because the design is totally different from standard equipment already available in industry.


UNOFFICIAL METHODS
1.ROTATING/STATIC DISK METHOD
Developed by late Eino nelson and described by Levy and Sahli.
In this method ,the drug is compressed in a non-disintegrating disc without excipients.
The disc is mounted in a holder so that only one face of the disc is exposed to the dissolution medium.exposed to the dissolution medium.
The holder and disc are immersed in medium and held in a fixed position as in static disc method and rotated at a given speed in rotating disc method.
Samples are collected at predetermined times.
Surface area of the drug through which dissolution
occurs is kept constant –intrinsic dissolution rate.

2.BEAKER METHOD:
Reported by Levy and Hayes(1960).
Dissolution medium, 250ml of 0.1N HCl at 37°C placed
in a 400ml beaker.
Agitation by three blade polyethylene stirrer, 5cm diameter and rotates at 60 rpm.
Stirrer immersed to a depth of 2.7 cm in medium and in the center.
Tablets are placed in a beaker and test was carried out.Tablets are placed in a beaker and test was carried out.
Samples are removed and assayed for the content.
3.FLASK STIRRER METHOD
Developed by Poole(1969).It includes RBF and a stirring element similar to that of beaker method.
RBF used to avoid the formation of moulds of particles in different positions on the flat bottom of a beaker.

4.PERISTALSIS METHOD:
To stimulate hydrodynamic condition of GIT tract in an in-vitro dissolution device.
It consists of rigid plastic cylindrical tubing fitted with septum and rubber stopper at both ends.
Dissolution chamber consists of a space between septum and lower stopper.
Dissolution medium is pumped with peristaltic action through Dissolution medium is pumped with peristaltic action through the dosage form.
5.ROTATING BOTTLE METHOD:
It consists of rotating rack to hold sample drug products in bottles and they are capped tightly & rotated in 37°C temperature bath.
Sample are decanted through a 40 mesh screen and residue are assayed.

6.DIALYSIS METHOD:
Cell consist of 32mm inflated membrane.
Plugged at the lower end by tight fitting cylindrical perspex box.
Upper end of the tube held by thin perspex ring inserted into the tube and secured by an elastic band.
The cell suspended , from the arm of the tablet disintegration apparatus and containing the dosage form in 150ml of distilled water at 37°C.water at 37°C.
The cell is raised or lowered 30times a min, into 150ml of distilled water at same temperature.
Agitation by slight flexing and stretching of the dialysis membrane as it enters and leaves the bath. Rotated at 60rpm.
.

7.DIFFUSION CELL
Static or flow through diffusion cells are used to characterize in-vitro drug release and drug permeation kinetics from a topical drug product e.g.: Ointment, cream or transdermal drug product.
The Franz diffusion cell is static diffusion system used to characterize drug permeation through skin model.
The skin is mounted on the Franz diffusion cell and the drug product is placed on the skin surface.product is placed on the skin surface.
The drug permeates across the skin into a receptor fluid compartment that may be sampled at various times.
This system is used for selection of appropriate formulation that has optimum drug delivery.

Diffusion cell


Pre-requisitePre-requisite

Instrument should be calibrated /Operational Checks ( document each time of use):
•Basket/shaft examination
•Paddle examination
•Vessel examination
•Vessel Temperature
•Vibration
Analytical balance must be calibrated Analytical balance must be calibrated
pH meter should be calibrated
Pipettes and other glassware's
Volumetric accuracy : ± 1%
Deareation of dissolution medium.
Lighting should be sufficient to perform visual observations.

Calibration of Dissolution Apparatus
Why ?
• To confirm suitability of the equipment and proper operation of the apparatus
How ?How ?
• Mechanical calibration (verification of physical parameters)
• Chemical calibration (Apparatus Suitability Test – USP)
When ?
• Before using new test equipment
• After relocation or major maintenance
• At regular intervals (Every 6 months)

Current Harmonized Physical Parameters and Tolerances USP Mechanical Calibration Parameters include:
Basket/Shaft Wobble (No significant wobble)
Vessel/Shaft Centering (2 mm from centerline)
Height check/Basket or Paddle Depth as measured at basket bottom or Paddle bottom (25 + 2 mm)bottom or Paddle bottom (25 + 2 mm)
No significant vibration
Rotational speed (+ 4%)
Vessel Temperature (37.0 + 0.5 C)
Basket Wobble (bottom rim) (+ 1mm)

USP Performance Verification Test (PVT) Official since 1978, USP Calibration with Prednisone and Salicylic
Acid has been the means of qualifying the dissolution apparatus.
Initially, the primary purpose was to indicate environmentaleffects on the apparatus and vibration since most other parameterscould be controlled by mechanical measurements
The original test was called “Calibration” which was not a trueindication of the test being performed, later changed to“PerformanceVerification” (PVT)
The PVT, has been responsible for detecting problems associatedwith dissolution apparatus that are found to be within mechanicaltolerances

Calibrator Tablets
1970’s : USP Calibrator Tablets Introduced
Disintegrating – 50 mg Prednisone (Upjohn)
Non Disintegrating – 300 mg Salicylic Acid (Hoffman LaRoche)
1997 : 50 mg Prednisone replaced with 10 mg Prednisone manufactured at University of Marylandmanufactured at University of Maryland
2004 : USP begins search for replacement for 10 mg Prednisone tablet
USP: Both Calibrators on a given apparatus (i.e. 4 calibration tests if instrument is used for paddle and basket methods)
JP, BP and EP: No calibrator tablets

Allowable Variations
A basket with gold coating 2.5 μm thick (0.0001 inch) is an allowable variation of the standard 40-mesh basket.
Some changes can be made to the compendial apparatus; for example, a basket mesh size other than the typical 40-mesh basket (e.g., 10-, 20-, or 80-mesh)basket (e.g., 10-, 20-, or 80-mesh)
Larger vessels accommodating up to two and four liters are now allowable variations in the USP. Such vessels are advantageous for poorly soluble drugs.
For example, a small-volume apparatus with mini paddles and baskets may be considered for low-dosage strength products.

De-areation Air bubbles can act as a barrier to the dissolution process
if present on the dosage unit or basket mesh and canadversely affect the reliability of the test results.
Furthermore, bubbles can cause particles to cling to theapparatus and vessel walls.apparatus and vessel walls.
Bubbles on the dosage unit may increase buoyancy, leadingto an increase in the dissolution rate, or may decrease theavailable surface area, leading to a decrease in the dissolutionrate.
Poorly soluble drugs are most sensitive to interferencefrom air bubbles, therefore, deaeration may be needed.

Degassing as per USP
Prepare dissolution media and
properly deaerate.
USP Method:USP Method:
Heat media to 41°C, vacuumfilter through 0.45μm filter,continue to pull vacuum for 5additional minutes

Common Degassing Methods
Acceptable Methods
USP Vacuum Filtration Method (default unless another approach is validated)
Helium Sparging*
Automated Degassing*
Unacceptable Methods
Nitrogen Sparging
Sonication
Automated Degassing*
Superheating*
Not Degassing At All*
*when validated against USP method

Method DevelopmentMethod Development

Dissolution Method Goals
A successful dissolution method will be:
Discriminatory
Robust and Rugged
Correlated to In Vivo Correlated to In Vivo
Transferrable
Controlled Variability

When to develop method? When to develop method?

