
Aubrey de Grey - Ending Aging
366
Transcript of Aubrey de Grey - Ending Aging



E N D I N G AGING


E N D I N G A G I N G
The Rejuvena t ion
Break th roughs That
Could Reverse Human
Aging in Our Li fe t ime
A u b r e y d e G r e y , P h . D . ,
w i t h M i c h a e l R a e
S T . M A R T I N ' S P R E S S N E W Y O R K

ENDING AGING. Copyright © 2007 by Aubrey de Grey. All rights reserved. Printed in the
United States of America. No part of this book may be used or reproduced in any
manner whatsoever without written permission except in the case of brief quotations
embodied in critical articles or reviews. For information, address
St. Martin's Press, 175 Fifth Avenue, New York, N.Y. 10010.
www. stmartins.com
LIBRARY OF CONGRESS CATALOGING-IN-PUBLICATION DATA
De Grey, Aubrey D. N. J . , 1 9 6 3 -
Ending aging : the rejuvenation breakthroughs that could reverse human aging
in our lifetime / Aubrey de Grey ; with Michael Rae.—1st. ed.
p. cm.
ISBN-13: 978-0-312-36706-0
ISBN-10: 0-312-36706-6
1. Longevity. 2. Aging—Molecular aspects. 3. Biotechnology. I. Rae, Michael.
II. Title.
QP85.D348 2007
612.6'8—dc22
2007020217
First Edition: September 2007
1 0 9 8 7 6 5 4 3 2 1

Aubrey dedicates this book as follows: "To the tens of millions
whose indefinite escape from aging depends on our actions today."
Michael dedicates this book as follows: "To the two tenders of the
flames that have inspired me throughout this work. To April Smith,
for erupting, like Athena, out of the secret depths of my mind,
raining Greek fire on my Manichee heart, reigniting smoldering
embers I had thought long extinguished, and opening up the
promise of a shared indefinite tomorrow; and to Dr. Aubrey de
Grey, for tirelessly and courageously bearing Promethean fire to a
world yet shivering under the winter of age-related death and decay,
kindling the sparks that we must fan into a blaze that will cast out
its obscuring darkness and melt its frozen grip."


Contents
Preface ix
P a r t One 1
1. The Eureka Moment 3
2. Wake Up—Aging Kills! 7
3. Demystifying Aging 16
4. Engineering Rejuvenation 32
P a r t Two 47
5. Meltdown of the Cellular Power Plants 49
6. Getting Off the Grid 77
7. Upgrading the Biological Incinerators 101
8. Cutting Free of the Cellular Spider Webs 134

Viii C O N T E N T S
Notes
Glossary
Index
341
363
379
9. Breaking the Shackles of AGE 164
10. Putting the Zombies to Rest 200
11. New Cells for Old 238
12. Nuclear Mutations and the Total Defeat of Cancer 274
P a r t T h r e e 309
13. Getting from Here to There: The War on Aging 311
14. Bootstrapping Our Way to an Ageless Future 325
15. War Bonds for the Campaign Against Aging 335

Preface
The biomedical revolution described in this book is still some way
off—at least a few decades, maybe more. Why, you may then ask, should
you concern yourself with it now?
The answer is simple: Once you know what I have to tell you, you'll
want to make it happen sooner, and some of you will put that desire into ac-
tion. The more people are aware of what has now become foreseeable in
the fight against our oldest foe, aging, the faster it will become acceptable
to "come out" as an ardent opponent of aging, and then unacceptable not
to. We aren't close enough to this revolution to put accurate timescales on
its arrival, but we are close enough that our action (or inaction . . . ) today
will affect the date at which aging is defeated.
In fact, we've been at that point for a few years now. It could, therefore,
be argued that I should have written this book sooner. Well, maybe I should
have—but there's a trade-off: with every year that has passed since I devel
oped the key concepts described here, progress has been seen in the labora
tory. Every step of this progress has strengthened the case that the overall
scheme will succeed, so the book as a whole is more compelling than it
could have been a year or three ago. In fact, without the diligent efforts of a

X P R E F A C E
large number of scientists within and beyond biogerontology, my plan for
defeating aging could not exist.
Another reason this book has only been written now is the usual one:
books don't write themselves, and I've been spending every waking hour
engaged in other work to further the anti-aging mission. Without doubt,
you would not have this book in your hands today if it were not for the dili
gent work of my research assistant Michael Rae, who dedicated much of
2006 to it: he can take credit for most of the text of Part 2.
Michael is not the only person without whom this book could not have
come to pass. Thanks to Peter Ulrich for painstakingly going over the fasci
nating history of patient work, inspired reasoning, and scientific serendip
ity behind the development of alagebrium. Any misunderstandings of this
story are Michael's. Special thanks go to our graphics team, who prepared
the illustrations: Bram Thijssen, Bryan English, Benjamin Martin, Tyler
Chesley, Zachary Bos, Hoyt Smith, and their coordinator, Jeff Hall. Addi
tionally, Michael and I received outstanding editorial help from Methuselah
Foundation volunteers Reason, Anne Corwin, and David Fisher. Our
agent, John Brockman, and his staff were tremendously efficient in shep
herding the book through the worldwide publication process, and our edi
tor at St. Martin's, Phil Revzin, also provided invaluable editorial input.
And finally, my work on this book has, as with all my contributions to the
crusade against aging, depended hugely on the unswerving intellectual and
emotional support of my beloved wife, Adelaide Carpenter.
I hope that this book will enjoy a wide readership; if it does, most read
ers will be nonbiologists and certainly nonbiogerontologists. Some, how
ever, will be people who do possess expertise in these areas. To that group
I would like to make clear at the outset that, in presenting SENS, the
"Strategies for Engineered Negligible Senescence," to a general audience, I
have not been able to delve into every nook and cranny of the relevant sci
ence, and you will surely identify aspects of SENS that, if what you read
here were all there was to it, would seem flawed. I merely remind you now
that this book is not all there is to SENS, and that, if you see what seems to
you to be a slam-dunk objection to what I say, you should consult my pub
lished academic work (and, preferably, consult me personally, too) before
dismissing it.
However, the above applies only to "errors" of omission, of course. Any
errors of commission are, I fully accept, my responsibility and mine alone.

Part One


T h e E u r e k a M o m e n t
Marriott Hotel, Manhattan Beach, California.
June 25, 2000.
Four o'clock.
In the morning.
It was 4 A . M . in California, but my body insisted on reminding me
that it was noon in Cambridge. I was exhausted from the intercontinental
flight and by a day spent in debate with some of the most influential per
sonalities in biogerontology, at an invitation-only brainstorming workshop
on ideas to combat aging. Evolutionary biologist Michael Rose was there.
So were calorie restriction researchers Richard Weindruch and George Roth,
nanotechnologist Robert Freitas, and several others. But I couldn't sleep:
On top of the mismatch between biological and geographical clocks, I was
frustrated at what I saw as the day's failure to make any real progress to
ward a concrete, realistic anti-aging plan. As I dozed and pondered, a ques
tion on the nature of metabolism and aging wormed its way into my brain
and wouldn't let go.
In my bleary irritation, I sat up, ran my hands over my beard, and be
gan pacing the room, turning over the quandary in my mind. "Normal" me
tabolism was just so messy, and the raging debates in the biogerontology
literature showed how difficult it was to determine what paced what: which
metabolic disruptions were causes of aging, and which were effects (or sec
ondary causes) that would simply disappear if the underlying primary
causes were addressed. How could we make a positive difference in such a
complex, poorly understood system? How could any meaningful change
1

4 E N D I N G A G I N G
made in metabolism not be like a butterfly flapping its wings—apt to cause
large, unwanted storms further down the line?
Then a second line of thought began to form in my mind—idly at first,
just as a notion. The real issue, surely, was not which metabolic processes
cause aging damage in the body, but the damage itself. Forty-year-olds have
fewer healthy years to look forward to than twenty-year-olds because of dif
ferences in their molecular and cellular composition, not because of the
mechanisms that gave rise to those differences. How far could I narrow
down the field of candidate causes of aging by focusing on the molecular
damage itself?
Well, I thought, it can't hurt to make a list. . .
There are mutations in our chromosomes, of course, which cause can
cer. There is glycation, the warping of proteins by glucose. There are the
various kinds of junk that accumulate outside the cell ("extracellular aggre
gates"): beta-amyloid, the lesser-known transthyretin, and possibly other
substances of the same general sort. There is also the unwholesome goo
that builds up within the cell ("intracellular aggregates"), such as lipofus-
cin. There's cellular senescence, the "aging" of individual cells, which puts
them into a state of arrested growth and causes them to produce chemical
signals dangerous to their neighbors. And there's the depletion of the stem
cell pools essential to healing and maintenance of tissue.
And of course, there are mitochondrial mutations, which seem to dis
rupt cellular biochemistry by increasing oxidative stress. I had for a few
years felt optimistic that scientists could solve this problem by copying mi
tochondrial DNA from its vulnerable spot at "ground zero," within the
free-radical generating mitochondria, into the bomb shelter of the cell nu
cleus, where damage to DNA is vastly rarer.
Now, if only we had solutions like that for all of this other stuff, I mused,
we could forget about the "butterfly effect" of interfering with basic metabolic
processes, and just take the damage ITSELF out of the picture.
Hmm.
Well, I thought, why the bloody hell not?
I went back over my list. Protein glycation? A biotech startup was al
ready running clinical trials using a drug that had been shown to break the
dysfunctional handcuffing of the proteins that this process caused. The
extracellular aggregates? Here again, animal studies had shown that you
could just remove the damage, in this case by vaccinating against the amy
loid plaque and letting immune cells gobble the stuff up. In theory, at least,
there were all kinds of ways to deal with cellular senescence, though I wasn't

T H E E U R E K A M O M E N T 5
sure which of them would ultimately pan out. Anyone who'd read a news
paper in the last year knew that scientists were hotly pursuing a way to deal
with the loss of cells: stem cells, cultured in the lab and delivered as a reju
venating cellular therapy. Lipofuscin? It was at this point in my survey that I
began to feel I might really be on to something, because just a year previ
ously I'd come up with a way to eliminate lipofuscin that, although ex
tremely novel, had already secured the enthusiastic interest of a few of the
top researchers in that area. I didn't have any radical new ideas up my sleeve
for cancer; it was going to have to rely (for now, at least) on other people's
ideas. But that was okay: after all, there was already a huge effort under way
to deal with it. And as for other problems arising from nuclear mutations, I
had recently come to the admittedly counterintuitive conclusion that they
were not in fact a major cause of age-related cellular dysfunction.
I went over my list again and again, and as I did so I became ever surer
that there was no clear-cut exception. The combination of my own idea for
eliminating intracellular garbage like lipofuscin; the idea I'd been champi
oning for a few years for making mitochondrial mutations harmless; and the
various other therapies being worked on by others around the world for
addressing glycation, amyloid accumulation, cell loss, senescent cells and
cancer—it seemed that this was really and truly an adequately exhaustive list.
Not necessarily totally exhaustive—there certainly might be other things go
ing wrong in the body—but very possibly comprehensive enough to give a
few decades of extra life to people who are already in middle age before we
start the treatments. And that was certainly a much more promising first step
than anything that had been suggested the previous day, or in the many con
ferences and articles that I'd devoured over the previous few years.
For decades, my colleagues and I had been earnestly investigating ag
ing in the same way that historians might "investigate" World War I: as an
almost hopelessly complex historical tragedy about which everyone could
theorize and argue, but about which nothing could fundamentally be done.
Perhaps inhibited by the deeply ingrained belief that aging was "natural"
and "inevitable," biogerontologists had set themselves apart from the rest
of the biomedical community by allowing themselves to be overawed by the
complexity of the phenomenon that they were observing.
That night, I swept aside all that complexity, revealing a new simplicity
in a complete redefinition of the problem. To intervene in aging, I realized,
didn't require a complete understanding of all the myriad interacting pro
cesses that contribute to aging damage. To design therapies, all you have to
understand is aging damage itself: the molecular and cellular lesions that

6 E N D I N G A G I N G
impair the structure and function of the body's tissues. Once I realized that
simple truth, it became clear that we are far closer to real solutions to treat
ing aging as a biomedical problem, amenable to therapy and healing, than
it might otherwise seem.
Grabbing a notepad, I jotted down the molecular and cellular changes
that I could confidently list as important targets for the new class of anti-
aging therapies that I would soon call SENS, the "Strategies for Engineered
Negligible Senescence." Each of them accumulated with age in the body
throughout life and contributed to its pathological decay at later ages. As
far as I could tell, the list was exhaustive, but I'd present it to my colleagues
and see if they could add to it. I rushed downstairs before breakfast to tran
scribe my scrawled notes onto a flipchart in the meeting room. I was burst
ing to present my new synthesis to my esteemed colleagues. But truth be
told, I already knew full well that at this first hearing they'd greet it with
blank stares. The paradigm shift was just too great.

2
W a k e Up — A g i n g K i l l s
How many lives do you think you could save, in your life?
This is not a trick question. But in order to make it even more precise,
I'm going to modify it a little. When we speak of saving lives, we mean giv
ing the beneficiaries of our action the chance to live longer than they could
otherwise have lived. However, when we ask in detail about the importance
of saving a life, we may not regard all lives equally. For example, saving an
eighty-year-old from drowning may give him only a few extra years of life
before he's likely to die of something else, whereas saving a child from
drowning gives him a probable seventy years or more of extra life. We may
also take into account the quality of life of the persons whose lives we
save—predominantly their health. So here's my modified question:
How many healthy, youthful years in total do you think you
could add to people's lives, in your life?
The ultimate purpose of this book is to show you that you could add
many more years than you may currently think. So many, in fact, that now is
the time to decide whether you want to. The way you can do this is by help
ing to hasten the defeat of aging. The specifics of how you can help—by

8 E N D I N G A G I N G
donating money or time to the Methuselah Foundation's Mprize fund or its
SENS research funding program—will be the topic of Chapter 15; in this
chapter I'll restrict myself to communicating the magnitude of what those
efforts can achieve in humanitarian terms.
I'll start with some numbers. Around 150,000 people die each day
worldwide—that's nearly two per second—and of those, about two-thirds
die of aging. That's right: 100,000 people. That's about thirty World Trade
Centers, sixty Katrinas, every single day. In the industrialized world, the
proportion of deaths that are attributable to aging is around 90 percent—
yes, that means that for every person who dies of all causes other than aging
added together, be it homicide, road accidents, AIDS, whatever, some
where around ten people die of aging. 1
And it's worse than that. Look again at my expanded question and
you'll notice a couple of adjectives: "healthy" and "youthful." Many people,
when thinking about the idea of adding years to life, commit the "Tithonus
error"—the presumption that, when we talk about combating aging, we're
only talking about stretching out the grim years of debilitation and disease
with which most people's lives currently end. 2 In fact, the opposite is true:
the defeat of aging will entail the elimination of that period, by postponing
it to indefinitely greater ages so that people never reach it. There will, quite
simply, cease to be a portion of the population that is frail and infirm as a re
sult of their age. So it's not just extending lives that I'll be telling you about
in this book: it's the elimination of the almost incalculable amount of
suffering—experienced not only by the elderly themselves, of course, but by
their loved ones and carers—that aging currently visits upon us. Oh, and
there's the minor detail of the financial savings that the elimination of aging
would deliver to society: it's well established that the average person in the
industrialized world consumes more health-care resources in his or her last
year of life than in an entire life up to that point, irrespective of age at death,
so we're talking about trillions of dollars per year.
In this book I will explain the scientific and technological basis for my
view that we can probably eliminate aging as a cause of death this
century—and possibly within just a few decades, soon enough to benefit
most people currently alive. But first, I need to get you interested—not just
in the sense of entertainment, the sense in which you might read a good
story, but in the sense of realizing that as and when this becomes possible it
will be rather a good thing. And I've been in this business long enough to
know that a description of the level of suffering that would be averted and
the number of lives that would be saved does not, on its own, convince

W A K E U P — A G I N G K I L L S ! 9
most people that it would be a good thing if aging were defeated. So I hope
you'll forgive me if I am blunt and to the point in this chapter, before I
move on to the science and technology that will get the job done.
Why Did I Write This Book?
I'm a scientist and technologist, and in an ideal world I would spend essen
tially all my time working on the scientific and technological details of my
life's goal, defeating aging. I wouldn't spend much time doing media inter
views, or giving public lectures—or even writing books. But there's some
thing about people's attitudes to aging that, for now, changes my priorities.
I call it the pro-aging trance. I'm going to start my discussion of the pro-
aging trance with a comparison.
Here in the United Kingdom, just as across the whole Western world,
there is a campaign of increasing ferocity against smoking. All cigarette pack
ets come with health warnings. Not just tiny inconspicuous health warnings
in cautious scientific language, either—warnings of the most hard-hitting and
in-your-face nature possible. The simplest and shortest consists of just two
words, typically printed in black on a stark white background:
Smoking Kills
And, slowly but surely, smoking is becoming less popular. Just like
drunk driving before it, smoking is becoming socially disreputable. It's a
long, hard road, though: not just because nicotine is addictive, but because
youngsters continue to take up smoking despite the social stigma increas
ingly attached to it.
It's that latter point, the continued influx of new young addicts, that is
my focus here. I've used smoking as my chosen analogy not in order to con
demn the smokers among my readers—not at all. No, my focus here is
something altogether less controversial, because the battle to protect young
sters from taking up smoking is one that virtually all adults, smokers or not,
support. My reason for mentioning it here is timeliness: this battle is still be
ing waged, so we can examine at close quarters the contradictions in our at
titudes, both as individuals and as a society, that make the battle so hard to
win. With specific diseases, there is no argument: the more we can do to de
feat them, the better. But with smoking, even though it causes some of those
self-same diseases, somehow society is itself subject to an addiction that robs

1 0 E N D I N G A G I N G
it of its rationality concerning new young addicts. We face every day the bru
tal disconnect between allowing cigarettes to be advertised and sold widely
and seeing how much they blight and shorten the lives of those who fall un
der their spell. And it's just the same, I claim, with aging.
There are two potential reasons why smoking is declining in popularity
and in public acceptability. One is that many people find it unattractive—
they don't like the smell (or, in more intimate contexts, the taste). But it's
hard to believe that this can be the main trigger for the rather recent change
of sentiment against smoking, because today's tobacco is surely no more
off-putting than tobacco of a century or three ago. Thus, I think it's clear
that the main reason so many people now disapprove of smoking is its
other downside, which was not much appreciated even half a century ago:
It's really rather bad for you, and also for those around you. Most of all, it
massively increases your risk of getting fatal lung cancer, which not only
shortens your life but also makes your declining years really miserable.
My goal with this book, as for all my outreach work, is to inject mo
mentum into a similar shift of public opinion concerning aging. I have been
aware for many years that most people do not think about aging in the same
way that they think about cancer, or diabetes, or heart disease. They are
strongly in favor of the absolute elimination of such diseases as soon as pos
sible, but the idea of eliminating aging—maintaining truly youthful physi
cal and mental function indefinitely—evokes an avalanche of fears and
reservations. Yet, in the sense that matters most, aging is just like smoking:
It's really bad for you. It shortens your life (see Chapter 14 for an assess
ment of just how much), it typically makes the last several years of your life
rather grim, and it also makes those years pretty hard for your loved ones.
So let's look a little more closely at why aging is so passionately defended.
The Motivation for the Pro-Aging Trance
First of all, let me be clear that I realize there's an immense gulf between
people's attitude to modest postponement of aging and their attitude to the
topic of this book, the genuine elimination of aging as a cause of infirmity
and death. The anti-aging industry is huge, despite the (shall we say) highly
variable ability of its products to do what they say they can do, and that can
only be because people are not very happy to see themselves falling apart, or
to be seen to be falling apart. Yet, the prospect of eventually being able to
combat aging as well as we can currently combat most infectious diseases—

W A K E U P - A G I N G K I L L S ! 11
essentially to eliminate aging as a cause of death, in other words—strikes
terror into most people: Their immediate (and, I must point out, often high-
pitched) reaction is to raise the specter of uncontrollable overpopulation, or
of dictators living forever, or of only a wealthy elite benefiting, or any of a
dozen other concerns.
Now, I'm certainly not saying that these objections are dumb—not at
all. We should indeed be considering them as dangers that we should work
to preempt by appropriately careful forward planning. No: what shocks me
is not that these concerns are raised, but the way they're raised. People who
are totally rational and open to discourse on any other matter approach the
topic of defeating aging with a resistance to debate that virtually defies de
scription. The determination with which people work to change the sub
ject, to relegate the conversation to an exchange of witticisms, or simply to
cast the opponent of aging as a deluded nincompoop has to be encoun
tered to be believed.
Perhaps you're wondering whether I've forgotten that I'm talking
about you here. But understand that I'm not castigating you at all, because
my remarks so far have dealt only with the logic of why aging should be
fought, and life is not all about logic. There is a very simple reason why so
many people defend aging so strongly—a reason that is now invalid, but
until quite recently was entirely reasonable. Until recently, no one has had
any coherent idea how to defeat aging, so it has been effectively inevitable.
And when one is faced with a fate that is as ghastly as aging and about
which one can do absolutely nothing, either for oneself or even for others,
it makes perfect psychological sense to put it out of one's mind—to make
one's peace with it, you might say—rather than to spend one's miserably
short life preoccupied by it. The fact that, in order to sustain this state of
mind, one has to abandon all semblance of rationality on the subject—and,
inevitably, to engage in embarrassingly unreasonable conversational tactics
to shore up that irrationality—is a small price to pay.
A Word About SENS Skepticism
This book is a description of SENS (Strategies for Engineered Negligible
Senescence), my "project plan" for defeating aging. I expect that this will be
many readers' first encounter with SENS, but others will have come across
it before. In particular, if you have had an interest in life extension for some
time, there's a good chance that you've already come across accounts of

12 E N D I N G A G I N G
SENS in the mainstream media. If so, you'll be well aware that, while many
highly credentialed gerontologists have applauded SENS, others have
greeted it with strong criticism—even derision. So far in this chapter I have
only addressed the flaws in people's reasons for feeling that the defeat of ag
ing might not be desirable. But in order to ensure that you read this book
with real care, and moreover that you then go out and do something to help
the anti-aging effort, I also need to make sure that you understand that the
defeat of aging is feasible. Therefore, I include here a brief account of where
the debate about SENS's chances of success currently stands.
I must first make sure you appreciate that it is the norm for radical new
concepts that receive a lot of attention to arouse a sharp division of opinion
among expert commentators. In many cases, the establishment detractors
are absolutely right and the upstart new idea really is misguided. Very often,
however, the detractors have failed to acquire—even avoided acquiring—a
detailed understanding of what they are criticizing and have been driven
more by vested interests than by scientific argument. If you are not a scien
tist you may feel that this is an unfair suggestion, but the intellectual and
emotional investment that senior scientists have made in their beliefs is a
powerful opponent to objectivity: all scientists acknowledge this problem
privately, if not publicly. It has been memorably summarized by a number
of the world's most eminent scientists over the years; for example, the
physicist Max Planck observed over eighty years ago that "science advances
funeral by funeral," and the biologist J. B. S. Haldane noted that "there are
four stages of acceptance: (i) this is worthless nonsense; (ii) this is an inter
esting, but perverse point of view; (iii) this is true, but quite unimportant;
(iv) I always said so."
Since I work on aging in order to hasten its defeat, and not in order to
become rich and famous, I am extremely keen to identify any major holes
in SENS so that, if they indeed exist, I can go back to the drawing board
without delay. To this end, I talk to my most prominent biogerontologist
critics all the time about SENS. I am invariably driven to the view that they
are indeed guilty of reacting to my conclusion (that SENS can totally defeat
aging) without studying the reasoning behind that conclusion—but I of
course appreciate that I, too, may be unable to be objective in this matter.
For this reason, and also because the speed of implementation of SENS de
pends greatly on both public and academic acceptance that it might work,
I have worked hard in recent years to generate unbiased evidence as to
whether SENS is sense or nonsense. In 2006, I achieved this rather deci
sively, with the assistance of the prominent magazine MIT Technology Re-

W A K E U P - A G I N G K I L L S ! 13
view. After publishing a rather negative portrayal of SENS in 2005, TR dis
covered that the mainstream gerontologists on whose opinions it had relied
in choosing to do so were unwilling to back up their assessment with any
scientific detail. TR then admirably put itself at risk of considerable loss of
face by organizing a prize challenge to settle the matter.3
In order to win the SENS Challenge, one or a group of credentialed bi
ologists had to write a demolition of SENS that I was unable to rebut to the
satisfaction of a panel of expert judges. The panel of judges had to be
demonstrably impartial, of course, with no connection either to me or to
my critics, but yet well versed in biotechnology; TR succeeded in appoint
ing a superb five-person panel including the biotech luminary Craig Venter.
TR put up $10,000 as a prize, and the Methuselah Foundation contributed
the same amount. A group of nine highly credentialed biogerontologists
obligingly submitted a coauthored entry, as did two other scientists inde
pendently. All three entries were unanimously and emphatically judged to
fall decisively short of a demonstration that SENS is not worth trying.
Now, I'm certainly not trying to say that this proves that SENS will in
deed deliver the defeat of aging: there's only one way to answer that, which
is to implement it and see what happens. But my critics made the stronger
claim that SENS is so implausible that there's no need to try to implement
it. That claim has been incontrovertibly refuted by the SENS Challenge
process. So, if you find someone still eager to tell you that SENS is
fantasy—especially someone who claims to have expert knowledge in this
area—you'll know, as TR now knows, that asking such people what they
think about SENS is a great deal less reliable than asking them what they
know about SENS. And after you've read this book—especially Part 2—
you'll be equipped to come to your own conclusions.
<**P Building a Case, Chapter by Chapter
I'm a fighter at heart; I would never have made my peace with aging, how
ever lost the battle seemed. But that's not the life everyone wants, and I re
spect that. Thus, I would probably not have written this book if I thought
we were still too far from defeating aging to have any real chance of success
within the lifetimes of anyone alive today. In the next chapter I'll describe
why aging is, in principle, just as amenable to modulation and eventual elim
ination as specific diseases are, and how an inappropriate way of looking at
aging has led most gerontologists to favor eventual therapeutic approaches

14 E N D I N G A G I N G
that I consider unlikely to bear fruit. Then, Chapter 4 provides an overview
of my scheme for defeating aging within (if all goes well) only a few decades.
That concludes Part 1 of the book. In Part 2, Chapters 5 through 12 elabo
rate on the individual components of that scheme. The book concludes with
Part 3, a trio of chapters covering what I predict will be the response of so
ciety to initial successes in the laboratory a decade or so from now, how the
advances of the next few decades will be progressively refined and aging
permanently kept at bay, and how you can already help to accelerate that
crusade.
Buried inconspicuously in that last paragraph was something that I
want to make sure you don't misinterpret: a tentative time frame. Yes, I
consider that if funding is sufficient we have a 50/50 chance of developing
technology within about twenty-five to thirty years from now that will, un
der reasonable assumptions about the rate of subsequent improvements in
that technology, allow us to stop people dying of aging at any age—
equivalent to the effect of today's antiretrovirals against HIV. There are
three big caveats in that statement, though. The first is that it's only a 50
percent chance. Any technological prediction as far in the future as twenty-
five to thirty years is necessarily very speculative, and if you ask me how
soon I think we have a 90 percent chance of defeating aging I wouldn't
even be willing to bet on one hundred years. But I think a 50 percent
chance is well worth shooting for—don't you? The second caveat is that ag
ing won't be totally defeated by the initial versions of this technology; we'll
have to carry on improving it at a reasonable rate in order to keep aging
permanently at bay. I will explain all the details of that in Chapter 14.
But the third caveat is perhaps the most important: the adequacy of re
search funding. I cofounded the Methuselah Foundation in order to ad
dress that problem: at present, the pace of most of the research avenues
that we need to pursue in order to combat aging adequately is limited by
funding. If you can help to change that—whether by giving money your
self, or by influencing friends, or by writing or broadcasting on the
subject—you'll be making as much difference to the speed with which ag
ing is overcome as if you were doing the science yourself.
There's a critical point about funding that I must emphasize here: the
pivotal role of relatively small amounts of money at this early stage in the
crusade. I've complained at length in this chapter about people's reluctance
to treat aging as the curse that it is, and I hope I'm making a difference to
that attitude by my outreach activities, but realistically I know that most
people are going to sustain their pro-aging trance for a while yet, and that

W A K E U P — A G I N G K I L L S ! 15
will severely limit the availability of either public or commercial funding for
life extension research. The point where that will really change—where the
global pro-aging trance will collapse like a house of cards—will, in my view,
be when middle-aged mice are rejuvenated thoroughly enough to extend
their healthy lives by a large amount. This is a milestone that I've termed
"robust mouse rejuvenation," or RMR. The amount of money needed to
achieve it is tiny compared to how much we'll then need to spend to get the
same result in humans; but when humanity as a whole is behind the effort,
willing to pay for it with taxation, there'll be ample funds available. It's
now, when private philanthropy is the only major source of funding for
such work, that the magnitude of that private philanthropy is so critical. I'll
elaborate on this in Chapter 13.
I've discussed in this chapter why aging is defended, but I haven't said
much about how—about the common objections to the prospect of indefi
nite life extension. In many of my writings and public presentations, and on
my Web site, 4 I do address the many questions that arise concerning how
society would be different in a post-aging world, and especially how we
would handle the transition to that world. This book does not address
those issues in detail; I've decided to deal here only with the practicality of
radical life extension. I hope you'll come away with a pretty good under
standing that the genuine defeat of aging is a feasible goal. Whether it's also
a desirable goal is a question that you'll then be able to consider more
seriously—even, dare I say it, more responsibly and conscientiously—than
you could if you still thought it was science fiction.

3
D e m y s t i f y i n g A g i n g
Aging has held us in a psychological stranglehold ever since we
realized it existed, and that stranglehold remains intact to this day. I dis
cussed in Chapter 2 the effect that this has on our willingness to think ra
tionally about how terrible a thing aging is, and I explained why this
irrationality used to have a valid psychological basis while there was no hope
of combating aging, and also why it is now such a formidable obstacle.
There's a complication, though. I've told you that we've recently reached
the point where we can engage in the rational design of therapies to defeat
aging; most of the rest of this book is an account of my favored approach to
that design. But in order to ensure that you can read that account with an
open mind, I need to dispose beforehand of a particularly insidious aspect
of the pro-aging trance: the fact that most people already know, in their
heart of hearts, that there is a possibility that aging will eventually be de
feated.
Why is this a problem? Indeed, at first sight you might think that it
would make my job easier, since surely it means that the pro-aging trance is
not particularly deep. Unfortunately, however, self-sustained delusions don't
work like that. Just as it's rational to be irrational about the desirability of ag
ing in order to make your peace with it, it's also rational to be irrational about

D E M Y S T I F Y I N G A G I N G 1 7
the feasibility of defeating aging while the chance of defeating it any time
soon remains low. If you think there's even a 1 percent chance of defeating
aging within your lifetime (or within the lifetime of someone you love), that
sliver of hope will prey on your mind and keep your pro-aging trance un
comfortably fragile, however hard you've worked to convince yourself that
aging is actually not such a bad thing after all. If you're completely convinced
that aging is immutable, by contrast, you can sleep more soundly.
The key qualification in what I've just said, of course, is the phrase
"while the chance of defeating it any time soon remains low." Once that
chance becomes respectable, you're better off doing your bit to increase it
further—not just the actual laboratory work, of course, but also agitating,
cajoling, helping others (not least those with influence over research fund
ing) to awaken from their own pro-aging trance. Conversely, if the chance
of aging being defeated is really tiny despite whatever you do, the cost-
benefit balance of abandoning your comfort zone may tip the other way, in
favor of applying the same irrationality to the existence of such a possibility
as you may be doing in respect of the pros and cons of aging.
Therefore, in this chapter I'm going to describe what aging is in practi
cal terms, so as to demystify it for you. By doing so I plan to show you that
the popular presumption that aging is a phenomenon unlike all other
health conditions, somehow beyond even the theoretical reach of medical
technology, cannot be reconciled with established fact. Thus, by the end of
this chapter I aim to have placed you in the awkward position of still want
ing to believe (for your own peace of mind) that aging is immutable and
thus not worth worrying about, but no longer actually being able to believe
that. From that point on, my task will be the relatively easy one of explain
ing why our chances of defeating aging in the foreseeable future are not just
non-zero, but high enough to justify my having broken your pro-aging
trance in the first place. Justify, because once your pro-aging trance is no
more, you—yes, you—can make a difference to how soon aging is defeated,
and the fulfilment you will derive from that effort will far outweigh any
comfort you may have found in your previous certainty that aging can never
be combated.
The Illusory Boundary Between Aging and Disease
It used to be the case that people died of aging, but, if you believe what's
written on death certificates, these days they rarely do. The phrase "natural

18 E N D I N G A G I N G
causes" was the accepted term for the cause of death when it occurred at an
advanced age and in the absence of clearly defined pathology. These days,
however, that's considered inadequately informative, and coroners or their
equivalent are encouraged to enter something more specific.1
We all know, however, that quite a few people do indeed die in that
way—not from a heart attack, not from pneumonia or influenza, not from
cancer, not even from a stroke, but just peacefully, often in their sleep, be
cause their heart simply stops. These relatively lucky people indisputably
die of aging.
That brings me to the first of several times in this book when I must en
gage in the unpleasant business of exposing a serious distortion of the facts
that has been perpetrated—often unintentionally, I realize—by a large
number of senior researchers in the field of biogerontology, the study of
how aging works. This distortion has by now been generally seen for the
awful error that it was, but the disastrous consequences for the field are still
being felt, and probably will be for many years to come. Through the
1950s, '60s, and 70s , while gerontology was making its big push for recog
nition as a legitimate biological discipline, rhetoric developed to the effect
that the infirmities of aging should be viewed as separable into two distinct
phenomena: on the one hand, age-related diseases, and on the other hand,
"aging itself." This distinction was publicly defended mainly on the basis
that everyone has aging, whereas no age-related disease is universal. The
motivation for this distinction, on the other hand, was purely pragmatic: by
ring-fencing their area of work intellectually, gerontologists hoped to ring-
fence it financially, too.
And ring-fence it they did, most notably with the creation (while Pres
ident Richard Nixon was paying limited attention, so it is said) of the Na
tional Institute on Aging. 2 So far, so good. However, it's not good enough.
All gerontologists know full well that it's no accident that age-related dis
eases are age-related: they appear at advanced ages because they are conse
quences of aging, or (to put it another way) because aging is no more and
no less than the collective early stages of the various age-related diseases.
Gerontologists knew this back then, too. Thus, they also should have seen
back then that, by trumpeting the short-termist rhetoric that "aging is not a
disease," they were constructing an immense obstacle for themselves in the
longer term: the response from policy makers that, well, if it's not a disease,
why should we spend money on combating it? The era of that backlash be
gan decades ago and shows no sign of ending. Gerontologists these days
point out over and over again that if we could just postpone aging even a

D E M Y S T I F Y I N G A G I N G 1 9
little bit we would derive far more health benefits than would result from
even the most decisive breakthroughs against specific diseases, but over
and over again their paymasters fail to get the message. 3 I maintain that it is
overwhelmingly the inaccurate rhetoric of gerontologists, resulting from
their misguided policy of previous decades, that has brought about such
entrenched resistance to a simple, obvious and (within the field) universally
agreed-upon truth about the potential value of postponing aging.
I told you just now that age-related diseases are merely consequences
of aging; now I'll tell you why we know that. In the process, I'll also tell you
why aging has the range of speeds that it does—within a single individual,
and between individuals, and also between species.
Why Aging Doesn' t Need a Timer
The fact that a fair proportion of people die of natural causes, rather than
of any specific disease, might at first sight imply that aging is a process inde
pendent of diseases: something that increases people's vulnerability to dis
ease (thus making diseases more common among the elderly) but that also
kills us itself if no disease does so first. This is only partly correct. The el
derly are indeed more vulnerable to infectious diseases, because one aspect
of aging is the decline of the immune system. However, most diseases of old
age have only a minor, if any, infectious component: they are mostly or
wholly intrinsic. Take cancer, for example. A few types of cancer affect
young people, but most types are never seen in people below the age of
forty or so (except for people with very rare congenital DNA repair defi
ciencies). Some cancers are caused by viral infections—the best known of
these is cervical cancer, caused by the human papilloma virus. But the ma
jor underlying cause of cancer is the simple accumulation over time of mu
tations in our chromosomes. Mutations are inevitable: they happen as a
purely intrinsic side effect of our biology. The time they most often happen
is when the DNA of our chromosomes is replicated during the process of
cell division. The accumulation of mutations is, therefore, part of aging,
and cancer is predominantly a consequence of aging—or, if you prefer, part
of the later stages of aging.
Sounds pretty simple, doesn't it? And yet, there is a pervasive
presumption—one shared even by some biologists—that aging is some kind
of mysterious phenomenon qualitatively different from any disease: some
thing that has eluded, and thus may forever elude, biological elucidation.

20 E N D I N G A G I N G
There are a few main reasons for this presumption, so I'll briefly describe
those reasons and why they're wrong.
The first is that aging proceeds so much more slowly than specific dis
eases. So slowly, in fact, that we hardly notice its progression, whereas we
are much more keenly aware of the more rapid development of conditions
like cancer or diabetes. This is a conspicuous difference, but in fact it's just
what one would expect, because aging is a downward spiral. The more we
age, the more our self-repair functions decline, so the less able our body is
to stop us aging, so we age faster and faster. So it's to be expected that the
late stages of aging, the diseases, would go faster than the earlier stages.
Another thing that confuses people about aging is that it proceeds at
very different rates in different species but at pretty similar rates in all
members of a given species. This might be thought to imply that there is
some kind of internal clock driving the process, which is set at different
speeds in different species. The inference is that this clock is somehow im
mune to biomedical intervention, because changing its speed would re
quire us to stop being human. But that's not correct either, for two reasons.
First, even if there were such a timer, we could in principle postpone the
later stages of aging without changing the speed of the timer itself—I'll be
elaborating on this below. And second, if there were such a clock, why
shouldn't it be amenable to biomedical intervention anyway? The fact that
organisms of the same species tend to age at the same rate is just one conse
quence of the fact that they're genetically very similar to each other. It says
nothing about what can or cannot be altered by biomedical technology.
Perhaps the most common reason for the belief that there is an "aging
clock" is the fact that the various outcomes of aging (including age-related
diseases) all tend to appear at more or less the same age in different indi
viduals within a given species. Surely this must mean that there is indeed a
central aging clock, which has ticked down enough to set these diseases on
their way, right? No—and, again, this is for two main reasons.
First, this is exactly what one would expect if the debilities of old age
were just the later stages of a multifaceted decay process, just so long as that
system has one key feature: a rich degree of interconnection of the various
chains of cause and effect. If lots of things are going quietly wrong through
out life, and their accumulation is feeding back on themselves and each
other to accelerate them, they'll necessarily all proceed at more or less the
same rate and all "go critical" (explode into clinically identifiable disease)
at about the same age. And that interconnectedness is, indisputably, indeed
present in aging.

D E M Y S T I F Y I N G A G I N G 21
Second, if we think about the evolutionary basis of aging for a moment
we can easily see that, even without much interconnection between the
chains of events that lead to the various diseases of aging, we'd still expect
to have them all emerge at roughly the same age. This is because, if we had
genes that defended against one particular cause of death so well that
everyone was dead from other causes before they died of that one, those
genes would not be protected by evolutionary selection and would accu
mulate random, mild mutations from one generation to the next. Over evo
lutionary time, therefore, the quality of those genes would thereby sink
down to the point where the disease they protected against occurred at the
same age as all other age-related diseases.
Another common but incorrect reason for thinking that aging is some
how special is that it is "universal"—it happens to everyone. Well, yes: If
you live long enough, you'll exhibit signs of aging. But this is only a corol
lary of my earlier point about rates—that aging is really slow compared to
age-related disease. Because age-related diseases progress from diagnos-
ability to death rather quickly, many people die of one such disease before
the others emerge, or at least while they are still too early-stage to have been
diagnosed. But if those people hadn't suffered the disease that killed them,
they'd have lived long enough to suffer others. In fact, all the diseases of ag
ing are universal in the sense in which the question ought to be asked:
namely, you'll definitely get them if you don't get something else first.
Thus, in concluding this section I hope to have convinced you that ag
ing is not something inherently mysterious, beyond our power to fathom.
There is no ticking time bomb—just the accumulation of damage. Aging of
the body, just like aging of a car or a house, is merely a maintenance prob
lem. And of course, we have hundred-year-old cars and (in Europe any
way!) thousand-year-old buildings still functioning as well as when they
were built—despite the fact that they were not designed to last even a frac
tion of that length of time. At the very least, the precedent of cars and
houses gives cause for cautious optimism that aging can be postponed in
definitely by sufficiently thorough and frequent maintenance.
The Corollary That Even Most Experts Overlook
Everything I've explained above is well known to biogerontologists, the
people who study aging. From the way that most biogerontologists go about
exploring how to postpone aging, however, you might think they didn't

2 2 E N D I N G A G I N G
know this at all. People who work to combat specific diseases explore the way
in which the disease progresses and look for ways to disrupt that pathway. In
gerontology, however, the predominant modus operandi for designing inter
ventions is to compare organisms that age at different rates—different
species, or individuals of the same species in different conditions—and to
come up with ways to copy or extrapolate those differences so as to make ag
ing happen more slowly. This is effectively an a priori capitulation, not even
trying to dissect and disrupt the process but rather treating it as a black box.
It's especially surprising when you bear in mind that biogerontologists cer
tainly do work hard to dissect the aging process in order to understand it—
just not in order to combat it. (Unfortunately, these two goals motivate
different types of dissection.) Rather, the most promising ways to postpone
aging are by disrupting the pathways underlying it, just as we do for specific
diseases. Thus, since aging is just the accumulation of damage, we should be
looking at ways to alleviate that accumulation. I'll return to this in greater de
tail in the next chapter and beyond.
Why Fixing Aging Is Easier than Fixing Similarly
Complex Machines
Now let's move on to another reason that people often give for clinging to
the belief that aging is inherently inaccessible to biomedical intervention. If
aging is just damage, and the body is just a complex machine, it stands to
reason that we can apply the same principles to alleviating the damage of
aging as we do to alleviating damage to machines. But people sometimes
point out that the body has a host of self-repair and self-maintenance pro
cesses, which machines basically don't have, hence we're not really ma
chines at all. Thus, they claim, the maintainability of machines is no basis
for confidence that the body is in principle similarly maintainable.
Well, I invite you to think about that logic for a moment. We have
built-in repair and maintenance machinery. Why on earth would that make
it harder to maintain our bodies in good working order? Clearly the oppo
site is the case: if our bodies are doing most of the job automatically, that
leaves less for us to do with biomedical technology.
Let me stress that I'm not saying the task is easy. The body is a great deal
more complicated than any man-made machine—and what's more, we didn't
design it, so we have to reverse-engineer its workings in order to understand
it well enough to keep it running. But that doesn't change the above logic: the

D E M Y S T I F Y I N G A G I N G 2 3
natural capacity for self-repair that we're born with is our ally in the anti-
aging crusade, not our enemy.
Postponed Aging in the Lab: No Longer Just Theory
By now I may have satisfied some readers that, indeed, aging is not a mysti
cal phenomenon beyond the reach of mere, um, mortals. I'm well aware,
however, that many people find theoretical arguments only modestly per
suasive, even if no holes in those arguments seem evident. Such people—
you, perhaps—feel altogether more comfortable with a conclusion if it is
backed up by hard evidence. You'll be pleased to discover, then, that for
several decades scientists have been finding ways to lengthen the lives of
various organisms in the laboratory. Best of all, they've done this not by ex
tending those organisms' period of declining vigor at the end of life, nor (by
and large) by keeping them immature for longer, but by extending the pe
riod of peak health and vigor between maturity and frailty.
One highly robust life-extension technique was discovered over twenty
years ago by a young Canadian researcher named Michael Rose, who is
now a professor at the University of California, Irvine. Rose is an evolution
ary biologist, and at that time he already had a thorough knowledge of the
ways in which evolution optimizes a species' longevity for its ecological
niche. He realized that it might be possible to breed longer lived organisms,
rather in the vein of the Howard families in Robert Heinlein's Lazarus Long
books, by maintaining them over many generations and only allowing those
with the longest lives (actually, strictly speaking the longest reproductive
lives) to contribute to the next generation. It would take many more gener
ations than Heinlein described, but Rose was working with fruit flies,
which reach maturity only a week after their own conception. And it
worked, spectacularly: Rose was eventually able to achieve average lifes
pans twice those in his starting population. 4
This approach, impressive though it was, has a fundamental and rather
relevant limitation—a limitation that has probably not escaped you. Specif
ically: it can't be applied to you, only to your great-great-great. . . great
grandchildren. Rose knew this, too, of course, and more recently he's been
working hard to identify the genetic, and thence molecular, basis for this
life extension with a view to eventual therapies that might work on those of
us unfortunate enough to be already alive. But thus far, all he has are long-
lived distant descendents of short-lived flies.

2 4 E N D I N G A G I N G
Luckily, other laboratory life-extension successes have not shared this
drawback. The first and best-known way to delay aging in the laboratory
was discovered way back in the 1930s by a researcher named Clive McCay,
working with laboratory mice. 5 It is called calorie restriction—or some
times dietary restriction, energy restriction or food restriction. It's an ex
traordinarily simple concept: If you feed rodents (or, in fact, a wide variety
of other animals) a bit less than they would like, they tend to live longer
than if they have as much food as they want. This is not simply because
such animals tend to overeat given the chance and become obese: animals
that "eat sensibly" and maintain a constant body weight throughout most
of their lives still live less long than those given less food.
The next researcher (not counting Rose) to take the postponement of
aging a major leap forward was a geneticist working with a third, almost
equally widely studied, model organism: the nematode worm Caenorhabdi-
tis elegans. His name is Tom Johnson. He was not, strictly speaking, the dis
coverer of the phenomenon I will describe here—that honor goes to one of
his coworkers—but he spearheaded the work on it for some years and that
work has become identified with him, so I'll focus on him for the moment.
What Johnson and his colleagues discovered and researched was a muta
tion in a single, identified gene, which on its own—without any of the sus
tained selective pressure employed by Rose—added at least 50 percent to the
youthful adult lifespan of his worms. 6 This was an immense breakthrough,
because single genes can be modified in the test tube and then introduced
into the body by gene therapy: either germline gene therapy, which affects
only the recipient's descendents, or somatic gene therapy, which affects the
organism that receives the treatment. Somatic gene therapy for humans is
still taking its baby steps, but there is widespread confidence that it'll work
well eventually. And human germline gene therapy raises ethical concerns
(though there are technical approaches to avoiding these). But as a proof of
principle, the postponement of aging by a single, defined genetic alteration
is vastly closer to clinical applicability than something accomplished by se
lection over many generations and affecting an unknown number of genes.
Perhaps because of this, and also partly because of the experimental
methods involved, Johnson's result initiated a massive surge in attempts to
identify genetic alterations to lab animals that would delay their aging. This
surge actually took a few years to get going, but when a second laboratory
(that of Cynthia Kenyon at the University of California San Francisco)
identified a mutation in a different gene, also in nematodes, that extended
their lives even more than Johnson's mutation did, the topic became one of

D E M Y S T I F Y I N G A G I N G 2 5
the hottest in the whole of biology.7 Kenyon and other top biogerontology
researchers have been able to publish nearly all their best work in the very
top few journals ever since—journals that scientists in most fields are lucky
to publish in even a couple of times in their whole career.
Johnson's and Kenyon's mutations were in different genes, but these
genes participate in largely the same range of metabolic processes. In partic
ular, they help to mediate an alternative developmental trajectory that nor
mal, nonmutant nematodes can follow, termed the dauer pathway. When a
nematode larva follows the dauer pathway, it suspends its development for a
period than can last much longer than the entire lifetime of a nematode that
follows the normal, non-dauer trajectory. What, you may ask, triggers this
developmental choice? And what "restarts" development and the resump
tion of the path toward normal nematode adulthood? Well, it just so hap
pens that the usual trigger for entry into the dauer pathway is starvation, and
that exit from dauer is stimulated by the presence of food. In other words,
the dauer pathway is neither more nor less than nematodes' extreme version
of rodents' response to calorie restriction.
Since Johnson's and Kenyon's breakthroughs, many other mutants
have been discovered—not only in nematodes but also in fruit flies and
mice—that have extended lifespans, and nearly all of these mutations have
also disrupted genetic machinery that mediates the sensing or metabolism
of nutrients. In general, the mutations confer a delay of aging at most equal
to that achievable by simply restricting calorie intake. 8 A few publications
have appeared in the past few years reporting life extension in mice caused
by reducing oxidative stress,9,10,11 but I am currently cautious about the re
producibility of these findings, because a huge number of other attempts to
postpone mouse aging in the laboratory in that way has failed.
At this point, therefore, I can point to a pretty compelling, double-
whammy argument that aging is worth trying to tackle. On the one hand we
should in principle be able to postpone aging by a large degree; moreover,
we have actually done so in the laboratory. This is surely great cause for op
timism that we will do so in the clinic in the not-too-distant future.
Isn't it?
Well, I would hardly have written this book if that were not indeed my
ultimate conclusion. However, the operative word here is "ultimate." Be
fore closing this chapter, I must explain why calorie restriction and its gene
tic emulation are not, in fact, pointers to the most promising route to
combating human aging.

2 6 E N D I N G A G I N G
Calorie Restriction and Its Emulation: A False Dawn
Do you know any perfectionists?
I do—and I always have, because my mother is one. I certainly
wouldn't be where I am today without my mother, and that includes her in
fluence on me as well as her sheer hard work and determination to give me
the best start in life. But there are certain ways in which her influence on
me was to show me a bad example, and her perfectionism is perhaps the
most extreme such case. I feel that in many ways it has prevented her from
achieving what she might have in her life, so I've never let myself become a
perfectionist—and I've certainly never regretted that.
What's wrong with perfectionism? We all know the main problem with
it: Perfectionism takes time. Most people are interested in getting things
done, and there are many circumstances in which a quick and dirty job is
the best policy, because the advantages of the "quick" outweigh the disad
vantages of the "dirty." There are certainly other circumstances in which
the balance is reversed, though—where a more painstaking approach is to
be preferred; hence, the ideal is to have good intuition and judgment for
how much attention to detail is appropriate in any particular case.
You may think that the above two paragraphs are a remarkably dra
matic digression, so let me surprise you by bringing my chain of reasoning
straight back to calorie restriction and its limitations in just a single sen
tence. The life-extending response to nutrient deprivation is neither more
nor less than the expression of an organism's genetically programmed intu
ition regarding the appropriate degree of attention to detail that it should
exercise with regard to its day-to-day molecular and cellular functioning—
and, because that's all it is, it's not amenable to substantial enhancement by
foreseeable biomedical technology.
Some elaboration is in order, so here goes.
I've explained, earlier in this chapter, that there are no genes for aging
in most species, simply because genes only survive if they confer enough
benefit (and thereby enjoy enough selective pressure for their survival) to
outweigh the constant stream of random mutations that all genes experi
ence over evolutionary time, and a gene can't confer any benefit if it only
mediates a process that would happen anyway. The only species in which
aging is actively driven by genetic machinery are those (such as salmon) in
which there is some reason to age and die rapidly—something that does not
happen by default to a machine that was running well for a long time

D E M Y S T I F Y I N G A G I N G 27
previously. Slow aging, the sort that we see in nearly all species, is the de
fault scenario, so no genes causing it can survive.
What we most certainly do have genes for, by contrast, is the panoply
of interacting processes that turns each of us from a single cell into a fertile
adult and that maintain our vigor and fertility until an age at which (in the
wild) we're very likely to have succumbed to starvation, predation, and so
on. Now, what does that have to do with perfectionism? Well, the reason
we have genes to keep us going until we're very likely to have been killed is
because the longer our fertile lives continue, the more progeny we'll have
time to have, so the greater the chance that our genes will be passed to fu
ture generations.
But what about the other end of our fertile life—the beginning? The
same applies: the sooner we achieve sexual maturity, the more offspring
we'll have time to produce before we die. But here's the problem: the be
ginning and end of fertile life are not independent of each other. Growth
from a single cell to a fertile adult is a process as complex as any known,
and mistakes always happen during its execution. You can probably see the
light at the end of this logical tunnel now: The organism has a choice be
tween doing a quick and dirty job of its growth, leading to early fertility but
sloppy construction, or a more perfectionist job that delays sexual maturity
but creates a more smooth-running machine in the end. And a more slop
pily constructed animal will on average live less long—partly because it
may be less able to defend itself against predators, famine, and such like,
but also because the molecular and cellular damage that it laid down dur
ing its headlong rush to maturity has effectively given it a head start in the
aging process. There's abundant evidence that this is not just a reasonable
idea but is also actually borne out in nature: for example, when you com
pare different species that are same size, the one that matures later tends to
be the longer-lived.
So now: What does this have to do with calorie restriction, dauers,
and the related genetic manipulations that I surveyed earlier in this chap
ter? Well, it's actually very simple. In a famine, there are two big problems
with passing on your genes. Firstly, gestation consumes a lot of energy,
which of course comes from food. And secondly, whatever offspring you
do succeed in having during a famine are very likely to die of starvation
before they can have their own offspring, which is no better for the sur
vival of your genes than if you hadn't had any offspring in the first place.
Thus, the advantage (in terms of your genetic heritage) of maturing

28 E N D I N G A G I N G
quickly is less during a famine than when food is plentiful. But wait: the
disadvantage of maturing quickly, namely the increased risk of death that
results from being sloppily constructed, is unaltered! In fact, that risk may
in some cases be amplified: If the duration of a particular famine is a large
fraction of the species's lifespan, the period late in life when the well-
constructed, late-aging animals are the only ones left to procreate will be
the only period when successful procreation can occur. In that case, the
benefit of being well constructed (i.e., the drawbacks of being sloppily
constructed) will be greater in a famine of that duration than when food is
plentiful throughout life.
Thus, famine shifts the happy medium toward favoring a more
painstaking development process. And since famines are unpredictable
events, occurring at irregular intervals, it's not possible for evolution to de
termine a species' ideal degree of perfectionism in advance: each individual
organism must have the ability to respond to its situation. Furthermore,
famines have always been like that, ever since organisms started eating other
organisms. It's therefore no surprise that, everywhere we look in nature, we
find the genetic machinery to respond to a famine early in life by slowing or
suspending growth.
You may know that nutrient deprivation in adulthood often has the same
effect to a milder extent, a phenomenon that doesn't seem to be explained by
what I've just told you. Indeed, there may not be such clear-cut evolutionary
reasons why adult-onset calorie restriction postpones aging at all. But there
don't need to be, because genetic programs that exist for one time or circum
stance are often activated unnecessarily in situations that are similar. Think,
for example, of the fact that startling someone causes a mild adrenaline rush,
something that exists to facilitate escape from life-threatening situations.
Finally I must explain why the logic I've outlined here implies that ma
nipulating these nutrient-sensing pathways isn't the most promising way to
postpone human aging. I actually have three reasons.
First, the degree of life extension that has been obtained thus far in
various species exhibits a disheartening pattern: it works much better in
shorter-lived species than in longer-lived ones. Nematodes, as I mentioned
above, can live several times as long as normal if starved at the right point in
their development; so can fruit flies. Mice and rats, however, can only be
pushed to live about 40 percent longer than normal. This pattern led me, a
few years ago, to wonder whether humans might even be less responsive
than that, and I quickly realized that there is indeed a simple evolutionary
reason to expect just such a thing. 1 2 It's a consequence of the fact that the

D E M Y S T I F Y I N G A G I N G 29
duration of a famine is determined by the environment and is independent
of the natural rate of aging of the species experiencing it.
Second, the adjustment of metabolism that organisms undergo when
food is scarce causes only a slowdown in the accumulation of molecular
and cellular damage, not a repair of damage that already happened. I've al
ready told you that the key "Eureka moment" in my development of SENS
was when I realized that repairing the damage of aging (before it progresses
into disease) might be simpler than preventing it—but even setting that re
alization aside, repair is bound to be preferable, simply because any feasi
ble therapy (whether to repair damage or to prevent it) will be only partial.
That's to say, repair therapies will repair some but not all damage, and pre
vention therapies will slow but not halt the accumulation of damage. Why
does this mean that repair is preferable? The logic is quite simple. In broad
terms, if you take a middle-aged person and halve the rate of their subse
quent aging, you'll double their remaining lifespan, but that might mean
adding only 20 percent to their total lifespan. By contrast, if you take that
same person at the same age and apply a therapy that halves their accumu
lated damage, and apply that same therapy periodically for the rest of their
life, you'll roughly double their total lifespan (because their accumulating
damage will only consist of the types of damage that your therapy can't re
pair), which means increasing their remaining lifespan (from the point
when you first applied the therapy) by a factor of maybe four or five! So
prevention-oriented approaches simply don't aim high enough.
But there's a third reason why I don't think nutrient sensing is the most
promising target for biomedical intervention in aging, and I would say it's
the most decisive. The reason it's been so incredibly easy to extend the lifes
pans of so many organisms by this one trick is because it's an evolved re
sponse to environmental conditions. The machinery that mediates that
response is fantastically complex and poorly understood, just like the rest
of our biology, but we can manipulate it easily despite that complexity, be
cause its initial step—the sensing of nutrient availability—is simple. Just as
you don't need to understand how your computer works to turn it on and
off, we also don't need to understand the process of how nutrient depriva
tion is translated into the adjustment of masses of interacting metabolic
pathways in order to turn that process on and off. But therein lies the show-
stopping problem. You may not need to understand how your computer
works in order to turn it on and off, but in order to make it do things that it
does not already contain the hardware and software to do, you have to un
derstand a lot more. And if the new functionality requires software that

30 E N D I N G A G I N G
hasn't yet been written or can't be installed, you have to understand a huge
amount more, enough to write that software yourself. The human body is, in
that sense, like a computer into which new software can't be installed—it's
very versatile, but that versatility cannot be extended by the same methods
that merely elicit the existing versatility. Therefore, we can be sure that there
is a fixed degree of life extension that can be achieved by manipulating the
nutrient sensing pathway—whether by calorie restriction (CR) itself, or by
drugs that trick the body into thinking it's being starved, or by genetic
changes that flip the same switch. As I explained a couple of paragraphs
ago, I think that ceiling is very modest, maybe only a two-to-three-year ex
tension; some of my colleagues think it may be as much as twenty to thirty
years—but it's still a ceiling. We will never be able to exceed that fixed de
gree of life extension by such means, however hard we try.
Not Good Enough—But Better than Nothing
I want to end this chapter on a positive note, though. Even though nutrient
sensing can only extend life by a fixed maximum amount, and even though
it may be a rather small amount, that's still better than nothing! Also,
there's a very general finding in laboratory life-extension experiments that
animals with some kind of mildly life-shortening genetic problem benefit
more from the therapy or regime than congenitally longer-lived individuals.
That's quite likely to apply to calorie restriction (CR) in humans, too—
which means that doing CR (or taking safe CR-mimicking drugs, as and
when they appear) may be a good insurance policy against unknown con
genital vulnerabilities. For these reasons, I strongly support the work that
many of my colleagues in biogerontology are doing to squeeze the most we
can out of that route to life extension.
But in closing, I want to bring you back firmly to the theme of this
chapter. Once upon a time, aging was a truly mysterious phenomenon, but
that time has passed. We can now reason about the aging of the human
body in just the same way, and with just the same confidence, as we can rea
son about the aging and decay of simple machines. We know why different
organisms age at different rates, whether that be because of different genes
or different environments. We know that our genes are our allies, not our
foes, in our war against aging—that they exist to postpone aging, not to
cause it, and we only age because those life-preserving genetic pathways are
not comprehensive.

D E M Y S T I F Y I N G A G I N G 31
Now—can you still tell yourself, with a straight face, that aging is too
mysterious to try to tackle? You may have just one straw to clutch at in your
effort to perpetuate your pro-aging trance: you may be telling yourself that
the devil is in the detail, detail that I have not yet provided. I'll be snipping
that straw in Chapter 4.

4
E n g i n e e r i n g R e j u v e n a t i o n
Let's briefly review what I've told you so far about aging. In a nut
shell, it's as follows:
• Aging is really bad for us, however much we like to forget the fact.
• Aging is not a mystery, and we can already postpone it a lot in the
• However, the techniques that have been so successful in the lab do
not seem promising for humans.
In this chapter I'm going to expand upon the "Eureka moment" that I re
lated in Chapter 1.1 will cover—in still broad, but somewhat more detailed,
terms—what each type of damage really is at the molecular and/or cellular
level, and also the broad strokes of how I think we can address that damage.
A Caveat: Why Prevention Is Usually Better than Cure
lab.
In Chapter 3,1 told you two heartening things about combating aging: firstly
that in principle it's no different than combating the aging of man-made

E N G I N E E R I N G R E J U V E N A T I O N 33
machines such as cars, and secondly that we've already discovered how to
postpone aging by a large factor in the laboratory. However, I then explained
that the second of these tidings of good cheer is actually going to be of only
very limited biomedical utility. Well, brace yourself, because I'm about to ex
plain that the first piece of good news is not so simple as it seemed, either.
I'll start with a rather more sobering thought about cars. Why are so
few of them maintained to an age far beyond that for which they were de
signed, even though we all know they can be?
There are two answers, one reassuringly inapplicable to the analogy
with human aging but the other very applicable indeed. The inapplicable
answer is: because their owners have the option of getting a new car. All
this says is that the chance that you will put in the effort and money to
maintain an old and declining machine depends on how much you are in
love with it. You may generally choose to junk your car when it starts to
malfunction because you're not very attached to it anyway, but if your
mother starts to malfunction and the wherewithal exists (even at a hefty
price) to repair her, it'll be a different matter.
The other answer is the problem: most people leave the serious
maintenance of their car until it's too late. It's obvious that the more
damage a machine sustains, the more work is needed to rectify that dam
age; but more than that, the technology needed to rectify it becomes
more and more sophisticated. When a car is really on its last legs, restor
ing it to full working order requires major attention—replacement of a
lot of parts, for example. And unlike the how-much-we-care argument
above, in this case the situation is absolutely the same for the human
body. The people who know this best are those who work not on the bi
ology of aging but on the medicine of aging: geriatricians. Geriatricians
try to help people whose aging has reached the point where physical or
mental function is appreciably impaired. They do their best to apply ex
isting medical technology to postpone the patient's further decline and
eventual death. But, as they know and as you also know, it's a losing bat
tle. The damage has already spun out of control: it's feeding back on it
self to accelerate the occurrence of additional damage, and the types of
damage that are occurring are becoming ever more numerous and var
ied. All the geriatrician can hope to deliver is a modest improvement of
the quality of life of the patient's last years, and perhaps a few months' to
a year's postponement of death. It's the age-old rule: Prevention is better
than cure.

34 E N D I N G A G I N G
But Only Usually . . .
But let's not leave it there. There's one thing about geriatrics that it has over
gerontology, and I hinted at it above: geriatricians use existing medical
technology. Why can they do that, when gerontologists can't?
The answer, when you think about it, is simple: To fix a problem that al
ready exists, you don't need to know how it arose. A car mechanic replacing
a car component doesn't need to know what type of corrosion wore through
a fuel line, or what size rock hit a windscreen; similarly, the geriatrician
doesn't need to know anything about free radical chemistry or cholesterol
metabolism in order to treat cardiovascular disease or diabetes. But by con
trast, preventing corrosion or shattered windscreens involves careful analysis
of the downstream side effects of salting roads and not clearing debris from
the highway; in the same way, the gerontologist needs to know a great deal
about extremely subtle and possibly hard-to-discover causal chains of events
in order to put "prevention is better than cure" into practice.
So, here we have two alternative approaches to postponing aging, one
preventative and one curative; I've explained a problem with each of those
approaches that makes them unpromising ways forward; and finally I've
pointed out that the problem that each approach has is not shared by the
other approach—preventing aging is soon enough but too complex, curing
the diseases of aging is simple enough but too late. Now, what does that say
by way of a possible way forward?
Worst of Both Worlds, or Best?
Well, I'll tell you what it said to me, that early morning in California.
Discussion during the day's roundtable-style sessions had focused on
the various theories of aging, and ways to prove or disprove them. This
mostly meant running through the multiple metabolic pathways that might
contribute to the development of aging damage. I had presented the case
that the production of free radicals by mitochondria—the tiny "power
plants" that extract energy from food and convert it into ATP, a form of en
ergy usable directly by the cell—is at the root of much of the aging process.
This was something that most of my colleagues suspected, but I had re
cently framed it in a novel way that reconciled some unexplained findings
in the field. I had confidence in my model, as it was my main specialist area
within gerontology at that time: a book-length treatment of it 1 had earned

E N G I N E E R I N G R E J U V E N A T I O N 35
me my Ph.D. But more important to me, it suggested a biomedical solution
to what I was convinced was a major cause of aging damage: With some
complex but foreseeable gene therapy, the connection between mitochon
drial free radicals and pathology could be severed, without the need to in
terfere with the mitochondria's normal energy-producing activity. (I'll say a
little more about this below, and lots more in Chapter 6.)
I had come to the conclusion that, in the best case, my mitochondrial
gene therapy proposal might (and I emphasize might) also slow the rate of
aging in humans attributable to most other causes by about 50 percent.
This would be a massive breakthrough, as it would lead to as good an ex
tension of healthy life as the most severe calorie restriction (even under the
CR optimists' scenario) but without CR's side effects. But I was far from
sure about that estimate, and in the wee hours of that morning, alone in a
hotel room, I was even less confident than usual, because I had spent all
day being reminded of just how many things go wrong in an aging body.
Many of these problems could be at least partly chalked up to the down
stream effects of the insidious, age-related increase in oxidative stress—the
imbalance between those substances in the body that tend to chemically
"need" electrons and substances that chemically "want" to donate them. I
believed my mitochondrial gene therapy proposal would nearly eliminate
this rise with age, but I couldn't be sure of just how much the rest of the ag
ing process would still go on without additional, targeted therapies—nor
what those therapies might be.
The candidates were numerous:
• Inflammatory enzymes essential to the immune system could also
oxidize cholesterol, particularly when there's a lot of it around,
contributing to atherosclerotic plaques.
• Our bodies' reliance on carbohydrates as a source of fuel exposes
us to the reactive chemistry of glucose, causing the "gumming up"
(glycation) of cellular proteins.
• Beta-amyloid, an aggregating protein, forms the basis of the "senile
plaques" in the brains of Alzheimer's patients. It is the result of ab
normal chopping-up of a normal precursor protein in the brain.
• The process of cell division gradually shortens each successive cel
lular generation's telomeres—the protective caps on the DNA dou
ble helix that serve the same function as the plastic bits on the end
of your shoelaces, preventing the chromosome from "fraying."
(See Chapters 10 and 12 for more.)

