
AusPAR Attachment 1: Extract from the Clinical … · Web viewHowever, while review of the data...
84
AusPAR Attachment 1 Extract from the Clinical Evaluation Report for enoxaparin sodium Proprietary Product Name: Crusia-AFT, Crusia-AFT Forte Sponsor: AFT Pharmaceuticals Pty Ltd First round CER: June 2016 Second round CER: November 2016
Transcript of AusPAR Attachment 1: Extract from the Clinical … · Web viewHowever, while review of the data...

AusPAR Attachment 1
Extract from the Clinical Evaluation Report for enoxaparin sodium
Proprietary Product Name: Crusia-AFT, Crusia-AFT Forte
Sponsor: AFT Pharmaceuticals Pty Ltd
First round CER: June 2016Second round CER: November 2016

Therapeutic Goods Administration
About the Therapeutic Goods Administration (TGA) The Therapeutic Goods Administration (TGA) is part of the Australian Government
Department of Health, and is responsible for regulating medicines and medical devices.
The TGA administers the Therapeutic Goods Act 1989 (the Act), applying a risk management approach designed to ensure therapeutic goods supplied in Australia meet acceptable standards of quality, safety and efficacy (performance), when necessary.
The work of the TGA is based on applying scientific and clinical expertise to decision-making, to ensure that the benefits to consumers outweigh any risks associated with the use of medicines and medical devices.
The TGA relies on the public, healthcare professionals and industry to report problems with medicines or medical devices. TGA investigates reports received by it to determine any necessary regulatory action.
To report a problem with a medicine or medical device, please see the information on the TGA website <https://www.tga.gov.au>.
About the Extract from the Clinical Evaluation Report This document provides a more detailed evaluation of the clinical findings, extracted
from the Clinical Evaluation Report (CER) prepared by the TGA. This extract does not include sections from the CER regarding product documentation or post market activities.
The words [Information redacted], where they appear in this document, indicate that confidential information has been deleted.
For the most recent Product Information (PI), please refer to the TGA website <https://www.tga.gov.au/product-information-pi>.
Copyright© Commonwealth of Australia 2017This work is copyright. You may reproduce the whole or part of this work in unaltered form for your own personal use or, if you are part of an organisation, for internal use within your organisation, but only if you or your organisation do not use the reproduction for any commercial purpose and retain this copyright notice and all disclaimer notices as part of that reproduction. Apart from rights to use as permitted by the Copyright Act 1968 or allowed by this copyright notice, all other rights are reserved and you are not allowed to reproduce the whole or any part of this work in any way (electronic or otherwise) without first being given specific written permission from the Commonwealth to do so. Requests and inquiries concerning reproduction and rights are to be sent to the TGA Copyright Officer, Therapeutic Goods Administration, PO Box 100, Woden ACT 2606 or emailed to <[email protected]>.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 2 of 62

Therapeutic Goods Administration
ContentsList of abbreviations__________________________________________________________5
1. Introduction_____________________________________________________________8
1.1. Drug class and therapeutic indication_________________________________________8
1.2. Dosage forms and strengths____________________________________________________8
1.3. Dosage and administration_____________________________________________________9
2. Clinical rationale________________________________________________________9
2.1. Guidance_______________________________________________________________________10
3. Contents of the clinical dossier______________________________________10
3.1. Scope of the clinical dossier___________________________________________________10
3.2. Paediatric data_________________________________________________________________11
3.3. Good clinical practice__________________________________________________________11
4. Pharmacokinetics_____________________________________________________11
5. Pharmacodynamics___________________________________________________11
5.1. Studies providing pharmacodynamic bioequivalence data________________11
5.2. Study ROV-RO20-2011-01 (Phase I)_________________________________________12
5.3. Evaluator’s overall conclusions on pharmacodynamics____________________23
6. Dosage selection for the pivotal studies___________________________32
7. Clinical efficacy________________________________________________________33
8. Clinical safety__________________________________________________________35
8.1. Studies providing evaluable safety data_____________________________________35
8.2. Evaluator’s comment on clinical safety______________________________________38
9. First round benefit-risk assessment_______________________________41
9.1. First round assessment of benefits___________________________________________41
9.2. First round assessment of risks______________________________________________41
9.3. First round assessment of benefit-risk balance_____________________________42
10. First round recommendation regarding authorisation_________42
11. Clinical questions_____________________________________________________43
12. Second round evaluation of clinical data submitted in response to questions______________________________________________________________________44
12.1. Overview of the clinical response.__________________________________________44
12.2. Sponsor’s response to first round questions_______________________________44
12.3. Evaluation of Study ROV-RO20-2015-01___________________________________48
13. Second round benefit-risk assessment____________________________57
13.1. Second round assessment of benefits______________________________________57
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 3 of 62

Therapeutic Goods Administration
13.2. Second round assessment of risks__________________________________________58
13.3. Second round assessment of the benefit-risk balance_____________________59
14. Second round recommendation regarding authorisation______59
15. References______________________________________________________________60
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 4 of 62

Therapeutic Goods Administration
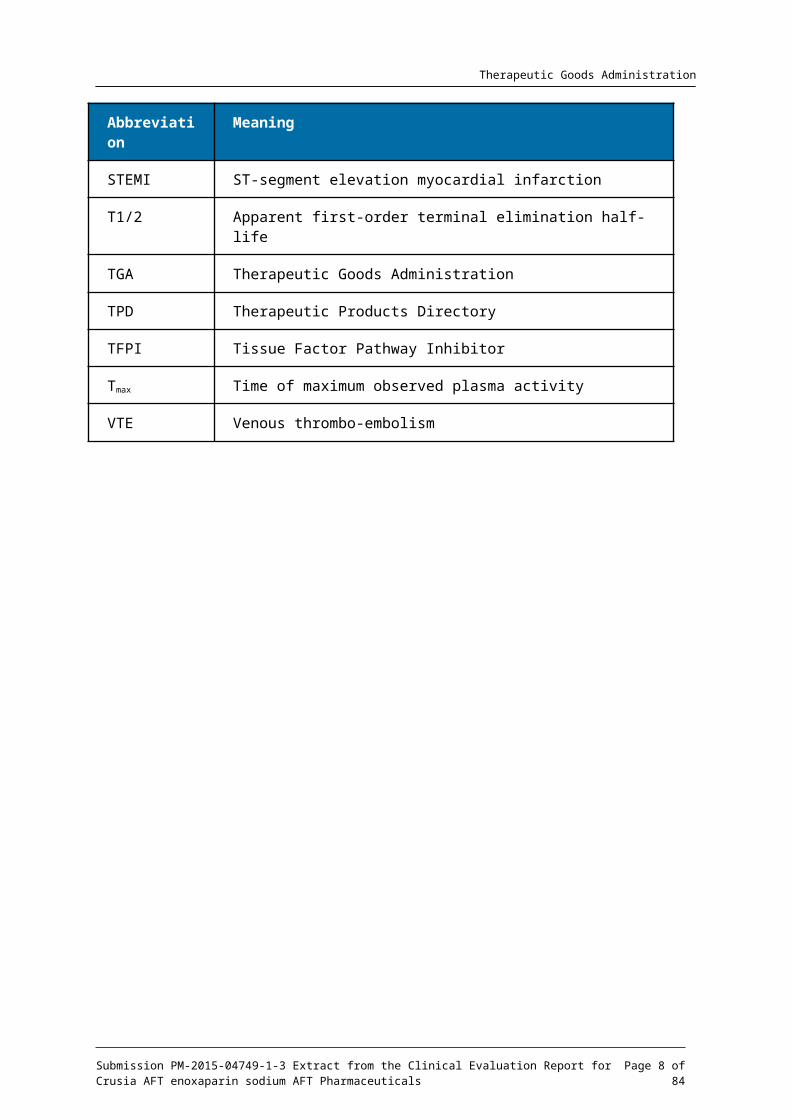
List of abbreviationsAbbreviation Meaning
AE Adverse event
Anti-Xa Anti-factor Xa
Anti-Xamax Peak effect for anti-Xa
Anti-IIa Anti-factor IIa
Anti-IIamax Peak effect for anti-IIa
ANOVA Analysis of Variance
AUEC Area under the effect curve
AUEC0-t Area under the effect curve from time zero to the last measurable value
AUECinf Area under the effect curve from the time zero point to infinity
BE Bioequivalence
BMI Body mass index
CER Clinical Evaluation Report
CHMP Committee for Medicinal Products for Human Use
CI Confidence Interval
CL/F Apparent clearance
CRF Case Report Form
CV Coefficient of Variation
ECG Electrocardiogram
EMA European Medicine Agency
EMEA European Medicines Evaluation Agency
EU European Union
FDA Food and Drug Administration
GCP Good Clinical Practice
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 5 of 62

Therapeutic Goods Administration
Abbreviation Meaning
GMR Geometric Mean Ratio
hr(s) Hour(s)
Kel Apparent first order terminal elimination rate constant
kg Kilogram(s)
IV Intravenous
LLOQ Lower limit of quantitation
Ln Natural logarithm
LSM Least square mean
m Metres
Min Minutes
mL Millilitre
Lμ Microlitre
N/A Not applicable
PD Pharmacodynamic
PE Pulmonary embolus
PK Pharmacokinetic
SAS Statistical Analysis System
SC Subcutaneous
SD Standard Deviation
STEMI ST-segment elevation myocardial infarction
T1/2 Apparent first-order terminal elimination half-life
TGA Therapeutic Goods Administration
TPD Therapeutic Products Directory
TFPI Tissue Factor Pathway Inhibitor
Tmax Time of maximum observed plasma activity
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 6 of 62

Therapeutic Goods Administration
Abbreviation Meaning
VTE Venous thrombo-embolism
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 7 of 62
Therapeutic Goods Administration
1. IntroductionThis submission is to register Crusia-AFT and Crusia-AFT Forte1 as biologically similar medicinal products to the approved products Clexane and Clexane Forte, produced by Sanofi-Aventis.
1.1. Drug class and therapeutic indicationEnoxaparin is a low molecular weight heparin (LMWH) with a mean molecular weight of approximately 4,500 daltons (3,500 to 5,500). The drug substance is the sodium salt. Enoxaparin sodium belongs to the Antithrombotic Agents pharmacological class, with ATC code B01AB05 enoxaparin. It is obtained by alkaline depolymerisation of heparin benzyl ester derived from porcine intestinal mucosa. Enoxaparin binds to anti-thrombin III leading to inhibition of coagulation factors IIa and Xa.
The proposed therapeutic indications for Crusia-AFT and Crusia-AFT Forte are:
Prevention of thromboembolic disorders of venous origin in patients undergoing orthopaedic and general surgery.
Prophylaxis of venous thromboembolism in medical patients bedridden due to acute illness.
Prevention of thrombosis in extracorporeal circulation during haemodialysis.
Treatment of established deep vein thrombosis.
Treatment of unstable angina and non-Q-wave myocardial infarction, administered concurrently with aspirin.
Treatment of acute ST-segment Elevation Myocardial Infarction (STEMI) as an adjunctive to thrombolytic treatment, including patients to be managed medically or with subsequent Percutaneous Coronary Intervention (PCI).
The proposed therapeutic indications being sought for Crusia-AFT and Crusia-AFT Forte are identical to those of the registered products Clexane and Clexane Forte.
1.2. Dosage forms and strengthsThe following Crusia-AFT dosage forms and strengths are proposed for registration:
Crusia-AFT injection syringe: 20 mg/0.2 mL; 40 mg/0.4 mL; 60 mg/0.6 mL; 80 mg/0.8 mL; and 100 mg/1 mL.
Crusia-AFT Forte injection syringe: 120 mg/0.8 mL; and 150 mg/1 mL.
The relevant registered and marketed Clexane dosage forms and strengths are:
Clexane ready-to-use pre-filled syringe: 20 mg/0.2 mL; 40 mg/0.4 mL; 60 mg/0.6 mL; 80 mg/0.8 mL; and 100 mg/1 mL.
Clexane Forte syringes ready-to-use pre-filled syringe: 120 mg/0.8 mL; and 150 mg/1 mL.
Comment: The proposed Crusia-AFT dosage forms and strengths are consistent with the Australian registered and marketed Clexane products.
1 At times referred to ‘Crusia’ only in this report.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 8 of 62

Therapeutic Goods Administration
1.3. Dosage and administrationThe proposed dosage and administration procedures for Crusia-AFT and Crusia-AFT Forte for each indication are identical to those approved for Clexane and Clexane Forte. Depending on the indication, the proposed routes of administration are by subcutaneous (SC) injection, intravenous (IV) injection, or extracorporeal administration.
2. Clinical rationaleThe sponsor states that Crusia-AFT has been developed to align with Clexane and Lovenox, both products of Sanofi-Aventis with enoxaparin sodium as their active ingredient. The sponsor states that, in accordance with current scientific thinking and the TGA adopted EU ‘Guideline on similar biological medicinal products’, similarity between the biosimilar and the reference product (Lovenox (USA)) has been established in a randomised, double blind, 2 way, crossover pharmacodynamic bioequivalence study in healthy volunteers (Study ROV-RO20-2011-01).2 In addition, two non-clinical bioavailability studies have been performed in rabbits in order to establish bioequivalence of the absorption profiles of the biosimilar and the reference product. These clinical and non-clinical studies were performed using Lovenox marketed in the USA as the reference product. In order to support registration in Europe and other territories where Lovenox is not registered, the sponsor states that an extensive state of the art analytical comparability exercise was performed to bridge Crusia-AFT, Lovenox (USA) and Clexane (Spain). In accordance with the TGA’s ‘Regulation of biosimilar medicines V2.0 (December 2015)’ guidelines, the sponsor considers Clexane (Spain) to be the Australian reference medicine.
Comment: The clinical data were limited to the single dose, pharmacodynamic (PD) bioequivalence (BE) study in healthy volunteers comparing the proposed enoxaparin product with the US marketed reference product (Lovenox). There were no clinical studies comparing the proposed enoxaparin product with the Australian marketed product (Clexane). The in vitro comparability exercise used Clexane obtained from the Spanish market, Lovenox obtained from the US market and the proposed enoxaparin product manufactured by Rovi. The submission included a number of justifications claimed by the sponsor to support its decision not to include the clinical studies requested by the Therapeutic Goods Administration (TGA) in pre-submission correspondence, and referred to in the TGA adopted ‘Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular weight heparins’.3 These justifications have been discussed in this clinical evaluation report (CER).
The sponsor indicates that the development of the proposed product (enoxaparin sodium solution for injection) is based on the published available data on the qualitative and quantitative composition of the reference medicinal product (Lovenox/Clexane). The sponsor states that the similarity between the proposed product and the reference product has been demonstrated by the in vitro 3 way state of the art comparability exercise (Rovi Enoxaparin sodium, Lovenox (USA), and Clexane (Spain)). The data indicate that all the steps of the manufacturing process take place at the Rovi Contract Manufacturing in Spain and [name redacted] as an alternative manufacturer for the secondary packaging. Both manufacturing sites are subcontracted manufacturing facilities of [information redacted] Rovi.
2 EMA/CHMP/437/04 Rev. 1: Guideline on similar biological medicinal products (2014)3 EMEA/CHMP/BMWP/118264/2007: Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular weight heparins (2007)
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 9 of 62

Therapeutic Goods Administration
2.1. GuidanceThere are currently no enoxaparin biosimilar medicines on the Australian Register of Therapeutic Goods (ARTG). The relevant TGA guidelines relating to the clinical evaluation of the submission include:
Regulation of biosimilar medicines (Version 2.0, December 2015)
Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular weight heparins (EMEA/CHMP/BMWP/118264/2007)
Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: non-clinical and clinical issues (EMEA/CHMP/BMWP/42832/2005/Rev 1)
Guideline on immunogenicity assessment of biotechnology derived therapeutic proteins (EMEA/CHMP/BMWP/14327/2006).
The key TGA adopted clinical guideline relating to the submission is considered to be the product specific guideline for biosimilar LMWH products (EMEA/CHMP/BMWP/118264/2007). The guideline provides nonclinical and clinical requirements for LMWH products claimed to be similar biological medicinal products to already marketed LMWHs. The guideline states that the major burden of demonstrating that two LMWHs are similar biological medicinal products is on a clinical trial, due to the high heterogeneity of LMWH, incomplete understanding of the mode of action of the product, and uncertainty about whether the pharmacodynamic markers are representative of the clinical outcome.
There is a draft revised EMA guideline relating to the nonclinical and clinical development of biosimilar LMWH products (EMEA/CHMP /BMWP/118264/2007 Rev 1). This draft revised guideline was released for consultation by the CHMP on 13 January 2013. The end of consultation (deadline for comments) for the guideline was 31 July 2013. The EMA has not released an overview of comments received on the draft guideline. Nearly three years has now elapsed since the end of the consultation period. The revised guideline has not yet been adopted by either the TGA or the EMA.
The clinical evaluation of the submission has been undertaken in the light of the TGA adopted LMWH biosimilar guideline (EMEA/CHMP/BMWP/118264/2007). The TGA has not yet adopted the revised LMWH biosimilar guideline (EMEA/CHMP/BMWP/118264/2007 Rev 1) nor has it rescinded the adopted guideline. Therefore, it is reasonable to infer that the adopted guideline still reflect the TGA’s current thinking on the clinical requirements for submissions to register a LMWH product claimed to be biosimilar to the Australian marketed product. While sponsors are not legally required to comply with TGA adopted guidelines, it is expected that they will adequately justify any deviations from the guidelines.
3. Contents of the clinical dossier
3.1. Scope of the clinical dossierThe submission consisted of an abbreviated clinical dossier consisting of one single dose pharmacodynamic (PD) bioequivalence (BE) study (Study ROV-RO20-2011-01) in healthy volunteers comparing the enoxaparin sodium product proposed for registration and the enoxaparin sodium product marketed in the USA (Lovenox). No other clinical studies were submitted.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 10 of 62

Therapeutic Goods Administration
In addition, the following were submitted: An Introduction; Quality Overall Summary; Nonclinical Overview; Clinical Overview; Nonclinical Written Summary; Summary of Biopharmaceutics and Associated Analytical Methods.
3.2. Paediatric dataNo paediatric data were submitted. The sponsor states that no paediatric data have been submitted to the EU. The sponsor indicates that the submission of paediatric data is not a requirement for similar biological medicinal products in the EU. The sponsor’s decision not to submit paediatric data is considered to be acceptable.
3.3. Good clinical practiceThe submitted PD bioequivalence Study ROV-RO20-2011-01 was conducted according to the International Conference on Harmonisation (ICH) guidelines for good clinical practice.
4. PharmacokineticsThere were no studies providing conventional pharmacokinetic data.
The TGA adopted LMWH biosimilar guideline states that:
‘Due to the heterogeneity of LMWHs conventional pharmacokinetic studies cannot be performed. Instead, the absorption and elimination characteristics of LMWHs should be compared by determining pharmacodynamic activities (including anti FXa and anti-FIIa), as surrogate markers for their circulating concentrations. In addition other pharmacodynamic tests such as Tissue Factor Pathway Inhibitor (TFPI) activity, as well as the ratio of anti-FXa and anti-FIIa activity should be compared. Assessment of these PD parameters will provide a fingerprint of the polysaccharidic profile’.4
5. Pharmacodynamics
5.1. Studies providing pharmacodynamic bioequivalence dataThe clinical data included one, single dose (100 mg SC), pharmacodynamic (PD) bioequivalence (BE) study (Study ROV-RO20-2011-01) in healthy volunteers comparing the test product (enoxaparin sodium Rovi injection 100 mg/mL) with the reference product (US marketed, Lovenox 100 mg/mL). The submission included an addendum to the final study report, which provided post-hoc analyses of the PD and AE data from the study. The submitted bioequivalence study has been fully evaluated. The study is summarised below in Table 1.
4 EMEA/CHMP/BMWP/118264/2007: Guideline on non-clinical and clinical development of similar biological medicinal products containing low molecular weight heparins (LMWH). Committee for Medicinal products for Human (CHMP) European Medicines Agency (EMA); London, UK.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 11 of 62