Discrimination
Discrimination in Dissolution simply means being able to tell the difference between good difference between good and bad formulations

Development of a Discriminating MethodThe procedure should be capable of distinguishing significantchanges in composition or manufacturing process that mightbe expected to affect in vivo performance.
Factors to consider:
Qualitative and quantitative excipient changes Qualitative and quantitative excipient changes
Manufacturing parameters:
Lubrication
Blend time
Compression force
Drying parameters

Steps for dissolution method development
Literature survey : Drug and drug product knowledge
Selection of Apparatus
Selection of dissolution medium
Optimization of dissolution parameters Optimization of dissolution parameters
Development of suitable analytical method for estimation of content
Validation of analytical method

Knowledge of Drug and Drug Product
Characteristics of the API e.g., particle size, crystal form,bulk density, solubility
Product composition e.g., drug loading /dose, and theidentity, type, and levels of excipients
Manufacturing process e.g., compression forces, Manufacturing process e.g., compression forces,equipment
Effects of stability storage conditions e.g., temperature,humidity
Incompatibility of the drug with certain buffers or salts

Dissolution Tester Choice Paddles and Baskets tend to be the choice for most solid
oral dosage forms.
If pH changes, greater/smaller volumes, or different agitation is needed then Apparatus 3 and 4 are often considered after exhausting Paddle and Basket testing
For Transdermals Apparatus 5-7 are the primary choices.
For semisolid dosage forms, the generally used apparatus include the vertical diffusion cell, apparatus include the vertical diffusion cell, immersion cell, and flow-through cell apparatus with the insert for topical dosage forms
A rotating bottle or dialysis tubes may have utility for microspheres and implants; peak vessels for eliminating coning; and modified flow-through cells for special dosage forms including powders and stents.

Agitation Rate Should be sufficient to allow for
media to interact with dosage form
Decreasing or increasing the apparatus rotation speed may be justified if to achieve an in-vitro–in-vivo correlation (IVIVC)
For better discrimination
Too much agitation can result innon-discriminatory profiles
•Baskets – 50-100 RPM
•Paddles – 25-100 RPM

USP.APPARATUS DESCRIPTION ROT.SPEED DOSAGE FORM
TYPE 1 Basket apparatus 50-100 rpm IDR,DR,ER
TYPE 2 Paddle apparatus 50-75rpm25-50rpm
IDR,DR,ERSuspension
TYPE 3 Reciprocatingcylinder
5-30 dips/min IDR,ER
TYPE 4 Flow through cell 2-50ml/min ER,Poorly soluble API
TYPE 5 Paddle over disk 25-50 rpm TRANSDERMAL
TYPE 6 Rotating cylinder N/A TRANSDERMAL
TYPE 7 Reciprocatingholder
30 rpm ER

The Ideal Dissolution Media
•Meets sink conditions
•Simple preparation
•Drug is Stable in media 24 hrs+
•Uses as little extras as possible possible
–Surfactants
–Alcohol
•Biologically relevant for site of dissolution in vivo
–IR typically in acid
–DR typically in acid, then neutral
–MR typically in neutral solution
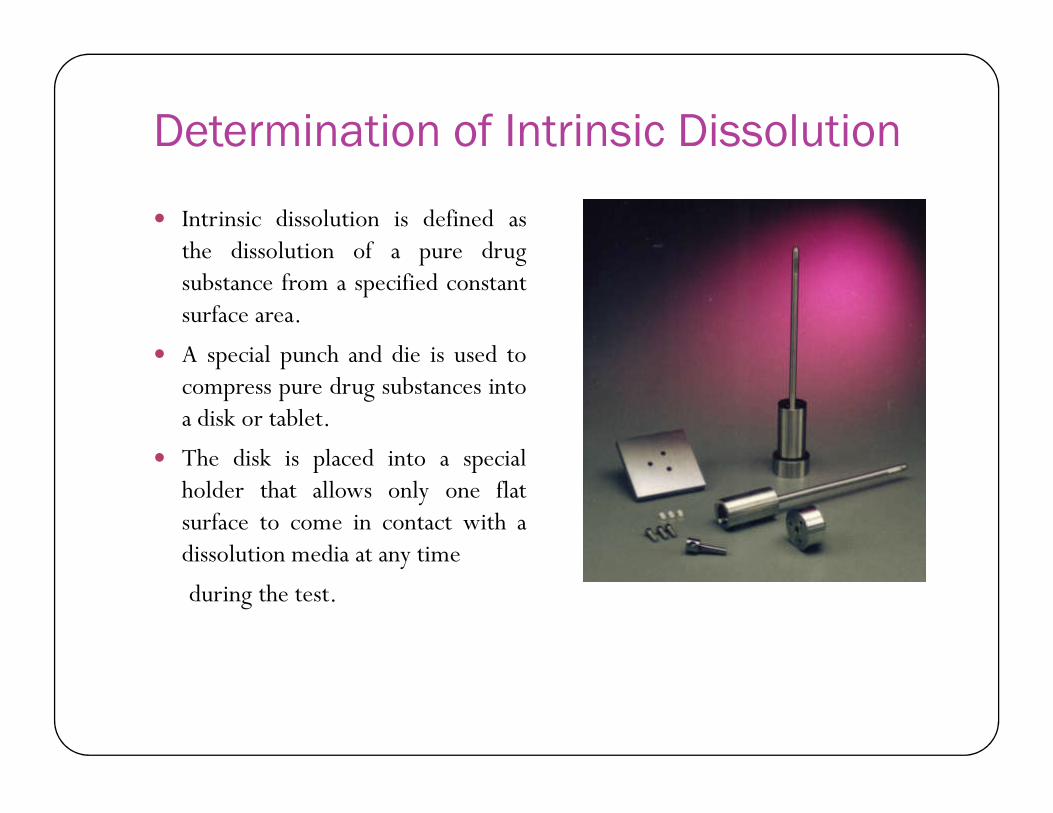
Determination of Intrinsic Dissolution
Intrinsic dissolution is defined asthe dissolution of a pure drugsubstance from a specified constantsurface area.
A special punch and die is used tocompress pure drug substances intoa disk or tablet.a disk or tablet.
The disk is placed into a specialholder that allows only one flatsurface to come in contact with adissolution media at any time
during the test.

Application of IDIntrinsic Dissolution
Intrinsic dissolution data is generally used in drugscreening but can provide helpful solubility informationfor method development.
Rates lower than 0.1 mg/min/cm2 generally mean that Rates lower than 0.1 mg/min/cm2 generally mean thatbioavailability will be determined by the dissolutionrate.
Rates higher than 1 mg/min/cm2 mean thatbioavailability will most likely be determined by thedrug permeability.

pH Dependent Solubility Solubility screen in multiple media (pH 1.2-7.5) should be
done to determine optimal solubility.
If needed, use as little surfactant as necessary.
Evaluate multiple surfactants (pay attention to grades and vendors)
Investigations of the stability of the drug substance should Investigations of the stability of the drug substance should be carried out, when needed, in the selected dissolution medium with excipients present, at 37°.
Effect of pH on solubility and stability need to be evaluated.