36 E N D I N G A G I N G
• Mutations in the cell's genetic database occur when, in the process
of creating needed copies of the DNA "instruction book" for the
new cell, the body's DNA-replicating machinery makes "typos."
• Tapping into the pools of stem cells (the primordial, unspecialized
cells that the body holds in reserve and causes to develop into par
ticular cell types as needed to replace cells lost to injury or disease)
gradually depletes what is, over a lifetime, a limited source of youth
ful cellular reinforcements.
The problem had me in its grip—and it wasn't just curiosity that was
keeping sleep at bay. While many of my colleagues viewed biogerontology
as a phenomenon to study for the sake of understanding it, I saw aging for
the humanitarian crisis that it is, the toll of tens of thousands of dead every
day ringing in my ears. Abandoning my first career in artificial intelligence
research, I had committed my life not just to alleviating the worst of the
morbidity and mortality of age-related disease, but to putting an end to the
entire horror show. I'd dedicated myself to the "engineering of negligible
senescence," as I had first termed the goal in my Ph.D. thesis—to the end
of aging.
But my inner dialogue that morning was leading to frustration, and
even some despair. Clearly, if real medical control of aging required cor
recting all of these potentially damaging metabolic processes individually,
real progress in anti-aging medicine might be like fighting the Hydra: no
matter how many heads you put down, more would spring up to take their
place. Normal metabolism is such an intricate, finely balanced web of reac
tions that tweaking one sends perturbations throughout the entire network,
usually creating new problems or negating the effect of the intervention by
eliciting a counterbalancing metabolic adjustment. For example, chronic
inflammation is a source of cellular damage. But if you interfere with in
flammation, you might impair immune defenses against pathogens. Equally,
free radicals—a by-product of your metabolism—cause oxidative stress
and damage over time. But crank up antioxidants, to defend against free
radicals, and you might help cancer cells to protect themselves against
chemotherapy drugs.
This process of dynamic metabolic adjustment is seen in the aging pro
cess, in fact. There are a number of aging changes that, while they might
have some pathological consequences, are not themselves forms of damage.
Putting it another way: they do not actually accumulate in the body's cells
and tissues; rather, they represent a shift in the equilibrium between creation

E N G I N E E R I N G R E J U V E N A T I O N 37
and destruction of the molecules involved. It seemed likely to me that such
changes, however harmful to the body's youthful functioning, were second
ary to something else. This meant that identifying and correcting that
"something else" would correct the maladaptive secondary change, render
ing moot the question of its contribution to the aging process. For instance,
the cell's ability to respond to many hormones and other signaling mole
cules tends to decline with age. But as we saw in Chapter 3, the logic of
evolution seems to dictate that this decline isn't programmed into the body.
It must therefore be secondary to some form of damage. Maybe the mem
branes of the cell lose their fluidity, impairing the ability of receptor mole
cules to change their shapes to pass on a signal. Maybe the machinery that
creates those receptor molecules becomes impaired. Whatever it is, identi
fying the damage itself would narrow the field of things that directly caused
such damage and that were thus at the root of aging.
And, come to think of it, there seemed to me to be far fewer kinds of
damage than processes that cause damage—hosts of different mutagens
and "pre-mutagenic" changes to DNA, for example, but only two types of
mutations: chromosomal and mitochondrial.
Well, I mused, that's a thought—just how many kinds of aging damage
ARE there? And are there similarly promising fixes for the rest of them?
There are mutations in our chromosomes, as I just mentioned; this sort
of damage causes cancer. I didn't (at that time—but see Chapter 12) have
any new proposals up my sleeve for that one; it was going to have to rely
(for now, at least) on other people's ideas. But there was no shortage of such
ideas: Cancer research is among the biggest fields in biomedicine.
What other problems could arise from nuclear mutations? It was
widely assumed that they were a major cause of age-related cellular dys
function, but I had been batting around a counterargument in my head for
some time—an argument that made me pretty sure that mutations not rele
vant to cancer would be irrelevant to aging within a currently normal life
time. Certainly, a noncancerous mutation in a single cell might make that
lone cell dysfunctional, but could it really substantially impact the tissue as
a whole? Clearly, if every cell in a tissue were misbehaving, a person would
be in trouble—but that can't be the case. Why not? Well, if it were that
easy for an average cell to pick up a mutation, then everyone would be rid
dled with cancer by the time they were adults, because it takes just one can
cerous cell to be allowed to grow to make a life-threatening tumor. What
that suggested was that nearly all cells are kept genetically intact into and
beyond a person's forties, and that the great majority of cells continue to be

38 E N D I N G A G I N G
so throughout the "normal" life span. In other words, in order to prevent
us from dying of cancer before puberty, our DNA maintenance machinery
has to be so good that mutations not relevant to cancer just don't happen
often enough to matter. Better yet, the exact same logic seemed to work for
what biogerontologist Robin Holliday had memorably termed "epimuta-
tions"—changes not to the DNA sequence itself, but to the structure of the
individual bases or the proteins around which the double helix is normally
wrapped. Epimutations can do great harm, because they change the rate at
which genes are decoded into proteins, but epimutations can cause either
cancer or other problems, just as bona fide mutations can, so the "cancer is
a bigger problem than anything else" conclusion applies to them too. I'll
tell you more about this line of reasoning in Chapter 12.
In addition to chromosomal mutations, there are mitochondrial muta
tions, which may be a major part of the problem caused by free radicals.
(Mitochondria are the only cell components that contain their own DNA
independently of our chromosomes.) Luckily, I thought, I believe I already
know a feasible solution to mitochondrial mutations. My solution was totally
unlike the problematic approaches that were being proposed by other re
searchers, and I felt that it was much more powerful. It didn't rely on
souped-up antioxidant defenses, an idea which was still being pursued not
only by vitamin salespeople but even by some biotech companies. (This de
spite the fact that biogerontology specialists had long ago concluded that
antioxidants are a dead end after they had failed, again and again, to affect
aging one whit. 2 A better demonstration that our ambivalence about aging
is only skin deep is hard to find.) Free radicals are just too reactive to be ef
fectively mopped up with vitamins, nor even with the novel free radical
scavengers that were coming out of pharmaceutical labs around this time
(with names like MnTBAP and EUK-134, synthetic versions of the antiox
idant enzyme superoxide dismutase). Or if not too reactive, they might be
too necessary—-it had recently become clear that cleaning up too many free
radicals would cause new headaches for the body. After millennia of expo
sure to their reactive chemistry, evolution has learned to harness free radi
cals as signaling molecules, 3 so a heavy-handed repression of the cell's
exposure to them would actually harm cellular metabolism, not aid it. The
body might even react to antioxidant supplements by reining in its natural
antioxidant defenses to compensate.
Trying to reduce free radical production was a job that many of my col
leagues considered to be the best way to slow down aging damage, but (for
the reason just given) actually pulling it off without seriously impairing the

E N G I N E E R I N G R E J U V E N A T I O N 39
organism's ability to carry on with life's many duties would be extremely
tricky. Not only that, most free radicals are produced in the mitochondria
in the process of making ATP from food energy, and trying to mess around
with that central feature of metabolism is bound to create side effects.
As I alluded to a few paragraphs ago, I had already proposed avoiding
these problematic approaches by a strategy I'll cover in detail in Chapter 6.
Briefly, the idea is to let metabolism proceed as normal—accepting that
some free radicals will be generated and some biomolecules damaged—but
to sever the link between free radicals and oxidative stress at its nexus. In
my Ph.D. thesis, I had argued that (contrary to the prevailing view at that
time) mitochondrial free radicals do not drive a systemwide rise in oxida
tive stress with age by damaging the rest of the cell directly. Instead, the
damage that they cause to the mitochondrial DNA causes the mitochondria
to enter a maladaptive state that spreads oxidative stress out beyond the
cell. This, I had reasoned, meant that scientists could solve the problem of
mitochondrial mutations by copying mitochondrial DNA from its vulnera
ble spot at "ground zero," within the free-radical generating mitochondria,
into the bomb shelter of the cell nucleus, where damage to DNA occurs far
less frequently. The proteins they encoded would have to be constructed in
a manner that induced the cell to transport them to mitochondria, but the
procedure to achieve that had been mostly understood for some time. In
this scenario, the nuclear copies would act as a "backup" for the mitochon
drial DNA: the mitochondria could operate as normal even if their DNA
became damaged, so they would not cause long-term harm to the organism
as a whole. Mitochondria would still suffer damage, but would not enter
the maladaptive state I just mentioned, so they would not cause the creep
ing, destructive slide into oxidative stress in the rest of the body.
Okay, two down (chromosomal and mitochondrial mutations); how
many to go? There is glycation, the warping of proteins by glucose. Well,
that seemed relatively easy, because it was well known in the field that
a biotech startup called Alteon was already running clinical trials using
a compound called ALT-711, which appeared to reverse the protein cross-
linking that this process caused. While the effect was weak, it was signifi
cant: the compound had a limited ability to restore some of the flexibility
that's lost to glycation with age in the heart and blood vessels, and also
showed promise for kidney damage in diabetics. It was proof-of-principle
that without interfering with glucose metabolism, you could allow the for
mation of protein cross-links but prevent the pathological results by undo
ing the damage after the fact. (This is an important and common theme, as

40 E N D I N G A G I N G
you will see—don't interfere with the process, but rather repair or clean up
the damage that has accumulated.) See Chapter 9 for lots more detail about
the glycation problem.
What else? There are the various kinds of junk that accumulate outside
the cell: beta-amyloid, the lesser-known transthyretin, and possibly other
substances of the same general sort. Here again, recent studies in the pri
vate sector—this time by a Californian company named Elan—had shown
that you could actively remove the problem, in this case by vaccinating mice
against the amyloid plaque and letting their immune cells gobble the stuff
up. The concept had shown such rapid success in the lab that it was already
close to clinical trials. Will I be telling you more about this, later in the
book? You bet—see Chapter 8.
We must also address the unwholesome goo that builds up within the
cell, such as lipofuscin. I started to get quite excited at this point, because
just a year previously, in Dresden in June 1999, I'd come up with a new
proposal to eliminate such material, involving the identification and engi
neering of enzymes from soil bacteria. (This was a classic case of someone
not immersed in their own experimental work being able to bring together
ideas from very distant disciplines to form a new approach to an existing
problem—a critical element of modern scientific progress, which has been
sadly neglected in many areas of medicine and biology.) The concept of us
ing soil bacteria to degrade long-lived organic material had been around for
decades, but not in gerontology, or even in any biomedical field. Rather, it
was a mainstay of environmental decontamination, where it is known as
"bioremediation." No one in gerontology had really even heard of it, let
alone seen its biomedical potential. If you're intrigued, well, you only have
to wait until Chapter 7.
Another item that must be added to the list is cellular senescence, the
"aging" of individual cells. Senescence, in this meaning of the word, is a
state of arrested growth in which the cell produces chemical signals dan
gerous to their neighbors. In theory, at least, there are all kinds of ways to
deal with cellular senescence, though I wasn't sure which of them would ul
timately pan out. Senescent cells express distinctive marker proteins, which
should allow them to be targeted for selective destruction. Alternatively,
once researchers tease out the damage or gene expression shifts that keep
cells locked in this abnormal, arrested state, it might be possible to restore
senescent cells to normal functionality. This was all still speculative, of
course, but Judy Campisi at Berkeley and others were already hot on the
trail. Chapter 11 will reveal all.

E N G I N E E R I N G R E J U V E N A T I O N 4 1
There's also the depletion of cells—of nondividing cells like neurons or
heart cells, which are not naturally replaced when they die, and also the
more paradoxical depletion of stem cell pools essential to healing and main
tenance of tissue. Anyone who'd read a newspaper in the previous few years
knew that scientists were hotly pursuing a way to deal with the age-related
loss of cells, including stem cells: more stem cells, cultured in the lab and de
livered as a rejuvenating cellular therapy. There were several viable ap
proaches to this, different ones probably suited to different conditions. One
was extracting the adult stem cells already in the patient, growing more of
them, and then reinfusing them into the patient. Another was harvesting
some of the more versatile embryonic stem cells that were already sitting,
waiting to be thrown out as medical waste, in fertility clinics all across the
world. The most complex was "nuclear transfer," in which a person's old,
specialized cells could be transformed into young, versatile stem cells again
via the environment of a woman's egg and a quick jolt of electricity. Re
searchers were already showing in animal models that these cells could be
used to cure age-related diseases and trauma, and there was every reason to
expect that the same techniques, once perfected, could be used to replace
cells lost to age-related decay.
What else? Er . . .
I couldn't think of any more categories of damage! Try as I might, I re
ally couldn't. There were a couple of other examples of molecular changes
that accumulated throughout life, but I had reasons to believe that they
were in the same boat as non-cancer-causing chromosomal mutations: they
might be harmful if we lived hundreds of years, but they very probably
weren't harmful in a normal lifetime. Other than that, everything I had
learnt about during my five years of study and conference-hopping seemed
to be covered.4
I stepped back for a moment and articulated the logic I had been de
veloping those past few hours. At root, I was addressing a simple question:
if geriatrics fails because prevention is better than cure, and gerontology
fails because our understanding of metabolism is so limited, then might an
intermediate target be the best of both worlds? Might it be possible to re
pair damage after it's been laid down (hence avoiding the need to under
stand the details of how it's laid down) but before it spirals out of control
(hence also avoiding the losing battle that is geriatrics)? See Figure 1.
I could only answer this question in the affirmative if I could make a
specific, extremely bold claim: that these intermediates, these proximate
side effects of metabolism that accumulate in the body throughout life,

42 E N D I N G A G I N G
Figure 1. The "engineering approach" that I conceived in June 2000, as an intermediate, best-of-both-worlds alternative to gerontology and geriatrics as a strategy to combat aging.
could all be either (a) ruled out of relevance to late-age pathology (as I felt
I could do for mutations that don't cause cancer) or (b) repaired or made
harmless by foreseeable therapies. If some could be repaired, and some
were definitely harmless or could be made so, but some fell into neither
such category, the idea would fail. Like any machine, the body is only as ro
bust as its weakest link, so partial maintenance will have little or no effect
on longevity.
But I went over my list again and again, and as I did so I became ever
surer that there was no clear-cut exception. The combination of my own
idea for eliminating intracellular garbage, the idea I'd been championing
for a few years for making mitochondrial mutations harmless, and the vari
ous other therapies being worked on around the world to address glycation,
amyloid accumulation, cell loss, senescent cells, and cancer . .. that was re
ally and truly an exhaustive list. Figure 2 shows my enumeration of the
problems and solutions that constitute the SENS (Strategies for Engi
neered Negligible Senescence) plan as it stands today.
As I mentioned above, there may well be other problems that will
emerge if we succeed in solving all of these and thereby live a great deal
longer. I felt, however, that my list might very well be comprehensive
enough to give a few decades of extra life to people who are already in mid
dle age before we start the treatments. And that was certainly a much more
promising first step than anything that my colleagues had reviewed the pre
vious day or in the many conferences and articles that I'd devoured over the
previous few years.
The California sun was rising, and with it my spirits. It was clear that
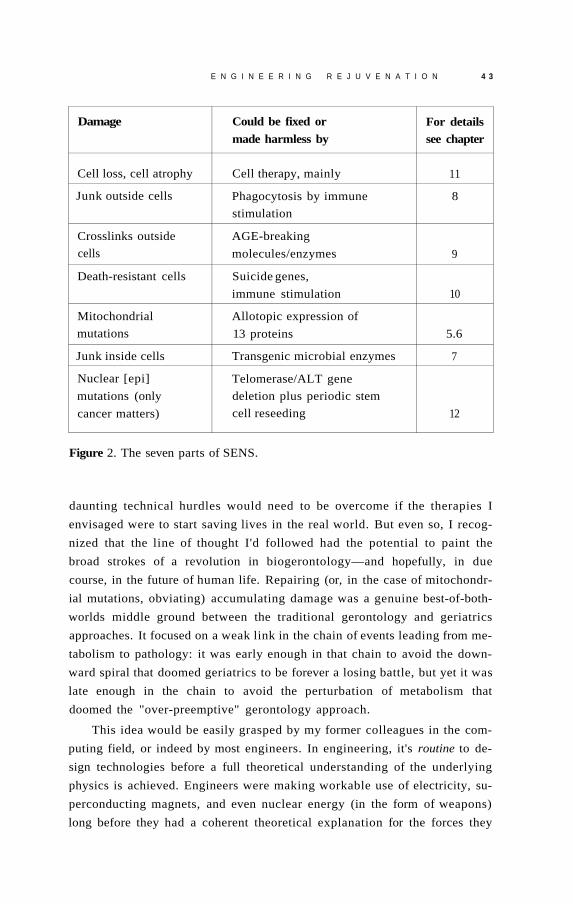
E N G I N E E R I N G R E J U V E N A T I O N 4 3
Damage Could be fixed or
made harmless by For details see chapter
Cell loss, cell atrophy Cell therapy, mainly 11
Junk outside cells Phagocytosis by immune
stimulation
8
Crosslinks outside
cells
AGE-breaking
molecules/enzymes 9
Death-resistant cells Suicide genes,
immune stimulation 10
Mitochondrial
mutations
Allotopic expression of
13 proteins 5.6
Junk inside cells Transgenic microbial enzymes 7
Nuclear [epi]
mutations (only
cancer matters)
Telomerase/ALT gene
deletion plus periodic stem
cell reseeding 12
Figure 2. The seven parts of SENS.
daunting technical hurdles would need to be overcome if the therapies I
envisaged were to start saving lives in the real world. But even so, I recog
nized that the line of thought I'd followed had the potential to paint the
broad strokes of a revolution in biogerontology—and hopefully, in due
course, in the future of human life. Repairing (or, in the case of mitochondr
ial mutations, obviating) accumulating damage was a genuine best-of-both-
worlds middle ground between the traditional gerontology and geriatrics
approaches. It focused on a weak link in the chain of events leading from me
tabolism to pathology: it was early enough in that chain to avoid the down
ward spiral that doomed geriatrics to be forever a losing battle, but yet it was
late enough in the chain to avoid the perturbation of metabolism that
doomed the "over-preemptive" gerontology approach.
This idea would be easily grasped by my former colleagues in the com
puting field, or indeed by most engineers. In engineering, it's routine to de
sign technologies before a full theoretical understanding of the underlying
physics is achieved. Engineers were making workable use of electricity, su
perconducting magnets, and even nuclear energy (in the form of weapons)
long before they had a coherent theoretical explanation for the forces they

44 E N D I N G A G I N G
were manipulating. Even in medicine, the effective use of treatments had his
torically often long preceded our mechanistic understanding of them. Salicy
lates from willow bark have been used as anti-inflammatory treatments for
centuries, and Bayer chemist Felix Hoffmann was even able to modify these
natural compounds to make them more palatable and less prone to upset the
stomach, yet the molecular basis for the action of the new wonder drug (as
pirin) would not be understood for seven decades. Of course, even more ef
fective drugs can often be designed from the ground up once the key
enzymes and genes upon which they might act are sequenced—but that level
of detail was not needed to get started in developing effective medicines.
Making this reorientation was dizzying—but once you accepted it, I re
alized, the whole project suddenly became tractable, and the way forward
clear. You could stop thinking of aging as a hopelessly complex theoretical
problem to solve, and get on with attacking it head-on, as an engineering
challenge that needed to be overcome. "Engineered negligible senescence,"
a phrase that I'd previously used offhandedly, suddenly presented itself as
the most precise description possible of the task ahead. 5
In fact, I realized, the problem might even be thought of in terms of the
way we prevent "aging" in other physical structures, such as houses or cars.
As I discussed in Chapter 2, evolution's priorities for nearly all organisms
stop them from living indefinitely without aging: mutations in the genes in
volved would not be removed by selection when the ageless organism was
eaten by predators or otherwise succumbed in just a tiny fraction of "for
ever." This is much like cars, which are designed to meet the opposing pri
orities of durability and low cost at some midway point that is acceptable to
the consumer. Thus, our bodies—like our other vehicles—are designed to
survive for a biological "warranty period": they are given enough robust
ness and self-repair capability to function at peak performance for as long
as they can reasonably be expected to stay alive in the wild, but no longer.
But of course, individual users of cars or of bodies may have very dif
ferent priorities from those of Detroit or of our "selfish genes." If you want
a car to last much longer than manufacturers of cheap cars typically intend,
you have two options. One is to pick up a better model in the first place:
buy yourself a Volvo instead of a Chevy Cavalier. This is all well and good
for cars, but it isn't an option for those of us who have only the genes we
were born with. And of course, even Volvos will still eventually break
down, only a few years after a more economical product would do.
That's why, when we want to keep a car on the road for an exception
ally long time, we actually choose the other option: we fix damage as it hap-

E N G I N E E R I N G R E J U V E N A T I O N 4 5
pens. Whether it's a poor laborer keeping his ancient VW bug running be
cause it's the only car that he'll ever be able to afford, or a wealthy collector
maintaining an old MG for the sheer love of it, we all know that a car can
be kept going more or less indefinitely with sufficient maintenance. We
don't have to keep the cars off the road in climate-controlled garages, and
we don't rely on the latest gasoline additive: we simply repair worn-out
parts when they begin to fail. As I saw then, and as I will describe in the
chapters ahead, the analogy to humans (at the cell, tissue, and organ level)
is strikingly exact.
The Devil Is in the Detail
At the end of Chapter 3,1 explained that the purpose of this chapter would
be to remove people's last hope for maintaining their pro-aging trance: the
belief that my breathlessness about the recent progression of aging from
mysterious to manipulable might be all talk and no substance. I hope I've
done that—but I've come up against the pro-aging trance often enough to
know it's sometimes very hard to break.
That's why, for most of the rest of this book, I'll be wading deep into
the fine scientific detail of the seven SENS categories and the remedies for
them. I know that most readers of this book will not be scientists, so this
may be intimidating. But Michael Rae and I have worked hard in Part 2 to
present cutting-edge science in a manner comprehensible to any educated
layman who's willing to put in the time to read it carefully. I therefore urge
you to dive in and learn in detail about the types of damage that comprise
aging and the foreseeable technologies that will, I am confident, enable us
to repair or obviate that damage comprehensively enough to avoid age-
related physical and mental decline indefinitely.




M e l t d o w n o f t h e C e l l u l a r
P o w e r P l a n t s -
The cellular components—"organelles"—known as
mitochondria play a large role in aging, hand in hand with
reactive chemicals known as free radicals. When I came into the
field in the mid-1990s, however, this role was not clearly defined;
evidence and interpretations were contradictory and awaiting
synthesis. In response, I developed what is now a widely
accepted mitochondrial free radical theory of aging. Read on: In
order to understand aging, you must understand a little of the
way in which our cells work.
In Chapter 4. I explained that there are seven major classes of life
long, accumulating "damage" that we must address if are to uncouple their
causes—the processes of life—from their eventual consequences, the pathol
ogy of aging, and thereby prevent those consequences. Six of those seven are
the subject of one chapter each in this part of the book—Chapters 7 through
12. But the first one I'm going to address, mitochondrial mutations, is going
to take two chapters. That's because the question of whether mitochondrial
mutations matter at all in aging is actually a really complicated one, and we
must do our best to answer it in order to know whether we need to worry
about them. Remember that in Chapter 4, I briefly noted that SENS does not
incorporate a plan to address mutations in our cell nucleus at all unless they
5

50 E N D I N G A G I N G
cause cancer, because non-cancer-causing mutations accumulate too slowly
to matter in a normal lifetime. (I will explain this logic much more thoroughly
in Chapter 12.) A lot of gerontologists feel that way about mitochondrial mu
tations. I disagree with them, so I need to tell you why. For each of the other
six SENS damage categories, by contrast, there's no argument: at least one of
the major pathologies of aging is clearly caused or accelerated by that type of
damage. So those six categories will only require one chapter each, focusing
mainly on the solution and with a relatively brief description of why there's a
problem to solve.
Free Radicals: A Brief Primer
Almost everyone has heard of free radicals by now. Their involvement in ag
ing is asserted so often and so confidently in popular press articles—
especially articles trying to promote the latest "antioxidant" nutritional
supplement—that you'd think the matter was done and dusted. As we'll
see, however, the exact roles played by free radicals in the aging process—
and the best ways to deal with the problems they cause—are a lot more
complicated, and more controversial, than these articles let on.
Free radicals in biology are, for the most part,1 oxygen-based molecules
that are missing one of the electrons in their normal complement. Electrons
are charged particles that surround the central nucleus of the atom, and they
occupy well-defined locations (you can think of these as distances from the
nucleus) termed orbitals. By their nature, molecules can only remain chemi
cally stable when each of the electrons in the orbitals of their constituent
atoms has a paired twin to complement it; an orbital with only one electron
is unstable. So when a molecule loses one half of an electron twosome, it be
comes chemically reactive until it gets that electron back. Usually, the free
radical's stability is restored when it tears an electron out of the nearest avail
able normal, balanced molecule—but with this electron stolen from it, that
second molecule generally loses its chemical stability, and will seek in turn to
restore its balance by a similar theft. It's a chain reaction.
Some unusual molecules—antioxidants—finesse this logic and are rel
atively stable even when they contain an unpaired electron. These mole
cules can "quench" free radical chain reactions. Until they do, however,
free radicals will tear their way through your body like biochemical vandals,
trashing whatever essential biomolecule they bump into: the structural pro
teins that make up your tissues, the fatty membranes that compartmental-

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 5 1
ize and facilitate your cells' various specialized functions, the DNA code
that holds the blueprints of the enzymes and proteins required by the cell,
and so on. In biology, function follows structure, so the ability of these mol
ecules to support metabolism and hold you together is impaired when they
are chemically deformed by this process.
This is obviously not good for you—and unfortunately, it's unavoid
able. Free radicals are part of being alive.
While popular press articles on aging often give the impression that free
radicals come mostly from environmental pollutants or toxins from an im
pure diet, the fact is that the overwhelming majority of the free radicals to
which your body is exposed are generated in your very own cells—in the mi
tochondria, our cellular "power plants." Mitochondria are one of several
types of "organelle," or self-contained cellular component that exists out
side the nucleus. Each cell has hundreds to thousands of mitochondria.
Man-made power plants take energy that is locked away in an inconvenient
form of fuel—such as coal, natural gas, the strong nuclear force which holds
atoms together, or wind—and convert it into a more convenient medium,
electricity, which you can use to run your blender or computer. In just the
same way, mitochondria convert a difficult-to-use energy source (the chemi
cal energy locked up in the glucose and other molecules in your food) into a
more convenient one: adenosine triphosphate, or ATP, the "universal energy
currency" that your cells use to drive the essential biochemical reactions that
keep you alive.
Mitochondria generate most of their cellular power using almost identi
cal principles to the ones used by hydroelectric dams—right down to the
turbines (see Figure 1). Using a series of preliminary biochemical reactions
(each of which generates a small amount of energy), energy from food in the
form of electrons is transferred to a carrier molecule called NAD+ (and a
similar one called FAD). These electrons are used to run a series of "pumps"
called the electron transport chain (ETC) that fill up a "reservoir" of protons
held back by a mitochondrial "dam" (the mitochondrial inner membrane).
The buildup of protons behind the "dam" creates an electrochemical
force that sends them "downhill" to the other side of the mitochondrial in
ner membrane, just as water behind a dam is drawn downward by gravity.
And just as a hydroelectric dam exploits the flow of water to run a turbine,
the inner membrane contains a quite literal turbine of its own called
"Complex V" (or the "F0/F1 ATP synthase") that is driven by the flow of
protons. The rushing of protons through the Complex V turbine causes it
to spin, and this motion is harnessed to the addition of phosphate ions

5 2 E N D I N G A G I N G
Figure 1. The F 0 / F 1 ATP synthase.

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 5 3
("phosphorylation") to a carrier molecule ( a d e n o s i n e diphosphate, or
ADP), transforming it to ATP.
Unlike hydroelectric dams, however, the use of chemical energy in food
to generate ATP via this system is a chemical reaction. As with the burning of
coal or wood to release energy, the powering-up of ADP into ATP consumes
oxygen, which is why we have to breathe to keep the whole system running:
oxygen is the final resting place of all those electrons that are released from
food and channelled through the proton-pumping electron transport chain.
Thus, the whole cycle is called oxidative phosphorylation (OXPHOS).
But while hydroelectric dams are (for the most part) environmentally
benign, mitochondria are in one key aspect more like conventional power
sources. Just like coal or nuclear power plants, mitochondria create toxic
wastes during the conversion of energy from one form into another. As the
proton-pumping complexes of the electron transport chain pass electrons
from one to the next, they occasionally "fumble" an electron here or there.
When this happens, the electron usually gets taken up by an oxygen mole
cule, which suddenly finds itself with an extra, unba l anced electron. (I just
mentioned that oxygen is also the sink for the electrons that are not
fumbled—that are properly processed by the mitochondria—but that pro
cess loads four electrons onto each oxygen molecule, not just one, so there's
no problem of electron imbalance.) Adding one electron, by contrast,
transforms benevolent oxygen into a particularly important free radical, su
peroxide. With your mitochondria generating ATP day and night continu
ally, the ongoing formation of superoxide is like having a constant stream of
low-grade nuclear waste leaking out of your local reactor.
Once scientists established that mitochondria were the main source of
free radicals in the body, it was quite quickly realized that these organelles
would also be their main target. Free radicals are so rabidly reactive that
they never travel far, attacking instead the first thing that they come
across—and the mitochondria themselves are at ground zero. And there are
plenty of potentially sensitive targets for these radicals in the mitochon
drion. Free radicals p r o d u c e d in the mitochondria are right next to the very
membranes and proteins on which ATP production depends, and also
within spitting distance of the mitochondrial DNA. What's that, you say?
Well, whereas other components of the cell have all their proteins coded for
them by the cell's centralized genetic repository in the nucleus, mitochon
dria have their own DNA for thirteen of the proton-pumping, ATP-
generating proteins in their membranes. 2 If that DNA is significantly
damaged, the mitochondrial machinery will go awry. Unfortunately, it's

5 4 E N D I N G A G I N G
clear that mitochondrial DNA does suffer a lot of self-inflicted damage,
taking as much as a hundredfold more initial oxidative "hits" than the cell's
central, nuclear DNA, and suffering many times more actual, enduring mu
tations with age.
Starting with a classic paper put out in 1972 by chemist Denham Har-
man 3 (who already had the distinction of being the father of the original
"free radical theory of aging"), researchers put these facts together with a
range of experimental findings and came up with several variations of a
"mitochondrial free radical theory of aging." Let's briefly survey that exper
imental evidence. 4
First of all, there was evidence from comparative biology. Slower-aging
organisms, relative to faster-aging ones of similar size and body tempera
ture, are always found to have slower mitochondrial free radical damage ac
cumulation. They produce fewer free radicals in their mitochondria; they
have mitochondrial membranes that are less susceptible to free radical
damage; and sure enough, they accumulate less damage to their mitochon
drial DNA. Calorie restriction (CR)—the only nongenetic intervention
known to slow down aging in mammals—improves all of these parameters:
it lowers the generation of mitochondrial free radicals, toughens their mem
branes against the free radical assault, and above all it reduces the age-
related accumulation of mitochondrial DNA mutations—the irreparable
removal or overwriting of "letters" in the genetic instruction book.
Leading toward the same conclusion from the opposite direction, CR
does slow down aging, yet it has no consistent effect on the levels of most
self-produced antioxidant enzymes. The enzymes that people examined in
this regard in the 1980s were ones that are found predominantly in the rest
of the cell, not in the mitochondria. This again suggests that free radical
damage outside of the mitochondria is not a directly important cause of ag
ing, since aging can in fact be slowed down (via CR) without doing the one
thing that would most directly interdict that damage.
Fast-forward, for a moment, to 2005. In that year, the most direct evi
dence so far on this point came to light. It involved mice that had been
given genes allowing them to produce extra amounts of an antioxidant en
zyme (catalase), specifically targeted to different parts of their bodies. 5 , 6
There was little to no benefit provided by giving these organisms catalase to
protect their nuclear DNA—the genetic instructions that build the entire
cell and determine its metabolic activity, except for those parts that the mi
tochondria code for themselves. And there was also no benefit observed
from targeting catalase to organelles called peroxisomes, which are involved

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 5 5
in processes that produce hydrogen peroxide (the molecule that catalase
detoxifies) and accordingly are already stoked up with the enzyme. Yet,
delivering catalase to the animals' mitochondria, which significantly re
d u c e d the development of mitochondrial DNA deletions, extended their
maximum lifespan by about 20 percent—the first unambiguous case of a
genetic intervention with an effect on this key sign of aging in mammals.
These mice were no more than a twinkle in their creators' eyes a de
cade ago, when I first addressed the question of mitochondrial oxidative
damage. But even back then, it seemed unassailable that free radical dam
age to the mitochondria was a key driver of aging. The question was: what
linked the one to the other?
That might sound like a stupid question, given that free radicals are ob
viously toxic, but it turned out to be decidedly tricky to come up with a co
herent, detailed, mechanistic explanation for the connection. Scientists
convinced that mitochondrial free radicals play a role in aging all begin with
the undeniable observation that free radicals spewing out from the mito
chondrion damage the membranes and proteins that it needs to generate
ATP, and also cause mutations in the mitochondrial DNA that codes for
some of those same proteins. But any such theory must explain how this self-
inflicted damage contributes to the progressive, systemic decay that consti
tutes biological aging. Until recently, nearly all such theories postulated the
existence of some form of mitochondrial "vicious cycle" of self-accelerating
free radical production and bioenergetic decay. 7 , 8
In these highly intuitive schemes, the mitochondrion is pictured to be
like a hydroelectric dam whose turbines become rusted, worn, or broken
by the forces to which they are subjected every day. Thanks to free radical
damage to their membranes, proteins, and DNA, mitochondria in cells
throughout the body would become progressively less able to pump pro
tons and to keep a lid on the "hot potato" chain of ATP synthesis with age,
leading to inefficient energy generation and increased production of free
radicals as more and more electrons escape from increasingly banged-up
transport complexes. Thus would begin the "vicious cycle," as more and
more renegade electrons tore into more and more mitochondrial con
stituents, leading to further damage to those same constituents, causing yet
more inefficient, dirty energy generation, and so on and so forth, spiralling
downward and ultimately starving the cell of energy and rendering it a haz
ardous waste site. See Figure 2.
Some version of this scenario is repeated in nearly all popular books and
articles about the role of mitochondria in aging, as well as most scientific

5 6 E N D I N G A G I N G
Figure 2. The "vicious cycle" theory of mitochondrial mutations accumulation. The key tenet of the theory is denoted by the asterisk: that typical mitochondrial mutations raise the rate of release of free radicals.
journal publications. Yet we've had evidence for nearly twenty years show
ing conclusively that it can't be right.
Everything You Know Is Wrong
The "vicious cycle" theory of mitochondrial decay paints a picture that
sounds seductively plausible, but it just isn't compatible with the data. While
many scientists remain oblivious to the glaring inconsistencies in these theo
ries even today, a few mitochondrial specialists and biogerontologists have
been pointing these problems out since the mid-1990s. So different were the
predictions of "vicious cycle" theories from the experimental findings that
many of these researchers went so far as to suggest that the findings simply

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 57
ruled out a role for mitochondria free radicals in aging—forgetting that such
a role might exist but not via a vicious-cycle mechanism.
The first problem with at least some versions of the vicious cycle the
ory had actually been pointed out decades earlier—just after Harman had
originally put forward the first version of the mitochondrial free radical the
ory, in fact—by none other than Alex Comfort. (Yes, that's the same Alex
Comfort who wrote The Joy of Sex. He was a true polymath: he was also a
controversial anarchist agitator, a poet, and a highly distinguished biogeron-
tologist.)
In 1974, Comfort pointed out that while each mitochondrion could
temporarily suffer progressively increasing damage to the proteins and
membranes that make up its ATP synthesis machinery from the ongoing
free radical barrage, no theory based on the idea that this damage would
get worse and worse with age could fly, for the simple reason that the cell is
constantly replacing and renewing those very components.
Periodically, old mitochondria are tagged for destruction in yet an
other type of organelle, the lysosome (the cellular "toxic waste incinera
tor," about which I'll be talking a great deal in Chapter 7). Then, to make
up for the loss of energy factories, the cell puts out a signal for the remain
ing mitochondria to replicate themselves. During replication, each mito
chondrion duplicates its DNA, and then that essential core "grows" itself
a new "body," including pristine new proton-pumping proteins and mem
branes. Whether you're five years old or fifty, any given mitochondrion in
your cells contains membranes and proteins that are on average only a few
weeks old. Thus, the newest and the oldest of these mitochondrial compo
nents are present in the same proportions in the very old as in the very
young. It just can't be that aging is driven by a progressive process of de
generation in components that undergo a continuous process of renewal.
However, this objection wasn't necessarily a problem for the more pop
ular versions of the vicious cycle theory: those that assert that free radical
damage to the mitochondrial DNA drives aging. Although mitochondrial
membranes and proteins are periodically replaced, all mitochondria inherit
their DNA directly from their "parent" power plants, which faithfully copy
out their DNA and pass it on to their "children"—and just as whole organ
isms pass on any mutations that they may harbor in their DNA to their off
spring, so errors in "parent" mitochondria appear in the next mitochondrial
generation. If the mitochondria that have damaged DNA are preferentially
destroyed by lysosomes, the effect will be as before—damage will be

5 8 E N D I N G A G I N G
removed as fast as it spreads—and that's what Comfort reasoned would be
occurring. But if there's no such bias in which mitochondria are and are not
destroyed, DNA damage will accumulate.
These theories received a superficial plausibility boost from studies
conducted in the 1990s, which showed that aging bodies do, indeed, accu
mulate cells populated with mutant mitochondria. However, the same stud
ies shot entirely new—and even more deadly—holes into mitochondrial
DNA-based "vicious cycle" theories.
For one thing, it was found that all of the mutant mitochondria in a given
cell contain the same mutation. This is exactly the opposite of what the "vi
cious cycle" would predict. If each mitochondrion individually decayed as a
result of a self-accelerating cycle of oxidative "hits" to its DNA, then each
one would display an unique, random profile of mutations. In the same way,
if one day—independently—two disgruntled librarians were each to snap,
going on automatic rifle rampages through the collections in their care, you
would naturally expect to see that the bullets would have hit different books,
even if the collections themselves were the same: one would have put a bullet
straight through the spine of a copy of Finnegans Wake, while the other
would have punched an off-center dot atop the " i" in Bridget Jones' Diary,
and so on, at random, until each mad bibliophile ran out of bullets.
Instead, wherever cells that contain defective mitochondria are found,
the mutants all harbor an identical DNA mutation. It's as if librarians across
the state had marched into work and each fired his or her guns exactly
once, in every case into copies of The Catcher in. the Rye. Random muta
tions happening continuously in each mitochondrion cannot reasonably re
sult in each of the damaged mitochondria in a cell containing the same
error in their DNA; a random mutation-creating process can't explain the
complete takeover of cells by such mitochondria, or the presence of other
cells that contain nothing but healthy ones.
In fact, it's even weirder than that, because the presence of mutant mi
tochondria turns out to be an all-or-nothing affair. That is: near enough, not
only do all of the mutant mitochondria in a given cell turn out to contain
the exact same mutation, but those cells that harbor any damaged mito
chondria are found to contain nothing but mutants—while the other cells
have nothing but pristine, youthful "power plants."
But wait: the findings are even more bizarre yet. While each cell is full
of mitochondria that all bear the same mutation, the mitochondria in dif
ferent cells contain different mutations. It's as if all of the librarians in
Delaware had pumped their local branch's copies of War and Peace full of

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 5 9
lead, while their colleagues in California had simultaneously displayed an
equally single-minded determination to purge their collections of Lady
Chatterly's Lover. It's a case in which even the most skeptical conspiracy-
theory debunker would be f o r c e d to admit that a "random act of violence"
was not a credible explanation for the crime scene.
The nature of these mitochondrial mutations also proved to be inconsis
tent with vicious cycle theories of mitochondrial priority in aging. The as
sumption had been that damage to the DNA blueprints for mitochondrial
proteins would most often result in minor defects in the instructions by
which those proteins are coded. The ensuing proteins would be close
enough to their proper structure to remain more-or-less capable of carrying
on, but would be dysfunctional, "fumbling" more electrons into free radi
cals and generating less ATP. Instead, the mutations that accumulate in cells'
mitochondria were found to be overwhelmingly deletions of large blocks of
DNA, which completely shut down the creation of all the mitochondrially-
encoded proteins.
The vicious cycle theory proposed that mitochondria would make pro
gressively less ATP and more free radicals as they accumulated more and
more of these defects in their DNA. Instead, it was found that mutant mi
tochondria produce essentially no free radicals, and that the change in each
mitochondrion could be attributed to a single, catastrophic event, rather
than to a "death from a thousand cuts."
Forget the Quality, Feel the Quantity
An additional finding seemed to leave no room for any way, vicious or oth
erwise, in which mitochondrial mutations could be involved in aging: very,
very few cells actually contain mutant mitochondria at all. The vast majority
of cells remain in perfect mitochondrial health well into old age. Yes, a tiny
proportion of older people's cells—about 1 percent—could be shown to be
completely taken over by mitochondria that all suffer from an identical de
fect in their DNA; but 99 percent of cells were fine. How could 1 percent
of cells matter?
Many biogerontologists concluded that these findings put the kibosh
on any theory that asserted that mitochondrial decay was important in ag
ing. If nearly every cell in the body still enjoys the same level of ATP output
as it had in its youth, and suffers no more free radical damage than it did in
its prime, how could a tiny proportion of cells that are low on power, but

6 0 E N D I N G A G I N G
whose mitochondria produce no more free radicals than their neighbors—
in fact, produce no free radicals at all—possibly have much negative effect
on the function of the tissue in which they reside or the organism as a
whole? To these scientists, the mitochondrial free radical theory of aging
seemed dead in the water.
This was where the field sat in the mid-1990s, when I first became
aware of the unsatisfactory state of aging science and decided to try to do
something to change the situation. When I saw the confused state of the
mitochondrial free radical theory of aging, I felt that the field looked ripe
for a new synthesis. On the one hand the evidence supporting the existence
of a central role for mitochondrial free radicals in aging seemed strong; on
the other hand, the standard vicious cycle theories simply could not be rec
onciled with the emerging results. It was into this fray that I stepped with
my first formal scientific papers in 1997 9 and 1998, 1 0 and where I have
made my most widely acknowledged contributions to biogerontology. My
key insights were, firstly, an explanation of how aging cells accumulate mi
tochondria that share one mutation in common instead of the random set
predicted by the vicious circle theory, and secondly, an explanation of how
only a small number of cells taken over by such mutant mitochondria could
drive aging in the body as a whole. Let's walk through these ideas one at a
time.
SOS: Survival of the Slowest
The vicious cycle theory had assumed that each mitochondrion would
slowly accumulate minor, random mutations over the course of its lifetime.
The fact that, in those cells where there were mutant mitochondria, all the
mutants shared the same mutation—and that the mutants had completely re
p l a c e d all healthy mitochondria in the cell—proved that assumption wrong.
The only reasonable alternative seemed to be "clonal expansion": the
idea that a single mitochondrion had originally gone bad, and that its prog
eny had slowly taken over the entire cell. Remember, mitochondria repro
duce themselves by splitting themselves in half, much like amoebae: the
original mitochondrion makes a copy of its DNA, and then forms two iden
tical genetic "clones" of itself. This means that each clone will contain ex
act copies of any mutations present in the original organelle. So it seemed
inescapable that the strange mitochondrial monocultures found in the all-
mutant cells were the result of one mitochondrion initially acquiring a mu-

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 6 1
tation, passing it on to its offspring, and then having its lineage somehow
outcompete all of its neighbors until it eventually becomes the only game in
town.
However, the idea that mitochondria with mutated DNA could some
how win a battle for dominance within the cell was itself a bit of a paradox.
After all, these mitochondria are defective, with one or more enormous
chunks blasted out of their DNA by free radicals or replication errors. While
it's true that once in a blue moon a mutation turns out to be beneficial—this
is, after all, what allows evolution to happen—it's supremely unlikely that it
would happen over and over again, such that random mutations occurring in
mitochondria in widely separated cells would turn out to be so beneficial to
the mitochondrion as to give it a Darwinian "fitness advantage" over its fel
lows. And indeed, the mutations in question were known to be deleterious:
they completely knock out the mitochondrion's ability to perform oxidative
phosphorylation, and thus turn off the great majority of their contribution to
the cellular ATP supply.
The "clonal expansion" explanation was also hard to reconcile with the
fact that many different mutations can cause a specific mitochondrial line
age to replace all other alternative lineages in the cell. That is: while the mu
tant mitochondria in a given cell all contain the same, specific mutation, a
second such cell often contains mitochondria that all harbor a completely
distinct mutation from the one that was found in the first. So it wasn't that
there is a single, specific mutation that gives the mutants their selective ad
vantage over their neighbors: numerous mutations, independently arising
in single mitochondria within widely separated cells, confer the same com
petitive edge. Was it really likely, I asked myself, that there were this many
advantageous, yet unrelated, mutations to be had?
Yet, these various mutations do have one thing in common. They aren't
mild mutations, damaging just one protein: all of them are of a type that
prevent the synthesis of all thirteen of the proteins that the mitochondrial
DNA encodes. This shared property, I felt, might be the key to how they
managed to take over the cell.
I set myself to thinking what would distinguish such mitochondria from
their healthy counterparts. They would not generate nearly so much ATP, of
course: only the small amount of cellular energy that gets p r o d u c e d in the ini
tial stages of extracting chemical energy from food, which was a fraction of
the total that a functioning oxidative phosphorylation system could churn
out. This would be unhealthy for the cell, certainly, but I realized that it
would have little negative impact on the mitochondrion itself, which normally

6 2 E N D I N G A G I N G
exported nearly all of the ATP that it p r o d u c e d anyway. So, while I couldn't
see how this r e d u c e d energy output could explain the selective advantage en
joyed by the mutants, I saw that—contrary to what one might initially
think—-it really wasn't a direct disadvantage relative to other mitochondria in
the cell that might hinder them from rising to dominance in the host.
The other thing that would set mitochondria with no oxidative phospho
rylation capacity apart from other mitochondria in a cell seemed more likely
to be advantageous: such mitochondria would no longer be producing free
radicals. Remember that mitochondrial produce free radicals when electrons
leak out of the regulated channels through which they pump protons into the
"reservoir" that drives the "turbines" of the mitochondrial inner membrane.
If you aren't feeding electrons into the pump system because the system itself
is missing, then clearly there will be no leakage—and no free radicals.
Not having to deal with constant free radical vandalism sounded like it
might be good for the mitochondrion—but it was not clear exactly how it
might lead to an actual competitive advantage vis-a-vis the healthy mito
chondria with which it was surrounded. True, its DNA would stop being
bombarded—but of course, by this time it would already be suffering from
a gaping hole in its DNA.
It was also obvious that the mitochondrion's inner membrane would no
longer suffer free radical damage—but again, it didn't seem that this would
matter in aging, since mitochondria are constantly having their membranes
torn down and r e p l a c e d anyway, either during replication or at the end of
their brief individual lives, when mitochondria with defective membranes
are sent off to the cellular "incinerator" in any case.
Now hang on a minute, I thought.
Like other researchers who had puzzled over this question, I'd been
trying to imagine some improvement to the mitochondria's function con
ferred by the mutation—the equivalent, in the microscopic evolutionary
struggle, to sharper teeth, faster running, or greater fecundity. But what if,
instead, the important thing about the mutation was not that it made its
carriers "better," but that it prevented them from being destroyed?
Recycling Cellular Trash
There's still a lot of work to be done to explain what exactly causes mito
chondria to be sent to the cellular garbage disposal system. Still, even as
early as Alex Comfort's criticism of the original mitochondrial free radical

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 6 3
theory of aging, it was widely believed that there was some selective process
that specifically targeted old, damaged organelles for destruction. This
could not be taken for granted, however. It was long believed that some
components of the cell are turned over by an ongoing process of random
recycling, in which the lysosome (strictly, a special sort of pre-lysosome
called an autophagosome or autophagic vacuole) simply lumbers about the
cell, swallowing a given number of various cellular constituents at random
each day, so that everything is ultimately turned over sooner or later.
It is now widely accepted that this isn't the way the lysosome works: the
engulfment of proteins and other cellular components is known to be a
highly directed process. In part, this is simply a matter of good use of scarce
resources. Imagine if, in order to ensure that old, decaying vehicles were
taken off the road (to improve the nation's overall air quality and greenhouse
gas emissions, eliminate the eyesore of old cars rusting on blocks, and bring
down the price of recycled steel), the government were to send its agents
wandering aimlessly through economically depressed neighborhoods to ran
domly select cars to be sent off to the scrap heap. Such a program would
achieve some of its goals, but it would hit too many fully functional vehicles
to make it viable, even setting aside the question of individual property rights.
But in some cases there is an even more powerful reason than mere ef
ficiency to be sure that specific organelles get sent to the scrap heap. Some
cellular components can become actively toxic to the cell if they aren't
quickly degraded once they've outlived their usefulness. Like the en
chanted broom in Fantasia that continues to fill the vat in the sorcerer's hall
with water until it overflows and floods the room, many proteins and or
ganelles are only useful for a limited period; when their job is done, they
must be "put away," and in the cell, this means torn apart for recycling. For
instance, producing a pro-inflammatory enzyme can be critical to mobiliz
ing an immune reaction against an invading pathogen, but leaving that en
zyme to keep generating inflammation after the invader has been defeated
would lead to a destructive, chronic inflammatory state with effects similar
to those of autoimmune diseases like rheumatoid arthritis or lupus.
It then occurred to me that there were very good reasons for the cell to
take care that its mitochondria were destroyed when their membranes had
suffered free radical damage. Recall that the inner mitochondrial mem
brane acts as a "dam" to hold back the reservoir of protons that powers the
energy-generating turbine of Complex V. Holes in that membrane would
be "leaks" in the dam, depleting the reservoir as ions simply seeped
through the holes without generating ATP. Evidence to confirm this basic

6 4 E N D I N G A G I N G
scenario, in the form of leaks created from damaged membrane molecules,
had actually been uncovered as early as the 1970s.11
This would make the "leaky" mitochondrion a serious drain on scarce
resources, as the electron chain would continue to consume energy from
food in a furious, futile attempt to refill the reservoir. Food-derived elec
trons would continue to be fed into the chain, which would use them to
keep pumping protons across the membrane, but these ions would leak
back across as fast as they were pumped "uphill," without building up the
electrochemical reservoir needed to create usable energy for the cell. This
would drain the cell of energy, turning nutrients not into ATP but instead
into nothing more useful than heat.
Moreover, the damage to the inner membrane might also allow many
of the smaller proteins of the mitochondrial inner space to be released out
of the mitochondrion and into the main body of the cell. If they continued
to be active, these components could well be toxic to the cell when released
from the controlled environment of the mitochondrion.
It would make sense, then, for the cell to have a system in place that
would ensure that mitochondria are hauled off to the lysosome for destruc
tion when their membranes become damaged by their own wastes. This pre
diction seems to have been fulfilled with the recent discovery of a specific
targeting protein that "tags" yeast mitochondria for lysosomal pickup. 1 2 We
still don't know for sure what makes the cell decide which mitochondria to
"tag," but it has now been shown that the formation of holes in the mito
chondrial membrane does send a signal that increases the rate at which these
organelles get sent to the scrapyard. 1 3
I had no doubt that this was all to the good: I'm all for the removal of
defective and potentially toxic components from the cell, and as usual na
ture has evolved an ingenious way to make sure that it happens. But I saw
that, ironically, large deletions in the mitochondrial DNA would actually al
low them to escape from the very mechanism that cells use to ensure that
damaged mitochondria get slated for destruction. When mitochondria suf
fer the mutations that have been shown to accumulate with aging, they im
mediately cease performing oxidative phosphorylation (OXPHOS)—and
with it, generating the resultant free radical waste. But r e d u c e d free radical
production, in turn, should lead to less free radical damage to their mem
branes. Don't forget that the prevailing vicious cycle theory proposed that
mitochondrial mutations proliferate by causing their host mitochondria to
make more free radicals than nonmutant mitochondria do. That, I saw, was
where the proponents of the vicious cycle theory had gone wrong.

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 6 5
Hiding Behind Clean Membranes
Having advanced this far, I immediately saw how the mutants gained their
advantage over their healthy counterparts. Even perfectly functional mito
chondria constantly produce a steady, low-level stream of free radicals,
leading to membrane damage. Every few weeks, this damage builds up to a
level at which the mitochondria are consigned to the rubbish tip, and then
the cell sends out a signal for a new round of mitochondrial reproduction to
replace the decommissioned "power plants."
But this process only weeds out mitochondria with damaged
membranes—which will overwhelmingly be power plants whose DNA is still
healthy enough to allow for the very electron transport that leads to the free
radical damage to their membranes in the first place. Mitochondria with in
tact membranes, but damaged DNA, would not show outward signs of their
internal injuries, and so would be passed over by the Angel of Death.
After a certain number of damaged mitochondria have been hauled off
to the lysosome, the cell will send out the signal for mitochondria to repli
cate. Some or all of the remaining mitochondria—the genetically healthy
and the mutants alike—will reproduce themselves, and because those mito
chondria that bear large DNA deletions will almost always have survived
the purge of outwardly damaged power plants, they will enjoy the opportu
nity to reproduce. But many of the membrane-damaged—but genetically
intact!—mitochondria will already have been removed before replicating
themselves. This will give the mutants a selective advantage over the non-
mutants: every time replication happens, more and more of them will have
survived a cull that has sent many of their genetically healthy competitors
to the garbage disposal unit.
This is exactly how evolution works in organisms, of course. Animals
that are slower runners, or less able to find food, or have poorer eyesight
are more vulnerable to death from predators, exposure, or disease, which
prevents them from successfully reproducing and passing on their genes.
Meanwhile, a disproportionately high number of better-adapted organisms
get the chance to breed, leaving behind progeny that carry their genetic
legacy into the future. Over time, the genes that are best adapted to the spe
cific threats in their environment come to dominate in the population.
In the cell, the threat to mitochondrial survival is the lysosome—a
"predator," which is supposed to ensure that only mitochondria fit to safely
support cellular energy production survive. What the mutant mitochondria
evolve (yes, evolve) is, in effect, camouflage that masks them from the eagle

6 6 E N D I N G A G I N G
eye of this predator. Thanks to their undamaged membranes, these highly
dysfunctional mitochondria appear healthy to the cellular surveillance sys
tem. Like the proverbial pharisees, their outsides are clean—but inwardly,
they are full of ravening and wickedness.
I concluded that this "camouflaged mutant" concept provided the first
consistent, detailed explanation for the takeover of cells by defective mito
chondria. I named it "Survival of the Slowest" (SOS), because it postulates
that the quiescent ("slow") mitochondria enjoy a fitness advantage in the
Darwinian fight for survival in the cellular jungle. See Figure 3.
But now, having explained how a small number of cells become a
monoculture of defective mitochondria, there remained a question which
might be thought to be more important—namely, how does that tiny frac
tion of the body's cells drive aging across the entire body? It wasn't long be
fore I had a good explanation for that, too.
The "Reductive Hotspot Hypothesis"
The old vicious cycle models didn't need to invoke any additional mecha
nism to explain how mitochondrial mutation could contribute to aging, be
cause they assumed that there would be an accumulation of increasingly
defective mitochondria in lots of cells with age. As more and more of their
mitochondria randomly went on the blitz, the cells would suffer more and
more oxidative hits, and be more and more starved for ATP, as the power
plants' efficiency sank with every new defect. It was a nice, common-sense
explanation for the role of mutant mitochondria in the aging of the body.
But, as we've seen, it was also clearly wrong. Most cells in the body sim
ply do not accumulate mutant mitochondria as we age: at most, about 1 per
cent of all of the trillions of cells in the body do so. Most cells and tissues
suffer no decline in production or availability of ATP, and far from increasing
free radical production, most of the mutant mitochondria that accumulate in
the body produce no free radicals at all, because their radical-generating elec
tron transport chains are simply absent.
It was hard to see how so few cells, containing mitochondrial mutants
that were not harming nearby cells in any obvious way, could possibly be
driving aging in the body. In fact, these findings were enough to make plenty
of biogerontologists talk about the death of the mitochondrial free radical
theory of aging. Yet, as we briefly discussed earlier in this chapter, the cir
cumstantial evidence that mitochondrial mutations somehow contribute to

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 67
Figure 3. The "Survival of the Slowest" model for mitochondrial mutation accumulation, (a) The proposed normal mode of turnover and renewal of non-mutant mitochondria; spots denote membrane damage. (b) The clonal expansion of mutations (denoted by X) resulting from low free radical damage to membranes and slow lysosomal destruction.
aging is too strong to dismiss. To reconcile the two sets of data would re
quire a truly novel solution to the puzzle.
I saw that any refined version of the mitochondrial free radical theory
of aging would have to do two closely related things. First, since so few cells
are taken over by these burned-out power plants, it would have to show
that cells harboring mutant mitochondria somehow spread toxicity beyond
their own borders. And second, it would have to explain the nature of that

6 8 E N D I N G A G I N G
toxicity, since the usual suspect—free radicals—appeared to have been
ruled out by the fact that the mitochondria in these cells would have their
normal free radical production turned off at the source.
I began by trying to work out just what cells that had been taken over
by mutant mitochondria were doing to survive in the first place. What were
they using as an energy source? Not only were these mitochondria unable
to perform the oxidative phosphorylation that provides their host cells with
the great majority of their ATP, but it was not at all obvious how they could
produce any cellular energy at all.
Upstream of a Blocked Dam
In normal cells, the initial metabolism of glucose from food is performed in
the main body of the cell through a chemical process called glycolysis. Gly
colysis generates a small amount of ATP, a breakdown product called pyru
vate, and some electrons which can drive oxidative phosphorylation in the
mitochondria. To shuttle electrons into the mitochondria for this purpose,
they are loaded onto a carrier molecule called NAD +. The charged-up form
of NAD + is called NADH.
The pyruvate formed during glycolysis is also delivered into the mito
chondria, where it is further broken down into another intermediate called
acetyl CoA. This process releases some more electrons, which are again har
vested for use in electron transport by "charging" NAD + into NADH.
Acetyl CoA is then used as the raw material for a complex series of chemi
cal reactions called the tricarboxylic acid (TCA) cycle (also called the Krebs
cycle or citric acid cycle), which liberates many times more electrons (again
leading to creation of NADH) than has been created in previous steps.
Finally, all of the NADH charged up via all these processes—glycolysis,
the breakdown of pyruvate into acetyl CoA, and the TCA cycle—is deliv
ered to the electron transport chain, which uses this electron payload to
generate the proton "reservoir" that drives the generation of nearly all the
cell's energy.
This was well-understood biochemistry, taught in its simple form to
students in middle-school science classes. But it's all predicated on being
able to feed these electrons into the electron transport chain machinery. So,
I asked myself, what would happen when that machinery was shut down, as
it is in mitochondrially mutant cells?
It seemed to me that the whole process might grind to a halt. Every

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 6 9
step along the way—from glycolysis to the TCA cycle—loads electrons
onto waiting NAD + "fuel tankers" for delivery to the electron transport
chain. There is, of course, only a limited supply of NAD + "carriers" avail
able to be charged up into NADH, but normally that isn't a big deal: there
are always plenty of these carriers available, because NADH is recycled
back into NAD + when it releases its electron cargo to the electron transport
machinery in the mitochondria.
But with that natural destination shut off, there is no obvious way for
NADH to relieve itself of its burden of electrons. (Similarly, you can imag
ine how, if all the refineries on Earth were suddenly decommissioned, the
taps on the world's oil wells would quickly need to be turned off. With
nowhere to deliver their oil for processing, fuel tankers could only be filled
once before their capacity would be taken out of circulation, and continu
ing to pump oil would lead to a logistical nightmare.) And since every step
of the process—glycolysis, intermediate metabolism of pyruvate into acetyl
CoA, and the TCA cycle—needs NAD + to proceed, a lack of NAD + would
be expected, at first glance, to lead to the deactivation of the entire process,
leaving no mechanism for the cell to produce even the small quantities of
ATP energy that result from these early processing steps.
In fact, I could see how mitochondrially mutant cells might be even
worse off than this. NAD + is required for a wide range of cellular functions
unrelated to energy production—and each time these functions utilize
NAD +, they not only reduce the pool of available NAD + , they also convert
it to yet more NADH, further upsetting the cell's metabolic balance. In
fact, some researchers believe that many of the complications of diabetes
are caused by an excess of NADH and a lack of NAD + , leading to disrup
tion of these various metabolic processes (although the imbalance of NAD +
and NADH in diabetics has different causes than the loss of OXPHOS
capacity).
Despite all this, however, cells that have been taken over by mutant mi
tochondria do survive, as is shown by their gradual accumulation with ag
ing. So they have to be getting ATP from somewhere. At the time I was
exploring this matter, the general presumption in the field was that these
cells could survive by shutting down the TCA cycle and relying entirely on
glycolysis for energy production. This is what happens in muscle cells as a
brief stopgap during intense anaerobic exercise, when the cell is working so
hard that it uses up all of the available oxygen and can't keep oxidative
phosphorylation going. Glycolysis would provide the cell with a small but
just adequate amount of ATP, and this school of thought suggested that the

70 E N D I N G A G I N G
cell could deal with the resulting small excess of NADH through a bio
chemical process that converts pyruvate into lactic acid—the biochemical
origin of the famous "burn" that weightlifters suffer during the very last
possible rep in their set.
But I realized that this theory didn't match the evidence. For one
thing, the expected rise in lactic acid didn't seem to happen. And even
more bizarrely, rather than a shutdown of TCA activity (as one would ex
pect because of the lack of the required free NAD +), enzyme studies had
strongly suggested that mitochondrially mutant cells have hyperactive TCA
cycles. So, I wondered, how do they keep this seemingly unsustainable
process going?
Learning from the Great Mr. Nobody
A big step toward understanding this phenomenon was achieved with the
creation of so-called p0 ("rho-zero") cells, whose mitochondria are com
pletely lacking in DNA. This condition renders cells functionally very simi
lar to cells that have been overtaken by mutant mitochondria, because the
deletion mutations found in these mitochondria actually shut down the
ability to turn any DNA instructions into functioning proteins. If your
DNA can't be decoded into usable blueprints, it might as well not be there,
like instructions on how to build a bridge that are written in a language you
don't understand. So having these DNA deletions puts mitochondria in just
the same condition as having no mitochondrial DNA at all.
One of the first things that scientists working with p° cells discovered
was that they did, in fact, quickly die—unless their surrounding bath of
culture medium contained one of a few compounds which are not normally
present in the fluid that surrounds cells in the body. Intriguingly, however,
some of these compounds are unable to enter cells, which meant that what
ever it was that these compounds do to keep cultured p° cells alive, it must
be something that can be accomplished from outside the cell. This fact
made little sparks go off in my brain, because I was looking for a way to ex
plain how cells that had been overtaken by a clonal brigade of mutant mi
tochondria could export some kind of toxicity outside themselves to the
body at large. Might these compounds rescue p0 cells by unburdening them
of this same toxic material?
And might this toxic material be none other than . . . e l e c t r o n s ?
I immediately drew a connection between the predicted excess of

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 7 1
NADH in cells that could not perform oxidative phosphorylation, and the
dependence of p° cells on the presence of the "detoxifying" compounds in
their medium. What the mitochondrially mutant cell needed to do was to
dump electrons, so as to recover some NAD+—and the "rescue" compounds
for p° cells were all electron acceptors, and they worked even if they were
kept outside the cell's boundaries. My hypothesis: Mitochondrially mutant
cells prevent a crippling backlog of unused electrons by exporting them
out of the cell, via a mechanism similar to that which is essential to the sur
vival of p0 cells in culture—and this export somehow spreads toxicity to the
rest of the organism.
The Safety Valve
To turn this idea into a reformulation of the mitochondrial free radical ag
ing theory, I needed clear answers to three questions. First, how were these
cells delivering electrons to acceptors that were located outside their own
membranes? Second, since the electron acceptors used in the p° culture
studies are normally not found in bodily fluids (or not at adequate concen
trations), what electron acceptors are available to do the same job for mito
chondrially mutant cells in the body? And third, could these processes
provide an explanation for these cells' systematic spread of toxicity
throughout the body, as seemed required in order to accept that they might
play a significant role in aging?
The first question turned out to have been answered already. For de
cades, scientists had known of the existence of an electron-exporting fea
ture located at the cell membrane that we today call the Plasma Membrane
Redox System (PMRS). While little was understood about its actual pur
pose in the body, its basic function was well established: it was known to
accept electrons from NADH inside cells and to transport them out of the
cell, thereby recycling the NADH to NAD +. This export allows even nor
mal, healthy cells to have better control over the balance of chemically oxi
dizing and reducing factors within their boundaries, and to keep tighter
control over the availability of NAD + and NADH for essential cellular bio
chemistry. In other words, it does exactly what mitochondrially mutant
cells would need to be able to do in order survive.
And the PMRS turned out to be an almost impeccable candidate for
the job. PMRS researcher Dr. Alfons Lawen, of Monash University in Aus
tralia, had by this time already shown (with no thought of its application to

7 2 E N D I N G A G I N G
aging, mind you) not only that the PMRS is able to deliver electrons to the
same membrane-impermeant electron acceptors that allow p° cells to sur
vive, but that PMRS activity is required for the survival of these cells. This
proved both that the export of electrons is a requirement for cell survival,
and that the PMRS is the dock that sets them loose into the ocean of sur
rounding bodily fluids.
The ability of mitochondrially mutant cells to recycle NADH back to
NAD + would allow them to carry out their normal cellular processes, and
without becoming so burdened with extra electrons as to create an internal
environment in which other critical cellular chemistry becomes impossible.
Moreover, I realized, this is a plausible explanation for the fact that these
cells have an unusually active TCA cycle. With oxidative phosphorylation
shut down, putting the TCA cycle into overdrive would allow the cell to
double its production of precious ATP from sugars (and to do many other
metabolic jobs in which the TCA cycle participates). The PMRS would
make this possible, by recycling the greatly increased amounts of NADH
that would be created and thereby providing the cell with the extra NAD +
required to keep the process going.
But the drastic increase in PMRS activity required to make the increased
TCA activity sustainable would make the surfaces of mitochondrially mu
tant cells positively bristle with electrons undergoing export, forming a
hotspot of electrically "reducing" pressure (i.e., an unstable excess of elec
trons). The next question, therefore, was onto what molecules the PMRS
was unloading the electron surfeit. None of the electron acceptors being
used to keep p° cells alive in culture existed in adequate concentrations in
the body to accomplish the task, so something else had to be performing
this essential role. For example, some of the burden might be taken up by
dehydroascorbate, the waste product of vitamin C that is created after it is
used in quenching free radicals, but there wasn't enough of that to deal
with the powerful "reductive hotspot" created by these cells.
At this point, an attractive candidate that could tie the story together
flashed into my mind: our old two-faced friend, oxygen.
Don' t Throw Your Junk in My Backyard, My Backyard . . .
Oxygen, of course, can and does absorb surplus electrons in its environ
ment. As we discussed above, this is exactly how mitochondria generate
free radicals during oxidative phosphorylation, as "fumbled" electrons

M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 73
slipping out of the electron transport chain are taken up by the oxygen dis
solved in the surrounding fluid. And oxygen is the only such molecule in
the body's bathing fluids that is present in sufficient quantity to be able to
sponge up the huge electron leak that mitochondrially mutant cells would
be predicted to generate.
This uptake of electrons by oxygen could, in an ideal world, be safe: the
PMRS might load four electrons onto each oxygen molecule, turning it into
water, just as the electron transport chain does with electrons that it doesn't
"fumble." But it was eminently possible that the PMRS would fumble some
electrons—maybe quite a lot of them. If so, it would generate large amounts
of superoxide radicals at the surface of mitochondrially mutant cells. The
consequences of this would clearly be bad. But actually, maybe not so bad:
one might think that the negative effects would mostly be confined to the
immediate locality. Like nearly all free radicals, superoxide is highly reactive,
and therefore short-lived. Either it would be dealt with by local antioxi
dants, or else it would attack the first thing with which it came into contact
(a neighboring cell's membrane, for instance)—but its aggressiveness would
be quenched in the process. Superoxide certainly couldn't remain a free
radical for long enough to reach the far corners of the body, as the implica
tions of the mitochondrial free radical theory required.
But what if, instead of attacking the components of a cell's immediate
neighbors, superoxide generated by the PMRS were to damage some other
molecule that was then stable enough to be carried throughout the body?
There was an obvious suspect: serum cholesterol, especially the LDL
("bad") particles—low-density lipoproteins, to give them their full name—
that deliver their cholesterol payload to cells all over the body.
Oxidized (and otherwise modified) cholesterol was already known to
exist in the body, and everyone now accepts that it's the main culprit be
hind atherosclerosis (a subject to which we'll return in Chapter 7). It was
quite plausible, I realized, that superoxide originating at the surface of mi
tochondrially mutant cells could be oxidizing LDL as it passed by, not only
because LDL is ubiquitous and therefore an easy target, but also because
the presence of loosely bound reactive metals such as iron ions would mul
tiply superoxide's potential virulence. One might think that the presence of
antioxidants—such as the vitamin E that's dissolved in LDL—would pre
vent this from happening, but researchers had already discovered that it
didn't. Not only is oxidized LDL found routinely in the body, but studies
using the most accurate available tests of lipid peroxidation had shown that
vitamin E supplements were unable to reduce the oxidation of fats in

7 4 E N D I N G A G I N G
healthy people's bodies. 1 4 In fact, the lack of antioxidant partners in the in
accessible core of the LDL particle means that when it is subjected to any
thing more intense than the most trivial free radical challenge, the particle's
vitamin E can actually accelerate free radicals' spread to its center through
a phenomenon called "tocopherol-mediated peroxidation." 1 5 (Tocopherol
is the technical name for vitamin E.)
I saw the light at the end of the logical tunnel now. The oxidation of
LDL would provide a very plausible mechanism to explain the ability of
mitochondrially mutant cells to spread oxidative stress throughout the ag
ing organism. Despite its ability to promote atherosclerosis when present in
excessive amounts in the blood, LDL cholesterol serves an essential func
tion in the body. Cells need cholesterol for the manufacture of their mem
branes, and LDL is the body's cholesterol delivery service, taking it from
the liver and gut (where it is either manufactured or absorbed from the
diet) out to the cells that need it.
But if its cholesterol consignment became oxidized by mitochondrially
mutant cells along the way, LDL would become a deadly Trojan horse, de
livering a toxic payload to whichever cells absorbs its cargo of damaged cho
lesterol. This would spread free radical damage into the incorporating cell,
as the radicalized fats propagated their toxicity through the well-established
chemical reactions that underlie the rancidity of fats. As more and more cells
were taken over by mutant mitochondria with age, more and more cells
would accidentally swallow oxidized LDL, and oxidative stress would grad
ually rise systemically across the entire body. See Figure 4.
The New Mitochondrial Free Radical Theory of Aging
I walked myself through the entire scenario again and again, and gradually
satisfied myself that I had indeed developed a complete, detailed, and con
sistent scenario to explain the link between mitochondrial free radicals and
the increase in oxidative stress throughout the body with aging. This model
answered all of my key questions and resolved all of the apparent para
doxes that had led so many of my colleagues to abandon the mitochondrial
free radical theory of aging entirely. Mitochondrial free radicals cause dele
tions in the DNA. These mutant mitochondria are unable to perform ox
idative phosphorylation, drastically reducing their production of both ATP
and free radicals. Because they are not constantly bombarding their own
membranes with free radicals, the cell's lysosomal apparatus will not recog-
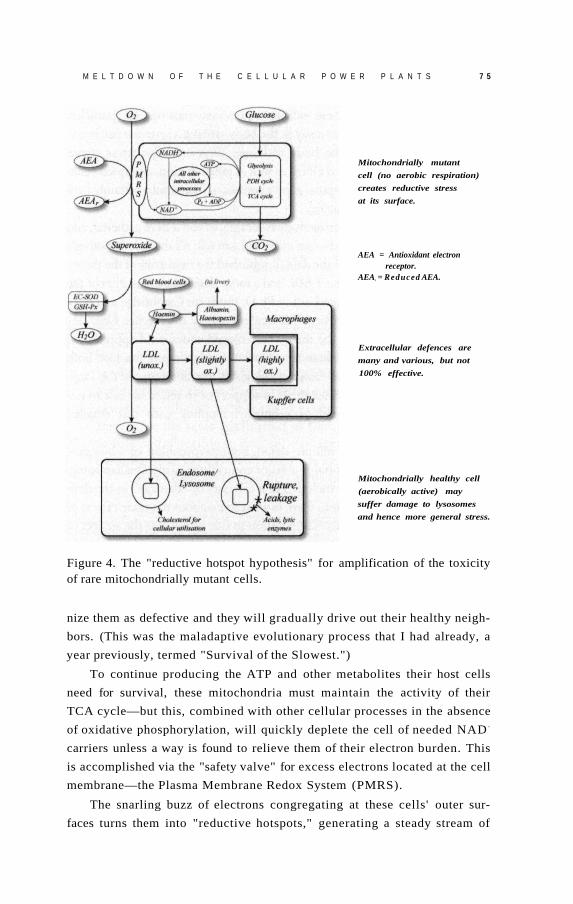
M E L T D O W N O F T H E C E L L U L A R P O W E R P L A N T S 7 5
Mitochondrially mutant
cell (no aerobic respiration)
creates reductive stress
at its surface.
AEA = Antioxidant electron receptor.
AEAr = Reduced AEA.
Extracellular defences are
many and various, but not
100% effective.
Mitochondrially healthy cell
(aerobically active) may
suffer damage to lysosomes
and hence more general stress.
Figure 4. The "reductive hotspot hypothesis" for amplification of the toxicity of rare mitochondrially mutant cells.
nize them as defective and they will gradually drive out their healthy neigh
bors. (This was the maladaptive evolutionary process that I had already, a
year previously, termed "Survival of the Slowest.")
To continue producing the ATP and other metabolites their host cells
need for survival, these mitochondria must maintain the activity of their
TCA cycle—but this, combined with other cellular processes in the absence
of oxidative phosphorylation, will quickly deplete the cell of needed NAD +
carriers unless a way is found to relieve them of their electron burden. This
is accomplished via the "safety valve" for excess electrons located at the cell
membrane—the Plasma Membrane Redox System (PMRS).
The snarling buzz of electrons congregating at these cells' outer sur
faces turns them into "reductive hotspots," generating a steady stream of

7 6 E N D I N G A G I N G
superoxide free radicals. These radicals contaminate passing LDL particles,
which then spread to cells far away in the body, driving a systemic rise in ox
idative stress throughout the body with age. With oxidative stress comes
damaged proteins, lipids, and DNA, as well as inflammation, disrupted cellu
lar metabolism, and maladaptive gene expression. This could certainly be a
central driver of biological aging.
The whole theory is unattractively elaborate, as you'll have gathered, and
as I immediately appreciated—but as far as I can tell, it's the only hypothesis
that can accommodate all of the data. I published the twin arms of the theory
in rapid succession in the late 1990s, and a more detailed presentation of the
integrated theory was accepted as my Ph.D. thesis for Cambridge and pub
lished in the Landes Bioscience "Molecular Biology Intelligence Unit" se
ries. 1 6 In the ensuing years the theory has enjoyed widespread appreciation
and citation in the scientific literature. Unfortunately, despite this fact, both
the popular press and many biogerontologists1 7 continue to cite the long-
disproven vicious cycle theories either to support or to refute the role of mi
tochondria in aging, instead of seriously grappling with this detailed
mechanistic account.
So now you know, in minute detail, my interpretation of why mito
chondrial mutations are probably a major contributor to mammalian aging,
and therefore why they are included as a category of "damage" in the defi
nition of SENS. The question, then, is what to do about the toxic effects of
mitochondrially mutant cells. The solution to this problem is the subject of
the next chapter.