Therapeutic Goods Administration
Table 1. Study ROV-RO20-2011-01 Pharmacodynamic bioequivalence study
Objective Design Treatment Subjects Objective
To determine the PD BE of the test and reference products.
Single dose, randomised, double blind, 2 way crossover. Duration approximately 6 weeks, including 30 day screening period.
Test: Enoxaparin sodium Rovi (100 mg/mL), 100 mg SC.
Reference: Lovenox (100 mg/mL), 100 mg SC; USA marketed.
Healthy volunteers (n = 42; 25 male, 17 female; mean age = 32.4 years (19, 45 years).
Demonstrate PD BE of the test and reference formulations based on anti-Xa and anti-IIa activity.
5.2. Study ROV-RO20-2011-01 (Phase I) 5.2.1. Design, objectives, location, and dates
5.2.1.1. Title
A ‘single dose, randomised, double-blind, two-way crossover bioequivalence study of enoxaparin (100 mg/mL) 100 mg subcutaneous injection manufactured by Rovi, Spain, and Lovenox (100 mg/mL) 100 mg subcutaneous injection manufactured by Sanofi-aventis, USA, in healthy volunteers’.
5.2.1.2. Location and dates
The study was undertaken at a single site in Austin, Texas, USA. The first subject was dosed on 19 January 2013 and the date of the last subject contact was 15 March 2013. The study report (Version 4.0) was dated 14 October 2014. The study was stated to have been conducted according to the International Conference on Harmonisation (ICH) tripartite Guideline for Good Clinical Practice E6 (R1). The sponsor was Laboratorios Farmacéuticos Rovi, S.A, Madrid, Spain.
5.2.1.3. Objectives
The primary objective of the study was to determine the bioequivalence (BE) of enoxaparin (100 mg/mL) 100 mg SC injection manufactured by Rovi, Spain and Lovenox (100 mg/mL) 100 mg SC injection manufactured by Sanofi-Aventis, USA in healthy volunteers.
The secondary objective of the study was to evaluate the safety and tolerability of enoxaparin (100 mg/mL) 100 mg SC injection manufactured by Rovi, Spain in healthy volunteers.
The exploratory objective of the study was to evaluate tissue factor pathway inhibitor (TFPI) activity after administration of the two enoxaparin (100 mg/mL) products.
5.2.1.4. Study design
Study ROV-RO20-2011-01 was a single dose, randomised, double blind, 2 period, 2 sequence crossover study in healthy volunteers. Subjects were screened up to 30 days before the study began and were admitted to the clinic on Day 1 of Period 1 for baseline assessments. Before dosing on Day 1 of Period 1, subjects were randomly assigned to a treatment sequence (AB, BA), with treatment A being the test treatment (Rovi; Spain) and treatment B being the reference treatment (Lovenox; USA). Subjects received a single SC dose of study drug on Day 1 of each treatment period. In subjects randomly assigned to Sequence AB, treatment A was administered in Period 1 and treatment B in Period 2. In subjects randomly assigned to Sequence BA, treatment B was administered in Period 1 and treatment A was administered in Period 2.
On Day 1 of Period 1, subjects received a single SC dose of assigned study drug after an overnight fast of at least 10 hours, and continued fasting for at least 4 hours after dosing.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 12 of 62

Therapeutic Goods Administration
Subjects remained in the clinic on Days 1 and 2 and were discharged on Day 3 of Period 1 after completion of all safety and PD assessments. There was washout between the two periods of at least 7 days, which is considered adequate to prevent PD carry-over effects. Enoxaparin doses of 73.8 mg to 132.6 mg have been reported to result in anti-Xa activity levels with first and second phase elimination half-lives of approximately 5 hours and 9 hours, respectively (Clexane/Clexane Forte PI).
Study subjects returned to the clinic no later than the evening before dosing (Day -1) in Period 2. The following assessments were performed at the time subjects re-entered the clinic: review of inclusion and exclusion criteria; urine drug screen; serum pregnancy test (for female subjects); and review of concomitant medications and adverse events (AEs). Negative urine drug screen and serum pregnancy test results were required in order for subjects to continue in the study. On Day 1 of Period 2, subjects crossed over to receive a single SC dose of the assigned study drug after an overnight fast of at least 10 hours and continued fasting for at least 4 hours after dosing. Subjects remained in the clinic on Days 1 and 2 and were discharged on Day 3 of Period 2 after completion of all safety and PD assessments. Subjects returned to the clinic on Day 7 of Period 2 for a follow-up visit. The total duration of the study was approximately 6 weeks, including the 30 day screening period.
The injection site for study drug administration in Periods 1 and 2 was alternated between the left and right anterolateral or left and right posterolateral abdominal wall in accordance with Lovenox prescribing information. The investigator was onsite for dosing and for at least 4 hours after dosing.
Blood samples were collected at the following time points for assessment of PD parameters of anti-Xa, anti-IIa, and TFPI activity before and after a single dose of study drug: Day –1 (Periods 1 and 2), before dosing (0 hour), and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 16, 20, 24, and 36 hours after dosing on Day 1 (Periods 1 and 2). PD calculations were based on actual sample times.
Safety parameters assessed throughout the study were AEs, clinical laboratory test results (haematology, coagulation, serum chemistry, and urinalysis), vital sign measurements (systolic and diastolic blood pressures, pulse rate, respiratory rate, and oral body temperature), 12-lead safety electrocardiogram (ECG) results, and physical examination findings.
The study schedule was provided.
Comment: The sponsor has confirmed that the test product used in this study (Rovi enoxaparin sodium; 100 mg/mL) is the product proposed for Australian registration. The BE study was conducted in accordance with the FDA (US) Draft guidance on enoxaparin sodium for demonstrating active ingredient sameness of test and reference of two products. However, this draft guidance has not been adopted by the TGA. [A copy of the draft FDA guidance is not provided in this document but was available from the FDA website].
5.2.2. Inclusion and exclusion criteria
The inclusion criteria included healthy male and females between the ages of 18 and 45 years, inclusive. The inclusion and exclusion criteria are considered to be acceptable for a single, SC dose enoxaparin PD bioequivalence study in healthy volunteers.
Participants could voluntarily withdraw from the study at any time. In addition, subjects could be withdrawn from the study at the investigator’s discretion for the following reasons: serious adverse events or intolerable adverse events; protocol violations; and symptoms or signs developing during the course of the study that were listed in the exclusion criteria. Other reasons that could result in a subject being withdrawn from the study included: at the request of the sponsor for safety concerns; at the request of the sponsor for clinical or administrative reasons; lost to follow-up; or death. The reasons for withdrawing a subject from the study were
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 13 of 62

Therapeutic Goods Administration
not limited to those expressly specified. Appropriate procedures were in place to follow-up patients who withdrew from the study.
5.2.3. Treatments
Enoxaparin (100 mg/mL) 100 mg SC injection manufactured by Rovi, Spain and Lovenox (100 mg/mL) 100 mg SC injection manufactured by Sanofi-Aventis, USA were provided by the sponsor as prefilled syringes. The reference treatment was Lovenox (100 mg/mL), which contains 10 mg enoxaparin sodium (approximate anti-factor Xa activity of 1000 IU) per 0.1 mL of sterile water for injection. The 100 mg/mL dose of enoxaparin was chosen as this is the dose specified in the FDA ‘Draft guidance on enoxaparin sodium’ for demonstrating active ingredient sameness of test and reference enoxaparin products. All doses of study drug were dispensed, administered, and recorded by clinic personnel, and any deviations from the dosing schedule were entered into the subject’s eCRF. Subjects remained ambulatory or seated upright for the first 4 hours after administration of study drug, but this procedure could be amended in the event of adverse events occurring during this time interval. Subjects were not to engage in strenuous activity at any time during the study.
Information about medications taken by the subject within the 30 days before the first dose of study drug and concomitant medications taken over the course of the study were assessed and recorded in the eCRF. Subjects were not permitted to use an investigational drug within the 30 days before the first dose of study drug. The use of prescription medications was prohibited for 4 weeks before dosing through to the completion of all study procedures. The use of any over the counter medication (including non-steroidal anti-inflammatory drugs, vitamins, herbal supplements, or dietary supplements) was prohibited within 2 weeks before dosing through to the completion of all study procedures, unless otherwise approved by the investigator and medical monitor. Subjects were not allowed to take anticoagulant medications (for example, aspirin, warfarin, LMWH, dabigatran, or rivaroxaban) within 7 days before screening or vitamin preparations containing vitamin C in excess of 250 mg per day within 3 days before screening. Any medications for the management of any AEs during the study could have been given at the investigator’s discretion.
5.2.4. Randomisation and blinding methods
The study was double-blinded in that the subjects and the laboratory staff were blinded. While the test and reference solutions for SC injection may have been identical in appearance, the prefilled syringes for both study drugs may not have been identical. Therefore, an unblinded pharmacist was responsible for dispensing the study drug in a manner consistent with maintaining the blind and an unblinded third party team member performed study drug administration. The investigator was responsible for maintaining the blind throughout the study. If a subject became seriously ill or pregnant during the study, the blind was to be broken only if knowledge of the administered study drug would have affected treatment options. The date, time, and reason for the unblinding were to be appropriately documented.
Comment: The randomisation method used to assign patients to treatment sequence AB or BA could not be identified in the submission. The population from which the healthy volunteers were selected for participation could not be identified. The sponsor is requested to provide information on these two matters (see Section 12: Clinical questions).
5.2.5. Pharmacodynamic analyses
The following plasma PD parameters were calculated for anti-Xa, anti-IIa, and baseline-adjusted TFPI using non-compartmental analysis: area under the effect curve (AUEC) from time 0 to infinity (AUEC0-inf); AUEC from time 0 to the last measured activity (t) (AUEC0-t); peak effect for anti-Xa (anti-Xamax); peak effect for anti-IIa (anti-IIamax); peak effect for TFPI (TFPImax); time of observed maximum measured plasma activity (Tmax); apparent first order terminal elimination half life (t1/2); apparent plasma clearance after extravascular administration (CL/F);
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 14 of 62

Therapeutic Goods Administration
mean residence time (MRT); and the ratio of AUEC0-t of anti-Xa to anti-IIa (RAUEC). The terminal phase related parameters were estimated when the data permitted.
Plasma samples from the venous blood samples were analysed for anti-Xa and anti-IIa using validated chromogenic methodologies. Anti-factor Xa activity, anti-factor II activity and TFPI were analysed using commercially available kits.
5.2.6. Statistical methods and sample size
5.2.6.1. PD population
The PD population included all randomly assigned subjects who received study drug (either the test or reference treatment) and had a sufficient number of valid bioanalytical results to facilitate calculation of the PD parameters. Missing data were not imputed. All PD summaries and statistical analyses were based on the PD population.
5.2.6.2. Plasma activity
The plasma activity values were not baseline-adjusted for anti-Xa and anti-IIa. If an activity value was less than the lower limit of quantification (LLOQ), the value was set to 0. Subjects with confirmed baseline values equal to or higher than the LLOQ of either anti-Xa or anti-IIa were excluded from the analysis. The LLOQ rule was not applied to the TFPI analysis since detectable baseline TFPI values were obtained for all subjects. Therefore, baseline adjusted TFPI values were calculated for all subjects.
5.2.6.3. PD parameters
Individual plasma activity data of subjects with sufficient plasma values over the LLOQ were used to derive the PD parameters of anti-Xa, anti-IIa, and baseline-adjusted TFPI using non-compartmental methods using Phoenix WinNonlin (Pharsight Corporation, St. Louis, Missouri) Version 6.2.1. The AUEC was estimated using the linear trapezoidal rule. The PD parameters were summarised using standard descriptive statistical methods.
5.2.7. Statistical analysis of PD data
The PD parameters AUEC0-inf, AUEC0-t, and peak effect for anti-Xa, anti-IIa, and TFPI activity from the test treatment were compared with those from the reference treatment. An analysis of variance (ANOVA) with fixed effects for sequence, period, and treatment, and random effect for subject nested within sequence was performed on the natural logarithms of AUEC0-inf, AUEC0-t, and peak effect for anti-Xa, anti-IIa, and TFPI activity to assess the differences between the test and reference treatments. The geometric mean ratio and 90% confidence interval (CI) for AUEC0-inf, AUEC0-t, and peak effect for the PD parameters of interest of the 2 treatments were calculated by the antilog of the mean difference and 90% CI of the log transformed values. Bioequivalence was concluded if the 90% CI of the ratio of the geometric LS means between the test and reference treatments for the PD parameters of interest were completely within the standard 80% to 125% BE interval.
Comment: The study specified that bioequivalence was to be concluded if the PD parameters for anti-Xa, anti-IIa, and TFPI met the pre-specified bioequivalence criteria. Based on the stated objectives of the study it is considered that the primary PD endpoints are anti-Xa activity and anti-IIa activity, and TFPI activity is a supportive endpoint. In addition, based on the presented study results it is considered that the RAUEC is a supportive endpoint. The TGA guideline relating to LMWH biosimilar products states that the selected PD margins should be ‘pre-specified and appropriately justified’. The sponsor provided a justification for the chosen BE margin of 80 to 125%, and this is discussed under ‘BD bioequivalence interval’ later in this section.
The sponsor states that the study meets the in vivo PD study design criterion required to support active ingredient sameness based on the FDA’s ‘Draft Guidance on Enoxaparin Sodium’. The draft guideline indicates that PD endpoints to be
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 15 of 62

Therapeutic Goods Administration
measured are anti-Xa and anti-IIa in plasma. The draft guideline states that the following PD parameters should be determined for anti-Xa and anti-IIa: peak effect (anti-Xamax, anti-IIamax), area under the effect curve (AUEC0-t and AUEC0-∞), Tmax, and t1/2. Equivalence is based on the 90% CI for the anti-Xa parameters; the 90% CIs for the geometric mean test/reference ratios of AUEC and anti-Xamax must fall within the BE limits of 80 to 125%. The anti-IIa data for the test and reference product are considered by the FDA to provide supportive evidence of active ingredient sameness. The draft FDA guideline includes no justification for the chosen equivalence interval of 80 to 125%. The draft FDA guideline has not been adopted by the TGA.
5.2.8. Sample size
36 subjects who completed the study provided at least 80% power to conclude PD BE, assuming that the mean ratio of the test versus reference treatments was between 0.9 and 1.1 and the intra-subject CV was less than 20%. The sponsor stated that point estimates of test/reference geometric mean ratio of 0.9 to 1.0 and an intra-subject CV of less than 18% have both been observed in the literature relating to biosimilarity studies comparing enoxaparin products.5,6 To allow for dropouts, the study planned to enrol 42 subjects (approximately equal numbers of men and women). A total of 42 subjects were included in the PD analysis for TFPI and 41 subjects for the PD analysis of anti-Xa and anti-IIa. Therefore, it can be concluded that the study was adequately powered.
5.2.9. Changes in the conduct of the study
The original protocol was dated 24 February 2012. There were 2 administrative letters and 3 protocol amendments to the original protocol. The protocol amendments were provided in the submission and are considered to be acceptable. There was 1 change from the planned analyses described in the Statistical Analysis Plan (SAP). Based on the SAP, subjects with confirmed baseline values equal to or higher than the LLOQ for either the anti-Xa or anti-II levels were excluded from the analysis. However, since most baseline values of TFPI were higher than the LLOQ of TFPI, a post hoc baseline-adjusted TFPI analysis was undertaken. In addition, RAUEC (ratio of AUEC0-t of anti-Xa to anti-IIa) was added to the statistical analysis.
5.2.10. Subject disposition
The disposition of the 42 subjects is summarised below in Table 2.
Table 2. Study ROV-RO20-2011-01 Subject disposition
Comment: All 42 subjects (100%) were included in the safety analysis. All 42 subjects were included in the PD analysis for TFPI. However, for anti-Xa and anti-IIa, only 41 subjects were included in the PD analyses, due to high baseline activity levels, or low post-baseline activity levels not meeting the LLOQ criteria.
5 Feng L et al. Bioequivalence of generic and branded subcutaneous enoxaparin: a single-dose, randomized-sequence, open-label, two-period crossover study in healthy Chinese male subjects. Clin Ther. 2009;31(7):1559-67.6 Kuczka K et al. Biomarkers and coagulation tests for assessing the biosimilarity of a generic low-molecular-weight heparin: results of a study in healthy subjects with enoxaparin. J Clin Pharmacol. 2008;48(10):1189-96. Epub 2008 Aug 20.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 16 of 62

Therapeutic Goods Administration
5.2.11. Protocol deviations
There were no admission criteria deviations. Minor deviations in PD sampling times occurred during the study (up to 40 minutes late). However, since calculation of the PD parameters was based on actual sampling times, these differences did not affect the results.
5.2.12. Characteristics of the subject population
The demographic and baseline characteristics of the subject population are summarised [in a table of the CSR, not included here]. The mean age of the total population (n = 42) was 32.4 years (range: 19, 45), with 59.5% (n = 25) being male and 40.5% (n = 17) being female. The majority of the 42 subjects were White (81.0%, n = 34), with the remaining subjects being Black or African American (16.7%, n = 7) or multiracial (2.4%, n = 1). No medical history findings at screening precluded any subject from entering the study. All serology results were negative at screening. All subjects tested negative for drugs of abuse and alcohol in urine at screening and Day –1 of both treatment periods. All female subjects tested negative for pregnancy at screening and Day –1 of both treatment periods. None of the subjects reported taking prior medications. One concomitant medication (Bactrim DS, single tablet) was reported for a spider bite. None of the subjects had a medical or surgical treatment procedure. All subjects received both treatments in the sequence assigned by the randomisation schedule.
5.2.13. Results
5.2.13.1. Anti-Xa activity
The statistical analysis of anti-Xa activity are summarised below in Table 3. The mean (CV) plasma PD parameters for anti-Xa activity are summarised in Table 4, and the mean plasma anti-Xa activity versus time on linear and semi-logarithmic scales are presented in Figure 1 (both below).
Table 3. Study ROV-RO20-2011-01 Statistical analysis of the plasma PD parameters for anti-Xa activity, PD population
Treatment A (Rovi Enoxaparin); Treatment B (Lovenox). Both treatments SC 100 mg (100 mg/mL).
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 17 of 62

Therapeutic Goods Administration
Table 4. Study ROV-RO20-2011-01 Mean (CV) plasma PD parameters for anti-Xa activity, PD population
Figure 1. Study ROV-RO20-2011-01 Mean ( SD) plasma anti-Xa activity versus time by treatment on linear and semi-logarithmic scales
For anti-Xa, a total of 41 subjects provided data to the PD analyses. One subject was excluded from the analyses for all 3 PD parameters for both treatments A and B because the anti-Xa baseline value was higher than the LLOQ. Another subject was also excluded from the analyses
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 18 of 62

Therapeutic Goods Administration
for all 3 PD for treatment B for the same reason. Another subject was excluded from the AUEC0-inf PD parameter for treatment A because the extrapolated AUEC was greater than 20%.
Comment: The study pre-specified that bioequivalence would be concluded if the 90% CI of the ratio of the geometric LS means for the anti-Xa parameters (AUEC0-t, AUEC0-inf, and anti-Xamax) were completely within the standard 80 to 125% BE interval. The pre-specified criteria were met. Therefore, it can be concluded that treatments A and B are BE based on the pre-specified PD criteria. The mean (CV) PK parameters for the test and reference products are similar, and the mean ( SD) plasma anti-Xa activity versus time curves for the test and reference treatments are virtually superimposable.
5.2.13.2. Anti-IIa activity
The statistical analysis of anti-IIa activity are summarised below in Table 5. The mean (CV) plasma PD parameters for anti-IIa activity are summarised in Table 6, and the mean plasma anti-IIa activity versus time on linear and semi-logarithmic scales are presented in Figure 2 (both below).
Table 5. Study ROV-RO20-2011-01 Statistical analysis of the plasma PD parameters for anti-IIa activity, PD population
Treatment A (Rovi Enoxaparin); Treatment B (Lovenox). Both treatments SC 100 mg (100 mg/mL)
Table 6. Study ROV-RO20-2011-01 Mean (CV) plasma PD parameters for anti-IIa activity, PD population
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 19 of 62