Sink condition and Solubility Sink condition : Volume of medium at least three times that required in
order to form a saturated solution of drug.
Solubility of the drug substance is usually evaluated by determining the saturation concentration of the drug in different media at 37° using the shake-flask solubility method (equilibrium solubility). Alternative methods for solubility determination may also be used.
In the absence of sink conditions, investigate methods to enhance solubility, e.g. use of a surfactant. If a surfactant is used, its concentration should be properly justified (e.g. typically <2% Sodium Lauryl Sulfate (SLS)).
In certain cases, it may be necessary to evaluate the solubility of the drug at room temperature.
The pH of the clear supernatant should be checked to determine whether the pH changes during the solubility test.

Rules of Thumb for Media Limits Surfactants below 1% tend to be accepted with appropriate checks that lower
limits aren’t acceptable
>1% require greater scrutiny, other surfactants usually
>1.5% tends to be very difficult to handle with automation
Alcohol is generally a last resort – unless doing a dose dumping study specifically.specifically.
Stay within pH 1.1 – pH 7.5 if at all possible
The use of surfactants needs to be justified by data that show lowsolubility in the aqueous media. The chosen concentration of surfactantalso needs to be justified by providing dissolution profiles in mediacontaining the surfactant at concentrations higher and lower than the chosenconcentration.

USP Surfactants

Use of enzymes The use of enzymes in the dissolution medium is permitted,
in accordance with general chapter Dissolution 711 ,whendissolution failures occur as a result of cross-linkingwith gelatin capsules or gelatin-coated products.
A discussion of the phenomenon of cross-linking and methoddevelopment using enzymes can be found in proposedgeneral information chapter Capsules–Dissolution Testing andRelated Quality Attributes 1094 .

Antifoaming Agents and antioxidants The hydrodynamics are influenced by the cylinder's
reciprocating motion and the resulting movement of thesample in the medium. The reciprocating motion of thecylinder and screen may cause foaming if the mediumcontains surfactants. Addition of an anti-foaming agent suchcontains surfactants. Addition of an anti-foaming agent suchas simethicone or n-octanol may be useful for avoidingfoaming from surfactants.
In some cases, antioxidants such as ascorbic acid may be used in the dissolution medium to stabilize the drug.

Media Cautions Be careful with water
–No buffering capacity
–Quality can differ b/w sites
–Quality can differ b/w DI systems, filters, etc.
Check pH before and after run to ensure buffering capacity is Check pH before and after run to ensure buffering capacity is acceptable
Beware of methods needing tight pH limits
Do not use SLS with Potassium Phosphate Buffers –Sodium Phosphate only

Bio-relevant Dissolution Medium Bio-relevant media is the media that represent the
conditions same as that of the in-vivo condition.
The fed and fasted state may have significant effects on the absorption or solubility of a compound.
Composition of media that simulate the fed and fasted Composition of media that simulate the fed and fasted condition is necessary to establish in-vivo in-vitro correlations.
This media reflect changes in the pH, bile concentration and osmolarity after meal intake and therefore have a different composition than that of typical compendial media.
Why the Need for Better Biorelevant Information Better IVIVC
Fewer Clinical Trials
Better Predictive tools
Shorter Development CyclesCycles

Choice of bio-relevant media is usually based upon: A mechanistic approach that considers the absorption site, if
known.
Whether the rate-limiting step to absorption is the dissolutionor permeability of the compound.
Fed and fasted states may have significant effects on theFed and fasted states may have significant effects on theabsorption or solubility of a compound.
These media are primarily used to establish in vitro-in vivocorrelation during formulation development and to assess potentialfood effects; they are not always intended for Quality Controlpurposes.

Selection of Dissolution medium Primary requirement for selection of dissolution media is
that, it should be able to reflect in vivo situations when it isused to establish an IVIVC.
For Class I and III drugs, use of simple aqueous media such asSGF without enzymes or SIF without enzymes isSGF without enzymes or SIF without enzymes isrecommended.
For Class II and III drugs, use of biorelevant media fordissolution testing is recommended. They are: 1) SGF plussurfactant 2) Milk with 3.5 % fat to stimulate fed statecondition 3)FaSSIF is used for poorly soluble drugs.

Temperature of Dissolution Medium
The standard temperature- 37±0.5 °C for oral dosage form .
Slightly increased temperatures such as 38±0.5 °C have been recommended for dosages forms such as suppositories.
Lower temperatures such as 32±0.5 °C are utilized for Lower temperatures such as 32±0.5 °C are utilized for topical dosage forms such as trans-dermal patches and topical ointments.
Media temperature readings must be taken at least twice during the dissolution test, at the start and end of a test.

Appropriate Test Duration Time points should be selected to adequately characterize the
ascending and plateau phases of the dissolution curve.
Typically:
Immediate release, 85% in <15 minutes, one time point
Immediate release, 15, 20, 30, 45, 60 minutes
For some products, including suspensions, useful information mayFor some products, including suspensions, useful information maybe obtained from earlier points, e.g., 5–10 min.
Delayed release,Acid 1-2 hours, Buffer +30 minutes
Extended release, minimum of three points
– Initial (1-2 hours) to show potential dose dumping
– Intermediate point to define similar in vivo profile
– Final point to show that essentially complete release (>80%) of thedrug is achieved.