G e t t i n g Off t h e G r i d
There are good reasons to believe that most present attempts to
modify metabolism to produce less damage to mitochondria
(and thereby to the body at large) are a poor use of resources.
Fortunately, a better path forward exists, promising far greater
results for the same application of time and money. It seems
possible and plausible to prevent damage to mitochondria from
harming us as we age—and scientists are already working on
many options for the first steps of this process.
In the previous chapter, I explained in excruciating detail my
views about the complex mechanisms whereby mitochondrial DNA dele
tions may act as a major engine of aging. I must now tell you that in a very
real sense, it simply does not matter if that hypothesis is correct or not.
This point applies equally strongly to the other SENS interventions,
and it is so central to the engineer's approach to anti-aging medicine—and
so enormously counterintuitive—that I must beg your indulgence if you
find me to be repeating it too often. We must all keep it in the forefront of
our minds when thinking about these problems. If our purpose were
simply to understand aging, then teasing apart the specific pathways that
lead to the accumulation of age damage would indeed be absolutely imper
ative. But that is not our purpose. Our purpose is to put an end to aging's
consequences: the daily descent into decrepitude, and subsequent deaths,
of tens of thousands of people.
6

7 8 E N D I N G A G I N G
Aging is a deadly pandemic disease, and I believe that our understand
ing of its mechanisms, while still highly imperfect, is now good enough that
we are in a position to intervene in it. We need only to identify the nature
of the damage itself—the accumulating lesions that are the source of age-
related loss of functionality in the organism—and then either to reverse
that damage, or to eliminate its threat to our health and life expectancy.
This goal should become the central focus of biogerontological work, and
the major target of biomedical funding generally.
The problem of mutant mitochondria is a case in point. Mitochondrial
DNA mutations are a form of molecular disorder that distinguishes the bi
ologically young from the biologically old, and there is powerful evidence
that they are deleterious. 1 So, whether mutant mitochondria take over their
host cells via "Survival of the Slowest" or through some other mechanism,
and whether these cells exert their toxic effects on the rest of the body by
way of the export of electrons through the PMRS or via a completely unre
lated process, the nature of the task at hand is ultimately the same. Our
therapeutic goal is clear: either to fix the mutations themselves, or to make
them functionally irrelevant. How to achieve that goal is the subject of this
chapter.
Before laying out my proposals to accomplish this goal, however, I
must first spell out why the appealing-sounding solutions that many
biogerontologists would propose are probably wastes of time and scarce
resources.
You Can' t Stop a Moving Train (Safely!)
I termed the "over-preventative" approach to combating aging the "geron
tology" approach because biogerontologists predominently favor it. By and
large, when my colleagues think seriously about actually doing something
about aging rather than just refining their understanding of it, their first in
stinct is to find some way to make metabolism run more cleanly. After all, ag
ing is the result of the accumulation of the deleterious by-products of our
metabolic processes; surely, the thinking goes, if we could just tweak or
dampen down those processes a little bit, we could reduce the exposure of
the organism to metabolism's reactive by-products, reduce the rate at which
our cells and tissues accumulate microscopic damage over time, and thus
slow down the gradual decay of our bodies into age-related frailty and accel
erating vulnerability to death.

G E T T I N G O F F T H E G R I D 7 9
This approach has a strong intuitive appeal, re inforced by the continu
ous stream of encouragement from supplement vendors and public health
authorities alike. We are constantly urged to clean up our lifestyles and
practice preventive medicine: surely, we think, it makes more sense to put
one's energies into attempting to interfere with the causes of aging and age-
related diseases than to try to undo an established molecular mess. But, as
we saw in Chapter 3, the causes of aging lie in the fundamental chemistry of
life, and our capacity to interfere beneficially with that chemistry is limited
by what the organism's underlying biology will accommodate.
I gave examples of this general principle in Chapter 3, but let's now
look at the more concrete case of intervention into the problem of mito
chondrial mutations. The obvious, old-school approach would be to try to
reduce the formation of mutant mitochondrial DNA by cutting back on the
bombardment of the mitochondrial DNA by free radicals. Just such a trick
has been pulled off with some success in mice 2 by giving them a copy of the
gene for the antioxidant enzyme catalase, specially targeted to their mito
chondria. Catalase breaks down the free radical-like molecule hydrogen
peroxide, turning it into harmless water before it can become more vicious
and do serious molecular damage. Animals that received the targeted cata
lase gene enjoyed a fifty-fold jump in the activity of the enzyme within their
mitochondria, preventing a great deal of mitochondrial DNA damage—
including some of the mutations that initiate the whole destructive process
of decay that I described in the last chapter.
Compared to the repeated, abject failures of dietary antioxidants to ex
tend lifespan, or the ambiguous 3 , 4 , 5 or negative 6 results of previous attempts
to hold back the free radical onslaught in mice using genetic manipulations,
these results are quite impressive. Mice given mitochondrially-targeted cata
lase gained a 20 percent extension not only of average, but of maximum
lifespan—the gold standard for data relevant to aging. And while no detailed
analysis of the level of age-related pathology in these animals has been pub
lished, we do know that they suffer less age-related heart degeneration and
fewer cataracts.
This being so, why should we not vigorously pursue the development
of a targeted boost in mitochondrial catalase for h u m a n s ? Well, in part this
goes back to the testability (and, thereafter, the possibility of full clinical
development) of this intervention. It suffers the same sorts of weaknesses
on these fronts that characterize all "preventive" anti-aging medicines, as
outlined in Chapter 3. Firstly, because there is no short-term disease against
which extra catalase could be tested as a cure, regulatory bodies won't let

80 E N D I N G A G I N G
clinical trials be performed using it, and will never approve it for human
use; and secondly, the timescales required to prove its effectiveness against
aging make it too expensive and risky for venture capital to touch it. That
probably means that no amount of agitation by scientists or the public will
actually put mitochondrially targeted catalase into the hands of clinicians to
save people's youth, health, and lives: the interests of those with the power
to fund or halt development are aligned against moving forward.
But even if these structural hurdles did not exist, I would still conclude
that there are more fruitful soils into which to plow our limited resources
for dealing with mitochondrial mutations. Boosting these mice's catalase
supply reduces the incidence of mitochondrial mutations—but it doesn't
eliminate or obviate them. Thus, it slows, but cannot treat, the progressive
accumulation of this form of aging damage. I think we can do a lot better
than this. My evaluation of the evidence indicates that it is not worth di
verting our intellectual and financial capital into an intervention that might
yield a 20 percent increase in lifespan (making the average person in a de
veloped country live into his or her late nineties), because the same bricks,
boards, and brains could be put to work in the development of an interven
tion that would not prevent this damage from happening, but would instead
render it harmless. My analysis suggests that this intervention might, in turn,
not only slash the rate of aging damage primarily caused by other mecha
nisms in half, but—when combined with a panel of similar therapies—
would ultimately give us an indefinite youthful lifespan. More on that in
Chapter 14.
In fact, if we were to eliminate, one by one, the other forms of molecu
lar damage that cause us to wither and to die as we age, but were to "only"
slow down the incidence of mitochondrial mutations by the degree seen
in the mitochondrially targeted catalase mice, we might find ourselves
stranded one step away from a future that stretches out further than our
eyes can see. When a d v a n c e d glycation endproduct (AGE)-breaking drugs
reverse the gumming up of our structural proteins; when careful exploita
tion of the immune system clears out the extracellular junk that impedes
cellular function; and when dormant cells that cause the decay of our im
mune system itself have been removed; at that time, with all the other key
SENS platform interventions in our hands, we would still have mitochon
dria that are playing a game of Russian roulette with their DNA. However
much more slowly they may be spinning the chamber, they could still end
up being the weak link in a chain that could otherwise give us indefinite
youthful lifespan.

G E T T I N G O F F T H E G R I D 8 1
We could apply these same objections to any approach to anti-aging
biomedicine based on prevention of aging damage instead of genuine re
mediation. But there is a more specific objection to the "catalase solution."
Relying on an extra dose of catalase to deal with mitochondrial mutations
could actually become harmful in important ways in a person whose body
had already been cleared of—or rendered immune to—all of the other
identified aging damage.
Catalase cleans up hydrogen peroxide, which can be damaging when it is
the result of imperfections in oxidative phosphorylation—as most of it is. But
evolution is, over the long term, an extremely clever engineer, and has
learned ways of making the best of a bad job, harnessing hydrogen peroxide
for its own purposes. While randomly spewing the stuff out of the cellular
power plants does us no particular good, our cells also generate some hydro
gen peroxide on purpose, for use as a chemical signal that regulates every
thing from glucose metabolism to cellular growth and proliferation.7 In this
way, damping down the level of hydrogen peroxide in a cell—even using a
technique designed to focus on the mitochondria—might well be expected
to interfere with the functioning of our complex intracellular machinery.
One of the most dramatic examples of this potential problem is the
need for hydrogen peroxide to reach the mitochondria themselves as part
of the signaling cascade that triggers apoptosis, or "programmed cell
death." Apoptosis is important during embryonic development as part of a
"remodelling" process that rids the nascent organism of excess cells that
are only required during specific phases of its growth. But its main role in
the body is similar to the self-destruct mechanisms built into James Bond's
Aston Martin or the Starship Enterprise: to give your cells a way to destroy
themselves if they have been hijacked by "enemy forces" (viruses or cancer,
for example) before they can threaten the integrity of the organism as a
whole. When the cells of our immune system detect a hijacked cell, they
bind to its surface, flipping a switch that signals its mitochondria to "blow
their tops" and destroy it. Hydrogen peroxide is a player in that signaling
system, and studies have shown that antioxidants—including catalase—
can block the proper activation of this apoptotic program.
Thus, the massive boosting of mitochondrial catalase activity that is re
quired to give these animals their partial protection from mitochondrial
DNA damage has a dark side. And while it's clear that the net effect of this
boost is good for them—as ev idenced by their extended lives and r e d u c e d
age-related pathology—the overall balance of risks and benefits would in
all probability be totally thrown off in an organism in which all other types

8 2 E N D I N G A G I N G
of aging damage had been eliminated. In this otherwise rejuvenated body, a
chronic dysregulation of cell signaling pathways would be a high price to
pay for lower oxidative stress.
Finally, there is reason to think that the catalase boost given to these
mice—which was performed in the mice while they were still early embryos,
rather than in adult organisms—might not even work if done in adult or
ganisms. Catalase genes only expressed the enzyme in some cell types, not in
others, and the scientists who performed the study suggested that this might
have been the result of a form of evolutionary selection during their devel
opment in the womb. The idea is that the extra catalase might be beneficial
in some kinds of cells but harmful in others, and therefore those cells that
were expressing a lot of the enzyme would tend to die off if they were of a
type in which the extra catalase was deleterious. Meanwhile, other cells of
the same type that did not express the new gene would survive, replicate,
and come to dominate. But performing the same trick in mature organisms
would short-circuit this internal evolutionary process, supplying the new
gene to all cell types indiscriminately, so that the benefits of the catalase gene
therapy in some cell types might be outweighed by negative effects else
where. This would mean that people unfortunate enough to have already
been born would not be able to reap any benefit from such therapy—and no
proposal to insert the gene into healthy developing infants, let alone em
bryos, is going to get past a medical ethics board.
None of this should dishearten us; it should simply remind us of the
need to focus our efforts elsewhere. As I outlined in Chapter 4, I advocate a
fundamentally different approach to dealing with age-related molecular
damage. Instead of trying to "mess with metabolism" in ways that might
prevent aging damage such as mitochondrial DNA deletions, it is my con
tention that we need to focus on developing anti-aging biomedicine that can
repair or render harmless any mutations that may occur in mitochondrial
DNA. While most people—whether laypersons or professional scientists—
tend to assume that this must be far more difficult to achieve than a preven
tive strategy, there are in fact several promising techniques sitting on the
drawing board that require no biotechnology more advanced than that
which would already be required to put catalase into the mitochondria—
namely, gene therapy. This suggests that we could actually have either type
of intervention in the clinic at about the same time. In fact, it really tells us
that the remedial technologies should be able to reach people suffering ag
ing damage sooner than preventive ones, for the regulatory and pragmatic
reasons I outlined earlier.

G E T T I N G O F F T H E G R I D 8 3
The Best Bet: "Head for the Bomb Shelter!"
As I discussed in the last chapter, the main reason for mitochondrial DNA's
unusual vulnerability to oxidative damage is its location: being next door to
a leaking nuclear reactor (the mitochondrial electron transport chain) is a
sure way to increase your risk of mutations, whether you're a growing child
or a tiny biological machine. Metabolism clean-up approaches try to make
this reactor run more cleanly (so that it produces less damaging waste), or
to install "pollution control" equipment that would catch more of its by
products before they do any harm (the cellular equivalent of the "smoke
stack scrubbers" on coal-fired power plants). My preferred approach is
completely different: that we should put these mutations "beyond use" of
harm. This could be accomplished by putting backup copies of the genes
that are currently housed in the mitochondria in the safe haven of the cell's
nucleus, far from the constant bombardment of free radicals from the mi
tochondrion itself. This solution is called "allotopic expression" of the pro
teins these genes encode—i.e., expression from a different (Greek allo-)
place (topos).
Let's be clear about this. Allotopic expression would do absolutely noth
ing to prevent the native mitochondrial genes from suffering mutations: free
radicals would hit the vulnerable mitochondria just as often, and mutations
would occur at exactly the same rate, as they did before. But with a nuclear
backup copy of these genes, any such mutations would be rendered func
tionally irrelevant, because the cell would be able to keep producing the pro
teins that the knocked-out genes in the mitochondrion had previously
encoded. These mitochondria would thus enjoy functional proton-pumping,
electron-transporting proteins, and would therefore behave exactly like mito
chondria with intact DNA, just as if they had not suffered mutations in their
"local" DNA. Electrons would continue to flow into the electron transport
chain from NADH; protons would continue to be pumped; free radicals
would continue to leak out of the system at random. The concept is illus
trated in Figure 1.
Not only that: because such mitochondria would continue to damage
their mitochondrial membranes, the cellular "incinerator system" (the lyso
some) would be able to tell when they got old and haul them off for de
struction. Therefore, the Survival of the Slowest mechanism that leads
mutant mitochondria to take over their host cells would never take place,
nor would cells be f o r c e d to shift into the abnormal metabolic state that
cells with mutant mitochondrial DNA require in order to deal with an

8 4 E N D I N G A G I N G
Figure 1. The concept of allotopic expression to obviate mitochondrial mutations.
imbalance in their NADH-to-NAD+ ratio. Thus, despite the fact that these
cells contain mutations in their mitochondrial DNA, they would not be un
loading their excess electrons into LDL, would not spread oxidative stress
to the rest of the body, and would make no more contribution to the aging
of the organism as a whole than cells with perfectly intact mitochondrial
DNA.
"But," I hear you ask, "what if one of these backup genes is itself mu
tated? Won't we be facing the same catastrophe?" Fortunately, no! There
is, in fact, no real risk of a functionally meaningful failure of this backup
system occurring, even over the course of a lifespan that has been dramati
cally extended by a full panel of SENS anti-aging interventions.
To understand why this is so, let's look at what would be required for
such a failure to occur. First, in order for a cell with an allotopic copy of a
mitochondrial gene to slide into Survival of the Slowest, it would have to
have suffered mutations in both copies of the gene: the mitochondrial orig
inal and the duplicate copy that we would have p l a c e d in the nucleus.
This is less likely to happen than it may initially sound. It is already un
usual for DNA located in the mitochondria to suffer permanent damage
(remember that as things stand, less than 1 percent of cells are overtaken by
mutant mitochondria), and the odds of a backup copy located in the nu
cleus being mutated are much lower. Aside from being better shielded from
free radicals because of its location (DNA housed in the nucleus is many
times less susceptible to mutations than its mitochondrial counterpart),

G E T T I N G O F F T H E G R I D 8 5
there are many more proteins encoded by genes located in the nucleus: tens
of thousands, versus only thirteen encoded by genes that are in the mito
chondria themselves. So even when a free radical does get into the nuclear
DNA, the odds of it damaging one of the allotopically expressed mitochon
drial genes are many times lower than the odds that it will hit some other
gene. Indeed, many such free radicals will not hit a gene (an instruction for
building a protein) at all, but one of the many stretches of nonfunctional
"junk" DNA. Therefore, the chances of both the mitochondrial copy and the
nuclear backup of the same gene being mutated are vanishingly small.
Moreover, while the unusual design of the mitochondrial DNA ensures
that large deletions in its structure wipe out its ability to synthesize any of
its proteins, the same will not occur in the nuclear case: only the protein for
the specific mutated gene will be affected. Of course, your mitochondria
can't function without all thirteen proteins, but we could help reduce the
risk of any actual shutdown of oxidative phosphorylation—and the result
ing clonal expansion of a mutant mitochondrion—by providing a double
or even triple set of functioning backup copies. 8
The "Mitochondriopathies"
One category of hurdle facing the clinical development of anti-aging bio-
medicines is structural: aging is not a recognized disease, so the FDA will
not allow trials to be performed on interventions claimed to cure it. This is
obviously a bucket of water thrown directly onto the heads of venture cap
italists who might otherwise be interested in investing in startups working
on a treatment for age-related mitochondrial DNA mutations. From the
perspective of getting effective anti-aging interventions into the clinic as
quickly as possible, allotopic expression has the advantage that it is already
being pursued as a treatment for a recognized group of diseases: the mito
chondriopathies.
These diseases are caused by defects in the mitochondrial DNA that
are inherited (or, more rarely, acquired through causes independent of the
aging process). These mutations lead to a failure of energy production that
causes a spectrum of dysfunctions in various organs, depending on the
exact disorder: the brain, heart, and muscles tend to be the most vulnera
ble, but damage can also extend to the liver, kidneys, lungs, and certain
glands. Because allotopic expression is a promising therapy for mitochon
driopathies, government funding (albeit not nearly enough) is already

8 6 E N D I N G A G I N G
available for work on its development; and once it is ready to move into
the clinic, there will be an incentive for venture capital to invest in its de
velopment, giving a clear route forward for near-term testing in FDA-
approved clinical trials.
In turn, once allotopic expression has been proven to be safe and ef
fective as a treatment for inherited mutations of the mitochondrial DNA,
we will be in an excellent position to make the small tweaks needed to
adapt it for use as a treatment for mutations acquired during the aging pro
cess. This parallel applicability is a feature of most of the anti-aging inter
ventions included in the SENS platform—and indeed, prototypical
versions of several of the proposed interventions are already in clinical tri
als today.
To Boldly Go Where Evolution Has Gone Before
The other hurdles facing allotopic expression are the more purely scientific
ones. Fortunately, as we will see, progress on these problems has been rapid
in the last decade. But it's better than that: evolution has been working on
a similar job for untold millennia.
In ages long past, the ancestors of the mitochondria occupying our own
cells were not just components of cells as they are today, but organisms in
their own right, which formed an I'll-scratch-your-back-you-scratch-mine
relationship with our one-celled ancestors. Because they were independent
organisms, these proto-mitochondria naturally had a full complement of
their own DNA—at least one thousand genes.
But precisely because the hazardous environment of the mitochondrion
put the genes housed there at extremely high risk of mutation, evolution has
been performing allotopic expression on mitochondrial genes since long be
fore humans appeared on the scene. Over the glacially slow timescales of
evolution, organisms have copied mitochondrial genes that code for mito
chondrial proteins into their cell nuclei, after which the original mitochon
drial genes became redundant components and mutated into oblivion.
And to give Mother Nature her due, evolution has gone a long way in
this direction. Out of more than one thousand original mitochondrial pro
teins, all but thirteen have had their genetic instructions moved into the
nucleus.
Starting in the mid-1980s, scientists started showing that they, too,
could perform allotopic expression of some mitochondrially coded pro-

G E T T I N G O F F T H E G R I D 8 7
teins using biotechnology, albeit initially only in yeast—a crucial series of
proofs-of-concept.
Obstacles, Evolutionary and Otherwise
But things get a lot trickier when we start trying to do the same thing with
the thirteen protein-encoding genes that are still located within the mito
chondria in human cells. The reason why evolution hasn't finished the job
for us already is a matter of some debate, but everyone agrees that there
must be some kind of "forces" holding the process back. Whatever those
forces are, this job is probably not going to be easy. What we have to do is
figure out what forces are keeping those genes in the mitochondrion, and
then devise ways of overcoming them. I have a d v a n c e d the case that there
are only two such forces that need concern us. 9
One, which doesn't apply to all organisms but does apply to us, is that
the DNA "languages" of mitochondria and of the cell nucleus have evolved
slightly different "dialects," so that an exact copy of a given mitochondrial
DNA sequence becomes indecipherable when it is dropped into the nu
cleus. This problem is called code disparity.
The case is rather like the changes that have occurred over time in the
writing of the letter "s" in English. Up until the nineteenth century it was
common for an "s" occurring in the middle of a word to be written in an
elongated fashion that looks much more like a modern "f" than an "s ."
Gradually, as writing became more widespread and irregularities in the
written language more standardized, the elongated " s" came to be r e p l a c e d
by the shorter, more curved version of the letter that we use today. Thus a
modern reader of an Enlightenment-era order to launch a naval attack
("Sail for the enemy") might mistake it for a command to "throw" the bat
tle ("Fail for the enemy"), and in other cases an instruction might be re
d u c e d to pure gibberish.
This disparity in the genetic codes of mitochondria and nucleus makes
moving mitochondrial genes that contain such discrepant lettering into the
nucleus a near impossibility for evolution. Indeed, all the genes still housed
in the mitochondria contain such quirks, and this fact alone can explain
why they haven't made the jump to the nucleus. But code disparity doesn't
pose any serious problem for biotechnology: with our outsider's under
standing of the discrepancies in the two codes, we can simply create the
new, allotopic gene using the nuclear version of the code (substitute " s" for

8 8 E N D I N G A G I N G
"f") and rest assured that it will be translated and turned into a protein just
like any of the other genes for mitochondrial proteins that are already
housed there. 1 0
The second problem appears to have been a somewhat less imposing
barrier to the evolutionary transmigration of mitochondrial genes to the nu
cleus, but is a much greater challenge for the anti-aging biotech engineer. It
is the repulsion by water ("hydrophobicity") of many of the mitochondrial
proteins whose genes are still located within the mitochondria themselves.
These proteins have sites in their chemical structures that have such a
"fear" (phobia) of water (hydro) that, like a phobic human, they will liter
ally curl themselves up into a ball in response to it.
Hydrophobicity is no problem for proteins when they are built from
DNA that is already located within the mitochondrion, because the final
three-dimensional shape of the protein is supposed to be twisted up, and
there are special enzymes that guide such proteins into the proper confor
mation. But it becomes a showstopper when the same protein is con
structed from DNA located in the nucleus. The genes for such proteins are
translated into their products in the main body of the cell, and the proteins
must then be moved from the fluid environment outside of the mitochon
drion, through the outer and inner mitochondrial membranes, and into
their final location in the mitochondrion's core.
Of course, the membranes won't just let proteins pass freely through
them, or the integrity of the mitochondrion—and its ability to preserve its
proton reservoir—would be compromised by the constant leak of material
into and out of its chambers. But mitochondria do need to be able to import
hundreds of proteins: for example, the dozens of the subunits of the elec
tron transport chain whose genes have already been moved successfully into
the bomb shelter of the nucleus by evolution. Mitochondria have therefore
evolved elaborate machinery that specifically moves (translocates) proteins
through these membranes. These are sensibly called the "Translocase of the
Inner Mitochondrial" membrane (TIM) and the "Translocase of the Outer
Mitochondrial" membrane (TOM), giving the whole system the glorious
name TIM/TOM complex.
The problem is that once a protein has been gnarled into a balled-up
Twister configuration by its repulsion to the water in the fluid compartment
of the cell, the cell becomes unable to jam it through the pores of the
TIM/TOM machinery—rather like trying to force a wildly bent-out-of-
shape coat hanger down a drainpipe. This would be not just a failure, but
actually counterproductive: not only would it prevent the newly allotopically

G E T T I N G O F F T H E G R I D 8 9
expressed proteins from getting where they have to go, it would also "clog
up" the TIM/TOM complex, disrupting the import of the many proteins
that were previously being naturally, successfully imported.
In fact, all of the thirteen proteins that are still coded directly in our
mitochondria are very hydrophobic. The instructions for several of those
proteins have never been moved into the nucleus in any species, and those
proteins are the most hydrophobic of all. This suggests that hydrophobicity
is indeed the biggest hurdle to the allotopic expression project. There are
several cases that seem to violate this rule in other species, but I have pub
lished detailed analyses 9 that explain why all of these apparent counterex
amples are misleading—why they would have happened in the course of
evolution despite the fact that hydrophobicity really is the most important
barrier to making the move.
So, if we are to obviate mitochondrial mutations via allotopic expres
sion (my preferred solution), then in addition to the relatively simple task
of editing the code in cases where the DNA "language" of mitochondria
and nucleus are mismatched, we will have to find ways of tweaking the pro
teins that, in their present form, can't be imported into the mitochondria.
When I first began contemplating this problem, I came up with a
workable-sounding but technically challenging way of dealing with it,
which I published in the journal Trends in Biotechnology in 2000. 1 1 I'll de
scribe this approach later on. The reason I'm putting off discussion of this
solution is that recent experiments suggest that we may not need to go to
the lengths that I then proposed in order to overcome the hydrophobicity
problem. There are at least two alternative ways to engineer these proteins
to make them importable—ways that appear to be much easier.
Pirating Mother Nature's Intellectual Property
The first solution to the hydrophobicity problem is to look around for cases
in which evolution has already done the yeoman's work for us—in other
species. We humans (and our evolutionary ancestors) have not yet enjoyed
the simultaneous good fortune of the right random mutations, the right en
vironment and the right selective pressures to pass nuclear versions of any
of these genes along to us as their descendents. But that doesn't mean that
the same happy confluence of circumstances has never occurred in other
species' evolutionary history. Natural selection has been working on the hy
drophobicity problem in many species independently of our own, and has

9 0 E N D I N G A G I N G
in several cases come up with workable solutions—solutions that we did
not inherit, simply because they occurred in a separate evolutionary line
age. By looking beyond our own mitochondrial DNA into the evolutionary
inheritance of other species, we might find viable solutions that we only
need to tweak slightly for use in our own cells.
Other species' mitochondrially encoded proteins are not identical to
our own, of course, but their structure is close enough to make it reason
able to believe that they could stand in for the originals if inserted into our
mitochondria, or at least show us how to modify the sequences of the hu
man counterparts to render them importable. If species could be found
whose genes for their versions of some of the thirteen proteins had sponta
neously moved into the nucleus, we would expect to be able to put those
same genes into our own cells' nuclei with only minimal modification.
Those proteins would be constructed in our cell bodies, imported into our
mitochondria, and take the place of the native version if and when muta
tions shut down the mitochondria's ability to do the job themselves.
This idea is not just a fancy of mine: it's been done in isolated human
cells already. Work on such a project began in 1998, shortly after I started
shouting about the importance of a discovery that had been made eight years
earlier, when the mitochondrial genetic library of the green alga Chlamy-
domonas reinhardtii was sequenced. When the proteins for which these crea
tures' mitochondrial genes code were identified by comparison with the
equivalent genes in other species, it was found that they are missing versions
of six of our thirteen hydrophobic electron transport protein genes.
In fact, of course, these genes are rather like your mysteriously "miss
ing" car keys: you haven't actually lost them, they just aren't where you
thought you'd left them. In these organisms, the changes needed to make
these proteins less hydrophobic have taken place, because the greatest bar
rier to the change—code disparity—was never erected. These algae are so
close to the "root" of the evolutionary "tree" that the disparity between the
DNA coding systems of the nucleus and the mitochondrion never took
place in them. Without that hurdle to leap, evolution has only had to ad
dress the much less challenging hydrophobicity problem—and it has done
so with some success. The genetic instructions for these proteins are now
housed in the algal cells' nuclei. The cell's machinery reads those instruc
tions, manufactures the proteins in the main body of the cell, and then the
mitochondria import them—the same thing that happens with most of our
own mitochondrial proteins.
At this point Dr. Mike King at Thomas Jefferson University, Philadel-

G E T T I N G O F F T H E G R I D 9 1
phia, comes into the picture. King was not originally interested in aging, but
in the inherited mitochondrial diseases (mitochondriopathies). Researchers
had long dreamed of gene therapy for these disorders, but there are im
mense technical challenges to putting genes directly into the mitochondria.
King thought that allotopic expression in the nucleus could provide a faster
route to a cure.
But the hydrophobicity problem loomed over this potential solution to
inherited mutations in the genes for the thirteen mitochondrially coded
proteins, just as it does for the plan for mitochondrial genes mutated dur
ing the aging process via free radical damage. When he heard about the ex
istence of nuclear-coded versions of some of these proteins in the algae in
the late 1990s, Mike saw that they offered a potential blueprint for replicat
ing the algae's tricks in human patients with mitochondrial diseases.
What followed was an astonishing surge of progress. In 1998, King be
gan a fruitful collaboration with Dr. Diego Gonzalez-Halphen of the De
partment of Molecular Genetics at the Autonomous National University of
Mexico, to identify and clone the algae's genes for the six analogous pro
teins. Within three years, these scientists had identified three of them. One
of these (ATP6, a component of the mitochondrion's Complex V "tur
bine") was of particular interest because inherited mutations in it cause two
rare but extremely serious disorders of the brain and muscular system in
humans: NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa) and mater
nally inherited subacute necrotising encephalomyopathy. In these diseases,
ATP synthesis is r e d u c e d by 50 to70 percent, leading to severe dysfunction
of the neuromuscular system. Thus, the identification of an importable ver
sion of the mutated protein held forth therapeutic potential for people suf
fering from these diseases, as well as for all of us as we age.
Picking up the ball, Eric Schon and his coworkers from the Depart
ment of Neurology at Columbia took the next step, inserting a cloned copy
of the algae's ATP6 gene into the nucleus of human cells whose mitochon
drial DNA harbored the same mutations that cause these neuromuscular
diseases in humans. The cells decoded the genetic instructions, turned out
the protein in the main chamber of the cell, imported it into the mitochon
dria, trimmed off its targeting sequence (a special string of amino acids that,
when appended to the "nose" of a protein, directs it into the mitochon
dria), inserted it into the electron transport chain, and apparently took the
place of the mutated protein, rescuing the cells from the destructive effects
of the mutation. 1 2
In other words, these researchers did exactly what I had been calling

9 2 E N D I N G A G I N G
for someone to do. They found, in an alien species, a nuclear gene for a mi
tochondrial protein whose human counterpart is located in the mitochon
drion itself; they inserted it into the nuclear DNA of human cells; and they
showed that those cells could use it in just the way that the algae do, restor
ing near-normal functionality to cells with otherwise disabling mutations.
There is still a lot of work ahead, of course. To turn this into a viable
therapy for people with inherited or age-related mitochondrial mutations,
we will have to do two things. One is to identify in other organisms, or en
gineer ourselves, genes that can be put in the nucleus for the rest of the mi
tochondrially encoded proteins, and show that they restore function to
mutant cells. And the other will be to do the trick in whole organisms bur
dened with these mutations: first in mice (a relatively simple task: genetic
manipulation of mice is now relatively routine), and then in grown humans
(a technology that we have not yet mastered, but upon which work is pro
ceeding with an intensity that may well yield safe, viable therapies in the
foreseeable future).
Borrowed Ideas, Novel Solutions
But of course, there is no guarantee that we will find all of the necessary
genes in other species. It would be smashing luck if we did, of course, but
it's entirely possible that many of the genes for electron transport chain
proteins have never been transferred from the mitochondria to the nucleus
in any species, or that the protein will have been so changed in the intricate
branching of life's evolutionary family tree that it will not work in human
cells. In that case, we'll just have to figure out for ourselves how to tweak
our existing genes to make the proteins they encode importable.
Even here, however, we'll be able to borrow tricks that we've learned
from other species. A remarkable example was reported in 2002, 1 3 when a
group at the University of Western Australia discovered that several legume
species—including the soybean and the common mung bean (Vigna
radiata)—are actually in an intermediate evolutionary state, having evolved
a nuclear copy of the mitochondrial gene for subunit two of the electron
transport chain component cytochrome c oxidase, but without having yet
discarded their original, mitochondrial copy.
The fact that these organisms survive while expressing the protein from
both sites at once is itself good news, as it relieves (to some extent, anyway) a
concern that some people have expressed about using allotopic expression as

G E T T I N G O F F T H E G R I D 9 3
a solution to mutations. The worry is that in cells with healthy mitochondrial
DNA and allotopic versions of the electron transport chain proteins, the ex
istence of two independent, working copies of the same gene might lead to
too many copies of the duplicate-coded proteins being produced, and that
this might somehow imbalance or overload the capacity of the mitochondria
to fit the various components together into working electron transport com
plexes. This would be like having two departments in the management of a
factory, each using an independent system to order components for their
product—a serious glitch in any "just-in-time" inventory system. The obser
vation that no such problem occurs in these organisms suggests that we may
not have much to worry about here—and that's good news.
But when other scientists compared the two versions of the legume elec
tron transport gene, they discovered something that makes me even more
bullish about our ability to move the full complement of mitochondrial elec
tron transport chain genes into the nucleus. The two versions of the protein
differ in twenty-five amino acids (the building blocks of protein) out of
hundreds—but only two of these differences are necessary to allow the nu
clear version to be imported into the mitochondria! This suggests that we
may only need to do some relatively minor fiddling with our thirteen proteins
of interest in order to make feasible their import into the mitochondria.
Again, I'm no longer just extrapolating from what evolution has
achieved in other organisms: progress in adapting these solutions to new
problems is definitely under way. Around the same time Schon's group ex
pressed ATP6 in human cells allotopically using the algal version of the
gene, they and another group also reported having developed different solu
tions to the challenge of engineering new, nuclear-coded versions of that
protein. The difference was that these new proteins were not taken
wholesale from another species, but modified from the mammalian original.
As with the success using the algal gene, these human-generated versions
were reported to rescue cells bearing inactive versions of ATP6 in their mi
tochondria. 1 4 , 1 5
Not long after this, a third group engineered a nuclear-coded version of
another mitochondrial gene named ND4, mutations in which cause one of
the mitochondriopathies, Leber's Hereditary Optic Neuropathy (LHON). 1 6
To do this, they first had to find solutions to the problems that had previ
ously been dealt with in the allotopic expression of ATP6. First, the DNA
code for the protein had to be altered to make the "spelling" compatible
with the nucleus, since ND4 suffers from the "code disparity" problem I
discussed earlier. Then, the researchers had to tag on a "targeting sequence"

9 4 E N D I N G A G I N G
copied from a completely different gene (aldehyde dehydrogenase) to guide
it into the mitochondria. Next, they had to figure out a way to get the gene
into the nucleus in the first place; this was accomplished using a trick bor
rowed from viruses that sneak their DNA into their infectees' nuclei. And fi
nally, they attached an additional sequence to the gene to allow it to be
picked up by the gene-decoding machinery of the nucleus, so that it would
be "read" and turned into a protein.
Given the need for so many alterations to the original gene, borrowed
from so many different inspirations, you might reasonably be concerned that
something would fail somewhere along the way. But no. The heavily modified
protein, custom-built out of the original by reasoned analysis of what would
be required to make it importable (rather than being copied in whole cloth
from one of our distant relatives), was successfully incorporated into human
cells bearing mutant ND4, which promptly began churning out functional,
mitochondrially targeted ND4. The allotopically expressed protein then
found its way into the mitochondria, took its place in the electron transport
chain . . . and promptly tripled the cells' ATP output, bringing it back to lev
els similar to those seen in normal, nonmutant cells. Not only that: when the
cells were subjected to conditions under which they were forced to lean heav
ily on OXPHOS for energy, these cells with the new allotopically expressed
mitochondrial electron chain subunits enjoyed three-fold better odds of sur
vival than mutant cells lacking the engineered gene.
The researchers boldly concluded that "Restoration of respiration by
allotopic expression opens the door for gene therapy of Leber Hereditary
Optic Neuropathy." I would add that it also puts a large foot into the same
door for the solution of the reductive hotspot problem. Doubts have re
cently arisen concerning the methods used to demonstrate rescue of the
treated cells in two of the above studies, but at least two others are still con
sidered clear-cut.
Inteins: Splitting the Difference
I am hopeful that these two approaches may be sufficient to allow us to
deal with the hydrophobicity issue; however, it is possible that we may have
to go further with some of the very hydrophobic proteins. One potential
solution—which I originally put forward before the above successes sug
gested that it might not be necessary—is to further modify the proteins in
question using inteins.

G E T T I N G O F F T H E G R I D 9 5
Inteins are sequences that are inserted temporarily into some proteins
when they are first synthesized, possibly to help the protein mature into its fi
nal form properly, and are then snipped out once they've served their pur
pose. In some cases, the two halves of a final, functional protein are
expressed separately, each with a complementary "semi-intein" at the end
where the two protein segments must finally be joined. When the two halves
of the final protein come together, the two semi-inteins are first bound to
gether, a little like the male-to-female electrical connectors on strings of
Christmas-tree lights. But then the united intein sequence is snipped out and
the two precursors of the final protein are permanently, directly fused to
gether into the final, completed structure.
We might be able to use inteins of this sort to help us deal with a hy
drophobic protein. One approach is to split the protein up close to a site in
its structure where it would otherwise curl up when exposed to water, and
put semi-intein "caps" on either side of the break. As an analogy, imagine
that you wanted to move a long piece of steel with a right angle in its center
(the protein) down a straight drainpipe (the TIM/TOM complex). As it
stands, the job is impossible. But if you could cut the piece of steel at or
near the place where it bends, and then attach a short segment to each of
the severed ends of the bar to show your coworker at the other end of the
pipe where to weld the two halves together again, you could easily drop the
two halves down the tube individually for reassembly at the other end.
Alternatively, whole inteins can actually be built right into the middle
of the two halves of the protein, creating one long structure with the intein
in its center. I'm afraid the way that this can be exploited is a bit harder to
analogize, but I'll try. Imagine if, instead of cutting your bent bar in half
where the bend occurs and sending each half down the drainpipe sepa
rately, you were to cut the bar, rotate it through a half-twist, and put in a
central joining segment, so that the final structure looks more like a stylized
lightning bolt than a right angle. You could then drop the "straightened"
bar (protein complete with intein) down the drainpipe (TIM/TOM com
plex) and then excise the joining segment (intein), allowing you to reassem
ble the bar into its proper configuration after intein removal.
This procedure is a good deal more complicated than switching a cou
ple of amino acids, so I'd rather not have to resort to it if one of the former
options will work. When I first came up with the idea of using inteins as a
solution to the hydrophobicity problem, I foresaw a whole series of poten
tially crippling technical hurdles that might have to be overcome to get it to
work, and I'm still not sure how easy that would be. Inteins would have to

9 6 E N D I N G A G I N G
be p l a c e d in just the right place, and be of an appropriate length, and this
would require a lot of fiddling. Also, natural inteins are designed to be
snipped out as soon as the protein that contains them has been con
structed, so mitochondrial inteins would need to be designed in ways that
prevent them from being removed until the complete protein has been im
ported into the mitochondria.
Another problem we might face is that of ensuring that, in the "semi-
intein" version of this approach, the protein segments are joined to their
proper partners. If we have used inteins to break up several proteins that
are imported simultaneously, or if one protein has been broken down into
more than two pieces (as seems likely to be necessary in at least a couple of
cases), the "exposed" ends of protein segments might be mismatched once
the inteins are removed. Fortunately, it appears that some such multiple in
teins do exist naturally and somehow "know" their proper partners, so this
may not really be a problem—and if it is, the alternative solution of putting
inteins into the hydrophobic stretches of these proteins is still available.
Yet another potential headache: segments of proteins could begin to fold
too early, either before joining to their "other halves" in a way that obstructs
access to the fusion site (so that the intended fusion with the mate is no
longer possible), or even right in the midst of the TIM/TOM machinery,
causing the exact problem that the use of inteins is supposed to prevent. And
finally, a protein's separate, newly inteinless segments might be altered by en
zymes while they wait to be joined to their "mates"; any such alterations
could render them nonfunctional, or prevent them from "hooking up."
The good news is that, despite all of these potential obstacles, at least
one split intein job has already been pulled off in cell culture: 1 7 Yoshio
Umezawa and coworkers took an unimportable green fluorescent protein
and in t roduced it into a cell's mitochondria by first adding on a borrowed
mitochondrial targeting sequence, and then adding inteins inserted into its
structure using a protein splicing system taken from algae, before finally
"infecting" the cell's DNA with the composite structure's code. The result
was that the protein was indeed expressed and imported into the cell's mi
tochondria, where it allowed the scientists to detect the presence of other
proteins that they were trying to direct into the organelle.

G E T T I N G O F F T H E G R I D 97
Not TINA but TATA
This string of successes indicates that we can expect rapid progress in ex
pressing the remaining mitochondrially coded proteins from nuclear genes
in reasonably short order, through some combination of the different tech
niques I've discussed here. Once we have figured out how to put all thirteen
of these proteins' genes into the nuclear "bomb shelter," we will be very
close to being able to negate the insidious effects of mitochondrial mutations
in aging, turning existing "reductive hotspots" into normal, healthy cells
and preventing the formation of new ones. When that is accomplished, we
will be able not only to stop but to reverse the slow upward creep in oxida
tive stress—and the accelerating spiral of molecular damage and metabolic
disruption—that we think is driven by these mutations today.
But, of course, the precedent of medical history tells us that some pit
falls may still lie in wait for this solution. One can make one's most edu
cated predictions, based on a sober consideration of the published science,
and still fail to anticipate an intractable roadblock. My biggest worry is
that, even after a bit of tweaking, the volume of TIM/TOM traffic required
to import the new allotopically expressed proteins will be so high, and its
rate of flow through the system so slow, that we will not be able to transfer
the full complement of proteins via this route even if we succeed with each
of them individually. I am especially concerned about this since some of the
proteins that have been allotopically expressed move quite poorly through
the TIM/TOM machinery; one of the examples we discussed above in
volves a protein that is only incorporated into the mitochondria at 40 per
cent of the efficiency of the native, mitochondrially-coded version.
Former prime minister Margaret Thatcher, the "Iron Lady," was fa
mous for intoning "There Is No Alternative" (TINA) to the neoliberal eco
nomic agenda. I have no intention of painting myself into an ideological
corner over my preference for allotopic expression: lives are at stake, not
just my ability to admit that I'm wrong. I will instead join the ranks of her
anti-globalization critics, who spilled into Trafalgar Square in their thou
sands chanting "Not TINA but TATA!"—that is, "There Are Thousands of
Alternatives."
Having made that bold declaration, I hasten to add that I think we can
safely trim down "thousands" to "a few, and even fewer that are viable." In
fact, quite a number of alternative fixes for mitochondriopathies have been
advanced, and at first glance these solutions might also be expected to treat
age-related mitochondrial DNA deletion accumulation. Unfortunately, I

9 8 E N D I N G A G I N G
predict that most of those solutions will be of no help in preventing the de
velopment of "reductive hotspots," because the mechanism whereby the
underlying mutations accumulate (mitochondrial free radical generation
and clonal expansion through the Survival of the Slowest) is quite distinct
from the mostly inherited problems in mitochondriopathy victims. Re
search toward cures for mitochondriopathies using these alternatives will
still doubtless yield techniques and insights that will in some way help us to
develop a cure (via allotopic expression or some other therapy), but I do
not believe that most of these will be adaptable as anti-aging biotechnology.
I shot down the major flawed proposals down in the 2000 article in Trends
in Biotechnology that I mentioned earlier. Two approaches exist that I feel
have much more promise, however.
One, a d v a n c e d by Drs. Rafal Smigrodzki and Shaharyar M. Khan of
the Center for the Study of Neurodegenerative Diseases at the University of
Virginia, starts with the ability to introduce into the mitochondria modified
versions, not of individual genes (as in the classical gene therapy approach),
but of complete copies of the entire mitochondrial DNA. They accom
plished this maneuver—which they have termed protofection18—using
boldly simple techniques that most scientists would have dismissed as un
workable. It's too early to say how versatile and repliable their method is,
though—it's too new to have been explored by other scientists.
The other very promising alternative to allotopic expression is to intro
duce genes for alternative versions of the mitochondrially encoded en
zymes, versions that don't quite work the way that the native ones do. The
enzymes in question already exist in—and could thus, in theory, be bor
rowed from—some lower organisms (yeasts and plants). They perform the
exact same electron transport activities of the enzymes that we partly en
code in our mitochondrial DNA, even though they themselves are not par
ticularly hydrophobic and are encoded in their species' nuclear DNA. The
problem—if it is one—is that they only do the electron transport, not the
proton pumping. But introducing them into our cells might be a fair trade
off, because while they would not restore the mitochondrially mutant cell
to its full capacity, they would prevent these cells from impairing the ability
of the rest of the body to get on with the business of life. This has actually
been documented in isolated human cells: when Complex I is chemically
inhibited, normal cells quickly die off, but cells given the relevant yeast
gene remain viable. 1 9
One problem with this proposal is that, if these proteins were to be ex
pressed in cells that had not suffered deletions, they would deny the cell of

G E T T I N G O F F T H E G R I D 9 9
much of its energy source. We would use one gene to bypass the first enzyme
in the electron transport chain (Complex I) and another to bypass the rest.
Neither of them pumps protons, so having both of them substituting for
much of the native, proton-pumping complexes in a mitochondrion would
seriously impair the buildup of a proton "reservoir" and, thus, ATP produc
tion. This could lead to anything from a mild functional impairment to a se
vere energy deficit—and it would extend through every cell in the body, not
just the "evil 1 percent" of cells with a population of mutant mitochondria.
This would leave us in a very bad way indeed, so we would need to find a
way to ensure that the genes for these "bypass enzymes" are expressed only
in cells whose mitochondria are no longer producing the native proteins.
There is no obvious way to do this at the moment, unfortunately. But luckily,
initial research indicates that these enzymes naturally possess a system that
activates them only when their proton-pumping counterparts are failing.
Even expressing one of the two "bypass enzymes" indiscriminately would
make cells less efficient at making ATP, so this is actually no surprise.
The Way Forward
Overall, the picture is a rosy one. We have not only a good idea of how mu
tations in the mitochondrial DNA contribute to the age-related decay of our
bodies, but a clear path forward to obviating the problem—even if our un
derstanding of the exact mechanistic link between mutations and pathology
turns out to be mistaken. Allotopic expression would allow our mitochon
dria to keep functioning normally even when their DNA acquired muta
tions; protofection, alternatively, could simply clear out the old mutant
DNA periodically, replacing it with a fully functional new set of genetic
blueprints; and the use of easier-to-handle enzymes that pump no protons
but keep electron metabolism harmless would at least keep mutant cells
from causing trouble outside of their own membranes.
Again, we will need to develop safe, effective, stable gene therapy that
works in grown humans in order to turn any of these interventions into a
real biomedical intervention against this aspect of the aging process, and
that will certainly be a challenge. But, again again, it's a challenge that sci
entists all over the world are already vigorously pursuing in order to treat
genetic disorders—ailments ranging from Huntington's disease, to inher
ited cancer risk, to familial Alzheimer's disease, to sickle-cell anemia. And
we can piggyback even more closely on research that is specific to the

100 E N D I N G A G I N G
mitochondriopathies—a much smaller field, but one that, for the moment,
still receives more serious funding and attention than work designed to
tackle the slow-motion global plague that is aging.
With the resources already being thrown at advancing gene therapy, we
can confidently predict that the clinical readiness of this enabling biotech
nology is foreseeable. I am therefore convinced that the major hurdle facing
speedy implementation of allotopic expression (or its alternatives) will not
be our ability to develop safe gene therapy for patients, but the dearth of
investment into the basic science of moving mitochondrial genes into the
nucleus.
Remember the positive result using inteins to import proteins into
mitochondria in culture? This achievement came about because scientists
were seeking a way to get rapid feedback on the results of a completely un
related project of interest to them. Imagine what could be accomplished
with resources specifically dedicated to developing allotopic expression for
the purposes of reversing aging damage!
How to bring about that change in research priorities is the subject of
Chapter 15; but now, let me take you on a tour of the next of the "Seven
Deadlies" of aging, and show you what we can do about it.

7
U p g r a d i n g t h e B i o l o g i c a l
I n c i n e r a t o r s
Just like our own households, cells generate garbage as an
inevitable result of their normal functioning. Again like
households, they are able to dispose of most of this waste—
though they recycle a proportion of it that would put
the most eco-friendly household to shame. But cells cannot recycle
quite all the junk they create, and the portion that escapes
destruction accumulates, to the cell's eventual detriment. Several
years ago I devised a new approach to this problem that
exemplifies, perhaps better than any of my other contributions to
this field, the value of the widely cross-disciplinary expertise
that is so rare in biology today.
Mary Shelley c o u l d n ' t ask for a more perfect scene, I thought, as I
sank my trowel into the scruffy graveyard sod.
A quick scan of the horizon at Coldham's Common would initially
make you think it was a nondescript, even rather dull little field in the heart
of England. But knowing its history transforms your view of the spot, open
ing the mind's eye to a bleak, windswept stretch of near wilderness,
dropped as if out of a Gothic horror novella into the midst of a plain
bounded by football grounds and parking lots, bisected by a railway line.
Though it is sometimes used for public events or cattle grazing, it spends

102 E N D I N G A G I N G
much of the year lonely and abandoned, its sole claim to fame arising from
its association with mass death.
In the late seventeenth century, the Great Plague swept its scythe
across England. When its icy fingers crept into Cambridge, the plague
claimed a third to a half of the residents—including sixteen of the forty
professors at the University—and sent the young Isaac Newton fleeing for
his life. In its wake, the survivors hastily plowed most of the plague's vic
tims anonymously under the unhallowed ground. Even before it became a
mass grave, the area bore the taint of association with infection and death:
its most enduring landmark is the remains of Cambridge's twelfth-century
Leper Hospital. As if to complete the cliche, on most days of the year Cold-
ham's Common is documentably several degrees colder than the cobbled
streets that surround it.
The scene, then, was complete: I, the "mad scientist" (complete with long
beard and pale, sunless skin), surrounded by Cambridge's Enlightenment-era
faux-Gothic castles and cathedrals, had hopped my way with an irrational
furtiveness over multiple fences into the last resting place of the mortal re
mains of untold scores of lives, and was now digging into the soil of a mass
grave, in hot pursuit of the secrets of Life and Death.
Victor Frankenstein, eat your heart out.
I must confess that the above account incorporates a small amount of
poetic license: the person who performed the above task was not I but a
graduate student in my University of Cambridge department, and actu
ally she retrieved the soil sample from Midsummer Common, not the
nearby Coldham's Common. But that's by-the-by. To understand what
she was doing there, let's take a detour out of the graveyard and into the
junkyard.
The Waste of Your Life
Whether we toss things into the garbage bin without a thought, or
painstakingly wash and sort our recyclables, we in the developed world
generate an astounding amount of waste material every day. When we de
cide that we don't need something anymore, or that it's too damaged or rot
ten to be worth our efforts to salvage, we simply stuff it into a bag or bin
and put it out for pickup, confident that the unsung heroes of the sanitation
department will take it away and out of our concern. Thanks to an efficient
waste-disposal infrastructure, a truly remarkable volume of waste material

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 103
can pass through our homes, workplaces, and streets—yet these places
remain clean, pleasant-smelling, and sanitary.
It wasn't always this way, of course. For most of the history of civiliza
tion, the streets of our cities were literal cesspools, into which the citizens
hurled their trash and human waste directly out of their windows without
care for what—or even who!—was below them. Most of us truly cannot
imagine what foul, malodorous, and dangerous places cities were until quite
recently. The toll of living in such a toxic environment can be seen in the
disparity of life expectancy for people living in different environments in
seventeenth-century England. An Englishman would typically live to be
thirty to forty years old if he lived in the countryside, but if he lived in
London, he could expect just twenty-one to thirty-four years of life.
Anyone who's lived through the kind of big-city garbage strike that
nearly paralyzed London in 1976 has an idea of just how vital a functioning
waste-collection system is for health and the carrying-on of the business of
daily life. In shockingly short order, trash can literally be piled ten feet into
the air, in precarious piles that fall apart in the wind or as new bags are
added to the crude structures. And the mountains of garbage are not
merely unsightly: Aside from the smell, the garbage attracts vermin, and
with it, disease—particularly when the contents of the trash bags begin to
spill onto the street because of attacks on the bags by animals, the elements,
or the putrefaction and liquefaction of its contents. Sidewalks become in
creasingly impassable, and even street traffic may be impeded. People be
come less willing to leave the house or go into shops. A strike that lasted
just nine days in 1968 came close to bringing New York City to its knees.
Well, something similar happens to your cells as they age—except that
in a sense it's worse. Rather than a temporary "interruption of service," ag
ing cells undergo a progressive degeneration of their waste management in
frastructure that would make the worst examples of inner-city decay look
like models of sanitation.
Two chapters ago, in discussing the process whereby mutant mitochon
dria "clonally expand" to replace all of their genetically healthy cousins in
the cell, I in t roduced you to the lysosome—an organelle that I called the
cellular "incinerator." Actually, "recycling center" would be a more precise
metaphor than incinerator, because a lysosome's job is not to out-and-out
destroy cellular wastes, but to break them down at the molecular level into
more basic components that can be used as raw materials for the biosyn
thesis of new cellular membranes, enzymes, and other important compo
nents of the cellular machinery. The incinerator metaphor is meant to

104 E N D I N G A G I N G
convey the extraordinary power of the lysosome's molecular-level dismem
berment of the materials that are thrown into it, as well as the chemical na
ture (burning is a chemical reaction, remember) of the lysosome's methods
of breaking down waste into its fundamental components.
While the cell actually has a variety of mechanisms for reprocessing
damaged cellular constituents, its lysosomes deal with some of the nastiest
of them, including the waste materials that are still left over after the other
cell waste-disposal systems have had a go at them but failed. In addition,
when those alternative waste disposal units themselves become worn-out or
damaged, it falls upon the lysosomes to break them (and, often, their semi-
digested contents) down. This chapter is about what goes wrong with the
cell's scrapyards of last resort and how we might avert that process.
» - * P Cleaning Up Life's Messes
Your cells, like your household, are constantly producing and consuming
goods and generating wastes of various kinds in the process. One sort of
waste is akin to packaging, or disposable pens, or tacky old bric-a-brac, the
possession of which has come to embarrass you. It may have served a pur
pose at one time, but today you have no further use for it and wish it gone.
Many cellular constituents are like this: Enzymes and signalling molecules
are "disposable," p r o d u c e d for temporary or even one-time use in response
to the immediate conditions in or around the cell, and they need to be de
graded once they've served their purpose.
Another type of waste is more like something for which you would still
have a use, except that it can no longer fulfill its purpose because it's been
broken. Just as you can turn a piece of your grandmother's china into a jig
saw puzzle, or make your shirt unwearable at work by spilling dark red
wine on it, so components of your cells—from the small (individual en
zymes) to the very large (entire organelles, like a mitochondrion)—can be
come incapable of performing their vital cellular function after suffering
molecular damage at the hands of free radicals and other products of the
dirty underside of metabolism.
And a third type of waste is genuinely toxic material. Just as a useful
substance (say, cottage cheese) can become a threat to your health through
chemical changes (such as being taken over by mold), so normal cellular
constituents sometimes become toxic to the cell through modification of
their structure, or through being present in excess. As surely as, upon en-

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 105
countering the new ecosystem growing in your cottage cheese, you seal the
container and drop it from shoulder level into the garbage, so, too, the cell
needs to eliminate similar threats to its functioning.
Recycling's Dirty Details
All of this waste (except, again, that which is destroyed by simpler machin
ery) is directed to the cell's lysosomes. Functioning lysosomes ensure that it
gets properly processed, removing toxic by-products of normal cellular
machinery, returning usable molecular building blocks to the cell from the
"slag" of the broken-down components, and making room for healthy, func
tioning cellular constituents.
So what exactly are these cellular incinerators? Lysosomes are
membrane-bounded organelles packed full of a variety of enzymes, each of
which evolved to target a "weak spot" in the chemical structure of a waste
product that will accumulate in our cells and kill us if it isn't broken down.
A lysosomal enzyme first binds to a waste product that carries the kind of
chemical structure that it evolved to destroy, and then twists its shape like a
tiny biological crowbar, physically tearing apart the target material's molec
ular joints. This is generally accomplished by a type of chemical reaction
called hydrolysis, which is why such enzymes are called hydrolases (hydro
being the Greek for water, as in "hydroelectric").
The exact chemical details of this process aren't terribly important for
our purposes, but you should be sure to remember one key point. In order
to break down a given waste product, the lysosome must have two things:
the right enzyme for the job (one that targets a vulnerability in the structure
of the specific waste product in question), and enough acidity in its interior
for the relevant enzyme to function. This latter is required because differ
ent levels of acidity cause enzymes and other proteins to assume slightly dif
ferent shapes, so that when the acidity in the lysosome is wrong, the enzyme
becomes "bent out of shape" and thus no more able to do its job than is a
flattened-out crowbar. Acidity is also required for the functioning of pro
teins that translocate some waste products into the lysosome in the first
place, so that a mild neutralization of the lysosome's acidity prevents junk
from even being delivered to the recycling center to begin with.
Lysosomal enzymes, like other cellular proteins, are created out of the
blueprints present in the nuclear DNA. They are then shipped into the
lysosome, though by machinery very different from the mitochondrion's

106 E N D I N G A G I N G
TIM/TOM complex. The extra protons that create the lysosome's acidity
are actively pumped out of the main chamber of the cell and into the lyso
some by an energy- (that is, ATP-) consuming pump located on its mem
brane (the vacuolar ATPase).
Incomplete Combustion
You won't be surprised to learn that bad things happen if your body fails to
produce a lysosomal hydrolase that is needed to break down a waste prod
uct being p r o d u c e d in some cell type—or if it produces a defective form of
the protein that doesn't do its job properly. In fact, this is precisely the de
scription of a group of rare but well-established genetic disorders known as
lysosomal storage diseases (LSDs).
There are about forty such diseases, but luckily only about one person
in 7,500 is born with any of them. Victims of all of these diseases suffer
from one or another type of failure of their lysosomal incinerators. Many of
them completely lack the gene for a lysosomal enzyme, or bear a mutated
copy of it, resulting in a misshapen and ineffective version of the hydrolase.
In other cases, the problem is that one of the specialized transport proteins
on the surface of the lysosomal membrane is missing or defective, so that
the lysosome can't bring the junk into itself to break it down.
No matter what their origin in a given patient, the result of such muta
tions is a deadly degenerative disease. Which organs a given mutation affects,
and how severely, varies from one LSD to another, depending on exactly
which missing or malfunctioning enzyme is at the root of the problem. This is
because different cell types produce different wastes at different rates, and
each particular waste exerts a distinct pathological impact on the cell if it isn't
degraded.
But in all cases, patients suffer pathology in major organs. In Gaucher
disease, the spleen swells up and anemia develops. There are two inherited
forms of Niemann-Pick disease. In the fast-acting version (Type A), the
liver and spleen enlarge and the nerves degenerate starting at birth, killing
its victims by age two or three. In the slower-acting variety (Type B), pa
tients may develop fatty, yellow nodules on their eyelids, neck, or back, and
an enlarged liver, spleen, and lymph nodes. And Hurler syndrome causes
facial features to twist up and bone deformities to occur, along with en
largement of the spleen and liver, joint stiffness, clouding of the eye, early-
onset dementia, and hearing loss.

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 107
The exact mechanistic links between the lack of effective waste dis
posal and particular pathologies have not all been worked out in detail, but
the basic picture is clear. The undegraded waste material accumulates in
the lysosome, causing it to swell up and take up too much room in the cell,
impeding the traffic of other materials in the main cell body. Meanwhile,
the acids and enzymes within the lysosomes are diluted, inhibiting their
ability to both import and break down other wastes for which the cell does
have the requisite enzymes, thereby setting up a vicious cycle.
There are also some cases in which it appears that toxic, undegraded
waste accumulates in the main body of the cell. This can be either because
it is not trafficked into the overburdened lysosome in the first place, or else
because the failing organelle starts to leak or even bursts, spewing its toxic
load—including the acids and enzymes that it carries, which are essential to
lysosomal function but potentially deadly to the rest of the cell.
Lysosomal Limitations and the Deadly Dregs
In addition to the terrible, early-acting pathologies that ravage the victims of
these genetic disorders, however, it's also long been known that undegraded
gunk builds up in the lysosomes of all of us as we age. Called lipofuscin (LIP-
oh-few-sin1) or popularly "age pigment," this noxious, accumulating goo is
a chemical hodge-podge of fatty and proteinaceous materials derived from
membranes, reactive metals like iron and copper, and a variety of other or
ganic molecules. It is easy to see with a microscope because it glows red
when exposed to light of a particular wavelength.
Lipofuscin is actually not a single, specific compound, but a catch-all term
for the mixture of stubborn waste products that refuse to be broken down af
ter they've been sent to the lysosome for degradation—materials so chemically
convoluted that the normal complement of lysosomal enzymes just doesn't
know how to deal with them. A combination of damage from free radicals and
from glycation (the random sticking-together of different "branches" of a sub
stance's proteins by reaction with the sugars in your blood and cells) twists
their structure back on itself like some demented child's molecular origami,
burying the vulnerable spots in their structure so that lysosomal hydrolases
can't get at them to break them down.2 As a result, these materials don't get
properly degraded—and because lysosomes are in most cases unable to ex
port them out of the cell, the material just accumulates, taking up more and
more room in the lysosomes of long-lived cells like the heart and the brain.