Therapeutic Goods Administration
Figure 2. Study ROV-RO20-2011-01 Mean ( SD) plasma anti-IIa activity versus time by treatment on linear and semi-logarithmic scales
For anti-IIa, 39 subjects contributed data to the PD analyses. AUEC0-inf was not included in the analysis because of the small sample size. 2 subjects were excluded from the analyses for both treatments because of unexpectedly low plasma levels of anti-IIa, with most of the plasma anti-IIa levels being below the LLOQ. Another subject was excluded from the PD parameters (AUEC0-t and anti-IIamax) for Lovenox, and one subject was excluded from both treatments because the baseline values of anti-Xa were equal to or higher than the LLOQ.
Comment: The results demonstrate that the 90% CI of the ratio of the geometric LS means for the two treatments for the anti-IIa activity PD parameters AUEC0-t and anti-IIamax are completely within the pre-defined 80% to 125% BE interval. The AUEC0-inf was not included in the analysis because of the small sample size for the two treatments (that is, n = 5 for Treatment A; n = 4 for Treatment B). The mean (CV) parameters for the test and reference products are similar, and the mean ( SD) plasma anti-IIa activity versus time curves for the test and reference treatments are virtually superimposable.
5.2.13.3. Baseline adjusted TFPI activity
The statistical analysis of baseline adjusted TFPI activity are summarised below in Table 7. The mean (CV) plasma PD parameters for baseline adjusted TFPI are summarised in Table 8, and the mean plasma baseline adjusted TFPI versus time on linear and semi-logarithmic scales are presented in Figure 3 (both below).
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 20 of 62

Therapeutic Goods Administration
Table 7. Study ROV-RO20-2011-01 Statistical analysis of the plasma PD parameters for baseline adjusted TFPI activity, PD population
Treatment A (Rovi Enoxaparin); Treatment B (Lovenox). Both treatments SC 100 mg (100 mg/mL)
Table 8. Study ROV-RO20-2011-01 Mean (CV) plasma PD parameters baseline-adjusted TFPI activity, PD population
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 21 of 62

Therapeutic Goods Administration
Figure 3. Study ROV-RO20-2011-01 Mean ( SD) plasma baseline-adjusted TFPI activity versus time by treatment on linear and semi-logarithmic scales
All 42 subjects contributed data to the PD analysis for the baseline-adjusted TFPI. However, AUEC0-inf was not estimated for 13 subjects who received treatment A and 12 subjects who received treatment B because no clear terminal elimination phase could be identified for these profiles.
Comment: TFPI activity following administration of the two treatments was an exploratory objective. The plasma data for TFPI activity used baseline-adjusted values as baseline levels were above the LLOQ. Consequently, these values would have been excluded from analysis based on the SAP, which specified that PD parameters should not be adjusted for baseline activity. The results for the baseline-adjusted TFPI activity PD parameters AUEC0-t, AUEC0-inf, and TFPImax met the pre-specified BE criteria, with the 90% CI intervals for the relevant ratios being entirely enclosed within the interval 80 to 125%. The mean (CV) parameters for baseline adjusted TFPI activity are similar for the test and reference products, and the mean ( SD) plasma baseline-adjusted TFPI activity versus time curves for the test and reference treatments are similar.
5.2.13.4. RAUEC
The statistical analysis of the RAUEC (ratio of AUEC0-t of anti-Xa to anti-IIa) is summarised below in Table 9.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 22 of 62

Therapeutic Goods Administration
Table 9. Study ROV-RO20-2011-01 Statistical analysis of the plasma PD parameters RAUEC, PD population
Treatment A (Rovi Enoxaparin); Treatment B (Lovenox). Both treatments SC 100 mg (100 mg/mL)
5.2.13.5. Post hoc analyses of the PD parameters
The submission included an addendum to the Study ROV-RO20-2011-01 clinical study report, which provided post hoc analyses of the PD data using a more stringent CI criterion for BE of 95%. The post hoc analyses of the PD data did not change the overall conclusions of the original PD data. The 95% CI for geometric LS mean ratios of the PD parameters of interest were within the pre-specified PD bioequivalence interval of 80 to 125%.
5.3. Evaluator’s overall conclusions on pharmacodynamics5.3.1. General conclusions
Study ROV-RO20-2011-01 in healthy volunteers satisfactorily demonstrated that the reference product (Rovi enoxaparin sodium 100 mg/mL; 100 mg SC) and the test product (Lovenox enoxaparin sodium; 100 mg SC) were bioequivalent, based on the pre-specified statistical analysis of the PD parameters for anti-Xa activity of AUEC0-t, AUEC0-inf, and anti-Xamax. The bioequivalence of the two products was supported by the pre-specified statistical analysis of the PD parameters for anti-IIa activity of AUEC0-t and anti-IIamax, but the data for AUEC0-inf were not included in the statistical analysis due to the small number of subjects in the two treatment groups. The PD bioequivalence analyses of the baseline-adjusted TFPI activity and the RAUEC supported the PD bioequivalence analyses of anti-Xa and anti-IIa activities. The post hoc analyses of the PD parameters using the more stringent criterion of a 95% CI were consistent with the pre-specified analyses using a 90% CI with the 80 to 125% PD BE interval.
5.3.2. Test and reference products
The test enoxaparin product used in Study ROV-RO20-2011-01 was the same product as proposed for registration, while the reference enoxaparin product (Lovenox; USA) was not the relevant Australian approved product (Clexane; Australia). There was no clinical study bridging Lovenox (USA) to Clexane (Australia).
The overarching TGA guidelines relating to the regulation of biosimilar products (Regulation of biosimilar medicines (Version 2.0 December 2015)) allow a reference medicine that has not been registered in Australia to be used for comparability studies if the following criteria are met:
1. The reference medicine must be approved for general marketing by a regulatory authority with similar scientific and regulatory standards as the TGA (for example, the EMA or US FDA).
2. A bridging study must be provided to demonstrate that the comparability studies are relevant to the Australian reference medicine.
The reference product (Lovenox) meets the first criterion as it is marketed in the USA. As regards the second criterion, because there was no clinical bridging study comparing the PD bioequivalence of Lovenox (USA) and Clexane (Australia) it will be a matter for the quality evaluator to determine if the in vitro comparability exercise bridge the two products.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 23 of 62

Therapeutic Goods Administration
5.3.3. Dose
The enoxaparin dose used in Study ROV-RO20-2011-01 was a single 100 mg SC dose administered to healthy volunteers. There were no data for other enoxaparin doses in healthy volunteers. The single 100 mg SC dose used in Study ROV-RO20-2011-01 can be considered to be representative of the higher enoxaparin doses used for treatment, but not of the lower doses used for prophylaxis. Based on limited clinical data, anti-Xa activity appears to be linear over the dosage range 20 to 80 mg in healthy male volunteers.7 The Clexane PI (Australia) indicates that maximum anti-Xa activity was 0.16 IU/L after 20 mg SC and 0.38 IU/L after 40 mg SC. The Lovenox label (USA) states that ‘enoxaparin pharmacokinetics appear to be linear over the recommended dosage ranges’. However, this statement is not found in the Clexane PI (Australia).
In discussing doses for SC and IV studies the TGA approved LMWH biosimilar guideline (EMEA/CHMP/BMWP/118264/2007) states that ‘the selected doses should be in the sensitive part of the dose-response curve and within the (recommended) dose ranges for the different indications’. There is no explicit requirement in the guideline for PK/PD studies at the high and low range of the approved SC doses for clinical use. However, the sponsor is requested to provide a formal justification for not undertaking a single dose PK/PD study in healthy volunteers comparing the proposed product with the Australian reference product (Clexane) at a low dose consistent with the use of enoxaparin for prophylaxis (for example 20 mg) (see Section 12: Clinical questions, below).
5.3.4. Strengths
The sponsor is seeking registration of 100 mg/mL and 150 mg/mL strengths of enoxaparin sodium for the proposed indications. Enoxaparin sodium 100 mg/mL solution for injection pre-filled syringe consists of a sterile solution of enoxaparin sodium in water for injections. The solution is filled in 0.5 mL (for filling volumes of 0.2 mL and 0.4 mL) and in 1.0 mL (for filling volumes of 0.6 mL, 0.8 mL and 1.0 mL). Enoxaparin sodium 150 mg/mL solution for injection pre-filled syringe consists of a sterile solution of enoxaparin sodium in water for injections. The solution is filled in 1.0 mL (for filling volumes of 0.8 mL and 1.0 mL).
No clinical PK/PD studies were submitted comparing the proposed product with the reference product for any proposed strengths other than 100 mg/mL. In pre-submission correspondence, the TGA requested the sponsor to provide a ‘scientific justification to support the extrapolation of the evidence to the registration of the other strengths in this submission not investigated in the PK/PD study’. The sponsor’s justification is provided immediately below.
‘The extrapolation of the evidence to the registration of the 150 mg/mL strength is supported because the concentration of enoxaparin is not relevant for the in vivo properties of the medicinal product, but for the convenience of the patient (less volume of injection).
To the best of the applicant's knowledge, the 150 mg/mL concentration of enoxaparin has never directly been studied in humans, but it is projected to result in anticoagulant activities similar to those of 100 mg/mL and 200 mg/mL concentrations at the same enoxaparin dose.
Please refer to the CHMP answer to Question 6 in the annexed copy of the scientific advice (Annex 1), where it is stated: ‘It is also agreed that separate clinical investigations using both concentrations are not necessary, as the only difference is the amount of API (see also answer to Question 2) and no clinical impact is expected in case the same dose is applied.’
The sponsor’s justification for a biowaiver is considered to be unsatisfactory. It is recommended that the sponsor provide a justification addressing the relevant criteria in the ‘Justification for
7 Frydman A et al. The antithrombotic activity and pharmacokinetics of enoxaparine, a low molecular weight heparin, in humans given single subcutaneous doses of 20 to 80 mg. J Clin Pharmacol. 1988 Jul;28(7):609-18.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 24 of 62

Therapeutic Goods Administration
not submitting biopharmaceutic data (15.9)’ in the ‘Australian Regulatory Guidelines for Prescription Medicines (ARGMP)’.
5.3.5. PD bioequivalence interval
The single dose PK/PD BE Study ROV-RO20-2011-01 used an equivalence interval of 80 to 125% to establish the PD BE of the two enoxaparin products. The TGA approved guidelines (EMEA/CHMP/BMWP/118264/2007) state that equivalence margins for PK/PD BE studies should be ‘pre-specified and appropriately justified’. In pre-submission correspondence, the TGA requested the sponsor to submit a scientific and clinical justification to support the chosen equivalence margins of 80 to 125%. The sponsor’s justification is provided immediately below.
‘In regards to equivalence margins for the clinical study, the TGA-adopted EU ‘Guideline on non-clinical and clinical development of similar biological medicinal products containing low-molecular-weight-heparins’ (EMEA/CHMP/BMWP/118264/2007), states that ‘The study should follow a strict equivalence design where equivalence margins have to be defined a priori and appropriately justified primarily on clinical grounds’.
There is no well-established consensus regarding the equivalence margins, therefore the justification must be based on evidence from previous trials. Nevertheless, the conventional equivalence margin beyond 80 to 125% for the primary parameters is the acceptance interval stated in the TGA adopted EU ‘Guideline on the investigation of bioequivalence’ (CPMP/EWP/QWP/1401/98 Rev. 1/Corr), as it is considered that differences in systemic drug exposure up to 20 % are not clinically significant. This range refers to the inter- and intra-individual biological variability that applies to the administration of any drug.
The symmetrical ± 20 % has to be in the log-transformed space for the bioequivalence tests to be valid. The statistical analysis requires a log-transformation of all concentration-dependent pharmacokinetic measurements; using base 10 or natural logarithms, for clinical, pharmacological and statistical reasons. Logarithmically transformed concentration-dependent PK parameters should be analysed in accordance with European guidelines.
Based on evidence from previous trials, the proportion of venous thromboembolic events was about 15.6%. Retention of at least 50% of the effect size gives a relative risk Delta of 1.53. A more conservative approach of at least 66% retention of the effect size gives a relative risk Delta of 1.33. Taking the Delta of 1.3, the necessary number of subjects to achieve a power of 80% would be 1,260 per group (total 2,520), or 1,520 per group (total 3,040) if we chose the more conservative 95% CI lower limit.
In the submitted bioequivalence Study ROV-RO20-2011-01, an analysis of variance with fixed effects for sequence, period, and treatment, and random effect for a subject nested within sequence was performed on the natural logarithms of AUEC0-inf, AUEC0-t, anti-Xamax, anti-IIamax, and TFPImax to assess the differences between the test and reference treatments.
The geometric mean ratio and 90% CI (and 95% CI in post hoc analysis in response to the CHMP scientific advice) for AUEC0-inf, AUEC0-t, and peak effect (anti-Xamax, anti-IIamax, and TFPImax) of the 2 treatments were calculated using the antilog of the mean difference and 90% CI (and 95% CI in post hoc analysis) of the log-transformed values.
In addition, RAUEC (ratio of AUEC0-t of anti-Xa to anti-IIa) was added to the statistical analysis. Bioequivalence was concluded if the 95% CI of the ratio of the geometric least squares (LS) means between the test treatment (enoxaparin 100 mg/mL, 100-mg SC injection manufactured by Rovi) and the reference treatment (Lovenox 100 mg/mL, 100-mg SC injection manufactured by Sanofi-Aventis) for AUEC0-inf, AUEC0-t, and anti-Xamax of anti-Xa activity were completely within the standard 80% to 125% BE interval and AUEC0-t and anti-IIamax of anti-IIa activity were completely within the standard 80% to 125% BE interval.
In addition, baseline-adjusted TFPI activity and RAUEC were considered as supportive secondary PD parameters for the biosimilarity assessment and therefore were also evaluated if the 95% CI
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 25 of 62

Therapeutic Goods Administration
of the ratio of the geometric least squares means between the test treatment and the reference treatment were completely within the 80% to 125% interval.
Bioequivalence was concluded considering that the 95% CI of the ratio of the geometric least squares means between the test treatment (Enoxaparin Sodium ROVI 100 mg/mL, 100 mg SC injection manufactured by ROVI) and the reference treatment (Lovenox 100 mg/mL, 100 mg SC injection manufactured by Sanofi-Aventis) for AUEC0-inf, AUEC0-t, and anti-Xamax of anti-Xa activity were completely within the standard 80% to 125% BE interval as per the ‘Guideline on the investigation of bioequivalence: London, 20 January 2010 (CPMP/EWP/QWP/1401/98 Rev. 1)’.
The sponsor’s justification is unsatisfactory. The TGA adopted bioequivalence guidelines referred to by the sponsor (CPMP/EWP/QWP/1401/98 Rev. 1/Corr) apply to conventional bioequivalence studies comparing plasma concentration exposures of chemical entities. The bioequivalence guidelines explicitly state that their ‘scope is limited to chemical entities. Recommendation for the comparison of biologics to reference medicinal products can be found in guidelines on similar biological medicinal products’. Therefore, the guidelines are considered not relevant to the investigation of the bioequivalence of enoxaparin products based on pharmacodynamic endpoints. It is considered that that the sponsor has not satisfactorily justified the use of the 80 to 125% interval to test the pharmacodynamic bioequivalence of the two enoxaparin products.
In the scientific advice provided by the CHMP to the sponsor the 95% CI was ‘strongly recommended as being relevant for demonstration of PD equivalence. This takes into consideration that in several instances the sensitivity of PD endpoints to detect differences is lower as compared to PK comparisons. In addition, a high robustness of the PD results is expected, when a waiver of a clinical Phase II study and approval of several clinical indications are to be based on a PD comparison. An acceptance range of 80 to 125% might be agreeable, but should further be clinically justified’. It is unknown whether the sponsor submitted further clinical justification to the CHMP relating to the 80 to 125% acceptance range.
The FDA ‘Draft Guidance on Enoxaparin Sodium’ specifies that PD equivalence is based on anti-Xa activity, with equivalence being established if the 90% CIs for the geometric mean test/reference ratios of the AUEC and anti-Xamax parameters fall within the BE limits of 80 to 125%. However, no justification is provided in the FDA document supporting the specified PD equivalence criteria.
5.3.6. Correlation between surrogate PD parameters and clinical outcomes
In pre-submission correspondence, the TGA requested the sponsor to submit data or a scientific justification to support the correlation between surrogate PD parameters (anti-Xa and anti-IIa) and clinical outcomes in patients. The sponsor’s justification is provided below.
‘The correlation of anti-Xa activity and clinical outcomes has been thoroughly researched since the beginning of use of enoxaparin in the clinical setting; a significant relationship between anti-Xa activity and clinical outcome (thrombosis and bleeding) has been observed in several studies.
Levine et al., (1989) confirmed the relationship between the anti-FXa levels and thrombosis and bleeding in patients undergoing total hip replacement (n = 163) who were given prophylaxis treatment once daily with enoxaparin.8 The incidence of wound haematoma was 5.3% when the maximum anti-FXa level was less than or equal to 0.2 IU/mL, but increased to 24.5% when the anti-FXa level exceeded 0.2 IU/mL (p = 0.0008). The incidence of postoperative thrombosis was low (6.3%) if the minimum anti-FXa level exceeded 0.1 IU/mL, but increased to 14.6% when less than or equal to 0.1 IU/mL, and to 18.8% if the
8 Levine M et al. The relationship between anti-factor Xa level and clinical outcome in patients receiving enoxaparin low molecular weight heparin to prevent deep vein thrombosis after hip replacement. Thromb Haemost 1989;6 2 (3):940-4.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 26 of 62