When to add dosage form?
Tablet introduction may beperformed manually orautomatically
Dosage forms may beintroduced simultaneously orintroduced simultaneously orsequentially but they must beintroduced into non-rotating media.
Evaporation cover shown withDosage Delivery Module(DDM)
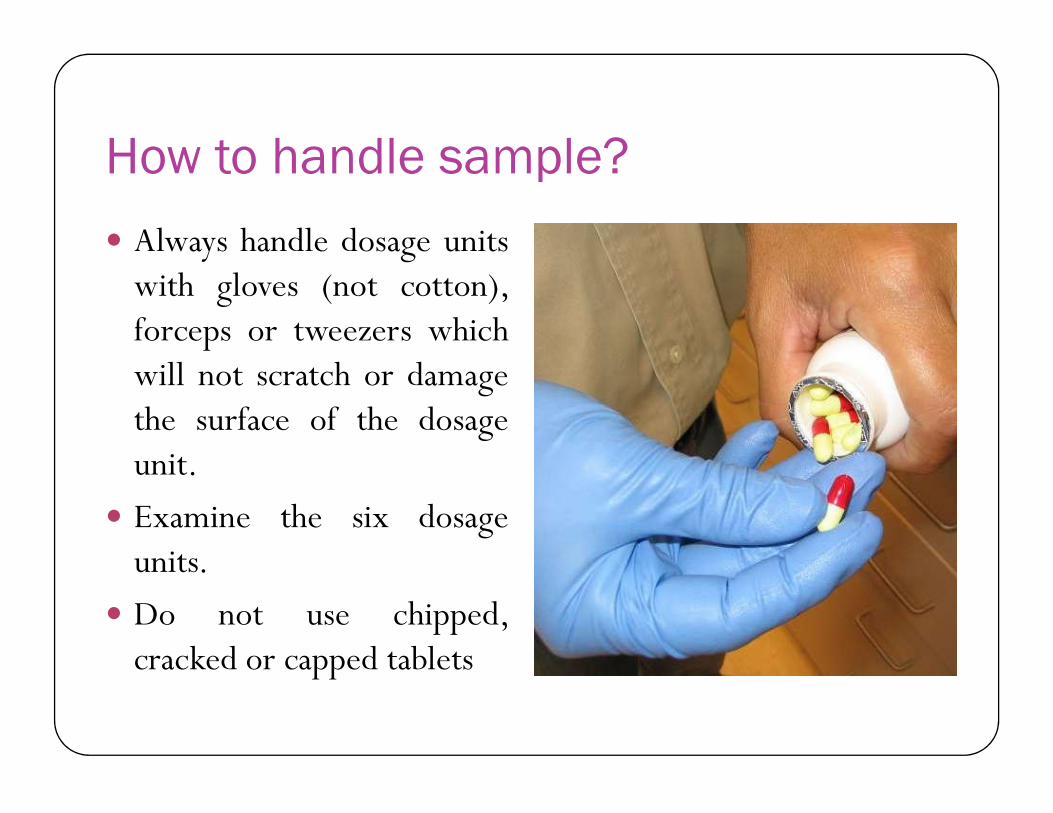
How to handle sample?
Always handle dosage unitswith gloves (not cotton),forceps or tweezers whichwill not scratch or damagethe surface of the dosagethe surface of the dosageunit.
Examine the six dosageunits.
Do not use chipped,cracked or capped tablets

Weighing of the sample?
Option: Record the dosageunit weights?
Weight is forinformation andinvestigation purposesonly.
Dosage units are to bechosen at random and maynot be selected ordiscarded based on weight.

Sinkers (USP Apparatus 2) In addition to sinking, floating dosage forms, sinkers may
assist in keeping a dosage form from sticking to the vesselinappropriately as in the case with some film coated tablets.
Sinkers Specifications must be adequately described inthe method to eliminate hydrodynamic variation associatedwith different sinker devices.
Sinkers may be fabricated by wrapping around a corkbore.
Should not be too tight – will restrict any disintegratingrelease mechanism
Too loosely, the dosage form may escape soon after the testbegins.



USP Recommendations about sinkers

Examples of typical observations Uneven distribution of particles throughout the
vessel
1. Particles cling to the sides of the vessel,
2. coning or mounding directly under the apparatus, e.g., below the basket or paddle,below the basket or paddle,
3. Particles float at the surface of the medium,
4. Film-coated tablets stick to the vessel

Examples of typical observations Air bubbles on the inside of the vessel or on the
apparatus or dosage unit
1. Air bubbles on the inside of the vessel or on the apparatus or dosage unit.
2. Sheen on the apparatus is also a sign of air bubbles.2. Sheen on the apparatus is also a sign of air bubbles.
This observation would typically be made when
assessing the need to deaerate the medium

Examples of typical observations Dancing or spinning of the dosage unit, or the dosage
unit being hit by the paddle.
Adhesion of particles to the paddle or the inside of thebasket, which may be observed upon removal of the
stirring device at the end of the run.stirring device at the end of the run.
Pellicles or analogous formations, such as transparentsacs or rubbery, swollen masses surrounding the capsulecontents.

Examples of typical observations Observation of the disintegration rate (e.g.,
percentage reduction in size of the dosage unit within a certain time frame).
Complex disintegration of the coating of modified or enteric-coated products. modified or enteric-coated products.
Whether the dosage form lands in the vessel center or off-center, and if off-center, whether it sticks there.
Time required for the complete dissolution of the capsule shell or for tablet disintegration.

Usual Remedies Visual observations
Changing any of the followingfactors:
1) Apparatus type,
2) Speed of agitation,
3) Level of deaeration,3) Level of deaeration,
4) Sinker type, or
5) composition of the medium.
For dosage forms that exhibitconing (mounding) under thepaddle at 50 rpm, the coning can bereduced by increasing the paddlespeed to 75 rpm

How To Sample Properly?
Filtration must occur at USP location and at appropriate time
±2 % from time point or 15 minutes (whichever less)
Halfway between top of paddle Halfway between top of paddle or basket and media
No closer than 1cm to vessel wall
Recommend not sampling close to shaft due to poor hydrodynamics

Acceptable Method Requirements
Low variability (<20% at initial time point,<10% at later points)
Complete Release (85%+ or Asymptote)
Proper understanding of dissolution release
Discrimination between batches -Challengedwith other formulations
Robust/Rugged/Reproducible results

SpecificationsSpecifications

Specification Parameters -Dissolution Dissolution is considered product-specific.
The method and limits should be appropriate for the proposed product.
It is useful to have the parameters (medium, apparatus, speed) in specs.specs.
Dissolution specs at release and shelf-life should be identical.
Surfactant use should be exceptional and appropriate.—not exceed 2% normally

Dissolution Acceptance Criteria
Q –Value :
Define as a percentage of drug content dissolved in a given time period.
It is commonly used in the USP for immediate release and It is commonly used in the USP for immediate release and delayed release dosage forms.
The quantity of Q is the amount of dissolved activeingredient specified in the individual monographexpressed as a percentage of the labeled content.

Q - Value For highly soluble and rapidly dissolving drug products (BCS
classes 1 and 3), a single-point dissolution test specification of NLT 85% (Q=80%) in 60 minutes or less is sufficient as a routine quality control test for batch-to-batch uniformity.
For slowly dissolving or poorly water soluble drugs (BCS class 2), a two-point dissolution specification, one at 15 minutes to include a dissolution range (a dissolution window) and the other at a later point (30, 45, dissolution range (a dissolution window) and the other at a later point (30, 45, or 60 minutes) to ensure 85% dissolution, is recommended to characterize the quality of the product.
For products containing water insoluble APIs, it is recommended to have a two tire dissolution limit. For example Artemether dissolution: NLT 40% in 1 hour and NLT 60% at the 3rd hour.

Immediate Release Forms – AcceptanceTable USP <711>
STAGE No. of Dosage units tested
Acceptance criteria
S1 6 Each unit is ≥ Q + 5%.
S2 6 Average of 12 units (S1 + S2) is ≥Q and no unit is < Q – 15%.unit is < Q – 15%.
S3 12(6+6+12=24) Average of 24 units (S1 + S2 + S3) is ≥ Q,not more than 2 units are < Q – 15% and no single unit is less than Q – 25%.
99
If a sample fails either Stage S1 or S2, proceed to the next stage and testthe number of units indicated.