108 E N D I N G A G I N G
First spotted in the nineteenth century, lipofuscin was largely ignored
by biomedicine until it became a hot—and controversial—topic in biogeron
tology in the 1970s. At that time, it seemed just obvious to many researchers
that lipofuscin must be bad for us: it slowly fills up our cells, (taking up as
much as 10 percent of the total space in aged primates' heart muscle cells,
for example), and the course of its accumulation tracks the cell dysfunction
seen in aging animals (including people). Indeed, the rate at which lipofus
cin accumulates in a given species' heart was found to be proportional to its
rate of aging, so that adolescent, middle-aged, and old monkeys of two dif
ferent species, with greatly different calendar ages but at similar stages in
their life cycle, have roughly the same level of lipofuscin clogging up their
cells. Many researchers outlined a mechanistic hypothesis of lipofuscin as a
contributor to aging similar to what happens in the LSDs: lysosomal fail
ure, waste accumulation, interference with cell trafficking, and the release
of toxic enzymes and acidity from ruptured lysosomes.
But the point was a contentious one. Many scientists believed that lipo
fuscin was benign, in part precisely because it is so hard to degrade: With
the reactive spots in their structure already balled up and stapled together
by previous free radical and glycation damage, lipofuscin is chemically
quite inert, so it doesn't interact with essential biomolecules the way that
free radicals or other toxic chemicals do. Also, speeding up the accumula
tion of lipofuscin in experimental animals by denying them adequate vita
min E did not shorten their lives, as one would expect of any manipulation
that accelerated a real cause of aging.
But those reports were strongly disputed by other experts in the field,
because it was not at all clear that the material whose accumulation was in
creased by vitamin E deficiency actually was lipofuscin. Much of what gets
referred to by this name in the older scientific literature is actually other, re
lated substances (often called ceroid) that share many of lipofuscin's prop
erties but are much easier for the cell to break down. It seemed likely that
vitamin E deficiency was increasing the production of this relatively
tractable material, while levels of "real" lipofuscin were unaffected.3 More
over, while ceroid accumulation was associated with a variety of diseases
(diseases in which normally degradable substances are not degraded—one
could think of them as nongenetic LSDs), it was not clearly related to "nor
mal" aging.
And so, the debate went 'round. As with many such cases in the early
days of biogerontology, the data were ambiguous, the definitions were im
precise, and there was little hope of a clear resolution.

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 109
I had, myself, remained an agnostic on the subject up to and including
the writing of the first draft of my doctoral thesis. But that began to change
in the spring of 1998, when I met Ulf Brunk, chair of pathology at Sweden's
University of Linkoping, at the Oxygen Radicals Gordon Conference in
Ventura, California. After listening to his presentation of his recent results, I
started taking lipofuscin more seriously as a potential nexus connecting the
intricately orchestrated chaos of metabolism to the pathology of aging.
Brunk had done some first-rate work in assessing the role of lipofuscin
in cultured heart cells—an important technical advance, especially because,
in the body, heart cells never divide. When a cell divides, its load of cellular
junk—including lipofuscin—is shared between the two daughter cells. Each
now has half as much, on average, and this will continue with each new gen
eration. If the junk is only being generated quite slowly, this dilution process
will fully balance the rate of creation of new junk and an unmanageable level
of junk will never accumulate. The quickly replicating skin cells that had
been used in much previous work did just this—they tended to dilute Out
lipofuscin and other cellular junk, and so could not replicate the real effects
of lysosomal buildup in critical nondividing cells like those of the heart and
brain. This had long been recognised, but heart cells had been found to be
notoriously hard to culture—in large part, it turns out, because of the high
levels of oxygen in the atmosphere. Cultured cells are still too often grown
under normal atmospheric air, despite the fact that our bodies ambiently
contain only about one seventh of air's concentration of oxygen.
By growing the longer-lived heart cells under more physiological levels
of oxygen, Brunk was able to show how increased oxidative stress—higher
levels of free radical damage, in other words—increased lipofuscin forma
tion. He also confirmed previous suspicions about how lipofuscin accumu
lation could impair the ability of the cell to recycle its used-up components.
And on top of that, he was also able to show that older heart cells accumu
late damaged mitochondria—which they would not do if the lysosome were
operating properly, since the disposal of defective cellular power plants is
one of their chief responsibilities in the cellular economy.
With his collaborator Alex Terman, Brunk outlined a "garbage catas
trophe" theory of aging, 4 in which accumulating lipofuscin inside the lyso
some dilutes the organelle's acidity and supply of enzymes. In this model,
lipofuscin also wastes a lot of the enzymes that the cell body produces, by
sucking them up without making effective use of them, thereby diverting
them away from the other, still-functional lysosomal contents against which
they could be put to effective use.

110 E N D I N G A G I N G
As the cell's lysosomes accumulate waste products that they aren't
equipped to handle, they become ever less able to break down materials
within them. As a result, the junk in question spends more time either out in
the main body of the cell, or even trapped inside the lysosome, before being
"incinerated." During that time, chemical alterations continue to occur in
the structure of these wastes, mangling them further and further and making
it more and more difficult for lysosomal hydrolases to reach the weak spots
in their structure. As a result, even the standard cellular junk that lysosomes
are, in theory, well equipped to degrade is no longer efficiently broken
down, but instead accumulates—which then further dissipates the hy-
drolytic enzymes and acidity of the lysosome. In their culture experiments,
Brunk and Terman showed that lipofuscin overload could even trigger cell
death, as lysosomes become loaded with the stuff and rupture.
The data underlying this model were compelling, and I liked it at once,
sneaking a short reference to it into my Cambridge biology thesis.5 But I
wasn't yet convinced that lysosomal failure was truly a significant contribu
tor to aging, because if the theory were right you would expect to find evi
dence connecting lipofuscin to actual age-related disease, and no such
evidence initially turned up when I went looking for it.
I quickly learned, however, that this seeming lack of data was more of a
communication breakdown than an information vacuum. Researchers tend
to get holed up in their narrowly specialized fields of study, and conse
quently they, too, rarely compare notes and observe the confluence of ob
servations in different fields of science (or even subfields within those
fields). I soon found that if I stopped specifically talking about "lipofuscin"
and began asking researchers about the importance of lysosomal dysfunc
tion in the diseases that they studied, I was suddenly inundated with evi
dence that the accumulation of junk that should be processed in the
lysosome was at the heart of the matter—but that this fact was being ob
scured by the use of specialist jargon in referring to those wastes.
Making the Link to Pathology
A t h e r o s c l e r o s i s
Just over a year after I was first exposed to Brunk's suggestive data, I was at
tending the biennial Gordon Conference on Atherosclerosis, at which I
found myself listening to a review on the complex processes that lead some-

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 111
one from having too much cholesterol in the blood, to having fatty plaques in
the arteries (atherosclerosis), to having diagnosable coronary heart disease
and eventually a heart attack. As I quickly learned, researchers had been
placing lysosomal failure at the core of the molecular events that underlie the
formation of atherosclerotic plaques for years before I began looking into the
issue—and they did so without ever mentioning "lipofuscin."
Most people visualise atherosclerotic arteries as being much like
clogged pipes. Greasy gunk (whether it's bacon fat or blood cholesterol)
simply accumulates on the inside of the tube, coating its surface and clog
ging it up, and blocking the passage of fluids—be those fluids the dishwater
in your sink or the blood in your arteries. In fact, however, we've known for
some time that the process is much more complicated than this. 6 Athero
sclerosis begins with a microscopic problem in the blood vessel wall. Many
things can cause or contribute to this, including friction from the passing
torrent of blood flow, or the force of high blood pressure, or infection; most
often, however, it's just the accumulation of our old friend LDL, low-density
lipoprotein, which has a tendency to get stuck there. The body responds to
this problem just as it does to any other injury: by secreting factors that in
flame the site in order to attract immune cells called macrophages.
Macrophages then infiltrate the damaged tissue to help it heal by cleaning
up the debris.
I didn't tell you very much about LDL in Chapter 5; now's the time for
more detail. Despite its bad reputation, cholesterol is actually a necessary
component of cell membranes. In fact, the so-called "bad" cholesterol
(LDL) in the blood is actually a carrier particle, designed by your body to
bring needed cholesterol to cells, and those cells in turn have specialized
receptors designed to allow them to ingest it for their internal use.
In order to reach most cells, the cells that comprise the walls of our
blood vessels must allow LDL to pass between them, and beyond into the
surrounding tissue. But when cholesterol is chemically modified—by expo
sure to free radicals (oxidized LDL) or reactions with blood sugar (glycated
LDL), for example—it becomes more prone to stick together and thus
more immobile. Because free radicals and blood sugar are (respectively) in
evitable by-products of, and necessary raw materials for, some of the most
fundamental metabolic processes in the body, they are ubiquitous. Hence,
LDL particles are constantly being subjected to their chemically disruptive
influence. Furthermore, enzymes that are designed to call in immune cells
when the vessel wall is injured also alter cholesterol in ways that make it
more toxic.

112 E N D I N G A G I N G
That's the main reason why having a high cholesterol level is bad for
you. The more cholesterol there is in your blood, the more contact it has
with these damaging agents, and the more toxic, modified cholesterol will
be coursing through your body.
So, when macrophages are attracted toward an inflammatory signal,
they find plenty of junk in need of removal. Initially, macrophages deal rea
sonably well with the gunk that they're internalizing, and they can often
successfully remove the detritus. But when an already compromised blood
vessel continues to be assaulted by high blood cholesterol levels, inflamma
tory signals created by excess body fat, or nasties from cigarette smoke, the
problem persists and macrophages hang around for longer.
As they take in more and more waste—particularly an excess of modi
fied LDL—macrophages begin to fall behind in their work. An increasing
percentage of the load of junk is not successfully processed, but instead ac
cumulates within the macrophages' lysosomes—or, just as bad, is puked
out of the lysosomes without being properly detoxified, forming droplets of
modified cholesterol in the cell body.
As this continues, macrophages eventually become the cellular coun
terparts to the obscenely bloated "Mr. Creosote" in the restaurant sketch
in Monty Python's The Meaning of Life. If you've seen this movie, you
will definitely remember the scene. Mr. Creosote comes into a fine French
restaurant already stuffed with food and badly nauseous, but is plied with
"moules marinieres, pate de foie gras, Beluga caviar, eggs Benedictine" and
sauces "rich with truffles, anchovies, Grand Marnier, bacon and cream" by
the perversely codependent and outrageously "French" maitre d' (John
Cleese).
Creosote becomes more and more ill as the meal progresses, but when
he finally attempts to summon up the will to stop eating he is cajoled into
having just one last "wafer-thin" after-dinner mint. When the spineless pa
tron swallows the mint, a look of helpless horror fills his face; as the maitre
d' runs for cover, Creosote literally explodes, his innards and lunch splatter
ing graphically over staff and guests alike.
Imagine your blood vessels to be such a restaurant, welcoming a steady
stream of customers just like Mr. Creosote in the form of macrophages that
come in to dine on modified cholesterol products. Imagine that they simply
refuse to leave when you're trying to close up, but continue to stuff them
selves until their "stomachs" (lysosomes) can't take any more—and then
keep going until it kills them, and your restaurant (blood vessels) becomes
their final resting place.

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 113
You now know, in essence, the genesis of the "foam cells" that accu
mulate in your vessel walls, forming "fatty streaks" as they become numer
ous enough to be seen with a microscope, and eventually developing into
full-blown, unstable atherosclerotic plaques—the scabbed-over messes
that ultimately form at the injury site, crammed with a miasma of clotting
blood, inflammatory signal molecules, and dead foam cells. Once this hap
pens, your days are numbered. It's only a matter of time until the pressures
within and without the plaque cause it to rupture, spewing its contents into
the bloodstream. This content is not a liquid but a horde of semisolid
chunks, and these chunks are rapidly swept from their origin in the major
arteries into progressively smaller vessels. They become stuck there, block
ing off the flow of blood—sometimes into the heart (causing a heart at
tack), sometimes the brain (causing a stroke).
So we now understand that lysosomal dysfunction is the key step in
the conversion of healthy macrophages into undead foam cells—and of
healthy blood vessel tissue into an atherosclerotic time bomb. This fact is
widely recognized, but unfortunately, nearly all researchers are pursuing
old-school, ultimately preventive treatments for the problem. Existing
anti-atherosclerotic drugs try to prevent macrophages from stuffing them
selves so badly, either by reducing blood cholesterol levels or by reducing
LDL's exposure to metabolically active agents (blood sugar, inflammatory
enzymes, and free radicals). Drugs currently in the pipeline seek to ap
proach the same problem from its flip side, by increasing the transport of
cholesterol out of the blood, cells, or organs before it gets a chance to do
its damage.
Meanwhile, basic researchers working in areas other than drug devel
opment are spending a lot of time trying to puzzle out exactly what causes
macrophages' lysosomes to fail, with the idea that, if they understood the
fine details of the process, they could design drugs that would interfere in
the relevant steps in the metabolic chain. Unfortunately, the evidence is
consistent with many interpretations, and the data are difficult to reconcile,
which has more-or-less stalled progress in developing therapies based on
this model.
For instance, some researchers focus on the fact that, in test tubes, oxi
dized cholesterol inhibits the necessary processing ("de-esterification") of
normal (unmodified) cholesterol in the lysosomes, slowing it down enough
to create a deadly backlog. Others think that modified LDL, like lipofus
cin, is itself undegradable, and dilutes out the factors needed for the lyso
some to degrade other materials as it accumulates. There is also evidence

114 E N D I N G A G I N G
that something in modified LDL (or some metabolic by-product of it) is
harmful to lysosomal function—such as the evidence (again in test tubes) that
the oxidized cholesterol variant 7-ketocholesterol (7-KC) interferes with the
activity of the membrane-bound ATPase enzyme. When this enzyme is im
paired, it can't maintain enough acidity in the cell's lysosomes to keep their
hydrolases working properly, so there are those who think that this is where
we need to look for a solution. And still others think that macrophages sim
ply take in too much LDL, so that its sheer volume overwhelms their pro
cessing capacity; if so, slowing uptake might also slow down the development
of the disease.
As yet, we don't know which school of thought is correct—and it's un
likely that we will resolve the question definitively any time soon, because
the conditions under which the relevant studies are carried out are so un
like what happens in the body. While the scientific debates continue, vas
cular disease caused by atherosclerosis remains the number one killer in
the developed world—and other problems that arise from the same failure
to process cholesterol may be related to a wide range of other age-related
diseases.
Fortunately, an intervention that came to me in a flash some years
ago 7—and that has since been worked out in greater therapeutic detail in
collaboration with others8 , 9—offers a solution that sweeps away the need
for this kind of detailed molecular map of the metabolic maze. This solu
tion does not rely on such detailed understanding of what causes lysosomal
failure in atherosclerosis. Instead, it provides a way for us to clean up the
lysosome itself, rather than the metabolic processes that overload it—and
in a manner that will work irrespective of what leads up to its initial failure.
But before we get into that, let's look at another fearsome disease of ag
ing that has lysosomal dysfunction at its heart: the decay of the brain.
Neurodegenerative Disease
Except in the case of stroke—which I've covered above, and which is more
of a one-off, traumatic injury than a degenerative process in itself—the
brains of people suffering from all the major neurodegenerative diseases
show evidence of inadequate lysosomal function. In most cases, the most
obvious pointer is the presence of clumps of a distinctive aggregated pro
tein material inside the brain cell: Lewy bodies in Parkinson's disease and
the boldly named "Dementia with Lewy Bodies" (DLB), aggregated hunt-

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 1 1 5
ingtin protein in Huntington's disease, and neurofibrillary tangles (NFT),
formed of aggregations of the protein tau, in Niemann-Pick and Alzheimer's
diseases. 1 0 Yet, because these aggregates are not located within the lyso
some, and are not themselves lipofuscin, the role of lysosomal dysfunction
in these diseases has been obscured—so again, people specifically looking
for a connection with "lipofuscin" can miss these data, obscuring the rela
tionship.
In several cases, however, there is more direct evidence of trouble at
the toxic waste dump. Some of the most remarkable such evidence has re
cently been found in the brains of Alzheimer's patients, in which the break
down of proteins through another of the main components of the cell's
recycling system (the proteasome) is badly impaired. In some victims, this
may be because mutations in the gene for a protein called ubiquilin cause it
to inhibit the activity of ubiquitin, a protein that "tags" proteins for break
down in the proteasome. Both neurofibrillary tangles in Alzheimer's and
Lewy bodies in Parkinson's disease are loaded with ubiquitin, yet the pro
teasome system seems incapable of picking these aggregated materials up.
The connection with the lysosomal apparatus is this: proteasomes that
are not doing their job put more pressure on the lysosomal system as the
defective proteasomes (and the material they have failed to destroy) are
sent to the lysosome, increasing lipofuscin formation. 1 1 At least some of the
waste that the proteasome fails to pick up—along with damaged protea
some units themselves—is ultimately sent to the lysosomes: this phenome
non has been definitively observed in the case of aggregates normally
degraded by the proteasome in Huntington's disease, and is probably
what's responsible for the finding of a lot of ubiquitin inside the lysosomes
of the neurons of Alzheimer's patients.
But the most dramatic hallmarks of abnormal trash disposal in
Alzheimer's disease are the signs of malfunction in the lysosomal system it
self. As a bit of background: One of the main ways in which cellular rub-
bish gets delivered to the cellular recycling center is through a process
called "macroautophagy," in which the waste in question is swallowed
whole by a membrane structure called an autophagosome or autophagic vac
uole (AV), which then hooks up with the lysosome and fuses with it. (If this
term rings a bell, that's probably because I briefly mentioned it in Chapter
5 as the way in which damaged mitochondria are delivered to the lyso
some.) The result, in effect, is a bigger lysosome, with a single combined
membrane that surrounds both the contents of the AV and the hydrolytic
enzymes (and acidity) of the original lysosome to digest that contents.

116 E N D I N G A G I N G
Recent studies show that this aspect of lysosomal function is in a very
bad way in the brains of Alzheimer's victims. 1 2 It has been known for some
time that the lysosomal system in the Alzheimer's brain is, like the protea
some, apparently both hyperactivated and inactivated: it's as if the neuron
were an unthinking driver of a car with a worn-out engine, trying unsuc
cessfully to compensate for its misfiring cylinders by pushing down harder
on the gas pedal. The new work suggests one major reason why they fail:
Their brain cells-—and especially the cells located in areas of the brain that
are most badly affected by the disease—are full of multilayered AV-based
structures that are a lot like Russian nesting dolls, with one AV contained
within another, larger one, which in turn is sequestered inside another, still
larger AV.
Some of these structures seem to form when AVs fail to fuse with lyso
somes, and hang around in the cell long enough to begin to take some
damage, ultimately becoming so badly degraded as to be recognized as
junk—at which point they are swallowed up into another autophagic vac
uole. Then, the cycle repeats itself, as the new AV itself fails to fuse. In other
cases, it appears that the AVs have fused with a lysosome, but that the lyso
some is so weak—or perhaps so immature—that it can't degrade the AV
contents.
It's a picture that reminds me of nothing so much as the infamous
Khian Sea, a ship hired by the city of Pennsylvania in 1986 to haul its incin
erator ash to an artificial island in the Bahamas for disposal. Unfortunately,
the Bahamian government had not given the operators of the Khian Sea
permission to dump its waste there. And so began a fourteen-year world
cruise of garbage, in which the ship traveled from port to port, attempting
to dispose of its load in different countries all over the world—first back up
the east coast of the United States, then back down south to the Caribbean
and South America, and ultimately wandering as far afield as Indonesia and
the Philippines.
Ultimately, the Khian Sea—renamed and reflagged—relieved itself of
its toxic burden by illegally dumping it into the Atlantic and Indian
Oceans. Sooner or later, peripatetic AVs can only be expected to discharge
their hazardous contents, too.
Scientists all agree about the basic facts: the major neurodegenerative
diseases are characterised by the presence of aggregated proteins and lyso
somal dysfunction in the brain, and it's clear to everyone involved that
there is some kind of connection between the clear failure of the cell's waste
disposal systems to deal with the aggregates and the diseases in which these

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 117
disruptions occur. The question is just what that connection is. Intuitively,
it makes sense that the aggregated junk sitting around in our brain cells
must be bad for them. Most scientists in the field share this intuition, and
indeed it's easy to show, in relatively crude test-tube experiments, that these
substances work mischief in brain cells to which they are added, including
the initiation of a vicious cycle in which the accumulation of aggregates dis
rupts normal neuronal function, leading to further lysosomal dysfunction
and protein aggregation.
But others have a different take on these phenomena. Surprisingly,
some scientists think that protein aggregates may in some sense be protec
tive. The idea is that while the aggregates themselves may in the long term
interfere with cell function by blocking cellular traffic with their sheer size,
the soluble, highly reactive units that make up the aggregate are a much
more immediate threat to the health of the cell. By handcuffing these units
together into a single cellular chain gang, the cell can keep them from at
tacking other cellular apparatus in their environment, preventing a deadly
short-term threat to cellular health.
And then there are those who view the aggregates as being more of an
epiphenomenon: a sign that something is wrong with the cell, but not an
actual contributor to pathology. In this model, undegraded protein de
posits are more like gunsmoke than actual guns or the bullets they fire: in
themselves they are more-or-less harmless, but their presence is an unmis-
takeable signal that you're in a crime scene. Perhaps, for instance, some
other contaminant is building up in the lysosome, preventing it from prop
erly incinerating cellular garbage, so that the aggregates build up—but the
aggregates themselves aren't the source of the problem or a major contribu
tor to cellular pathology. This is still a bad thing to have happening, of
course, because cells rely on a functional lysosome—both to break down
benign cellular constituents that are past their useful life in order to make
use of their building blocks for future cellular construction projects, and to
destroy genuinely toxic wastes. But the source of the problem is to be
found elsewhere than the obvious piles of trash cluttering about the main
body of the cell.
For example, Alzheimer's patients may have more defective mitochon
dria in need of recycling than healthy people do, putting demands on the
lysosome that it just can't satisfy; once the lysosome fails, other components
may form the observed aggregates, but it's still the dysfunctional mitochon
dria that started the ball rolling downhill. But again, it's pretty hard to es
cape the conclusion that the resulting protein clumps constitute cellular

118 E N D I N G A G I N G
"speed bumps" that must eventually cause the cell some serious problems
of their own.
Unfortunately, there is substantial evidence—both in neurodegenera
tive disease and in aging—to support each of these positions. "Unfortu
nately," I say, because I feel it is paralysing researchers in their quest for
cures. Researchers spent much of the 1990s in entrenched holy wars be
tween the "BAPtists" (named for "Beta-Amyloid Protein") and the
"Tauists" (named for the tau-based neurofibrillary tangles or NFTs), each
of which expended considerable effort in trying to prove their favored can
didate to be the primary problem in Alzheimer's disease. ("What's beta-
amyloid?" I hear you cry. You'll learn plenty about that in Chapter 8.)
Today, there is a similar feud simmering over the different interpretations
of the role of protein aggregates generally in neurodegenerative disease.
And in old-school thinking—in which the goal is to find drugs that will
shut down the metabolic processes that lead to a disease outcome, or at
least perturb that aspect of the pathway that causes the most harm—issues
of this kind must be definitively resolved in detail before we can even begin
to design treatments for humans, since interfering with metabolic pathways
is a risky business that can only lead to harm if the process that you're
blocking turns out to be an innocent bystander.
Even more so than with atherosclerosis, then, traditional medical ap
proaches to neurodegenerative disease are, with respect to protein aggre
gates, at a standstill because of inadequate understanding of the link
between the junk in question and the disease itself.1 3 Again, however, I have
a solution in mind that sweeps aside the need to resolve these ambiguities.
Macular Degeneration
While I don't wish to tease you, I do want to go over the critical role of unde
graded aggregates in a third important aspect of aging before finally revealing
my proposed therapy for all diseases involving lysosomal failure—including
aging itself. This third age-related problem is age-related macular degenera
tion (AMD).
There is some relief from suspense in this section, inasmuch as there is
no controversy about the involvement of aggregates in AMD. This is a clas
sic case of how biochemical cycles with which we absolutely cannot dis
pense lead to the destruction of the systems in which they are embedded.
Vision, like all of life's processes, is ultimately mediated by a carefully con-

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 119
trolled, complex chemical chain reaction, and our conscious perceptions
correspond in a one-to-one fashion with the particular electrochemical
phenomena that this cascade triggers in our brains. In order to perceive an
object, the energy from the light that reflects off it and into the lens of our
eyes must be translated into the chemical signalling language that corre
sponds to our subjective "sight" of the object.
For our purposes, the important step in this translation process—
important because fatal to the cells that suffer it, and hence to our eyesight—
is the (nearly) perpetual cycle of a derivative of vitamin A between two
forms.14 The rods and cones of your eyes contain the "storage" form of this
compound (ll-cis-retinal), which is chemically transformed into an "acti
vated" derivative (all-trans-retinal) when it absorbs energy from incoming
light. This activated form is used as a signal to turn on the electrochemical fir
ing of the optic nerve, which carries the signal to your brain; then, normally,
an enzyme converts it back into its "storage" form, readying it for the next
burst of incoming light.
But any system that relies on chemically unstable components always
runs the risk that their reactive chemistry will spill over the tight controls of
the system they're meant to serve. In this case, all-trans-retinal can react
with some of the fatlike molecules that make up the cell membrane, lead
ing through a complex series of steps to the formation of a stubborn end
product called A2E. This compound is completely resistant to digestion
within the lysosome, so it's a major source of undegraded junk in the lyso
somes of these cells. Over time, so much A2E is p r o d u c e d and absorbed
into the lysosome without being degraded that it can take up as much as
one fifth of the total cell volume in the cells that accumulate it. These un
fortunate cells make up the retinal pigmented epithelium (RPE) of the
eye—a part responsible for maintaining the function of the light-sensing ar
eas of the retina.
But again, because of the specialist terminology in use (A2E, rather
than "lipofuscin"), the role of lysosomal inadequacy has been—and you
will pardon the unfortunate pun!—obscured .
Toxic Waste Problem—Toxic Waste Solution
By the morning that I was throwing some clothes into a duffel bag for the
1999 Society for Free Radical Research meeting in Dresden, I had come to
see lysosomal inadequacy—and the resulting accumulation of cellular

120 E N D I N G A G I N G
waste products—as perhaps the key step linking the mitochondrial
mutation-driven rise in oxidative stress with age with the actual pathology
of aging. Remember from Chapter 5 that, by then, I had a scheme for how
mitochondrial mutations in a few cells could propagate toxins to mitochon
drially healthy cells elsewhere. What I didn't explain in Chapter 5, not least
because when I developed the Reductive Hotspot Hypothesis I didn't
know it, was just how these "toxins" are toxic—what harm they might do
to the cells that ingest them.
But now, a year later, this mystery was beginning to resolve. It was
clear, once one got over one's attachment to the term "lipofuscin," that the
failure to dispose of specific waste products was at the root of the most ter
rible diseases that accompany biological aging: atherosclerosis, age-related
macular degeneration, and neurodegenerative diseases like Alzheimer's. It
was just that the kind of waste that was linked to a given disease was specific
to the cell type and the particular diagnosis.
As it happened, Ulf Brunk was again presenting his data in Dresden.
As I listened to his talk and contemplated his slides—the telltale red glow
of lipofuscin choking cells, his computer-generated diagrams illustrating
his and Terman's "garbage catastrophe" theory—I saw that it was a waste
of time to argue about whether lipofuscin contributes to "aging" in the nar
row sense. Clearly, we needed a way to solve this problem if we were to pro
tect our bodies from age-related pathology. But I also became convinced
that it would not be enough to try to prevent the accumulation of this junk
"upstream" by obviating mitochondrial mutations. We were also going to
have to deal with the junk directly.
But how? With the recalcitrant materials in question being so multifar
ious, and with the metabolic pathways, chemical identities, and even spe
cific role in pathology of these materials being still largely unknown, it
seemed that no classic "magic bullet" approach—one small molecule to
match one therapeutic target—would work. And simply putting the lyso
some into overdrive wouldn't really solve the problem: While, as later ani
mal studies would show, 1 5 , 1 6 simply souping up lysosomal activity or
topping up their existing enzyme supply could slow down the progression
of lysosomal storage diseases, such approaches could not ultimately stop
those diseases. It is the very nature of the problem that the body simply
does not have the enzymes to degrade the really ugly junk—and thus, that it
will choke up your cells, steal your mind, blind you, and clog your arteries
later, if not sooner.
Ulf's talk ended, and with a hundred other scientists I got up and

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 121
streamed into the outer hall for the coffee break. The red glow of lipofuscin
on the slides had gotten me to thinking of the stuff as the toxic waste that it
is, and of the aging cell as a tiny contaminated environmental site equipped
with a woefully inadequate waste management system. So the job of restor
ing the cell to health was really a kind of environmental cleanup job, and
what was needed was a biomedical superfund project to develop new reme
diation technologies capable of dealing with materials that had so far
evaded the lysosome's capacities.
Superfund!
It suddenly occurred to me that this was more than a metaphor. (If
you've never heard of Superfund, be patient—I'll explain shortly.) There
were actual land sites all over the planet that should be very badly contami
nated by lipofuscin, because their soil has been seeded with the stuff for
generations. I speak, of course, of graveyards. Think about it: hundreds of
bodies put into the ground—sometimes en masse, as happened throughout
Europe during the horrors of the Plague, and more recently following acts
of genocide in Rwanda and elsewhere. These soils should be chockablock
with aggregates from their inhabitants' decaying bodies.
Yet, to my knowledge, there was no accumulation of lipofuscin in
cemeteries—and if there were, we certainly ought to be aware of it, because
lipofuscin is fluorescent. Months later, when I was discussing the issue with
fellow Cambridge scientist John Archer, he would put the disconnect suc
cinctly: "Why don't graveyards glow in the dark?"
Soil microorganisms struck me as the most likely explanation. Bacteria,
fungi, and other microbes normally play a role in turning our remains into
compost, of course, but it was not so immediately obvious that they would
be able to digest something so resistant to enzymatic action as lipofuscin.
And yet, I recalled, we'd known for decades that soil microbes display an
astonishing diversity in their choice of food.
Scientists became interested in this phenomenon in the 1950s, when it
was noted that the levels of many hard-to-degrade pollutants at contami
nated sites were present at much lower levels than would have been ex
pected. A big part of the explanation turned out to be the rapid evolution
of quickly reproducing organisms like bacteria. Any highly energy-rich
substance represents a potential feast—and thus, an ecological niche—for
any organism possessing the enzymes needed to digest that material and
liberate its stored energy. The presence of high levels of such a material
therefore creates a powerful evolutionary "pull," driving the evolution of
the necessary enzymes in microorganisms that come into contact with it.

122 E N D I N G A G I N G
And this is especially so if the substance is not easy to break down, because
then the chances are good that most of the other organisms in the vicinity
will not have enzymes capable of this degradation.
It was proposed in 1952 that these forces might well be so strong as to
guarantee that, given enough time, evolution would find a way to create mi
crobes with the capacity to digest anything we throw at them that is both
carbon-based and rich enough in energy to be a worthwhile fuel source.
This was given the immensely memorable name "the microbial infallibility
hypothesis." While it has turned out to be a bit of an overstatement—no
one has yet discovered the microorganism that can eat Teflon, for instance—
studies over the next few decades tended to confirm the general principle.
U.S. Geological Survey scientists collected case studies showing that mi
croorganisms were breaking down significant amounts of a variety of or
ganic chemical pollutants in wastewater. Oil spills, chlorinated solvents,
pesticides—you name it: soil bacteria learned how to digest almost any
thing that was thrown at them, leaving only harmless residues like carbon
dioxide and water.
Scientists' first attempts to harness this power failed, because they were
trying to invent organisms to order, imitating what nature was already do
ing very well. But eventually, researchers realized that they just weren't as
smart (nor, more accurately, as fast) as the forces of nature. Out of these ob
servations developed bioremediation: the exploitation of evolution's ability
to generate novel digestive capacities in microorganisms for the intentional
cleanup of contaminated environments. "Superfund" was the name of a
U.S. government initiative to stimulate and commercialize bioremediation
research.
Sipping my coffee in Dresden, my mind brought this train of thought
full circle, feeding back into my original musing on the lysosome as an in
adequate toxic waste disposal system. The lysosome already deals with the
cell's waste products using enzymes to break them down into their con
stituents. But it is not equipped with the capacity to deal with every possible
waste material. This is just what you'd expect from evolutionary theory. Re
member, again, as we discussed in Chapter 3, that evolution only designs
your body to last as long as your environmental niche will allow it to last. In
the Paleolithic environment in which we evolved, that meant about three
decades—far less time than it takes lipofuscin or atherosclerotic cholesterol
aggregates to build up to life-threatening levels.
For this reason, evolution has never bothered to equip the lysosome
with enzymes designed to deal with these wastes, because it has never had

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 123
a good reason to do so. But, as we've seen, it seems very likely that evolu
tionary forces have pushed soil microorganisms to develop these capacities
in order to exploit a new fuel source—an issue, for them, of day-to-day sur
vival. Not only do evolutionary theory and the "microbial infallibility hy
pothesis" predict this, but it also seems to be confirmed by the absence of
large accumulations of lipofuscin in mass grave sites: were it not so, all such
locations would have an eerie glow about them, instead of such a phenom
enon being confined to cheesy horror flicks.
Suddenly it came together. The imaginative spark of metaphor had
fallen upon the very concrete fuel of data in the oxygen-rich environment
of evolutionary theory, and a fire began to burn in my brain. These two ob
servations implied that we could perform a sort of medical bioremediation,
in which we would identify the soil bacteria that already clean up our un
degraded junk after we have died, determine the enzymes that allow them
to do it—and then deliver these enzymes into the lysosomes of people who
were still alive to benefit from it. Figure 1 gives a graphical depiction of this
cycle.
These enzymes would give new powers to our cellular recycling centers,
allowing them to process materials that presently go undegraded within us—
not just preventing, but reversing their pathological accumulation. Our
brains would be cleared of neurofibrillary tangles; the dying macrophages in
our arteries would gain new life, letting them clear out the oxidized LDL tox
ins and allowing the necrotic vessel tissue to finally heal; the blind would see.
And aging cells all over our bodies, choking on their own filth, would be
come clean and new again.
Figure 1. Medical bioremediation, exploiting the microbial enzymes that turn dead people into decomposed people, may retard many of the processes that turn young people into old and eventually dead people in the first place.

124 E N D I N G A G I N G
Normally, when new ideas come to me, I give myself a few days to try to
punch holes in them before bouncing them off anyone else. But this time I
felt supremely confident. Taking my nose out of my coffee, I scanned the
hall for Dr. Brunk. I spotted him across the room: chubby, graying, earnest
but with a mien of compassion that made you think of him as an aging so
cial crusader. In a few purposeful strides I was confronting him.
"Listen, Ulf," I said quickly, "I've just had the most fabulous idea . . ."
A Quick-and-Dirty Test
I was a little disappointed by Brunk's reaction, though I was not sure how
much of what I was seeing was a reflection of his assessment of the feasibil
ity of the entire scheme versus its inherent audacity or my jumbled, hot-
off-the-fire delivery. Perhaps it was just Nordic caution. Whatever it was, it
was clear that, while not dismissive of the proposal, Brunk was clearly not
experiencing spontaneous combustion from the white heat of my brain
wave.
Still, I pressed him for his thoughts on possible ways to perform pre
liminary tests of the idea. The first thing, we agreed, would be to test the
sturdiness of the very foundation of the castle that I had just constructed in
the air: the idea that soil microorganisms are, indeed, routinely digesting
lipofuscin in corpses. Fortunately, there was a reasonably straightforward
way to execute such a test: collect some soil microorganisms from a site
likely to be "enriched" in human remains, and then see if they could break
down lipofuscin in a test tube.
It turned out that what you might think to be the easy part—getting
the lipofuscin needed to test the abilities of graveyard bacteria—was actu
ally almost impossible to pull off in the real world. There are only small
amounts of lipofuscin in most of our cells, and the tissues in which there's
more (such as the heart) are not so readily available, so to get a useful
quantity of the stuff would be a challenge. But Brunk said that he could
whip me up a batch of an excellent substitute: the synthetic lipofuscin
used by people working in his field, which is created by merely exposing
mitochondria to enough ultraviolet radiation to induce cross-linking of
their membrane proteins. The resulting recalcitrant gunk has the same flu
orescence spectrum as the real thing, and seems to also have the same
physical and chemical properties—which is as you'd expect, since most
experts think lipofuscin largely is the remains of unsuccessfully degraded

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 125
mitochondria, damaged by the effects of free radicals and left festering in
the lysosome.
The next thing would be to gather microorganisms from soil that had
been exposed to a large supply of lipofuscin, to look for the ones that, in
my hypothesis, had been responsible for breaking the stuff down. My mind
had already leapt ahead to the fact that John Archer—the man who would
later make the "glowing graveyard" quip—was working on bioremediation
at Cambridge. As such, he was well-versed in the techniques used by scien
tists in the industry to isolate and culture bacterial strains capable of di
gesting classical toxic waste materials, and to identify and clone the
genes involved in producing the enzymes responsible for that capacity. If I
could enlist his help, we could do the same thing for lipofuscin-digesting
hydrolases.
Tomb Raider
Fortunately, John was immediately fascinated by the whole idea, and agreed
to give it a go. So it was that his graduate student would find herself, like a
good mad scientist, at Midsummer Common in the twilight of a late sum
mer day, digging into the soil of an ancient mass grave with a trowel, seek
ing not bodies but tiny, mysterious creatures imbued with the power to
turn the most stubborn, aggregated junk in our bodies into compost.
The sense of being immersed in a Gothic horror novella lasted only a
moment. Having scooped up the soil and brought it back to the lab, John
and his student isolated the microbes and put them in petri dishes with the
synthetic lipofuscin as their only potential food supply. Then, we waited to
see if the force of natural selection would reveal the existence of strains that
were capable of surviving on a diet of pure lipofuscin.
Almost immediately, the microbes that we had isolated began to give
off the characteristic red glow of lipofuscin under the fluorescing light of
the specialized microscopes. This was not yet a success, because all it meant
was that the microbes were engulfing the material; they weren't necessarily
digesting it. But it was not long before clear differences began to emerge
among different strains. Most of the colonies of microbes were in a state of
growth arrest, failing to thrive for lack of nourishment. But a few of them
were clearly enjoying a ghoulish feast: their numbers were expanding rap
idly as their hydrolytic enzymes slowly broke the stubborn goo down into
usable components, tearing apart its complex organic chemical bonds to

1 2 6 E N D I N G A G I N G
release the stored energy. Within short order, we had a sample of microor
ganisms equipped with enzymes that could digest lipofuscin in the same
way that the enzymes in your stomach digest a steak.
The hypothesis had been confirmed. The next challenge would be to
move those enzymes into our own lysosomes. No one has done this yet, and
indeed there is a sense in which the task sits just where it was when I fin
ished my work with John Archer. Fortunately, however, we do not have to
create an entirely new field of medicine in order to get going with this idea
(which I will from here on call "LysoSENS"). That's because the funda
mental biotechnology required to pull it off is already in clinical use. Pio
neering physicians have been introducing "foreign" lysosomal enzymes
into patients for several years—not in aging, but in the lysosomal storage
diseases.
Cleaning Out the Drain
Lysosomal storage diseases, the syndromes that we now know to be the re
sult of mutations in genes that code for our normal complement of lysoso
mal enzymes, had been known for decades before researchers figured out
what was causing them. Once their origins became clear, however, a way to
treat most LSDs became apparent: enzyme replacement therapy (ERT—
don't confuse the abbreviation with estrogen replacement therapy). In indi
viduals lacking an enzyme for some common metabolic waste, undegraded
cellular waste products build up within the lysosome (and also outside it in
the main body of the cell), and cellular dysfunction inevitably results.
Therefore, it was reasoned, if the right enzyme could be delivered to the
lysosome, the cellular recycling center would return to normal function, the
piled-up garbage would be broken down, cells would return to health, and
victims would be able to lead normal lives.
After a few decades of work, victims of three of the most common
LSDs are now being successfully treated with such therapies. There are, for
instance, about four thousand people now living normal lives despite hav
ing Gaucher's disease, thanks to regular injections of the lysosomal enzyme
that their cells are unable to produce for themselves. The drug develop
ment process has been reasonably clear, although technically challenging.
In one disease after another, scientists have identified the enzyme whose
absence causes the disorder; modified it in various ways to allow it to be in
jected, taken up by cells, and delivered to the patient's lysosome, where

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 127
they function exactly like the same enzyme does in the rest of us when it is
produced by our own cells; and watched as symptoms have disappeared,
lives have been extended, and victims have been enabled to live the life that
the rest of us take for granted.
Of course, the same fundamental problem faces all of us in the case of
diseases of long-term lysosomal failure: we will all ultimately suffer from
age-related "lysosomal storage diseases" (such as age-related neurodegener
ative disease, macular degeneration, and atherosclerosis), even though only
a tiny proportion of the population is stricken with the currently recog
nized congenital ones (Gaucher's disease and the like). While the exact ori
gins of the two kinds of LSDs are different (rare genetic mutations in genes
for lysosomal hydrolases that are otherwise part of the species' standard
evolutionary legacy in the congenital LSDs, versus never having evolved
the enzymes needed to break down neurofibrillary tangles, A2E, etc., in the
age-related diseases), the molecular natures of both congenital and age-
related LSDs are essentially the same—and as anti-aging bioengineers, that
is quite sufficient to let us do our job, which is to clean out the accumulat
ing molecular damage. To arrive at that destination, we will need to address
a series of specific challenges. Fortunately, in all cases we have options
available with which we already have experience, or for which the solutions
are clearly in sight and under development by researchers in other fields of
biomedicine.
First Challenge: Identifying Suitable Enzymes
Our "grave robber" raid on Midsummer Common proved that the en
zymes exist to degrade the highly cross-linked remains of mitochondria—
believed to be the single largest contributor to lipofuscin. However, we still
don't know exactly what enzyme or series of enzymes is doing the job.
Moreover, enzymes that degrade this synthetic lipofuscin will not be enough:
we also need to identify other enzymes that will deal with wastes clogging
up lysosomes in a variety of tissues and associated with various disease
states.
This doesn't necessarily mean enzymes that will degrade any known
aggregate, such as neurofibrillary tangles. As we discussed above, the messes
that we see are not necessarily the ones causing the problems: they may be
the gunsmoke rather than the gun itself. For instance, it's possible that
some other junk is actually responsible for backing up the system, and thus

128 E N D I N G A G I N G
that the aggregates that pile up like so much trash in the street are simply
what results when the lysosome can't keep up with its normal load. In fact,
the problem might not even be the presence of undegradable materials: in
several diseases, some researchers have presented evidence to suggest that
the problem is a substance (for instance, A2E in macular degeneration) that
directly inhibits the activity of the pump responsible for keeping lysosomes
acidic enough for their enzymes to work.
Fortunately, we again don't need to trouble ourselves with this. Once
again, our job is not to tease apart the minutiae of metabolism, but to clean
up age-related damage. To do that, we can follow the example of the biore
mediation experts: throw enzymes at the problem until the problem is
solved (normal lysosomal function is restored), and then identify the en
zyme that did it. That's a simple task in principle—but when the bioreme
diation field first got going thirty years ago, it was a long slog to actually
narrow down which of hundreds of enzymes in a strain of microorganisms
were responsible for breaking down the wastes of interest.
One of my reasons for optimism, therefore, is the fact that so much
progress was made in those days—and today, we have much more sophisti
cated molecular tools available to do the job. One is a method called mo
lecular fingerprinting, but that term is a little bit misleading. It suggests
a process of finding a clear, unique identifier of an individual—like a
fingerprint—and then finding the individual that bears that same identifier.
Instead, molecular fingerprinting is based on the fact that the members of a
closely knit family of organisms tend to carry genes with broadly similar se
quences, and that (similarly) genes for a range of enzymes with broadly
similar functions within such a community also tend to have a similar
stretches of code.
This allows us to winnow our way down to the genes (and, therefore,
enzymes) of interest from either of two angles. One option is to focus on a
class of enzymes for whose encoding genes we are searching (in this case,
hydrolase enzymes), and then to look for genes that match the overall pat
tern and are expressed in large amounts when the parent organism is feast
ing on the contents of the dysfunctional lysosomes. The other option is to
identify, within a community of organisms, which specific ones are thriving
best when only given those contents as nourishment—and which therefore
carry the genes encoding those enzymes most effectively tearing their
contents down.
Another powerful tool at our disposal is DNA microarrays, or gene
chips. These are tools that identify, in real time, which genes in an organism

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 1 2 9
are being actively expressed at a given moment. So, if we can isolate strains
that are doing well on a diet of lysosomal detritus, we can sequence their
genetic libraries, and then test which of those genes are being used inten
sively when they are feasting on the stuff.
We can also use techniques that allow us to "knock out" (simply re
move) specific genes in such strains, and then retest them. When we knock
out a given gene in a strain of microbe and see that the mutants starve on a
diet that was previously their version of a gourmand's wet dream, we can in
fer that the gene in question encodes a protein that is critical to the se
quence of processes involved in digesting such materials. We can then
identify these genes, and see whether the enzymes they encode are the cru
cial ones that will fill in the weak spot in our human hydrolytic arsenal.
Second Challenge: Getting Them to the Cells
Once we have enzymes in hand that will do the job, we'll have to find ways
of getting them into the cells that need them. Not every cell type will be con
fronted with the same kinds of waste: as we've seen above, particular disease
states are characterized by specific aggregated waste products, and (as is es
pecially likely in the case of A2E in macular degeneration) this is the result
of the particular metabolic pathways that produces them. How much we
have to do to address this challenge will depend on exactly how we're going
to get them into the body at all—for which there are again multiple options.
Right now, for instance, doctors treat L S D patients by intravenously in
jecting modified forms of their missing lysosomal enzyme. Of course, the
enzyme does the patient no good when it's just floating around in the
bloodstream, and it might even do some harm if it were active (since it
might start attacking functional proteins), so the enzymes are tweaked in
ways to ensure that they go where they're needed. First, they are targeted to
the right cells. In Gaucher's disease, for instance, macrophages are espe
cially vulnerable to the lack of the enzyme that causes the disease. There
fore, the enzyme is hitched to targeting molecules that are already recognised
by macrophages as passports to entry. The same trick might, therefore, be
used to target enzymes needed to clear away the substances that cause
macrophage lysosomes to fail in atherosclerosis.
This method has the advantage of being relatively simple to implement
in the short term, and indeed of already being in use for a recognized dis
ease (so that we have a large body of practical, clinical experience with the

130 E N D I N G A G I N G
basic technique on which to draw). It does, however, face a variety of limi
tations. Most notably, there's a big difficulty in using it to move enzymes
past the protective blood-brain barrier, which is a highly effective shield de
signed to guard your brain against exposure to the many potentially toxic
substances floating around in your blood. Obviously, injecting enzymes that
cannot reach the brain will seriously limit their benefits—and represents an
almost complete barrier against their use for age-related neurodegenerative
disease. Even today, some Gaucher's patients develop neurological compli
cations as a result of their enzyme deficiency, and injected hydrolases are of
little help to such patients.
Fortunately, scientists are making progress in coming up with ways to
move proteins across the blood-brain barrier—and in the future, we can ex
pect to have much more powerful delivery systems. In the relatively short
term, we should be able to develop a form of cell therapy, involving seeding
the patient with cells that produce the needed enzyme and secrete it into the
bloodstream or into the fluid bathing surrounding cells. 1 7 This would there
fore act like a biological version of the nicotine patch, providing a continuous
dose of the enzyme. This might be extremely useful: right now, LSD victims
rely on regular injections of heroic amounts of their needed enzyme, and it's
possible that the sheer quantity of (several) enzymes required to combat all
types of lysosomal failure due to aging or age-associated disease might make
injection impractical. One reason why so much enzyme is needed is that
some of these enzymes are proteases—that's to say, they break down
proteins—but enzymes are proteins, so proteases in the lysosome actually de
stroy themselves and each other.
And, of course, the ideal would be to modify our own cells using so
matic gene therapy, introducing DNA to instruct the relevant cells to pro
duce the very enzymes that they need to stay healthy. This, and the cell
therapy option too, are still a long way away from the anti-aging clinic—but
again, fortunately, the major hurdles will be tackled first for a variety of
better-recognized diseases, from sickle cell anemia to the severe combined
immunodeficiency disease that creates "bubble babies." Thus, we can ex
pect to ride on their coattails to a certain extent. Indeed, once somatic gene
therapy is available for use in treating relatively common genetic disorders,
it will doubtless be seized upon by LSD researchers as a way to replace
genes for the lysosomal hydrolases missing in their patients. Again, the spe
cific use of gene therapy to provide better solutions for LSD victims will be
a useful source of information and collaboration to develop a gene therapy
version of the LysoSENS project.

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 131
Third Challenge: Getting Them to the Lysosome
This is similar to the second challenge, above: lysosomal enzymes do us no
good—and might conceivably even cause us some problems—if they wind
up (or, in the case of gene therapy, are synthesized) inside the cells where
they're needed but are not then localized in the lysosome where the junk
accumulates and where the acidity is available to let them do their work.
Again, one potential solution is already in use in the LSDs: the use of mol
ecules of the sugar mannose 6-phosphate, which is recognized and taken
up—along with its cargo—by the lysosome.
But again, we are also in the process of learning to hide a few other
cards in our sleeves. We might be able to use a backdoor solution, by turn
ing a targeting system that is currently used to ensure wastes get delivered
to the lysosome into a method of sending enzymes into the heart of the same
system. (This system is called chaperone-mediated autophagy.) Lysosomes
would take the enzyme up just as if it were any of numerous classes of cel
lular detritus, but would instead incorporate a hydrolase capable of pre
serving and restoring its normal functioning.
We might also be able to take advantage of targeting systems already in
use in the organism from which we originally isolated the enzymes in ques
tion. Bioremediation typically uses bacteria as the microorganism of choice
because they digest their food quickly. Fungi, by contrast, are usually viewed
as too slow-acting and slow-growing to provide viable solutions for oil slicks
or contaminated chemical spill sites. But in a slowly accumulating, low-
volume toxic waste problem like age-related lysosomal failure, these issues
would be less of a problem. The advantage of using fungi is that they—like
us, and unlike bacteria—have a lysosome-like structure of their own, called
the vacuole, which shares many of the key characteristics of the human
equivalent (including, for instance, the need for an acidic internal milieu to
work properly). Enzymes taken from such sources, then, might come al
ready equipped with a range of features that would be useful for the
LysoSENS project in humans.
Fourth Challenge: Potential Side Effects
Even once we have ways to deliver useful enzymes to the lysosomes of af
fected cells, we will still face the key challenge of preventing the interven
tion itself from causing us harm. One potential issue is that the enzymes in

132 E N D I N G A G I N G
question might, as suggested earlier, also be active elsewhere than in the
lysosome. One reason to expect that this won't pose a major challenge is
the fact that, as already mentioned a few times, lysosomal enzymes typically
require a very acidic environment to function properly, so they will proba
bly be nearly inactive in the main body of the cell.
We might also, however, further modify the enzyme so that it only be
comes active after it has been taken up by the lysosome. One possible such
modification would be to attach an extended sequence of amino acids that
would prevent the enzyme from being active, but that would be cleaved off
by enzymes present and active in the lysosome, liberating the active form
for duty at its destination. This concept sounds really tricky, but it is already
used by the cell for the safe delivery of some members of the standard hu
man lysosomal enzyme complement, so the technique should be adaptable
without overmuch heroics.
Another potential worry is that the enzyme might cause an immune re
action, just as any "foreign" protein might. But experience with the LSDs
suggests that this will not be as big a problem as one might at first expect.
Remember that, for a person who was born unable to produce the hy-
drolytic enzymes that the rest of us take for granted, these proteins are
every bit as "foreign" as the microbial hydrolases will be to all of us. Nor
mally, we learn to be tolerant of the proteins of our own bodies because our
immune system is exposed to them early on in our prenatal and childhood
development, allowing it to recognize them as "self." Having never been
exposed to such proteins in early life (because without the gene, the pro
tein can't be constructed), LSD victims lack immune tolerance to them.
And in such patients, immune reactions do occur. But, reassuringly, they
are always mild, and they taper off with time. This seems to be because en
zyme replacement therapy delivers enzymes to the lysosome in a way that
does not allow the cell to hack them up and to display them on its cell sur
face, alerting a suspicious immune system to their presence.
Moreover, even if the experience with the newly introduced enzymes is
not the same (for instance, if we find reasons to use gene therapy and
chaperone-mediated autophagy rather than ERT), we aren't necessarily
stuck. Dampening down an excessive immune response is a necessary part
of many medical procedures, from organ transplants to over-the-counter
allergy medications, and we are getting better at it all the time. We might
also eventually be able to produce the protein within the bone marrow, as
has already been done with some lysosomal enzymes; this might also help
to induce tolerance, because of the role of bone marrow cells in immunity.

U P G R A D I N G T H E B I O L O G I C A L I N C I N E R A T O R S 1 3 3
Inside, Outside
As you can see, there are quite a few hurdles to be overcome before we will
be able to use novel hydrolytic enzymes to clear out the junk in our cells,
preventing or reversing many of the most debilitating health problems of
old age. But, as I've shown, perfectly plausible solutions to all of these
problems seem to exist that are either already in use in treating the recog
nized (congenital) LSDs, or else have clear routes to implementation that
are the subject of intense study by researchers the world over. Identify the
enzymes we need, and a first-generation therapy might look much like ERT
for lysosomal storage diseases today: expensive, inconvenient, and limited
in its scope, but lifesaving. And as we go on, we will progressively improve
the therapy, making it more comprehensive and advancing its safety and ef
ficiency in lockstep with the advance of gene therapy and other enabling
technologies that will also be exploitable in LSD treatment.
As in previous cases, the pursuit of this solution will depend on an inter
disciplinary synthesis of research performed in areas that have, ostensibly, lit
tle to do with aging, and original work done by scientists dedicated to the
goal of adapting existing technologies to the novel problems associated with
the aging process. What is clearly needed is to get private and public capital
devoted to the latter half of the equation, which suffers from a serious lack of
investment of dollars and brainpower, and without which the greatest killer
of all in the modern world will continue to cripple, torture, and kill our fellow
human beings in enormous new cohorts every day.
Let me now turn from the junk within our cells to some of the aggre
gated junk coating our cells, exploring how it is harmful, what can be done
about it, and how its threats to your health—and its therapeutic solutions—
are intimately tied up with the lysosomal failure problem that we've been
exploring here.

C u t t i n g F r e e o f t h e C e l l u l a r
S p i d e r W e b s
Our cells—and thus our bodies—are progressively damaged
by protein-derived junk that gathers over the years in the
space between cells. Alzheimer's disease is perhaps the best-
known condition associated with this, but there are others that
are equally fatal. However, there is a way forward for medical
science and our health: recent and very promising research demon
strates that science can turn our own immune systems against
this dangerous material.
In the previous chapter, I talked about the junk that accumulates
inside our cells with age—how it contributes to the biological aging pro
cess, and what can be done to get rid of it. In this chapter, the focus is on
the garbage that accumulates on the outside of our cells and tissues, en
meshing them in webs of damaged proteins, impairing their function, and
contributing to aging and age-related disease.
Most of the junk that we'll be discussing is amyloid of one kind or an
other. When I say "amyloid," of course, almost everyone thinks of beta-
amyloid protein (also called "amyloid beta"), which accumulates as the
waxy "senile plaques" that cluster around the brain cells of people with
Alzheimer's disease. But many other, less-well-known diseases (amyloi-
doses) are also rooted in abnormal protein aggregates of this type. Most
8

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 135
amyloids are cell-snaring chains of molecules that begin their existence as
healthy proteins already present naturally in our blood, or in the fluid
bathing our brains. A wide range of proteins can become amyloids under
the wrong circumstances, including immunoglobulin light chain, a key com
ponent of the antibodies in your immune system; the protein transthyretin,
which is responsible for carrying around thyroid hormones in your blood;
and a small protein (islet amyloid polypeptide, or IAPP—also referred to as
amylin) that helps your body regulate its blood sugar levels in association
with insulin.
What turns these proteins into snares that squeeze the life out of cells
and organs is how they are folded. Misfolded proteins are just what they
sound like: proteins that have become twisted out of their proper configura
tion in ways that cause them to undergo toxic interactions with each other, or
with other constituents of the cell. The ones that cause amyloid diseases con
tain sites within their structure that, if exposed, readily stick to other proteins
of the same sort, causing them to link together one after another in a sinister,
self-assembling daisy chain. These sticky sites are normally kept safely tucked
away within the complex folding of the protein's three-dimensional architec
ture, precisely to prevent such interactions from happening. Misfolding ex
poses such sites, initiating the spinning of a cell-choking web.
Many of the amyloid diseases result from the victims having faulty genes
that produce defective versions of these proteins. In some such disorders,
the underlying mutation introduces fatal flaws into the structure of the pro
tein itself, causing it to open up at inappropriate locations, exposing the crit
ical "sticky" site in its structure. Others involve mutations in enzymes that
normally chop up the protein into functional units as it emerges from the
cell's protein-assembly machinery. These mutations cause the enzymes to
cut too close to the critical site, again unleashing it from the restraining in
fluence of the rest of the protein's normal conformation. Another route to
congenital amyloidosis is errors in "chaperone" proteins whose job it is to
direct the emerging (and potentially amyloidogenic) protein to assume a
safe, nonamyloidogenic final shape.
But in addition to these inherited protein-misfolding diseases, there are
also universal amyloidoses—ones that are the result not of mutations, but
of the fundamental vulnerabilities that proteins face in the course of their
critical jobs in the molecular maelstrom of cellular biochemistry. With free
radicals, sugars (sugars? Oh yes. See Chapter 9) and vibration constantly
acting on them, proteins are bound to get bent out of shape now and again
in ways that open them up to becoming the seed of an amyloid fibril. Once

136 E N D I N G A G I N G
one such protein is formed, it can sometimes twist other proteins out of
shape as it grabs at them, exposing another site and forming the nucleus of
an ever-expanding fibril chain. One example of this happening in fast-
forward is seen in people with kidney failure, when the body ceases to pass
out beta-2-microglobulin in the urine. Beta-2-microglobulin is normally a
perfectly safe protein that helps the body to distinguish its own cells as
"self" from "nonself" cells of bacteria or other organisms. But without reg
ular excretion, levels of this protein begin to climb to abnormal levels, and
they eventually reach so high a concentration that they start to sponta
neously glom together, forming amyloid deposits.
Indeed, Cambridge professor Chris Dobson, who has spent his academic
life looking into protein misfolding diseases, says that "conditions could be
found in which seemingly any protein could form amyloid fibrils [emphasis
mine] . . . although the propensity to form such structures under given cir
cumstances can vary greatly from one protein to another."1 Over time, these
fibrils build up to potentially pathological levels, coiling around our cells and
organs, choking them off like so much bindweed.
Mind-Forg'd Manacles
Most researchers now believe that the horrors of Alzheimer's disease can
mostly be traced to abnormal processing of an otherwise healthy molecule
called amyloid precursor protein (APP). Everyone's brain produces APP,
and it is required for some essential function in our bodies. Ironically, in
fact, properly processed APP actually appears to be required for many of
the key activities of healthy neurons, such as their ability to rewire them
selves in response to new learning and to grow out the branching "electric
cables" (neurites) that allow them to talk with one another.
When things go right, APP is produced in the main body of the cell
and then sent for further processing to alpha-secretase,2 a type of enzyme
called an endoprotease. The result is the creation of two molecules, one of
which remains in the membrane of the neuron's neurites, while the other is
released into the fluid inside the cell. APP cannot form the evil beta-
amyloid when it is processed by alpha-secretase. After this, one of the frag
ments is chopped further, by a distinct enzyme named gamma-secretase.
APP only become dangerous when, instead of being trimmed down by
alpha-secretase, it is mistakenly cut up by a different, but related, enzyme
called b e ta - s ec re ta se . 3 Beta-secretase, like APP, is not a villain: it has a proper

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 137
place in the cellular "factory," as part of another, distinct cellular assembly
line from the one that handles APP. In that assembly line, beta-secretase
makes essential trims in the structure of other proteins that bear some mo
lecular resemblance to APP itself. But if beta-secretase performs the same
action on APP, it chops it in the wrong place. This distorts the protein's
shape and creates a molecule with a totally different action within the cell.
It's as if beta-secretase were an overly helpful laborer who, while cross
ing the factory floor on the way back from lunch, had seen some APP lying
on a stopped assembly line and mistaken it for a part that he or she nor
mally works on. Seeing no alpha-secretase around, and presuming to know
what the half-finished product needs, beta-secretase steps in to do alpha-
secretase a favor by taking care of a little bit of its workload. After giving it
a few whacks of its molecular hammers, beta-secretase tosses the APP
fragment—now subtly misshapen—back onto the line, where it eventually
reaches gamma-secretase. And because gamma-secretase is a busy enzyme,
it's too caught up in its work to notice the change, and proceeds to splice
and dice the distorted APP fragment just as it would if alpha-secretase had
made the proper modifications. Beta-amyloid is the product of this mis
taken sequence—the sequential cleavage by beta- and gamma-secretase
rather than by alpha and gamma.
When processed appropriately, the middle APP component (between
the sites of cleavage of alpha- and gamma-secretase) assumes a shape simi
lar to a stretched-out coiled spring—a conformation called an alpha helix.
But thanks to beta-secretase's molecular meddling (and gamma-secretase's
unwitting cooperation), this fragment loses its normal shape—and, just as
would happen if you were accidentally to cut into a tightly drawn spring
with a pair of wire cutters, the improper slicing of APP causes the fragment
to jump backwards on itself, creating a shape more like a bent hairpin (a
beta-sheet) that gives beta-amyloid the fatal molecular stickiness that char
acterizes amyloid proteins.
Once released by gamma-secretase, individual fragments ( m o n o m e r s )
of beta-amyloid initially float free in the brain. But they soon come into
contact with other monomers, and their "stickiness" causes them to glom
together into larger—but at this stage still free-floating—units called
oligomers. These fibrous strands, in turn, get stuck to one another to form
still longer strands, which eventually grow so large and complex that they
can't stay dissolved in the brain's fluids, but precipitate out into the spaces
between neurons to form the notorious plaques. Under a microscope, these
mind-snaring webs can be seen extending to the neurons' caretaking support

138 E N D I N G A G I N G
staff (the glial cells), and down to the neurites (the wiring system that I
mentioned earlier).
Some people produce unusually large amounts of beta-amyloid be
cause they have inherited mutations that either cause their bodies to pro
duce too much of APP itself (thus increasing the odds that the problematic
enzymes will come across molecules of it and mistakenly hew their struc
ture), or else encode defective secretase enzymes that are not so good at do
ing their selective jobs as the more common varieties. But because everyone
has both APP and the enzymes that can sometimes turn it into beta-
amyloid, we all produce beta-amyloid, and given a constant output of the
stuff, some fraction of that amyloid precursor is bound to get snipped in
the wrong way now and again. Once that happens, it's only a matter of time
before enough of it builds up to form Alzheimer's-type plaques-—and in
deed, all of us have at least some plaque in our brains by the time we reach
late middle age.
Thus, like other aging damage, beta-amyloid plaques simply accumu
late over time, and it's reasonable to think that neurological impairment oc
curs when a critical threshold is reached. This is probably why most cases
of Alzheimer's disease are not inherited, but instead occur sporadically in
the population: the underlying biochemistry is just part of the kind of or
ganisms we are, living in the kind of universe that we do. Lifestyle risk fac
tors and most genetic predispositions merely determine how early on in our
lives the process begins to impair our intellects and identities. 4 This is also
why, apart from a very small number of inherited, early-onset cases, almost
no one in early middle age or younger gets Alzheimer's . . . and why the
prevalence of the disease doubles every five years beyond age sixty-five, so
that victims pile up with age like the grains of rice on the Emperor's chess
board in the old fable. Our brains are slowly being enmeshed in beta-
amyloid plaques—it's just a matter of when we reach the threshold beyond
which our brains can't keep up sufficient function to carry on the lives and
identities that we have spent so many years creating. Barring some radical
new therapy, each and every one of us will be struck down by Alzheimer's
dementia if something else doesn't kill us first.
"o Captive, Bound and Double-Ironed . . ."
Beta-amyloid also causes brain damage and death in many people who
never develop Alzheimer's disease. This is because, in addition to building

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 139
up in the neurons of the brain, beta-amyloid also clings to the interior sur
faces of its blood vessels. The resulting condition, called cerebral amyloid
angiopathy (CAA), is a crusting-up of these pipelines of oxygen and nutri
ents, weakening them and reducing their ability to flex in response to the
surging flow of the pulse. This leaves them vulnerable to bursting open in a
bleeding stroke.
CAA is certainly more common in people with Alzheimer's (about a
quarter of all patients have it as a complication), but as we age it becomes
an increasingly serious issue in people not struck by the latter disease. Just
5 percent of us have CAA in our seventies, but after the age of ninety over
half of us are suffering from the disease, and it is responsible for about 15
percent of all bleeding strokes in people over the age of sixty.
But there's more: beta-amyloid is just one mangled protein among
many. Less well known, and less recognized as causes of death and disabil
ity, are a variety of other age-related amyloidoses that also don't seem to be
related to an inherently malformed protein, but to the healthy version being
damaged in the rough-and-tumble of its biochemical environment. Senile
cardiac amyloidosis is one example. As you might guess, this disorder is
most clearly characterized by amyloid fibrils building up in the heart, al
though it damages the lung, liver, and kidneys, too. This buildup interferes
with the regular beating of the heart, and can cause heart failure. The fibrils
are made up of transthyretin—the rickshaw driver for thyroid hormones
that I mentioned earlier on—and while it can arise from a mutated version
of the protein, it can also result (at a slower rate) from damage to the form
that most of us carry.
As the name implies, senile cardiac amyloidosis is a strongly age-related
disease—first showing up in people over the age of seventy, and found at
pathological levels in about a quarter of people over ninety. As in the case
of Alzheimer's disease, if we all lived long enough without something else
killing us first, each of us would wind up with the lives squeezed from our
hearts by this form of aging damage. The disease is known to be a common
contributing cause of death in the "oldest old," such that about half of
people over ninety years old have diagnosable senile cardiac amyloidosis at
autopsy.
Much earlier on in life, nearly everyone gets some degree of amyloido
sis of the aorta, the main blood vessel leading out of the heart. Two differ
ent proteins are involved, one of which builds up in the innermost layer of
the aorta in an astounding 97 percent of people over the age of fifty, while
the other accumulates deeper into the middle of the vessel wall in about a

140 E N D I N G A G I N G
third of these cases. This amyloidosis is not currently recognized as a cause
of specific pathology or death, but again it seems that this is only a matter
of stepping over a fatal threshold that we don't reach in a normal lifespan
today because we die of other things first.
Add them all up, and amyloid deposits of any of several misfolded pro
teins in the heart are significant contributors to death in the elderly, causing
abnormal heartbeat, weakening of the muscle of the heart, "blackouts" of
the electrical activity that keeps it beating, and heart failure.
And those are just deposits of the cardiovascular system. Every one of
us becomes riddled with microscopic amyloid deposits across multiple tis
sues in the body by the time we hit our eighties. Its toll isn't widely appre
ciated because the very old are autopsied so rarely, and you just can't see
the deposits without opening a body up. This lack of curiosity about death
in the very old of today is just another example of our routine acceptance of
the massive toll of aging processes in people who have enjoyed only—yes,
only—a few score years of life.
Moreover, while the evidence is still preliminary, amyloidosis of one or
gan system or another appears to become an increasingly critical factor in
the snuffing-out of people at the extremes of the current "natural" longevity
range. Some of this evidence comes from Japan, where the presence of a
few centenarian "hotspots" has made it an exception to the widespread
pattern of voluntary ignorance about what kills the oldest old. An autopsy
study carried out at Aichi Medical Center in Japan from 1989 to 1995
found brain-wide CAA in 16 of their 19 centenarian patients. 5 Unfortu
nately, this study was restricted to the central nervous system, so it did not
provide any information on what other amyloid diseases might have rid
dled these long-lived humans' bodies or to what extent such diseases may
have contributed to their deaths. 6
Even more suggestive is the early evidence coming out of the important
effort by the newly launched Supercentenarian Research Foundation (SRF) 7
to autopsy as many "supercentenarians" (the extremely rare people who
live beyond the age of 110) as can be identified and convinced to donate
their remains to science after their deaths. Of the six who have thus far
been examined, four were felled by some form of amyloid disease (the other
two deaths were cancer victims). 8
Again, we don't yet know what the pathological consequences of many
of these deposits may be, but it seems awfully likely that they are doing us
harm—so by the engineer's definition, they're aging damage, since they're
not found in the young. Hence, you can bet your life that I want to clean

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 141
them up along with the ones that have already been exposed as culprits in
specific age-related diseases.
Alzheimer's, Amyloids, Aging
In a perverse way, then, the fact that Alzheimer's is such a widespread and
obviously terrible disease has aided the cause of general anti-aging biomed-
icine. The attention to this specific age-related curse—together with the
widespread professional and public belief that amyloid beta deposits are a
major factor in its development and progression—has driven scientists to
attack this particular form of amyloid as a therapeutic target in its own
right, and that work has opened up the strong possibility that a similar
strategy can form the basis of foreseeable therapies for amyloid-type extra
cellular damage generally. As with other cases that we've discussed in pre
vious chapters, the existence of a recognized disease that is caused by aging
damage has "legitimized" research into ways to clean it up—and, as this re
search bears fruits in new cures for these diseases, anti-aging biotech will
be able to hitch a ride to develop treatments for aging damage itself.
Alzheimer's is an especially good example of this phenomenon, be
cause it is both so utterly fearsome and so common in our parents and
grandparents (in contrast to rare and rapidly fatal disorders like the mito-
chondriopathies or the lysosomal storage diseases). As the sheer number of
Alzheimer's victims explodes as the population's biological age creeps up
ward, victims' families and loved ones have organized politically. There are
now thousands of people in the United States and elsewhere who are de
manding—and getting—enormous government investments of intellectual
and financial capital into the quest for a cure. (Indeed, for better and for
worse, Alzheimer's research now consumes over half of the National Insti
tute on Aging's budget.)
Once the view that beta-amyloid was the key to the disease became
dominant, Alzheimer's specialists (possibly because they were not
biogerontologists?) began thinking about this particular form of extracellu
lar junk along the same damage-reversal lines that underlie the engineering
approach to age-related damage generally. All that currently available treat
ments for Alzheimer's disease can do is improve the symptoms of the disease:
sadly, no existing therapy has the power to check the ongoing degeneration
of the brain itself (see sidebar, "Alzheimer's treatment today"). This does
mean that users of these drugs are better off at any given point than they

142 E N D I N G A G I N G
would be without them, but the underlying progression of the disease con
tinues unabated with every passing day. Functionally, even modern
Alzheimer's drugs perform the way ibuprofen and antidepressant medica
tions do for diabetics—in providing merely superficial relief from the nerve
pain that often accompanies the disease. While the pain may indeed dimin
ish, diabetics' nerves themselves continue to be savaged by the "carameliza-
tion" chemistry of that disease. (See Chapter 9 for SENS's main answer to
late-onset diabetes.)
A L Z H E I M E R ' S T R E A T M E N T TODAY
Currently, the most widely used treatments for Alzheimer's dis
ease are the cholinesterase inhibitor drugs, such as donepezil
(Aricept), rivastigmine (Exelon), and the herb galantamine
(Reminyl/Razadyne), which attempt to bolster brain functioning
by boosting levels of some of the signalling molecules involved in
some of the flagging aspects of memory. Of course, such a treat
ment is purely palliative, with no effect on the underlying disease
process. A recent study9 suggests that these drugs are even less ef
fective than is implied by their inability to prevent the brain-
ravaging effects of the disease: It appears that while the drugs
boost scores on standardized tests of some aspects of brain func
tion, they have no effects on the kind of real-world functionality
whose loss forces families to institutionalize victims.
There was hope, for a while, that a more recently introduced
drug called memantine (Namenda) would at least slow down
the progress of Alzheimer's disease, by shielding neurons against
the damaging effects of another signalling molecule (glutamate)
that can kill brain cells when present in excess. A recent trial1 0 sug
gests that this isn't so. The study compared people who had al
ready been on memantine in a six-month placebo-controlled
trial, and who were then allowed to continue taking it for an addi
tional six months, to people in the same trial who had originally
been taking the placebo but who were given the real deal for
those subsequent six months. If memantine were really slowing
down the underlying disease process, you'd expect that people
who started on the drug earlier would have been in better shape

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 1 4 3
than those who'd had to wait through the first six months on the
sugar pills, because they would not have been suffering the full ef
fects of brain degeneration for the first six months and thus would
have more intact brains later on. But instead, it was found that the
patients who started taking the drug later quickly caught up, in
terms of their improvement over their baseline condition, with
those who had been getting it all along. This suggests that me-
mantine's effects are only on the immediate symptoms of the dis
ease.
That's good news, in a sense, for those who start taking me
mantine at a more advanced disease state, because it means
that they haven't lost anything by waiting to get started. However,
the bad news is that no one taking memantine can expect that it
will actually stop their minds slowly dying from the tangled mess in
their brains.
It's actually not clear that blocking glutamate's effects on neu
rons would be an entirely good thing in any case. As with so many
things in the finely tuned network of metabolic pathways, gluta-
mate is a molecule with two faces. While it can stimulate brain cells
to death when it's present in excess, it's also a key chemical sig
nalling molecule in the brain, required for the normal storage and
retrieval of memories. This suggests the possibility that memantine
may cause problems in the laying down of new memories, even as
it preserves the brain cells that store the old ones. There is no direct
evidence of such an effect yet, but it still isn't clear that the drug
helps much, either: While this trial did seem to show a benefit from
memantine, there were no statistically significant benefits com
pared to sugar pills in the two other major trials of the drug.
In any case, even a drug that could slow down the rate at
which brain cells are lost would be unable to prevent— let alone
reverse—the degeneration of the brain, since the underlying
damage that has already occurred is left unrepaired.
As the evidence supporting a central role for beta-amyloid in the develop
ment and pathology of Alzheimer's built up, a new hope emerged. Scientists
began talking seriously about the idea that, by making beta-amyloid itself
the target for new medical interventions, they would be able to develop new

144 E N D I N G A G I N G
treatments that would treat the disease instead of merely providing crutches
to a crippled—and rapidly deteriorating—mind. Once researchers had the
tools they needed, in the form of engineered mice whose brains produced
variations on human beta-amyloid that led to the formation of brain plaques
and dysfunctions of brain and memory, they could start work on testing
therapies that would target beta-amyloid directly.
New Targets, Old Rifles
But simply believing that that beta-amyloid is the main villain in the
Alzheimer's story doesn't tell you what to do about it. Thus, it's no surprise
that academic labs and pharmaceutical companies around the world have
been working on quite a variety of anti-amyloid strategies, each hoping to
make a Nobel-quality breakthrough or market a blockbuster new drug
with a desperate—and ominously, inexorably expanding—"target market."
Predictably, however, when scientists began thinking about how to
tackle the beta-amyloid plaque problem, many of them first turned to clas
sically preventative strategies typical of the old-style gerontological ap
proach to aging. Recall that beta-amyloid, like other amyloid proteins, is
formed from an essentially healthy protein—amyloid precursor protein
(APP). That protein is found in long strands woven through brain cell
membranes, and while its exact function is unknown, it's at least harmless
as long as it remains intact and in place. But occasionally, the beta-secretase
enzyme mistakenly latches onto the APP protein and chops it at an unin
tended place; gamma-secretase innocently follows suit, not recognising the
fatal flaw in the misprocessed APP; and beta-amyloid—with its exposed,
sticky binding sites—is released to wreak its havoc in the brain.
With this in mind, one of the first ideas for an anti-amyloid therapy was
to create drugs that would dampen down the activity of these enzymes,
thereby cutting down the production of beta-amyloid. This in turn would
reduce plaque formation, and thus either slow down or prevent the emer
gence of the disease.
First out of the gate was a drug that interfered with gamma-secretase ac
tivity. Animal studies showed that even a single dose of the drug could reduce
levels of soluble, pre-plaque beta-amyloid in both the brain and the plasma,
and it was duly moved through the development pipeline into the "Phase II"
trials that are designed to give preliminary evidence of a drug's efficacy and
safety in a moderate-sized population of people with the disease.