Therapeutic Goods Administration
anti-FXa level was less than or equal to 0.05 IU/mL. Regression analysis revealed that there was a statistically significant relationship between anti-FXa level and wound haematoma (p = 0.002) and anti-FXa level and thrombosis (p = 0.03).
Montalescot et al., (2004) found a correlation between anti-Xa activity and efficacy outcome; the patients were treated with enoxaparin for acute coronary syndrome, and those with anti-Xa values below 0.5 IU/mL had 3 fold increase in mortality versus patients with anti-Xa values within the target range (0.5 to 1.2 IU/mL).9
Desjardins et al., (2004) investigated the relationship between plasma coagulation parameters thromboprophylaxis, and incidence of venous thromboembolism (VTE) in a controlled, multicentre, double blind, randomised study; 224 acutely ill medical patients were administered either 20 or 40 mg of enoxaparin.10 The correlation of the higher anti-Xa activity reached with the 40 mg dose of enoxaparin and the low incidence of VTE confirmed the relationship of anti-Xa and thromboprophylaxis.
Also, Sagedal et al., (1999) demonstrated the relationship of anti-Xa plasma levels and anticoagulant efficacy for thromboprophylaxis during haemodialysis, stressing that an anti-Xa above 0.4 IU/mL is correlated to anticoagulant effect.11
Furthermore, anti-Xa activity correlates not only with the antithrombotic efficacy but also with the risk of bleeding complications.
Barras et al., (2009) developed a population PK/PD model using data obtained from patients treated for pulmonary embolism, DVT, acute coronary syndrome or atrial fibrillation who were allocated to either a dose-individualised or conventional dosing arm in a prospective randomised controlled trial (n = 118); anti-Xa data was collated, and the risk for bleeding and bruising modelled; the authors concluded that the occurrence and severity of bleeding are a function of cumulative enoxaparin AUC.12
In addition, Lim et al., (2006) have compared the risk of major bleeding and anti-Xa levels in patients receiving LMWHs who had severe renal insufficiency (creatinine clearance (CrCI) approximately 30 mL/min) with those without severe renal impairment (CrCI > 30 mL/min).13 In 12 studies involving 4,971 patients under LMWH therapy, the odds ratio for major bleeding was 2.25 (95% CI, 1.19 to 4.27) in patients with a CrCI approximately 30 mL/min compared with those with a CrCI > 30 mL/min. Enoxaparin at a therapeutic dose was associated with a further increase in major bleeding in patients with a CrCI approximately 30 mL/min (8.3% versus 2.4%; odds ratio, 3.88; 95% CI, 1.78 to 8.45), but this was not observed when the dosage of enoxaparin was empirically reduced (0.9% versus 1.9%; odds ratio, 0.58; 95% CI, 0.09 to 3.78). Patients with a CrCI of 30 mL/min or less who are treated with standard therapeutic doses of enoxaparin have elevated levels of anti-Xa and an increased risk for major bleeding.
A population PK/PD model built by Sanofi (former Aventis) using data of clinical trials on enoxaparin in acute myocardial infarction considered the AUC of anti-Xa plasma levels as exposure of enoxaparin and bleeding events as a binary variable; a logistic regression
9 Montalescot G et al. Anti-Xa activity relates to survival and efficacy in unselected acute coronary syndrome patients treated with enoxaparin. Circulation 2004 Jul 27;110(4):392-8.10 Desjardins L, et al. Correlation of plasma coagulation parameters with thromboprophylaxis, patient characteristics, and outcome in the MEDENOX study. Arch Pathol Lab Med 2004; 128(5): 519-26.11 Sagedal et al. A single dose of dalteparin effectively prevents clotting during haemodialysis. Nephrol Dial Transplant 1999;14:1943-7.12 Barras M et al. Modelling the occurrence and severity of enoxaparin induced bleeding and bruising events. Br J Clin Pharmacol 2009;68:700-11.13 Lim W et al. Meta-analysis: Low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144: 673-84.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 27 of 62

Therapeutic Goods Administration
analysis showed that a higher exposure of enoxaparin yields a higher incidence of all bleeding and major bleeding events.14
Enoxaparin showed their anticoagulant effect measuring the inhibitory effect of enoxaparin upon thrombin generation using the measurement of anti Xa and anti-IIa activity.
The pharmacokinetic data of anti-IIa activity for enoxaparin need to be submitted as supportive evidence of comparable therapeutic outcome’ (Gadiko et al., 2012).15
Overall, it is considered that the sponsor’s justification has failed to establish a clear correlation between surrogate PD parameters (anti-Xa and anti-Xa levels) and clinical outcomes. In Barras (2013), the author comments that monitoring anti-Xa assays in patients being treated with LMWH is controversial ‘as there is a poorly defined therapeutic range in different clinical settings and with different dosing regimens’.12 He also states that the ‘evidence for all therapeutic ranges originates from studies in arterial disease. Few data exist that define a separate range for venous disease’. Similarly, in a review of the anti-Xa range for LMWHs it was stated ‘that while the AFXa ranges for therapeutic levels of [low molecular weight heparins] are relatively well defined in the literature, prophylactic ranges are much less clear, thus making it difficult to interpret current research data’ (Wei and Ward, 2015).16 Comments on the relevant studies referred to by the sponsor in support of its justification are provided below.
In Levine et al. (1989), the authors conclude that the findings suggest that when enoxaparin is administered SC once-daily, the 12 hour anti-Xa level should not exceed 0.2 IU/mL to minimise bleeding and levels greater than 0.05 IU/L should be obtained to maximise efficacy (that is, prevent post-operative thrombosis).8 The study is considered to provide support for an association between anti-Xa levels and the post-operative incidence of wound haematoma and the post-operative incidence of thrombosis in patients receiving prophylaxis with enoxaparin following total hip replacement.
In Montalescot et al., (2004) 803 patients with unstable angina/non-ST-segment elevation myocardial infarction (UA/NSTEMI) were treated with enoxaparin 1 mg/kg twice daily and followed up for 30 days.9 The 30-day mortality rate was significantly associated with low anti-Xa levels (< 0.5 IU/mL), with a > 3-fold increase in mortality compared to the patients with anti-Xa levels in the target range of 0.5 to 1.2 IU/mL (p = 0.004). Multivariate analysis revealed low anti-Xa activity as an independent predictor of 30 day mortality at least as strong as age, left ventricular function, and renal function. In contrast, anti-Xa activity did not predict major bleeding complications within the range of anti-Xa levels observed in this study, with the mean SEM anti-Xa levels being 0.91 0.01 IU/mL for patients without major bleedings and 0.83 0.01 IU/mL for patients with major bleedings (p > 0.05).
In Desjardins et al., (2004) 224 acutely ill medical patients were randomised to receive enoxaparin 20 mg (n = 73), 40 mg (n = 83) or placebo (n = 68) by SC injection once daily for 10 4 days.10 The objective of the study was to investigate the relationship between plasma coagulation parameters and patient characteristics, including renal function, thromboprophylaxis, and incidence of venous thromboembolism (VTE) in the Medenox study population (that is acutely ill medical patients treated with enoxaparin for prophylaxis of VTE). In this study, there was no significant difference in anti-Xa and anti-IIa activities in patients with or without VTE or in patients with and without bleeding. Despite the median anti-Xa levels in the enoxaparin 40 mg group being almost twice as high as in the enoxaparin 20 mg group, the
14 FDA Clinical Pharmacology Biopharmaceutics Review. Lovenox (Enoxaparin sodium) Injection Sanofi-Aventis U.S. Inc. NDA: 022138 Approval Date: 5/16/200715 Gadiko C et al. Pharmacokinetic parameters to be evaluated for selected Low Molecular Weight Heparins in Bioequivalence Studies. Int J Pharm Sci Res. 3(11); 4065-4072.16 Wei M and Ward S. The anti-Factor Xa range for low molecular weight heparin thromboprophylaxis. Hematol Rep. 2015 Nov 23; 7 (4): 5844.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 28 of 62

Therapeutic Goods Administration
authors comment that the large overlap of individual values among groups precludes the use of anti-Xa activity as an indicator of clinical antithrombotic efficacy in medical patients. However, the authors state that a correlation between anti-Xa activities and VTE cannot be excluded.
The sponsor states that the ‘correlation of higher anti-Xa activity reached with the 40 mg dose of enoxaparin and the lower incidence of VTE confirmed the relationship of anti-Xa and thromboprophylaxis’. However, this statement is not supported by the study data or the authors’ conclusions regarding the data. Examination of a figure (see Figure 4, below) from the study shows higher median anti-Xa levels at Day 10 in the enoxaparin 40 mg group compared to the enoxaparin 20 mg group, but the anti-Xa levels associated with both doses are overlapping.
Figure 4. Distribution of anti-Xa activity according to thromboprophylaxis group
From Desjardins L, et al. Correlation of plasma coagulation parameters with thromboprophylaxis, patient characteristics, and outcome in the MEDENOX study. Arch Pathol Lab Med 2004; 128(5): 519-26. Note: Shaded boxes, interquartile range; thick line, median.
In Sagedal et al., (1999) a single dose of dalteparin effectively inhibited coagulation in the bubble trap or dialyser in 84 dialysis sessions in 12 patients on chronic haemodialysis.11 Anti-Xa activity above 0.4 IU/mL after 4 hours of dialysis inhibited clotting during dialysis. This was normally achieved with an initial bolus dose of 70 IU/kg. Over the dalteparin dose range 25 to 90 IU/kg inter-subject variability was marked for each individual dose. For example, at a dose of 50 mg IU/kg anti-Xa levels ranged from approximately 0.15 IU/mL to 0.7 IU/mL. In addition, there was significant overlapping of anti-Xa levels over the dalteparin dose range. Therefore, the use of anti-Xa activity as an indicator of clotting during haemodialysis following dalteparin is considered to be unreliable.
In Barras et al., (2009) a population PK/PD model using nonlinear mixed effects techniques was developed to describe the occurrence and severity of bleeding or bruising as a function of enoxaparin exposure.12 PK and PD data were collected during a prospective randomised control trial from subjects (n = 118) treated for pulmonary embolism, deep vein thrombosis, acute coronary syndrome or atrial fibrillation who had been allocated to either a dose-individualised or conventional dosing arm. A total of 103 subjects had a bleeding and bruising assessment beyond baseline and were used to develop the PD model. Of the 103 subjects included in the PD analysis, 27 had a major event, 40 had a minor event and 36 had no event. Anti-Xa concentrations were sampled using a sparse design and the size, location and type of bruising and bleeding event during enoxaparin therapy were collected daily. A total of 349 anti-Xa concentrations was collected during the study with a mean of 3 (range: 1, 4) samples per subject, with sampling primarily occurring in the first 48 hours of therapy (93% of concentrations).
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 29 of 62

Therapeutic Goods Administration
The study found a two-compartment model with first-order input and linear elimination was the best structural model to fit the data. Between-subject-variability (BSV) was included on clearance (CL), central volume of distribution (Vc) and absorption rate (Ka), with Vc and CL allowed to co-vary. Residual variability was described by an additive error model. The authors stated that their study demonstrated that increasing cumulative area under the concentration time curve (cAUC (hr x IU/mL)) and subject age best describe the occurrence and severity of bleeding and bruising events. The relationship was described graphically in the publication (see Figure 5, below).
Figure 5. Plot of empirical (right column) and model-predicted (left column) probability (P) of event severity versus cumulative area under the concentration time curve at median age for bleeding events
From Barras M et al. Modelling the occurrence and severity of enoxaparin induced bleeding and bruising events. Br J Clin Pharmacol 2009;68:700-11. Note: N = number of subjects in each dosage group; white segments = no bleeding events; gray segments = minor bleeding events; and black segments = major bleeding events.
Barras et al., (2009) commented that data from their study were obtained from subjects administered treatment doses for a mean SD duration of therapy of 3.5 2.3 days and, therefore, the current model can only be used in circumstances that are similar to the study. In addition, the authors identified the following limitations to their study. First, traditional binary data analyses require extensive data and a total of 63 events may be considered small. To assess confidence in the results the authors bootstrapped the PD data and added 90% CIs to the model predictions. The corresponding plots showed that both cAUC and age are good predictors of bleeding, with age being a stronger predictor than cAUC. Second, independent data would be necessary to evaluate this model, but such data were not available at the time of the study. Third, therapeutic failure such as re-infarction was not modelled. Overall, this is considered to be a good quality population PK study. However, the authors’ comments relating to the limitations of the study should be noted.
In Lim et al., (2006) a meta-analysis demonstrated that non-dialysis dependent patients with creatinine clearance levels ≤ 30 mL/minute treated with standard doses of enoxaparin had elevated levels of anti-Xa and an increased risk of major bleeding.13 The study found that standard weight-adjusted enoxaparin dosage was associated with a 2 to 3 fold increased risk of major bleeding events in patients with severe renal insufficiency (creatinine clearance ≤ 30 mL/min) versus patients without renal insufficiency. The authors concluded that weight adjusted doses of LMWH may reduce the risk for bleeding events in patients with a creatinine clearance ≤ 30 mL/min, but this needs to be further evaluated. This was a good quality study, but the results are limited to patients with creatinine clearance levels ≤ 30 mL/min. The authors’ commented that the findings need further evaluation.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 30 of 62

Therapeutic Goods Administration
Perusal of the FDA document referred to by the sponsor suggests that the relevant population PK/PD study is DMPK/FR 2409. This study used data from the TIMI11A trial in patients with unstable angina or non-Q wave myocardial infarction to model the relationship between exposure to enoxaparin and the incidence of major and all haemorrhagic events. Major bleeding was defined as clinically overt haemorrhage resulting in a fall of ≥ 3 g/dL in haemoglobin or a retroperitoneal, intracranial, or intraocular haemorrhage. All other episodes of bleeding were considered minor haemorrhages. Patients were assigned a binary variable (0 or 1) according to whether they experienced haemorrhagic events while on treatment. A logistic regression was conducted to relate the probability of experiencing haemorrhage to drug exposure or other covariates. The logistic regression results showed that higher exposure to enoxaparin (AUC anti-Xa IU x h/mL) resulted in a higher incidence of major and all haemorrhagic events.14
5.3.7. IV route of administration
In Australia, Clexane is approved for administration as a single IV bolus dose of 30 mg (plus a 1 mg/kg SC dose), in conjunction with a fibrinolytic, to initiate treatment of acute ST-segment elevation MI, and as an IV 0.3 mg/kg bolus dose for patients being managed with percutaneous coronary intervention if the last Clexane dose was given more than 8 hours before balloon inflation. Clexane is also approved in Australia for patients undergoing repeated sessions of haemodialysis to prevent thrombosis in the extracorporeal blood circuit by injection of 1 mg/kg into the arterial line of the dialysis circuit at the start of the session. The relevant TGA adopted EU guidelines (EMEA/CHMP/BMWP/118264/2007) indicate that if the originator enoxaparin product is also licensed for the intravenous of intra-arterial route then an additional PK/PD clinical study should be performed by the IV route.
In pre-submission correspondence, the TGA requested the sponsor to submit an IV study to support the comparability of the PK/PD properties of the proposed product with the reference product or submit a scientific justification for the lack of an IV study. The submission did not include an IV PK/PD comparability study, but the sponsor provided a justification for not doing so. The sponsor’s justification is provided below:
‘Early research into comparative PK of enoxaparin compared to unfractionated heparin (UFH) after IV and SC administration of 40 mg indicated that the mean absolute bioavailability, as measured by SC/IV ratio of anti-Xa activity, is 91% and the anti-Xa half life was exactly the same for both routes of administration (275 min).17 This is further supported with information in the Australian Product Information for Clexane, which states that ‘After injection of Clexane by the subcutaneous route (SC), the product is rapidly and completely absorbed. The absolute bioavailability is over 90%’. The comparative PK study (Bara et al., 1985) also demonstrates that the PK parameters related to the elimination behaviour of enoxaparin administered by IV or SC route do not change.
The use of a 100 mg dose in the Lovenox versus proposed product enoxaparin BE trial (Study ROV -RO20-2011-01) will allow [sufficient PD values to be obtained] in order to calculate parameters related to the terminal elimination, and consequently to make valid conclusions on biosimilarity of [the] proposed product and the reference LMWH for IV administration.
Therefore, it is not considered necessary to develop any specific clinical program to support the IV route of administration as the SC administration provides more than 90% bioavailability and extrapolation from this route is feasible.
The CHMP agreed with this approach in the scientific advice received for the proposed product, which was also supported by BfArM. Specifically, the CHMP scientific advice states: ‘Regarding approval of also the intravenous administration route, subcutaneous PD data might indeed suffice as the more sensitive route to demonstrate PD equivalence. If PD
17 Bara L et al., Comparative pharmacokinetics of a low molecular weight heparin (PK 10 169) and unfractionated heparin after intravenous and subcutaneous administration. Thromb Res. 1985 Sep 1;39(5):631-6.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 31 of 62

Therapeutic Goods Administration
biosimilarity is concluded via the subcutaneous route, which is subject to a more complex pharmacokinetics intravenous administration, a relevant difference via direct administration is not expected’.
The key issue is whether PD bioequivalence following IV administration can be predicted from the PD bioequivalence data following SC administration obtained from Study ROV-RO20-2011-01. In essence, the sponsor considers that it is not necessary to develop a specific clinical program to support the IV route as SC administration provides more than 90% bioavailability for anti-Xa and the terminal half life of anti-Xa is the same following both SC and IV administration. However, while review of the data from Bara et al., (1985) in healthy subjects aged 21 to 29 years confirms the values given by the sponsor for anti-Xa activity for absolute bioavailability (91%) and terminal half life (275 minutes after IV and SC administration), the data from the study show that the absolute bioavailability of anti-IIa activity following enoxaparin 40 mg (SC/IV) is only 19%.17
In a later study, the absolute bioavailability for anti-Xa following a 1.5 mg/kg dose of enoxaparin (SC and IV) in non-obese subjects was also higher than the absolute value for anti-IIa (106% versus 85%, respectively) (Sanderink et al., 2002). This study also showed that the terminal half life of anti-Xa in non-obese subjects was similar following SC administration on Days 1 and 4 (4.85 and 4.60 hours, respectively), while the terminal half life of anti-IIa was longer following SC than IV administration (2.75 versus 1.46 hours, respectively). The lower absolute bioavailability of anti-IIa compared to anti-Xa suggests that there is some selectivity in absorption or pre-systemic biotransformation of those sections of the enoxaparin molecule responsible for anti-Xa and anti-IIa activities.
It is considered that PD bioequivalence of the test and reference enoxaparin following IV administration based on anti-IIa activity cannot be accurately predicted from the SC data. Therefore, it is considered that an IV PK/PD study is required to adequately characterise the PD bioequivalence of the test and reference products following IV administration.
5.3.8. Other matters
The submission did not include a bioequivalence study in patients. This is acceptable as the guideline (EMEA/CHMP/BMWP/118264/2007) has no requirement for bioequivalence studies in patients. The submission did not include a repeat dose bioequivalence study. This is acceptable as the guideline (EMEA/CHMP/BMWP/118264/2007) does not specify repeat dose bioequivalence studies in either healthy volunteers or patients. However, it is interesting to note that the International Society on Thrombosis and Haemostasis (ISTH) statement on biosimilar LMWHs recommends that ‘Phase I clinical trials in human volunteers should be performed using a low prophylactic and a high therapeutic dose over 5 to 7 days each’.18
6. Dosage selection for the pivotal studiesNo pivotal Phase III efficacy and safety studies were submitted.
7. Clinical efficacyThe TGA adopted LMWH biosimilar guideline (EMEA/CHMP/BMWP/18264/2007) states:
‘Since a clear correlation between surrogate PD parameters (anti FXa or anti FIIa) and clinical outcome has not been established, a similar biological medicinal product containing LMWH should show equivalent efficacy and safety to a reference product
18 Harenberg et al. Update of the recommendations on biosimilar low-molecular-weight heparins from the Scientific Subcommittee on Control of Anticoagulation of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2013; 11: 1425-5.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 32 of 62

Therapeutic Goods Administration
approved in the EU. This therapeutic equivalence should be demonstrated in at least one adequately powered, randomised, double-blind, parallel group clinical trial. In theory, this could be done either in the setting of prevention of venous or arterial thromboembolism, or in the setting of treatment of venous thromboembolism. However, the most sensitive model to detect potential differences in efficacy between the new LMWH and the reference product should be selected’.
The guideline recommends demonstration of efficacy in the prevention of venous thromboembolism (VTE) in patients undergoing surgery with a high VTE risk. The guideline states:
‘Demonstration of comparable efficacy and safety in surgical patients at high risk for VTE as recommended may allow extrapolation to other indications of the reference medicinal product if appropriately justified by the applicant’.
In pre-submission correspondence, the TGA requested the sponsor to provide a ‘therapeutic equivalence study (adequately powered, randomised, double blind, parallel group clinical trial with pre-specified equivalence margins) in the most sensitive model to detect potential differences in efficacy between the proposed enoxaparin product and the reference product. Preferably, the trial should be in patients in the setting of prevention of venous thromboembolism in patients undergoing major orthopaedic surgery with high VTE risk such as hip surgery’. This request is consistent with the TGA adopted LMWH biosimilar guideline (EMEA/CHMP/BMWP/18264/2007).
The sponsor did not comply with the TGA’s request for a therapeutic equivalence study. No clinical efficacy and safety studies were submitted. The sponsor’s response to the TGA’s request is provided below.
‘As stated in the TGA-adopted, EU Guideline on similar biological medicinal products (CHMP/437/04 Rev 1):
‘In specific circumstances, a confirmatory clinical trial may not be necessary. This requires that similar efficacy and safety can clearly be deduced from the similarity of physicochemical characteristics, biological activity/potency, and PK and/or PD profiles of the biosimilar and the reference product. In addition, it requires that the impurity profile and the nature of excipients of the biosimilar itself do not give rise to concern. It is recommended to discuss such simplified approaches with Regulatory Authorities’.
In this case, the manufacturer of the proposed product enoxaparin sodium has discussed with EMA the necessity of a therapeutic equivalence study and has been advised in the scientific advice that, ‘The CHMP believes that it could indeed be acceptable to waive a pre-approval Phase III efficacy/safety trial’.
Because the analytical tools for the characterisation of complex molecules such as enoxaparin have greatly improved since the approval of the TGA-adopted EU guideline on Low Molecular Weight Heparins (EMEA/CHMP/BMWP/118264/2007), deriving data from only PK/PD trials should be considered acceptable provided that the analysis of important molecule characteristics does not reveal differences which would contradict an assumption of biosimilarity and biosimilarity can convincingly be established based on nonclinical studies and clinical PD studies as well.
The sponsor comments that the manufacturer discussed with the EMA whether a therapeutic equivalence study was necessary, and received advice from the CHMP that it might be possible to waive the requirement for a pre-approval Phase III efficacy/safety trial. In the opinion of the manufacturer, a confirmatory clinical efficacy and safety study would not provide any additional data to support similarity to that already obtained from the ‘comprehensive physiochemical characterisation, the nonclinical comparability studies and the Phase I healthy volunteer study’. The manufacturer comments that, ‘preliminary analysis of the biosimilar version of enoxaparin
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 33 of 62

