
Autophagy as a neuronal survival mechanism in ischemic stroke · hemorrhagic cerebral accidents...
7
Rom J Leg Med [26] 333-339 [2018] DOI: 10.4323/rjlm.2018.333 © 2018 Romanian Society of Legal Medicine 333 FUNDAMENTAL RESEARCH Autophagy as a neuronal survival mechanism in ischemic stroke Bogdan-Ioan Coculescu 1,2,3 , Gheorghe Manole 3,4 , Elena Claudia Coculescu 5,* , Ecaterina Ionescu 5 , Olivia Popoviciu 5 , Cristina Maria Stocheci 6 _________________________________________________________________________________________ Abstract: Based on the histological examination of the encephalic fragments collected during the necropsy of 38 patients who died from ischemic stroke, the authors admit that the "ischemic tolerance phenomena" that are described as particular to transient ischemic strokes are also present in the peripheral areas of the "core" of a cerebral infarction. is marginal area called the penumbra area has the characteristic that the ischemia did not exceed the critical threshold so that neuronal necrosis can occur. As a result, the metabolism of these neurons proceeds under the conditions of a critical supply and becomes responsible for the characteristics of the nucleo-cytosolic environment. Consequently, modified intracellular environments induce the alteration of their "form" that is histochemically expressed, nucleocytosolic, changing, among other things, the coloration, hence the name of "red neurons". Etiopathogenic, the changes are based on the installation of oxidative stress, but also the possibility of autophagy installation, as a form of neuronal survival, expressed clinically by reducing the sensory-motor deficit of the patient. Key Words: stroke (AVC), red neurons, neuronal hypoxia, oxidative stress, autophagocytosis. 1) Center for Military Medical Scientific Research, Bucharest, Romania 2) “Titu Maiorescu” University, Faculty of Medicine, Bucharest, Romania 3) “Bioterra” University, Faculty of General Nursing, Bucharest, Romania 4) Clinical Hospital Colentina, Clinical Department, Bucharest, Romania 5) “Carol Davila” University of Medicine and Pharmacy, Faculty of Dental Medicine, Bucharest, Romania * Corresponding author: “Iuliu Hatieganu” University of Medicine and Pharmacy Cluj-Napoca, 23 Gh. Marinescu street, Cluj-Napoca, Romania, Tel: +40742138148. E-mail: [email protected] 6) University of Pitesti, Faculty of Sciences, Pitesti, Romania All the authors have equal contribution at this paper INTRODUCTION According to the World Health Organization (WHO), the stroke is defined by the persistence of a neurological deficit over 24 hours, and, in 87% of the cases, its etiology is regional ischemia. Moreover, hemorrhagic cerebral accidents themselves begin with cerebral ischemia. is is because the development of the cerebral hematoma in a non-stretch space (the cranial box) induces the effect of compression exerted on the stroma. e mechanism responsible for setting up the neurological deficit is the degree of neuronal mass reduction, although neuroglycemia (oligodendrogliles, microglia, astrocytes) are also affected by cerebral infarction. In the periphery of the center/"core" of a cerebral infarction, irrespective of its etiology (thrombosis or embolism), there is a pathogenic reduction in local hemodynamics, which provides critical irrigation for the nervous cells. e consequence is the generation of histological lesions, in this area designated by the term "ischemic penumbra". Identifiable histological lesions are superposable to those who define "low flux" cerebral infarction [1]. If, in order for regional necrosis to be installed in a cerebral infarction, it is necessary for the regional flow to be reduced to values less than 10% of the normal, in the peripheral area of the hypoperfusion this is close
Transcript of Autophagy as a neuronal survival mechanism in ischemic stroke · hemorrhagic cerebral accidents...

Rom J Leg Med [26] 333-339 [2018]DOI: 10.4323/rjlm.2018.333© 2018 Romanian Society of Legal Medicine
333
FUNDAMENTAL RESEARCH
Autophagy as a neuronal survival mechanism in ischemic stroke
Bogdan-Ioan Coculescu1,2,3, Gheorghe Manole3,4, Elena Claudia Coculescu5,*, Ecaterina Ionescu5, Olivia Popoviciu5, Cristina Maria Stocheci6
_________________________________________________________________________________________ Abstract: Based on the histological examination of the encephalic fragments collected during the necropsy of 38 patients who died from ischemic stroke, the authors admit that the "ischemic tolerance phenomena" that are described as particular to transient ischemic strokes are also present in the peripheral areas of the "core" of a cerebral infarction. This marginal area called the penumbra area has the characteristic that the ischemia did not exceed the critical threshold so that neuronal necrosis can occur. As a result, the metabolism of these neurons proceeds under the conditions of a critical supply and becomes responsible for the characteristics of the nucleo-cytosolic environment. Consequently, modified intracellular environments induce the alteration of their "form" that is histochemically expressed, nucleocytosolic, changing, among other things, the coloration, hence the name of "red neurons". Etiopathogenic, the changes are based on the installation of oxidative stress, but also the possibility of autophagy installation, as a form of neuronal survival, expressed clinically by reducing the sensory-motor deficit of the patient. Key Words: stroke (AVC), red neurons, neuronal hypoxia, oxidative stress, autophagocytosis.