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 1 4 5
That was in 2001. I'm writing this in early 2007, and to date there has
been total silence about the results of the trials of this first gamma-secretase
inhibitor. We may never know what happened, but we may be able to
guess. Even as the drug was being tried in humans, greater understanding
of the role of gamma-secretase in the body emerged. A question that had
long hung over the enzyme-inhibition approach was what, exactly, the en
zyme was supposed to be doing in the body.
Many harmful mutations lurk in isolated pockets of the human family,
but we all have gamma-secretase in our brains—and, as I discussed in Chap
ter 3, evolution does not design us to suffer horrible diseases. Although
gamma-secretase has the unfortunate long-term side effect of beta-amyloid
production, scientists always had in the back of their minds the acknowl
edgement that it had to serve some useful purpose, too. And, sure enough,
researchers discovered while the trial was still progressing that gamma-
secretase operates on several proteins in the body—including Notch receptor
1 (NOTCH1), a protein with critical functions. By this I mean really critical:
They include the activation of stem cells that renew damaged muscle tissue,
the growth of new blood vessels, and the maturation of some kinds of im
mune cells.
So, what happens to these important biological processes, when you
start interfering with an enzyme that's needed to keep them going? Animal
models, using either a "knockout" of the gamma-secretase gene or an alter
native gamma-secretase inhibitor drug, showed that dampening down the
enzyme clearly prevented immune cells from developing in both the bone
and the thymus, reducing the number of these cells and causing pathology
in the gut. It seems highly plausible that the reason for the stony silence on
the human trials is a similar profile of side effects.
However, some researchers are still chasing after therapies based on
the same basic strategy. In 2002, researchers at Eli Lilly presented animal
data showing that LY450139, the code name for a new gamma-secretase in
hibitor, lowered beta-amyloid levels in Alzheimer's mice without interfer
ing with NOTCH1. As this chapter was being drafted in 2006, the results
of an early human trial in seventy Alzheimer's patients came in, showing
that the drug reduced levels of beta-amyloid by 38 percent without appar
ently causing any serious side effects. By April 2006, the company had part
nered with several universities and hospitals and was gearing up to perform
a larger clinical trial to see if it could actually affect the disease. Elan Phar
maceuticals is also continuing to research a gamma-secretase inhibitor.
But deactivating NOTCH1 is far from the only potential concern with

146 E N D I N G A G I N G
these drugs. Remember, gamma-secretase is also an essential partner in the
normal, non-amyloidogenic metabolism of APP into products that appear
to be essential to the functioning of neurons. It's unlikely that you can get
away with ratcheting its normal activity down by force with no negative im
pact on the very brain function that researchers are desperately trying to
preserve. It may just take longer than the brief six weeks over which the
new drug's first safety test was performed. The new trials are just starting to
recruit patients at this writing; we'll see how they fare in the clinic, and
keep our fingers crossed for the people taking them.
Other scientists are pursuing a slightly different version of the same ba
sic strategy. Some are developing drugs that inhibit beta-secretase, or that
turn up the activity of alpha-secretase, with whose normal processing of
APP beta-secretase interferes. Because such drugs are still in the early stages
of development, we don't yet know what their side effects will be, but again
it seems unlikely that the activity of an enzyme produced normally through
out the body—and especially in the brain—can be altered without cost. For
instance, one specter that already looms over the beta-secretase inhibitors
(based on animal studies) is that they may make users more vulnerable to
some psychological disorders. While very young animals with their beta-
secretase genes knocked out seem to be physiologically more or less normal,
they are timid and don't like to explore their environment, and seem to run
through serotonin (the chemical messenger whose metabolism is modulated
by drugs like Prozac) abnormally quickly.
Freeing the P r i s o n e r s . . . or Letting Loose the Inmates?
Other scientists are pursuing an alternative approach that is, superficially,
more in line with the engineering principles that I have advocated.
Namely: to ignore the formation of beta-amyloid itself, and instead focus
in on the process whereby beta-amyloid becomes aggregated into neuron-
enmeshing plaques. A surprisingly large number of drugs and even herbal
concentrates—from extracts of the spice turmeric (used in curries) to cus
tom drugs with the highly marketable moniker beta-breakers—either inter
fere with the glomming-together of beta-amyloid into plaque fibrils, or
even break existing aggregates a p a r t . . . in a test tube. Most of these com
pounds never worked out once they got beyond the petri dish and into a liv
ing, breathing organism, but a few have been shown to reduce the plaque
burden in animals genetically engineered to produce large amounts of

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 147
beta-amyloid, and a few of those are now going into clinical trials in people
with Alzheimer's disease.
Here, however, we once again run into the problem of over-reliance on
our hypotheses about what biochemical processes "cause" the disease. In the
other amyloidoses, the connection between fibril and pathology is pretty ob
vious. Indeed, you can dramatically extend life expectancy in patients with
several kinds of amyloidosis by "simply" replacing the amyloid-strangled or
gan with a transplanted one free of fibrils.
But the reality is that, despite a decade of intense research into the
"amyloid hypothesis" of Alzheimer's disease, we still don't understand the
mysterious metabolic underpinnings of the disease. Even among the major
ity of researchers who are convinced that beta-amyloid is the key to
Alzheimer's, the consensus around the detailed mechanistic role of the pro
tein in the disease is as shallow as it is wide. Controversy continues to rage
about what exactly links it—the protein itself, and/or the plaques that form
from it—to the decay of brain and body that we see in its victims. Thus, the
premise that beta-amyloid plaques cause Alzheimer's—or link the underly
ing metabolic defect(s) to disease—is still not enough in itself to tell you
what should be done about them.
If anything, the balance of evidence is that it may not be the plaques
themselves that impair neurological functioning the most, but the soluble
beta-amyloid oligomers: short chains, made up of just a few single beta-
amyloid molecules ("monomers") linked together in the same way that
plaque fibrils are, but whose small size allows them to remain dissolved in
the fluid bathing the cells of the brain rather than precipitating out into de
posits. In cultured cells and in experimental animals, beta-amyloid oligomers
derived from human nerve cells clearly disrupt neuronal function and in
terfere with normal memory, in ways that are not observed with either the
beta-amyloid plaques that they form or the lone "links" of beta-amyloid
monomers of which they are composed. After being microinjected with
human-derived oligomers, rats become confused and forgetful. Beta-
amyloid monomers don't have the same effect, and while giving animals or
brain cell cultures chemicals that clear all forms of beta-amyloid (oligomers
and monomers) out of the fluid bathing the neurons prevents the negative
effects of exposure to a mixture of oligomers and monomers, an enzyme
that selectively degrades free beta-amyloid monomers while leaving the
oligomers intact provides no protection.
Likewise, the rats' memory function is largely recovered a day after in
troduction of the oligomers, when they have been cleared out by the animals'

148 E N D I N G A G I N G
natural protective systems. This again suggests that the oligomers, rather
than the plaques that they form, are the guilty parties in cognitive function.
And indeed, preliminary studies suggest that the oligomers act as the mo
lecular equivalent of dust in the eyes of neurons, interfering with their abil
ity to receive signals from other neurons and pass the signal on to their
internal machinery. Adding to the uncertainty, one group have reported a
mouse model in which abundant plaques form but no neurological deficits
result.
Another series of experiments strongly suggests that the plaques are
the result, rather than the cause, of the widespread death of neurons in the
Alzheimer's brain. 1 1 This study looked in detail at the location of both sol
uble amyloid species and plaques in the brains of people who had died of
the disease. Soluble amyloid was found accumulated in still-intact cells
within their lysosomes (the cellular garbage disposal units discussed exten
sively in the last chapter). Areas with very high levels of amyloid showed ev
idence of the rupturing of neurons, with beta-amyloid and the lysosomes'
digestive enzymes dispersed outward from a central locus in a pattern that
suggests nothing so much as a bomb blast. And wherever plaques were
found, the researchers also found the remnants of a destroyed neuron's cell
nucleus in the debris.
The strong suggestion was that the cells had been trying to dispose of
the toxic amyloid oligomers by dumping them into the lysosome. Remem
ber, again from the last chapter, that the Alzheimer's brain shows clear evi
dence of dysfunction of these organelles. Moreover, a lot of the beta-amyloid
found inside brain cells is actually clustered in and around lysosomes, and
the aggregates themselves are in fact naturally taken up and degraded in mi
croglia (the immune cells of the brain), but at a rate that is too slow to keep
up with plaque formation.1 2
This suggests that the burden of amyloid may eventually overcome the
capacity of lysosomes to dispose of it, leading to the death-spiral of dys
function outlined previously—and eventually to the death and rupture of
the cell, during which the beta-amyloid and lysosomal enzymes spew forth
from the dying neuron, creating plaque deposits like so much slag from a
bombed-out building.
However, the innocence of plaques in Alzheimer's disease can't be as
serted any more confidently than their guilt. The beta-breakers that have
been shown to be effective in animal models (as opposed to just test tubes)
do restore memory function as they break up the plaques. And again, a
mere glance at the snarling webs of amyloid-enmeshed cells of the

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 1 4 9
Alzheimer's brain defies the observer to accept the notion that the plaques
are harmless.
One theory that may reconcile these conflicting conclusions is the idea
that, at least in the short term, the plaques' most damaging effect on the
brain is to act as reservoirs for the beta-amyloid oligomers. You may have
done simple experiments involving solutions in junior high-school science
class, in which a substance is dissolved at very high concentrations in a glass
of water. Eventually, levels reach so high a concentration that the water
can't hold any more, and a crystal precipitates out of the solution.
But the teacher may have explained to you—or even demonstrated—
that the crystal is not a static entity, but exists in a state of "dynamic equi
librium," with some dissolved material continually precipitating out onto
its surface to build it up even as some of its existing surface molecules are
continually dissolving out into solution. The volume of crystal will remain
constant at a given solution concentration, as the rate of dissolution out of
the crystal remains equal to the rate of precipitation into it. But of course, if
you add more water or dissolved material into the solution, the equilibrium
shifts accordingly.
The same thing, more or less, may be happening with amyloid plaques.
As the concentration of beta-amyloid monomers and oligomers increases,
they aggregate into plaques, which keeps the level of dissolved oligomers
lower than it might otherwise be, thus effectively reducing their potential
toxicity to local neurons. But when the level of oligomers dissolved into the
fluid is reduced, the aggregated oligomers dissolve back into solution,
maintaining their signal-jamming influence in a toxic steady state.
This potentially creates a real dilemma for therapies designed to deal
with beta-amyloid. Using drugs like beta-breakers to prevent amyloid fib
rils from forming, or to break existing plaques apart, would free up beta-
amyloid oligomers that would otherwise have been sequestered in the
plaque mass—which would actually expose neurons to more oligomeric in
terference than just leaving the plaques alone or even letting them build up.
In fact, both things would happen at once—and this could well have
parallels in the Alzheimer's brain. It seems very likely to me that beta-
amyloid plaques play—or eventually would play—a similar role in the
brain of the Alzheimer's patient to what, say, transthyretin deposits do in
the hearts of people struck by senile cardiac amyloidosis: you can't look at
the mess of a victim's brain and not suspect that the plaques have been
choking neurons to death. While the evidence is strong that any inter
vention which results in an increase in neuronal exposure to soluble

150 E N D I N G A G I N G
oligomers of beta-amyloid can be expected to damage the brain, simply
leaving the plaques to grow larger and larger as the diseased neurons con
tinue to produce beta-amyloid (and/or to rupture and spill unprocessed
beta-amyloid out of their lysosomes) surely sets the brain up for an even
greater problem further down the road.
These intriguing theoretical questions are the kind that excites the cu
rious minds of scientists; moreover, answering those questions seems to
many such scientists to be the natural way of identifying and developing
treatment for this terrible disease. As for other kinds of aging damage, how
ever, I believe that this assumption is flawed. Let me say it again: We do not
need to understand in detail how aging damage accumulates, or by what
mechanism it wreaks its havoc, in order to undo that damage. However ex
citing an intellectual challenge it may be to sort out the exact pathway that
leads from the fatal nips and tucks on APP, to beta-amyloid formation, to
plaques, lysosomal dysfunction, cognitive impairment, and neuronal cell
death, the bottom line in terms of the biomedical challenge is that we have
here a material that is clearly accumulating and altering the composition of
our aging and diseased bodies. When I see that, I say: it must go.
You can probably guess, by now, what sort of anti-amyloid therapy I
prefer. While the remaining concerns about the exact role of plaques and
oligomers in the disease process make simply breaking apart the aggregated
beta-amyloid a potentially risky strategy, that doesn't rule out a solution
based on removing the plaques in whole cloth. That would eliminate the
source of the problem, no matter what step in the formation, metabolism,
or aggregation of the constituent material is in fact the key to its toxicity.
Such an intervention would be classic anti-aging engineering as I've envis
aged it—if it could be done.
Fortunately, we should know very soon. A potential solution is already
undergoing clinical trials.
" Immune" from Plaque: The Beta-amyloid Vaccine
As I mentioned earlier, researchers found evidence some time ago that mi
croglial cells—the immune cells of the brain—slowly eat up and digest away
beta-amyloid deposits from nerve cells. Unfortunately, it was clear that the
rate of clearance was not nearly high enough to keep up with the pace of
deposition in Alzheimer's patients. But researchers guessed that this natu
ral defense mechanism could be stimulated to greater throughput. The ob-

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 151
vious way to do this would be as we do for other targets of the immune sys
tem: with a vaccine.
The vision: inject patients with beta-amyloid, and the silent sentinels of
the immune system would be roused up in defense, seeing the protein as a
foreign invader. The same forces that your body marshals against chicken
pox or influenza would be mobilized for an all-out war on the brain-
choking protein, churning out antibodies specific to it and inducing the
brain's microglial cells to go on a search-and-destroy mission against brain
beta-amyloid.
This was an even more exciting idea than it might at first appear, be
cause an anti-beta-amyloid vaccine would be expected to have an even
greater impact against Alzheimer's disease than had the vaccines used
against earlier epidemics. Such vaccines had allowed us nearly to wipe out
diphtheria, polio, and measles in developed countries by preventing new
cases from emerging. The promise of a vaccine targeting beta-amyloid was
that it would actually cure all but the most advanced cases of the disease.
Armies of activated microglia, their appetites for beta-amyloid whetted,
would actually consume and thereby remove the existing beta-amyloid de
posits. Once those choking fibrils were removed, the brain's normal
structure—and thus, function—would be restored. 1 3
Critically, this approach should work no matter what theory of the link be
tween beta-amyloid and memory loss turned out to be correct. (It would not be
the whole solution if other hallmarks of Alzheimer's, such as the intracellular
neurofibrillary tangles, were also contributing to disease progression—but
SENS incorporates solutions to those other aspects too, as you've seen.) The
vaccine cleared out the plaques—and also, it shortly turned out, the more solu
ble oligomers, even inside the nerve cell itself14—not by breaking them apart
into their constituent elements, but by causing immune cells to internalize and
digest them.1 5 Because no soluble beta-amyloid is released by such an ap
proach, there is no risk of doing new damage as levels of the soluble form rise:
beta-amyloid just goes away, clearing neurons of its malign influence no matter
what its exact mode of toxicity might be.
It turned out to be a relatively easy matter to test this concept in ani
mals engineered to develop a version of Alzheimer's disease. While vac
cines against viruses must be carefully modified to ensure that they are
close enough to the real thing to set off the immune system's alarms, but yet
different enough that they don't actually infect people with the disease, no
grand feat of molecular engineering was used in the first test of the concept:
mice were simply injected with aggregated human beta-amyloid.

152 E N D I N G A G I N G
It worked smashingly well from the outset. Plaques quickly regressed
from the mice's brains. The swelling and dysfunction of the neurites
(the branches that transport chemical messengers from one nerve cell to
the next) faded away, leaving healthy, functional units. The dense, inflam
matory overgrowth of supporting cells around the neurons retreated. 1 6
And memory function—evidenced by the animals' abilities to find hidden
platforms in flooded mazes—became more like that of younger, healthy
animals . 1 7 , 1 8
The company coordinating this work, Elan Pharmaceuticals, obtained
results in many different models of engineered mice, each with a different
mutation in the processing of APP. Best of all, the treatment appeared to be
quite safe. Contrary to what had been feared, the immune attack on the
beta-amyloid around the animals' brain cells did not cause collateral dam
age to the fine network of supporting cells onto which the plaques were
glued. Likewise, some had feared that the immune attack on beta-amyloid
would be so aggressive that it would punch holes in the protective shield
that protects the brain from the toxins in the bloodstream, triggering a
flood of foreign substances into the brain from the rest of the body; but lit
tle evidence of such an effect was found. The FDA was so impressed with
the results that it quickly gave Elan the nod to move their vaccine—code-
named AN-1792—into placebo-controlled clinical trials.
Nearly 400 patient volunteers were recruited, of whom 300 received
the amyloid vaccine and 72 were given injections of saline solution as a
placebo control. Their baseline condition was determined, their mental
state and functionality were assessed on a battery of neuropsychiatric and
clinical tests, and a regimen of periodic injections was instituted.
Twelve months later, disaster struck.
A Fire in the Brain
Just a few months after this trial had begun, some patients started exhibit
ing serious side effects. Of more than 300 patients recruited from 28 clini
cal centers across Europe and North America, about one in 15 developed
meningoencephalitis, a life-threatening swelling of the brain, apparently as a
result of an overreaction of the immune system inside the brain itself.19
As soon as the side effect was discovered, the trial was halted, sending
researchers scrambling to try to figure out what had gone wrong. The prob
lem came as a complete shock. The vaccine had been tested in mice with a

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 153
wide range of genetic abnormalities, each leading to Alzheimer's-like
plaque formation from a different defect in the synthesis or metabolism of
APP, and no such side effect had been observed—this despite the fact that
scientists had been much more aggressive in their treatment protocols with
mice than they would dare to be with human patients.
How such a crisis could occur, after such careful preclinical testing, has
been documented extensively in the media and the academic literature, and
I think it's too much of a digression to describe here. The important point is
that researchers rapidly homed in on the problems with the first vaccine—
and, as we shall see, on how those problems can be overcome.
Snatching the Ore from the Smelter 's Ashes
The first trial proved catastrophic for some patients, and it was terminated
before science could assess the full range of effects of the vaccine—
positive and negative. But despite—and in a way, because of—the con
straints imposed by this serious adverse reaction, researchers were
desperate to squeeze every available bit of data that they could from the
trial, to make up for its human and financial cos ts . 2 0 , 2 1 , 2 2 Through careful
sifting of the data, scientists managed to collect some preliminary informa
tion suggesting that, despite the horrors of inflamed brains in a few pa
tients, immunization with beta-amyloid fundamentally does work as a
therapy in humans. And when combined with further animal studies, the
findings also suggested ways to avoid this side effect (and others) in future
vaccines—some of which are either under development, or even in clinical
trials, already.
In the immediate aftermath of the study's sudden termination, infor
mation about the patients was still scattered throughout the twenty-eight
independent clinical centers at which the patients had received treatment.
Preliminary assessments of the readily available information were not
promising: researchers saw little evidence of improvement in the memory
or other cognitive functions of people in the trial. But as the dust settled
and researchers began to collect and analyze volunteers' full medical rec
ords, a new pattern emerged. 2 3 A given vaccine (any vaccine) does not do
the same to all its recipients: some people create a stronger immune re
sponse to it than others. By separating out those participants whose blood
work showed that they had mounted a substantial antibody response to
the injected beta-amyloid by the time the study was shut down (fifty-nine

154 E N D I N G A G I N G
volunteers) from the rest (who had not), scientists were able to show that
those who had responded well to the vaccine had, apparently, fared better.
This finding took time to emerge because it was initially obscured by
the statistics. When the researchers looked individually at each of the cog
nitive tests that volunteers had been administered, they could find no dif
ferences that passed the test of statistical significance. However, an
integrated analysis of the whole battery of tests suggested that people
whose immune systems had responded to the challenge suffered less de
cline during the study period than had people administered the placebo—
a difference that was only beginning to become clear at the twelve-month
mark when the trial was halted. Of particular interest was the fact that the
emerging difference was most apparent for a composite score on the mem
ory tests. And most suggestively, there did seem to be a sort of "dose-
response" effect, with the greatest improvements in scores on overall
memory, immediate and delayed memory, a nine-component memory
score, and possibly a test of "executive function" (that is, higher-order
brain functions involved in governing ourselves in a goal-directed fashion
in the beginning and over time) in those whose antibody responses were
strongest.
These results were all the more impressive considering the likelihood
that most of the active responders were suffering from low-level brain in
flammation and "ministrokes" triggered by the vaccine—a possibility sug
gested not only by autopsy findings and animal studies, but by the fact that
headaches and confusion were reported as side effects so much more often
in subjects who had received vaccination than in those who had received
the placebo. Indeed, studies in breeds of Alzheimer's mice that had beta-
amyloid deposits on their vasculature, and were thus vulnerable to micro-
hemorrhages in response to the vaccine, still showed some cognitive
improvements, despite the direct damage their brains suffered from the
tiny bleeds. If your brain function is being preserved in spite of a quiet,
chronic attack on it by your own immune system, the implication is that
something else about your underlying clinical condition has improved even
more. A vaccine that would clear out beta-amyloid (as this one appears to
do) without causing the inflammatory side effects would therefore be ex
pected to yield a much more robust improvement in the workings of the
mind.
Another interesting finding came when researchers looked at the levels
of the protein tau, which is the major constituent of neurofibrillary tangles,
in the fluids bathing the central nervous system (the cerebrospinal fluid, or

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 155
CSF). Even though they only had about ten subjects in each group with
good enough data to compare, the medical teams did observe that the vac
cine responders had lower levels of tau than did the patients receiving
placebo. While it's very indirect, this might be a sign of reduced rates of
brain cell death, since high levels of tau in these fluids are associated with
the death of neurons in people with the disease.
What about evidence on the intended effects of the vaccine—its ability
to actually clear out beta-amyloid plaques? Unfortunately, it's not yet rou
tine for scientists to look into the brain to see the plaque burden in people's
brains before they've died, although new imaging techniques to do just that
have been developed and are being tested for accuracy now. What can eas
ily be done is to analyze levels of beta-amyloid in the cerebrospinal fluid. In
animal studies, vaccination against beta-amyloid almost always results in an
increase in CSF levels of beta-amyloid—a finding usually interpreted as a
sign that the vaccine is helping the animals to clear the stuff out of their
brains and then transport it elsewhere in the body for disposal. This effect
was not seen in the small number of trial volunteers for whom scientists
had samples that they could compare. But we have another, more direct
source of evidence on the subject: the three vaccine responders who died
over the course of the trial. While one must again be cautious in reading too
much into the results observed in just three people's brains, autopsies of
these volunteers by independent groups did show a dramatic reduction in
levels of plaques in key regions of each of them as compared with control
subjects.
Moreover, pathologists examining these brains found microglia in
close proximity to many of the plaques that remained, suggesting that the
vaccine was working as scientists had always hoped: the activated immune
system had successfully mobilized microglia to clear out the deposits of
beta-amyloid. This conclusion was further bolstered by a study—only com
pleted after the end of the human trial—showing that old Caribbean green
monkeys given beta-amyloid vaccination exhibited huge (66 percent) re
ductions in beta-amyloid levels in the brain, and a complete absence of
plaques, and that the dense tangling-up of neurons' supporting glial cells
usually seen in human Alzheimer's patients was considerably reduced.24
This is an important piece of supporting evidence, because these monkeys
develop some Alzheimer's-type pathology naturally as they age, and are
much closer relatives to us than any mouse. The researchers who reported
this finding are currently working to develop ways to assess the monkeys'
cognitive function.

1 5 6 E N D I N G A G I N G
The Next Beta-amyloid Vaccine
All of this tantalizing, albeit inconclusive, information has once again
erected the banner of beta-amyloid vaccination as a true cure (or at least a
major component of a cure) for Alzheimer's disease. We've learned enough
about both the potential of the vaccine and the reasons for the deadly brain
inflammation to develop a variety of new approaches to the basic strategy,
one or more of which will almost certainly prove effective without putting
its recipients at risk.
The key to designing a safe vaccine is, of course, to avoid enraging the
immune cells that attacked the original subjects' brains in the first trial even
as the microglia were loyally cleaning out the amyloid plaques. There are
several ways that this might be done, and each of them now enjoys support
in animal models of the disease—supporting their efficacy while providing
evidence that they will not have a deadly immune side effect.
One relatively straightforward approach that has already entered clini
cal trials is something called passive vaccination. Unlike a conventional ac
tive vaccine, in which the patient receives the offending agent itself (in this
case, beta-amyloid) in order to signal the immune system to produce its
own antibodies in response, passive vaccination involves directly providing
the very antibodies that are desired, bringing out the immune response that
the same antibodies elicit when they are produced by the body. The advan
tage of this approach is that it would allow scientists to choose which anti
bodies would be circulating throughout a patient's body. These antibodies
might be chosen, or even custom-made, to activate the type of response
that sends the microglia out to break down and digest the amyloid de
posits 2 5 without the risk of eliciting the undesired antivasculature response.
The main disadvantage of this approach is that it would not induce the
kind of semipermanent immune vigilance we've come to expect from vacci
nation against diseases like mumps or polio, but would instead require pa
tients to receive regular reinjections of the antibodies to keep their
amyloid-fighting supplies topped up. But even this apparent inconvenience
has an upside: because the recipient's immune system isn't put into a long-
term state of vigilance against beta-amyloid, physicians can stop treatment of
an individual patient—or even stop an entire trial—at any time in case of side
effects, without the fear that the body will remain in a destabilized
autoimmune-like state of the kind elicited by the original vaccine.
Another approach is to manufacture an active vaccine composed of only
part of the beta-amyloid molecule. When scientists looked at the immune

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 157
response to active vaccination with the whole beta-amyloid molecule, they
found that a mixture of different antibodies was being produced. Only a
small number of those antibodies were of a type that harms the vasculature—
and, significantly, these antibodies were only observed in humans adminis
tered the original vaccine, and not in the mice or monkeys that received the
same vaccine and had not suffered the tragic assault on the brain. These anti
bodies were sensitized to only one segment of the total beta-amyloid protein,
located in the middle of the molecule. By contrast, in mice, monkeys, and hu
mans, most of the antibodies were of types that would mobilize the immune
system against a completely different segment of the beta-amyloid molecule,
located on its far end.
Since it's reasonably clear that the antivasculature response is not only
unnecessary (since it isn't seen in the animal models with active or passive
vaccination, yet these procedures dramatically clear out amyloid and im
prove memory) but is extremely harmful (due to its role in brain inflamma
tion), it seems highly likely that a vaccine based on the above principles will
work fine and be free of the risk of brain inflammation so long as it reacts
only to this key subsection of the protein. Such a vaccine would induce the
desired immune response on an active basis, without sending the body's
immune system on a misguided mission against the very brain in whose de
fense it was aroused.
To date, several vaccines have already been developed based on this
principle. They work by binding the key segment of the beta-amyloid pro
tein to other proteins or antigens, or by combining it with appropriate im
mune stimulants, or by joining several such segments together into a sort of
beta-amyloid molecular pincushion, all in an effort to maximize the anti
body response to the vaccine without initiating the overreaction. These
vaccines have all been shown to reduce levels of beta-amyloid, and often of
plaques—some of them using very convenient delivery systems, such as
transdermal patches similar to the ones widely used for nicotine treatment,
or else the kind of nasal spray now used for quick decongestant relief of
stuffed-up noses.
And even this has not exhausted scientific creativity. As this book was
in preparation, for instance, researchers at the University of Texas' South
western Medical Center reported that injecting animals with the DNA for
the most toxic form of the beta-amyloid proteins, smuggled under the skin
inside tiny gold microparticles, led to production of the protein and to the
body responding with a rigorous and apparently safe antibody response.
The result: after receiving eleven injections over the course of several

158 E N D I N G A G I N G
months, the Alzheimer's mice enjoyed a 60 to 77.5 percent reduction of
plaque burden. 2 6
As I mentioned, the most well-studied passive vaccine is already in
mid-stage clinical trials orchestrated by Elan Pharmaceuticals. And while
this vaccine was the first out of the gate, the race to develop a clinically ef
fective and safe vaccine to defeat beta-amyloid is very much on. Indeed, the
question today seems to be not so much whether vaccination against amy
loid will work, but which of these many ingenious strategies will ultimately
prove to be most effective and safe. Surveying the progress made so far, we
can say with a high degree of confidence that we should soon be able to
harness the ancient powers of the immune system to cut our way through
the mind-stealing webs of beta-amyloid, redeeming the captive minds that
they bind.
Amyloid Vaccination: Beyond Alzheimer's
I've spent so much time discussing the Alzheimer's beta-amyloid vaccine
that you may well have forgotten how I got started: by noting that beta-
amyloid is just one famous example of a class of extracellular aggregating
proteins ("amyloids") associated with aging and with age-related diseases.
And no, I'm not repeating the mistake of the National Institute on Aging in
this regard, which is to throw nearly half of all of its resources every year
into Alzheimer's research, at the expense of its real mandate, which is (or
ought to be) to find ways to treat aging itself rather than one particular age-
related disease. Instead, I've been guiding you through the vaccine's
progress simply because it is in such an advanced state of clinical develop
ment. There's every reason to think that we will be able to exploit the same
sort of immunological strategy to tackle most of the other age-related disor
ders caused by coatings of extracellular junk.
Next to beta-amyloid vaccination for Alzheimer's disease, the immuno
logical amyloid-buster in the most advanced state of development is a vac
cine for systemic Ah amyloidosis, also known as primary amyloidosis. I
briefly mentioned this form of amyloidosis early on in this chapter. This is
the most common form of amyloidosis in the United States and some other
industrialized countries—it strikes two to three thousand Americans annu
ally. It's the result of overproduction by a specific cell type, plasma cells, of
immunoglobulin light chain (L—thus "AL," for "Amyloidosis Light-
chain"), a component of a class of antibodies.

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 1 5 9
However, I'm not going to spend much time on AL amyloidosis here,
because it's not an age-related amyloidosis and the amyloid involved may
differ in important ways from ones that are age-related. The main differ
ence is that it's laid down so extremely fast that it may not have the same
degree of problematic cross-linking as age-related amyloids. The main
thing I do want to tell you about AL amyloidosis concerns an immunother
apy protocol for it that shows promising signs of being transferable to age-
related amyloidoses.
The existing therapies for AL are decidedly inadequate. Until recently,
the standard intervention was a regimen of high-dose chemotherapy de
signed to kill off the originating plasma cells, often combined with bone
marrow transplants to replace some of the other blood cells that are de
stroyed in the process. More recently, a new chemotherapeutic drug called
I-DOX has been used, after the serendipitous discovery that it could accel
erate removal of systemic AL amyloid plaques through an as-yet unknown
mechanism that apparently does not involve suppressing plasma cells. But
even this new therapy tends to cause blood disorders, and both these treat
ments are only helpful for people with soft-tissue deposits, extending no
benefit to the more serious cases involving the heart or kidneys. They're
also not terribly successful at saving lives, with a mortality rate of about 40
percent.
At the turn of the millennium, scientists in Dr. Alan Solomon's lab at
the Human Immunology and Cancer Program at the University of Ten
nessee Graduate School of Medicine developed a new animal model for
AL, created by simply injecting mice with human light-chain amyloid ex
tracted from the livers or spleens of patients who had died of the disease.
The AL quickly began forming amyloid "tumors" in the animals, the size of
which varied with the amount of material the animals were administered: at
higher doses, the amyloid masses on their backs grew so large that they
could be felt by hand. 2 7 , 2 8
As they explored the effects of the disease on the animals, Dr.
Solomon's group demonstrated that antibodies against a segment of the
unique "beta-sheet" conformation of the light-chain amyloid fibrils were
partially breaking down the deposits, making them more susceptible to im
mune attack. When they took amyloid tumors out of mice, subjected them
to such antibodies and then returned the tumors into the mice, the animals
cleared them out twice as quickly as new tumors of similar size. Clearance
was also speeded when the original, unaggregated amyloid extracts were
pretreated with antibodies before being injected. Even immune-deficient

160 E N D I N G A G I N G
mice cleared out the deposits more quickly when they were given antibod
ies along with the amyloid extracts. 2 9 One such antibody—an immunoglob
ulin G1 antibody that they named 11-1F4—was found to have the strongest
"homing instinct," rapidly converging on tumors composed of either of the
two major classes of human light-chain amyloid fibrils, whether in a test
tube or in mice bearing them. Just as important, the antibody was found to
target the amyloid specifically, without infiltrating tissues anywhere else in
the body or samples of human tissue. And it also worked on pre-formed AL
amyloid.
A Jack-of-All-Amyloids?
As I mentioned, the reason I'm telling you about 11-1F4 is not because of
its effects on AL amyloidosis. The one disadvantage of Solomon's model of
systemic AL amyloidosis was that it's an imprecise model of the human dis
ease, with little spread of the deposits to major organs. Noting this, his
group sought ways of testing it against other amyloid diseases, such as amy
loid protein A amyloidosis (AA—also termed "inflammatory" or "second
ary" amyloidosis), the most common amyloid disorder outside the United
States. 3 0 AA has the advantage of being easy to induce as a full-blown dis
order in many strains of mice: you simply inject them with chemicals like
silver nitrate that induce a strong inflammatory response, resulting in over
production of serum amyloid A (SAA), a protein produced by the liver dur
ing times of inflammation. Like immunoglobulin light chain, SAA is only
partially degraded by the body's macrophages, resulting in the release of
sticky, half-digested SAA fragments that tend to clump up and accumulate
in the kidney and liver. This is the genesis of the disease in both mice and
humans, so the amyloid disease that results when mice are given these in
flammatory chemicals very closely mimics its human counterpart.
Surprisingly, the University of Tennessee team found that the 11-1F4
antibody also reacted to AA amyloids from mice—and that it cleared them
out of affected mice . 3 1 , 3 2 In fact, it was very nearly as effective against AA
deposits as it was against the original AL target, with the average organ
amyloid burden dropping by over three-quarters in liver and spleen alike!
This may be because the molecular architecture underlying the different
fibrils' stickiness is similar, leading to a similar antigenic profile. It may also
be related to the long-established fact that extracts of AL amyloid deposits
accelerate the development of AA in response to inflammation in mice. If

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 161
so, maybe the aggregating properties of the different amyloids are such that
they can interact, with one serving as a sort of crystallizing center around
which other amyloids gather. Think of a small deposit of cooked-on food
on the surface of a pot. If the stain isn't immediately scrubbed out, it be
comes increasingly stubbornly imbedded on its surface by future cooking,
and then begins to catch food particles from subsequent meals, slowly ex
panding into a larger and larger stain that is ever more difficult to remove.
Even more remarkably, the University of Tennessee team then went on
to show that 11-1F4 could also react with, and remove, amyloids based on
transthyretin (TTR), the thyroid-hormone transporting protein whose ag
gregation causes senile cardiac amyloidosis, and which is a cause of death
in so many of the oldest humans among us today. If passive immunization
with 11-1F4 can reverse the course of both of these major forms of amyloi
dosis, we are well on our way to clearing out some of the most important
sources of extracellular junk in the population at large—as well, potentially,
as other forms of amyloidosis that are less common causes of death only be
cause our lives are currently so brief.
This is exciting research, and the next step for the antibody is clearly to
test it out in humans with the corresponding amyloidoses. For this to be
done, the antibody must first be "humanized." Remember, while the light-
chain amyloids that the antibodies remove are derived from humans, the
agent that's doing the removal is a mouse antibody, which would probably
not interact well or safely with the human immune system. To overcome
this problem, Dr. Solomon's team "chimerized" the antibody, combining
its antigen-seeking "business end" with a human "handle." The resulting
antibody still recognized both light-chain amyloid and amyloid protein A
aggregates, and even cleared them out of mice just as the original vaccine
had done. The results are so promising that the National Cancer Institute's
Drug Development Group has arranged for large-scale pharmaceutical pro
duction of the new antibody, with the intention of moving it into prelimi
nary human clinical trials.
Open Possibilities
There is every reason to believe that this kind of immune-based, vaccina
tion approach to amyloids, demonstrated in animal models of Alzheimer's
disease and three human amyloidoses (and now in clinical trials for the for
mer), will also work in other cases of cellular bindweed. Take, for example,

162 E N D I N G A G I N G
amylin, or "islet amyloid polypeptide," whose amyloid-inducing properties
I briefly mentioned early on in this chapter. Amylin aggregates accumulate
on insulin-producing beta cells in the pancreases of nearly all people with
type 2 (late-onset, non-insulin-dependent) diabetes. Either the aggregates
or the soluble oligomers of which they're composed appear to play some
role in the gradual dying-off of beta cells that occurs as the disease pro
gresses, 3 3 leading to the failure of the body to produce enough insulin to
keep up with the incessant surges of sugar that accompany every meal.
No one has yet tried to develop a vaccine to remove these deposits, but
the feasibility of such an approach is suggested by the fact that amylin fib
rils have been identified inside macrophages harvested from areas adjacent
to the amylin deposits, where amylin accumulates without being fully de
graded. Moreover, amylin fibrils are engulfed by and accumulate within
macrophages exposed to them under test-tube conditions. 3 4 All of this sug
gests that that the immune system mounts an attack against this form of ex
tracellular junk just as it does against amyloid beta and the junk responsible
for secondary amyloidosis—in which case, there is every reason to think
that this attack could be strengthened with a vaccine similar to those cur
rently in the pipeline for those other amyloidoses. The therapeutic promise
of such an approach would be even greater if it were combined with a
souping-up of the macrophages' lysosomes with enzymes that are more
able to digest the amylin fibrils—-a job that cries out for the use of the
LysoSENS approach that I discussed in the last chapter.
Other forms of amyloidosis could also fall before an infusion of tar
geted antibodies or other vaccines. And while the focus for drug develop
ment today is on treatments for specific amyloid-based diseases, this same
research can be bootstrapped into the SENS agenda. Once it has proven its
efficacy in Alzheimer's disease, senile cardiac amyloidosis, and type 2 dia
betes, the spinoff technology will allow for the rapid development of vac
cines for more obscure amyloid deposits that today may go nearly
unno t iced except in people with a hundred candles or more to illuminate
their birthday cakes.
The fact that these therapies have moved so quickly from the labora
tory into clinical trials (remember, results in mice with the first beta-
amyloid vaccine were reported in 1999, and it was in clinical trials by 2001)
suggests that we will be able to move even more rapidly in the future, when
the first anti-amyloid vaccines have passed through clinical trials and have
been successfully used in doctors' offices all over the world.
Eventually, I envisage a protocol to keep our bodies clear of extracellular

C U T T I N G F R E E O F T H E C E L L U L A R S P I D E R W E B S 1 6 3
junk in which we might take a regular sequence of anti-amyloid vaccines,
not unlike the standardized series now given in regular succession over the
course of our childhood. The timing and frequency of administration of a
given vaccine would depend on how quickly its target builds up to levels
that impair function: we would get a "booster shot" of some every few
years, while others would be administered only a few times in each century
of a greatly expanded lifespan. Each time we took one of these vaccines,
our cells and organs would once again live and function free of a specific
species of molecular bindweeds, returning them to the literally unbound
potential of youth.

9
Year after year, ongoing chemical processes are shackling the
structural proteins of your body together, holding them back
from their vital jobs. Eventually, this leads to a familiar (and
ultimately fatal) range of age-related disabilities and diseases—
especially in the kidneys, heart, eyes and blood vessels. What if
we could break these chemical shackles, and thereby allow those
proteins to get back to work, as they did when you were young?
Scientists are making progress toward drugs that could
achieve just such a goal.
You're in the last hours before the big holiday feast, and the atmos
phere is heavy with the smells and emotional charge of the season. It's
been a long day in the kitchen—the oven running continuously, the ma
tron of the house trying to keep cool by leaving a window left slightly
ajar—and at last the hurry and stress are giving way to a more expectant,
eager sort of tension. The potatoes are mashed, the cranberry sauce has
been spooned into serving dishes, the sweet potatoes are being kept hot in
the oven, pumpkin pie is cooling on the windowsill. . . and now, a single
component of the meal dominates the cook's attention and the appetites of
her family.
Every fifteen minutes, like clockwork, for the last hour and a half, the
B r e a k i n g t h e S h a c k l e s o f
A G E

B R E A K I N G T H E S H A C K L E S O F A G E 1 6 5
turkey has been lovingly basted with its own fat, and perhaps a little honey;
now, to perfect the feast, the broiler is flipped on, to glaze its surface.
All the time that the turkey has been in the oven, complex chemical
processes have been imperceptibly proceeding—and now they accelerate.
Down at the molecular level, the high heat causes the sugars and fats to at
tack the proteins in the bird's skin. Molecular bonds are forged; new chem
ical products arise and are broken down; neighboring proteins are tied
together in shotgun marriages, tightening the outside surface of the turkey
and coating it with thick, gooey chains of linked proteins, fats, and sugars.
Finally, the deed is done. Mom flips the oven off and dons her oven
mitts as she calls to Dad to get the carving knife. The family looks on her
handiwork with eager eyes, gazing with hunger and appreciation at the
darkened, crispy, sticky, slightly toughened surface that the chemical mael
strom has made of the turkey's skin. Dinner is served.
I'm sure that you and your family have played out a similar script at
Christmas—or at Hanukkah, browning the latkes or sufganiyot. But, in a
profound sense, you have as much in common with the feast as you do with
the family.
Every day of your life, the same processes that are involved in the
browning of meats and other glazed or fried foods are insidiously at work
in your body. In your arteries. In your kidneys. In your heart, your eyes,
your skin, your nerves. At this very moment, in all your tissues, the sugar
that provides your body with so much of its energy is also performing some
unwanted chemical experiments, caramelizing your body through exactly
the same processes that caramelize onions or peanut brittle. Slowly but
steadily, unwanted bonding by sugars and fats handcuffs your proteins, in
activates your enzymes, triggers unhealthy chemical signals in your cells,
and damages your DNA. Aging you.
Make that: AGEing you. And I'm not just reminding you of my nation
ality by adding that final e.
The Way We AGE
The body relies on sugar as a key energy source. But, like any fuel, sugar can
only be "burned" by our cells because it is chemically reactive—and, again
like other fuels, that volatility can make it dangerous to work with. A d v a n c e d
Glycation End-products (or "AGEs," as they're appropriately called) are the
end results of the complex chemical processes through which the structure

166 E N D I N G A G I N G
of proteins is warped by sugars and other fuels. This same chemistry is the
cause of the "browning" you see when you roast a turkey, caramelize a
sauce, or pop a slice of bread into a toaster. AGEs accumulate in your tis
sues, leading to gradual loss of function, then disease, and ultimately an
early grave. AGEs transform the supple grace of youth into a "crusty" old
age, through exactly the same chemical processes by which they form the
crust on a loaf of bread.
The many chemical reactions, intermediates, and stable end products
of AGE chemistry have been the subject of an enormous amount of re
search, first in the food technology and chemical sectors and more recently
in biomedicine. Scientific study began with work by a food chemist named
Maillard in the 1910s and '20s, but it took until the 1980s for role of AGEs
in the complications that ravage the diabetic body to become a hot topic in
diabetes and aging research. Even now it's clear that we've only begun to
understand the furious promiscuity of this biochemistry and its impact on
the aging or diabetic body.
Though the details needn't concern us here, you will need to grasp the
outlines of how AGEs form if you are to understand the various strategies
that have been employed in the search for a way to shield us from their fos
silising influence. In the best-understood pathway (the main stream of the
Maillard reaction—see Figure 1a), a molecule of sugar opens its structure
and glues onto ("glycates") a protein molecule, forming a Schiff base. This
structure is relatively unstable, so the Schiff base will often spontaneously
fall apart. Sometimes, however, it will collapse into a more stable structure
called an Amadori product. Amadori products are much longer-lived than
Schiff bases. (This fact has long been exploited in a lab test that measures
levels of glycated hemoglobin or HbA1c, an Amadori product in red blood
cells, as an indicator of the average amount of sugar that has been present
in the blood over the course of the previous few weeks.)
Relatively stable though they may be, however, Amadori products are
still subject to the biochemical hurly-burly around them. They can there
fore be put through any number of further chemical transformations, such
as rearrangement or degradation of their basic structure, forcible insertion
of water molecules or removal of amino groups, or attack by free radicals.
Many of these changes lead to the formation of even more stable structures,
either directly or via highly reactive intermediate compounds such as
oxoaldehydes. These structures are stable enough, in fact, to be called "end
products"—they are the advanced glycation end products, or AGEs.
For our purposes, the important outcome of these processes is the

B R E A K I N G T H E S H A C K L E S O F A G E 167
Figure 1. The ways we AGE. (a) The "chemical" (Maillard) pathway; (b) The "metabolic" (triosephosphate) pathway; (c) Sources of methylglyoxal.

168 E N D I N G A G I N G
Figure 1. (continued).
formation of AGE cross-links, a subset of AGEs in which proteins that are
already working with one arm tied behind their backs because of glycation
become shackled to a second, neighboring protein.
AGEing happens much more quickly in people with diabetes than in
the rest of the population, partly for the simple reason that diabetics' blood
sugar levels are higher: In any chemical reaction, a higher concentration of
an active agent will tend to increase the rate of its interactions with its tar
gets, provided those targets are plentiful. But AGE cross-links also accu
mulate in people with normal blood sugar levels, and it's quite clear that
they are responsible for much of the pathology and increased vulnerability
to the insults of daily life that accompany "normal" aging.
Browning to Death
The cross-linking of proteins is similar, at both the molecular level and the
functional level, to the processes that cause windshield wipers to lose their
flexibility. For people who don't have diabetes, the most life-threatening lo
cus of the ensuing stiffening of the tissues is the cardiovascular system.
AGE cross-links slowly impair the youthful elasticity of your heart and
blood vessels, making them rigid and unyielding. The resulting hardening
of the arteries is in large part responsible for the increase in systolic blood
pressure that everyone suffers with age. (Systolic pressure is the first of the
two numbers that you get from a blood pressure reading, like the "110" in
"110 over 80.") Meanwhile, the AGEing of your heart impairs its capacity
either to contract to pump blood through your body, or to expand in order
to fill up with that blood in the first place. The combination of these two

B R E A K I N G T H E S H A C K L E S O F A G E 1 6 9
factors increases the workload on the heart, ultimately leading to one of
several forms of heart failure if nothing else kills you first. The same lack of
plasticity also means that your blood vessels become less able to withstand
the constant surges of blood that course through them: they become brittle
and eventually break under the pressure like old rubber bands, one poten
tial result of which is a bleeding stroke.
And the damage caused by AGE cross-links extends well beyond the
cardiovascular system. They shackle proteins all over the body, accumulat
ing with age in tissues as diverse as the tiny blood vessels in your eye and
the supporting myelin sheaths of your nerves. Everywhere they occur, AGE
cross-links impair the functioning of those proteins, contributing to age-
related dysfunction, disability, and death. In your eyes, they accumulate on
the crystallin proteins that make up the structure of the lens. AGEd lens
proteins stop allowing light to pass through them, leading to the brown
pigmented spots in the lens that we know as cataracts. The combination of
this browning with several effects at the cellular level is why age and dia
betes are the major risk factors for this, the single greatest cause of vision
loss worldwide.
And that isn't the only way in which AGEs contribute to vision loss.
Elsewhere in the eye, AGEs contribute to diabetic retinopathy (vision loss
in diabetics linked to damage to the fine blood vessels feeding the light-
absorbing tissues at the back of the eyeball), to age-related macular degen
eration, and possibly also to open-angle glaucoma.
The kidney, too, suffers badly from AGE assault—again, especially
among people with diabetes. Diabetic damage is the single biggest cause of
kidney failure in the United States, and a third of all patients who find
themselves in the dialysis ward got there because of their diabetes. Indeed,
the severity of kidney disease in diabetics tracks the level of renal AGEs,
which cross-link the proteins of the kidneys' biological filter material and
trigger an inflammatory process that leads the body to overcompensate by
growing too much replacement tissue, in a sort of out-of-control wound-
healing response. The net effect of these two processes is a buildup of
something similar to scar tissue in the kidney, which accumulates to levels
that literally squeeze the tiny blood vessels where filtration is supposed to
occur, reducing the amount of filtering surface available and leading to in
efficient screening of materials in the blood—as if you had glued a coffee
filter paper back on itself before running the machine, leading to a ground-
filled mess when the water starts to back up.
AGEs also contribute to diabetic neuropathy, the debilitating damage

170 E N D I N G A G I N G
to the nerves that is suffered by so many diabetics. The severity of this dis
ease can vary, but the most common symptom is an unremitting version of
the experience one has after a temporary, pressure-induced reduction in
blood flow to the hands or feet (i.e., when the extremity is said to be
"asleep"): a sensation of "pins-and-needles," pain, or numbness in the af
fected limbs, along with some loss of control or clumsiness in their use.
People with diabetic neuropathy also lose some of the unconscious control
by their nervous systems of functions such as the regulation of the heart's
rhythm, the digestive process, the bladder, and erectile function; they also
often suffer dizziness, and nausea that may extend to vomiting. Whether
AGEs play any role in similar, more subtle defects in nerve function with
age in otherwise healthy people is unclear, but it seems likely.
Comparisons of the rates of accumulation of cross-links in the tissues
of slower- and faster-aging species, and of slower- and faster-aging individ
uals within a species, suggest that AGEing plays an important role in aging
per se, not just in specific diseases or the complications of diabetes. Both
the rate of age-related buildup of one of the more easily measured AGEs
(pentosidine) and the related toughening-up of the proteins in skin or tail
tissue are inversely associated with the maximum lifespan of different
mammalian species. This means that the more slowly a species ages, the
more slowly its collagen is stiffened by AGEs (see Figure 2). Likewise, calo
rie restriction—which is, as I've mentioned in previous chapters, the best-
studied way to slow down aging in mammals—slows these processes down;
and in fact, higher rates of tissue AGEing have been shown to predict early
death in individual calorie-restricted animals. 1 In our own species, studies
show that even within the "normal" range (i.e., at values well below those
typical of people with diabetes), higher blood levels of either glucose itself2
or of the Amadori product HbA1c 3 are associated with a higher risk of
death from all causes.
A drug that would slow down or reverse the accumulation of AGEs
would thus help people with a wide range of diseases and disabilities. It
could potentially improve, or even cure, problems as wide-ranging as the
gradual increase in blood pressure over a lifetime; the terrible kidney,
Figure 2. Maximum life span as a function of AGE formation rate. Redrawn.4

B R E A K I N G T H E S H A C K L E S O F A G E 171
nerve, and visual complications of diabetes; and several forms of heart fail
ure. And it could also help us to address a major contributor to aging itself.
This idea didn't just pop into my head recently, of course: a variety of
schemes to reduce the tissue AGE burden have been explored over the
years, and many of them have even reached relatively advanced clinical trial
status. Yet, despite years of work, none of these treatments has been shown
to be safe and effective enough to find its way into the drug arsenal of any
developed country. The obstacles that have plagued their development and
limited their usefulness represent yet another case study in the problems of
trying to deal with age-related damage by tinkering with the complex bio
chemistry of life.
Listening to Parmenion
"Sugar P i l l s "
The fact that AGE cross-links are often ultimately the result of sugar mole
cules acting like a glue, gumming up our tissue proteins, immediately sug
gests one possible solution to the problem: just lower people's blood sugar
levels, and you'll reduce the formation of Schiff bases (see Figure 1) in their
bodies and thereby lower their AGE burden. Of course, this has long been
the major focus of diabetes management, and in the 1990s, two massive and
widely cited scientific studies—the Diabetes Control and Complications
Trial (DCCT) and the UK Prospective Diabetes Study (UKPDS)—were
hailed as the clearest proof yet of the effectiveness of this strategy when
taken to its limits. These two studies showed that when diabetics take strict
steps (aggressive use of blood-sugar-lowering drugs and regular feedback
in the form of frequent blood sugar testing) to keep their blood sugar un
der very tight control, they are at greatly reduced risk of developing the ma
jor complications of the disease. The DCCT, in particular, showed that—as
compared with the standard of care at that time—a regimen of intensive
blood sugar control could reduce a diabetic's risk of developing nerve dis
ease by close to two-thirds, diabetic kidney disease by about half, and dia
betic retinopathy by an astounding three-quarters.
The results of these two studies were trumpeted around the world—by
their government sponsors, by patient advocate organizations, and by phar
maceutical companies looking to boost sales of glucose-lowering drugs. The
plan was to encourage doctors to prescribe these drugs to patients whose

172 E N D I N G A G I N G
blood sugar control was in the range that made them safe by previous stan
dards but demonstrably at risk based on the new data, and also to increase the
doses taken by people with worse control who were already using the drugs.
The benefits that would accrue to patients as a result of such a surge in
drug use seemed to be clear-cut: people with diabetes all over the world
would enjoy miraculous improvements in the quality and length of their
lives through dramatic reductions in their risk of blindness, nerve damage,
and kidney failure. But when scientists actually assessed the overall quality
of life of people who had undergone the intensive therapy regimens in the
trials, the results were surprisingly gloomy. Despite the fact that more ag
gressive treatment had reduced the risk of all major diabetic complications,
the intensive-therapy patients enjoyed no improvement in their net well-
being as compared to people who had been assigned to standard care. 5 , 6
Many factors probably contribute to the lack of clear-cut benefits from
aggressively lowering blood sugar levels. While diabetic complications
clearly have a negative impact on quality of life, the drugs used to lower
blood sugar also come with costs that are not included in the sticker price.
People on such medications tend to gain weight, which reduces their qual
ity of life—both directly, and by increasing the risk of other diseases such as
osteoarthritis. Many patients also find that sticking with the rigid schedule
of injections and finger-prick tests required to keep up with these regimens
imposes real restrictions on their lives, which some studies report con
tributes to depression, frustration, isolation, or troubles at work.
And finally, constantly trying to push blood glucose even into the "nor
mal" range carries with it the risk that blood sugar levels will drop too far,
leading to a "hypoglycemic crisis" whose consequences can range from
dizziness to a coma. This is of particular relevance to normal aging. If push
ing blood sugar levels down is a mixed bag for diabetics, you can see that it
would be a decidedly dubious solution to the AGE problem for the rest of
us, in whom the wiggle room between our normal blood sugar levels and a
hypoglycemic crisis is much smaller, making the potential benefits more
limited and the risks higher.
And even if we could safely bring our blood sugar down to the lowest
possible safe level, we'd be quite far from a complete solution to the AGE
problem. All of us must maintain some level of glucose in our blood as an
energy source, and some percentage of that glucose will always wind up re
acting with tissue proteins, leading to cross-linking.
And on top of that, not all AGEs are even derived from glucose. Blood
fats ( tr iglycerides) can also cause the cross-linking of proteins, particularly if

B R E A K I N G T H E S H A C K L E S O F A G E 173
there's a high level of oxidative stress; this is the chemistry that underlies
the browning of a turkey skin as it roasts, even without a sweet, syrupy
slather on its surface. As with blood sugar, diabetics usually have high
triglyceride levels, and even many nondiabetic people would benefit from
having their triglyceride levels brought down; but triglycerides also resem
ble blood sugar in being indispensable to normal function, so there's only
so far that such a strategy can be safely pursued.
Less Is More . . . Is Worse
And that's not all: attempts to control levels of both these early precursors
of AGEs, even by nonpharmacological means, can have perverse metabolic
consequences.
For instance, one established effect of very low-carbohydrate diets of
the Atkins type is to bring down both triglyceride levels and the body's to
tal exposure to carbohydrates, so some advocates have hypothesized that
these diets would reduce a person's AGE burden. Unfortunately, it turns
out that the metabolic state that these diets induce (the notorious "ketosis")
has the unfortunate side effect of causing a jump in the production of the
oxoaldehyde methylglyoxal, a major precursor of AGEs that is also, ironi
cally, produced within the cells of diabetic patients when they are f o r c e d to
take in more glucose than they can immediately process (see Figures 1b and
1c). A recent study tested the size of this effect in healthy people who suc
cessfully followed the first two phases of the Atkins diet for a month, and
who had the ketones in their urine to prove that they were sticking to the
diet. These previously healthy people suffered a doubling of their methyl
glyoxal levels, leading to concentrations even worse than those seen in
poorly controlled diabetics. 7 Like other oxoaldehydes, methylglyoxal is far
more chemically reactive than blood sugar (up to 40,000 times more reac
tive, in fact), and is known to cause wide-ranging damage in the body, of
which AGE cross-links are but one example. This potentially makes the
Atkins diet a recipe for accelerated AGEing, not a reprieve from it.
"Radical" Proposal—-Lukewarm Results
Even before the counterintuitive results of the DCCT came out, it was obvi
ous that a blood-sugar-lowering strategy would not be a complete solution

174 E N D I N G A G I N G
to the AGE problem. The body needs blood sugar and fats as fuels, and yet
no level could be so low as to eliminate all cross-link formation: at best, low
ering the concentration of glucose and triglycerides would delay the in
evitable. So some scientists turned their attention elsewhere, to
cross-link-inducers whose harmful role in the body is less ambiguous.
One such AGE-prevention strategy is the use of high doses of antioxi
dants to bring down free radical levels. As you can see from Figure l a , free
radicals can accelerate the conversion of some AGE precursors into certain
specific full-blown cross-links—a phenomenon called glycoxidation. Based
on the effect of adding free radicals to proteins and sugars in test-tube ex
periments, glycoxidation can be predicted to hit diabetics with a double
whammy, because in addition to the excessive levels of blood glucose and
fat, the impaired metabolic state of diabetes also causes an overproduction
of free radicals in victims' cells. Put the two factors together and you have a
potentially synergistic interaction. Also highlighting the importance of free
radicals in AGE formation is the fact that birds have sky-high blood glu
cose levels that would rapidly kill a human, yet generally live around ten
times longer than mammals of the same size; part of how they get away with
this is probably by having really good control of oxidative stress.
If glycoxidation were a major reason for diabetics' high levels of AGE,
then sopping up their excess burden of free radicals with antioxidants might
considerably reduce the cross-linking of their tissue proteins, resulting in
longer life expectancy and reduced risk of crippling complications. And
many studies carried out in laboratory rodents have supported this expecta
tion: dosing them with various free radical quenchers typically reduces the
cross-link burden in their tissues considerably, cutting back on the incidence
and severity of diabetic kidney, nerve, and even retinal damage.
When antioxidants were tried as an anti-AGE therapy in humans,
however, the results were disappointing. The effects on AGE levels and
symptoms were minor or nonexistent—and even when benefits were ob
served, the effect was almost exclusively confined to the most severe cases
of the disease, with more typical diabetics getting no relief. 8 , 9 , 1 0 We now
know that there are a couple of major reasons for this. First, human dia
betes causes a much less severe increase in oxidative stress than is suffered
in the rodent version of the disease, as can be seen by comparing the levels
of molecules damaged by free radicals in the two species' skin. This lower
free radical load makes glycoxidation a less important factor in human dia
betics' AGE chemistry, and thus weakens the potential benefit of reducing
its impact.