Delayed Release Forms – Method AUSP <711> USP Delayed Release Dosage Forms – Method A:
Media is 750 mL of 0.1N HCl
Samples are removed for analysis after 2 hours ± 2%
Within 5 minutes of withdrawing the acid stage sample aliquots, add 250 mL of 0.20-M tribasic sodium phosphate,
•
aliquots, add 250 mL of 0.20-M tribasic sodium phosphate, adjusting to pH + 0.05 at 37 °C while stirring at the specified rate.
Dissolution continues for 0.75 h (or per monograph) or less.
Sample aliquots are then analyzed with Q being the total % dissolved for both acid and buffer stages.
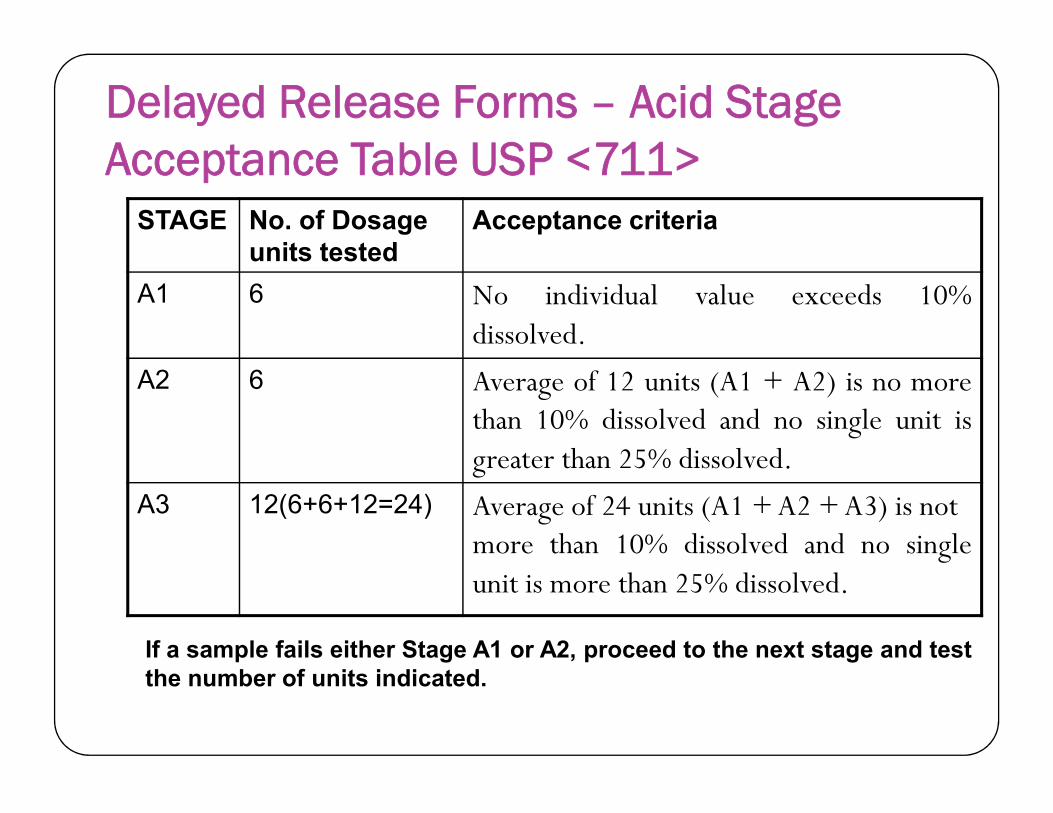
Delayed Release Forms – Acid Stage Acceptance Table USP <711>
STAGE No. of Dosage units tested
Acceptance criteria
A1 6 No individual value exceeds 10%dissolved.
A2 6 Average of 12 units (A1 + A2) is no morethan 10% dissolved and no single unit isthan 10% dissolved and no single unit isgreater than 25% dissolved.
A3 12(6+6+12=24) Average of 24 units (A1 + A2 + A3) is notmore than 10% dissolved and no singleunit is more than 25% dissolved.
101
If a sample fails either Stage A1 or A2, proceed to the next stage and testthe number of units indicated.

Delayed Release Forms - Buffer Stage Acceptance Table USP <711>
STAGE No. of Dosage units tested
Acceptance criteria
B1 6 Each unit is ≥ Q + 5%.
B2 6 Average of 12 units (B1 + B2) is ≥ Q and no unit is < Q– 15%.– 15%.
B3 12(6+6+12=24) Average of 24 units (B1 + B2 + B3) is ≥ Q, not morethan 2 units are < Q – 15% and no single unit is less thanQ – 25%.
102
If a sample fails either Stage B1 or B2, proceed to the next stage and testthe number of units indicated.

Delayed Release Forms – Method B USP <711> USP Delayed Release Dosage Forms – Method B:
Media is 1000 mL of 0.1N HCl
Samples are removed for analysis after 2 hours ―
USP Delayed Release Dosage Forms – Method B:
Drain the acid from the original vessel and replace with
•
Drain the acid from the original vessel and replace with
1000mL pH6.8 phosphate buffer pre equilibrated at 37°C
or switch the dosage form to a second vessel containing the phosphate buffer

Disssolution specification – Case study -1 Ethambutol hydrochloride 400mg tablets. The applicant
claimed USP standard for the product.
Bioequivalent of the product is accepted as per BCS class 3 based biowavier
The applicant set the dissolution specification limits as below: The applicant set the dissolution specification limits as below:
NLT 80% (Q) in 45min at release
NLT 75% (Q) in 45min at shelf life, which is in line with the requirement of USP monograph.
Is it acceptable? What limits should be applied?

Disssolution specification – Case study -1Answer: not acceptable
The dissolution limits at release and shelf life should be the same.
A limit of NLT 80% (Q) in 15min should be set for A limit of NLT 80% (Q) in 15min should be set for both release and shelf life as for the BCS class 3 biowaiver.

Comparison of Dissolution Profiling

Why Profiling / Comparision?
It reflects its release pattern under the selected condition sets. i.e. either sustained release or immediate release of the formulated formulas.
For optimizing the dosage formula by comparing the dissolution profiles of various formulas of the same drug.of various formulas of the same drug.
Dissolution profile comparison between pre change and post change products for SUPAC (scale up post approval change ) related changes or with different strengths, helps to assure the similarity in the product performance and green signals to bioequivalence.
Modify formulation as per Brand Leader.