1) Center for Military Medical Scientific Research, Bucharest, Romania2) “Titu Maiorescu” University, Faculty of Medicine, Bucharest, Romania3) “Bioterra” University, Faculty of General Nursing, Bucharest, Romania4) Clinical Hospital Colentina, Clinical Department, Bucharest, Romania5) “Carol Davila” University of Medicine and Pharmacy, Faculty of Dental Medicine, Bucharest, Romania* Corresponding author: “Iuliu Hatieganu” University of Medicine and Pharmacy Cluj-Napoca, 23 Gh. Marinescu street, Cluj-Napoca, Romania, Tel: +40742138148. E-mail: [email protected]) University of Pitesti, Faculty of Sciences, Pitesti, Romania All the authors have equal contribution at this paper
INTRODUCTION
According to the World Health Organization (WHO), the stroke is defined by the persistence of a neurological deficit over 24 hours, and, in 87% of the cases, its etiology is regional ischemia. Moreover, hemorrhagic cerebral accidents themselves begin with cerebral ischemia. This is because the development of the cerebral hematoma in a non-stretch space (the cranial box) induces the effect of compression exerted on the stroma. The mechanism responsible for setting up the neurological deficit is the degree of neuronal mass reduction, although neuroglycemia (oligodendrogliles,
microglia, astrocytes) are also affected by cerebral infarction. In the periphery of the center/"core" of a cerebral infarction, irrespective of its etiology (thrombosis or embolism), there is a pathogenic reduction in local hemodynamics, which provides critical irrigation for the nervous cells. The consequence is the generation of histological lesions, in this area designated by the term "ischemic penumbra". Identifiable histological lesions are superposable to those who define "low flux" cerebral infarction [1]. If, in order for regional necrosis to be installed in a cerebral infarction, it is necessary for the regional flow to be reduced to values less than 10% of the normal, in the peripheral area of the hypoperfusion this is close

334
Coculescu B.I. et al. Autophagy as a neuronal survival mechanism in ischemic stroke
to the value of 25-80 ml/100g tissue/minute [2]. Nervous cells from the ischemic penumbra area, carrying out their metabolism in such a supply regime, develops excess production of reactive oxygen and nitrogen species (ROS/RNS), leading to redox imbalance [3,4]. It induces nucleo-cytosolic changes, "marking" the nervous cell with a defective supply. The neuron, which has become "labeled apoptotic", is forced to selectively remove the organites that have become inoperable in order to maintain its quality control. Taking into account the various modes of cell death (apoptosis, necrosis and oncosis) it can be stated with certainty that: a. The first two modes of neuronal death are specific to neurons in the central area/"core"of the cerebral ischemic center [5, 6]. b. The only biological process that allows, in the dynamics of its development, the survival of a cell subjected to hypoxia, is autophagia. This is because only autophagy, by the removal/digestion of damaged organites, recycles the released compounds allowing for neuronal survival, thereby compensating for nutrient deprivation because of bad irrigation [7]. Otherwise, the neuron undergoes the process of premature-induced apoptosis that follows the pathway of mixed autophagous forms: necrotic autophagy/autophagic necrosis and/or necropsy/necroptosis. We think that the other way in which an "apoptotic-labeled" cell can survive, through proteasome involvement, can not explain how large protein complexes and worn organites are destroyed in the "apoptotic-labeled" neuron. This is because it is scientifically proven that the proteasome ensures, through ubiquitin/ubiquitin ligases mediation the possibility of protein degradation only up to oligopeptides and very rarely to the amino acid stage [8-10]. In terms of the clinical progression of the patient suffering from an ischemic stroke, the two possible ways of evolution of neurons located in the ischemic penumbra area have different prognostic significance: neuronal survival is expressed by the recovery of sensory-motor deficits and the failure of autophagia leads to premature apoptosis, accentuating the sensory-motor deficit. Of the three modes in which autophagy - macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) - can be carried out at the nerve cell level, the last way to recycle cellular components damaged by the action developed by ROS is a much more complex way and does not appear to be specific to nerve cells. The efficiency of the first two types of autophagy stems from the proteolysis and degradation of cellular organites with the help of acid lysosomal hydrolases [9]. The distinction between the two neuronal destructive processes consists in the way the formation of digestive vacuole/autolyzosomes are formed: a. In macroautophagy, cellular organites/
material "marked" for destruction surround themselves with a double membrane forming an autofagosome. Endoplasmic reticulum (ER), trans-Golgi and endosomes contribute to the formation of the membrane, and its structure is similar to all membranes [11, 12]. The autophagosome that possesses the capacity to travel through the neuroplasm under its own power, reaches the level of a lysozyme through chemotherapy, with which it fuses [13]. The result is the formation of the autolysosome, the content of which is "digested" by acid lysosomal hydrolases, especially cathepsins [14, 15]. b. In microautophagy, cellular components damaged to form autolysosome are absorbed through a lysosomal membrane invagination.