B R E A K I N G T H E S H A C K L E S O F A G E 175
But another, more general reason for the lack of efficacy of antioxi
dants as an anti-AGE therapy is the sheer riotous promiscuity of the highly
reactive precursors of AGE cross-links. One highly revealing animal
study 1 1 illustrates the point. Diabetic rodents were given diets fortified with
different antioxidant supplements (vitamins or green tea extracts), and the
impact on the animals' AGE burden was assessed by comparison with both
healthy animals and diabetics given unsupplemented chow. To tease out the
biochemical pathways involved, scientists measured levels of substances
produced at several different steps across the spectrum of the glycoxida
tion process, from initial glycation events to the creation of specific AGE
cross-links—some of which are created by glycoxidation, and others by
straightforward glycation, i.e., without the involvement of free radicals.
As previous rodent studies had shown, antioxidant treatment did exert
some benefits on diabetic complications. Equally predictably, the treat
ments had no effect on the initial glycation of proteins, since the window of
opportunity for free radicals to work mischief in AGE chemistry opens up
further along in the process (Figure 1a). But the researchers got a surprise
when they began looking at actual cross-links. Antioxidant supplements
had no effect on the levels of those AGEs whose formation doesn't require
free radicals, of course—but the intervention actually increased the levels of
the two glycoxidation-derived AGEs, so that diabetic animals receiving
green tea extracts actually wound up with more total cross-linking than
those who simply suffered the "natural" course of the disease.
This remarkable result yet again illustrates the hopeless complexity of
the tangled skein that is metabolism. The precursors of these AGE cross
links don't simply disappear when they aren't hit by free radicals—they
have to go somewhere—and when much of the excess oxidative stress was
relieved by antioxidant supplementation, these precursors began to build
up until they spilled over into one of the alternative pathways of cross-link
formation. It was the same effect you see in traffic jams when a main traffic
artery is cut off: a few drivers may indeed just turn their cars around and
go home, but most of them turn off onto the local side streets, creating
secondary traffic congestion in hitherto sleepy residential neighborhoods.
Collateral Damage
The best-understood pathways of AGE cross-linking are fundamentally
random events, not too far removed from what happens in the browning of

1 7 6 E N D I N G A G I N G
food, or in a test tube. The fuels of metabolism, dissolved in your blood or
in the fluid inside your cells, randomly bump into tissue proteins; depend
ing on factors like temperature, concentration, and the presence of transi
tion metals and free radicals, a series of chemical events may occur; and if
they happen in just the right order, an AGE cross-link will form.
But some AGEs result more directly, from the regulated activity of
metabolic processes. One recently identified example is the enzyme
myeloperoxidase, which is used by macrophages to kill bacteria by gener
ating toxic hypochlorous acid. It has been shown that hypochlorous acid,
in the presence of the protein building-block serine, can itself induce
AGE-type cross-linking, independent of the usual fuel chemistry of sugars
and fats. 1 2
If myeloperoxidase were only ever activated to kill bacteria, it might be
a relatively unimportant source of AGEs in people living in the developed
world who don't have chronic infections (although the number of such
people is much higher than is generally appreciated). But, as we saw in
Chapter 7, macrophages don't just attack bacteria: they also become
aggravated—and crank up their myeloperoxidase activity—in their short
sighted efforts to clear trapped cholesterol from your arteries. Some scien
tists now believe that myeloperoxidase is probably a major contributor to
the high levels of AGE found in the atherosclerotic foam cells of nondia-
betic people.
While reducing excess myeloperoxidase activity might be desirable at
the sites of atherosclerotic plaque, we probably could never lower its activ
ity pharmacologically without also impairing our ability to defend ourselves
against bacteria. As people with AIDS know, when your immune system is
suppressed, you're not just at risk from relatively rare bacterial killers like
tuberculosis: you can be felled by infections that most of us shake off be
fore we have even the beginnings of symptoms. Moreover, and surprisingly,
one study found that animals bred to produce something like human ather
osclerosis, but lacking the ability to produce myeloperoxidase, showed
more severe atherosclerosis than animals with normal activity, again illus
trating the frustrating complexity of metabolic processes. 1 3
The Drug That Failed
Okay, so trying to lower the levels of the ultimate AGE precursors, like
glucose and fat, is difficult—and also unsafe, because they are essential

B R E A K I N G T H E S H A C K L E S O F A G E 177
biological fuels. Soaking up free radicals and sequestering transition metals
is of limited effect because there are so many alternative routes to AGE
formation.
But an overview of Figure 1 may suggest a much more attractive target:
oxoaldehydes. For one thing, these reactive compounds are present at
much lower concentrations than blood sugar or triglycerides, meaning that
you would only have to knock out relatively few molecules to lower the to
tal level in the body by a significant proportion: methylglyoxal, for instance,
is several thousand times less concentrated in the blood than glucose. On
top of this, oxoaldehydes are very virulent molecules (as I mentioned ear
lier, methylglyoxal is up to 40,000 times more prone to attacking tissue pro
teins than glucose is), so that each molecule you take out of circulation is
much more likely to translate into the prevention of a cross-link in the wait
ing. Oxoaldehydes also play their role in cross-link formation relatively late
in the process, leaving fewer alternative pathways by which an AGE might
form if they could be soaked up. And in contrast to sugars, which are es
sential molecules for which there is a limit beyond which lowering their
concentration in the blood becomes life-threatening, oxoaldehydes are fun
damentally toxic molecules, so that one should be able to reduce their con
centration drastically without doing any harm to the body.
Thus, if trying to lower AGE formation with antioxidants or blood-
sugar medications is like launching a wide-sweeping crackdown on an entire
crime-plagued neighborhood, a drug that mops up oxoaldehydes would be
like springing a carefully targeted police raid on known members of a brutal
criminal gang.
For a long time, a drug called aminoguanidine (trade name Pimagidine)
seemed poised to fulfil this promise, revolutionizing the treatment of dia
betes and perhaps landing the first serious punches on the Mike Tyson that
is biological aging. The drug enjoyed a lot of buzz in the scientific literature
on diabetes, as well as in some of the commentary on life extension in pop
ular magazine articles and Internet discussion groups, because its clearest
mechanism of action was precisely its ability to mop up oxoaldehydes.
Over the course of many years, researchers put aminoguanidine to the
test—first in test tubes filled with AGE precursors and mixtures of cata
lysts, then in cell cultures, and eventually in animal studies. And at nearly
every juncture, hopes for the drug continued to rise. In diabetic rats, it low
ered AGE formation in the cells of the retina and reduced the maladaptive
overgrowth of the blood vessels feeding them. In dogs, it prevented the loss
of the retinal blood vessel cells and the associated accumulation of dead

178 E N D I N G A G I N G
blood vessels through which blood had stopped flowing. It also kept both
species' hearts and blood vessels more flexible.
Somewhat less consistently, aminoguanidine showed promise against
other complications of diabetes. It reduced the total level of kidney tissue
that was so cross-linked as to be indigestible by strong acids, and prevented
much of the thickening of the kidney's filtration machinery that accompa
nies diabetes in rats—although it was unable to affect the course of the dis
ease in dogs. Further, diabetic rodents (but not baboons) given the drug
exhibited less loss of blood delivery to nerves, and improved ability to con
duct nerve impulses.
Most important for those of us looking for interventions against AGEs'
role in the degenerative processes of aging, aminoguanidine even seemed
to reduce heart and kidney AGE levels (and the resulting loss of those or
gans' function) in animals that were suffering from purely age-related AGE
accumulation rather than diabetes.
After a few small human studies designed to test for any obvious toxicity
seemed to go well, the company that had patent control over aminoguani
dine for use as a drug for diabetes attracted the attention of the biotech giant
Genentech, which partnered with them to launch two full-scale clinical trials.
Each one was to involve about six hundred people with the beginnings of di
abetic kidney disease, in medical centers spread out all over North America.
The first trial (ACTION I) recruited people with type I (autoimmune) dia
betes; the other trial (ACTION II) was to involve patients with the more
common type II (late-onset) diabetes, which usually develops as a result of
lifestyle, albeit sometimes overlaid on genetic vulnerability. Ambitiously, pa
tients in both trials were to be well medicated to control both blood sugar
and blood pressure before being put on aminoguanidine, so that differences
between the groups would be entirely the result of the test drug's direct anti-
AGE effects.
But when ACTION I was completed in 1996, the best spin that one
could put on the results would be to say that they were disappointing. On the
positive side, risk factors like blood pressure, LDL ("bad") cholesterol, and
triglycerides went down in people who had received the drug. And some
crunching of the data suggested that the drug might improve some indicators
of kidney function. Plus, in a tiny subgroup who had been tested before and
after the trial, diabetic damage to the retinas seemed less serious in patients
taking aminoguanidine than those taking placebo—though these observa
tions were suspect, because they were made as an afterthought after the trial
had been shut down rather than being part of its original design. 1 5

B R E A K I N G T H E S H A C K L E S O F A G E 1 7 9
What the study was supposed to show was a direct effect on kidney
health, as measured by a standard laboratory test for kidney function—and
the data just weren't strong enough to support that conclusion. The raw
numbers looked, on their face, better in aminoguanidine users than in the
placebo group, but the difference was so small relative to the number of pa
tients in the trial that it seemed likely to be a statistical fluke, like getting
"heads" in six out of ten coin tosses instead of the expected five.
Worse: while the benefit attributable to aminoguanidine was dubious,
the risks associated with the drug seemed undeniable. Along with signs of
an overactive (and possibly damaged) liver and strange flu-like symptoms
that went away when they stopped using the drug, a few people taking
aminoguanidine developed signs in their blood of an autoimmune disorder,
which—in three patients taking the higher dose—was associated with a
form of highly inflammatory kidney disease that leads to complete loss of
kidney function in a matter of just weeks or months. Two of the three pa
tients who developed the disease progressed to end-stage kidney failure.
Fortunately, this apparent side effect was caught early in the trial, and the
safety committee accordingly introduced a monitoring program, after
which no one was allowed to progress into clinical signs of the disease.
A FATAL R E S E M B L A N C E
Even now, we still don't know for sure what caused aminoguani-
dine's severe toxicity, which had not been observed in animal stud
ies. But there's a good guess—and if it's correct, it makes
aminoguanidine yet one more case study in how trying to repress
the dark side of metabolic processes so often has repercussions.
What made aminoguanidine a promising AGE-blocking drug
was its mechanism: the sopping-up of oxoaldehydes. These sub
stances are in a class of chemical compounds called carbonyls:
organic molecules with a carbon atom double-bonded to an oxy
gen atom. This structure makes many carbonyls highly biologically
active, which is why oxoaldehydes are such relentless shacklers of
bodily proteins. Of course, metabolism relies on the harnessing of
highly active compounds to carry out the biochemistry of life—
and so it's hardly surprising that many essential biological mole
cules also feature prominent carbonyl groups.

180 E N D I N G A G I N G
The problem is that aminoguanidine can't necessarily tell one
carbonyl-bearing molecule from another. That might be expected
to cause it to sop up some essential carbonyl-bearing molecules
along with toxic ones like oxoaldehydes. In fact, we know that it
does so in at least one case: vitamin B6. As a result, animals given
aminoguanidine can easily develop a deficiency of this vitamin
indistinguishable from simply putting a subadequate supply of it in
their diet.
Damningly, a blood pressure drug called hydralazine, which
brandishes the same carbonyl-trapping hydrazine surface that
aminoguanidine's business end uses to neutralize oxoaldehydes, is
well known to cause a lupus-like autoimmune disorder, the first sign
of which is the appearance in the blood of the same antibodies
observed in the aminoguanidine users in ACTION I.
If a drug as promising as aminoguanidine can't safely prevent enough
AGE damage to improve the health of diabetics, you can be sure that it
won't do much for the basically healthy among us. Because the concentra
tions of blood sugar and fats are much lower in people without diabetes,
the buildup of AGE is much slower, and thus harder to slow down to a de
gree that causes a measurable change in their health. Thus, it would take a
lot longer for any potential benefits to accrue, whereas the risks remain at
the same high levels for each individual year of use.
Indeed, a study published after aminoguanidine's withdrawal from clini
cal development 1 7 appears to show that even the initial reports of a reduction
in age-related AGE cross-linking in nondiabetic rodents were specific to the
strain of rat used in early studies (which is particularly susceptible to kidney
disease). Other strains showed little or no benefit from lifelong aminoguani
dine administration.
These are just a few illustrations of the known or anticipated ways in
which the mechanisms underlying cross-link formation undermine our
ability to prevent AGEing. This nightmare of biochemical complexity is so
elaborate as to cause even the most dedicated puzzle enthusiast to snap
pencil in two and go to bed in frustration; it should raise serious doubts
about the wisdom of continuing to invest resources in seeking new ways to
interfere with such a poorly understood, multiply-branching network of
pathways (Figure 1). In the pell-mell of the body's biochemistry, a certain

B R E A K I N G T H E S H A C K L E S O F A G E 181
quantity of AGEs is simply inevitable, and trying to prevent enough cross-
linking from happening to have a real impact on the stiffening of our tis
sues, without somehow disturbing essential metabolic processes, may
ultimately be futile.
If you've read the preceding chapters of this book, you probably have a
pretty good idea of the sort of strategy I'd like to see used to deal with the
problem of AGEs, whether in diabetics or in "normal" aging. Don't mess
with blood sugar. Don't try to block free radicals. Don't go chasing after
ways to outsmart metabolism. Don't try to prevent AGEs from forming at
all. No, the anti-aging engineer's solution should be to allow metabolism to
proceed in its infamously messy way, and then to remove full-blown AGEs
themselves before they build up enough to impair tissue function, robbing
us of youthful flexibility of heart and sinew and increasing our risk of death
and disability.
In this case, however, I am not playing the role of visionary, so much as
of cheerleader. At least two companies have developed such drugs and
tested them in animals. One of them has undertaken several clinical trials
already.
A SENS Serendipity
In the decade since Drs. Tony Cerami and Peter Ulrich had first suggested
that the cross-linking of proteins by glycation might be the link (pun in
tended) between high blood sugar levels and the complications of diabetes,
they had spent a lot of their time working on ways to do something about it.
They had played key roles in the development and early testing of
aminoguanidine, but well before the failure of ACTION I they knew that
much stronger molecules would be required to help two specific groups of
people with quite different AGE-inducell disabilities. On the one hand, di
abetics whose disease had progressed so far that they had already suffered
a lot of cross-linking would be quickly approaching the threshold at which
their total level of cross-linking would begin to result in disability and
death, and would therefore require much more effective interventions than
would people only in the early stages of the disease. And on the other hand,
many people who suffer with AGE-derived diseases such as hypertension
and heart failure have normal blood sugar levels. In these people, the pre
cursors of AGEs are present at much lower levels and are thus are much
harder to intercept: it's like trying to shoot down a single bird in the sky,

182 E N D I N G A G I N G
whereas taking on AGE precursors in diabetics is like sighting one of the
incredible flocks that once blacked out the sun in the early days of Europe
an colonization of the Americas, into which hunters could fire off buckshot
without even bothering to aim.
So in late 1991, Cerami arranged for a summit at their labs at the Pi-
cower Institute for Medical Research in Manhasset, New York. The meet
ing brought together himself, Ulrich, several other Picower staffers, and
scientists working for a company Cerami had helped form named Alteon,
to brainstorm on new ways to inhibit AGE formation.
Analysis of what was believed about the chemistry and products of
these reactions had already led many researchers to conclude (correctly) that
reactive carbonyls (like oxoaldehydes) would be important potential sources
of AGE cross-links, and thus targets for anti-AGE drugs. This was exactly
the rationale for the development of aminoguanidine. Ulrich saw that, theo
retically, a lot of the body's AGEs might be formed from a class of reactive
carbonyls known as Amadori diones and the related Amadori-ene-diones.
These molecules would form when Amadori products broke down: car-
bonyl groups would join hands across the gulf of adjoining proteins, result
ing in an alpha-dicarbonyl link—specifically, a type of alpha-dicarbonyl
called an alpha-diketone link. On chemical grounds, such a link would not
be expected to remain intact for long—but it wouldn't just disappear, either.
Most likely, Ulrich thought, it would rearrange into a more stable, final
structure—a molecular marriage which only death would part.
If that were true, Ulrich could see one potential path to the develop
ment of novel anti-AGE interventions. The body has enzymes that break
down at least some kinds of dicarbonyl compounds, and many of these en
zymes share in common the incorporation of the vitamin thiamine. Re
search by Ukrainian scientists in the mid-1980s had shown that molecules
in the same chemical family as thiamine (called thiazolium compounds)
break linkages of the same chemical type, albeit embedded in organic
chemicals rather than as AGEs in tissue proteins. The inclusion of thiamine
in so many of these enzymes, combined with the mechanism of other thia
zolium compounds, suggested that thiamine was the essential feature to all
of them, like the common head shape of different brands and sizes of
Phillips screwdrivers. The active core of the incorporated thiamine would
get an electrochemical grip on the carbonyls in the enzyme's target mole
cule, whereupon the enzyme would twist its shape, opening up and tearing
the bonds apart.
Ulrich wanted to design a new molecular "tool" that would do the

B R E A K I N G T H E S H A C K L E S O F A G E 1 8 3
same splitting job with the dicarbonyl bonds in Amadori diones, eliminat
ing their cross-link-forming potential. Starting with the concept of a
thiamine-like "business end," the assembled scientists began throwing out
suggestions on how different kinds of molecular "levers," "swivels," and
"sprockets" might behave, predicting their interactions with Amadori
diones from their structures. Ulrich stood at a blackboard, drawing out
their proposals.
Finally, they came up with a basic template of a class of molecules that
might be expected to cleave the kinds of bonds present in the Amadori-
diones that they believed would probably be found in the body. Then they
threw together a variety of specific variations on the theme by tacking on
various "limbs" to the core "backbone" structure.
At the end of a marathon session, the Alteon scientists took the results
of their work back with them for preliminary testing. At Alteon, Dr. Jack
Egan assigned several junior scientists to synthesize test quantities of each
of their various candidate molecules, and also to cook up large batches of
some model Amadori diones. From there it was a straightforward series of
simple experiments: pipette small quantities of the candidate molecules
into test tubes full of the AGE precursors, and see if they could inhibit
their conversion into more permanent structures.
As it turned out, however, "straightforward" did not mean "quick."
After having run dozens of tests at different concentrations and still not
having nearly exhausted the range of experiments they wanted to do, Egan
was looking for a faster way to run the experiment. In collaboration with
scientist Sara Vasan, he devised an alternative method. He wasn't sure this
new method would work, but they would certainly save a lot of time and ef
fort if it did.
At first, the new procedure seemed to work fine: a lot of the work was
moved from the old protocol to the new one, and soon they had accumu
lated a broad enough sample of data to expect that the answers they
needed would be buried somewhere inside their mountain of notes. Vasan
gathered the results together and began writing them up for internal analy
sis and possible publication.
And at first, it seemed that they had their results: the test tubes con
tained varying amounts of AGEs, suggesting that the compounds had in
hibited their formation from their precursors to varying degrees. But a few
of the results seemed to be wildly out of step from the main body of work,
with the levels of the expected reaction products being well in excess of
what could be accounted for by the small variations in concentrations and

184 E N D I N G A G I N G
other factors as compared with other, similar tests using the same com
pound. The chemistry just didn't make sense.
Embarrassed at having to ask her supervisor, and afraid that she had
simply overlooked something or that she or one of her coworkers had im
properly performed the experiments, Vasan showed the results to Egan. He
agreed that the results didn't make sense, and they began going back to the
original lab notebooks to double-check the results.
It didn't take long to see that the outliers were coming quite consis
tently from experiments using the new, faster protocol. Egan and Vasan
went back over the protocol, looking for a flaw—the kind of "garbage in,
garbage out" error that makes an accounting program tell you that you owe
twice your annual income in taxes. Eventually, they found a mistake in the
last few steps in the initial production of the model Amadori diones them
selves. At one key point, the correct procedure is to put a halt to the reac
tions occurring in the test tube, preserving the compounds that have been
produced. Instead, the protocol was allowing further reactions to occur,
generating alpha-diketones instead of freezing them at the precursor phase.
They had, in effect, been "overcooking" their biochemical soup, generating
mature alpha-diketone linkages and leaving few or no intact Amadori
diones available against which Ulrich's carbonyl-busters could be tested.
But while this was clearly a mistake, Egan and Vasan doubted that it
was the only one, because it couldn't fully account for the anomalous re
sults of the inhibition tests. The results of those experiments had initially
looked right, because after the inhibitors were mixed in with their test com
pounds, their quick-and-dirty assay methods had detected the remnants of
shattered Amadori diones floating like molecular flotsam in the test tubes.
But how would such chemical debris have been produced if there had been
no Amadori diones present for the thiazolium inhibitors to destroy?
It was then that it hit them. The explanation was staring them in the
face; indeed, the chemistry would've been obvious, if they hadn't walked
into the experiment with a preconceived understanding of the reactions
that they would be observing. Because they could see the broken carbonyl
groups that would be expected to be left behind after the thiazolium com
pounds had torn apart the Amadori-diones that they thought were present
in the test tubes, Egan and Vasan had been assuming that the presence of
those broken bonds meant that the inhibitors were doing what they had
been designed to do. But what if they were doing something else entirely?
What if the carbonyl groups that they were detecting in the final samples
were the remnants of alpha-diketone links that had been produced in error

B R E A K I N G T H E S H A C K L E S O F A G E 1 8 5
during the generation of the test compounds, and then torn apart by their
model drugs?
Egan felt no "Eureka!" moment of insight, however—and not only be
cause he still hadn't figured out how those alpha-diketones could have per
sisted long enough to be attacked by Ulrich's model drugs. No, his mood was
neither intellectual satisfaction nor ongoing curiosity, but a sinking recogni
tion that they had been wasting their time. The Amadori diones would have
to be resynthesized, probably using the original, time-consuming protocol,
and the inhibition assays run over again.
There was no sense in covering things up. Egan contacted Cerami and
explained the situation, apologizing for the wasted time and emphasising
that it was all going on Alteon's bill.
Egan initially thought nothing of the questions that Cerami asked
about the experiment: scientists are nothing if not inquisitive. But it did be
gin to seem odd as the doctor pressed him for more and more arcane de
tails of the procedure, the reasoning underlying his conclusions, and even
speculations about how the thiazolium compounds might have reacted
with the alpha-diketones. Unfortunately, Egan had no real idea how such
an interaction might have led to his observations: he was a bench scientist,
not a medicinal chemist. What a relief, he thought: Cerami's curiosity seems
to have gotten the better of his frustration with this setback.
Cerami hung up the phone and leaned back in his chair, his mind rac
ing. Was he missing something? Did he dare to believe what Egan was
telling him—or to accept the implications?
Trying to calm his trembling fingers, he dialed Peter Ulrich. Impa
tiently, he waited for his partner to pick up the phone. Finally, the click of a
lifted receiver. "Ulrich," said the familiar voice at the other end.
"Peter?" Cerami said, keeping his voice steady. "Can you explain to me
how one of these compounds could break an AGE cross-link?"
Reverse-Engineering Serendipity
Over the course of the next few weeks, Ulrich worked backward from
Egan's protocol and Vasan's results, developing a tentative scenario under
which mature AGE compounds based on an alpha-diketone linkage could
be broken by their new thiazolium compounds. Finally, he thought he had
the chemistry right.
If the result held, then they were really on to something. Thanks to the

186 E N D I N G A G I N G
laboratory flub, Picower and Alteon were at the center of not one, but two
breakthroughs in AGE biochemistry: one theoretical, and one of enormous
potential medical significance. First, the result implied that alpha-diketone
AGEs might be stable enough to persist in the body long enough to con
tribute to tissue stiffening without any further chemical alteration. And sec
ond: they had unwittingly designed a class of molecules that would not
merely prevent AGE from forming, but actually buzzsaw their way through
them.
The biomedical implications were startling. Imagine being able to take
patients whose bodies were already extensively riddled with cross-links,
and to give them a drug that would break the AGE apart. AGEd tissues
would be rejuvenated. Arteries would dilate outward in response to the
pulsing tide of blood; hearts would fill with the incoming flow; even skin
could become flexible again. It was just the solution that one would dream
of for advanced diabetic cases, or for people whose AGEs had built up be
cause of time, not high blood sugar. The market would be enormous.
The hard-nosed chemist in Ulrich brought him back from this vision to
the steps that lay between him and its realization. For starters, Alteon
would have to run the previous experiments again, monitoring the reac
tions at each step to provide evidence to support the theoretical chemistry
that he had outlined to explain the original result. Additionally, everything
that they had done thus far was with an AGE that had been cooked up in a
beaker: he still didn't know whether any alpha-diketone AGE (let alone the
specific molecule that Vasan had accidentally produced) ever actually
formed in the body at levels sufficient to impair tissue function. And then
there was the question of whether his test compounds would be able to re
produce in human subjects what they were doing under glass: the body's
detoxification machinery might metabolize them into inactive forms, or it
might be impossible to take enough of the stuff to have any effect.
The first few questions were answered by a more careful, intentional
repetition of Egan's and Vasan's initial experiments, which seemed to con
firm all his hopes. The results of these studies were consistent with the
hypothesis—that the predicted AGE was in fact formed from the model
Amadori diones, and that it persisted long enough to react with his thia
zolium compounds, which did indeed appear to sever the cross-link at the
alpha-diketone bridge. And based on the observed results, one particular
thiazolium compound—a chemical known as N-phenacylthiazolium bro
mide (PTB)—was an especially effective wrench with which to pull these
AGEs apart.

B R E A K I N G T H E S H A C K L E S O F A G E 187
But the fact that PTB severed a bond in an artificial AGE didn't prove
that it would break any of the cross-links that actually tie up the arteries,
hearts, and other organs of aging and diabetic humans. At this point, it was
time for Cerami, the more medically minded member of their tag-team, to
get more actively involved. The two decided to put PTB through a graded
series of increasingly challenging tests using more and more lifelike model
systems, working their way step by step from single cells up to functional
and molecular investigations in living, breathing animals.
The Manacles Fall Away
These necessary studies were again farmed out to people working under
Jack Egan at Alteon, whose lab scientists first confirmed PTB's ability to
cut through AGE using isolated, cross-linked proteins and tissues. With
each successful jump over an experimental hurdle, their optimism grew,
until they were ready to move their work into the living laboratory of dia
betic laboratory rodents. When Egan's team injected the animals with their
new compound, the results were again positive: levels of glycated proteins
bound to the animals' red blood cells dropped by over a third in the first
week, and kept dropping, going down to half of the original level after
three weeks and to just 40 percent by the end of the month. It really looked
as though they were on to something.
With that evidence to hand, Alteon scientists began giving PTB injec
tions to rodents with hearts, kidneys, and arteries hardened by AGEs, ac
cumulated over a normal healthy lifetime or in the fast-forward mode of
diabetes. Here the real excitement began to build, as PTB continued to live
up to expectations, restoring supple performance to cardiovascular systems
that had previously lost their youthful flexibility, instead of just slowing
down an inevitable decay as aminoguanidine had done. Structurally, the tis
sues of treated animals were softer and more elastic, stretching out like rub
ber bands fresh out of the pack, and readily melting away when doused
with digesting chemicals; functionally, their hearts were expanding to fill
with incoming blood like new balloons, and blood coursed through their
arteries without the large backward-rippling "echoes" of pulse that are
characteristic of old blood vessels.
They did have one problem, though, which was that PTB is too unsta
ble to succeed as a drug for human use: by the time a pill had made its
way through digestive system and the complex chemistry of the body's

188 E N D I N G A G I N G
drug-metabolizing processes, too little would be present to have a mean
ingful therapeutic effect. But Ulrich was not going to give up on so promis
ing an agent, and with a bit of work he and the Alteon chemists were able
to develop a variant on its basic structure that was not only more stable but
more active: 4,5-dimethyl-3-(2-oxo-2-phenylethyl)-thiazolium chloride. For
convenience, Alteon first shorthanded this mouthful to ALT-711 (because it
was ALTeon's 711th compound); later, the compound would be rebranded
to the more marketable alagebrium.
A drug with the ability to cleave AGEs that had already formed in the
body would have applications in diabetes as well as in a wide range of dis
eases of aging, but regulators only approve drugs for one specific indication
at a time. Wanting to carve out as exclusive a niche for the drug as they
could, Alteon strategists set their sights on developing alagebrium for con
ditions that were not already being successfully treated with existing medi
cines, and that would be expected to respond uniquely well to their new
treatment.
One of these diseases was isolated systolic hypertension (ISH), the kind
of high blood pressure in which a person's systolic reading (again, this is
the first of the two numbers that you get from a blood pressure cuff, like
the "110" in "110 over 80") is high, even though their diastolic pressure
(the second number) is fine. Systolic pressure is a measure of how much
pressure is applied to the artery wall by the surge of blood into the vessel as
the heart contracts, whereas diastolic pressure is the baseline pressure in
the arteries at rest (technically, at "diastole."). Hormonal and other factors
can actively tighten up the blood vessel, keeping the pressure inside the ar
tery high even during diastole; such effects raise blood pressure irrespec
tive of the intrinsic flexibility of the artery as a tissue. But when systolic
pressure is high despite a normal diastolic pressure, it's a sign that the ves
sel itself has become stiff, unable to expand to accommodate the incoming
rush of blood from the heart.
This nonatherosclerotic "artery hardening" is not just a concern in
people with a diagnosis of isolated systolic hypertension. As people push
past middle age, arterial stiffness becomes an increasingly powerful predic
tor of heart disease and heart attack, and indeed comes to override many
conventional risk factors like cholesterol and blood pressure when it comes
to risk of actual cardiovascular events (heart attacks and strokes). FDA and
other regulators don't recognize this "normal" effect of the aging process as
a "disease" for which they'll approve a drug, so Alteon knew that they
could never get official sanction for the use of alagebrium to treat these

B R E A K I N G T H E S H A C K L E S O F A G E 1 8 9
people; but they also knew that, once the drug had been proven to buzz-
saw through the constricting manacles of AGEs in the artery, restoring
flexibility and opening up the vessels to the systolic flow, they could vastly
expand the market for it by quietly encouraging its unapproved ("off-
label") prescription to untold thousands of aging people with age-related
arterial stiffening.
Another disease whose victims don't get much benefit from existing
drugs and would be expected to respond more specifically to an AGE-
breaker is diastolic heart failure (DHF). The more common, systolic form
of heart failure occurs when the heart's lower, pumping chamber loses the
strength to push out enough of the blood that it receives from the upper
chamber to keep the body supplied with oxygen and nutrients. But about
a third of heart failure patients have a perfectly normal capacity to pump
blood; their problem is that the same chamber can't expand sufficiently
well to take in the required volume of blood in the first place, so that the
body's needs remain unmet even after it squeezes out nearly all of the load
that it first takes in. The result is the same—the body's tissues are starved
for blood—but the cause is different, and treatments that admirably ad
dress systolic heart failure leave the bodies of DHF patients still crying out
for critical fuels. While the underlying loss of the heart's filling capacity
can be the result of a variety of factors, many cases of the disease are asso
ciated with AGE stiffening of the heart. Again, an AGE-breaking drug
would be uniquely suited to restoring healthy functionality to these peo
ple, and trials showing that it could restore the elasticity of old hearts
would also spark interest in its use by large segments of a "healthy" but
rapidly aging population.
Alagebrium proved its mettle quickly, doing everything that PTB could
do and more. Studies showed that alagebrium in the drinking water of lab
oratory animals could deliver the same kind of restoration of heart and ar
tery flexibility that PTB had elicited only via injection, and more easily. And
there were things that alagebrium could do that PTB had never been able
to pull off. For example, PTB had cleaved some of the AGEs that had ac
cumulated in the kidneys of diabetic rodents, but not enough to restore the
organ's functionality. Treat the same animals with alagebrium, and not only
does their kidney collagen become more soluble, but also the fibrotic dam
age to their kidneys recedes, and the organs get better at filtering proteins
out of the blood, preventing their spillover into the urine.
And rodents were only the first order of mammals to benefit from
alagebrium. Alteon and their collaborators soon proved that alagebrium

190 E N D I N G A G I N G
could rejuvenate the hearts and blood vessels of dogs and monkeys. These
studies were much more informative about the prospects for alagebrium as
a true anti-aging drug than anything that had come before, for a couple of
reasons. First, they were carried out in animals that were undergoing "nor
mal" aging, whereas the rodent alagebrium studies had used severely dia
betic animals. Second, dogs and nonhuman primates enjoy longer lives,
and the extra years give the forces of aging more time to induce the same
kinds of pathological cardiovascular system changes that are observed in el
derly humans, making them better models of human disease from a clinical
and theoretical point of view.
As in elderly humans, older dogs' heart chambers stretch less to ac
commodate incoming blood than do those of younger animals, leading to
reduced filling with blood and a simultaneous increase in the pressure
within it. In other words, old dogs suffer from mild diastolic heart failure.
When older animals were given a moderate dose of alagebrium for a
month, their hearts became about 42 percent more flexible, as demon
strated by an increase in the volume of blood taken up in the absence of
any increase in blood pressure inside the chamber. The contrast was even
more marked when the volume of blood delivered into the heart pump
chamber was increased using drugs: just weeks before, this treatment had
even further widened the performance gap between old and young dogs in
cardiac flexibility, but after alagebrium treatment their hearts were nearly
as elastic as those of the young controls. 1 8
The results seen in our fellow primates were even more striking. 1 9 In
2001, scientists from Alteon—working in collaboration with researchers
from the National Institute on Aging (NIA) who were studying the effects
of aging and calorie restriction on nonhuman primates, as well as with NIA
specialists in cardiovascular medicine—published the results of a study on
the effects of alagebrium in the cardiovascular systems of rhesus monkeys.
Their test group was old, but "healthy" as biologically old monkeys go—
and in particular, free of diabetes.
At the beginning of the study, the monkeys' arterial flexibility was as
sessed, as was the degree to which their heart chambers ballooned outward
during their blood-filling (diastolic) phase, as a measure of the flexibility of
the tissue. The monkeys then received alagebrium every other day for three
weeks, after which their tissues were retested every few weeks for the next
nine months.
Surprisingly, there was no measurable effect on blood pressure—
systolic or diastolic. But three weeks after treatment, and even more pro-

B R E A K I N G T H E S H A C K L E S O F A G E 191
foundly at the six-week mark, their cardiovascular systems' tissues had
clearly undergone a restoration to more youthful elasticity. Using one rough,
easy-to-administer test of arterial flexibility, their arteries had become
an astounding 60 percent more pliable; a more direct assay revealed a
25 percent improvement. At the same time, their hearts were also opening
up more easily: they were taking in 16 percent more blood during the dias
tolic phase, and other measures of cardiac function at least partially de
pendent on improved diastolic filling likewise improved after alagebrium
treatment.
Alagebrium wasn't expected to prevent new bonds from forming be
tween sugars and proteins, so it was no surprise that the withdrawal of the
drug was followed by the loss of these gains once the gradual molecular
manacling of the monkeys' tissues was no longer being counteracted by an
even more rapid breaking of those bonds. Within a few weeks of the peak
of their alagebrium-inducell return to more youthful suppleness, the mon
keys' arteries were once again as stiff as they had been in the initial run-up
to the study. Their hearts held onto their gains a little longer than the arter
ies, but then they too began tending to fall back into their old recalcitrance.
Quitting the drug didn't leave the monkeys any worse than they had been
to begin with—but it was clear that the AGE links being broken by alage
brium could be quickly reforged. The implication is that users of alage
brium would have to take the drug on an almost continual basis in order to
keep enjoying their newfound arterial plasticity.
But that didn't much dampen anyone's spirits. The results of these
studies marked a clear landmark in the development of alagebrium. Toxic
ity was low; no serious side effects had been observed; and the promise of a
new treatment for stubborn diseases was clear.
It was time for human trials.
From Darkness, Light
The first human alagebrium trial, published in the prestigious American
Heart Association journal Circulation in 2001 , 2 0 looked like the tentative
beginning of something big. Seventy-three older men and women with
signs of vascular stiffening had their blood pressure and arterial flexibility
assessed and were then p l a c e d at random into one of two groups. For two
months, two-thirds of the patients took alagebrium in pill form; at the same
time, the remainder received a look-alike pill with no active ingredient as a

192 E N D I N G A G I N G
placebo control. At the one-month mark, and again at the end of the trial,
their parameters were reassessed.
The results were not altogether clear, allowing for a range of interpre
tations, but the study was understood to be preliminary by its very nature,
and most researchers were willing to give the drug the benefit of the doubt
based on the remarkable results achieved in the animal models. Systolic
and overall blood pressure had gone down in both groups, probably be
cause of an unusually strong "placebo effect" in the group getting the
dummy pill: the influence of the power of belief on the actual state of the
body, which is a notoriously important confounder in hypertension studies.
Whatever the reason, the result was that the drug conferred no clear ad
vantage in blood pressure results. At the same time, the arterial stiffness of
the people taking alagebrium seemed to have improved in comparison to
the placebo group using two different measures, but there were technical
objections to the method used in one of these assays and the nature of the
comparison between the groups also made its results less than decisive.
Subsequent trials, however, have provided results that, in aggregate, al
low us to draw firmer conclusions—and, unfortunately for Alteon, they
suggest that alagebrium will never be approved by regulators for clinical
use. Over a thousand patients with systolic hypertension, diastolic heart
failure, systolic heart failure (with and without an associated, compensatory
overgrowth of the heart's main pumping chamber) and even erectile dys
function, as well as some healthy individuals, have been treated with alage
brium in early clinical t r i a l s , 2 1 , 2 2 , 2 3 , 2 4 and while the results provide enough
evidence to suggest that the drug is safe and is breaking AGEs in these pa
tients, the effect is clearly insufficient to have much impact on actual func
tion. The results on diastolic function in the heart have not been impressive;
the benefits in improved arterial flexibility have not been clear-cut; and lit
tle, if any, effect on hypertension per se has been observed. Often the main
objective of the trials has not been achieved, with the benefits mostly ac
cruing in less-important markers of the disease process that are not clearly
linked to clinical outcomes (cardiovascular disease, heart attack, or stroke).
Moreover, the benefits that have been observed have not been cleanly tied
down to any particular dose of the drug. This is paradoxical because one
might well expect that, in a drug that breaks AGEs, benefits would increase
with the dose: more drug ought to mean more broken AGEs and therefore
more youthful cardiovascular systems.
To date, the sum of the data from the animal studies clearly suggests
that alagebrium can break AGEs; the question is why this benefit is not

B R E A K I N G T H E S H A C K L E S O F A G E 1 9 3
translating into improved vascular and cardiac health in human users as
they do in so many other species.
Some critics hold that alagebrium is not actually an AGE breaker after
all, but an AGE inhibitor, just as Ulrich and his colleagues originally de
signed it to be. There is a certain superficial plausibility to this view, but
these arguments can't stand up against the irrefutable fact that, in animal
studies, alagebrium doesn't just slow down the development of complica
tions in diabetic rodents on prevent the AGE-related tissue stiffening of the
cardiovascular system in normally aging dogs and monkeys: it reverses
them. A drug that only inhibited the cross-linking of tissues would be able
to reduce the rate at which new cross-links would form, and thereby slow
down the degeneration of cross-linked tissues—but it would not have the
kind of rapid restorative effects that have been elicited by alagebrium.
The fact that the tissues of alagebrium-treated animals become inflexi
ble again so quickly after withdrawal of the drug also seems to weigh in
against the suggestion that the drug is actually just reducing AGE forma
tion, since the underlying cross-links are clearly reforming much more rap
idly than they did over the many years that were required for their initial
buildup. This observation suggests that the severing of the alpha-diketone
bridge in these AGEs exposes a highly reactive carbonyl group, which soon
sticks itself back onto an adjacent proteins. Because of the ongoing break
ing of other cross-links, users of the drug keep ahead of this problem in
"two steps forward, one step back" fashion—but take it away, and they un
dergo a rapid return to their old, AGEd state.
What about the inability of researchers to find alpha-diketone cross
links in the body? The reason is almost certainly that these structures are,
ironically, a bit too easy to destroy. The difficult thing about designing an
AGE-breaker drug is not that there's any lack of chemicals that can break
apart a given cross-link; the problem is to come up with something that
won't also tear normal, healthy proteins to shreds in the process. The com
mon ways of finding AGEs in the body involve soaking a tissue sample in
strong acids and examining whatever's left over. This technique catches the
most extremely hard-to-destroy AGEs, such as pentosidine, but wipes out
all trace of more delicate cross-links.
It was long suspected, and has in recent years been confirmed, that the
crudeness of such assays introduced serious distortions in AGE research.
In the last decade, new methodologies have been developed to uncover
AGE cross-links in tissue through a painstaking process of breaking down
the normal, healthy chemical bonds in a tissue almost one by one, leaving

1 9 4 E N D I N G A G I N G
behind only abnormal chemical linkages such as AGEs. Using these tech
niques, researchers have proven that the AGEs we previously thought to be
the most abundant are actually just the most resistant to the chemical
carpet-bombing that had previously been used to drive them out of hiding.
The most readily-assayed AGEs (like pentosidine) are in fact relatively rare
in the body (and therefore make little contribution to the overall state of tis
sue stiffening), while other cross-links that are much more common (and
therefore impose a much larger total protein-shackling burden on living tis
sues) remained invisible to our testing methods.
I believe that this is the explanation for our inability, thus far, to iden
tify the molecular targets of alagebrium. The predicted structure of alpha-
diketone cross-links is such that they would be relatively easy to break
apart: indeed, you'll recall that this is why Peter Ulrich originally didn't
think that they would even hang around for long enough to be worthwhile
pursuing as a drug target.
In turn, the unfortunate difference in the functional impact of alagebrium
treatment in human patients, as compared with lab rodents, dogs, and mon
keys, may be the result of alpha-diketone cross-links simply being a much
more common kind of AGE in those species than in our own. It's clear that
there are differences in the metabolic pathways underlying AGE formation
amongst the species. For instance, as we saw above, diabetic rats' bodies
suffer much more oxidative stress than ours do in response to the disease.
This should affect not only how AGEs are generated, but which specific
cross-links are formed: structures whose formation involves free radicals
will probably be a much bigger burden in rat tissues than in human ones.
Another reason to think that alpha-diketone cross-links may be less im
portant to our own species than to others is the simple fact that we're much
longer-lived than those other organisms. Long-lived, recalcitrant AGEs like
pentosidine are very hard for the body to break down, so they tend to ac
cumulate pretty linearly with age: the result is that, while longer-lived or
ganisms accumulate them more slowly than shorter-lived ones, they wind
up with higher absolute levels of such AGEs by the time they end their
lives, simply because they've had many more years to accumulate them. So,
if you look at Figure 2, you'll see that at fourteen years of age, an extremely
"elderly" dog has about forty units of pentosidine in a milligram of its col
lagen, while a minipig of the same age—but with half of its maximum life
expectancy still ahead of it—has only fifteen units. A monkey could possi
bly live for forty years, and at age ten it has only accumulated five units of

B R E A K I N G T H E S H A C K L E S O F A G E 1 9 5
pentosidine. A human, with a maximum life span of more than hundred,
has fewer units still. Yet by age sixty, when AGE cross-links are beginning
to weigh in seriously on a person's chances of surviving for another year,
human skin is burdened with some fifty units of pentosidine cross-linking
its proteins per milligram of collagen—more than any of the shorter-lived
species had time to accumulate.
Now: remember that, in contrast to a supremely tenacious cross-link like
pentosidine, alpha-diketone cross-links—the kind broken by alagebrium—
are predicted to be relatively fragile as AGEs go, so the level of these cross
links is an equilibrium between rather rapid formation and breakage. Like
all AGEs, the decline of metabolic control of fuel as we age would lead
to an increase in the rate of its formation with age—but its relative ease
of elimination should allow the body to largely keep on track of this in
crease, leading to a much slower rate of accumulation than the stubborn
pentosidine.
The net result of this would be that, late in life when AGE-inducell
stiffening is becoming rapidly fatal, the contribution that alpha-diketone
cross-links would make to the total burden of AGE (and thus, to loss of
needed flexibility) in a tissue would be less in a long-lived species like ours
than in a monkey or a dog (let alone a mouse), for the simple reason that we
would have accumulated so much more of the more resistant types of AGE
than shorter-lived creatures ever get the opportunity to do. Thus, an alpha-
diketone breaker like alagebrium would, even if highly effective at its spe
cific molecular task, leave a much greater burden of other cross-links
behind than would be the case in model organisms, resulting in much less
effective restoration of youthful tissue plasticity.
Alagebrium and Beyond
So where does this leave alagebrium in the SENS agenda? Clearly, the lack
of clear-cut clinical benefits in humans indicates that this drug itself will not
play a major role in the reversal of cross-link damage in our tissues. Its
value, instead, is as a proof-of-principle: it shows us that AGEs can be
cleaved in the body, and tissues regenerated, long after metabolism has
done its dirty work in binding our proteins together. What will be required
is a new generation of AGE-breakers that slice through more abundant
AGEs, and thus free us of the structures that are really holding our tissues

1 9 6 E N D I N G A G I N G
immobile. Such agents will yield the same kind of benefits in humans that
alagebrium does in animals—benefits first demonstrable in diabetes, ISH,
and diastolic heart failure, and ultimately in aging itself.
It's important to stress that no single drug will totally save us from tis
sue cross-linking. As we've seen, glycation leads to the formation of many
different AGEs, each with a distinct structure. Drugs that will sunder any
given AGE will probably leave most others untouched: no one molecule
will be able to sever all of these distinct intermolecular linkages. Therefore,
as we saw with amyloids in Chapter 8, we will need to develop a range of
drugs, each of which cleaves either one specific cross-link structure or at
most a small family of similar ones.
But the eventual need to develop drugs to break a number of distinct
AGE structures doesn't mean that we can't effectively stop AGEs from
contributing to the aging of our hearts, arteries, and other tissues until we
have a solution for all such cross-links in hand. The insight underlying the
"engineering" school of anti-aging biotech tells us that we don't have to
solve all of our problems at once to intervene in the process: We can "reju
venate as we go," taking one challenge at a time.
To see why this is so, remember that the molecular damage underlying
aging begins accruing while we are still in our mothers' wombs, yet we re
main youthful well into our thirties: it takes many decades of these insults
before the amount of damage is sufficient to exert a functional impact on
our bodies. Until this threshold level is reached, a given form of aging dam
age is essentially harmless to us in itself.
Therefore, to restore more youthful flexibility to a tissue, we do not
need to sever all the various kinds of AGE cross-links in our bodies, but
only the ones that make the greatest contribution to the stiffening of our
tissues. For practical purposes, rejuvenation will be effected as soon as we
can cleave a sufficient proportion of the AGE structures in our bodies to
keep the total amount of cross-linking beneath the threshold level that ac
tually impairs tissue function.
Once we have a drug that breaks a given kind of cross-link, we will be
free of its baleful influence. The solution, by its nature, will not be once and
for all: we will undergo a new course of treatment once every few years or
decades, taking the drug for a few weeks or months—long enough to break
enough AGEs to leave our tissues as flexible as they were in our youth.
These cross-links will immediately begin to build up again, of course—but
we have the luxury of letting them do so until that critical threshold level is
approached again. (Note that this scenario assumes that the AGE-breaker

B R E A K I N G T H E S H A C K L E S O F A G E 197
leaves the broken AGE in a chemically inert state, which alagebrium evi
dently doesn't do to alpha-diketones; if the broken AGEs are reactive, the
drug will have to be taken continuously.) However, the first effective AGE-
breaker will not solve the entire problem of AGEs. It will probably reach
the clinic first by virtue of targeting the most abundant AGE, but it will
surely not break all AGE species. Thus, other AGE cross-links will con
tinue forming in our bodies unabated, albeit at a slower pace. These cross
links will first reach pathological levels once our lives have been extended
for long enough that they stiffen our tissues on their own as much as they
and the first-targeted AGE jointly do in a currently normal lifetime.
So, yes, we will need to identify these AGEs as well, and to develop
treatments that target them. But the important point from an engineering
perspective is simply that this doesn't emerge as an actual biomedical prob
lem until the first wave of anti-aging (including anti-AGEing) treatments
has extended our lives quite a bit on its own. The first breaker of an abun
dant AGE will buy us the time to identify such AGEs and to develop new
treatments that will free us from them in turn.
Eventually, we will develop a lifelong regimen of AGE-breakers not
unlike the childhood vaccine schedule today, under which we will receive a
series of specific cross-link-severing drugs, each administered on its own
cycle of years, decades, or perhaps even centuries, based on the rates and
sites of their targets' formation in the body. But to achieve the first great
leap forward in restoring the suppleness of youth to AGEd tissues, we need
only prioritize the development of cross-link breakers that will carve their
way through the cross-links that cause us clinical problems within a
presently normal lifespan.
The above section only discusses AGEs, but similar logic applies right
across the SENS spectrum. I'll be discussing it in more detail in Chapter 14.
Know Your Enemy
Today, for the first time in history, we are in the position to design such
drugs on a rational basis. Just over a decade ago, when Peter Ulrich and the
Alteon chemists were doing the work that ultimately led to the develop
ment of alagebrium, they were working in the dark. They didn't even know
for sure that the types of AGE that they were targeting actually existed in
the body—they were simply guessing, from studies carried out in test tubes,
that such links might form and contribute to stiffening of living tissues. But

198 E N D I N G A G I N G
the new enzyme-based methods that I mentioned earlier now allow us to
slowly take tissues apart at the molecular level, layer by layer, revealing the
presence and levels of pathological cross-links in our body, exposing our
previously hidden opponents to the light of science.
Researchers who have taken the time to develop and apply these new,
painstaking procedures to aging human tissues have given us reliable tar
gets on which to fix our crosshairs: a complex structure called glucosepane,
which was only identified using these new techniques in 1999, 2 5 and prob
ably another AGE called K2P, which is prominent in the lenses of our eyes
and possibly other tissues. 2 6 Glucosepane is the single most important con
tributor to the body's AGE burden known to date, tying up as much as one
out of every five molecules of the key structural protein collagen in old,
nondiabetic humans. Glucosepane levels are around one hundred times as
high as that of any other AGE that had previously been found in collagen
or the lens. A drug that could free our tissues from these AGE shackles
would have a much larger impact on total cross-linking burden than alage
brium does, and would thus bring our tissues much closer to their full
youthful flexibility and functionality.
Today, while we will still use the same sort of blackboard molecular en
gineering that Ulrich and his copanelists did in the early '90s to design new
AGE-breakers, we can fashion molecular bolt-cutters that are precisely
tooled to break specific AGEs, whose exact molecular structures are in our
possession, and whose presence in our bodies and biomedical significance
as major contributors to the total cross-linking of our tissues with age are
certainties.
We also have the advantage of having faster screening tools in our pos
session. We can use software to simulate the behavior of AGE-breakers, and
to automate the generation of variations on a core molecular theme. We can
put robotics to work to synthesize thousands of ampoules of candidate drugs,
and use mechanized, high-throughput techniques to assay their effects
against a known culprit in the loss of elasticity in our hearts and other tissues.
Being able to look at glucosepane's exact chemical structure gives us an
other advantage in developing its nemesis that Ulrich no doubt wishes he
had had so many years ago. The hard part of developing a glucosepane-
breaker for clinical use is not identifying chemicals that can destroy it: as we
saw earlier, the acids we used in our old AGE-assaying techniques did a re
grettably excellent job of it. The problem is to create molecules that will do
it selectively, without damaging healthy biomolecules that share the same
vulnerability exploited by the would-be drug. Having reconstructed glu-

B R E A K I N G T H E S H A C K L E S O F A G E 1 9 9
cosepane's molecular identity, biochemists can now see just how wide is the
latitude they have in designing molecular shears for it.
Fortunately, this latitude may be very wide indeed. The structure of
glucosepane is so different from any functional structures in ourselves or
other mammals that a drug that selectively targets them should be harmless
to any molecule that is supposed to be found in our bodies.
As I've explained, the resulting glucosepane-breaker should be the first
in a series of AGE-cleaving drugs that we will need to unbind our proteins
from their molecular shackles. Then we will have achieved for the first time
in humans what was observed just a few years ago in dogs and monkeys
treated with alagebrium. Old hearts will open wide again, free to fill with
life-giving blood. Hardened old arteries will once again readily expand in
response to the surge of the blood of life. The stiffened, inflexible tissues of
the aged will move with the suppleness of youth. The absurdity and outrage
of a body tying itself into molecular knots will come to an end, and we will
bend and flex within and without as children in the jungle gym of life.

10
P u t t i n g t h e Z o m b i e s t o R e s t
As we age, we build up an increasing collection of "death-
resistant" cells in our tissues. This is one part of our
biochemical program to avoid cancer: shutting down the activity
of potentially cancerous cells before they can cause trouble.
Unfortunately, rather than simply remaining silent and harmless,
such cells still manage to damage surrounding tissue through
chemical signals gone awry. But by taking a leaf from the
world of new, targeted cancer therapies, we can foresee the
development of safe, effective methods to remove these senescent
cells from the picture.
So far, I've mostly been talking about specific forms of damage that
happen at the molecular level to our cells and their components, and how
we can restore functionality to our cells and tissues by undoing, or render
ing harmless, that damage. But there are a few cases where the aging body
accumulates cells that are damaged in such a way that they don't just stop
contributing to the economy of the body, but actually become toxic to the
system that supports them.
I've already discussed one such case, back in Chapter 5: cells that have
been taken over by mutant mitochondria. When mitochondria lose the
ability to process fuels, as a result of mutations to their internal DNA, what
ultimately causes us harm is (in my view) not the resulting failure of these
organelles to carry out their job. Rather, it's the maladaptive way that their

P U T T I N G T H E Z O M B I E S T O R E S T 2 0 1
host cell alters its metabolism in order to survive that failure. This meta
bolic alteration keeps such cells limping along by dumping oxidative stress
outside their membranes and on to far-flung areas of the body.
At first glance, one might think that the best thing for the body to do
with such cells would be to kill them off, thereby saving the rest of the body
from their toxic influence. But the nature of the specific cells that develop
this problem makes any attempt at simple removal fraught. The most no
table case, arguably, is skeletal muscle. The design of muscle means that de
stroying a single muscle cell-like structure will snap the entire fiber in
which it's embedded. Loss of muscle cells to aging (rather than to disuse) is
already a major source of age-related frailty; we can't afford to add to that
problem by killing off more of them in self-defense.
So in this particular case, as I described in Chapter 6, what seems to
make the most sense is to find a way to preserve and restore normal meta
bolic activity in affected cells in the face of their colonization by mutant mi
tochondria, instead of killing them.
However, there are plenty of other cases in which the costs of destroying
a toxic cell are negligible, and the benefits clear and direct. Everyone is fa
miliar with one such case—cancer—and no one disputes that destroying
cancer cells is unambiguously positive. I won't be discussing cancer in this
chapter, however (except for the applicability beyond cancer of some exist
ing anti-cancer treatments), because that disease poses such unique chal
lenges that I've devoted a whole separate chapter (Chapter 12) to it. Instead,
I'll focus on three cell types that pose a much less catastrophic threat than
cancer, but that still make a substantial collective contribution to age-related
descent into illness, frailty, and death. From what I can see, there is no rea
son to attempt to rehabilitate these cells: it seems best that they, like cancer
cells, be destroyed. I'm choosing to discuss them together because of the
similarity of the threats that they pose and of the strategies that I advocate
for dealing with them.
Attack of the Clones
The decline of the immune system is one of the most deadly effects of ag
ing. Infections that young people shake off as mere inconveniences are
commonly fatal in the biologically old. Influenza, for instance, puts 114,000
Americans into the hospital each year, and flu and flu-related illnesses claim
the lives of about 51,000. But the disease burden is dramatically skewed

202 E N D I N G A G I N G
Figure 1. Aging vulnerability to pneumonia and flu. (a) Hospitalization rates by age, Connecticut, 1993-1997 (females). Redrawn.1 (b) Death rates by age. Redrawn from CDC data.2
toward people who have previously suffered the ravages of aging (see Fig
ure 1). Deaths from influenza and influenza-associated pneumonia are al
most unheard-of in adults until the seventh decade, after which rates climb
exponentially. In the United States, over 90 percent of all deaths from the
two diseases are in people over sixty-five years of age.
We could, of course, do something about the death toll through vacci
nation. But not much: between 30 percent and 75 percent of older people
fail to respond to flu shots, compared with just 10 percent of young adults.
Add to this the facts that we sometimes vaccinate against the wrong strain
of the flu, limiting the effects of even successful vaccination. There are var
ious reasons for the weakening of the immune system that happens with ag
ing, some of which are ultimately downstream effects of aging elsewhere in
the body (like the systemic increase in free radical stress spread by mutant
mitochondria). But one of the most profound—and unexpected—factors
underlying our inability to mount a defense against infections that young

P U T T I N G T H E Z O M B I E S T O R E S T 203
people shrug off without ever suffering a sniffle is, believe it or not, a form
of immunological overcrowding.
State of the Forces Report
There are two main branches to the immune system. One is the so-called
innate immune system, whose "innateness" comes from the fact that its job
is so general that it doesn't have to "learn" to identify a specific enemy. Its
job is similar to that of regular soldiers on patrol in a demilitarized zone,
trying to maintain order but unsure of who might be the enemy, ready to
confront anything suspicious-looking that they happen upon. There seem
to be very few changes in the innate immune system with aging, and those
that have been reported appear to be secondary to other aspects of aging
and age-related disease (and we'll be discussing a few of those later on), or
to factors that are common in the elderly but not a result of biological aging
at all, such as vitamin and mineral deficiencies.3
The other main branch is the adaptive immune system. The adaptive
immune system is more like a division of highly trained special forces units,
each of them expert in waging targeted, tactically sophisticated warfare
against specific enemies. This branch is responsible for the ability of the im
mune system to learn about invaders—and, thus, for the effectiveness of
vaccines.
Within the adaptive immune system are the B cells and T cells. B cells
are mostly responsible for defending us against pathogens like bacteria and
parasites that are purely foreign to the body, and that can therefore be tar
geted directly for destruction. B cells recognize specific markers (antigens)
on the surface of such an invader that reveal it as foreign, and churn out an
tibodies to them. I talked about antibodies back in Chapter 8, in discussing
vaccination as a way of clearing out amyloids: they destroy alien cells by
binding to the antigens of the organisms they're intended to fight, acting
like homing beacons that attract "missiles" fired by other components of the
immune system, or blocking receptors and other proteins that are needed
for the pathogen's survival.
By contrast, cytotoxic T cells (also called CD8 cells because of the char
acteristic receptor they bear) are responsible for rooting out the enemy
within: cells that are native to the body but that have now been turned
against it, such as cancer cells or cells hijacked by viruses. (There are other
types of T cell in addition to CD8 cells—more on them later.) CD8 cells

2 0 4 E N D I N G A G I N G
also use antigens to target their foes, but because the targets are hiding in—
or in the case of cancer, as—the body's own cells, CD8s don't get the
chance to catch the pathogens' calling cards on their own surfaces, and
can't target the invaders directly. Instead, CD8 cells pick up antigens on the
surfaces of host cells that have been infected by a pathogen, or more often
on other cells in the immune system that act like reconnaissance agents,
scooping up copies of the antigen left in the wreckage of cells destroyed by
the enemy and reporting their findings back to CD8 cells to alert them to the
threat. Having spotted enemy colors, CD8 cells seek out and destroy the in
fected host cells, eliminating their threat to the rest of the body.
The problem, once again, begins with the body's need to balance com
peting priorities in its metabolic processes—and to do so in the face of lim
ited resources. On the one hand, it's critical that the immune system be able
to identify and fight off infectious agents that it's never seen before, so it
needs to have a reserve of CD8 cells that are ready to respond to new
threats, "learn" about their key antigens, and then mount an attack; these
are called naive CD8 cells. On the other hand, the process of ferreting out
an enemy that you don't recognize takes time, during which an invader
could gain a life-threatening foothold in the body, so we also have a com
plement of memory CD8 cells—veterans of old immunological battles,
which remember the enemy that they defeated and stand ready to identify
and fight them off again.
B a l a n c e d Budgets
This would be fine if we could keep on hand as many T cells as we might
like, including plenty of naive cells and large contingents of memory cells
specific to each of the many pathogens that our body has racked up in its
rogues' gallery over the course of the years. But producing and maintaining
these armies is a resource-intensive investment, and as with everything else,
the body's "budget" for the immune system is limited. To avoid going into
deficit on its "military" spending, the body maintains a strict policy of bal
a n c e d budgets—a limited amount of "immunological space" (as it's been
called) for the entire T cell population in aggregate. The immune system
ruthlessly maintains a cap on the total number of naive and memory cells
combined in the body at any given time, although the specific makeup of that
population is in constant flux, shifting dynamically as the body responds to
the threat of the moment.

P U T T I N G T H E Z O M B I E S T O R E S T 2 0 5
When this system is working well—as it does in most young people—it
is the very model of the kind of flexible, low-cost, highly mobile, well-
trained army that many of today's generals and world leaders dream of con
structing. During an infection with a particular pathogen, there is a rapid
redeployment of forces to meet the threat on the ground. Whether it's mem
ory cells mobilizing against an enemy that they've seen before, or naive cells
uncovering and mounting an attack on a brand-new threat, CD8 cells ap
propriate to the enemy at hand expand their numbers, dividing rapidly in a
process called clonal expansion, and then fan out, identifying and destroying
cells bearing the foreign protein markers against which they specialize. (This
use of the term "clone" is one of several in biology; it must not be confused,
let me stress, with the popular nonscientific use of the word. I'll have more
to say about the various meanings of "cloning" in the next chapter.)
But once an enemy has been defeated, maintaining huge numbers of
CD8 cells whose only mission is to wage war on a foe that has just been
driven away would be a waste of limited resources. With the body's iron dis
cipline on its immunological budget, it can't afford to have so much of its
army be specialized for combating just one opponent if that enemy isn't ac
tually in the process of waging a campaign. So the body initiates a rapid and
massive scaling back of these cells, ordering the bulk of the veterans to en
gage in a carefully orchestrated self-destruct program (apoptosis), after which
it can rebalance its deployment of forces to a more generic defensive posture.
But a few veterans of the recent conflict are kept on after the cessation of hos
tilities as memory cells, on the lookout for signs of a renewed attack from the
invaders that they know so well. The small numbers required to maintain the
body's vigilance against a known enemy make this expense quite tolerable, so
that the cost of keeping these cells on the payroll never puts significant strain
on the "budget" of the immune system. That's the plan, anyway.
Old Soldiers Never Die . . .
Unfortunately, this model of fiscal and military discipline only works well for
infections that can be totally eliminated from the body. It begins to break
down when the body faces enemies that it can fight to a standstill but not
quite wipe out entirely. One class of such enemies is viruses of the herpes
family: not just the infections commonly called "herpes" (herpes simplex of
the mouth or genitals), but also Epstein-Barr virus (the one that usually
causes glandular fever), varicella zoster (which causes chicken pox), and

2 0 6 E N D I N G A G I N G
most especially a little-known infection called cytomegalovirus (CMV). All
these viruses can be beaten back enough to put an end to active, sympto
matic disease, but they are never completely defeated. A few copies of the
virus continue to lurk hidden in some hard-to-reach corner of the body, dor
mant and out of sight of the immune system, waiting for the day when the
tissue or the body as a whole is in such a weakened state that they can flare
up again. In fact, the very name "herpes" is taken from the Greek herpein,
"to creep," in reference to their ability to sneak about the body while they
await conditions favorable to their reactivation.
You may never have heard of CMV, even though the odds are good that
you are carrying it (up to 85 percent of adults over the age of forty do).
That's because CMV rarely causes a recognizable illness, even briefly:
about half of those undergoing CMV infection or reactivation suffer no
symptoms at all, while the other half are afflicted only with hard-to-
diagnose, nonspecific complaints such as general malaise, fever, and sweats.
But new research is showing us how CMV (and probably some other
viruses) can also cause serious long-term harm to those of us who only suf
fer mild and transient activation and reactivation of the virus. Because the
body can never quite consolidate its victory against these viruses, anti-
CMV memory cells get called up to active duty again and again, and over
successive iterations they gradually begin to ignore the apoptotic signal that
is supposed to scale back their forces at the cessation of hostilities. There
are various theories as to why this might happen, but I think it's most likely
to be part of a complex adaptation to protect us against uncontrolled cell
division (i.e., cancer) in these cells. 4 Whatever its origin, the inability to re
call these veteran troops progressively weakens the immune system's ability
to fight other infections, new or old. The iron limitations on the "immuno
logical space" or "military budget" ensure that, when the body can't cull
unneeded T cells specific to CMV or other infections, it has to make up the
numbers with other immunological soldiers. As a result, the numbers of
naive cells available to keep the body ready to face new threats, and of
memory cells for other pathogens, dwindle to dangerously low levels. 5
. . . They Just Fade Away
It's bad enough that death-resistant anti-CMV veterans refuse to take their
scheduled retirement, preventing needed redeployments and the hiring of
new recruits. But the situation is actually worse than this. These problem-

P U T T I N G T H E Z O M B I E S T O R E S T 2 0 7
atic clonal expansions don't just refuse to make room for other soldiers to
do their job: like crippled or aged fighters, these weakened (the immunolo-
gist's term is anergic) T cells can't even carry out their own duties. 6
One of the most important factors crippling these anergic T cells ap
pears to be the loss of a key cell-surface receptor called CD28—an effect
that has been observed in humans 7 and animals. 8 T cells are alerted to the
presence of enemy forces by antigen-presenting cells (APCs), the immune
system's reconnaissance teams, which identify enemy combatants' antigens
through direct encounters with them or by digging through the rubble of
old battlegrounds (the remains of cells ravaged by them). When T cells lose
CD28, APCs can't recognize them to alert them to the danger, and their in
telligence report gets filed away unread. CMV-specific CD8 cells are un
usually susceptible to this loss.
Another problem with anergic CD8 cells is that, having staked out a
huge territory for themselves and thereby squeezed out other T-cell popula
tions, they simultaneously lose the ability to reproduce themselves. Memory
T cells normally express a receptor called KLRG1, which is there to keep
them from proliferating when no infection is present. But healthy cells bear
ing KLRG1 are able to reproduce when a threat is actually present. Usually.
Anergic CD8 cells have KLRG1 present on their surfaces,9 but they also
have another marker on their surfaces called CD57, which is lacking in nor
mal memory cells. 1 0 When CD57 is present at the same time as KLRG1, the
cells' ability to reproduce themselves is locked down tight, so that they still
can't transform their reserve division into a full-fledged army when their
sworn enemy is swarming over the ramparts.
Also contributing to this cellular "infertility" is the fact that the same
cells have short telomeres-—the long stretches of nonsense DNA that cap
our chromosomes' ends. Anergic T cells hit this problem because, unlike
most immune cells, they have lost the effective activity of the enzyme telom-
erase, which is required to renew telomeres. Most cells don't express telom-
erase, but it's essential to the healthy functioning of CD8 cells because they
are called upon to expand their numbers quickly and frequently over the
life span in response to new infections. So the lack of strong telomerase ac
tion is a further mechanism of the enfeeblement of these cells. See the side
bar, "Backgrounder on Telomeres and Telomerase," for more information,
which will be amplified in Chapter 12.

2 0 8 E N D I N G A G I N G
B A C K G R O U N D E R ON T E L O M E R E S AND T E L O M E R A S E
Every time a ceil divides, it must make a new copy of its DNA.
The enzyme responsible for doing this—called the DNA
polymerase—is a bit like a molecular monorail train, zipping for
ward along the "guide rail" provided by the DNA strand that it is to
replicate. As it travels along the "guide rail," the polymerase en
zyme makes a letter-by-letter copy of the strand beneath it, spool
ing the new, replicated strand out to the side as it goes.
DNA polymerase has a fundamental shortcoming, however.
For reasons whose details needn't concern us here, the machin
ery never quite manages to replicate the entire strand of DNA. A
small amount of a chromosome's DNA material is therefore lost
with every round of cell division, leaving the copied strand shorter
than the original. Eventually, the end of the chromosome is eaten
away.
A second problem that our cells have to solve regarding our
chromosomes is that they often break, due to radiation and other
stressors. The cell needs to repair such breaks. But what it must
scrupulously avoid doing is stitching two intact chromosomes to
gether end-to-end, mistaking the unjoined ends of the chromo
some for the unjoined ends of a broken chromosome. Thus, it
needs a way to recognize that the bona fide end of a chromo
some is not just one side of a chromosome break.
Telomeres are half of Nature's solution to both these prob
lems. Telomeres contain no genetic information—they are ex
tremely boring DNA consisting of many copies of a short
sequence—and they are present at the ends of all our chromo
somes. If this repeated sequence gets a bit shorter during succes
sive rounds of cell division and DNA copying, no harm is done until
it is mostly eroded. The other half of the solution is the enzyme
telomerase, which is able to add copies of that sequence to the
end of a DNA strand. This solves both problems: cells expressing
telomerase can compensate for the telomere shortening that oc
curs during cell division, and cells with or without active telom
erase can avoid stitching chromosomes end-to-end because the

P U T T I N G T H E Z O M B I E S T O R E S T 209
break-repairing machinery recognizes the distinctive telomeric se
quence and leaves it alone.
Humans and some other species have made ingenious use of
the telomere/telomerase system to protect themselves against
cancer. Cancer can only kill us by its cells dividing a lot; this is im
possible without telomerase, because without a way to renew the
telomere, it will slowly erode away, the chromosome ends will be
come indistinguishable from chromosome breaks, and the cancer
cell will be stopped in its tracks by a joining-together of some of its
chromosomes. Humans therefore turn off their telomerase genes
as thoroughly as they dare, so that a lot of mutation is needed to
turn telomerase on again and thereby allow a cancer to divide
often enough to kill us.
Although there's less research on it, old CMV carriers also suffer from
the expansion of defective CD4 cells—the "T-helper" cells that help other
immune cells to ramp up their counteroffensive when pathogens first in
vade. Outwardly healthy older carriers of CMV infection have the same
large clonal expansions of CMV-targeting but CD28-lacking CD4 cells as
are seen in their CD8 populations, leading to the same crowding-out of
other T-cell specialists and lack of responsiveness to activation by antigen-
presenting cells. 1 1
As with their CD8 cousins, CD4 cells stripped of CD28 can't respond
to antigen-presenting cells by deploying CD8 and other immune cells to
face the threat. Put this together with the inability of those very CD8 cells
to attack their targets effectively, and CMV would be left to run rampant,
generating yet more clonal expansions and wider immune dysfunction.
Clonally expanded anti-CMV CD8 cells are anergic (ineffectual) in
other ways, too. When they are first infected with their species' version of
CMV, young mice produce very effective CD8 cells targeting the virus,
which recognize at least twenty-four proteins specific to it; but after the in
fection becomes chronic, their anti-CMV forces become restricted to
clones that recognize an average of only five such proteins. 1 2 And older
CMV-infected humans' anergic CD8 cells mount a weaker response to the
threat than do younger infectees' cells, producing significantly lower
amounts of interferon gamma, a key chemical messenger responsible for
ramping up the T-cell response to the vi rus . 1 3 , 1 4

2 1 0 E N D I N G A G I N G
When Bad Generals Lead Good Armies
The failure of anergic T cells to clear out CMV infections then probably
leads to many of the other failures of immune function commonly seen in
the frail elderly that can't be chalked up to any direct effect of aging of the
cells in question. Some of these effects might be expected to flow from
changes in the production of cytokines by such cells, which influence the ac
tivity of many other soldiers in the adaptive and innate immune systems, but
others exert much more lasting changes than just problems in signaling.
Notably, it's now widely accepted that the aging of T cells is responsi
ble for the age-related losses in effectiveness seen in our B cells—the im
mune cells that produce antibodies to foreign antigens, which flag pathogens
for destruction by other cells. B cells rely on signals from CD4 (T-helper)
cells to mature and to develop antibodies, so it was only a matter of time
before someone confirmed that old T cells cause declines in the develop
ment and effectiveness of B cells, independent of the aging of the B cells
themselves. 1 5 , 1 6 , 1 7 Unfortunately, no one has (to my knowledge) yet directly
looked to see if these effects are due to the effects of CMV-inducell clonal
expansion that are so central to other aspects of T-cell aging, so we don't
know how much the specific phenomenon of anergic T cells contributes to
these declines. I'd be very interested in the results of such experiments.
Moving beyond the mechanistic studies and molecular biology, the real
impact of the creeping takeover of the immune system by anergic CD8
clones on the health of people bearing them is also becoming clear as sci
entists begin to study its influence. Animal studies show that age-related
clonal expansion of specific CD8 populations reduces the variety of T cells
present in their bodies and compromises their ability to mount an effective
immune defense. 1 8 The parallel in humans can be seen in findings such as a
poorer CD8 response to flu shots 1 9 and a blunting of the reinforcement of
T cell immunity to Epstein-Barr virus that can otherwise occur later in
life, 2 0 in people with clonal expansions of anti-CMV memory cells.
Taking the Full Toll
If the cost of anergic T-cell clones to the body were limited to the increase in
deaths and disabilities that can be directly chalked up to infectious disease,
that would be plenty enough reason to want to do something about them.