Requirement for Comparative dissolution testing
Two or more products or batches containing the same APIare compared
The strength of products / batches may or may not be the same (depending on purpose of test)
The dissolution conditions are similar, e.g.
Apparatus, medium, volume, rotation speed & temp.• Apparatus, medium, volume, rotation speed & temp.
• Minimize possible experimental differences in conditions
Samples are taken at the same time points and the data (dissolution profiles) compared

How to Compare? Aim: To show similar in vitro dissolution under physiologically
relevant experimental pH conditions.
Advisable to investigate more than one batch of test and referenceproducts; must include bioequivalence batches.
Investigate within pH 1-6.8 (normally pH 1.2, 4.5 and 6.8) and QC media
(if different).
Water may be used as an additional medium, especially when the APIWater may be used as an additional medium, especially when the APIis unstable in buffered media to the extent that data is unusable.
Additional investigations may be required at pH values in which thedrug has minimum solubility.
Use12 units to enable statistical evaluation
For each condition, present comparative dissolution profiles(mean valuesvs.time) together with statistics (max, min, mean, RSD; f2 similarity factor ifcalculated; individual values)
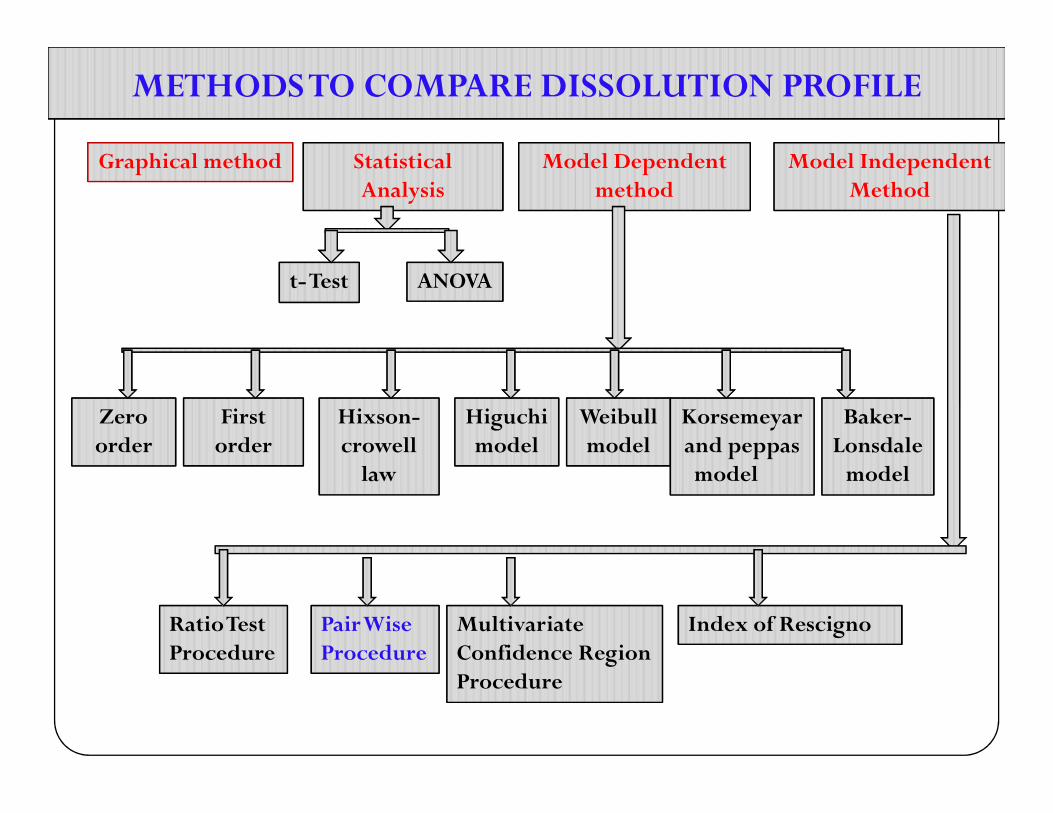
METHODS TO COMPARE DISSOLUTION PROFILEMETHODS TO COMPARE DISSOLUTION PROFILE
Graphical methodGraphical method Statistical Analysis
Statistical Analysis
Model Dependent method
Model Dependent method
Model Independent Method
Model Independent Method
t-Testt-Test ANOVAANOVA
Zero Zero First First Hixson-Hixson- Higuchi Higuchi Weibull Weibull Korsemeyar Korsemeyar Baker-Baker-Zero order Zero order
First orderFirst
orderHixson-crowell
law
Hixson-crowell
law
Higuchi model
Higuchi model
Weibull model
Weibull model
Korsemeyar and peppas model
Korsemeyar and peppas model
Baker-Lonsdale
model
Baker-Lonsdale
model
Ratio Test ProcedureRatio Test Procedure
Pair Wise ProcedurePair Wise Procedure
Multivariate Confidence Region Procedure
Multivariate Confidence Region Procedure
Index of RescignoIndex of Rescigno

Paired Wise Procedure DIFFERENCE FACTOR (f1) & SIMILARITY FACTOR (f2)
The difference factor (f1) as defined by FDA calculates the % difference between 2 curves at each time point and is a measurement of the relative error between 2 curves.
n
TtRtf1 = × 100
where, n = number of time points
Rt = % dissolved at time t of reference product (pre change)
Tt = % dissolved at time t of test product (post change)
n
t
t
Rt
TtRt
1
1

The similarity factor (f2) as defined by FDA is logarithmic reciprocal square root transformation of sum of squared error and is a measurement of the similarity in the percentage (%) dissolution between the two curves
f2 = 50 ×
100
1
log )(1
1
5.0n
r
TtRtwtn 1rn

Guidance for Industry
A specific procedure to determine difference and similarity factors is as follows:
1. Determine the dissolution profile of two products (12 units each) of the test (postchange) and reference (prechange) products.
2. Using the mean dissolution values from both curves at each time interval, calculate the difference factor (f1 ) and similarity factor (f2) using the above equations.equations.
3. For curves to be considered similar, f1 values should be close to 0, and f2 values should be close to 100. Generally, f1 values up to 15 (0-15) and f2 values greater than 50 (50-100) ensure equivalence of the two curves and thus, of the performance of the test (postchange) and reference (prechange) products.
4. This model independent method is most suitable for dissolution profile comparison when three to four or more dissolution time points are available.