MATERIAL AND METHODS
Brain fragments were sampled from the lesion area of a number of 38 patients who died from ischemic stroke and who underwent necropsy. More precisely, from its peripheral area ("ischemic penumbra area"), located some distance from the necrotic lesion center. In establishing sampling areas in relation to the ischemic outbreak, consideration was given to: - (speciality) expert literature records showing that histological changes at the level of the nervous tissue are extremely different from one area to another, depending on the size of the ischemic outbreak, the vessels concerned, the collateral circulation and the presence of possible anastomoses, as well as the general state of the cerebral vascular system; - the objective of the study, the identification of histological lesions in the perilesional zone (the ischemic penumbra). The biological material taken was subjected to fixation in a 10% neutral formalin solution and then to paraffin inclusion, for processing and microscopic examination. The 10% formalin fixative solution was obtained from formaldehyde (commercially available 40% formalin) according to the following formula:
Formalin 40%..............................................1 partDistilled water..............................................9 parts
Calcium bicarbonate was added to the solution in order to obtain a neutral pH (7.2-7.4). The addition was necessary in order to neutralize the formic acid that spontaneously forms in the concentrated solutions. The predominant use in medical practice of the fixation method using formalin is motivated by the cheap cost, the high penetration power in the biological material taken without altering the structure or affinity of its coloring and the reduced toxicity for the medical staff. Additionally, fixing with 10% formalin gives two other advantages: - the use of the material taken for immunohistochemical studies as well; - it preserves the biological material for a long
Romanian Journal of Legal Medicine Vol. XXVI, No 4(2018)
335
time, without altering the cellular or infracellular structures. Knowing the influence exerted by the quality of the fixation of the biological material in the histological and histochemical interpretation, in order to achieve a good fixation, the procedural norms were strictly observed. After fixation, in order to ensure a good biological material for the histological examination, we resorted to the classic histological technique of paraffin inclusion [16]. Histological preparations were colored with hematoxylin-eosin, the most commonly used method to highlight tissue, and tricromatic, with green light (the Goldner-Szeckeli method).