P U T T I N G T H E Z O M B I E S T O R E S T 2 1 1
But there's considerable evidence that anergic CD8 cells contribute to age-
related morbidity and mortality from causes with no obvious immunological
link.
For starters, when you throw an influenza or influenza-induced pneu
monia attack onto an aged body, you wind up with shocking long-term
consequences that can greatly accelerate other disease process and hasten a
person's slide into helplessness and the grave. 2 1 A significant body of evi
dence shows that influenza in the elderly increases deaths from unexpected
sources like heart attacks, strokes, and seemingly unrelated respiratory dis
orders; it also worsens the course of congestive heart failure.
Also, the fact that it takes biologically old people so long to recover
from the flu, when overlaid on the general frailty induced by other aspects
of aging, probably contributes to serious, often permanent functional de
cay and disability. A bout of influenza often lays an older person in a hospi
tal bed for as much as three weeks, and studies show that for each day that
they spend "resting" this way, elderly people lose up to 5 percent of their
muscle power and 1 percent of their aerobic capacity. But no one thinks of
influenza or immunological aging when they see an elderly woman struggle
to open the doors at the mall, or slip on the ice and break her hip.
There are other age-related diseases in which anergic T-cell clones ap
pear to play an important role, but where the evidence is not nearly so
clear-cut. One is osteoporosis. Older women who have suffered an osteo
porotic fracture have been found to carry higher levels of anergic CD8 cells
than matched women with no bone disease, and there is a molecular basis
for thinking that defective CD8 cells are actually a cause, rather than an ef
fect, of the underlying thinning of the women's bones. 2 2
Additionally, albeit more speculatively, even the course of atherosclero
sis could be affected by the creeping "clonalisation" of the T-cell popula
tion, by leading to a state of chronic inflammation that could hasten a heart
attack. In support of this hypothesis, patients with coronary artery disease
have higher levels of anergic CD8 cells than otherwise matched healthy
people—a fact that is independently related both to CMV infection and
also to the presence of the disease itself.2 3 Thus, the weakening of the im
mune system appears to be both facilitating and also the result of arterial in
fections, which may in turn be the itchy trigger finger toying with the
loaded gun of atherosclerotic arteries.
As I said, the evidence for many of these downstream effects of anergic
T-cell clones is still not conclusive. But a couple of remarkable studies now

2 1 2 E N D I N G A G I N G
coordinated through the European Union T-CIA (T Cell Immunity and
Ageing) project have gone some way towards giving us a clearer picture of
the total cost, in deaths, of this driver of immunological aging, whatever
may ultimately wind up written on the death certificate.
These researchers hunted through two cohorts of Sweden's "oldest old"
(people in their eighties 2 4 and nineties, 2 5 , 2 6), selecting only people who were
particularly healthy compared to most people of their chronological age: free
of preexisting serious diseases of the heart, brain, liver, or kidney; without di
abetes or cancer or signs of existing, active infection or chemical markers of
inflammation; and not taking any drugs that have significant effects on the
immune system, including recent vaccination. The European team found
that even amongst these relatively healthy but old people, a few are silently
suffering a complex of immunological defects (the "immune risk pheno-
type") including several forms of aging damage that can be caused by CMV
infection—not least, the clonal expansions of anergic anti-CMV CD8 cells.
The facts that the resulting study population was healthy but very
calendar-old (by today's standards), and that some did and some did not
harbor anergic T-cell clones, allowed the T-CIA team to study their effects
"cleanly," in a population where its presence could really be said to predict,
rather than follow, preexisting disease, over the course of the next two years.
It was hardly a surprise that having the immune risk phenotype in
creased these people's chances of death—but the size of the effect was a
shock. The effect was especially powerful in the population in their
nineties, amongst whom its presence could predict 57 percent of the deaths.
This, remember, from the immunological aging damage induced by a virus
whose active infection state is passed through without any notice in many
people, and even in the rest of us usually causes only low-level malaise and
fevers.
It's important to see the full implications of this finding. The impact of
having the immune risk phenotype was seen at the level of all-cause mortal
ity, not just in risk of death from infectious disease. While pathogens do
claim many very biologically old people's lives, such deaths can't entirely
account for the result.
Slash-and-Burn for New Growth
As more and more evidence has accumulated fingering anti-CMV CD8 cell
clones in the age-related enfeebling of the immune system, immunologists

P U T T I N G T H E Z O M B I E S T O R E S T 2 1 3
have begun to see the hopeful side of the phenomenon. If so much of the
aging of the immune system is indeed the result of this overreaching expan
sionism, then preventing or reversing it should (respectively) protect or re
store a youthful immune system in chronologically aged people. Vaccines
would again be as effective in people at presently advanced ages as they
were in their youth, and the enormous burden of suffering caused in the
old by infections that young people escape after an unpleasant day or two
home from work or school would be lifted.
One option for prevention, advocated by many immunologists, is vacci
nation against CMV. Even before we understood that CMV infection is a
central driver of the age-related weakening of the immune system, a 1999 re
port on the sluggish pace of development of new vaccines put out by the In
stitute of Medicine (IOM) of the National Academy of Sciences ranked the
pursuit of an effective anti-CMV vaccine as the highest priority item on the
list, based only on the then-known lifetime human and financial costs of
the virus. The U.S. National Vaccine Program Office would later agree, call
ing for more government dollars to go into CMV vaccine research. Today,
confronted with the strong evidence condemning CMV infection as a major
reason for the aging of the immune system, many immunologists are sound
ing the call for such investments even more loudly.
Though the merits of this case seem strong, it's worth noting that this
strategy is fundamentally preventative. While it can reduce the risk of CMV
infection, and possibly improve the immune response of existing infectees
to the virus, vaccination can't eliminate it—and it certainly can't reverse the
accumulated effects that a lifetime of CMV infection has had on the im
mune system. Thus, a CMV vaccine might save a relatively small number of
babies from suffering tragic birth defects, and prevent the deaths of many
AIDS and transplant patients, but it would do little for the many millions
of people already suffering with the chronic infections and continuous vul
nerability of having immune systems worn down by clonally expanded an
ergic CD8 cells.
Other proposals, which would at least have the potential to undo some
aspects of immunological aging, involve trying to remedy the defects of the
existing anergic T cells using gene therapy. The idea is that by delivering
copies of genes for proteins that are either missing or underactive in these
cells (such as those for the CD28 receptor or telomerase), we could restore
their effectiveness at doing their particular job, and prevent their suppres
sive effects on other T-cell populations. While there is some merit to these
proposals, there are also limits to their likely effectiveness and a lot of

2 1 4 E N D I N G A G I N G
uncertainties to their path to clinical development. And in the case of
telomerase, we'd still face the big worry that has to be taken seriously when
contemplating the introduction of telomerase into any cell, let alone one
that we know is wracked with aging damage: cancer. I'll talk about this
problem much more in Chapter 12, but here's a brief taster: because cells
require a minimum telomere length in order to keep reproducing them
selves, and because each cell division shaves off a little nub from the cell's
telomeres, cells with potentially carcinogenic mutations require a way to re
new their telomeres if they are going to go on to become full-blown malig
nancies. Nearly all cancer cells accomplish this by wrenching out the
self-imposed parking brake from their telomerase genes. Do we really want
to introduce this gene into defective cells—or worse, into "bystander" cells
in which telomerase should never be turned on, should some of the gene
therapy vector "infect" them too?
No: the solution here is not to try to rehabilitate these cells, but to get
rid of them. Older CMV infectees appear to have no shortage of functional
T cells targeting cells infected by the virus: it's just that these cells are sup
pressed by the crowding influence of huge populations of anergic ones.
And remember that even if we could restore all of these defective T cells to
their full immunological power, they would still cause problems so long as
they continue to sprawl out over precious, limited immunological real es
tate, preventing the retention of both naive and memory cells needed to
protect us from other pathogens.
The solution to this is conceptually simple. Remove the anergic T-cell
clones, and immunological space will be opened up for healthy cells of
other types and specificities to move in—and the repressive effects of the
anergic clones on their healthier anti-CMV cousins will be lifted.
The problem, of course, is how to purge anergic T cells from our sys
tems while leaving behind all (or, at least, nearly all) the healthy memory and
naive cells that we're trying to liberate from the former's repressive domina
tion. While oncologists can to some extent increase the effectiveness—and
decrease the toxicity—of drugs or radiation by applying them as narrowly as
possible to a relatively large lump at a fairly well-defined spot in the body, we
can't do the same against anergic T cells, which are spread all over the body
rather than being concentrated in one place. The same feature rules out sur
gery: tumors can often be removed (or at least beaten back) with the knife,
with varying degrees of safety and clinical benefit, but we will not be in a po
sition to pluck individual anergic T cells from the body one-by-one for the
foreseeable future.

P U T T I N G T H E Z O M B I E S T O R E S T 2 1 5
But even though the cancer therapies of the recent past don't offer a
good model for the development of the required biotechnology, the most
exciting of the currently available and imminent treatments for cancer sug
gest a path to therapies that would indeed selectively eliminate the burden
of cells that will not die.
Smells like Gleevec
Even if no one you know has cancer, there's a good chance you've heard
about Gleevec (a.k.a. STI—571 or imatinib), Iressa (ZD1839 or gefitinib),
Herceptin (trastuzumab), and others less famous or still working their way
through the approval process. These so-called "targeted cancer therapies"
have been rightly hailed as breakthroughs; even the language of "miracles,"
although absurdly overused in popular books on health, seems justified to
many people who have seen tumors disappear from their own bodies or
from those of their loved ones, without the horrific side effects associated
with radiation and chemotherapy. Even so, these drugs are not completely
without side effects—no drug that "messes with metabolism" can be. Her
ceptin, for example, targets a growth receptor called HER-2: by tying up
HER-2, it prevents the excessive growth of cancer cells that get their
growth-stimulus fix by producing too much HER-2 on their surfaces. But
other, healthy cells rely on a low level of HER-2 stimulation to proliferate
normally. Because of this, Herceptin users can suffer deadly congestive
heart failure—a side effect that recent research has also uncovered in a
small number of users of Gleevec, which was thought to be an extremely
clean drug precisely because it only targets an abnormal form of a growth-
signal transducer. 2 7
In the same way, interfering with anergic T cells' resistance to apop
tosis might lead to their death, but this still leaves open the question of
how to undo this resistance without killing needed cells elsewhere in the
body.
I am confident that we can perform reverse-engineering, to adapt the
new targeted cancer therapies—and even newer ones that are now in vari
ous stages of clinical development—to develop the ability to create "smart
bombs" that will destroy anergic T cells (and also the other kinds of toxic
cells that we'll be discussing later on) with minimal harm to healthy ones. 2 8
We can foresee the ability to couple carefully chosen toxins to molecules
that home in selectively on the tell-tale signatures of anergic clones and

2 1 6 E N D I N G A G I N G
thereby to directly, decisively kill them flat out instead of just interfering
with their metabolism.
Light Kills Vampires
One cancer treatment that suggests ways to take out anergic T cells is photo-
dynamic therapy (PDT). PDT starts with a drug that, when illuminated with
laser light, either heats up greatly or produces a massive burst of free radicals.
Drugs exist that have this feature and are also taken up selectively by cancer
cells, allowing oncologists to cause a lot of photosensitizing drug to accumu
late in the target cells while avoiding much uptake by normal cells. By them
selves, PDT drugs are harmless, having no effects as long as the patient is
kept away from the light. Similarly, low-energy red laser lights is harmless to
people who have not received such drugs: the rays pass harmlessly through
the body. But when such a laser beam penetrates cells that contain a photo-
dynamic drug, the agent's photosensitizing properties are revealed in a sear
ing targeted blaze of heat or a maelstrom of free radicals that destroys the
tumor cells while leaving all but their very close neighbors unharmed.
The first PDT drug, Photofrin, was approved in industrialized coun
tries as a treatment for advanced lung, digestive tract, and urinary tract can
cers in the early 1990s, and more advanced versions are now in use
clinically or are in late stages of development. The most interesting of these,
Pc-4, accumulates more in some kinds of cancer cells than in healthy ones
because it dissolves well in fats, and these particular cancers have an un
usually high fat content. Once it enters the cell, certain features of Pc-4's
structure allow it to insert itself into the cancer cell's energy factories, the
mitochondria of which I've said so much in Chapters 5 and 6. Turn on the
laser, and the free radical bombardment begins, either taking the cell out
cleanly via radical-induced apoptosis, or at worst leaving some debris as the
cell dies the nasty way instead, when the free radicals tear through the cell,
cross-linking its proteins, turning its lipid membranes rancid, and wracking
its DNA with mutations.
The Molecular Swiss Army Knife
On the frontiers of medicine, we are now seeing the emergent use of nan-
otechnology—engineering performed at the molecular level—to destroy

P U T T I N G T H E Z O M B I E S T O R E S T 2 1 7
cancer cells selectively, again providing us with a road map toward the devel
opment of a targeted therapy for toxic cells such as anergic T-cell clones. One
such technology is dendrimers: tiny particles with exquisitely complex
branching structures that extend outward like bushes, forming a spherical
shape (see Figure 2). Dendrimers' branches are engineered in a way that al
lows us to bind a wide range of molecules to them. This makes them like nan-
otechnological Swiss Army knives: several useful tools can be united into one
compact little package. Dendrimers can carry one molecule to target a given
cell type, one or more deadly drugs or other poisons to kill target cells once
they're located, and (if desired) a molecule that will allow researchers or doc
tors to track the progress of the whole package as it moves through the body.
One dendrimer under experimental development combines folic acid
(yes, the vitamin) with the established anti-cancer drug methotrexate and a
fluorescent compound called fluorescein. The folic acid is there to target the
dendrimer to cancer cells. Many cancers suck up massive amounts of this
vitamin because it is required for the production of new DNA—and be
cause the cell needs to create a whole new copy of its DNA blueprints every
time it divides, cancers have high metabolic requirements for folic acid to
support the feverish pace of their proliferation. To keep their reserves of
the vitamin topped up, many cancer cells "learn" to sprout veritable forests
of folic acid receptors on their surface.
Figure 2. An "unloaded" dendrimer.

2 1 8 E N D I N G A G I N G
Figure 3. Targeted dendrimer technology against cancer growth in mice. Redrawn. 2 9
This dendrimer was tested in mice that had been injected with a hu
man nasopharyngeal cancer line. Their tumors had grown quickly, reaching
a plateau at about fifty days. Giving one group of animals a low dose of
plain methotrexate had hardly any effect at all on the growth of the tumors
(see Figure 3). A dose more than four times as high (the "medium dose" in
the figure) reduced the growth rate quite significantly, but didn't actually
benefit the animals much: half of them were dead of either the tumors or
the side effects of the drug within thirty-nine days. Increasing the dose by a
further 50 percent (the "high" dose) brought cancer growth down to al
most nothing—but without doing the animals any good, because the drug's
toxicity caused the rapid loss of a third of the animals' body weight, and
again either allowed or caused the death of half of them just over a month
into the experiment.
But look at what was achieved with the same drug targeted using a
dendrimer! When targeted using this new technology, a dose of methotrex
ate equivalent to the lowest untargeted methotrexate dose was as effective
at slowing tumor growth as a dose of plain methotrexate over four times
higher. Moreover, the dendrimer-targeted methotrexate appeared to have
very low toxicity. 3 0
In a follow-up study, the same group compared the effects of the low-
dose methotrexate to those of the same dose delivered using the targeted
dendrimer, this time for an extended period of ninety-nine days. Left un
treated, the cancer-bearing mice began dying quickly—about fifty days
into the experiment—and low-dose methotrexate improved survival only

P U T T I N G T H E Z O M B I E S T O R E S T 2 1 9
modestly. But by the end of the ninety-nine-day study, three out of eight of
the animals getting the dendrimer-targeted drug were still alive—and im
pressively, one of these animals was completely cured of its cancer by day
thirty-nine. Again, the dendrimer-targeted drug was nontoxic.
Switch the Hacksaw with the Toothpick
The beauty of dendrimers, like Swiss Army knives, is that they are so readily
customized. Researchers are experimenting with dendrimers bearing many
different targeting molecules and cancer-killing agents. One very clever ap
plication under development is a "dendrimerized" version of a one-two an
ticancer punch known as boron neutron capture therapy (BNCT)—a neat
idea that was first proposed over fifty years ago, but that until now no one
has ever quite managed to make work.
The idea behind BNCT is similar to photodynamic therapy. First, you
inject the patient with a form of the mineral boron. Once enough of the
mineral has accumulated in cancer cells, you flood them with low-energy
beams of neutrons. The boron itself is harmless, and so (more or less) is the
neutron beam. But when incoming neutrons hit the form of boron used in
BNCT, it absorbs them into its atomic nucleus and suddenly becomes ex
tremely unstable, releasing radioactive alpha particles. Alpha particles have
enough energy to nuke the cell in which the original boron is located and a
few of its neighbors, but they run out of energy quickly and the low-energy
leftovers are harmless. Thus, the boron-loaded cells are killed but no wide
spread damage ensues.
The trick, of course, is to find a way to target the boron selectively to
cancer cells, so that when you flip on the neutron beams you don't destroy
healthy brain and other tissue. Scientists have been trying to turn BNCT
into a viable clinical therapy for a rare, extremely aggressive, and hard-to-
treat brain cancer called glioblastoma multiforme since 1951, with some
limited success, but they've never achieved good enough responses to jus
tify using it as a standard treatment for the disease.
But scientists have recently reported very promising results in an ani
mal model of glioblastoma multiforme that was treated with a BNCT using
dendrimer-targeted boron. Human glioblastoma cells bearing a mutated
version of the epidermal growth factor receptor (EGFR) called EGFRvIII
that is implicated in the majority of these cancers were implanted into the
brains of rats. The researchers then heavily loaded their dendrimers with
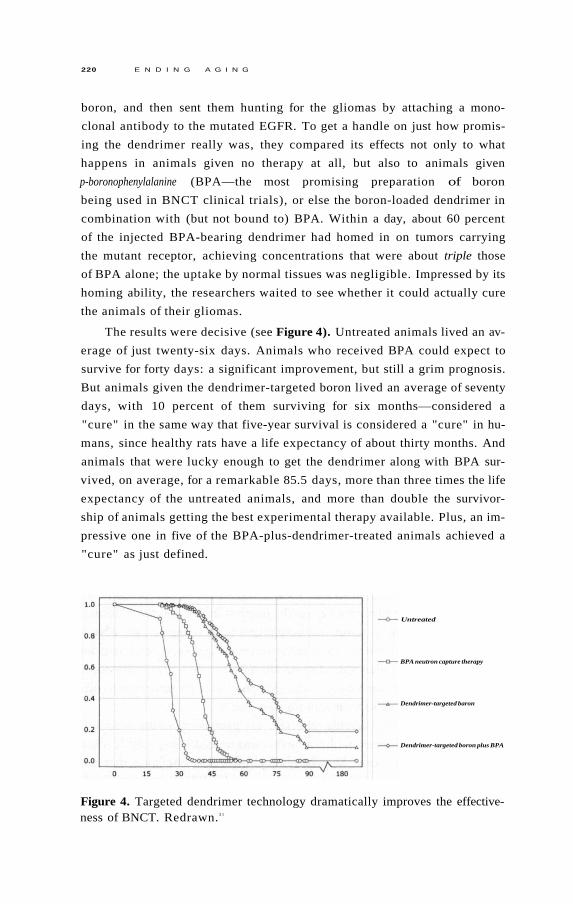
220 E N D I N G A G I N G
boron, and then sent them hunting for the gliomas by attaching a mono
clonal antibody to the mutated EGFR. To get a handle on just how promis
ing the dendrimer really was, they compared its effects not only to what
happens in animals given no therapy at all, but also to animals given
p-boronophenylalanine (BPA—the most promising preparation of boron
being used in BNCT clinical trials), or else the boron-loaded dendrimer in
combination with (but not bound to) BPA. Within a day, about 60 percent
of the injected BPA-bearing dendrimer had homed in on tumors carrying
the mutant receptor, achieving concentrations that were about triple those
of BPA alone; the uptake by normal tissues was negligible. Impressed by its
homing ability, the researchers waited to see whether it could actually cure
the animals of their gliomas.
The results were decisive (see Figure 4). Untreated animals lived an av
erage of just twenty-six days. Animals who received BPA could expect to
survive for forty days: a significant improvement, but still a grim prognosis.
But animals given the dendrimer-targeted boron lived an average of seventy
days, with 10 percent of them surviving for six months—considered a
"cure" in the same way that five-year survival is considered a "cure" in hu
mans, since healthy rats have a life expectancy of about thirty months. And
animals that were lucky enough to get the dendrimer along with BPA sur
vived, on average, for a remarkable 85.5 days, more than three times the life
expectancy of the untreated animals, and more than double the survivor
ship of animals getting the best experimental therapy available. Plus, an im
pressive one in five of the BPA-plus-dendrimer-treated animals achieved a
"cure" as just defined.
Figure 4. Targeted dendrimer technology dramatically improves the effectiveness of BNCT. Redrawn. 3 1
Untreated
BPA neutron capture therapy
Dendrimer-targeted baron
Dendrimer-targeted boron plus BPA

P U T T I N G T H E Z O M B I E S T O R E S T 2 2 1
Other targeting molecules, tumor types, and cancer-killing agents have
been successfully treated by dendrimers in experimental models. These
first-generation devices are turning out to be a very effective, versatile way
of creating specific, lethal missiles for seeking out tumors that express
known cell-surface receptors as hallmarks—and they offer promise for
anergic T cells, too.
PRO-Suicide Counseling
Today, gene therapy is a routine practice in mice, used to do everything
from testing experimental gene-based therapies, to investigating the effects
of turning genes on and off in an organism, to creating new models of hu
man disease by modifying animal cells to be more like human ones. Gene
therapy for humans is still highly experimental, but it's clearly only a matter
of time before we master it: the need for cures for congenital diseases, and
the potential utility of gene therapy in medical challenges as wide-ranging
as rheumatoid arthritis, trauma, dental tissue engineering and AIDS (to
name just a few), is providing the impetus for the basic and clinical science
needed to bring it into our therapeutic armory. 3 2
One option that gene therapy will furnish us with is the ability to build
a new suicide mechanism into our T cells that would cause them to self-
destruct should they ever turn anergic. Scientists have for some time been
able to introduce into mice (and other laboratory animals) genes that will
only turn on in the presence of a particular factor, such as an antibiotic, UV
light, a sugar, or even a signalling factor like calcium. This allows us to turn
such genes on and off at will, simply by administering the relevant factor.
The ability to introduce a gene that is only expressed when researchers
want it to be has been a powerful new tool for studying those genes' effects.
But these techniques are also now being turned to medical purposes. If, in
stead of designing these genes to be turned on in response to externally
supplied factors, we instead make their activation dependent on the pres
ence of a particular protein whose internal synthesis is diagnostic of a cell
that we want to be rid of, then we have yet another way to selectively target
cells for destruction.
Just as with the other targeting technologies I've discussed, the first
work in this direction has been in the cancer field. As I've noted, the one
absolute requirement for a cancer cell to threaten us is that it have a way to
keep renewing its telomeres: otherwise, its furious growth will grind to a

2 2 2 E N D I N G A G I N G
halt when it reaches the end of the telomeric line, which is long before it
can meaningfully threaten our health. Usually, this is accomplished by acti
vating the repressed gene for the telomerase enzyme—a gene that all of our
cells contain, but which is turned off in healthy cells most or all of the time.
So by "infecting" the cells of a patient with a "suicide gene" that would
be activated in the presence of high levels of telomerase, cancer cells could
be killed from within. This would eliminate the need to target a drug or the
immune system to the offending cell: every cell would hold within it the
seeds of its own destruction should it ever turn to the dark side.
In principle, we could generate a literal "suicide gene" that would de
stroy the cell in the presence of the tell-tale protein. Indeed, this has already
been done in animal models of cancer, using genes that regulate apopto
sis; 3 3 anergic T cells are resistant to apoptotic signaling, but this resistance
might be overcome by bombarding them with insistent messages to shrivel
and die. But there's an even better alternative that's under more advanced
development. This uses the somewhat more readily controlled—and there
fore safer—technique of installing the gene for a protein that is largely
harmless in itself, but which activates an inactive form of a deadly drug—a
so-called "prodrug."
Prodrugs are substances that are inactive and harmless until they are
metabolized in some way, whereupon they are chemically transformed into
a pharmacologically active product. Most prodrugs are activated by en
zymes in our livers and are then released in active form to the rest of the
body, but others act more like molecular "sleeper agents," going about the
body unobtrusively, minding their own business and blending in with their
environment, until a prearranged signal is given—and their hidden pur
pose suddenly becomes revealed in the form of a precision strike on their
target.
Several antiviral drugs, such as the herpes drug ganciclovir (Cytovene/
Cymevene), work somewhat along these lines. Ganciclovir stops viruses
from using the DNA-replication machinery of their host cells to reproduce
themselves. It does this by interfering with the action of the virus's unusual
version of thymidine kinase (TK), an enzyme that is required for the syn
thesis of DNA.
Thymidine kinase's job is to make thymine (a "letter" in the DNA
code's "alphabet") available to be added onto the new DNA chain, by join
ing thymidine to phosphate molecules taken from the "energy currency
molecule" ATP. Ganciclovir acts like a molecular impersonator on a mission
to sabotage an enemy factory. It first uses its strong structural resemblance

P U T T I N G T H E Z O M B I E S T O R E S T 2 2 3
to thymidine to fool the viral TK into thinking that it is that molecule.
Duped, TK hands over thymidine's rightful phosphate group to the drug.
Ganciclovir then uses its shiny new phosphate group to perpetuate its
identity theft, presenting its phony credentials to the cell's DNA synthesis
machinery, which unknowingly inserts it into the emerging DNA strand in
thymine's place. At this point, the sabotage of the hijacked equipment is
accomplished, because while the machinery can slide ganciclovir onto the
DNA strand, it can't add any further genetic letters onto ganciclovir once
it's in place. Without the ability to copy its DNA, the virus can't replicate
itself, and its expansion campaign is brought to an abrupt end; all that's left
is for the immune system to besiege and ultimately destroy the cells in
which the remaining virus is holed up.
If ganciclovir were as good at tricking the version of the TK enzyme
used by our own cells as it is against the viral version, it would potentially
be a very effective cancer-killer: again, cancer can only survive by keeping
up the insane pace of its growth, and turning this off by shutting down its
DNA synthesis capacity quickly tames tumors. But, of course, such a
drug would come with some pretty serious side effects, because it would
shut down the growth of normal cells at the same time. This might make
it an acceptable therapeutic bargain for cancer—the effect would wear
off after the drug was withdrawn, allowing patients to recover—but it
would make it totally unacceptable for its present use as a herpes treat
ment.
In fact, however, ganciclovir is rather poor at mimicking the human TK
enzyme, and its effects are thus mostly restricted to turning off virus
replication—though it does have some negative impact on the body's abil
ity to regenerate its blood cells and on the production of sperm. But a team
of Japanese and American scientists recently realized that they could in
principle use the viral TK/ganciclovir combination to shut down cancers if
they could introduce the enzyme into cancer victims' cells using gene ther
apy, but turn it on exclusively in cancer cells.
As I've already indicated, there is one obvious way to distinguish can
cer cells from normal ones which could provide a mechanism for control
ling the activation of viral TK: active telomerase. By designing a version of
the viral TK gene that would be attached to a "trigger" (promoter ) that
would turn the gene on only in the presence of telomerase, the researchers
realized that they could set the enzyme to work in a cancer patient's malig
nant cells, while leaving it dormant almost everywhere else in the body.
At this point the flow chart of what they were designing was beginning

2 2 4 E N D I N G A G I N G
to look like the biotech equivalent of one of those exceedingly convoluted,
multistep devices that players build up in the board game "Mousetrap."
The scientists would set the "trap" by first seeding a copy of the gene for
the viral TK enzyme, complete with its special telomerase "trigger," into
every cell in the patient's body. The patient would then swallow some gan
ciclovir tablets, which would penetrate all of his or her cells indiscrimi
nately.
In most cells, the drug would have no effect, because nearly all cells
have their telomerase enzyme firmly turned off. But when ganciclovir en
tered a cancer cell, the trap would be sprung. The cancer's abundant telom
erase enzyme would flip on the viral TK enzyme; the TK would scoop up
the ganciclovir, adding on the phosphate group needed by DNA "letters"
for insertion into the emerging DNA copy strand; the next time it reached
for the relevant "letter," the DNA-copying machinery would grab the
phosphorylated ganciclovir by mistake, jamming it into the "letter's" place
on the strand. At that point, you would almost hear the cry of "Mousetrap!"
as the DNA synthesis machines seized up, cell division came to a screech
ing halt, and the cancer shut down. See Figure 5.
Figure 5. How ganciclovir enables viral thymidine kinase to kill mammalian cancer cells.

P U T T I N G T H E Z O M B I E S T O R E S T 2 2 5
It was a crazy, convoluted solution—but it worked in the test-tube
against liver, kidney, pancreas, and thyroid cancer cells. Moreover, the
setup proved largely harmless to normal rat thyroid and human skin cells. 3 4
So the team took the next step in bringing something off a lab bench and
into a clinic: a careful study in laboratory animals.
The researchers first cooked up two batches of customized viral TK
genes: one with the telomerase-activated "on" switch, and another with a
switch that could be expected to be flipped in healthy and cancerous cells
alike. They then slid these genes into viruses from the same family as the
common cold, allowing them to literally infect animals with the gene con
structs. They first tried these constructs out on healthy animals, to see what
the potential was for side effects. As expected, animals that got viral TK un
der the control of the nonselective promoter suffered nasty liver damage
upon injection with ganciclovir, while putting in the same gene under a
telomerase promoter appeared to be basically harmless, since there were no
telomerase-expressing cancer cells present to activate it.
At this point, the researchers decided that their experimental therapy
was ready to hit the next stage: a test in animals that had been injected with
implanted human thyroid carcinoma cells. Giving such animals a copy of
the viral TK driven by a promoter that did not rely on the presence of
telomerase to activate it brought tumor growth to a complete standstill—
but, as expected, it also prevented normal cells from reproducing them
selves, leading to nasty liver damage.
But when scientists tried the selective targeting of viral TK to cancer cells
by using the telomerase promoter, ganciclovir shut down tumor growth just
as completely as it had when the TK was controlled by the nonselective
promoter—and without the latter's toxic effects. The apparent safety of this
highly selective intervention is all the more convincing when you remember
that the effect can be turned on and off at will, by administering or with
drawing the ganciclovir.
Know Your Enemy
We've seen that biologists can be just as creative as weapons engineers
holed up in the Skunk Works at finding new ways to target and kill cancer
cells selectively, i.e., leaving healthy cells unharmed. It is foreseeable that
the same methods in use or under development against cancer could be
used to target anergic T cells. In this case, we're blessed with an enemy that

2 2 6 E N D I N G A G I N G
is walking around with a bull's-eye painted right on its chest. The same dys
functional receptor profile that strips anergic T cells of their ability to recog
nize their target antigens (absence of CD28) and to proliferate in response
to infection (presence of KLRG1 and CD57), possibly along with some
other markers (such as reduced levels of CD154, implicated in the failure of
old T cells to support B cell development) already allows scientists to iden
tify these cells, and could also be used to target such cells for destruction.
Unleashing the wrath of the immune system against its oppressors would
be poetic justice, but vaccination (either passive or active) against these cells
might be tricky. For one thing, their most prominent antigenic feature is the
lack of a cell-surface protein (CD28), and while we could target the combina
tion of KLRG1 and CD57, it's not yet clear whether all unwanted cells ex
press these two proteins, nor whether other, desirable cells do. As it happens,
immunologists already do identify anergic cells using a combination of im
mune proteins—but these could not easily be used for vaccination purposes.
Also, after all, the problem that we're looking to resolve is characterized by a
poor response to vaccination, so an "anergic T-cell shot" might only be effec
tive in relatively immunologically "young" people. So while this approach is
promising for many kinds of toxic cells, it may be less so for CD8s.
But that still leaves us with a lot of options. Dendrimer-based targeting
approaches seem the most straightforward, because they allow for the tar
geting of cells via multiple identification criteria, and also because they can
introduce any number of poisons into the cells that they select, from out
right toxins to boron for BNCT.
And while using vaccines to target anergic T cells might be problematic,
there could be another way to use the immune system as a proxy army, coop
erating with it to restore the sovereignty of the immune system's government-
in-exile. Remember that anergic CD8 cells first become a problem because
they stop listening to the apoptotic order to scale back their forces that's sent
out after their target pathogen has been routed from the body's frontiers.
They are able to ignore these orders because they produce high levels of bcl-
2, a protein that blocks apoptotic signalling.
This suggests the possibility of restoring normal apoptotic signaling in
such cells by delivering "antisense RNA" for bcl-2's blueprints to them-—
strips of genetic material matched to the transcribed DNA instructions for
the protein, preventing the encoded bcl-2 from actually being produced in
the cell. With bcl-2 production brought down to normal or nearly nonex
istent levels, anergic CD8 cells would finally hear their curtain call and
bow out.

P U T T I N G T H E Z O M B I E S T O R E S T 2 2 7
Would purging the immunological "space" of anergic T cells be
enough to rejuvenate the immune system completely? I can't say for sure,
because it hasn't been done—and, as I am well aware, the body is an in
credibly complex machine whose parts have not yet all been identified, let
alone their purposes and interactions. Existing research tells us pretty
clearly that the direct and indirect immunosuppressive effects of these cells
are powerful enough that a thorough spring cleaning of them will pro
foundly improve T cell-mediated immunity, and also very probably the
functioning of other aspects of the immune system that T cells support and
govern. But we'll only know just how profoundly once we've done it.
I can tell you right now that there is at least one aspect of immune aging
that the removal of anergic T cell clones will not address: thymic involution.
The thymus is a gland located just behind your breastbone. It's where im
mune cells first produced in your bone marrow go to learn to become T
cells. As we age, the thymus loses cells and shrinks away, and in the process
its output of naive T cells plummets. This, of course, imposes further limits
on the body's ability to respond to new threats.
In principle, there is a fairly straightforward way of dealing with this,
however: stem cell therapy. This is foreseeable biotechnology, as we will see
in the next chapter (see the sidebar there, titled "Rebuilding the Thy
mus"). 3 5 The accomplishment of such a goal would entail significant ad
vances in the stem cell field, including mastering the art of turning
embryonic stem cells into the progenitors of the different cells of the body,
and then engineering new tissue to rejuvenate the old one, renewing old tis
sues with pristine new cells—but these are just the same problems that are
being solved quite rapidly for tissues all over the body, so there is ample
reason for optimism.
That's all I have to say about the immune zombies; now it's time to look
at some other types of supernumerary cells.
Deadly Combat in the Battle of the Bulge
The second kind of toxic cell that we'll want to rid ourselves of is excess fat
tissue—most important, the so-called visceral fat that surrounds your inter
nal organs, as opposed to the subcutaneous fat that lies under your skin all
over the body. It's widely believed that, as people get older, they just "natu
rally" become more resistant to the effects of the hormone insulin, whose
job it is to move carbohydrates and amino acids into fat and muscle cells.

2 2 8 E N D I N G A G I N G
This change causes a range of threatening metabolic changes, the most ex
treme of which manifest in people with full-blown type II ("adult onset")
diabetes. It's also common wisdom that older people "naturally" enter into
a more inflammatory state, with the body slowly burning up from within
because of an excessive production of inflammatory signal molecules.
When you take in more calories than you expend, your body hangs on
to them rather than allowing them to go to waste. This is not the result of
perversity on the part of evolution, but a survival strategy: until very re
cently (by evolutionary standards) there was a good chance that quite soon
you'd be in a period of famine, when those stored calories would be your
lifeline. If your body isn't under the kinds of challenges (like weight-
bearing exercise) that signal the body to build up metabolically expensive
muscle or bone tissue, it will take the easy way out by storing the calories as
fat. But in an environment where feast is never followed by famine, and
where exercise is almost entirely a voluntary affair, we fail to shed that extra
fat tissue, and it slowly accumulates as we age. Because this accumulation is
a difference of aging versus healthy young bodies, it qualifies as "aging
damage" under my engineering definition, even though it might not be
considered as such from a purely theoretical point of view.
It's long been known that this damage—in the form of being over
weight or obese—puts you at greater risk of diabetes, heart disease, and
various other ailments, but it's only recently become clear why and how.
You may have heard that fat stored in different parts of the body has differ
ent health implications: having an "apple shape" (fat centralized in your
midsection, as in a "beer belly") puts you at great risk for diabetes and
heart disease, while having a "pear shape" (fat clumped on your bottom or
thighs) is unsightly but much less hazardous to your health. To the extent
that there is some biomedical basis for this distinction, it lies in the differ
ence in the locations of visceral and subcutaneous fat. Visceral fat is most
visible around your middle because it clumps around major internal organs
like the liver and kidneys. By contrast, subcutaneous fat just lies under your
skin—and of course, there's skin all over your body, though there are more
prominent depots for this fat type in some places than others.
Recent studies have found that nearly all observed age-related insulin
resistance, and much of the age-related pro-inflammatory signaling shift,
can be attributed to the accumulation of excess visceral fat, which precedes
and predicts the development of all the elements of the metabolic storm
known as syndrome X: insulin resistance, low HDL ("good") cholesterol,
and high blood pressure, triglycerides (blood fats), and blood sugar.

P U T T I N G T H E Z O M B I E S T O R E S T 2 2 9
Even more tellingly, when you compare people of different ages, you find
that the difference in insulin effectiveness between young and old disappears
when you account for the difference in fat, and visceral fat in particular. 3 6 , 3 7
In striking studies at the Albert Einstein College of Medicine, aging an
imals have had most of their resistance to insulin and other hormones re
versed by highly invasive surgery that scrapes out most of their visceral fat,
making their body compositions similar to much younger animals, or to an
imals of the same age that had been subjected to calorie restriction (among
whose benefits are dramatic reductions in age-related insulin resistance and
inflammatory signaling). 3 8 This latter result was especially striking because,
when the scientists looked at the distribution of fat in the calorie-restricted
animals, they found that they actually had more subcutaneous fat than their
young counterparts, but less visceral fat—and their insulin effectiveness
was almost the same.
Further pinning the blame on visceral fat, a recent study confirmed
that undergoing liposuction—whose invasiveness and risks of trauma are
kept low by removing only the relatively easy-to-access but cosmetically sig
nificant subcutaneous fat, while leaving the much harder-to-remove vis
ceral fat behind—does not improve the insulin resistance associated with
the original obesity. 3 9 On the other side of the same coin, several studies
have now shown that putting overweight people on low-calorie diets or ex
ercise programs significantly improves their insulin resistance quite early
on—well before it has had a chance to impact their overall weight by much,
but after it has had time to reduce their level of visceral fat, which (fortu
nately) is the first thing to go when energy needs aren't being met.
The reasons for all of this have become clear as scientists have increas
ingly come to understand the nature of fat itself. Fat tissue was once con
sidered to be just inert storage space, like carrying around a spare tank of
gas on your derriere. Instead, we now know that it is a metabolically active,
dynamic tissue that secretes and responds to a range of hormonal and other
signaling molecules. We also now appreciate that fatty tissue is not com
posed only of "fat cells" (ad ipocy tes) , but is a mixture of different cell types,
including supporting connective tissue, nerves, and blood vessels, as well as
immune cells—notably, macrophages. In fact, adipocytes are actually derived
from the same precursors as macrophages, and secrete many of the same
immune-system regulating molecules, such as the coagulation-enhancing en
zyme plasminogen activator inhibitor-1, and pro-inflammatory signaling
molecules (cytokines) such as tumor necrosis factor alpha, monocyte
chemoattractant protein-1, and interleukin-6.

2 3 0 E N D I N G A G I N G
As the size of the fat depot increases, adipocytes begin pumping out
more and more of these inflammatory molecules, some of which promote
infiltration of the tissue by macrophages and some of which signal the pre
cursor cells that give rise to both adipocytes and macrophages to take the
path toward the latter instead of the former. Macrophages, in turn, produce
even more inflammatory messenger-molecules, creating a self-reinforcing
inflammatory feedback loop.
The most exciting finding in the emerging science of fat in the last ten
years has been the discovery that these signaling molecules not only cause a
potentially pathological increase in systemic inflammation, but also in
crease the body's resistance to insulin. This conclusion is supported by
studies showing that isolated muscle and fat cells become insulin-resistant
when bombarded with the very inflammatory mediators that adipocytes
and macrophages produce, and that chubby lab rodents' insulin resistance
is relieved by aspirin treatment, in part via blocking the effect of cytokines.
And the relationship doesn't just hold up under the artificial conditions of
the laboratory: during sepsis (the inflammatory storm generated in response
to severe infection), human patients often exhibit very severe insulin resis
tance as part of the immune response.
Evidently, these and related questions will keep an army of basic and
clinical researchers in the diabetes field busy for decades, resolving para
doxes, isolating deeply intertwined metabolic pathways, and double-
checking their results in different models. But for engineering purposes,
fortunately, we don't have to wait to have the results of these investigations:
we just need to observe the presence of damage, and fix it.
Doing It the Old-fashioned Way
In this case, of course, there are two very simple, inexpensive solutions that
don't involve advanced biotechnology: diet and exercise. But unfortunately,
as decades of research and centuries of anecdotal experience have shown,
most of us find it very difficult to lose weight once we put it on. As little as
a 100-calorie daily energy imbalance—about what you get in a single
medium-sized biscuit—accounts for the standard weight gain that creeps
up on the average person in the decades between high school and middle
age, and while it's easy to load it on, it's hard for most people to shed those
extra pounds and keep them off. The situation is not nearly as dire as is of
ten made out—studies show that about one overweight person in five

P U T T I N G T H E Z O M B I E S T O R E S T 2 3 1
successfully achieves long-term weight loss, and research is clarifying what
it takes to get there—but the metabolic consequences of excess visceral fat
are far too deadly, and the magnitude of the current obesity epidemic far
too staggering, to leave the cure of this form of aging damage to self-help
programs or public health measures designed to remediate our present
"toxic food environment." 4 0 Realistically, unless we are ready to leave the
fate of millions to a sudden wave of greater personal or political responsi
bility, we must look for biomedical solutions to visceral fat.
Fat for the Fire
One option that we might pursue is to shrink away visceral fat by causing it
to burn off its excess stored energy. Scientists have of course been trying to
develop drugs to do this for decades, but so far the only moderately effec
tive such drugs are amphetamines, and their side effects and addictiveness
clearly don't fit our remit.
For several years, the appetite-regulating hormone leptin seemed to of
fer the chance of shrinking fat and maintaining insulin sensitivity. An ex
tremely rare genetic mutation that leads to a congenital lack of leptin makes
both rodent and human victims monstrously obese, and injecting these
mice with leptin leads to dramatic weight loss. Causing these rodents to
produce more leptin inside their fat cells (using genetic engineering) makes
them eat 30 percent to 50 percent less food, leading to more insulin sensi
tivity and an almost complete disappearance of their body fat. Moreover,
the effects are stronger than can be accounted for by their newfound light
dining habits alone. 4 1 Fat cells of animals with the extra leptin gene were
expressing other genes that activate mitochondria, turning them into little
"fat burning machines." 4 2 Paradoxically, however, while injections of leptin
lead to rapid fat loss in both normal and obese rodents, the same level of
leptin that would peel away the pounds in lighter mice circulates naturally
in the bodies of their fat cousins, necessitating a much higher level of leptin
to achieve similar weight loss. This is in part because leptin is, ironically,
produced by the very fat cells whose swelling with stored energy it was sup
posed to inhibit, so that chubbier rodents naturally produce more leptin,
not less. And indeed, after drugs giant Hoffmann-La Roche invested a for
tune to develop a way to mass-produce human leptin in genetically modi
fied bacteria, they found the hormone to be a miserable failure as a
weight-loss treatment. 4 3

2 3 2 E N D I N G A G I N G
This led scientists to speculate that overweight members of both
species come to suffer "leptin resistance" in the same way that they suffer
insulin resistance: levels of the hormone are high, but cells stop responding
properly to its signals to turn off the appetite and turn on fat-burning. Re
cent studies by Roger Unger, the researcher who had originally raised
hopes for leptin by showing its powerful effects in mice, show how this can
happen at the molecular level. When you overfeed mice a high-fat, high-
calorie chow, their fat cells scale back the expression of the genes that tell
the cell how to build leptin receptor "doors" on their surfaces, so that the
cell stops hearing the signal. 4 4 Similarly, unpublished studies of the fat tis
sue of grossly overweight people show that their expression of the leptin
receptor gene is consistently so low as to be undetectable, while lean, young
people may have anywhere from quite low to extremely high levels of gene
expression at any given time. 4 5
Unfortunately, as with leptin itself, the path to using leptin receptor
gene therapy as a way to reverse the negative effects of visceral fat is not a
clear one. Mice with the extra leptin gene may have stayed slim in the face
of an overly rich diet, but they did not fully escape its consequences: they
still suffered the same "ectopic" (mislocalized) fat infiltration of their livers,
muscle, and heart as did mice lacking the extra leptin receptors eating the
same chow, and their insulin resistance—the key negative effect of excess
visceral fat that we need to address—was just as bad.
While we might find some way of avoiding some of this by turning the
gene on and off again, thereby restoring our insulin sensitivity, it seems un
clear how we would deal with the ectopic fat. We might expect that as soon
as we turned the gene off the calorie imbalance that had led to the initial
overgrowth of fat cells would begin again; while we would shrink fat cells
back down with each round of therapy, we would have no way to eliminate
the cells themselves, and over the course of a greatly extended life a visceral
cavity full of large numbers of even relatively small fat cells might still lead
to metabolic mayhem.
Really Tr imming the Fat
No—what seems most likely to solve the problem of visceral fat is not to try
to tame it, but to cull it: actually to remove a substantial number of the
bloated, excess cells. The same sorts of cell-specific targeting of cancer or
anergic T cells we've been discussing seem likely to be applicable to visceral

P U T T I N G T H E Z O M B I E S T O R E S T 2 3 3
fat too; and while, unlike in those other cases, no specific markers of vis
ceral fat cells have been identified, we don't need to be nearly so specific.
Unlike cancer or anergic T cells, some fat—including some visceral fat—is
not only metabolically harmless, it's necessary for carrying on the business
of life. Aside from being a spare tank of metabolic fuel that we draw upon
and refill every day, those metabolic factors we've been talking about—
energy-regulating hormones, inflammatory peptides, and others—also have
healthful uses. As with all of metabolism, this was built into us by evolution
for our benefit. As anti-aging engineers, it's not our job to interfere with
it—just to prevent the damage that it causes in aging bodies.
Night of the Living Dead
The last class of toxic cells that I want to address in this chapter are so-called
"senescent" cells. 4 6 They got this name because of the (rather dubious) anal
ogy drawn between these cells and aging humans by their codiscoverer, Dr.
Leonard Hayflick, then of the Wistar Institute in Philadelphia. These cells,
like the others that we've been discussing, begin their lives as normal con
stituents of the skin, joints, and other tissues. They are normally quiescent,
not dividing regularly, but they remain capable of reproducing themselves
on demand, as part of their normal function (unlike "postmitotic" cells,
which lose the ability to divide ever again once they reach their mature form
and are only r ep l aced by new cells coming from the body's stem cell pools—
if they're r ep l aced at all).
The defining feature of senescent cells is that they, like postmitotic
cells, have lost the ability to divide. Hayflick observed, contrary to the
dogma of the day, that cells from these tissues would not keep reproducing
in the petri dish indefinitely: they would seem normal for several successive
periods of replication, but would then suddenly enter into a twilight state
in which they didn't die, but became in various ways abnormal. Their ap
pearance became blotchy, their shapes irregular. They failed to form the
neatly whorled colonies of mutually adhering cells that were the norm in
younger cultures. And above all, they stopped reproducing themselves.
The use of the word "senescent" to describe these cells is, however, a
bit misleading. When people hear about these cells, they often assume that
cellular "senescence" is the ultimate fate of all of the cells in the body with
age, and that the entry of "young" cells into this senescent state is the un
derlying cause of aging. Moreover, the term evokes an image of these cells

2 3 4 E N D I N G A G I N G
as somnambulant old has-beens, sleepwalking through the remaining days
of the rest of the body's life, not contributing anything to the organs in
which they reside but also not doing us any positive harm. Their only
downside, we might think, is a crime of omission: inability to replenish ag
ing organs.
In fact, senescent cells are generally held to be extremely rare even in
very aged people. 4 7 However, their possible role in aging has turned out to
be far more complex—and far more active—than we at first imagined.
The most obvious characteristic of senescent cells is, as mentioned, their
loss of the ability to reproduce. But, like worn-out lechers, senescent cells
desperately try to stimulate themselves into activity—pumping out sub
stances that, though essential to their healthy function back when they were
contributing members of a healthy tissue, may promote the development of
cancer when present in excess. Various mechanisms are involved.
For a start, some of the most common signal molecules overproduced
by senescent cells are chemical messengers like epidermal growth factor that
directly spur cell division in their neighbors.
As a second example, many senescent cells also overproduce protein-
digesting enzymes such as matrix metalloproteinases (MMPs), which are
the "demolition teams" of tissue remodelling. These enzymes perform the
essential function of clearing away the old, damaged "scaffolding" in which
cells are embedded in a tissue, making space for new growth. But, just as
having the outside wall of your house knocked down during a renovation
would leave you vulnerable to burglary, so an excessive or uncontrolled
MMP activity can enable cancerous cells to escape from the restraints of
the tissue in which they were originally embedded, and/or to work their
way into new tissues far removed from the original cancer site—the metas
tasis process.
And most recently, scientists have found yet another way that senescent
cells potentially roll out the welcome mat for nascent cancers: by churning
out dangerous overdoses of vascular endothelial growth factor (VEGF) 4 8
and stromal cell-derived factor 1 (SDF1), 4 9 which promote the growth of
new blood vessels.
As you can see, the picture turns out to be a lot more complicated than
we had once thought. As I'll discuss later on, the senescence phenomenon
is probably an evolved response to DNA damage, helping to prevent that
damage from becoming cancerous. From the point of view of that one cell,
this is an extremely effective short-term protection against cancer, because
it shuts down the cell proliferation that is the very heart of the disease. But

P U T T I N G T H E Z O M B I E S T O R E S T 2 3 5
it seems likely that the "unlifestyle" that a senescent cell leads ultimately fa
cilitates cancer progression in the long term by destabilising its neighbors.
So—you guessed it—they've got to go.
Putting the Zombies to Rest
One way to eliminate senescent cells is to make them no longer senescent. In
cell culture experiments, this has been achieved in a variety of ways, such as
by relengthening exhausted telomeres with telomerase or by depleting pro
teins associated with senescence. But reversing senescence would run the
risk of cancer, because senescent cells typically get that way as a response to
potentially carcinogenic changes to the cell, such as damaged DNA, hyper
active cancer-promoting genes, or (again) very short telomeres, which pro
mote a mutagenic state. Restoring proliferative capacity in such cells could
potentially take us out of the frying pan of senescence and into the fire of
cancer.
Similarly, approaches based on turning off the dangerous metabolic
abnormalities of senescent cells carry risks, because other, healthy cells de
pend upon these same pathways for normal function. Chronically jamming
growth signals, enzymes, and inflammatory messengers might well prevent
senescent cells from watering the seeds of cancer, but it would also lead to
"crop failures" of cells all across the body.
As usual, the engineering approach to this dilemma is to rewrite the
rulebook. We will maintain the body's evolved capacity to shut down cells
in danger of going cancerous, by leaving the metabolic regulation of senes
cence as it is. Indeed, as we'll see in Chapter 12, we will ultimately need to
transform the body in ways that will make it almost immune from cancer by
ensuring that all cells run out of steam long before they would threaten us
with uncontrolled cell growth. But we will eliminate the threat posed by
those cells that do senesce by eliminating the cells themselves.
Silver Bullets
The first significant development in this area came in 1995, in a lab at the
Lawrence Berkeley National Laboratory headed by Dr. Judith Campisi,
one of my coauthors on the original SENS scientific manifesto. Campisi
and coworkers found that a relatively easy, reliable test for the activity of an

2 3 6 E N D I N G A G I N G
enzyme called senescence-associated beta-galactosidase (SA-beta-gal) could
identify senescent cells not only in petri dishes, but in skin samples taken
from older humans.
Unfortunately, SA-beta-gal is not a perfectly selective marker for senes
cence. As subsequent studies showed, the enzyme does occur in nonsenes-
cent cells—usually at very low levels, but sometimes in high concentration.
It turns out that, contrary to the simple interpretation of the Campisi lab's
findings, this enzyme is actually identical to one that is normally found in all
of our lysosomes—the cellular waste incinerators, whose clogging (you'll
recall from Chapter 7) underlies many of the worst pathologies of aging.
The change into the senescent state does not suddenly trigger the secretion
of SA-beta-gal into the main body of the cell out of nowhere: instead, it ap
pears that there is always some low level of SA-beta-gal floating around in
even normal cells, as can be detected with techniques that assess the level of
the enzyme itself in the cell—but the level is so low that its activity is barely
(if at all) detectable by the methods that Campisi's lab initially used, which
are unfavorable to the enzyme's functioning. 5 0 , 5 1 , 5 2
But as a cell goes through round after round of replication—thereby
drawing ever closer to senescence—its SA-beta-gal levels rise. 5 3 Probably
this is because the cell begins to overproduce the enzyme in response to the
stresses of aging—notably, the need for more lysosomes as they become less
and less effective at doing their job (and also as their cell division rate,
hence garbage dilution rate, slows and the job thereby becomes intrinsically
more challenging). Eventually the level gets so high that its activity is no
ticeable even under suboptimal conditions.
SA-beta-gal activity is, notably, found at abnormally high levels in cells
taken from tissues where cells are under stress, due to inflammatory dis
eases that fuel cell proliferation (such as chronic hepatitis C, atherosclerotic
plaques, and venous ulcers). Most interesting is the finding that levels of the
enzyme shoot up in cells undergoing "cr is is ," 5 4 , 5 5 a period in which cells
that have somehow escaped senescence are still undergoing cell division
and the erosion of their telomeres. Such cells usually just run out of steam,
but occasionally they undergo a mutation that removes the clamp from
their telomerase genes, making full transformation into malignancy almost
inevitable.
What's emerging, then, is a picture of SA-beta-gal as an enzyme that
appears at high level in the main bodies of cells undergoing some kind of
stress that may ultimately threaten their neighbors. This might mean that
by using high levels of SA-beta-gal as an identifier for the destruction of

P U T T I N G T H E Z O M B I E S T O R E S T 2 3 7
senescent cells, we would simultaneously take out some useful "targets of
opportunity."
However, we may be able to establish a system of double-checks, to
help us weed out more genuinely senescent cells while leaving more inno
cent (but suspicious-looking) cells unmolested. This is because, in addition
to SA-beta-gal, senescent cells also produce abnormally high levels of other
molecules involved in the programmed senescence response. Senescent ba
boon skin cells, for example, contain an activated form of the protein ATM
kinase, which responds to DNA damage by activating several tumor sup
pressor genes, including the famous p53. Senescent cells also exhibit high
levels of p53, as well as the binding protein (53BP1) by which its gene in
teracts with ATM kinase, and p21, a senescence regulator that works under
p53's command. 5 6 Some senescent cells also contain high levels of p l 6 , the
other main regulator of the process. Levels of this protein, for reasons as
yet unknown, also climb slowly with age in nonsenescent cells, making it an
unreliable marker for senescence when taken in isolation; but it—like these
other features—could still potentially be used as part of a double-checking
mechanism, with multiple proteins being used to distinguish genuinely
senescent cells from those expressing only one of them for unrelated rea
sons. 5 7
This chapter has focused on the accumulation of toxic cells with age,
and the foreseeable biotechnology with which we should be able to purge
ourselves of those cells as part of our platform for the rejuvenation of our
bodies—restoring the immune system, alleviating metabolic mayhem, and
protecting our cells from being goaded on to cancer. In the next chapter,
I'll look at the opposite problem: cell loss with age, and the scientific—and,
just as importantly, political—hurdles that we face in bringing forward the
ability to renew our tissues with fresh, new replacements.

11
N e w C e l l s fo r O l d
Throughout our lives, we gradually lose cells vital to our
continuing health. Many fatal diseases of aging—such as
Parkinson's disease—are caused by the loss of populations of
cells responsible for one or another crucial function in the body.
Fortunately, therapies based upon stem cell research offer the
possibility of recreating our missing cells, good as new; politics
stands in our way as much as any remaining scientific obstacles.
After the enormous effort that had been required to organize the
conference, it was an incredibly rewarding moment to see the man who was
revolutionizing stem cell biology take the podium in front of a packed
crowd of colleagues.
It was the second conference I'd run at Cambridge focusing on scientific
progress toward the reversal of human aging, so the pressure was on for me
to top the success of the first. I'm on the board of the International Associa
tion for Biomedical Gerontology (IABG)—one of the few biogerontological
societies in the world with an explicit brief to pursue the development of bio
medical solutions to aging—and a couple of years earlier I had volunteered to
spearhead their tenth conference. I knew at the time what I was getting into.
The society would provide little logistical assistance beyond networking op
portunities, so I would have little help beyond the support (moral and other
wise) of my beloved wife Adelaide, and that suited me perfectly. With the
formal authority of a society already on the progressive wing of the biogeron-

N E W C E L L S F O R O L D 2 3 9
tology community, I wanted to push the envelope a little further, and being
left to my own devices meant that I would not have to debate my priorities
with a committee.
Despite the society's mandate, previous IABG conferences had tended
to be dominated by the same kind of presentations that I saw at every
biogerontology conference I attended (and I try to get to most of them): ba
sic science, geriatric medicine, and work in model organisms that the re
searchers hoped might someday be translated into a pill to slow down aging
in humans. I took on the enormous and exhausting work of running this
conference because it would give me the opportunity to highlight work that
could contribute to a panel of interventions designed to reverse aging.
IABG 10—the meeting that would be, in retrospect, the first in a series
of SENS conferences—was an enormous success. I am saying so myself, but
I am making no boast of my own: the enthusiasm with which my colleagues
thanked me for my efforts at the end of the week was ubiquitous and unmis-
takeably genuine. Attendees were surprised and excited by what they had
heard, not only on its own merits but because most of it was completely
novel to them. This was to be expected: while a typical biogerontology con
ference invites a roster of speakers almost entirely drawn from within the
biogerontological community, I had introduced a strong interdisciplinary el
ement, bringing in researchers working in cancer, diabetes, stem cells, and
other fields, whose work would in my mind be critical to the development of
effective anti-aging biomedicine but who were almost entirely unknown to
researchers prone to pegging themselves in the "biogerontology" slot.
At the same time, those presenters had the opportunity to mix with re
searchers in whose laboratories the degenerative processes of aging were, if
not being reversed, certainly being dramatically delayed in mice and other
model organisms. This was work that often hardly raised an eyebrow
amongst biogerontologists, who were immersed in a field in which it had
been taking place since the first calorie restriction experiments nearly seven
decades previously, but which amazed the experimental oncologists and tis
sue engineers that I had brought in to show the biogerontologists what
they'd been missing.
IABG 10 was so successful in meeting my academic goals, and the re
quests from my colleagues that I run a sequel were so obviously sincere, that
I felt sure that I could harness its momentum to make it the de facto inaugu
ral meeting of an ongoing series of academic conferences on SENS science
at Cambridge. From then on, however, I knew that the effort would be en
tirely my own: I could not rely on the support (nor brook the interference,

2 4 0 E N D I N G A G I N G
little though it had been) of the IABG or any other society. Challenging as
the job of directing such events was, I knew it would be worth it.
On the other hand, I also knew that I had set my own bar quite high
with the first conference, and that some of my colleagues would be less in
clined to attend a conference not run under the aegis of a recognized
biogerontological society. This was all the more so when I was the organiz
er, because a whispering campaign against my credentials as a scientist had
been initiated by some of my genuinely well-intentioned but old-school
gerontological rivals shortly after the first conference. So if I wanted to get
people to show up to SENS2, and to have the series continue, the quality of
the conference lineup would have to be top-notch despite the opposition. I
would have to meet an ambitious standard—and I wanted to overachieve.
The Master Cells: Accept No Substitutes
I knew that I would once again want to devote a whole session to embryonic
stem cells (ESCs)—the primordial "master cells" from which our mature
cells spring, and which play a critical role in our development into complex
multicellular organisms from the simple ball of cells that is an early embryo.
Thanks to the tragic confusion of the science of ESCs with the ethical, le
gal, and religious disputes around the status of the embryo in the abortion
debate, ESCs are the one plank of the SENS platform with which you can
not help but be familiar. You have doubtless heard that, with the right kind
of biochemical stimulation, embryonic stem cells can be coaxed into be
coming any kind of cell in the body: nerve, muscle, heart, kidney, the lot.
These resulting, "differentiated" cells can then be used to repair or to
replace cells and tissues that are lost to—and whose loss is a central patho
logical feature of—multiple debilitating, often nearly untreatable diseases,
including many of the worst scourges of aging. ESCs will be needed to de
velop full cures for Parkinson's disease, spinal cord injuries, juvenile dia
betes, amyotrophic lateral sclerosis ("Lou Gehrig's disease"), heart attack
damage, some cancers, and other devastating conditions—including aging
itself. Indeed, under the purely pragmatic, engineering definition of aging
that clarifies so much about what needs to be done to keep our bodies age
less indefinitely, the net loss of cells is itself a form of aging damage. This
makes it a central target of SENS.
However, because media coverage of the issue focuses on the political
firestorm rather than on the real, hopeful medical story of ESCs' enormous

N E W C E L L S F O R O L D 2 4 1
potential as medicine, you may still not be clear on the key differences in
basic biology and therapeutic potential between ESCs and adult stem cells.
There are also important differences between ESCs derived from embryos
being stored in fertility clinics and those that can be custom-made for each
patient out of his or her own mature cells by fusing them with egg cells (a
technique known as somatic cell nuclear transfer, SCNT, to which we shall
return). For this reason, I will spend a little time disentangling these issues.
True ESCs are found only in very early-stage embryos called blasto
cysts, which are the very primitive balls of cells that are formed within just a
few days after sperm meets egg. The embryo only remains in this stage of
development very briefly; it has developed much further by the time the
embryo is implanted in the womb. It is from the blastocyst that every cell in
the mature organism must be derived, yet the blastocyst itself has none of
these differentiated cells: no neurons, no heart cells, no insulin-producing
beta-cells, and so on. So for the embryo to go on to transform itself into an
organism with the complex structure of a human being, its cells need the
ability to transform themselves into each and every one of those mature
cells—a power called pluripotency.
Adult stem cells, on the other hand, are much more limited in their abil
ities, and also with good reason. These cells emerge in the late stages of de
velopment, and are retained in particular tissues during life as a reserve to
replenish cell stores. They thus hold on to only the limited repertoire of
possible fates that is relevant to their role in that particular tissue. Thus,
blood stem cells can become oxygen-carrying red blood cells or any of the
many blood-borne immune system cells, but (despite what has been
claimed—see later on in this chapter) they cannot form either neurons or
heart muscle cells: if asked, a blood stem cell would doubtless indignantly
reply, "That's not my job." They're there to fulfill a specific role in the body,
and to do it well, but not to be on reserve to heal all damage everywhere.
This more limited range of developmental flexibility (or "plasticity") is
called mul t ipo tency . 1
Indeed, there are many areas of the body for which there are no adult
stem cells dedicated for use in repair—and, as you might expect, these in
clude the areas that suffer the worst cell loss during aging. This is the situa
tion, for instance, in much of the brain. For many years, it was believed that
the entire brain loses cells over the course of normal aging, and that there
was no way for the body to replace these losses. This dogma was overturned
a few years ago, largely due to the work of Fred Gage and his coworkers at
the Salk Institute, who showed that the brain does indeed harbor stem cells

242 E N D I N G A G I N G
capable of renewing some parts of the brain. This has led to a swing, in the
popular imagination, to the impression that the entire brain has the inbuilt
capacity, through its adult stem cells, to keep the entire brain young and
functional.
In fact, however, that impression is also wrong. Only a small number of
areas in the brain produce stem cells capable of developing into new neu
rons: a sub-subsection of the hippocampus called the subgranular zone of
the dentate gyrus, and a part of the subventricular zone, where neurons are
created to supply the olfactory bulb (the area of the brain that processes the
sense of smell). There's evidence that some of these cells do attempt to re
pair areas of the brain damaged by age-related disease, but there's little ev
idence that they're much help. After a stroke, for instance, a few of the stem
cells formed in the subgranular zone do change their normal habits and mi
grate toward the site of damage, but over 80 percent of them die within a
few weeks, and the remaining cells replace only about 0.2 percent of the
cells destroyed by the incident. 2
Why do we maintain the capacity to replace neurons in some areas of
the brain and not others, like the cerebral cortex where our long-term
memories are stored, or the frontal lobe where our ability to make and stick
to plans for our future is centered? Most likely, it's because the olfactory
bulb and the dentate gyrus are the only places where evolution has encoun
tered the need for a regular influx of new cells within the brain's "biological
warranty period." Both those areas have short-term functions that require
the regular renewal of their cell populations. There is no built-in popula
tion of adult stem cells to deal with cell loss induced by the ravages of aging
and age-related neurological diseases such as Alzheimer's and Parkinson's.
As you'll have realized if you still remember Chapter 3, this is because,
while these disorders have their seeds in molecular damage that occurs
throughout life, that damage does not reach a threshold where function is
impaired sufficiently to affect Darwinian fitness in a short, Paleolithic hu
man lifespan.
Another example of a tissue in which cells die but are not naturally replaced is the thymus, a key organ in the immune system which acts to "ma
ture" precursor cells into T cells. Its regeneration using stem cells is at an
early stage of development, so there's not much to tell you yet, but a proof
of concept exists in a rare but very serious congenital disease—see the side
bar "Rebuilding the Thymus." I've described the immune system generally,
and T cells in particular, at some length in Chapter 10, so you might want
to refer back to that chapter while reading the sidebar.