The following recommendations should also be considered:
The dissolution measurements of the test and reference batches should bemade under exactly the same conditions.
The dissolution time points for both the profiles should be the same (e.g., 15, 30, 45, 60 minutes).
The reference batch used should be the most recently manufactured prechange product.
A minimum of three points required for comparison of profile. Only one measurement should be considered after 85% dissolution of both the Only one measurement should be considered after 85% dissolution of both the
products. To allow use of mean data, the percent coefficient of variation at the earlier
time points (e.g., 10 minutes) should not be more than 20%, and at other time points should not be more than 10%.
The mean dissolution values for R can be derived either from (1) last prechange (reference) batch or (2) last two or more consecutively manufactured prechange
batches.
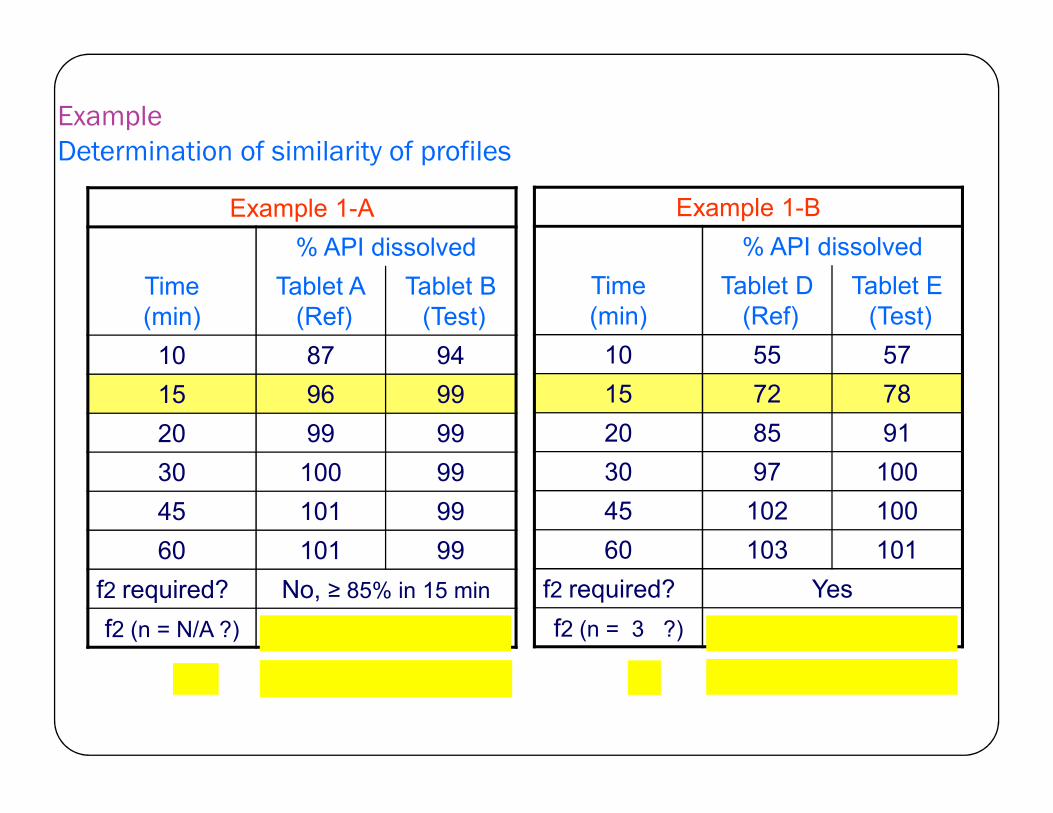
Example Determination of similarity of profiles
Example 1-B
% API dissolved
Time(min)
Tablet D(Ref)
Tablet E(Test)
10 55 57
15 72 78
Example 1-A
% API dissolved
Time(min)
Tablet A(Ref)
Tablet B(Test)
10 87 94
15 96 99 15 72 78
20 85 91
30 97 100
45 102 100
60 103 101
f2 required? Yes
f2 (n = 3 ?) 64 (similar)
15 96 99
20 99 99
30 100 99
45 101 99
60 101 99
f2 required? No, ≥ 85% in 15 min
f2 (n = N/A ?) profiles similar

ExampleDetermination of similarity of profiles (cont.)
Example 1-D
% API dissolved
Time(min)
Tablet A(Ref)
Tablet Y (Test)
10 87 55
15 96 72
Example 1-C
% API dissolved
Time(min)
Tablet X(Ref)
Tablet Y(Test)
10 29 34
15 38 41 15 96 72
20 99 85
30 100 97
45 101 102
60 101 103
f2 required? Yes
f2 (n = 3 ?) 31 (not similar)
15 38 41
20 47 50
30 63 64
45 80 79
60 95 91
f2 required? Yes
f2 (n = 6 ?) 74 (similar)

Comparison to reference medicinal product Immediate Release Tablets/Capsules
> 85% dissolved within15 minutes : test and reference similar without any further calculationcalculation
≤ 85 % dissolved within15minutes: calculate f2 similarity factor
Modified Release Preparations
Calculate f2 similarity factor

Comparison to reference medicinal product
Prolonged Release Preparations
Minimum of 3 time points, but may be prudent to do more
Particularly important if desired release profile not uniform Particularly important if desired release profile not uniform
(e . g. immediate release outer coat and prolonged release core)
If only3 time points: expected to mirror final specification timepoints (20-30%(dose dumping),50%(defines profile),>80%)
More time points =↑confidence of bioequivalence
e.g. for once daily preparations(1,2,4,8,12,16,20 & 24 hours)

Alternative methods to dissolution testing
In ICH Q6A permits use of disintegration testing as a surrogatefor conventional Compendial dissolution tests, provided
highly soluble drug substances
intrinsic rate of solubilization is rapid intrinsic rate of solubilization is rapid
overall drug release rate is dominated by cohesive propertiesof the formulation

DT in place of dissolution?ICH Q6A decision trees #7 can be used to assess the proposed dissolution criteria, however:
Highly soluble throughout physiological pH range.
Solubility at 37°C ± 0.5°C, dose + solubility < 250 ml, pH 1.2 - 6.8.
For considering /accepting DT in place of dissolution: all the considerations For considering /accepting DT in place of dissolution: all the considerations should be carefully assessed: highly soluble and very rapidly dissolving, plus significant supporting development data – including
when DT is more discriminating or
has a demonstrated relationship to dissolution, robustness of the formulation/manufacturing process have been demonstrated wrt DT, etc.
Dissolution may not be necessary or proposed as a skip test.

Example
APIs with good solubility at gastric pH levels may begranted BCS Class I and III classification i.e. may becharacterized by disintegration testing alone.
In liquid filled capsules, drug dissolved in solubilisation aidsoffering a true mechanism for drug release is likely to be therupture of the capsule use disintegration as a surrogate forthe QC dissolution test

Bio vaiver (In vitro equivalence testing)
The term biowaiver is applied to a regulatory drug approvalprocess where the efficacy and safety part of the dossier(application) is approved based on evidence of equivalence otherthan through in vivo equivalence testing.
A biowaiver can be applied only for products which meetrequirements on pharmaceutical similarity, as well as similarity inrequirements on pharmaceutical similarity, as well as similarity incomparative dissolution tests.
A BCS-based biowaiver has become an important andcost-saving tool in approval of generic drugs.
The bio-relevance of the BCS properties and the in vitro releaseare best expressed through a correlation between in vitro and invivo data.

BCS Classification
Class 1 Class 2 Class 3 Class 4
Highly Poorly Highly Poorly Highly Soluble
Poorly Soluble
Highly Soluble
Poorly Soluble
Highly Permeable
Highly Permeable
Poorly Permeable
Poorly Permeable

‘High solubility
The definition of ‘high solubility’ refers to the highest dosestrength of an immediate release product, which has to besoluble in 250 ml or less of aqueous media over the pHrange of 1 – 7.5,. Solubility measurements should berange of 1 – 7.5,. Solubility measurements should beperformed at 37 °C using a stability indicating, validatedmethod.

High permeability
The classification regarding high permeability refers to the extent of absorption in humans, i.e. high permeability is concluded if the extent of absorption in humans reaches at least 90 % of an orally administered dose. reaches at least 90 % of an orally administered dose. This conclusion may be based on either pharmacokinetic studies in humans (e.g. mass balance, or absolute bioavailability studies) or intestinal permeability methods like e.g. in vivo intestinal perfusion studies in humans or validated in vitro permeation studies across a monolayer of cultured epithelial cells.