RESULTS
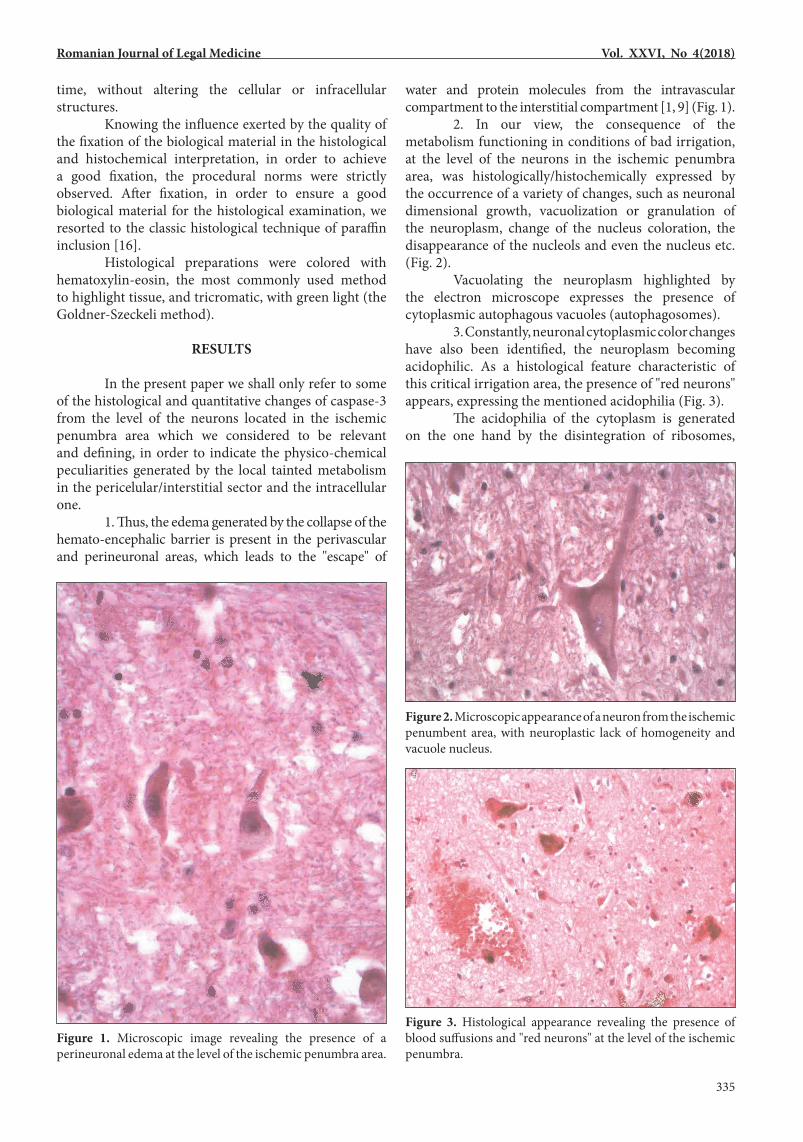
In the present paper we shall only refer to some of the histological and quantitative changes of caspase-3 from the level of the neurons located in the ischemic penumbra area which we considered to be relevant and defining, in order to indicate the physico-chemical peculiarities generated by the local tainted metabolism in the pericelular/interstitial sector and the intracellular one. 1. Thus, the edema generated by the collapse of the hemato-encephalic barrier is present in the perivascular and perineuronal areas, which leads to the "escape" of
water and protein molecules from the intravascular compartment to the interstitial compartment [1, 9] (Fig. 1). 2. In our view, the consequence of the metabolism functioning in conditions of bad irrigation, at the level of the neurons in the ischemic penumbra area, was histologically/histochemically expressed by the occurrence of a variety of changes, such as neuronal dimensional growth, vacuolization or granulation of the neuroplasm, change of the nucleus coloration, the disappearance of the nucleols and even the nucleus etc. (Fig. 2). Vacuolating the neuroplasm highlighted by the electron microscope expresses the presence of cytoplasmic autophagous vacuoles (autophagosomes). 3. Constantly, neuronal cytoplasmic color changes have also been identified, the neuroplasm becoming acidophilic. As a histological feature characteristic of this critical irrigation area, the presence of "red neurons" appears, expressing the mentioned acidophilia (Fig. 3). The acidophilia of the cytoplasm is generated on the one hand by the disintegration of ribosomes,
Figure 1. Microscopic image revealing the presence of a perineuronal edema at the level of the ischemic penumbra area.
Figure 2. Microscopic appearance of a neuron from the ischemic penumbent area, with neuroplastic lack of homogeneity and vacuole nucleus.
Figure 3. Histological appearance revealing the presence of blood suffusions and "red neurons" at the level of the ischemic penumbra.

336
Coculescu B.I. et al. Autophagy as a neuronal survival mechanism in ischemic stroke
of the endoplasmic reticulum and of Nissl corpuscles, and on the other by the appearance in the hyaloplasm of numerous ergastic structures of protein nature with eucinophilic tinctorial affinity [16].