Therapeutic Goods Administration
sodium Rovi has showed similarity to the original drugs, Clexane and Lovenox. All of them demonstrate sameness: 1) in weight-average molecular weight and weight distribution; 2) in 1H NMR spectra and signals areas; 3) in HSQC NMR spectra and monosaccharide compositional analysis by HSQC NMR; and 4) in values of in vitro anti-FXa and anti-FIIa activities both drug substance and drug product’. In further support of a waiver from the CHMP, the manufacturer referred to the outcome of the study in healthy volunteers demonstrating bioequivalence of the two enoxaparin products based on PD outcomes (Study ROV-RO20-2011-01), and the correlation between anti-Xa activity and clinical outcomes established in the literature.
The manufacturer acknowledged to the CHMP that a confirmatory therapeutic clinical equivalence trial ‘could potentially overcome some of the uncertainties that enoxaparin sodium Rovi is biosimilar to Clexane’. However, the manufacturer outlined the difficulties in conducting a suitably powered therapeutic equivalence study comparing the incidence of venous thromboembolic events between the two enoxaparin products. These included, no well established consensus regarding the equivalence margin, large number of patients (1,260) required to adequately power the study based on a relative risk delta of 1.33, and the use of invasive venography (‘gold standard’) to detect outcomes of proximal and distal DVT. The manufacturer stated that a study of the required size would take years to recruit, ‘especially when there is a lack of interest by investigators to take part in biosimilar trials’. The sponsor commented that a multinational, multisite study of ‘such long duration…will be a real challenge for any sponsor. Moreover, given the variability of standard of care and methods of assessment across countries which could also change of the years, data integrity could be compromised. Therefore, one may question the scientific value of such a study and whether it is ethical to conduct such a study’.
The sponsor’s comments relating to the difficulties of undertaking a suitable therapeutic equivalence study are unconvincing. The challenges in undertaking an appropriately designed study are not insurmountable. As regards the sponsor’s comments regarding the scientific value and ethics of conducting a therapeutic equivalence study, it is considered that one would have to be certain that, based on the totality of the submitted data, the two products were biosimilar in order to scientifically and ethically justify not undertaking such a study. The sponsor has not demonstrated a clear correlation between the surrogate PD markers (anti-Xa and anti-IIa) and clinical outcomes. Therefore, it is considered that a clinical study is required to establish the therapeutic equivalence of the two products and to provide reassurance that the safety data are comparable.
In its response to the manufacturer, the CHMP ‘acknowledged that analytical tools for characterisation of complex molecules such as enoxaparin have greatly improved and stated that it believed ‘that it could indeed be acceptable to waive a pre-approval Phase III efficacy/safety trial’. However, ‘such a scenario would only be acceptable if: 1) comparisons on ‘Chemistry, Manufacturing and Controls’ (CMC) level are performed with state-of-the-art and analysis of important molecule characteristics does not reveal differences which would contradict an assumption of biosimilarity; and 2) biosimilarity can convincingly be established based on nonclinical studies and clinical PD studies as well. In this context, it has to be clearly stated that the recommendation to conduct a Phase III trial is not meant as a 'rescue' of failure to show similarity in early development phases’.
The problem with the CHMP’s criteria to waive the requirement to submit a Phase III efficacy and safety study relates to the previously discussed lack of a demonstrated correlation between the PD parameters and clinical outcomes. Therefore, it is considered that even if the CHMP criteria were satisfied this would not remove the requirement for a Phase III clinical study to be submitted demonstrating the therapeutic equivalence of the 2 enoxaparin sodium formulations.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 34 of 62

Therapeutic Goods Administration
8. Clinical safety
8.1. Studies providing evaluable safety dataNo Phase III clinical efficacy and safety studies were submitted. The only clinical safety data in the submission related to the single dose bioequivalence study in healthy volunteers. The data from this study are summarised below.
8.1.1. Study ROV-R020-2011-01
All 42 subjects (healthy volunteers) in this study received a single 100 mg SC dose of the proposed enoxaparin product (Rovi) and the reference product (Lovenox, USA).
8.1.1.1. Brief summary of adverse events
12 of the 42 subjects (28.6%) reported 24 TEAEs:
4 subjects reported 6 TEAEs after receiving Rovi and no TEAEs after receiving Lovenox;
4 subjects reported 4 TEAEs after receiving Lovenox and no TEAEs after receiving Rovi; and
4 subjects reported 14 TEAEs after both treatments, including 6 TEAEs after Rovi and 8 TEAEs after Lovenox.
In summary, the same number of subjects (n = 8, 19%) reported 12 TEAEs after receiving Rovi and Lovenox. All TEAEs were mild in severity and resolved by the end of the study. The TEAEs reported in the safety population are summarised below in Table 10. The addendum to the CSR included a post hoc statistical analysis of the TEAEs for the two treatments. This analysis showed no statistically significant differences between the two treatment arms for any of the events.
Table 10. Study ROV-RO20-2011-01 Treatment emergent adverse events, safety population
System Organ Class, Preferred Term; Number of patients, (%)
Enoxaparin (Treatment A) (N = 42)
Lovenox (Treatment B) (N = 42)
Overall (N = 42)
Total number of TEAEs 12 12 24
Number of subjects with at least 1 TEAE 8 (19.0) 8 (19.0) 12 (28.6)
Nervous system disorders 3 (7.1) 4 (9.5) 7 (16.7)
Headache 2 (4.8) 2 (4.8) 4 (9.5)
Dizziness 1 (2.4) 1 (2.4) 2 (4.8)
Disturbance in attention 0 1 (2.4) 1 (2.4)
General disorders and administration site conditions
4 (9.5) 4 (9.5) 6 (14.3)
Injection site haematoma 2 (4.8) 3 (7.1) 4 (9.5)
Fatigue 0 1 (2.4) 1 (2.4)
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 35 of 62

Therapeutic Goods Administration
System Organ Class, Preferred Term; Number of patients, (%)
Enoxaparin (Treatment A) (N = 42)
Lovenox (Treatment B) (N = 42)
Overall (N = 42)
Injection site erythema 1 (2.4) 0 1 (2.4)
Injection site irritation 0 1 (2.4) 1 (2.4)
Injection site swelling 0 1 (2.4) 1 (2.4)
Thirst 1 (2.4) 0 1 (2.4)
Gastrointestinal disorders 4 (9.5) 0 4 (9.5)
Nausea 2 (4.8) 0 2 (4.8)
Dry mouth 1 (2.4) 0 1 (2.4)
Sensitivity of teeth 1 (2.4) 0 1 (2.4)
Immune system disorders 0 1 (2.4) 1 (2.4)
Seasonal allergy 0 1 (2.4) 1 (2.4)
Injury, poisoning, and procedural complications 1 (2.4) 0 1 (2.4)
Arthropod bite 1 (2.4) 0 1 (2.4)
Renal and urinary disorders 0 1 (2.4) 1 (2.4)
Dysuria 0 1 (2.4) (2.4)
The total number of adverse events counted all TEAEs for subjects in the safety population. Subjects could have more than 1 TEAE per system organ class or preferred term. At each level of subject summarisation, a subject was counted once if the subject reported 1 or more events. Treatment-emergent AEs were summarised by treatment at onset of the event. Adverse events were coded using the Medical Dictionary for Regulatory Activities, Version 15.0. Percentages were based on the number of subjects in the safety population within each treatment and overall. Treatment A = Enoxaparin (100 mg/mL) 100 mg subcutaneous injection manufactured by Rovi, Spain; Treatment B = Lovenox (100 mg/mL) 100-mg subcutaneous injection manufactured by Sanofi-aventis, USA.
Overall, headache and injection site haematoma were the most commonly reported TEAEs (9.5% each). Headache was reported by the same number of subjects after receiving Rovi and Lovenox (2 subjects, 4.8% each). Injection site haematoma was reported by 2 subjects (4.8%) after receiving Rovi and 3 subjects (7.1%) after receiving Lovenox. Injection site irritation and swelling were each reported by 1 subject (2.4%) after receiving Lovenox and no subjects after receiving Rovi. Injection site erythema was reported by 1 subject (2.4%) after receiving Rovi and no subjects after receiving Lovenox.
Overall, 8 of 42 subjects (19.0%) reported 17 TEAEs related to study drug. The same number of subjects reported TEAEs related to study drug after receiving enoxaparin manufactured by Rovi and Lovenox (5 subjects, 11.9% each; 8 and 9 TEAEs, respectively). All TEAEs of headache and injection site haematoma, injection site irritation, injection site swelling, and injection site erythema were considered to be related to study drug.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 36 of 62

Therapeutic Goods Administration
Death, serious adverse events (SAE), and other significant adverse events
There were no deaths or SAEs reported in the study, and no subjects discontinued the study due to AEs.
8.1.1.2. Clinical laboratory data
The standard range of clinical laboratory tests was undertaken. Clinical laboratory testing (haematology, coagulation, serum chemistry, and urinalysis) was performed at screening, Day -1 (Period 1 only), and Day 3 (Period 2 only) and at the follow-up visit (Day 7 of Period 2 only).
Overall, mean haematology, serum chemistry, and urinalysis results were within the reference ranges at all time-points assessed and the mean values observed after dosing were similar to those observed at Baseline. There were no treatment-related trends observed in the mean clinical laboratory results over time. No comparative clinical laboratory data for the two formulations could be identified in the submitted data.
Haematology parameters in which shifts from normal at Baseline were observed in 2 or more subjects were:
Basophils/leukocytes: 2 subjects (4.8%) shifted from normal at Baseline to high at Period 2 Day 3/early termination.
Haematocrit: 3 subjects (7.1%) shifted from normal at Baseline to low at Period 2 Day 3/early termination; 4 subjects (9.5%) shifted from normal at Baseline to low at Follow-up.
Haemoglobin: 4 subjects (9.5%) shifted from normal at Baseline to low at Period 2 Day 3/early termination; 4 subjects (9.5%) shifted from normal at Baseline to low at Follow-up.
Serum chemistry parameters in which shifts from normal at Baseline were observed in 2 or more subjects overall were:
Albumin: 2 subjects (4.8%) shifted from normal at Baseline to high at Follow-up.
Urinalysis parameters in which shifts from normal at Baseline were observed in 2 or more subjects overall were:
Blood: 7 subjects (16.7%) shifted from normal at Baseline to abnormal at Period 2 Day 3/early termination; 5 subjects (11.9%) shifted from normal at Baseline to abnormal at Follow-up.
Clarity: 3 subjects (7.1%) shifted from normal at Baseline to abnormal at Period 2 Day 3/early termination.
Ketones: 6 subjects (14.3%) shifted from normal at Baseline to abnormal at Follow-up.
Leukocyte esterase: 2 subjects (4.8%) shifted from normal at Baseline to abnormal at follow-up.
Nitrite: 2 subjects (4.8%) shifted from normal at Baseline to abnormal at Period 2 Day 3/early termination.
Protein: 2 subjects (4.8%) shifted from normal at Baseline to abnormal at Follow-up.
The majority of individual laboratory results were within the reference ranges and none were reported as TEAEs.
8.1.1.3. Vital signs, physical examination, 12-lead electrocardiograms (ECGs)
Mean vital sign measurements observed after dosing were similar to those observed at Baseline in both treatments. There were no treatment related trends observed in mean vital sign measurements over time. No individual vital sign value was reported as a TEAE by the investigator.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 37 of 62

Therapeutic Goods Administration
Physical examinations were performed only at Screening and Day -1 of Period 1. None of the physical examination findings precluded any subjects from entering the study.
Electrocardiogram measurements were obtained only at Screening and Day –1 of Period 1. None of the abnormal findings were considered clinically significant by the investigator or precluded any subjects from entering the study.
8.2. Evaluator’s comment on clinical safety No post-marketing safety data relating to the proposed enoxaparin product were submitted, as at the time of the application Crusia had not been approved for marketing in any country. The clinical safety data provided in the submission are limited to the single-dose data in healthy volunteers from the PD BE Study ROV-R020-2011-01. In this study, the safety data indicated that both enoxaparin products were well tolerated when administered to a small number of healthy subjects. However, no meaningful conclusions regarding the clinical safety of the proposed enoxaparin sodium product can be drawn from Study ROV-R20-2011 for the following reasons:
1. Based on the ‘rule of three’s’ the number of subjects (n = 42) is too low to reliably identify adverse drug reactions associated with the proposed product occurring with an incidence of less than 7%.
2. There were no single dose safety data in patients.
3. There were no repeat dose safety data in either healthy volunteers or patients.
Overall, no assessment of the clinical safety of the proposed enoxaparin sodium product can be made from the submitted clinical data.
The TGA adopted LMWH biosimilar guidelines (EMEA/CHMP/BMWP/118264/2007) state:
‘Even if the efficacy is shown to be comparable, the similar biological medicinal product may exhibit a difference in the safety profile. Pre-licensing safety data should be obtained in a number of patients sufficient to determine the adverse effect profiles of the test medicinal product. Care should be given to compare the type, frequency and severity of the adverse reactions between the similar biological medicinal product and the reference products. Usually, comparative safety data from the efficacy trial will be sufficient to provide an adequate pre-marketing safety database’.
The guidelines also state:
‘For the detection of the immune-mediated type of Heparin-induced Thrombocytopenia (HIT Type II) monitoring of platelet count and an adequate diagnostic procedure in patients developing thrombocytopenia and/or thromboembolism (HITT) during the trial has to be performed’.
In pre-submission correspondence, the TGA asked the sponsor to provide ‘comparative clinical safety data between the proposed product and the reference product, which could be provided from the previous therapeutic equivalence study.’ The submission did not include the requested clinical safety data. The sponsor’s justification for not submitting the requested follows:
‘The incidence of bleeding of LMWH in general and enoxaparin in particular is between 0.5% and 5% during clinical trial for prevention of thromboprophylaxis of patients undergoing hip or knee arthroplasty and it depends on several factors (e.g. standardisation of bleeding, hospital setting, patients involved in clinical trials), so that in many cases clinical trials were underpowered to find differences between those anticoagulants used.19 That is the case of the study to evaluate the comparative effect of two enoxaparins
19 Dahl O et al. A critical appraisal of bleeding events reported in venous thromboembolism prevention trials of patients undergoing hip and knee arthroplasty. J Thromb Haemos 2010;8: 1966-75.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 38 of 62

Therapeutic Goods Administration
(Sanofi-Aventis branded enoxaparin versus Eurofarma enoxaparin, a generic version) as prophylaxis for VTE following major abdominal surgery, where no statistically significant differences between the two groups were detected.20
The incidence of HIT is estimated at 0.2 to 0.4%, although it depends on several factors: individuals (platelet counts, previous exposition to heparin/LMWH), type of heparin/LMWH, type of patient (surgical, medical), kind of intervention (prevention or treatment of DVT/pulmonary embolism).21 HIT is understood to be a result of a non-specific oligosaccharide interaction with endogenous chemokine PF4. These interactions are largely dependent on oligosaccharide molecular weights and charge densities.22
Rauova et al. demonstrated that the formation of PF4-heparin complexes is dependent on heparin polymer length.23 Analytical comparative studies to quantify these complexes constitute supporting evidence of similarity. Qualitative and quantitative characterisation of impurities, as well as the non-clinical immunogenicity study performed by the sponsor with the proposed product enoxaparin sodium, Clexane and Lovenox, provide further assurance that the risk of immunogenicity of the biosimilar product is comparable to the reference product.
Moreover, the sponsor considers it not necessary to assess, in a clinical study, the incidence of HIT associated with the proposed product enoxaparin sodium because it has been shown that the proposed product has similar quality as the reference enoxaparin, for example similar disaccharide building blocks and sequence of oligosaccharide, and hence a similar propensity for PF4 complex formation, as well as similar incidence of HIT.
Furthermore, in the submitted bioequivalence Study ROV-RO20-2011-01, there were no unexpected safety findings in the 42 healthy adult subjects participating in the single-dose crossover biopharmaceutic study. No serious adverse effects (AEs) were reported. Enoxaparin has a well-established safety and tolerability profile, as described in published literature. The data do not indicate a higher frequency or more severe AEs with the proposed product enoxaparin sodium compared with the reference product’.
The sponsor’s justification for a waiver is not supported for the following reasons:
1. As previously discussed, the sponsor’s justification for undertaking a therapeutic equivalence study is not supported.
2. In Dahl et al., (2010) the authors conclude that randomised ‘VTE prevention trials report markedly different rates of major bleeding despite similar patient populations and doses and durations of anticoagulant prophylaxis and were underpowered to detect modest differences in patient-important bleeding events. Standardization of bleeding definitions and reporting seems desirable’.19 There is nothing in the conclusions of Dahl et al., (2010) relating to the author’s appraisal of the literature that would preclude the sponsor from undertaking a comparative safety study of the proposed enoxaparin product and Clexane (Australia).
3. In Gomes et al., (2011) the authors compared the effect of two enoxaparin products (Sanofi-Aventis branded enoxaparin versus Eurofarma enoxaparin, a generic version) as prophylaxis for VTE following major abdominal surgery.20 The study randomised 200 patients in a 1:1 ratio to either 40 mg of branded enoxaparin or generic enoxaparin
20 Gomes M, et al. Generic versus branded enoxaparin in the prevention of venous thromboembolism following major abdominal surgery: report of an exploratory clinical trial. Clin Appl Thromb Hemost 2011; 17(6): 633-9.21 Kelton J and Warkentin T. Heparin-induced thrombocytopenia: a historical perspective. Blood 2008; 112: 2607-16.22 Newman P et al. Heparin-induced thrombocytopenia: IgG binding to PF4-heparin complexes in the fluid phase and cross-reactivity with low molecular weight heparin and heparinoid. Thromb Haemost 1998; 80: 292-7.23 Rauova L, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005;105:131-8.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 39 of 62