Requirement
Comparative in vitro dissolution investigations should ensure that no less than 85 % of the labeled amount is dissolved within 30 min in each of the required media: 0.1 N HCl, pH 4.5 and 6.8 buffers. Regarding experimental requirements, reference is made to the US Pharmacopoeia and the US-FDA guidance for industry on Dissolution Testing of Immediate Release Solid Oral Dosage Forms (August 1997) 3. Oral Dosage Forms (August 1997) 3.
Resulting profiles should be compared using the similarity factor (f2), unless 85% or more of the labeled amount dissolves within 15 min from both products.
The latter case would allow the conclusion that the investigated products are similar without requiring any further statistical calculations.

Restrictions
BCS based waivers are not applicable for the initial in vivo bioavailability characterization for NDAs.
Other restrictions of application include:
1) narrow therapeutic index (NTI) drug; and 1) narrow therapeutic index (NTI) drug; and
2) drug products intended to be absorbed in the oral cavity.
3) Similarly, prodrugs and excipients require special consideration. In case of prodrugs, whether to measure the prodrug or the drug for permeability determination will depend on where the conversion occurs.
4
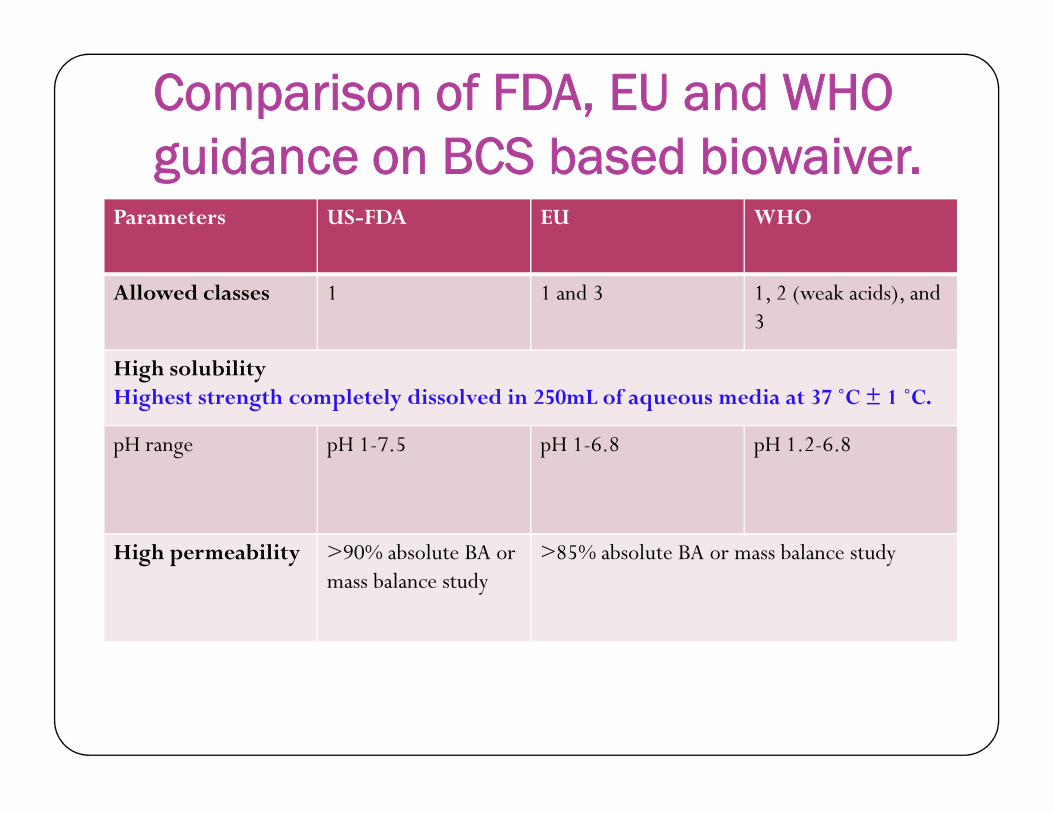
Comparison of FDA, EU and WHO guidance on BCS based biowaiver.
Parameters US-FDA EU WHO
Allowed classes 1 1 and 3 1, 2 (weak acids), and 3
High solubilityHighest strength completely dissolved in 250mL of aqueous media at 37 ˚C ± 1 ˚C.Highest strength completely dissolved in 250mL of aqueous media at 37 ˚C ± 1 ˚C.
pH range pH 1-7.5 pH 1-6.8 pH 1.2-6.8
High permeability >90% absolute BA ormass balance study
>85% absolute BA or mass balance study

Comparison of FDA, EU and WHO guidance on BCS based biowaiver.Parameters FDA EU WHO
Rapid dissolution
Media (studies should beconducted at 37 ±
900 mL or less aqueousmedia (0.1N HCl
900 mL or less aqueousmedia (pH 1.0-1.2
900 mL or less aqueousmedia (pH 1.2conducted at 37 ±
1 C)media (0.1N HClorSGF; pH 4.5 buffer; andpH 6.8 buffer or SIF)
media (pH 1.0-1.2buffer, usually 0.1NHCl or SGF; pH 4.5buffer; and pH 6.8buffer or SIF)
media (pH 1.2HCl solution; pH 4.5acetate buffer; and pH6.8 phosphate buffer)
Apparatus (APP) USP APP I - 100 rpmUSP APP II - 50 rpm
Paddle APP - 50 rpmBasket APP - 100 rpm
Paddle APP - 75 rpmBasket APP - 100 rpm

Comparison of FDA, EU and WHO guidance on BCS based biowaiver.Parameters FDA EU WHO
Rapid dissolution
Criteria >85% in 30 min in 3 media
Class 1: >85% in30 min in 3 media
Class 1: >85% in 30 minin 3 media (Rapid)in 3 media 30 min in 3 media
(Rapid)Class 3: >85% in 15min in 3 media (VeryRapid); or, >85% in30 min and similardissolution profile toRLD (Similarly Rapid)
in 3 media (Rapid)Class 2: >85% in 30 minin pH 6.8 medium andsimilar dissolutionprofile in 3 mediaClass 3: >85% in 15 minin 3 media (Very Rapid)
Restrictions Narrow therapeutic drugsOral products intended to be absorbed in the oral cavityModified release drug products

CONCLUSION: In vitro dissolution is the best available tool today which can at
least quantitatively assure about the biological availability of drug.
Great Scope for New Method Development / Pharmacopoeial monographs
Systematic and scientific Approach is needed. Systematic and scientific Approach is needed.
Academic standards / Research ?????? Basic infrastructure / facilities / sophisticated instrument /
positive efforts / attitude and honesty is the need of time to improve the quality of research.

Guidelines Referred BCS Guidance (Waiver of In Vivo Bioavailability and
Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System”); August 2000
IR Dissolution Guidance (Dissolution Testing of Immediate Release Solid Oral Dosage Forms); August 1997
IVIVC Guidance (Extended Release Oral Dosage Forms: IVIVC Guidance (Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations); September 1997
General BA/BE Guidance (Bioavailability and Bioequivalence Studies for Orally Administered Drug Products - General Considerations); 2003
WHO, US FDA and EMEA guidelines
ICH guidelines

Thank you


