DISCUSSIONS
Anatomo-functionally, the ischemic penumbra area comprises the area of the nervous tissue that provides the transition from the maximal ischemic lesion focal point to the relatively cerebral parenchyma integrity. If at the center of the ischemic focal point there is necrosis, expressing an existing major imbalance between nutrient and oxygen intake and the optimal regional need, the penumbra is characterized by a deficient supply of oxygen and nutrients [17, 18]. For this area, irrigation that is at a critical level pathogenically confers the characteristic of "risk" cells on neurons and glial cells [9, 16]. This is a determinant of the way in which nerve cells will evolve, with the possibility of preserving viability due to neuroplasticity, but also of cell death. In fact, the path of evolution is imposed by the degree of irrigation improvement, but also by the duration of hypoxia persistence [9]. In our view, the blood irrigation deficit in the ischemic penumbra area recognizes a complex pathogenic mechanism: - presence of the vasogenic/interstitial edema that exerts vascular compression, diminishing regional flow, concurrently slowing the exchanges at the level of the membranes of the nerve cells that structure the site; - generation, local as well as by diffusion, from the central ischemic area, of products such as organic acids, or kalicreinic compounds which, acting synergistically with some cytokines, become strong vasodilating factors; - alteration of the glutamic acid-glutamate conversion process with its concentration increase, exacerbating the existing oxidative stress; - synthesis deficit of the macro-active compounds as a result of the permeability of the mitochondrial membranes [6]. The presence of hypoxia at the level of the nerve cells located in the ischemic penumbra area generates several classes of pathogenic mechanisms whose intensity decides the survival or premature apoptosis. a. The consequence of the development of neuronal metabolism following the anaerobic path is the appearance of oxidative stress, which acts pathogenetically on structural and functional neural components. The existing ROS/RNS [19, 20] will determine the direction of evolution of a neuron located in the ischemic penumbra area, toward survival or apoptosis, by its influence on the expression of some of the functional proteins [9]. The path of neuronal evolution is decided by a double mechanism via: - acting as a stimulus for triggering the stress
response signal transmission paths, activating signaling pathways, primarily by inducing direct oxidative alterations to sensitive redox signaling proteins; - its intervention in the remodeling of chromatin; death signals and survival signals, respectively, act at the level of transduction, of transcription or of execution control. In support of these assertions, there is evidence of partial regulation by means of a redox mediated mechanism of the post-translational modifications of signaling proteins, such as phosphorylation [21] (Fig. 4). An important control of survival and plasticity control of synapses/neurotransmitters between factors with a modular structure is also developed by the Egr-1/zif 268 protein. This, by coupling to DNA domains, allows the activation or repression of transcription. The action is a result of the interaction of the transducer signal: PI3K/Akt (Figs 4 and 5) [22]. In this signaling process, what is important is the involvement of Akt, which induces the inhibition of FOXO proteins [11]. Figure 5 shows that the way/path of evolution of the neuron subjected to critical hypoxia, from the penumbra to survival or apoptosis, is dependent on the opposing action of the two enzymes involved: lipid phosphatase PTEN and phospho-inositide -3-kinase, respectively. The latter leads to survival, via activation of protein kinase B/Akt [22]. Concretely, the transcription suppression action developed by the Egr-1 protein via the p53 protein consists in blocking the cell cycle. This prevents replication of DNA alterations, reducing genetic instability, which allows cells to perform important repair functions before initiating a new cell cycle. The apoptosis induced by this protein is directed to the elimination of "marked" cells (Fig. 4) [23]. b. Mitochondria are (mitochondrion is) the main organite(s) responsible for: maintaining redox homeostasis, producing energy and triggering apoptosis, depending on the received signaling. Control of intracellular redox equilibrium is a primary function of the same p53 protein mentioned earlier [24]. The p53 protein orchestrates the mitochondrial redox signaling by coordinated control of at least two effector keys: the superoxide anion acting as MnSOD capture agent and the ROS generator p66shc [24]. The latter is the molecule that also intervenes in the regulation of cell survival. Its action seems to be expressed only after the formation of the trifactorial connection complex: p53-MnSOD-p66shc [24]. The intraneuronal presence of oxidative stress generates changes in the permeability of the mitochondrial membrane pores, with consequences both on the matrix composition in the intermembrane space and on the deficient synthesis of the macroergic compounds. Constantly, the mitochondria with such lesions/defective mitochondria are subject to mitophagy, a process that may also be of interest to those who are integrated. The process is selective, autophagous [25].