Therapeutic Goods Administration
once daily for 7 to 10 days post-operatively as prophylaxis for VTE following major abdominal surgery. No statistically significant differences between the 2 enoxaparin groups were detected. In all, 2 patients in the branded enoxaparin group experienced DVT (2.1%) compared to no patients in the generic group. The authors conclude that ‘this exploratory trial suggests that the generic LMWH is probably as safe and effective as the branded enoxaparin (Lovenox, Brazil) in the prophylaxis of VTE in this population’. There is nothing in the conclusions of Gomes et al., (2001) relating to their exploratory trial that would preclude the sponsor from undertaking a comparative safety study of the proposed enoxaparin product and Clexane (Australia).
4. The sponsor refers to a number of matters relating to the association between treatment with enoxaparin and immune-mediated heparin induced thrombocytopenia (HIT Type II), including the physicochemical similarities of the proposed and reference products and the results of the nonclinical immunogenicity study. The sponsor appears to be of the opinion that the risk of immunogenicity of the proposed and reference products is comparable, due to the similar physicochemical properties of the two products and the data from the nonclinical immunogenicity study. The sponsor also appears to be of the opinion that the proposed and reference products have a similar propensity for PF4 complex formation as well as a similar incidence of HIT Type II, due to the similar disaccharide building block sequence for the oligosaccharides of the two products. The assessment of the nonclinical immunogenicity study is a matter for the nonclinical evaluator and the assessment of the disaccharide and oligosaccharide characteristics of the two products is a matter for the quality evaluator.
5. While it is acknowledged that the incidence of HIT Type II associated with enoxaparin is low, this does not preclude a safety study of the proposed enoxaparin product and Clexane (Australia) being undertaking. It is not a requirement that a comparative safety study be specifically powered to detect HIT Type II. However, the study could reasonably include comparative assessment of AEs of thrombocytopenia and platelet counts. Information relating to the incidence of HIT Type II and other severe but uncommon immunogenic events (for example anaphylaxis and anaphylactoid reactions) associated with the proposed enoxaparin product is only likely to emerge from post-marketing pharmacovigilance.
6. It is considered that the safety data from the single dose PD BE Study ROV-RO20-2011-01 cannot be used as a surrogate for a clinical safety study comparing the proposed enoxaparin product and Clexane (Australia) [see the 3 reasons given at the start of the evaluator’s comments on clinical safety, above].
7. There has been a published report of a patient in the USA developing two life-threatening haemorrhages within 4 months of initiation of treatment with a generic enoxaparin product, while there had been with no complications with 4 years previous treatment with branded enoxaparin.24 There has been a reported communication identifying four cases of enoxaparin induced skin necrosis in the initial 18 months after switching from branded to generic enoxaparin.25 The authors commented that they had not observed any cases of this condition for several years raising a ‘concern of a greater risk of heparin-induced skin necrosis with the generic formulation’. While the number of reported adverse events associated with a generic enoxaparin following switching from a branded enoxaparin is low, the occurrences point towards the need to undertake comparative clinical safety studies when evaluating generic and branded enoxaparin products.
24 Kaffenberger B and Bekaii-Saab T. Clinical and Applied Thrombosis/Hemostasis. 2012;18(1):104-106.25 Gucalp A et al. Skin necrosis induced by generic enoxaparin. American Journal of Hematology. Letter to the editor. Published online 24 January 2013.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 40 of 62

Therapeutic Goods Administration
9. First round benefit-risk assessment
9.1. First round assessment of benefitsIt is not possible to assess the benefits of Crusia-AFT and Crusia-AFT Forte based on the submitted data. The submission did not include a therapeutic equivalence study comparing the efficacy and safety of the proposed enoxaparin product with the Australian enoxaparin reference product (Clexane). Furthermore, there were no clinical studies exploring the PD effects of switching from Crusia to Clexane or vice versa. The sponsor seeks a waiver from the requirement to submit a therapeutic equivalence study. However, it is recommended that the justification for a waiver be rejected. It is considered that the sponsor has not satisfactorily demonstrated a clear correlation between surrogate PD parameters (anti-Xa and anti-IIa) and clinical outcomes. Therefore, the PD bioequivalence data from the single dose study in healthy volunteers comparing the proposed enoxaparin product with the US enoxaparin reference product (Lovenox) cannot be extrapolated to patients with the clinical conditions.
9.2. First round assessment of risksIt is not possible to assess the risks of Crusia-AFT and Crusia-AFT Forte based on the submitted data. The submission did not include clinical efficacy data in patients with any of the clinical conditions for which registration of Crusia-AFT and Crusia-AFT Forte are being sought. The sponsor justified the absence of therapeutic equivalence studies on the basis that it considered that the comparative molecular analysis data, nonclinical PD data and clinical PD bioequivalence data supported the essential similarity of Crusia and Clexane. Therefore, the sponsor argued that a bridging therapeutic equivalence study comparing the two products administered SC for the prevention of venous thromboembolism (VTE) in patients undergoing surgery with high VTE risk (for example, major orthopaedic surgery) was not required. Consequently, the sponsor considered that no other clinical studies for other indications supporting the clinical efficacy and safety of Crusia administered by SC injection were required. In addition, the sponsor considered that the PD bioavailability of Crusia following IV administration could be estimated from the comparative PD bioequivalence study of Crusia following SC administration. Therefore, a therapeutic equivalence study comparing Crusia and Clexane administered as an initial IV dose for the treatment of acute STEMI, in conjunction with a fibrinolytic agent was not required.
However, it is considered that the sponsor should submit a clinical therapeutic equivalence study comparing Crusia and Clexane administered SC for the prevention of venous thromboembolism (VTE) in patients undergoing surgery with high VTE risk (for example, major orthopaedic surgery). The sponsor has not demonstrated a clear correlation between surrogate PD parameters (anti-FXa and anti-FIIa) and clinical outcome. If comparable efficacy and safety of Crusia and Clexane administered SC for the prevention of VTE had been demonstrated in surgical patients at high risk of the condition, then the sponsor would have been in a position to justify extrapolation of the results of this study to other indications. In the absence of a bridging study, there are no clinical data supporting the efficacy and safety of Crusia for any of the proposed indications for which the product is to be administered by SC injection. In addition, as previously argued in this CER, the PD bioequivalence of Crusia and Clexane following IV administration based on anti-IIa activity cannot be predicted from the SC data. Therefore, it is considered that a therapeutic equivalence study comparing Crusia and Clexane administered as an initial IV dose for the treatment of acute STEMI, in conjunction with a fibrinolytic agent, is required to support approval for this indication.
It is considered that the safety data from the single-dose study in healthy volunteers comparing the proposed enoxaparin product with the US enoxaparin reference product (Lovenox) cannot be meaningfully extrapolated to patients with the medical conditions of interest. Comparative safety data from a submitted efficacy trial would have been sufficient to provide an adequate
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 41 of 62

Therapeutic Goods Administration
pre-marketing safety database (LMWH biosimilar guidelines (EMEA/CHMP/BMWP/118264/2007)). However, the sponsor elected not to submit such a study and the justification for a waiver is considered to be unsatisfactory. The sponsor’s justification for a waiver for submitting clinical safety data has been examined and is considered to be unsatisfactory.
Other risks that have not been adequately addressed in the submission relate to the absence of PK/PD bioequivalence data relating to the low dose of Crusia proposed for prophylaxis (that is, 20 mg), the absence of PK/PD bioequivalence data relating to the higher strength of Crusia (that is, 150 mg/mL), the absence of a satisfactory justification for the 80% to 125% PD equivalence interval used in Study ROV-RO20-2011-01, and the lack of any immunogenicity data from a therapeutic clinical efficacy and safety study.
9.3. First round assessment of benefit-risk balance As it is not possible to assess the benefits or risks of Crusia-AFT and Crusia-AFT Forte based on the submitted data, it is not possible to assess the benefit-risk balance of the products for the proposed usage. Therefore, for regulatory purposes the benefit-risk balance of Crusia for the proposed indications is considered to be unfavourable.
10. First round recommendation regarding authorisationIt is recommended that the application to register Crusia-AFT and Crusia-AFT Forte be rejected for the following reasons:
1. No clinical efficacy data relating to any of the proposed indications have been submitted. The sponsor’s justification for not submitting at least one adequately powered, randomised, double-blind, parallel group clinical trial establishing therapeutic equivalence of the proposed enoxaparin sodium product with the Australian registered reference product (Clexane) is considered to be unsatisfactory. It is considered that efficacy in the target patient populations for the proposed indications cannot be inferred from the pharmacodynamic bioequivalence data from the single-dose study in 42 healthy volunteers (Study ROV-RO20-2011-01). It is considered that the sponsor has not satisfactorily established a correlation between surrogate PD parameters (anti-Xa and anti-IIa) and clinical outcome. The absence of a clinical therapeutic equivalence study precludes the known efficacy and safety data for Clexane being safely extrapolated to Crusia. The sponsor’s justification for not providing a therapeutic equivalence study is considered to be unsatisfactory for the reasons provided above in Section 5: Pharmacodynamics.
2. No clinical safety data relating to any of the proposed indications have been submitted. Comparative safety data from an efficacy trial would have been sufficient to provide an adequate pre-marketing safety database. However, the sponsor elected not to submit an efficacy trial. The sponsor’s justification for not submitting clinical safety data is considered to be unsatisfactory for the reasons provided above in Section 8: Safety
3. Other clinical limitations of the submitted data include:
a. No pharmacodynamic bioequivalence studies comparing the proposed enoxaparin sodium product with the Australian reference product (Clexane) were submitted. No clinical studies were submitted bridging the data for Lovenox (USA) used as the reference product in Study RO-RO20-2011-01 to Clexane (Australia). Therefore, there are no clinical data establishing the PD bioequivalence of the proposed formulation (Crusia) with the Australian reference product (Clexane). This raises doubts about the relevance of the submitted PK/PD bioequivalence Study ROV-RO20-2011-02 to Australian clinical practice.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 42 of 62

Therapeutic Goods Administration
b. No single dose IV pharmacokinetic bioequivalence study in healthy subjects comparing the proposed enoxaparin sodium product to the Australian reference product (Clexane) was submitted. The sponsor’s justification for a waiver of the requirement for such a study is considered to be inadequate. The PD bioequivalence of Crusia and Clexane following IV administration based on anti-IIa activity cannot be predicted from the SC data.
c. No adequate justification has been provided for selecting 80 to 125% as the pharmacodynamic bioequivalence interval in Study ROV-RO20-2011-01. The sponsor’s justification was based on the bioequivalence guideline relating to conventional chemical entities (CPMP/EWP/QWP/1401/98 Rev. 1/Corr). This guideline specifies the use of plasma drug concentrations (that is Cmax, AUC) to establish bioequivalence rather than PD outcomes. Furthermore, this guideline expressly states that its scope is limited to chemical entities.
d. No adequate justification has been provided for not submitting a PD bioequivalence study with the 150 mg/mL strengths of Crusia and Clexane 150 mg/mL. Consequently, no conclusions can be made about the PD bioequivalence of Crusia and Clexane presented in the higher strength formulations (that is, 150 mg/mL).
e. No low dose, single dose, SC pharmacodynamic bioequivalence study in healthy volunteers comparing the proposed enoxaparin sodium product to the Australian reference product (Clexane) was submitted. Consequently, no conclusions can be made about the PD bioequivalence of Crusia and Clexane at the clinically relevant lower prophylactic SC dose of 20 mg.
11. Clinical questionsQ1) What randomisation method was used to assign patients to treatment sequence AB or BA in Study ROV-RO20-2011-01?
Q2) What population was the healthy subjects participating in Study ROV-RO20-2011-01 drawn from?
Q3) The sponsor is requested to provide a formal justification for not undertaking a SC, single dose PD BE study in healthy volunteers comparing the proposed product with the Australian reference product (Clexane) at a dose of 20 mg (that is, a low dose consistent with the use of enoxaparin for prophylaxis).
Q4) The sponsor’s is requested to submit a justification addressing the relevant criteria in the ‘Justification for not submitting biopharmaceutic data (15.9)’ in the ‘Australian Regulatory Guidelines for Prescription Medicines (ARGMP)’ for not submitting pharmacodynamic bioequivalence studies for the proposed enoxaparin product at strengths other than 100 mg/mL.
12. Second round evaluation of clinical data submitted in response to questions
12.1. Overview of the clinical response. The sponsor’s response to TGA questions relating to the first round clinical evaluation report (CER) included individual responses to each of the four questions raised in the first round CER and submission of an additional Phase I PD equivalence study (Study ROV-RO20-2015-01). The additional Phase I PD equivalence study was designed to demonstrate the PD equivalence of
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 43 of 62

Therapeutic Goods Administration
enoxaparin (100 mg/mL) manufactured by Rovi (Spain) to Clexane (100 mg/mL) manufactured by Sanofi (EU) in healthy volunteers. The sponsor’s response to the first round CER questions and accompanying clinical evaluator’s comments are provided below followed by evaluation of the additional Phase I PD equivalence study.
12.2. Sponsor’s response to first round questions12.2.1. Question 1
‘What randomisation method was used to assign patients to treatment sequence AB or BA in Study ROV-RO20-2011-01?’
12.2.1.1. Sponsor’s response
The sponsor indicates that a paper randomisation schedule was generated by a validated computer program. The randomisation schedule was provided to the clinical site’s unblinded pharmacist. In addition, the randomisation method used in [the new] Study ROV-RO20-2015-01 was the same as that used for Study ROV-RO20-2011-01
12.2.1.2. Clinical evaluator’s comment
The response is acceptable.
12.2.2. Question 2
‘What population was the healthy subjects participating in Study ROV-RO20-2011-01 drawn from?’
12.2.2.1. Sponsor’s response
The sponsor indicates that the volunteers for Study ROV-RO20-2011-01 were selected from a normal healthy volunteer population recruited through advertisements in newspapers, as well as on the clinical laboratories website (based in Austin, Texas, USA). The sponsor indicates that similar methods were used to recruit healthy volunteers for the new Study ROV-RO20-2015-01 by the Contract Research Organisation and Clinical Site (based in the Netherlands).
12.2.2.2. Clinical evaluator’s comment
The response is acceptable.
12.2.3. Question 3
‘The sponsor is requested to provide a formal justification for not undertaking a SC, single dose PD BE study in healthy volunteers comparing the proposed product with the Australian reference product (Clexane) at a dose of 20 mg (that is, a low dose consistent with the use of enoxaparin for prophylaxis)’.
12.2.3.1. Sponsor’s response
The sponsor responded that the TGA adopted LMWH biosimilar guideline (EMEA/CHMP/BMWP/118264/2007) states that ‘the selected doses should be in the sensitive part of the dose-response curve and within the recommended dose ranges for the different indications’. The sponsor considers that the selection of a dose of 100 mg for the reference and the test enoxaparin fulfils this requirement and testing of a 20 mg dose is unnecessary for the following reasons:
1. There is no explicit requirement in the aforementioned guideline for PK/PD studies at the high and low range of the approved SC doses for clinical use.
2. A dose of 20 mg will not allow for comparisons to be made regarding the PD parameters of anti-IIa activity, since plasma anti-IIa activity after SC administration is approximately ten-fold lower than anti-Xa activity. The mean maximum anti-IIa activity reaches 0.13 IU/mL
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 44 of 62

Therapeutic Goods Administration
and is observed approximately 3 to 4 hours following subcutaneous injection of 40 mg, while it is not detectable at the 20 mg dose level when using the conventional amidolytic method.
3. The absorption rate and bioavailability patterns are consistent and predictable, because enoxaparin has a constant t1/2β, irrespective of dose, with the AUC increasing linearly with dose. The anti-Xa activity is linear over the recommended full dosage range, including prophylactic and therapeutic doses. In a clinical trial performed in 16 healthy male volunteers, the pharmacokinetic profile of enoxaparin at single SC doses of 1.0, 1.25, 1.50 and 2.0 mg/kg is characterised by a linear relationship between dose and extent of SC resorption, and exhibits stable biodynamic profile as previously described for lower doses (0.30 to 1.20 mg/kg). [The] linearity of enoxaparin across the full range of doses, from 20 mg up to 100 mg, [has been] demonstrated based on published data and data from Rovi’s bioequivalence Study ROV-RO20-2015-01 submitted with its response.
Therefore, the sponsor considers that the PD bioequivalence demonstrated for Crusia-AFT 100 mg versus Clexane 100 mg manufactured by Sanofi (from EU market) can be fully extrapolated to the full range of recommended dosage. This approach is also consistent with the CHMP opinion stated in the corresponding EMA Scientific Advice report dated 21 November 2013 (Procedure No. EMEA/H/SA/2647/1/2013/III).
The correlation provided the sponsor between the dose of enoxaparin and anti-Xa activity, both as peak (Amax) and area under curve (AUC) activity, across the dose range (20 to 100 mg) using data obtained from a published source (Noble et al, 1995) and the sponsor’s bioequivalence trial, Study ROV-RO20-2015-01, is provided below in Figure 6.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 45 of 62

Therapeutic Goods Administration
Figure 6. Linearity data from Study ROV-RO20-2015-01
12.2.3.2. Clinical evaluator’s comment
The sponsor’s response is acceptable. The sponsor provided appropriate references supporting its position. The graph based on data from Noble et al., (1995) and from Study ROV-RO20-2015-01 showed a linear relationship between Clexane dose over the range 20 mg to 100 mg and both mean Amax and AUC0-t anti-Xa activity. Graphs based on data from Noble et al., (1995) showed a linear relationship between Clexane dose over the dose range 20 mg to 80 mg and both mean Amax and AUC0-t anti-Xa activity.26 In addition, data demonstrating a linear relationship between over the therapeutic dose range for Clexane of 60 mg to 100 mg and both mean Amax and AUC0-t anti-Xa activity were also presented.
12.2.4. Question 4
‘The sponsor is requested to submit a justification addressing the relevant criteria in the ‘Justification for not submitting biopharmaceutic data (15.9)’ in the ‘Australian Regulatory Guidelines for Prescription Medicines (ARGMP)’ for not submitting pharmacodynamic
26 Noble S et al. Enoxaparin. A reappraisal of its pharmacology and clinical applications in the prevention and treatment of thromboembolic disease. Drugs. 1995; 49(3):388-410.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 46 of 62

Therapeutic Goods Administration
bioequivalence studies for the proposed enoxaparin product at strengths other than 100 mg/mL’.
12.2.4.1. Sponsor’s response
AFT provides the following justification in line with 15.9 of the Australian Regulatory Guidelines for Prescription Medicines (ARGPM) for not submitting pharmacodynamic bioequivalence studies for the proposed enoxaparin product at strengths other than 100 mg/mL:
1. Pharmacokinetic characteristics of the drug substance, such as permeability (or absolute bioavailability), linearity, first-pass effect (if any) and its significance
All presentations from both enoxaparin concentrations, Clexane (100 mg/mL) and Clexane Forte (150 mg/mL), currently marketed in Australia share the same Product Information (Australian PI), and interestingly this does not contain any information related to pharmacokinetic characteristics for each strength. This is also the case for the Summary of Product Characteristics for ‘Clexane Forte Syringes’ (UK document) and for Clexane pre-filled syringes (UK document), in which the wording of Section 5.2 (‘Pharmacokinetic properties’) is exactly the same for both products.
No published data can be found studying the 150 mg/mL concentration of the enoxaparin in humans. Although not studied clinically, the 150 mg/mL concentration of enoxaparin sodium is projected to result in anticoagulant activities similar to those of 100 mg/mL and 200 mg/mL concentrations at the same enoxaparin dose (US prescribing information for Lovenox). When a daily 1.5 mg/kg SC injection of enoxaparin sodium was given to 25 healthy male and female subjects using a 100 mg/mL or a 200 mg/mL concentration [similar] PK profiles were obtained (US prescribing information for Lovenox).
2. Clinical consequences of any potential differences in bioavailability of the products under consideration (for example, increased dose leading to toxicity or decreased dose leading to lack of efficacy)
According to the findings above, it is highly unlikely that there might be any potential differences in bioavailability of both products under consideration that could increase the dose leading to toxicity or decrease the dose leading to lack of efficacy.
3. Margin between the minimum effective and minimum toxic plasma concentration
As per the information provided above, the sponsor considers that no differences can be expected from Crusia-AFT and Crusia-AFT Forte in the margin between the minimum effective and minimum toxic plasma concentration. Therefore, the sponsor requests a biowaiver for Crusia-AFT Forte given that the concentration of enoxaparin is not relevant for the in vivo properties of this medicinal product, but for the convenience of the patient (less volume of injection).
12.2.4.2. Clinical evaluator’s comment
The sponsor’s response is satisfactory. However, the sponsor might also have referred to the rapid and complete absorption after subcutaneous injection of enoxaparin resulting in a high absolute bioavailability of ‘over 90%’, and the linear relationship between Clexane over the dose range of 60 mg to 100 mg and both mean Amax and AUC0-t anti-Xa activity. Both of these factors would support a decision not submit pharmacodynamic bioequivalence studies for the proposed enoxaparin product at strengths other than 100 mg/mL.
It is also noted that in the Summary of Biopharmaceutic Studies and Associated Analytical Methods, that the sponsor claims a waiver for the in vivo bioequivalence studies of the enoxaparin Rovi 20 mg (2,000 IU), 40 mg (4,000 IU), 60 mg (6,000 IU), 80 mg (8,000 IU), 120 mg (12,000 IU), and 150 mg (15,000 IU) solution for injection in pre-filled syringes. The sponsor considers that the concentration of enoxaparin is not relevant to the in vivo properties of enoxaparin, but for the convenience of the patient (that is, less volume of injection).
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 47 of 62

Therapeutic Goods Administration
Furthermore, the sponsor comments that the CHMP agreed in their Scientific Advice that ‘separate clinical investigations using both concentrations [100 mg/mL and 150 mg/mL] are not necessary, as the only difference is the amount of API (...) and no clinical impact is expected in case the same dose is applied’ provided that the manufacturing process for both strengths is adequately validated and their respective stability data are available. The sponsor states that the qualitative composition of the different strengths is the same and the composition of the strengths are quantitatively proportional (i.e., the ratio between the amounts of the sole excipient water for injection to the amount of active substance is the same for all strengths).
12.3. Evaluation of Study ROV-RO20-2015-0112.3.1. Study title
‘A single dose, randomised, double blind, 2 way crossover study for the demonstration of pharmacodynamic equivalence of enoxaparin (100 mg/mL) 100 mg subcutaneous injection manufactured by Rovi (Spain) to Clexane (100 mg/mL) 100 mg subcutaneous injection manufactured by Sanofi (EU) in healthy volunteers’.
12.3.2. Objectives
The primary objective was to demonstrate the PD equivalence of enoxaparin (100 mg/mL) 100 mg subcutaneous (SC) injection manufactured by the sponsor (Madrid, Spain) to Clexane (100 mg/mL) 100 mg SC injection manufactured by Sanofi (European Union (EU), Paris, France) in healthy volunteers.
The secondary objectives were to evaluate the safety and tolerability of enoxaparin (100 mg/mL) 100 mg SC injection manufactured by the sponsor (Spain), in healthy volunteers.
12.3.3. Design
The study was a single-dose, randomised, double blind, 2 period, 2 sequence crossover trial. Subjects were screened up to 30 days before first administration of the study drug and were admitted to the clinic on Day -1 of Period 1 for baseline assessments. Before dosing on Day 1 of Period 1, subjects were randomly assigned to one of two treatment sequences according to the randomisation code generated by the clinical organisation that undertook the study. The study was double-blinded in that the subjects, study team members and laboratories were blinded.
The two treatment sequences were: AB comprising treatment A (Rovi) in Period 1 and treatment B (Sanofi) in Period 2; and BA comprising treatment B (Sanofi) in Period 1 and treatment A (Rovi) in Period 2. On Day 1 of Period 1, subjects received a single SC dose of enoxaparin manufactured by Rovi or Clexane manufactured by Sanofi after an overnight fast of at least 10 hours. Subjects continued fasting for at least 4 hours after study drug administration. Subjects were confined to the clinic on Days 1 and 2 and discharged on Day 3 of Period 1 after completion of all scheduled assessments. The washout period between administrations of study drug in each period was at least 7 days. The process was repeated in Period 2 with the alternative enoxaparin to that in Period 1 being administered. Subjects returned to the clinic centre on Day 7 of Period 2 for a follow-up visit. In addition, there was a follow-up telephone call on Day 15 of Period 2. The total duration of the study was approximately 6 weeks, including the 30-day screening period.
The study included satisfactory criteria for discontinuation from the study and adequate procedures for follow-up. Subjects who discontinued were not replaced.
12.3.4. Locations and dates
The study was undertaken at a single-site in the Netherlands from 25 September 2015 (first screening) to 1 December 2015 (last follow-up). The final version of the report (version 2.0) was dated 10 October 2016. The study was sponsored by Laboratorios-Farmaceuticos Rovi, S.A.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 48 of 62

Therapeutic Goods Administration
(Rovi), Madrid, Spain. The study was conducted in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonisation (ICH) E6 Guideline for Good Clinical Practice (GCP) and with the European Union Clinical Trial Directive (EU CTD).
12.3.5. Inclusion and exclusion criteria
A total of 46 healthy male or female subjects were planned to be enrolled with the goal of 40 subjects completing the study. The inclusion criteria included healthy male and female subjects aged between the ages of 18 and 45, inclusive. The BMI was required to be between 18 and 30 kg/m2, inclusive. Subjects were required to have no clinically significant abnormalities in medical history, vital sign measurements, or physical examination findings. Subjects were required to be non-smokers or to have quit smoking at least 6 months before the first dose of study drug. The inclusion and exclusion criteria have been examined and are considered to be satisfactory for Phase I, single dose crossover studies in healthy volunteers administered a LMWH.
12.3.6. Treatments
As discussed above, the two treatments used in this single-dose, crossover study were enoxaparin manufactured by Rovi (test treatment A) and Clexane (EU) manufactured by Sanofi (reference treatment B) and administered as 100 mg by SC injections. The injection site for study drug administration in Periods 1 and 2 was alternated between the left and right anterolateral or left and right posterolateral abdominal wall in accordance with the Clexane (prescribing information). The pre-filled syringes for the two study drugs were not identical. Therefore, an unblinded pharmacist was responsible for dispensing the study drugs in a manner consistent with maintaining the blind and a dedicated unblinded team member performed study drug administration to maintain the blind. Procedures were specified for unblinding treatment in the case of emergency.
Subjects were not permitted to use an investigational drug within 60 days before the first dose of study drug. The use of any prescription drugs (with special attention to antiplatelet or anticoagulant medications) or over-the-counter medication that may affect coagulation was prohibited within 4 weeks before dosing through the completion of all study procedures. Any other over the counter medication was prohibited within 2 weeks before dosing through the completion of all study procedures, unless consistent with the investigator’s criteria (for example, paracetamol). Any medications for the management of any AEs during the study could be given at the discretion of the investigator.
Strenuous activity was not allowed at any time during the study. Subjects were instructed to avoid bruising or falls from the first study drug administration until 14 days after the last study drug administration. The use of alcohol was not allowed from 48 hours before the first dose of study drug up to the follow-up visit. Male subjects were required not to donate sperm from first administration of the study drug until 3 months after the follow-up visit. Male and female subjects were required to use an effective method of birth control while they were participating in this study and for 120 days after the last dose of study drug.
12.3.7. Pharmacodynamic endpoints and statistical analyses
12.3.7.1. Primary PD parameters endpoints
The primary PD parameters/endpoints were:
AUEC0-inf, AUEC0-t, and Amax for anti-FXa; and
AUEC0-t and Amax for anti-FIIa activity.
An analysis of variance (ANOVA) with fixed effects for sequence, period and treatment, and random effect for subject nested within sequence was performed on the natural logarithm transformed values of the primary and secondary endpoints to assess the differences between the test and reference treatments. The geometric mean ratio and corresponding 95% CI for
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 49 of 62

Therapeutic Goods Administration
AUEC0-inf, AUEC0-t, Amax of the two treatments were calculated by the back-transformation of the mean difference and 95% CI of the log-transformed values. AUEC0-inf values obtained after extrapolation of more than 20% were excluded from the analysis.
Pharmacodynamic equivalence (biosimilarity) was concluded if the 95% CI of the ratio of the geometric least squares (LS) means between the test treatment and the reference treatment for AUEC0-inf, AUEC0-t, and Amax of anti-FXa activity, as well as AUEC0-t, and Amax of anti-FIIa activity, were completely within the 80.00% to 125.00% interval.
12.3.7.2. Supportive secondary PD parameters/endpoints
The secondary PD parameters/endpoints were:
AUEC0-inf, AUEC0-t, and Amax of baseline-adjusted tissue factor pathway inhibitor (TFPI) levels; and
AUEC0-t ratio of anti-FXa activity and anti-FIIa activity (RAUEC).
The geometric mean ratio and corresponding 95% CI for secondary PD parameters were calculated as described above for the primary PD parameters. The PD equivalence criteria for the secondary PD parameters were the same as described above for primary PD parameters.
12.3.8. Sample size
40 subjects completing the study were considered to provide at least 80% power to conclude biosimilarity, assuming that the geometric mean ratio of the test versus reference treatments was between 0.9 and 1.1 and the intra-subject CV was less than 18%. Biosimilarity was to be concluded when the 95% CIs for the geometric mean ratio of the primary PD parameters lay within the 80.00% to 125.00% equivalence interval. The assumed and intra-subject CV and geometric mean ratios between 0.9 and 1.0 had been observed in the literature and in previous crossover studies with enoxaparin. To allow for dropouts, 46 subjects were enrolled.
12.3.8.1. Analysis sets
Randomised set: All subjects who were assigned a randomisation number. This set was used to summarise disposition.
Pharmacodynamic set: All randomised subjects who received study drug (either the test or reference treatment), and had sufficient number of valid bioanalytical results to facilitate calculation of the PD parameters. This could exclude subjects whose derived PD parameters were considered invalid due to relevant missing values, at the discretion of the pharmacokineticist, or if any other problem occurred during sampling or bioanalytical laboratory analysis, which could invalidate the measurements. The PD set was used for the summary of PD activity results and PD parameters.
Sensitivity set: All subjects in the PD set with less than 3 missing anti-FXa activities in the planned 2 to 6 hours post-dose interval.
Safety set: All randomised subjects who received at least 1 dose of study drug (either the test or reference treatment). The safety set was used for the safety data summaries and for baseline characteristic summaries.
12.3.8.2. Blood sample collection times for analysis of PD parameters/endpoints
Blood samples were collected at the following time points for assessment of anti-FXa, anti-FIIa, and TFPI activity before and after a single dose of study drug: on Day -1 of Periods 1 and 2, before dosing, and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 16, 20, 24, and 36 hours after dosing on Day 1 of Periods 1 and 2.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 50 of 62

Therapeutic Goods Administration
12.3.9. Subject disposition
A total of 90 subjects were screened, and 46 were randomised and treated in the study. 3 subjects were withdrawn during the study: 1 subject withdrew her consent on Day 2 of Period 1 because of vein problems (haematoma and pain at site of venepuncture); 1 subject 35 was withdrawn from the study due to AEs of syncope, dizziness, pharyngitis, headache on Day -1 of Period 2; and 1 was withdrawn from the study because of a protocol deviation (inclusion error due to BMI being greater than the allowed upper limit). A total of 43 subjects completed the study as per protocol. All randomised subjects who received at least 1 dose of study drug were included in the safety set. A total of 45 subjects received enoxaparin manufactured by Rovi and 45 subjects received Clexane. The sensitivity set included 38 randomised subjects, with the 8 excluded subjects comprising the 3 previously mentioned withdrawn subjects and an additional 5 subjects. The disposition of the subjects in the study is summarised below in Table 11.
Table 11. Subject disposition
Number of subjects n (%)
Screened volunteers 90
Screening failures 40
Approved but not dosed (reserve)
4
Randomised subjects 46 (100)
Dosed subjects 46 (100)
Completed subjects 43 (93)
Completed Period 1 45 (98)
Completed Period 2 43 (93)
Withdrawn subjects 3 (7)
Reasons for withdrawal
Adverse event 1 (2)
Withdrawal by subject 1 (2)
Protocol deviation 1 (2)
Safety set 46 (100)
PD set 43 (93)
Sensitivity set 38 (83)
12.3.10. Major protocol deviations
All protocol deviations were considered prior to the assignment of subjects to the PD set. During the study, there were 8 important protocol deviations across 4 subjects (1 subject with 3
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 51 of 62

Therapeutic Goods Administration
deviations, 2 subjects with 2 deviations each and 1 subject with a single deviation). The important protocol deviations (grouped by subject) were:
1. Some blood samples on Day 2 of Period 1 were collected 81 minutes later than planned due to problems with blood sampling with some samples not being collected.
2. Some blood samples on Day 2 of Period 1 and on Day 1 of Period 2 were not collected due to problems with blood sampling. The same subject had a glass of wine after completion of the first period, which was not allowed according to the protocol. However, this was 5 days prior to admission in the second period and therefore the subject was allowed to participate in Period 2 of the study.
3. One subject was enrolled in the study without meeting eligibility criteria due to BMI being above the upper limit of the protocol specified range. The sponsor decided that this subject should be discontinued from the study, but could be included in the safety set but not in the PD set.
4. Some blood samples on Day 2 of Period 2 were not collected due to problems with blood sampling.
12.3.11. Demographics and other baseline characteristics
A total of 46 subjects (33 male, 13 female) with a median age of 25 years (range: 18, 44 years) participated in the study. The median weight of the 46 subjects was 76.0 kg (range: 47.8, 93.0 kg), the median height was 178 cm (range: 150, 197 cm) and the median BMI was 23.6 kg/m2 (range: 19.0, 31.1 kg/m2). The majority of subjects were White (n = 39, 85%), with the remaining subjects being Black (n = 4, 9%), Asian, Multiple or Other (1 patient each, 2%).
There were no clinically significant findings with regard to medical history or previous medication. Drug and alcohol screen results were negative for all subjects at screening and each admission. The results for the serology parameters and the occult blood test were negative at screening for all subjects. For females, the pregnancy test results were negative at screening and each admission. All subjects except 1 (BMI above upper limit of pre-specified range) complied with the inclusion and exclusion criteria.
12.3.12. PD results for the primary endpoint analysis
The statistical analysis for the plasma PD parameters for anti-FXa activity are summarised below in Table 12.
Table 12. Statistical analysis of plasma PD parameters for anti-FXa activity, PD set
Parameter Treatment Geometric
LS Means
Treatment comparison
Estimate 95% CIs CV (%)
Amax (IU/mL) A 0.974 A/B 100.1 94.6, 105.9 13.0
B 0.973
AUEC0-t (h*IU/mL)
A 8.05 A/B 103.8 99.8. 108.0 9.1
B 7.75
AUEC0-inf
(h*IU/mL)A 8.91 A/B 104.2 100.0, 108.6 8.8
B 8.55
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 52 of 62

Therapeutic Goods Administration
Treatment A: Enoxaparin (100 mg/mL) 100 mg SC injection, manufactured by Rovi (Spain); Treatment B: Clexane (100 mg/mL) 100 mg SC injection, manufactured by Sanofi (EU). Note: A linear mixed effects model was applied to the natural log-transformed PD parameters with sequence, period and treatment as fixed effects and subject nested within sequence as a random effect.
The statistical analysis for the plasma PD parameters for anti-FIIa activity are summarised below in Table 13.
Table 13. Statistical analysis of plasma PD parameters for anti-FIIa activity, PD set
Parameter Treatment Geometric LS Means
Treatment Comparison
Estimate 95% CIs CV (%)
Amax (IU/mL) A 0.181 A/B 103.3 94.7, 112.6 20.2
B 0.175
AUEC0-t (h*IU/mL)
A 1.08 A/B 103.5 90.9, 117.9 30.5
B 1.04
Treatment A: Enoxaparin (100 mg/mL) 100 mg SC injection, manufactured by Rovi (Spain); Treatment B: Clexane (100 mg/mL) 100 mg SC injection, manufactured by Sanofi (EU). Note: A linear mixed effects model was applied to the natural log-transformed PD parameters with sequence, period and treatment as fixed effects and subject nested within sequence as a random effect.
Sensitivity set, FXa activity
The sensitivity set consisted of all subjects in the PD set with less than 3 missing anti-FXa activities in the planned 2 to 6 hours post-dose interval. Thus, all subjects that had 3 or more missing data points within the time interval of 2 to 6 hours post-dose were excluded from the sensitivity set. The sensitivity analysis set included 38 subjects compared to 43 subjects in the PD set. The statistical analysis for the plasma PD parameters for anti-FXa activity in the sensitivity set are summarised below in Table 14.
Table 14. Statistical analysis of plasma PD parameters for anti-FXa activity, PD set
Treatment A: Enoxaparin (100 mg/mL) 100 mg SC injection, manufactured by Rovi (Spain); Treatment B: Clexane (100 mg/mL) 100 mg SC injection, manufactured by Sanofi (EU). Note: A linear mixed effects model was applied to the natural log-transformed PD parameters with sequence, period and treatment as fixed effects and subject nested within sequence as a random effect.
Comment: The statistical analysis of anti-FXa activity showed that the 95% CIs of the ratios of the geometric LS means for the PD parameters Amax, AUEC0-t and AUEC0-inf were all enclosed completely within the pre-specified PD equivalence interval of 80.00% to 125.00%. The key PD parameters for anti-FXa activity were comparable, and the
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 53 of 62

Therapeutic Goods Administration
Tmax was 4.00 hours for both treatments, and inter-subject variability was moderate for the parameters for both treatments. The mean plasma anti-FXa versus time-concentrations curves were virtually superimposable for the two treatments. The sensitivity analysis for anti-FXa activity for the primary PD parameters showed similar results to the primary analysis in the PD set.
The statistical analysis of anti-FIIa activity showed that the 95% CIs of the ratios of the geometric LS means for the PD parameters Amax and AUEC0-t were both enclosed completely within the pre-specified PD equivalence interval of 80.00% to 125.00%. The key PD parameters for anti-FXII activity were comparable, with the tmax being reached at 4.50 hours for both treatments. The mean plasma anti-FXa versus time-concentrations curves (linear scale) were virtually superimposable for the two treatments, while the corresponding curves (log-linear scale) separated at about 12 hours. The mean terminal half-life values for both treatments were similar (4.88 hours, Treatment A and 4.77 hours for Treatment B), but inter-subject variability for this parameter was high for both treatments.
Based on the pre-specified criteria, the PD equivalence of enoxaparin manufactured by Rovi and Clexane manufactured by Sanofi has been established in terms of the primary PD parameters Amax, AUEC0-t and AUEC0-in for anti-FXa activity and the primary PD parameters Amax and AUEC0-t for anti-FIIa activity.
12.3.13. PD results for the secondary parameters/endpoints
The statistical analysis for the plasma PD parameters for baseline adjusted TFPI levels are summarised below in Table 15.
Table 15. Statistical analysis of plasma PD parameters for anti-FIIa activity, PD set
Parameter Treatment Geometric LS Means
Treatment Comparison
Estimate 95% CIs CV (%)
Amax (ng/mL) A 207 A/B 104.1 95.6, 113.4 19.9
B 199
AUEC0-t
(h*ng/mL)A 913 A/B 105.9 99.1, 113.1 15.2
B 863
AUEC0-inf
(h*ng/mL)A 910 A/B 108.4 102.1, 115.2 9.0
B 839
Treatment A: Enoxaparin (100 mg/mL) 100 mg SC injection, manufactured by Rovi (Spain); Treatment B: Clexane (100 mg/mL) 100 mg SC injection, manufactured by Sanofi (EU). Note: A linear mixed effects model was applied to the natural log-transformed PD parameters with sequence, period and treatment as fixed effects and subject nested within sequence as a random effect.
The RAUEC is the ratio AUEC0-t of anti-FXa to anti-FIIa. The mean RAUEC for anti-FXa/anti-FXII activity in the PD set for Treatment A (enoxaparin) was 7.92 (CV% = 34.9%) and for Treatment B (Clexane) was 8.00 (CV% = 39.8%). The geometric LS means for RAUEC for anti-FXa/anti-FXII activity in the PD set for Treatment A (enoxaparin) and Treatment B (Clexane) were 7.46 and
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 54 of 62