Romanian Journal of Legal Medicine Vol. XXVI, No 4(2018)
337
c. Under physiological conditions, cellular proliferation or cellular structure rebuilding is modulated by ROS concentration, the synthesis of which is dependent on the growth factor-induced activation on enzyme complexes, such as NADH-oxidase [4, 26]. The deficient synthesis of the neural growth factor, as well as other cytokines, will delay both cellular regeneration and
intracellular communication at the level of the penumbra area. This is because the information transmitted by the growth factor insufficiently activates FOXO proteins that are at the interface of crucial cellular processes, orchestrating the gene expression programs that regulate apoptosis, cell cycle progression, and resistance to oxidativity. The action developed by FOXO transcription factors is selective in that: - the induction of apoptosis by FOXO only targets damaged or abnormal cells, thus conferring the possibility of longevity conservation to the cell; - regulating the different genes generating apoptosis or survival also depends on the cell type. Thus, the results of some studies admit that at the neuronal or lymphocyte level it is possible for FOXO to induce apoptosis by activating the transcription of FasL and the Bim protein, a pro-apoptotic Bap-2 member of the family [11]. This explains clinically the possible evolution towards accentuating the sensory-motor deficit of the patient with ischemic stroke. d. When histological changes are introduced at the neuronal level, a last pathogenic mechanism by which the presence of hypoxia acts at the level of the nerve cells located in the ischemic penumbra, is the induction of mutations of the genes that encode the synthesis of functional intraneuronal proteins [26]. This may affect components such as neurofibrils, storage vesicles of neurotransmitters, and even neurotransmitters themselves. Cumulatively, their action leads to the alteration of the intraneuronal transport of the informational message [7]. Equally, gene mutations lead to the alteration of the receptor synthesis processes at the level of the presynaptic neuroleme, thereby compromising the transmission of information at the synaptic level [9]. The study of Figure 4 shows that the redox imbalance modulates the transcription of the coding genes for functional protein synthesis by a transcription factor control, and the main protein involved in these processes is, as previously mentioned, the p53 protein. Synergistically, other transcription factors involved in neuronal survival participate as well, including: AP-1, Nrf-2, HIF, and the kappa B nuclear transcription factor (NF-kB) [21, 28]. The triggering of the autophagy signal for neural repair is done by the PUMA protein action, and the modulation of the process is done by NF-kB and FOXO3a [9, 29]. Physiologically, the NF-kB responsible for the development of redox-sensitive actions is located in the cytoplasm, inactivated by kappa B inhibitors (IkB), with which it forms an inactive dimer. Under oxidative stress conditions occurs the activation of IkB-kinase (IKK) which, through the phosphorylation of IkB, leads to NK-kB release and IkB degradation after ubiquitin labeling, at the proteasome level [21, 30, 31]. Free, the NF-kB of the cytosol is translocated in the nucleus, joining regions of the DNA and interfering with the transcription
Figure 4. The mechanisms mediated by ROS/RNS which induce post-translational modifications of signaling proteins [21].
Figure 6. Mechanism of nerve cell survival by activating the NF-κB transcription factor [21].
Figure 5. The mechanism of induced survival or apoptosis of the neuron by the synergistic action of Erg-1 and p53 [22, modified].

338
Coculescu B.I. et al. Autophagy as a neuronal survival mechanism in ischemic stroke
of the target operative genes at this level (Fig. 6). As a result of NF-kB binding, through the RHD domain in its structure, at the level of DNA nucleotides and specific operator genes modulation, we reach an increased expression of: - members of the Bcl-2 anti-apoptotic family such as Bcl-xL, XIAP (X-linked inhibitor of apoptosis protein XIAP), also known as inhibitor of apoptosis protein 3 (IAP3), and A1 / Bfl-1 [21, 32, 33]. - cFLIP (c-FLIP= cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein) with caspase-8 inactivation action [33]. - caspase inhibitors, such as IAPs (IAPs = inhibitor of apoptosis proteins), with direct action to inhibit caspase activation; - TRAF1 factor (TNF receptor associated factor) associated to the TNF receptor; - GADD45, multifunctional protein that is regulated by p53, whose role is to inhibit JNK-mediated apoptosis; - modulation of gene expression of some antioxidants such as the ferritin heavy chain (FHC) [21, 35]. By acting synergistically with the protein-1 (AP1) activator, NF-kB contributes to the regulatable increase of antioxidant gene expression, MnSOD and catalase, enzymes involved in maintaining redox homeostasis [11, 36 quoted by 21]. In the quantitative regulation of these two constitutive enzymes of the stress-resistance system, it acts synergistically with NF-κB and the protein-1 (AP1) and FOXO [5] activator transcription factors. In conferring resistance to oxidative stress, FOXO transcription factors interfere by facilitating the repair of damaged DNA by stimulating the expression of genes, such as GADD45 and DDB1 [11]. Drawing conclusions on the role developed by the NF-kB transcription factor at the neuronal level, experimental studies have shown that high levels of ROS maintain its inactivation, generating apoptosis, and moderately elevated concentrations lead to NF-KB
activation and cell survival by inhibiting apoptosis and redox rebalancing [21].