Therapeutic Goods Administration
7.44, respectively, and the point estimate for the geometric mean ratio A/B was 1.07 (CV% = 31.1), with a 95% CI of 87.9 to 114.5.
Comment: Pharmacodynamic equivalence of enoxaparin manufactured by Rovi and Clexane was demonstrated after administration of 100 mg as a single SC injection based on the secondary PD parameters Amax, AUEC0-t and AUEC0-inf for TFPI levels and the secondary PD parameter RAUEC (AUEC0-t ratio of anti-FXa activity to anti-FIIa activity).
12.3.14. Safety
12.3.14.1. Exposure
A total of 46 subjects were exposed to study drug. 44 subjects participated in 2 treatment periods and received 2 doses of study drug (that is 100 mg enoxaparin Rovi (Treatment A) and 100 mg Clexane-Sanofi (Treatment B)). 2 subjects received only 1 dose of study drug (that is, 1 subject received 100 mg Clexane-Sanofi in Period 1 and 1 received 100 mg enoxaparin manufactured by Rovi in Period 1. A total of 45 subjects received enoxaparin-Rovi and 45 subjects received Clexane-Sanofi.
12.3.14.2. Adverse events (high-level overview)
The overview of treatment-emergent adverse events (TEAEs) is summarised below in Table 16. There were no marked differences between the two treatment arms in high level AE profiles.
Table 16. Overview of treatment emergent adverse events
Number (n) and percentage (%)
Enoxaparin-Rovi (n = 45)
Clexane-Sanofi (n = 45)
Total (n = 46)
At least 1 AE 23 (51%) 19 (42%) 32 (70%)
At least 1 treatment related AE
7 (16%) 6 (13%) 10 (22%)
At least 1 mild severity AE 22 (49%) 19 (42%) 32 (70%)
At least 1 moderate-severity AE
3 (7%) 2 (4%) 5 (11%)
Withdrawn due to AE 1 (2%) 0 (0%) 1 (2%)
12.3.14.3. Treatment-emergent adverse events (irrespective of relationship to treatment)
During the study, 32 subjects (70%) reported a total of 81 TEAEs. The number of TEAEs and percentage of subjects reporting TEAEs was comparable between enoxaparin-Rovi (23 subjects (51%) reported 44 TEAEs) and Clexane-Sanofi (19 subjects (42%) reported 37 TEAEs). TEAEs by SOC reported in ≥ 5% of subjects in either treatment arm, enoxaparin-Rovi versus Clexane-Sanofi, respectively, were: general disorders and administration site conditions (31% (n = 14) versus 20% (n = 9)); nervous system disorders (18% (n = 8) versus 22% (n = 10)); respiratory, thoracic and mediastinal disorders (11% (n = 5) versus 2% (n = 1)); gastrointestinal disorders (7% (n = 3) versus 4% (n = 2)); infections and infestations (7% (n = 3) versus 0%); and injury poisoning and procedural complications (0% versus 7% (n = 3)).
The percentage of subjects reporting TEAEs in the SOCs of respiratory, thoracic and mediastinal disorders, infections and infestation and injury, poisoning and procedural complications were statistically significantly different between enoxaparin-Rovi and Clexane-Sanofi with p-values of
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 55 of 62

Therapeutic Goods Administration
0.0184, 0.0140 and 0.0140, respectively. The pairwise comparisons were tested using a Fisher’s exact test with significance indicated by p < 0.05.
TEAEs reported in ≥ 2% of subjects in either of the two treatment arms, enoxaparin-Rovi versus Clexane-Sanofi, respectively, were: headache (16% (n = 7) versus 13% (n = 6)); vessel puncture site haematoma (11% (n = 5) versus 2% (n = 1)); fatigue (7% (n = 3) versus 2% (n = 1); and catheter site haematoma (0% versus 7% (n = 3)). The percentage of subjects reporting TEAEs was statistically significantly different for vessel puncture site haematoma (p = 0.0184) and catheter site haematoma (p = 0.0140). None of the other pairwise comparisons for TEAEs were statistically significantly different (p ≥ 0.05). The pairwise comparisons were tested using a Fisher’s exact test with significance indicated by p < 0.05. No hypersensitivity AEs were reported.
12.3.14.4. Treatment-emergent AEs related to treatment
There were no notable differences between the two treatment arms in the number and percentage of subjects reporting treatment-related TEAEs (PTs). There were no statistically significant differences between the two treatment arms in the percentage of subjects reporting TEAEs grouped either by SOCs or presented as PTs. The pairwise comparisons were tested using a Fisher’s exact test with significance indicated by p < 0.05. Treatment-related TEAEs reported in the two treatment arms are summarised below in Table 17.
Table 17. Treatment related treatment emergent adverse events, safety analysis set
N = number of subjects exposed per treatment; n = number of subjects that experienced the adverse event. e=number of times the adverse event occurred. %=the percentage of subjects that experienced the adverse event per treatment: (n/N)*100%. Adverse events are classified according to MedDRA v18.1. A relationship to the study drug classified as ‘Suspected’ is categorised as related. A P-value of less than 0.05 indicates a statistically significant difference between treatment A and B at the significance level of 0.05. P-value is based on Fishers exact test.
12.3.14.5. Deaths, SAEs, and other significant AEs
No deaths or SAEs occurred during the study. There was 1 withdrawal due to TEAEs during the study. The TEAEs resulting in withdrawal from the study were syncope, dizziness, headache, pharyngitis and pyrexia in 1 subject reported on Day -1, Period 2, with all events being considered by the investigator to be unrelated to treatment.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 56 of 62

Therapeutic Goods Administration
12.3.14.6. Clinical laboratory, vital signs, ECG changes and physical examination
There were no notable trends observed in clinical laboratory, vital signs, ECG or physical examination changes during the study.
Comment: The study met its primary objective of demonstrating PD equivalence of enoxaparin-Rovi and Clexane-Sanofi. The primary statistical analysis showed that the 95% CIs of the geometric mean ratios for the primary PD parameters/endpoints for anti-FXa and anti-FIIa were enclosed entirely within the pre-specified equivalence interval of 80.00% to 125.00% (that is Amax, AUEC0-t and AUEC0-inf for anti-FXa activity and Amax and AUEC0-T for anti-FIIa activity). These results were supported by the secondary statistical analysis, which showed that the two formulations were bioequivalent based on the PD parameters/endpoints for baseline adjusted TFPI levels (that is, Amax, AUEC0-t and AUEC0-inf) and for the RAUEC for anti-FXa and anti-FII activity (that is, 95% CIs for the relevant geometric mean ratios were enclosed entirely within the interval 80.00% to 125.00%).
The use of single 100 mg SC doses of enoxaparin-Rovi and Clexane-Sanofi in healthy volunteers in order to investigate the PD equivalence of the two formulations is considered to be acceptable. In describing the rationale for the study, the protocol indicated that the study had been conducted in compliance with the European Medicines Agency (EMA) Guideline on non-clinical and clinical development of similar biological medicinal products containing low molecular weight heparins (EMA/134870/2012). The protocol stated that these guidelines present ‘the current view of the CHMP on the nonclinical and clinical requirements for demonstration of comparability of two LMWH containing medicinal products’. It is noted that this guideline, which has been adopted by the TGA, states that equivalence margins for the PD parameters should be ‘pre-specified and appropriately justified’. While the PD equivalence margins were pre-specified an appropriate justification for the use of margins could not be identified in the protocol or study report.
There were no notable differences in the safety profiles of the two formulations following single 100 mg SC doses. However, the data are too limited to draw clinically meaningful comparisons relating to the safety of the two formulations.
13. Second round benefit-risk assessment
13.1. Second round assessment of benefitsAfter consideration of the responses to clinical questions and the additional PD equivalence Study ROV-RO20-2015-01 provided by the sponsor in its response to TGA questions, it is still not possible to assess the benefits of Crusia-AFT and Crusia-AFT Forte for the proposed usage. Neither the original submission nor the sponsor’s response included a therapeutic equivalence study comparing the efficacy and safety of the proposed enoxaparin product with the Australian enoxaparin reference product (Clexane). Furthermore, there were no clinical studies exploring the PD effects of switching from Crusia to Clexane or vice versa. The sponsor seeks a waiver from the requirement to submit a therapeutic equivalence study. However, it is recommended that the justification for a waiver be rejected. It is considered that the sponsor has not satisfactorily demonstrated a clear correlation between surrogate PD parameters (anti-Xa and anti-IIa) and clinical outcomes. Therefore, the PD bioequivalence data from the two single-dose studies in healthy volunteers comparing the proposed enoxaparin product with the US enoxaparin reference product (Lovenox) and with the EU reference product (Clexane) cannot be extrapolated to patients with the clinical conditions proposed for approval.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 57 of 62

Therapeutic Goods Administration
13.2. Second round assessment of risksAfter consideration of the responses to clinical questions and the additional PD equivalence Study ROV-RO20-2015-01 provided by the sponsor in its response to TGA questions, it is still not possible to assess the benefits of Crusia-AFT and Crusia-AFT Forte for the proposed usage. Neither the original submission nor the sponsor’s response included clinical efficacy data in patients with any of the clinical conditions for which registration of Crusia-AFT and Crusia-AFT Forte are being sought.
The sponsor justified the absence of therapeutic equivalence studies on the basis that it considered that the comparative molecular analysis data, nonclinical PD data and clinical PD bioequivalence data supported the essential similarity of Crusia and Clexane. Therefore, the sponsor argued that a bridging therapeutic equivalence study comparing the two products administered SC for the prevention of venous thromboembolism (VTE) in patients undergoing surgery with high VTE risk (for example, major orthopaedic surgery) was not required. Consequently, the sponsor considered that no other clinical studies for other indications supporting the clinical efficacy and safety of Crusia administered by SC injection were required. In addition, the sponsor considered that the PD bioavailability of Crusia following IV administration could be estimated from the comparative PD bioequivalence study of Crusia following SC administration. Therefore, a therapeutic equivalence study comparing Crusia and Clexane administered as an initial IV dose for the treatment of acute STEMI, in conjunction with a fibrinolytic agent was not required.
However, it is considered that the sponsor should submit a clinical therapeutic equivalence study comparing Crusia and Clexane administered SC for the prevention of venous thromboembolism (VTE) in patients undergoing surgery with high VTE risk (for example, major orthopaedic surgery). The sponsor has not demonstrated a clear correlation between surrogate PD parameters (anti-FXa and anti-FIIa) and clinical outcome. If comparable efficacy and safety of Crusia and Clexane administered SC for the prevention of VTE had been demonstrated in surgical patients at high risk of the condition, then the sponsor would have been in a position to justify extrapolation of the results of this study to other indications. In the absence of a bridging study, there are no clinical data supporting the efficacy and safety of Crusia for any of the proposed indications for which the product is to be administered by SC injection. In addition, as previously argued in this document, the PD bioequivalence of Crusia and Clexane following IV administration based on anti-IIa activity cannot be predicted from the SC data. Therefore, it is considered that a therapeutic equivalence study comparing Crusia and Clexane administered as an initial IV dose for the treatment of acute STEMI, in conjunction with a fibrinolytic agent, is required to support approval for this indication.
It is considered that the safety data from the single dose PD equivalence studies in healthy volunteers comparing the proposed enoxaparin product with the US enoxaparin reference product (Lovenox) and the EU enoxaparin reference product (Clexane) cannot be meaningfully extrapolated to patients with the medical conditions of interest. Comparative safety data from a submitted efficacy trial would have been sufficient to provide an adequate pre-marketing safety database (LMWH biosimilar guidelines (EMEA/CHMP/BMWP/118264/2007)). However, the sponsor elected not to submit such a study and the justification for a waiver from the requirement to submit clinical safety data is considered to be unsatisfactory.
Other risks that have not been adequately addressed in the submission relate to the absence of a satisfactory justification for the 80% to 125% PD equivalence interval used in Studies ROV-RO20-2011-01 and ROV-RO20-2015-01, and the lack of any immunogenicity data for Crusia-AFT or Crusia-AFT Forte from a therapeutic clinical efficacy and safety study.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 58 of 62

Therapeutic Goods Administration
13.3. Second round assessment of the benefit-risk balanceAs it is not possible to assess the benefits or risks of Crusia-AFT and Crusia-AFT Forte based on the submitted data and the additional data provided with the sponsor’s response to the first round clinical evaluation report, it is not possible to assess the benefit-risk balance of the products for the proposed usage. Therefore, for regulatory purposes the benefit-risk balance of Crusia for the proposed indications is considered to be unfavourable.
14. Second round recommendation regarding authorisation
It is recommended that the application to register Crusia-AFT and Crusia-AFT Forte be rejected for the following reasons:
1. No clinical efficacy data relating to any of the proposed indications have been submitted. The sponsor’s justification for not submitting at least one adequately powered, randomised, double blind, parallel group clinical trial establishing therapeutic equivalence of the proposed enoxaparin sodium product with the Australian registered reference product (Clexane) is considered to be unsatisfactory. It is considered that efficacy in the target patient populations for the proposed indications cannot be inferred from the pharmacodynamic bioequivalence data from the two, single-dose studies in healthy volunteers (Studies ROV-RO20-2011-01 and ROV-RO20-2015-01). It is considered that the sponsor has not satisfactorily established a correlation between surrogate PD parameters (anti-Xa and anti-IIa) and clinical outcome. The absence of a clinical therapeutic equivalence study precludes the known efficacy and safety data for Clexane (Australian registered product) being safely extrapolated to Crusia. The sponsor’s justification for not providing a therapeutic equivalence study is considered to be unsatisfactory for the reasons provided in the conclusions in Section 5 of this document.
2. No clinical safety data relating to any of the proposed indications have been submitted. Comparative safety data from an efficacy trial would have been sufficient to provide an adequate pre-marketing safety database. However, the sponsor elected not to submit an efficacy trial. The sponsor’s justification for not submitting clinical safety data is considered to be unsatisfactory for the reasons provided in the conclusions in Section 8 of this document.
3. Other clinical limitations of the submitted data include:
a. No pharmacodynamic bioequivalence studies comparing the proposed enoxaparin sodium product with the Australian reference product (Clexane) were submitted. No clinical studies were submitted bridging the data for Lovenox (USA) used as the reference product in Study ROV-RO20-2011-01 to Clexane (Australia) or for Clexane (EU) used as the reference product in Study ROV-RO20-2015-01 to Clexane (Australia). Therefore, there are no clinical data establishing the PD equivalence of the proposed formulation (Crusia) and the Australian reference product (Clexane). This raises doubts about the relevance of the two PD bioequivalence Studies ROV-RO20-2011-01 and ROV-RO20-2015-01) to Australian clinical practice.
b. No single dose IV PD equivalence study in healthy subjects comparing the proposed enoxaparin sodium product to the Australian reference product (Clexane) was submitted. The sponsor’s justification for a waiver of the requirement for such a study is considered to be inadequate. The PD equivalence of Crusia and Clexane following IV administration based on anti-IIa activity cannot be predicted from the SC data.
c. No adequate justification has been provided for selecting 80 to 125% as the PD equivalence interval in Studies ROV-RO20-2011-01 and ROV-RO20-2015-01. The
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 59 of 62

Therapeutic Goods Administration
sponsor’s justification was based on the bioequivalence guideline relating to conventional chemical entities (CPMP/EWP/QWP/1401/98 Rev. 1/Corr). This guideline specifies the use of plasma drug concentrations (that is, Cmax and AUC) to establish bioequivalence rather than PD outcomes. Furthermore, this guideline expressly states that its scope is limited to chemical entities.
15. ReferencesBarras M et al. Modelling the occurrence and severity of enoxaparin induced bleeding and bruising
events. Br J Clin Pharmacol 2009;68:700-11.Barras M. Anti-Xa assays Australian Prescriber. 2013;36:4. Dahl O et al. A critical appraisal of bleeding events reported in venous thromboembolism prevention
trials of patients undergoing hip and knee arthroplasty. J Thromb Haemos 2010;8: 1966-75.Desjardins L, et al. Correlation of plasma coagulation parameters with thromboprophylaxis, patient
characteristics, and outcome in the MEDENOX study. Arch Pathol Lab Med 2004; 128(5): 519-26.FDA Guidance Department of Health and Human Services, Food and Drug Administration, Center for
Drug Evaluation and Research (US). Draft Guidance on Enoxaparin Sodium. Feng L et al. Bioequivalence of generic and branded subcutaneous enoxaparin: a single-dose,
randomized-sequence, open-label, two-period crossover study in healthy Chinese male subjects. Clin Ther. 2009;31(7):1559-67.
Frydman A. Low-molecular-weight heparins: an overview of their pharmacodynamics, pharmacokinetics and metabolism in humans. Haemostasis. 1996; 26 Suppl 2:24-38.
Gadiko C et al. Pharmacokinetic parameters to be evaluated for selected Low Molecular Weight Heparins in Bioequivalence Studies. Int J Pharm Sci Res. 3(11); 4065-4072.
Gomes M, et al. Generic versus branded enoxaparin in the prevention of venous thromboembolism following major abdominal surgery: report of an exploratory clinical trial. Clin Appl Thromb Hemost 2011; 17(6): 633-9.
Gucalp A et al. Skin necrosis induced by generic enoxaparin. American Journal of Hematology. Letter to the editor. Published online 24 January 2013.
Harenberg et al. Update of the recommendations on biosimilar low-molecular-weight heparins from the Scientific Subcommittee on Control of Anticoagulation of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2013; 11: 1425-5.
Kaffenberger B and Bekaii-Saab T. Clinical and Applied Thrombosis/Hemostasis. 2012;18(1):104-106.Kelton J and Warkentin T. Heparin-induced thrombocytopenia: a historical perspective. Blood 2008;
112: 2607-16.Kuczka K et al. Biomarkers and coagulation tests for assessing the biosimilarity of a generic low-
molecular-weight heparin: results of a study in healthy subjects with enoxaparin. J Clin Pharmacol. 2008;48(10):1189-96. Epub 2008 Aug 20.
Levine M et al. The relationship between anti-factor Xa level and clinical outcome in patients receiving enoxaparin low molecular weight heparin to prevent deep vein thrombosis after hip replacement. Thromb Haemost 1989;6 2 (3):940-4.
Lim W et al. Meta-analysis: Low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144: 673-84.
Montalescot G et al. Anti-Xa activity relates to survival and efficacy in unselected acute coronary syndrome patients treated with enoxaparin. Circulation 2004 Jul 27;110(4):392-8.
Newman P et al. Heparin-induced thrombocytopenia: IgG binding to PF4-heparin complexes in the fluid phase and cross-reactivity with low molecular weight heparin and heparinoid. Thromb Haemost 1998; 80: 292-7.
Noble S et al. Enoxaparin. A reappraisal of its pharmacology and clinical applications in the prevention and treatment of thromboembolic disease. Drugs. 1995; 49(3):388-410.
Sagedal et al. A single dose of dalteparin effectively prevents clotting during haemodialysis. Nephrol Dial Transplant 1999;14:1943-7.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 60 of 62

Therapeutic Goods Administration
Sanchez-Pena et al., Anti-factor Xa kinetics after intravenous enoxaparin in patients undergoing percutaneous coronary intervention: a population model analysis. Br J Clin Pharmacol. 2005 Oct 60(4): 364-373.
Rauova L, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 2005;105:131-8.
Wei M and Ward S. The anti-Factor Xa range for low molecular weight heparin thromboprophylaxis. Hematol Rep. 2015 Nov 23; 7 (4): 5844.
Submission PM-2015-04749-1-3 Extract from the Clinical Evaluation Report for Crusia AFT enoxaparin sodium AFT Pharmaceuticals
Page 61 of 62

Therapeutic Goods AdministrationPO Box 100 Woden ACT 2606 Australia
Email: [email protected] Phone: 1800 020 653 Fax: 02 6232 8605https://www.tga.gov.au


