CONCLUSIONS
Functional recovery in ischemic strokes depends equally on the extent of the area in which the neurons were necrosed, as well as on the survival capacity of the neurons located in the ischemic penumbra area. As neurons in the penumbra area perform their functions under critical irrigation conditions, they are "apoptotically marked" by the deterioration of some of its components (mitochondria, reticulo-endothelial system, Golgi apparatus, etc.). Their structural change determines the functional inefficiency of the neurons as a whole. Autophagy, as a biological process capable of removing these "damaged" structures, recycles the components, providing the compounds needed to produce energy, but also to the renewal the cellular/survival components. The morpho-functional refurbishment of the neuron located in the ischemic penumbra area [37] will be clinically expressed by the alleviation of the sensory-motor deficiency of the patient with ischemic stroke. Running in excess, autophagy becomes a pathogenic contributor, explaining the worsening of sensory-motor deficits in the disease. From the point of view of medical practice, the present study provides an additional argument regarding the therapeutic re-evaluation guide for ischemic stroke, which should aim above all at ensuring the survival of nerve cells in the ischemic penumbra area. This is because the prolongation of hypoxia of the neurons at the level of the penumbra area leads to an increase of the redox disorder and consequently leads to apoptosis induced prematurely by accentuating the functional sensory-motor deficit of the respective patient.
Conflict of interest. The authors declare that there is no conflict of interest. Acknowledgement. All authors have contributed equally to this paper.
References1. Manole G. General clinical pathophysiology. Vol. 2 (The pathophysiology of organs), National Company Printers, Coresi S.A. Publishing:
Bucharest, Romania, 2003, pp. 181-215; ISBN 973-570-253-3 [In Romanian].2. Opriș L. Brain imaging by magnetic resonance. Solness Publishing, Timișoara, Romania, 2004, pp. 230-232, ISBN 973-847-280-6 [In
Romanian].3. Bălăeţ C, Coculescu BI, Manole G, Bălăeţ M, Dincă VG. Gamma-glutamyltransferase, possible novel biomarker in colon diverticulosis: a
case-control study. J. Enzyme Inhib. Med. Chem. 2018, 33(1):428-432.4. Bălăeţ C. Coculescu BI, Bălăeţ M, Manole G, Dincă GV. Haemolytic anaemia and hepatocitolysis associated with hypermagnesaemia by
repeated exposures to copper–calcium fungicides. J. Enzyme Inhib. Med. Chem. 2018, 33(1):184-189.5. Azevedo SP, Polegato FB, Minicucci FM, Paiva RAS, Zornoff MAL. Cardiac remodeling: concepts, clinical impact, pathophysiological
mechanisms and pharmacologic treatment. Arq. Bras. Cardio. 2016, 106(1):62-69.6. Schaper J, Elsasser A, Kostin S. The role of death in heart failure. Circ. Research 1999, 85(9):867-869.7. Glick D, Barth S, Macleod FK. Autophagy: cellular and molecular mechanisms. J Pathol. 2010, 221(1):3-12.8. Vigliano CA, Cabeza Meckert PM, Diez M, Favaloro LE, Cortes C, Fazzi L, Favaloro RR, Laguens RP. Cardiomyocyte hypertrophy, oncosis,
and autophagic vacuolization predict mortality in idiopathic dilated cardiomyopathy with advanced heart failure. J Am Coll Cardiol. 2011, 57(14):1523-31.

Romanian Journal of Legal Medicine Vol. XXVI, No 4(2018)
339
9. Manole G. Neural suffering in severe obstruction syndrome of the internal carotid artery. In Cerebral circulatory insufficiency; M.E. Pătruț (coord.), Medical Publishing House, Bucharest, Romania, 2008, pp. 35-93 [In Romanian].
10. Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Bio. 2009, 10(2):104-15.11. Carter EM, Brunet A. FOXO transcription factors. Current Biology 2007, 17(4):R11-R114.12. Webb AE. Brune A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014, 39(4):159-169.13. Burman C. Ktistakis NT. Autophagosome formation in mammalian cells. Semin Immunopathol. 2010, 32(4):397-413.14. Česen MH, Pegan K, Spes A, Turk B. Lysosomal pathways to cell death and their therapeutic applications. Exp Cell Res. 2012, 318(11):1245-
51.15. Levine B, Mizushima N. Virgin. H.W. Autophagy in immunity and inflammation. Nature, 2011, 469 (7330):323-335.16. Stocheci C.M. Cerebrovascular accidents in Arges County - clinical, histological and immunohistochemical study. Ph.D. Thesis, Doctoral
School - University of Medicine and Pharmacy of Craiova, Romania, 2014 [In Romanian].17. Băcanu I. Neuroplasticity, the continuous metamorphosis of the brain. Practica medicală 2012, 5(3-4):126-127 [In Romanian].18. Gavriliuc M, Luchianciuc R. The phenomenon of ischemic preconditioning at the patients with ischemic and hemorrhagic stroke (Review
of literature). In: Scientific Annals of IP USMF “Nicolae Testemiţanu”. 14th Ed. Kishinev, Moldova: CEP Medicine, 2013, Vol. 3: Current issues in internal medicine, pp. 483-488 [In Romanian].
19. Coculescu BI, Dincă GV, Bălăeţ C, Manole G, Bălăeţ M, Stocheci CM. Myeloperoxidase, a possible biomarker for the early diagnosis of cardiac diastolic dysfunction with conserved ejection fraction. J Enzyme Inhib Med Chem. 2018, 33(1):1292-1298.
20. Coculescu BI. Dincă GV, Manole G, Purcărea VL, Oproiu AM, Stocheci CM. Serum concentration of hsCRP - possible marker for therapy evaluation in left ventricular dysfunction with preserved ejection fraction. Rev de Chimie (Bucharest) 2018, 69(10): 2881-2884.
21. Trachootham D, Lu W, Ogasawara MA, Rivera-Del Valle N, Huang P. Redox regulation of cell survival. Antioxid. Redox Signal 2008, 10, (8):1343-1374.
22. Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002, 193(3): 287-292.23. Ursu P. Redox-sensitive transcription factors from superior eucaryocytes. Available online: http://www.academia.edu/13978362/Factori_
transcrip%C5%A3ionali_redox-sensibili_de_la_eucariotele_superioare (accessed on 12 May 2018) [In Romanian].24. Pani G, Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxid. Redox Signal 2011, 15, (6):1715-27.25. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 2011, 12(1):9-14.26. Essers MA, Weijzen S, de Vries-Smits MA, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by
oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23(24):4802-12.27. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kB signaling. Cell. Res. 2011, 21(1):103-15.28. Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator
that binds to the tandem NF-E2/App1 repeat of the beta-globulin locus control region. Proc Natl Acad Sci USA 1994, 91(21):9926-30.29. Sun YK, Webb AE. Neuronal function of FOXO/DAF-16. Nutr Healthy Aging. 2017, 4(2):113-126.30. Gilmore TD, Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006, 25(51):6680-4.31. Kaltschmidt B, Kaltschmidt C, Hofmann TG, Hehner SP, Dröge W, Schmitz ML. The pro- or anti-apoptotic function of NF-kappaB is
determined by the nature of the apoptotic stimulus. Eur J Biochem. 2000, 267(12):3828-35.32. Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM, Vaux DL. The anti-apoptotic activity of XIAP is retained upon
mutation of both the caspase 3- and caspase 9-interacting sites. J.cell Biol. 2002, 157(1):115-124.33. Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, The CE, Gray DH, Tai L, Lew AM, Villunger A, Strasser A, Herold MJ.
Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. 2017, 24, 3, 534-545.
34. Safa, A.R. c-FLIP, a master anti-apoptotic reglator. Exp. Oncol. 2012, 34(3):176-84.35. Maeda T, Hanna AN, Sim AB, Chua PP, Chong MT, Tron VA. GADD45 regulates G2/M arrest, DNA repair, and cell death in keratinocytes
following ultraviolet exposure. J Invest Dermatol. 2002, 119(1):22-26.36. Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002, 3(3):221-7.37. Moldovan C, Dobrescu L, Ristoiu V, Firtat B, Dinulescu S, Brasoveanu C, Ion M, Dobrescu D, Gheorghe R, Pascalau AM, Pogarasteanu M,
Coculescu BI, Oproiu AM. Experimental measurements in the acquisition of biosignals from a neuronal cell culture for an exoprosthesis command. Rev de Chimie (Bucharest), 2018, 69(10):2948-2952.















![Imaging of Hereditary Hemorrhagic Telangiectasia · Spinal and cerebral vascular malformations are mani-festations of underlying vascular dysplasia [12]. These lesions represent abnormal](https://static.fdocuments.us/doc/165x107/5ed59c731b7fdd786a1b540e/imaging-of-hereditary-hemorrhagic-telangiectasia-spinal-and-cerebral-vascular-malformations.jpg)