N E W C E L L S F O R O L D 2 4 3
R E B U I L D I N G THE T H Y M U S
The promise of using stem cells to treat thymic involution can
be seen in recent advances in treating babies with DiGeorge
syndrome—a genetic disorder whose victims are born with a vari
ety of defects, including having a thymus gland that is underdevel
oped, or in some cases completely absent (the latter being called
"complete DiGeorge"). Complete DiGeorge has, until recently, of
ten been a very near-term death sentence: with no ability to pro
duce T cells, these babies would die of infections that are trivial to
the rest of us, within a few months of leaving their mothers' wombs.
The obvious way to solve the problem of a missing thymus is
transplantation, but that's a tricky business: to do its job, the tissue
needs a very good blood supply and plenty of oxygen saturation,
which is difficult to achieve without the natural penetration of tiny
blood vessels. There have also long been problems with rejection
and graft-versus-host disease: perversely, sometimes a few of the
child's bone marrow cells will "spontaneously" transform into dys-
regulated T cells that don't recognize either the child's own anti
gens or the thymus tissue donor's. This leads to a ferocious attack
on both, usually killing the child; moreover, often the donor's T cells
would turn on the transplant recipient's foreign tissues in an
equally deadly, reciprocal attack.
Recently, surgeons and immunologists at Duke University de
veloped a protocol using very thin slices of tissue to ensure maxi
mum transfer of oxygen, which are engrafted into the child's thigh
to give it a generous, readily accessed supply of blood, along with
a novel immune-suppressing drug that targets T cells specifically.
The intervention is still experimental, but it's become progressively
better through new innovations and now seems relatively success
ful. In a 2004 report, the Duke team found that five of the six pa
tients receiving the new therapy were still alive fifteen to thirty
months later, a greatly improved survival rate.
If, instead of using transplants of foreign tissue, we could take
the child's own stem cells, coax them into becoming thymus cells,
and engraft them, we would eliminate the need for risky immune
suppression. Then, if we could encourage these cells to grow in a

2 4 4 E N D I N G A G I N G
scaffold in which we could build up a complex organ structure, in
cluding a proper blood supply, we could abandon the highly un
satisfactory replacement of an organ with a wafer-thin tissue slice
in favor of a real organ "transplant." We may never actually be
able to do this in DiGeorge syndrome, for the simple reason that
we don't have enough time—but if a foreign tissue implant can
generate viable T cells and increase survival in babies born with
no thymus, then I can only see promise in delivering a person's
own cells, taught to become T cells and if necessary coaxed and
structured into a more complex tissue, to an existing but atrophied
organ, to restore it to youthful functionality,
Similarly, in the heart, cells exist, which some researchers have called
"cardiac progenitor cells" or similar names; but, while these cells can be
nudged into showing some stem-cell-like molecular signatures in a test
tube, they have not been shown to form heart cells in the body. Indeed,
some closely related stem cells found elsewhere in the body (mesenchymal
stem cells) have the same hallmarks but definitely cannot become heart
cells. Whatever the ultimate truth of the matter, what we do know is that
neither these nor any other cells in the body step in to heal the massive
damage wrought to the heart muscle by being starved of oxygen during a
heart attack—as any cardiologist or heart-attack survivor can sadly attest.
Again, the reason for this lies in the cold statistical analyzes effectively per
formed by natural selection after generations of genetic dice-rolling in a
premodern environment: heart attacks don't kill twentysomethings, so by
evolution's calculus it's not worth investing in a repair system that will al
most never be used before its owner is killed by something else.
In the first days of the political debate around embryonic stem cells,
some very respected laboratories issued reports of ESC-like flexibility in
adult stem cells—of blood-forming cells spontaneously transmuting them
selves into liver and brain cells, and perhaps most promisingly of such cells
being injected into the hearts of rats given simulated heart attacks, forming
new heart muscle tissue, and restoring functionality to the organ. These re
ports were taken so seriously that several groups began early clinical trials
in humans, in which stem cells have been derived from the bone marrow of
heart attack victims and then injected into their ravaged cardiac tissue.
But independent laboratories have been unable to confirm these

N E W C E L L S F O R O L D 2 4 5
claims. Instead, what may be happening is that the cells are indeed being
incorporated into the tissues in question, but are doing so by fusing with
the existing c e l l s . 3 , 4 , 5 , 6 , 7 , 8 , 9 , 1 0 There may be some limited benefit to this: the
process of fusion may support the surviving cells in damaged tissues, either
by secreting growth factors needed during repair, or by helping new blood
vessels to grow into the tissue. 1 1 But, while such effects may help to keep a
disintegrating ticker beating for a short while more, it cannot substitute for
actually rebuilding heart tissue, either for heart attack victims or for the
aged humans whose hearts we wish to rejuvenate.
Indeed, recently the New England Journal of Medicine published the
results of the first trials of bone marrow stem cells as a treatment for human
heart attack victims that were large enough to give meaningful information
about actual clinical outcomes in the patients (as opposed to just collecting
safety data and early reports of physician and patient experience). One of
these trials 1 2 found no benefit, and the other two 1 3 , 1 4 reported what the
Journals summarizing editorial described as "small, [statistically] signifi
cant, but clinically uncertain improvements" 1 5 in treated patients com
pared to those receiving dummy injections. They reported no evidence
either way on the subject of the cells actually transforming into heart mus
cle cells, but the animal studies mentioned above have at this point dashed
previous hopes of such an effect.
Contrast these weak effects with the results of an animal study using em
bryonic stem cells to treat an induced heart attack. Eighteen sheep were sub
jected to such an assault, and then allowed to decline for two weeks. During
this time, scientists harvested ESCs and nudged them to begin making the
transition into becoming heart muscle stem cells. Before the embryonic stem
cells had completed their developmental journey, the researchers seeded
these cells onto the hearts of half of the group, while for comparison the re
maining nine animals were left to slide further down the road to disability.
Where the benefits of adult stem cells had been dubious, the healing
influence of ESCs was undeniable (see Figure l a ) . The cells took hold in
the damaged hearts and were shown to transform into mature heart cells,
and the animals exper ienced a dramatic recovery. In the two weeks since
their matched relations had been given the ESC treatment, the control
group's hearts had lost an additional tenth of their blood-pumping ability.
By contrast, animals who had received the cardiac-committed stem cells
enjoyed a 6.6 percent improvement in pumping capacity.
And if you dig into the details of the study, you find even more reason
to be optimistic about the potential for ESCs as a therapy for the heart. For

2 4 6 E N D I N G A G I N G
2 weeks after
infarction, but
before
transplantation
Controls ESC-transplanted
Figure la. Restoration of the heart's pumping ability by embryonic stem cells, (a) Controls vs. ESC recipients, (b) Controls, ESCs plus immunosuppressive drugs, and ESCs alone. Redrawn. 1 6
one thing, the scientists in this study waited until two weeks after the ani
mals suffered their heart attack to do anything about the damage to their
hearts, and it was during this period that the bulk of the degeneration of
the animals' hearts' pumping capacity occurred. Early intervention,
whether with stem cells or even with more conventional medical duty-of-
care, might have prevented a lot of this decline, potentially leading to much
better outcomes after ESC treatment.
Second, the ESCs that were used in this study weren't even derived
from sheep, but from mice—an important point to which we will return
later. While the cells clearly did their job—maturing into heart cells, unit-
1 month
after cell
transplantation
2 weeks after
infarction, but
before
transplantation
1 month
after cell
transplantation

N E W C E L L S F O R O L D 2 4 7
ing with the native tissue, and restoring significant functionality to the ani
mals' hearts—it still seems reasonable to think that using cells that were ac
tually from their own species would have yielded a better metabolic and
functional match, and, therefore, better outcomes.
And third, the average improvement in the ESC-treated group actually
conceals a very positive variation in response to ESCs within the group. Be
cause of the possibility that their immune systems might reject the mouse-
derived ESCs and spoil the experiment, five out of the nine treated animals
had been given immune-suppressing drugs. It turned out that the drugs
were unnecessary: the researchers took slices from all animals' hearts after
the study was over, and there was no evidence of inflammation or attack by
immune cells in the hearts of the animals given ESCs, no matter whether
they were dosed with immunosuppressive drugs or not.
This is positive news in and of itself, but there was even better news to
follow. The reported 6.6 percent recovery of heart pumping capacity in
ESC-treated animals was a pooled result, including animals that did and
did not receive immunosuppressive drugs. When the researchers broke
down the results according to whether animals received these drugs or not,
they found that the immunosuppressed animals had actually responded
more weakly to ESC treatment than ones whose immune systems were left
to carry out their business. The sheep in the ESC-only group healed 25 per
cent more scar tissue from their original heart attacks than the drug-treated
animals, and their hearts recovered over twice as much pumping capacity:
about a 9 percent versus roughly a 4 percent gain (compared, again, to a 9.9
percent further loss of functionality in animals not receiving ESCs—see
Figure lb ) . So, in evaluating the prospects for human use of ESCs, we
should look at the stronger results available from an ESC-only approach,
rather than the weaker results from pooling these animals together with
those given immunosuppressants.
After this study was published, the first head-to-head comparison of
ESC versus adult stem cell therapy for heart damage similar to that en
dured during a heart attack were reported; the results showed clearly the
superiority of the ESCs, which transformed into heart muscle cells, achieved
long-term incorporation into the animals' heart tissue, and improved the
animals' heart function, while the bone marrow stem cells had no signifi
cant effect.17
And this is only the beginning of the biomedical promise of these
amazingly versatile cells. Embryonic stem cells have been used to cure ani
mal models of some of the most fearsome diseases human beings suffer,

2 4 8 E N D I N G A G I N G
Controls ESC + Immune- ESC Alone Suppressing Drug
Figure lb.
such as juvenile diabetes, 1 8 spinal cord injuries, 1 9 , 2 0 multiple sclerosis
(MS) , 2 1 cerebral palsy, 2 2 stroke, 2 3 , 2 4 Parkinson's disease, 2 5 a form of paraly
sis caused by a virus that induces a standard mouse model of ALS, 2 6 and—
very recently—macular degeneration (the form of blindness caused by the
loss of light-sensing cells in the center of the eye's retina). 2 7 All of these are
diseases where a person's native, adult stem cell supply fails even to begin
to replace the cell loss caused by the disease.
Of course, none of these therapies has made its way into the clinic—
yet. But there's every reason to think that they will lead to dramatic im
provements in our ability to treat these patients. The balance of preliminary
evidence from human trials using fetal cells or cells derived from stem-cell
tumors (not true ESCs) in Parkinson's disease and stroke victims, for in
stance, already shows a lot of promise that can only be expected to improve

N E W C E L L S F O R O L D 2 4 9
with the use of actual stem cells, and recently a study using ESCs in a mon
key model of Parkinson's has confirmed their ability to transform into the
required type of neurons, engraft into the appropriate area of the brain,
and relieve many of the symptoms of the disease. 2 8 These are exciting times.
Why We Need Them
Because the horizons for the ultimate fate of ESCs as differentiated cell
types are wide open, and because of their ability to proliferate indefinitely
(unlike adult stem cells, whose replication capacity tends to be more lim
ited), the scientific consensus acknowledges the greater therapeutic poten
tial of ESCs over that of adult stem cells. There are certainly therapeutic
uses for adult stem cells; indeed, the only stem cell-based therapies currently
in clinical practice are things like bone marrow transplants, which use adult
stem cells taken from a donor or from the patient's own body. But the oft-
repeated claims by social-conservative lobby groups that adult stem cells
can effectively treat "70 diseases" or "more than 65 diseases" have rightly
been called "patently false" and the accompanying information on one
prominent such group's Web site "pure hokum" in the editorial mentioned
earlier from the normally diplomatic New England Journal of Medicine.
As things stand, only embryonic stem cells hold the potential—both in
terms of the range of cells required, and in terms of the sheer quantity of
cells needed to create large tissue grafts and in some cases even whole
organs—that will be needed to make young bodies from old ones. And
need them we will. In addition to cells lost to heart attacks and neurode
generative diseases, the truth is that we are losing cells—and the function
ality that those cells provide—from our tissues on a continuous basis.
Parkinson's disease, for example, is the result of the loss of neurons in the
brain that produce dopamine, a chemical messenger involved in fine con
trol of the muscles. You get a clinical diagnosis when you have lost about
half of these neurons, impairing this control enough that parts of your body
begin an involuntary rhythmic shaking and your face turns into a staring
mask with a fixed blank or even hostile expression. But all of us are losing
dopamine-producing neurons every day to aging; people with Parkinson's
just lose them more rapidly, reaching the clinical threshold earlier. Without
the ability to replace these cells, we'll all develop the disease eventually (if,
as the refrain goes, something else doesn't kill us first).
And it's happening all over your body, and not just for the kind of in-

2 5 0 E N D I N G A G I N G
trinsic metabolic reasons that are most precisely termed "aging." You are
permanently losing cells every day to molecular damage caused by the re
active by-products of normal metabolism, and even after we undo such
damage using the foreseeable biotechnologies of the SENS platform, we
will still need to reverse these losses if we are to build ageless humans. Plus,
we also lose cells to other causes. We all regularly destroy some naturally ir
replaceable cells to minor bumps on the head, moments of oxygen depriva
tion, and the apoptosis ("programmed cell death") imposed on cells by the
body when it senses that they are doing more harm than good.
Whether these latter cell losses are a part of "aging" is debatable, but
fortunately it's not an issue that we need to resolve in order to get moving
on the restoration of old and dysfunctional bodies to the full health and
functionality of youth. Replacing these missing cells will play an essential
role in anti-aging biomedicine, no matter what the causes of their attrition
or their relationship to "aging" in the abstract. Progressive cell loss repre
sents a change away from the healthy ideal of youth, and therefore an anti-
aging engineer should work toward fixing it, just as any engineer will work
to restore machinery back to the state in which it functions best.
Throwing Away the Key to the Medicine Chest
Adult humans have adult stem cells, not embryonic ones: again, true ESCs
only exist in blastocysts. Thus, getting a supply of ESCs for use as cellular
medicine involves somehow deriving such cells from early-stage embryos.
Fortunately, there is a quite generous—and heretofore almost untapped—
supply of such cells that is already being produced by an existing industry:
in vitro fertilization (IVF) in fertility clinics.
The chances of any given IVF embryo being successfully implanted
and then carried to term as a result of the procedure are still relatively low,
so fertility clinics routinely create several embryos from the sperm and eggs
supplied by either would-be parents or their donors. That way, they have a
supply of embryos available for multiple attempts, without requiring
women to undergo multiple rounds of the expensive, very unpleasant, and
modestly dangerous hormonal treatments required to extract eggs from
them. Typically eight such embryos are left over after every round of IVF,
with the result that there were 400,000 surplus embryos frozen in storage in
American fertility clinics alone as of 2002. At least 16,000 of these are
unclaimed by any donor, an additional 45,000 have a similarly murky sta-

N E W C E L L S F O R O L D 2 5 1
tus, 2 9 and nearly none of the others will ever actually be used in fertility
procedures. These embryos are ultimately discarded, or become sufficiently
decayed that they cease to have any potential to form a baby.
This is what makes the debate around the use of embryos from fertility
clinics such a frustration to doctors and scientists. These embryos are
slated for destruction no matter what we do with them: there is no chance
that the vast majority of them will ever be implanted in a womb and un
dergo the additional development needed to make a baby. The opponents
of ESC research and therapy have proposed preventing their disposal by
implantation into volunteers who would carry them to term for adoption,
but even in that scenario there is no realistic prospect that even one percent
of such embryos would be diverted from the rubbish tip. Once created, the
fate of those blastocysts that are not actually implanted into a woman is
sealed; the only question is whether scientists will be allowed to use their
cells for research and as cures.
Actually, the insertion of these cells into the midst of the abortion debate
is even more artificial than this makes it sound. Blastocysts are so primitive a
stage in embryonic development that they have not yet made the biochemical
"decision" to become a distinct human being. This is part of why they have
the full flexibility to become any type of cell in the human body—and also
why the confusion of stem cell technology with the abortion debate is so eth
ically misguided. At this early stage, for instance, an embryo could still divide
into two separate cell populations, each of which can go on to become a sep
arate, unique person. Indeed, this is exactly what happens when identical
twins are formed. Since this ball of cells can still go on to become either one,
or two, or even more different people, clearly the unified cell mass that pre
cedes this separation does not embody the identity, the essence, or the soul of
any single, personal human being. And while we can stand in justified awe of
the potential for life (or lives) locked up in these cells, that should not cloud
our ethical vision into thinking of this potential as morally being even in the
same ballpark as the actual lives of patients that need its cells for medicine,
when it is closest to that of skin cells in a petri dish.
The Nicodemus Solution
Powerful though embryonic stem cells derived from embryos left over from
IVF may be, however, they do have one potential disadvantage hanging
over their medical use. Cells derived from such embryos will, by definition,

2 5 2 E N D I N G A G I N G
be immunologically alien to the patient's own cells, making them a target
for attack by the immune system. Thus, the same kinds of problems that
currently plague conventional organ transplantation—the horrors of rejec
tion, graft-versus-host disease, and the dangers of living with an immune
system turned off artificially with drugs to preserve the transplant—might
possibly be an issue in embryonic stem cell transplants, too.
So far, the evidence suggests that we will be able to manage this issue
with little hassle in many cases. Much of our confidence on this front de
rives from recent experience in actually using ESCs in experimental treat
ments for various diseases. Most such studies have just assumed that
rejection would be a problem, and have preemptively taken steps to pre
vent it, either by using animals with defective immune systems, or by ad
ministering immunosuppressive drugs. But more recently, some studies
have been performed using ESCs without taking such steps, and the results
suggest that there may have been nothing to worry about in at least some
cases. In the sheep heart-attack study I mentioned earlier and in several ro
dent s tudies, 3 0 , 3 1 ESCs taken even from another species have incorporated
themselves into the "patient's" native tissues and provided substantial re
generative benefits, with no rejection issues.
Such results may mean that ESCs' state of development is so early and
tentative that they may not even distinguish themselves with enough anti
gens to create a problem across the species barrier—let alone the barrier
between individual humans. Additionally, it now appears that ESCs pro
duce their own, very localized immunosuppressive signalling molecules
that selectively protect them from immune attack, and even trigger any at
tacking killer T cells to undergo self-destruction (apoptosis). 3 2 Because
these mechanisms involve either direct cell-to-cell contact or factors se
creted and used very close to the stem cells themselves, this local immune
shielding system is free from the systemwide side effects of taking immuno
suppressive drugs.
Moreover, in some specific applications the risk of rejection will be low
to begin with, because the tissues where we'll be delivering the cells are
substantially shielded from the immune system. A lot of the nervous sys
tem, for instance, is largely inaccessible to immune attack (which is how the
virus that causes shingles can hide out there for years after being purged
from the rest of the body).
We can also lower the risk of rejection by providing patients with ESCs
from isolates ("lines") that are a match for all of the major antigens
involved, which we could readily do in many cases if we are allowed to pick

N E W C E L L S F O R O L D 2 5 3
and choose our stem cell lines from among the embryos currently slated for
destruction. It has been calculated that a bank of just 150 donor embryos
randomly selected from the existing stockpile could do this perfectly for
one patient in five, provide a probably usable match for almost two in five,
and allow for a long-shot match for almost 85 percent of potential patients—
and if we were able to choose specific immunological combinations out of
the surplus instead of choosing embryos at random, just ten such donations
could give grade-A matches for nearly 40 percent of patients and good
matches for over two-thirds. 3 3
But we can't yet rule out the possibility that rejection may present a
barrier to our effective use of ESCs in human medicine for aging and dis
ease. In that case, the good news is that technology exists that already al
lows us to generate embryonic stem cells that are a perfect immunological
match for animals as complex as cattle and monkeys, and several scientific
teams say they're on the verge of being able to do the same thing for hu
mans. I've already mentioned it: somatic cell nuclear transfer (SCNT). In
SCNT, doctors begin by taking a mature cell from the patient's body (a "so
matic cell"), by for example swabbing the inside of the cheek, and then turn
hack its clock, releasing it from the strictures of a mature, differentiated
complexity and transforming it into a patient-specific embryonic stem cell.
This biological miracle is accomplished by a technique that is incredi
bly simple. The metamorphosis occurs in an egg cell, provided by a donor.
This cell's nucleus is removed to make way for the one from the patient's
cell. With a biochemical boost or a zap of electricity, the two become one,
and the egg begins dividing just as it would if it had been fertilized, kick-
starting the production of embryonic stem cells created from a patient's
own genetic instructions, creating a perfect immunological match (see Fig
ure 2). The cells can then be used for medicine just as any ESC would be,
but with absolutely no fear of rejection.
Actually, you may well already have heard of this advanced biomedical
research under a name more popular with the media: therapeutic cloning.
While this term is perfectly scientifically accurate, it has generated an enor
mous amount of confusion about the nature and purpose of SCNT, splash
ing political napalm onto the heated fires burning in legislatures and online
chat rooms surrounding stem cells. Let me try to extinguish those flames.
To a scientist, the word "clone" means simply a set of genes, cells, or or
ganisms that are identical to one another at the DNA level because they are
derived from a single ancestor. We've used the word in this strict scientific
sense in the "clonal expansion" of T cells, and the "monoclonal antibodies"

2 5 4 E N D I N G A G I N G
Figure 2. How SCNT ("therapeutic cloning") works.
that are currently used to treat some cancers and will probably be used as
part of our panel of engineering solutions to aging. Similar uses of the word
occur when scientists speak of a "clone" of common bacteria bearing a
gene that turns them into tiny biological factories for the production of in
sulin for diabetics, or even when gardeners talk about a "clone" of strawberry
plants.
But say "clones" to even highly educated people who don't work in a
few disciplines of biology and biomedicine, and you evoke images of a sea
of indistinguishable, zombielike drones, enslaved to technocrats or created
for other sinister purposes. That this confusion is corrupting the debate
about this potentially essential life-saving technique can be seen starkly in a
speech delivered to the Canadian Parliament on February 27, 2003, during
debates surrounding Canadian legislation to regulate stem cell research.
Mr. James Lunney, a Conservative party member of Parliament for the
Nanaimo-Alberni riding on Vancouver Island, began by saying that "[I]f
we took one of [the speaker of Parliament's] cells, extracted the nucleus
and put it into an ovum, one could stimulate it electrically and allow it to
grow." So far, so good. But then Mr. Lunney rocketed off into a grotesque
but all-too-common flight of misunderstanding: "The so-called therapeutic
clone would be to take the immature model of Mr. Speaker and extract an
organ, if he needed one, killing the clone in the process. That is so-called
somatic nuclear cell transfer or therapeutic cloning." Similar outrageous

N E W C E L L S F O R O L D 2 5 5
confusions have been perpetrated on the floors of the U.S. Congress and
elsewhere in the course of the stem cell debate.
SCNT doesn't involve making clones of people at all. It involves mak
ing blastocysts—balls of cells that, as we've seen, have not yet even made
the necessary steps to decide whether they will become one, two, or more
people. True, these blastocysts could in principle be used to make babies if
they were implanted into a woman the same way as is done with blastocysts
produced through IVF, but this is a potential, not a fact. When blastocysts
are created by SCNT for therapeutic purposes, no egg is fertilized by a
sperm; no new, unique DNA identity is created; no embryo is implanted in
an uterus; no pregnancy results. Biomedical SCNT creates cell life, but not
human life: renewed cells, not new people. They certainly have no organs
that we could harvest—including, importantly, no brain, nor even the be
ginnings of nerve cells. We no more "kill" a blastocyst produced by SCNT
when we derive stem cells from them than we "kill" a vat full of replicating
skin cells when we throw it out at the end of an experiment. Fundamentally,
SCNT would be the basis for therapies that cure you with your own cells,
restored to the potential they had in their first moments of existence by the
power of the stimulated human egg.
Because they derive from the patient's own DNA, SCNT cells are an
exact genetic match to those in your own body, and are treated as "self" by
your immune system. 3 4 Whatever may emerge from further research with
ESCs derived from surplus embryos left over from IVF, SCNT cells offer a
virtual guarantee of freedom from the specter of rejection, graft-versus-
host disease, and a lifetime spent on toxic immunosuppressive drugs.
In preliminary, preclinical research, the new regenerative powers of
cells derived from SCNT have already shown their promise. In animal
models, SCNT medicine has already been used to cure many of the devas
tating conditions for which human treatments must still be found, such as
Parkinson's disease, 3 5 heart attack damage, 3 6 and the animal equivalent of
the "bubble baby" syndrome (SCID)—rescuing not mere weanlings, but
fully developed, adult organisms that had suffered with the disease for their
entire lives. 3 7 As we've seen, the ESCs taken from more conventionally gen
erated blastocysts have worked some of the same marvels—but some of
these studies suggest that, even where rejection doesn't happen, SCNT may
still provide some advantages. And indeed, the results tend to downplay the
therapeutic potential of SCNT, because in these studies the scientists have
not actually derived the cells from each animal individually so as to provide

2 5 6 E N D I N G A G I N G
a perfect match (as we would do for human patients), but have used one
line of cells to treat an entire colony of close cousins.
In the Parkinson's study, for instance, the researchers coaxed SCNT-
derived cells to produce neurons suited for use in several areas of the central
nervous system (forebrain, midbrain, hindbrain, and spinal cord) and re
sponsible for a broad range of functions. Some of them were the kind that
produce the neurotransmitter dopamine, which is involved in fine motor con
trol; as I noted, it's the loss of these cells that causes Parkinson's disease. Oth
ers were cells whose central functions involve another neurotransmitter
called choline, and whose loss is characteristic of Alzheimer's disease. They
were also able to derive cells that secrete other neurotransmitters in the brain
( se ro ton in and GABA); that carry movement-control signals from the spinal
cord to the muscles (and whose degeneration is central to motor neuron dis
eases); and that act as "support" cells to neurons proper, nourishing and pro
tecting them. This was a much wider range of mature cells than had been
successfully derived from previous protocols using conventional ESCs.
The team then put these cells to the test in the Parkinsonian rodents
(whose dopamine-producing cells had been knocked down to less than a
third of their healthy numbers by a toxin), comparing their effects to ESCs
derived by the conventional route. Dopamine-producing neurons derived
from either protocol formed solid, stable grafts, and improved behavior in
their recipients, and there was no sign of rejection of either type of ESC.
But even though the SCNT-derived cells came from recipient animals'
cousins rather than their own, individual bodies, these cells performed bet
ter than conventional ESC cells, forming larger grafts in their brains, with
double the number of transplanted nerve cells surviving eight weeks after
transplantation, and the final graft sites containing about 50 percent more
dopamine-producing neurons.
And it appeared that they might have been somewhat more effective
at restoring function, too. Because all the damage had been inflicted on
one side of the brain, chemically stimulating the remaining dopamine-
producing cells in the brain caused an imbalance in their motion, with the
larger number of intact cells on one side of the brain sending out stronger
signals to the legs they control than the more damaged side does. The result
was that the animals began to veer to one side, rather like what happens
when you push a shopping cart that has a damaged wheel on one side of it.
This "rotational behavior" is a key test of the function of the damaged part
of the brain. Treating these animals with dopamine-producing cells derived
either from conventional ESCs or ESCs created using SCNT reduced this

N E W C E L L S F O R O L D 2 5 7
aberrant motion by more than 70 percent, with a hint that the SCNT-
derived cells were more effective (see Figure 3).
Because the range of neurons and supporting cells produced using
these protocols was so broad, the researchers who performed the study be
lieve that their technique could also be used to treat multiple sclerosis and
other "demyelinating" disorders (in which the myelin sheath essential to
the correct function of neurons is damaged or destroyed), Huntington's
Figure 3. Embryonic stem cells, and especially SCNT-derived cells, restore normal motion in a Parkinson's disease model.

258 E N D I N G A G I N G
disease, amyotrophic lateral sclerosis (Lou Gehrig's disease), and other mo
tor neuron diseases.
There remain technical hurdles to overcome in developing SCNT, but
theoretical objections to their ultimate use in human medicine continue to
fall. There have been concerns about the mitochondria in these cells, for one
thing: SCNT is achieved by replacing the DNA in an egg cell with a patient's
DNA, but this still leaves the egg's own energy factories providing the juice to
keep things going. Many researchers have therefore been concerned that the
resulting cells would be dysfunctional due to a mismatch between these mi
tochondria (created originally from the egg donors nuclear and mitochondr
ial DNA) and the final cell, or that the patient's body might reject the cells
based just on the immune-sensitive parts of the transplanted mitochondria.
So far, however, it doesn't seem likely that this mitochondrial mismatch is go
ing to trouble us. Aside from the fact that these cells have been incorporated
successfully into the patient-animals' bodies without any signs of rejection in
studies so far, a very careful study that looked all the way down at specific
proteins that are used to monitor for mitochondrial "foreignness" found that
the cells were accepted as completely native by all available measures.
Similarly, there are concerns that the "epigenetics" of SCNT-derived
cells—the "scaffolding" around the genes in the DNA code, which regulates
the expression of those genes—would be abnormal, leading to cancer or
dysfunction. Again, however, while this has been a problem with using em
bryos created using the nuclear transfer technique for making cloned ani
mals, it has not yet appeared to prevent stem cells derived from SCNT from
functioning properly when transplanted as a treatment into animals. And
indeed, early in 2006, scientists from the Whitehead Institute for Biomedical
Research in Cambridge, Massachusetts, reported that stem cells derived
from SCNT have identical patterns of being transcribed and turned into
proteins as do ESCs created by conventional IVF-type fertilization, with any
differences attributable to the genetic differences among the animals donat
ing the cells rather than the kind of cell involved. 3 8 Moreover, it appears that
the very process of generating stem cells from SCNT blastocysts of necessity
imposes a kind of "survival of the fittest" of its own, with any epigenetically
inappropriate cells collapsing under their own dysfunctionality; this may ex
plain a large part of why it's been so difficult to get a high yield of such cells
from a given blastocyst. This might pose problems for anyone actually look
ing to use nuclear transfer to clone a person (a point that should itself relieve
those concerned about such a use of the technique), but it appears that

N E W C E L L S F O R O L D 2 5 9
epigenetic problems will only make the use of SCNT for medicine more dif
ficult, not prevent its safe and effective use.
Another open question is where we l l get all the eggs we l l need to use
SCNT widely as an anti-aging therapy. The supply is limited by the number
of women prepared to donate their eggs, and the hormone treatments and
moderately invasive surgery needed are likely to continue to keep the num
bers of such women down for some time. Many people also raise ethical
concerns about offering money or other inducements to solicit more
donors, especially for a procedure which is not without risk.
Even this, however, may yet be overcome on a technical basis instead of
a sociopolitical one. One option would be to take the eggs from other
species. Such an approach would not be without technical hurdles: notably,
the presence of mitochondria in such eggs that are not just from a different
person, but from a different species, might make the cells unable to create
and sustain an energy supply. With my background in mitochondrial biol
ogy, I recently proposed a solution to this problem should it arise. 3 9 My so
lution results in the mitochondria of the eventual ESCs being derived from
the eventual recipient of the cells, just like the nucleus, so it also avoids the
"intra-species" mitochondrial problem I mentioned above.
Another alternative may be to mass-produce egg cells bioengineered
from more common cell types, such as skin. Canadian researchers recently
reported 4 0 having used skin cells from fetal pigs to produce cells which
look—based on gene expression patterns, cellular structure, and some
functional abilities—an awful lot like egg cells. Whether these cells have
the full range of functions of egg cells remains to be seen, but they—or a
more developed version of them—might have the same power to reset the
clock in mature somatic cells that conventional eggs do. This would mean
that we can bioengineer an almost unlimited source of eggs: human fetal
skin tissues, which contain nineteen billion such cells per square inch. Such
huge numbers would allow us to avoid entrapment in the battlefields of the
culture wars, if we can simply reach agreement on the use of tissues from
stillbirths rather than aborted babies.
Frozen Embryos, Frozen Science
This brings me back to my second SENS scientific conference. At the time,
you could still smell the ozone in the air from the second in a pair of scientific

2 6 0 E N D I N G A G I N G
lightning strikes from a previously obscure group of Korean scientists from
Seoul National University, headed by veterinarian Hwang Woo-Suk. A few
years after Dolly the sheep, Hwang had claimed to have cloned a cow, and
more recently a dog, but his fame came when he announced in the winter
of 2004 that he had achieved the world's first derivation of fully fledged hu
man ESCs using SCNT. This proclamation rocketed him to international
fame, but it was just the beginning: a little more than a year later, in the
months leading up to SENS2, he reported a dramatic improvement on the
technique. In his first report, Hwang had only been able to derive a single
stem cell line from the 242 eggs that had been donated—and this line had
been taken from an egg fused with DNA taken from the egg donor herself,
which was of very limited biomedical use. Now, Hwang was saying that he
had created eleven human lines using only 185 eggs, and using DNA taken
from completely different people, including potential patients of both gen
ders and many age groups.
Everyone in the field, as well as the popular press, hailed this result as
a phenomenal breakthrough, and I was far from alone in seeing its poten
tial for treating not only age-related disease, but aging damage more
broadly. I knew that I would want someone to present not only Hwang's
results—with which most attendees would be at least peripherally familiar,
due to the enormous press coverage—but what they would mean for scien
tists working in the field.
Hwang's result was clearly going to have an enormous galvanizing ef
fect on stem cell research. Because of the political climate in which it had
taken place, the impact of the announcement was far greater than could be
accounted for by the purely technical breakthrough (great as it might have
been) of actually being able to make customized stem cells for healing pa
tients. Stem cell research had been stymied for years by President George
W. Bush's notorious decision, in the summer of 2001, to limit federal gov
ernment funding of stem cell research to work done using lines created
prior to the morning when he announced the policy.
That decision reversed a policy accepted under the Clinton administra
tion, but not yet implemented, that would have plowed NIH funds into
ESC research using lines derived either from IVF clinics or from work orig
inally performed with private funds. It came not out of science but from the
political maelstrom of the abortion debate, and the antiabortion stance
held by President Bush and by his key constituency of the Christian Right.
And while it is not accurate to call that executive fiat a "ban" on ESC re
search, it created an enormous chill over the entire field—and not just

N E W C E L L S F O R O L D 2 6 1
because of the direct effects of cutting off funding for research performed
on nearly all available ESC lines.
The most obvious problem was the stranglehold it put on direct U.S.
federal government funding for embryonic stem cell work. The administra
tion holds the purse strings of a remarkably large share of U.S., and even
global, basic research in science, with US$20 billion in research-related
funding coming out of the National Institutes of Health alone each year.
Bush and his political allies would argue that their policy provides scientists
with plenty of opportunity to work with stem cells because of the availabil-
ity of the approved lines, but that claim ignores the actual state of the lines
in question.
The White House originally announced that their policy would allow
scientists to work with as many as seventy-eight robust stem cell lines, but
when senators put the question to NIH Director Elias Zerhouni, he admit
ted that only nineteen of these lines were actually viable, available in prac
tice (as opposed to locked up by intellectual property restrictions and
similar constraints), and ready for use in stem cell work. By 2004, this num
ber was still no higher than twenty-one. In preliminary research presented
at the National Academy of Science on October 12, 2004, fourteen of the
lines tested by Carol Ware at the University of Washington were found no
longer to grow well and to be hard to separate because of the outmoded
way in which ESC lines were derived and cultured at the time. One such
line was actually withdrawn from scientific use because of this finding. 4 1
A supply of just twenty-odd lines also fails to represent the genetic di
versity of humanity well, so it's hard to verify whether a given finding is a
quirk of that line or, say, of people of a given race. A supply of hundreds of
viable ESC lines is the likely minimum for healthy progress in this field of
science. Indeed, the present situation is worse than this: because of their
age, these lines are accumulating mutations that could skew the results of
research performed with them because they no longer even represent the
stem cells of those original donors. It also means that we can't study the stem
cells of people with particular diseases, or how experimental drugs affect
those processes—studies that would best be done with SCNT, which
would let us take the cells of people already known to suffer with a given
disease and wind back their clocks to the first moment of their primitive ex
istence as blastocysts. Researchers could then watch the cells as they under
went differentiation into the cells most affected by the disease and then the
late-phase changes that happened as their abnormal metabolism inter
sected with aging processes that happen even in healthy people's cells.

2 6 2 E N D I N G A G I N G
And not only are the cells of very limited use for basic research: every
one working in the field recognises that these cells will never be usable for
actual therapies either. All the lines that are both approved and available
are useless for clinical purposes because they were originally cultured using
feeder cells taken from mice—supporting cells needed to secrete factors
and provide structural support that is essential to keeping them in their pri
mal, unspecialized state. Contact with these cells has tainted them in vari
ous ways: one study 4 2 found that their cell surfaces contained a sugar that
the body's immune system recognises as foreign and attacks, and it's widely
expected (though not yet proven) that they may also contain mouse cell
proteins and even viruses.
It's only been in the last couple of years that scientists have developed
new techniques that first allowed for the propagation of human ESC lines
from human feeder cells, and most recently ESCs generated using no
feeder cells at al l . 4 3 And, again, it's at least possible that nothing short of
custom-made ESCs produced specially for the patient with SCNT will fully
address the potential problem of rejection. Thus, only ESC lines derived
well after the 2001 line-in-the-sand can actually be used as medicine for dis
ease and for the full repair of aging damage in the future.
The policy also ripples out well beyond the labs of people actually
working with the approved lines. For one thing, the restriction on provid
ing money to unapproved stem-cell work is so aggressively enforced that
the NIH has to snatch away grant money awarded even for research com
pletely unrelated to ESCs, if any of that money goes into facilities or equip
ment also used for work on "banned" ESC lines. You could be testing a
cancer drug on rats and have your funding pulled if someone else on cam
pus were sharing the use of gene-expression array equipment with you and
were using it for ESC work using lines not approved under the August
2001 decision. That makes it enormously difficult for anyone to carry out
work on ESCs other than the sanctioned lines on most university cam
puses, or in essentially all government research centers. Laboratories in
which sanctioned work does take place must expensively duplicate and
track equipment, all the way down to sticking colored dots on petri dishes
and other glassware, and must generally work in ways that degrade effec
tiveness.
The policy also erects enormous roadblocks even to privately funded
research—research that, in theory, is not the target of the Bush policy. Sci
entists in industry are first trained in universities, and when those universi
ties can't carry out ESC work, because of a mixture of lack of federal

N E W C E L L S F O R O L D 2 6 3
dollars and the handcuffing of non-ESC work carried out using shared
equipment with work on "forbidden" lines, young researchers don't get
trained in the techniques of working with stem cells, let alone get the op
portunity to perform original research that would advance the field. This,
of course, means that such researchers aren't available for hire by private
firms even if they had the money to bring them on.
And naturally, the political uncertainty swarming around stem cell
work makes potential investors reluctant to pour money into companies fo
cused on developing ESC-based cures. At the time, it seemed possible that
the United States would follow other countries in making aspects of this
research—such as SCNT—not merely ineligible for government funding
but actually a criminal act. Investors will stomach most risks, but not politi
cal risk, and so they abandoned private-funded ESC research for a number
of years.
It briefly seemed likely that the Bush administration's policy would be
quickly brought down by political pressure. Even a very conservative selec
tion of polls shows that the majority of people in the United States and else
where are in favor of a fairly open policy on scientific access to ESCs. Even
in surveys in which the question is posed flat-out with no mention of po
tential human benefits, the majority of people say that they support deriv
ing stem cells from surplus embryos from fertility clinics for scientific
research. 4 4 When the question mentions the potential for human treat
ments, this proportion climbs into the 70 percent range. And most people
even support SCNT research—a fact that fills me with optimism about
their future acceptance of other anti-aging therapies.
So, in the August heat, with the controversy raging and President
Bush's popularity resting on unsteady ground, it seemed possible that pub
lic opinion would mobilize against the restrictive policy, and scientists
would within reasonably short order be enabled to work with ESCs from a
wide range of sources.
Then, the planes hit the World Trade Center.
In a month, everything had changed. Where ESC research had been
front-and-center on the national stage in August, it was off the radar screen
for almost everyone in late 2001, replaced by the immediate fear of terror
ism. As pressure and scrutiny from the wider public melted away, those
whose organization, resources, and ideological investment were strong
enough to continue to push their agenda on the subject even in the shadow
of the ruins of the Twin Towers suddenly became the only voices pushing
legislators—and in this case, because of the conflation of the science with

2 6 4 E N D I N G A G I N G
the abortion debate, that meant almost entirely forces opposed to ESC re
search. Anti-abortion groups, who are well organized and well funded,
made their case to Washington as strongly as ever, without the usual bal
ancing force of either the public at large or of their usual opponents: pro-
choice and civil liberties groups had no particular stake in stem cell science,
and the latter had their hands full with presenting the case for preserving
Constitutional rights in the face of the threat of terrorism. And while pa
tient advocacy groups might have stood in opposition to blockades on re
search, such groups were nascent at the time, and lacked the support from
pharmaceutical companies that often sustains them, since in this case the
companies had no vested interest in promoting the groups' cause.
Feeling deferential to a newly popular wartime president, handed
plenty of unba lanced misinformation by the anti-abortion activists on the
religious right that made up that president's base of support (and had
played a significant role in their own sweep into power), and with a leader
ship dominated by representatives with a social-conservative worldview of
their own, the Republican-dominated Congress substantially raised the
threat against stem cell science. Parallel bills introduced by Sam Brown-
back in the Senate (S 245) and Dave Weldon and Bart Stupak in the House
(HR 234) sought to ban all forms of "human cloning"—including SCNT
performed entirely for scientific or medical purposes.
These measures would not only deny federal funding to, but criminal
ize, the creation of blastocysts using SCNT—imposing actual jail sentences
on scientists performing the work. They would also have imprisoned scien
tists doing any scientific research using ESC lines derived from SCNT; in
the original texts of these bills, this went so far as to threaten both doctors
and patients with prison time if they administered or accepted cures using
SCNT-derived stem cells. Some language even suggested that people who
went abroad to receive treatment with SCNT stem cells could be penalized
for it on their return to the United States.
But in the coming months, as the public slowly began to raise their
heads out of their foxholes, proresearch and patient activist forces began to
countermobilize. They were greatly helped by the voices of prominent pa
tients suffering with diseases likely to benefit from SCNT and those close
to them, including Michael J. Fox (Parkinson's), Kevin Kline (whose son
has juvenile diabetes), Christopher Reeve (spinal cord injury), and, most
powerfully, Nancy Reagan (whose husband, the former president, died of
Alzheimer's disease). A bipartisan coalition favoring expanded embryonic
stem cell access—and in many cases the full legalisation of biomedical

N E W C E L L S F O R O L D 2 6 5
SCNT—began to form, including such prominent anti-abortion Republi
cans as Orrin Hatch, Strom Thurmond, Arlen Specter, John McCain, and
ultimately then senate majority leader Bid Frist, and apparently extending
even to Bush's own secretary for Health and Human Services, Tommy
Thompson. Meanwhile, prominent scientific organizations (including the
National Academy of Sciences, the American Medical Association, the As
sociation of American Medical Colleges, and even the National Institutes
of Health itself), as well as multiple disease-specific charities (such as the
Juvenile Diabetes Research Foundation, the American Association for Can
cer Research, the Lance Armstrong Foundation, and the American Diabetes
Association) endorsed research using new ESC lines and the advancement
of work on SCNT.
Hatch's coalition introduced legislation to legalise SCNT for scientific
and medical research, while banning use of the technique as a means of
cloning people. They also introduced legislation to allow access to surplus
embryos from fertility clinics as a source of stem cells. Slowly, more and
more legislators from both sides of the aisle signed on to the pro-research
side of the debate. For the next several years, the two forces fought to a
standstill, with both bills repeatedly introduced and defeated. This created
a legal and scientific limbo that ultimately served the anti-research camp's
agenda: the few scientists working on SCNT remained out of prison but
without access to funding, potential private investors continued to wait out
the political uncertainty, and the President's restrictions on ESC research
remained in place.
False Dawn
Then suddenly, in 2005, came Hwang's announcement of relatively high-
yielding techniques for creating individually tailored ESCs. The news acted
like a juggernaut, smashing through barriers both scientific and political.
Technically, the ability to make viable, customized embryonic stem cells tai
lored to individual patients was a massive breakthrough. Politically, it not
only reenergized pro-research forces, but applied a new source of pressure
on politicians. Stem cell advocates had long argued that, if the government
continued to keep a tight lid on ESC research, the science would be done
elsewhere: the United States would simply suffer a brain drain, as Ameri
can scientists moved to more hospitable climes to pursue their vital work
and as foreign graduate students (already chafing under new security

2 6 6 E N D I N G A G I N G
restrictions) refused offers from American universities. Now, the prophesy
began to come true. The Korean government was ready to back their new
scientific star's work with significant resources; countries as far-flung as the
United Kingdom, Israel, Sweden, and Singapore began establishing them
selves as well-funded hubs for ESC research; and reports of prominent sci
entists packing their bags started appearing in the media.
The forces of competition began to work their usual magic. Individual
U.S. states, fearful of being left behind, began putting up bills to fund stem
cell research within their own borders. Federal politicians not strongly ideo-
logically committed against ESCs—including many free-market-oriented
Republicans—became increasingly willing to challenge the agenda of the
antiresearch ideologues. A couple of years earlier, fifty-eight senators—most
of them Democrats, but with substantial support from key Republicans—
had signed a letter asking Bush to rescind his policy; a little over a month af
ter Hwang's announcement, 206 House members followed suit.
I knew that highlighting these advances, and the opening that they af
forded to researchers, would be a great way for me to further my confer
ence's mission to promote anti-aging biomedical research. Short of Hwang
himself, the best person to present the opportunities was Gerald Schatten, a
stem cell researcher at the University of Pittsburgh who had been working
with Hwang for the last two years, had used his veterinary techniques to
clone a monkey, and had signed off on the paper announcing the new SCNT
lines in Science. I asked him to present their results and outline the royal
road to accessing patient-specific ESCs through Hwang's team at Seoul Na
tional University: a "World Stem Cell Hub" that would generate SCNT cells
to order using Hwang's established facilities and exper ienced technicians.
I was delighted when Schatten accepted my invitation—but I was pos
itively overjoyed when, not long after, he came back with another e-mail
saying that he'd like to bring along a friend. Hwang himself had expressed
an interest in presenting at SENS2, he said; he realized that it was short no
tice, but would I be willing to let him share Schatten's own half-hour slot in
the conference? I, of course, offered instead to give Hwang his own half-
hour talk as a featured presenter in the session on stem cells and regenera
tive medicine. I'd have been happy to do this even if it had required
throwing out my original schedule and starting from scratch, begging for
giveness from presenters as I shuffled them around at so late a stage in the
planning of a very packed conference schedule; but fortunately I didn't
have to do this, as another presenter had recently been f o r c e d to pull out.
With hardly any shuffling, Hwang was confirmed.

N E W C E L L S F O R O L D 2 6 7
So it was that, with great pleasure on my part and keen attention from
hundreds of my colleagues, Hwang mounted the lecture podium of Cam
bridge's Fitzpatrick Lecture Hall.
Of course, as you know full wed, unless you spent much of the winter
of 2005-2006 in a cave in Nepal, it was all a sham. Within months of elec
trifying my scientific audience in September, Hwang had been exposed as a
fraud.
Bad Wizards and Bad Men
First there were ethical questions about the sources of Hwang's eggs; then,
questions about the viability of four of the eleven stem cell lines that he had
submitted to Science. And then, reporters looking at photographs presented
with Hwang's data began to notice some suspicious resemblances between
allegedly unique stem cell lines. Hwang brushed these off as the result of a
confusion with the Science production staff about which of the numerous
photos that he had submitted were to be used as figures in the article.
The case against Hwang quickly picked up momentum. Scientists re
viewing the paper's data n o t i c e d suspect similarities in the genetic profiles
of the various lines' cells. Then Schatten requested that his name be retro
spectively removed from the paper's authorship because of "allegations
from someone involved with the experiments that certain elements of the
report may be fabricated." And on December 15, one such collaborator
came forward with the flat statement that nine of Hwang's eleven lines were
flat-out fakes, sharing identical DNA with one another, and claiming that
Hwang himself had admitted the fraud.
As each doubt was raised, Hwang would protest his innocence, variously
blaming errors, contamination, and the incompetence of others for each of
them—even going so far as to claim that one former collaborator had
"switched" some of his lines. But, eight days after his former collaborator
claimed fraud, he proffered his resignation to Seoul National University—
which was refused, on the grounds that he was now the subject of an internal
investigation. He was suspended in February, dismissed in March, and in
dicted in May for fraud, embezzlement, and violation of bioethics legislation.
The fallout of these revelations was felt at many levels. There was, of
course, enormous outrage at the fraud, and great disappointment that
Hwang's breakthrough turned out to be flimflam. And it was a political fiasco,
exploited by anti-ESC campaigners to cast aspersions on the entire field.

268 E N D I N G A G I N G
But Hwang's fraud had also set the progress of the entire field back by at
least a year—an eternity in science. The Bush restrictions on ESC research,
amplified by the looming threat of criminalization for SCNT from the Amer
ican Congress, had kept all but a few research teams from working to perfect
nuclear transfer for human patients. Hwang's claims had further diminished
the incentive to put resources into the goal: No one wanted to reinvent the
wheel already spinning in Korea, and private firms would no longer have a
competitive edge as the first creators of patient-tailored ESCs, using in-house
methods that could be kept exclusive as trade secrets or through the patent
process.
One private firm, A d v a n c e d Cell Technology (ACT), had been coura
geously soldiering on with the work, producing a great deal of quality ESC
and SCNT science (much of it, admittedly, overplayed in the media)—this
despite constantly lurching from one financial crisis to the next because of in
vestors skittish over the legal climate of their research. In late 2001, they fa
mously announced the first "cloned" human blastocyst,4 5 although the DNA
that was transferred into the egg cell came from the donor's own body—in
fact, from cells that normally enshroud the egg itself—and the resulting blas
tocysts couldn't develop beyond a six-cell ball. They spent much of the next
two years perfecting that technique, issuing many publications (most of them
on research done in cattle) charting their progress toward teasing out the rea
sons for the low yield of viable blastocysts in SCNT techniques, and working
steadily to perfect the technique for human biomedical use.
In late 2003, insists ACT scientific director Robert Lanza, they were
very close to resolving the sticking points in their technique, and could
shortly have generated the first viable stem cells tailor-made for patients or
for research on specific diseases. With Hwang's eleven-cell-line announce
ment, however, investors began pulling their bets out of what was perceived
to be an "also-ran" in the race—a blow turned into a haymaker by ACT's
simultaneous setback of losing their main source of human eggs. Cells were
put into deep-freeze, and ACT's human SCNT work shut down.
Equally infuriating is the case of Professor Alison Murdoch and Dr.
Miodrag Stojkovic, of the Newcastle Centre for Life, a fertility clinic and
research center in Newcastle-upon-Tyne in Britain. These researchers man
aged to create the first human SCNT blastocysts using DNA taken from
the cells of a person other than the egg donor. 4 6 Like the ACT cells, these
blastocysts were not fully viable, but in the United Kingdom's more
research-friendly environment, the group had received formal approval
from regulatory bodies to pursue work using SCNT-derived stem cells, giv-

N E W C E L L S F O R O L D 2 6 9
ing them the green light to perfect their technique. But their publication
came on the heels of Hwang's, and compared to his explosive success, the
creation of only three fused cells that actually began dividing, and no actual
stem cell lines, seemed to be of no relevance to the progress of the field.
They, too, promptly shut down their research on the technique—a decision
that Murdoch says cost them at least a year.
It was the same story elsewhere. SCNT research teams in Sweden, and
also at three American universities that had raised enough private or state
money to set up stem-cell research centers with elaborate financial firewalls
separating them from federally funded research elsewhere on the same
campuses, either dropped their efforts altogether or put them on hold
while waiting to see whether the Korean team's plans would make their ef
forts redundant.
But if Hwang's shot, heard 'round the world, turned out to be a backfire
at best, it still served to wake up a lot of people. All over the globe, and es-
pecially in the United States, researchers began thinking seriously again
about what they could do once they had access to the Korean team's expert
ise. It promised stem cells with the miraculous flexibility of the blastocyst,
but perfectly matched to patients suffering with the worst of the nightmares
of aging: Parkinson's disease, stroke, scarred and weakened hearts, eyes
blinded by the death of light-sensing cells choked in their own waste, limbs
withering away as the electrical sparks stopped flowing through nerve cells
or muscle cells snapped one by one under the force of their own molecular
decay. Labs began contemplating grant proposals. Young science students
turned back to stem cell research as an exciting career prospect. The dawn
was false—but its rays woke the slumbering forces of science all the same.
Today, after the collapse of Hwang's house of cards, and in defiance of
the Bush administration's politically driven, morally misdirected obsti
nacy, the field is undergoing a renaissance. Murdoch and ACT have fired
up their programs again. Teams are in hot pursuit of successful SCNT
techniques, and the research and cures that they will enable, all over the
world. Cutting-edge work is occurring at the Center for Regenerative
Medicine at Edinburgh University, Scotland (taking over clinical research
from the veterinary work that created Dolly the sheep); at the Karolinska
Institute in Sweden; at Shanghai Second Medical University, China; and at
several privately funded centers in the United States, including the Harvard
Stem Cell Institute, the University of California at San Francisco, and
UCLA's Institute for Stem Cell Biology and Medicine.
The legal climate is shifting, too. In addition to China, Great Britain,

270 E N D I N G A G I N G
and Sweden, SCNT is already explicitly legal in Singapore, Belgium, Japan,
Spain, and Israel. And it remains legal in the United States, despite the ef
forts of Senator Brownback and his allies: the Brownback-Weldon bill has
twice faded to make it through Congress, though it was reintroduced in
2005 as S 658/HR1357, the Human Cloning Prohibition Act of 2005. More
excitingly, Orrin Hatch's bipartisan Stem Cell Research Enhancement Act,
which would have opened up ESC research using lines derived from IVF
surplus embryos, passed both the House and the Senate, and was only
blocked from becoming law by a Presidential veto—the first of Mr. Bush's
six-year administration. Hatch's pro-SCNT bill is also back in play, al
though no vote is imminent.
Meanwhile, individual U.S. states are moving ahead, doing their best to
sidestep the funding and regulatory vacuums at the federal level. SCNT re
search has been legalized in California, Connecticut, New Jersey, Rhode Is
land, Illinois, and Massachusetts, and although several other states have also
specifically prohibited all SCNT work, many more are permitting work done
using surplus IVF blastocysts. A ballot initiative in Missouri that would con-
stitutionally protect scientists' ability to conduct ESC and SCNT research,
and patients' ability to access cures based on these techniques that are avail-
able anywhere else in the nation, narrowly passed in November 2006.
The states are also digging up the funds needed to get work going in
side their own biotech sectors. The most famous of these is California's
Proposition 71, a ballot initiative that established the California Institute
for Regenerative Medicine and gave it a bond-funded budget of $3 billion
to fund ESC work including (but not exclusive to) SCNT research. The ac
tual disbursement of these funds has so far been bogged down by anti-
research legal actions and some legitimate questions about oversight and
ethics, but legal rulings have been almost uniformly favorable, and Gover
nor Arnold Schwarzenegger has recently stepped in with a bridging loan,
to start the Institute's mandated dollars flowing toward development of
SCNT- and other ESC-based cures.
And if California is the most famous case, it is far from the only one—
or even the first. That honor goes to New Jersey, which in early 2004 be
came the first state to earmark state funds for ESC research. Starting in
December 2005, New Jersey has put out a total of $5 million in grants
awarded to seventeen research institutions for research on stem cells from
embryos and other sources, and has set up the $23-million New Jersey Stem
Cell Institute. Similar initiatives are starting up in Connecticut, Illinois,

N E W C E L L S F O R O L D 2 7 1
Maryland, Massachusetts (despite a faded veto attempt by the state's gover
nor), and the state of Washington.
Meanwhile, efforts have been under way in the private sector, despite
the lack of support, adverse regulation, and a climate of uncertainty that
sends all but the steeliest of investors off in search of other opportunities.
ACT is one prominent example; another is Geron Corporation, a biotech
company most famous for its work on the "youth enzyme," telomerase
(which it is now seeking to manipulate in order to shut down cancers by
turning off the telomerase taps—see Chapter 12 for much more on this).
Geron has perfected methods of raising human ESCs without feeder cells,
and is testing six different lines in animals; excitingly, it expected to be
ready to begin the earliest stages of human trials using neural stem cells for
spinal cord injuries in the spring of 2007.
Some scientists are also seeking purely technical ways to liberate sci
ence from the phony moral dilemma surrounding the use of blastocysts for
research into, and as cures for, human disease. There are multiple proposed
ways to create ESCs from blastocysts without eliminating the potential for
these cell balls to ultimately become human lives. One is parthenogenesis, a
label taken from the technical term for "virgin birth." In this technique, the
genes in an egg cell (which naturally contains only half of a full comple
ment, as it is designed to be augmented by those in the sperm cell at fertil-
ization) are doubled up on themselves, thereby generating a complete set of
DNA instructions; this allows the egg cell to behave enough like a blasto
cyst to produce ESCs for the egg donor, without actually fertilizing the egg.
Another approach is to take stem cells from IVF clinics' embryos that have
defects preventing them from going forward to make a fetus, or to actually
induce such defects into a patient's DNA before making a blastocyst from it
using SCNT, in order to eliminate even its potential to form a human life.
Recently, ACT introduced yet another option: using stem cells derived
from a single cell plucked from the blastocyst, while leaving the rest of them
in place as a potentially viable embryo (as is already sometimes done for
genetic testing of IVF embryos before implantation). 4 7 Yet a fourth possi
ble option is to coax adult stem cells into behaving more like embryonic
stem cells, using growth factors and other chemical messengers, instead of
using the inherent renewing power of the egg to do the same thing. 4 8
I have no doubt that such work is valuable—but primarily because it
will tell us more about stem cell biology. The lessons learned will allow us
to manipulate ESCs and SCNTs more capably when the legal environment

2 7 2 E N D I N G A G I N G
finally unleashes the scientific racehorses, champing at the bit to bring the
promise of these cells to fruition. The specific techniques involved will
probably not, and most certainly should not, be necessary to bring cures to
patients with "official" diseases or to regenerate human bodies deprived by
the aging process of their capacity to self-heal. Their perceived necessity is
a purely political construct, unrelated to scientific reality or underlying hu
manitarian need. The real need is to free scientists from misguided interfer
ence with the quest to turn the enormous potential of embryonic stem cells,
including patient-specific cells created by fusing a patient's cells with an
egg, into therapies for the sick and the old.
Fortunately, this is one area where nearly all my colleagues already es
sentially agree with me—and not just in biogerontology, but across the
medical and basic biological science world. The grant-review scientists at
the National Institutes of Health would be delighted to disburse funds to
promising ESC and SCNT research around the nation, if their hands were
not bound by their president's executive orders. Everyone not under the
blinders of a mi sp laced sense of moral responsibility to a ball of cells recog
nises the need for ESC research guided by responsible ethics and regula
tion, not artificial restrictions created out of confusion, fear, and the grasp
of political opportunity.
Certainly, there are scientific hurdles to be overcome before ESCs can
be used directly as medicine. We have to develop much more reliable tech
niques for deriving stem cells, transforming them into the kinds of cells that
we need, and making them play the same full range of roles that the corre
sponding cells growing under the guidance of sophisticated developmental
programs within our bodies already do. The nascent field of regenerative
medicine is already making surprising progress even using cells and tissues
grown from patients' adult cells: tissue engineers are moving from cells to
organs, seeding cells into a biodegradable scaffold that guides them to form
into a structurally appropriate engineered tissue and then melts away, leav
ing functioning tissue behind. 4 9 Human patients have been given functioning
urethras, starting with cell-free structural tissue taken from cadavers and
seeded with the patient's cells, which have lasted seven years. Functioning
bladders have been engineered and transplanted into Beagle dogs, and rab
bits have received, and successfully used, engineered erectile penis tissue.
And in the most ambitious work to date, cattle have been given simple kid
neys created using SCNT, with DNA taken from an ear clipping. The reju
venated cells were expanded, growing to fill in every cranny of a complex
biodegradable kidney scaffold, and the resulting organ implanted. The ar-

N E W C E L L S F O R O L D 2 7 3
tificial kidneys were functional, filtering the blood and producing a fluid
with close chemical similarity to normal urine.
But the fundamental impediment to the dream of new cells to replenish
bodies worn by the years or by disease is a political one—and so must its so
lution be.
Action Now for Science and Medicine
Nearly all the anticipated readers of this book are citizens of democratic
states. A few live in countries that have already given their scientists the
green light to pursue cures with ESCs within careful ethical and regulatory
frameworks. If so, congratulations. You can help to lead the race for cures
forward further by lobbying your politicians to increase funding for such
research.
But probably the single largest share of my readership will be in the
United States—the country that still makes the greatest contributions to
world scientific progress, where young scientists still flock to chase the ad
vancement of human capacity, and where government and industry funding
could, if unleashed, have the greatest impact on the field. The National In
stitutes of Health need to have the brakes taken off their funding power,
and to be allowed to step down on the accelerator—hard.
Your voice can help the scientific process along in a way that the sci
entists themselves can't match. Write letters, join lobby groups, educate
yourself about local issues and your Congressional representatives' posi
tions; then vote for pro-stem-cell ballot initiatives and research-friendly
politicians. Excellent background information and tools to help you sup
port favorable legislation are available from the Coalition for the Advance
ment of Medical Research (CAMR) at http://www.CAMRadvocacy.org.
You can bring forward the date when animal studies become clinical
cures—and when, eventually, the old grow young, their bodies renewed by
their own rejuvenated cells and tissues. Scientists need your help now to
bring medicine out of the lab and into the lives of suffering patients.
When you and your loved ones need their help, you will want to know
that you have done everything you could to support their lifesaving work.

12
N u c l e a r M u t a t i o n s a n d t h e
T o t a l D e f e a t o f C a n c e r
Apart from the small amount in our mitochondria, all our DNA
is housed in the nucleus of our cells. Like mitochondrial DNA,
it accumulates damage throughout our life, and this can
theoretically lead to innumerable health problems. However, I
believe that in practice only one of those problems—cancer—
arises within what we currently consider a normal lifetime.
Thus, if we could really thoroughly defeat cancer, nuclear
mutations would be harmless. The most audacious component of
SENS is just that—a way to defeat cancer altogether.
In a few places in earlier chapters, particularly Chapter 10, I've
whetted your appetite with regard to telomeres and telomerase. I know you
have that appetite, because when someone asks me what I do and I say I
work on combating aging, the commonest response (apart from the just
slightly predictable "Hurry up!") is "Ah, telomeres." And indeed, telom
eres and telomerase play a very prominent role in SENS. But not the role
that most of you are probably expecting.
As I've stressed throughout these chapters, the "engineering" ap
proach to combating aging is fundamentally different from conventional
thinking about aging and what to do about it, in that it focuses on the actual
damage that the aging organism accrues, rather than the metabolic processes
that cause that damage to accumulate.
This operational definition of aging makes the problem tractable. In

N U C L E A R M U T A T I O N S A N D C A N C E R 2 7 5
the old-school, "gerontological" approach, the number of potential con
tributors to the aging process is legion, and getting control of all of them is
a paralyzingly daunting task. It requires us to have a detailed understanding
of an enormous number of complex pathways, interference with any of
which is both difficult and bound to cause unwanted side effects as the nor
mal function of those pathways is perturbed.
Anti-aging engineering sets us largely free of these problems. We leave
metabolism to carry out its necessary but messy work, and find ways to
undo or render harmless the relatively small number of fixed changes—
molecular damage, in other words—that occur in the actual structure of
the aging organism as a result of those processes. We are left with a field of
just seven classes of damage to deal with—classes for which solutions are
foreseeable, and whose repair is unlikely in itself to cause any negative side
effects. All that we intend to remove is initially inert but eventually patho
genic (pathology-causing) damage—aspects of the aged body that the
young organism does just fine without.
There is, however, one apparently gargantuan hole in this logic, and
that is the question of damage to the DNA code in the cell's nucleus (as op
posed to the DNA held in the mitochondria, which I discussed back in
Chapters 5 and 6). Whereas mitochondrial DNA is responsible only for the
production of the energy factories in which it is housed, nuclear DNA is
the master blueprint from which our entire biological structure is built up
and maintained over time. The proteins 1 that it encodes not only make up
essential structural features of the body, from the lens of the eye to the
pumping muscles of the heart and the miles and miles of arteries that carry
blood to our cells, but also include tiny enzymatic machines that do every
thing from detoxifying poisons to budding up fatty membranes and carry
ing chemical signals from one cell to the next. Damage the DNA, and you
corrupt the code of our genetic program, or render perfectly good genetic
instructions unreadable by the machinery that transcribes them into orders
to be sent out to the body's protein-making "factories."
And your genes do suffer accumulating damage over time. The DNA
in the nucleus is subject to a continuous assault on its structure. Each cell's
nuclear DNA takes about a million damaging "hits" every day, caused by
everything from ultraviolet radiation and environmental toxins to the free
radical by-products of its metabolic processes. And even brand-new DNA
isn't necessarily pristine: when the cell replicates itself, errors perpetrated
by the machinery that copies the cell's genetic information often create
production flaws of varying degrees of seriousness.

2 7 6 E N D I N G A G I N G
Much of this mischief is quickly fixed by the cell's elaborate quality
control system for DNA, but some of it is irreparable by its nature. Some
other damage is potentially reparable, but becomes indelible if the cell di
vides before repairs are made. Such permanent changes are mutations, and
while mutations that occur elsewhere than in sperm and egg cells (and their
progenitors) will not be passed on to the organism's progeny, they will be
perpetuated in the cell in which they occur and in any of its "descendents."
On top of damage to nuclear DNA itself, there is damage to the so-
called epigenetic structures of our chromosomes—the "scaffolding" that is
anchored to our DNA. Epigenetic structures contribute important informa
tion by determining which genes are turned on in a cell and which are
turned off, allowing the same overall DNA to be used to create cells as di
verse as liver, heart, and kidney cells. Because of this, changes in the epige
netic scaffolding of a cell's DNA ultimately have the same range of functional
effects on the cell as changes to the genes themselves: By turning on genes
that should be turned off (or vice-versa), or increasing or decreasing their
activity, these "epimutations" change the complement of proteins produced
by the cell. Because they are operationally equivalent in terms of their im
pact on cell function, I'd allow myself a little terminological sloppiness to
avoid belaboring the point with extra verbiage. From here on, I'll mostly be
using "mutations" to refer to both these kinds of genetic damage—true mu
tations and epimutations.
Because they occur on an occasional, random basis and are permanent,
mutations accumulate with age—and therefore, they qualify as "aging dam
age" by the definition e m b r a c e d by the anti-aging engineer. The implica
tion, then, is that we will have to either fix them or render them harmless if
we're going to keep the body from progressively declining into pathology
over time.
It Is Broke. We Can't Fix It
Having read this far, you may be expecting that I'll propose a fix for muta
tions similar to those I've suggested for AGE cross-links, unwanted cells, or
lysosomes: just get rid of the junk. But a moment's reflection should show
that this can't be done. Damaged genes may be dysfunctional, but we can't
afford to just do without them: a cell with anything other than a functional
gene is still damaged. And that presents a daunting challenge—so daunting,
in fact, that for a time I despaired of its solution, and feared that mutations

N U C L E A R M U T A T I O N S A N D C A N C E R 2 7 7
would act as ship-smashing cliffs to any ark that we might build to survive
the deluge of metabolism and emerge into an ageless future.
So, you may think, If we can't afford to destroy defective genes, can't we
repair them instead? Unfortunately, this is a technical near-impossibility in
the foreseeable future, for the simple reason that there are so many differ
ent genes in the nuclear DNA. The human nucleus has two copies of nearly
all our genes; the aggregate size of one copy (the "haploid genome") is
about three billion "letters" of DNA. Exactly how much of this is really ac
tual instructions for building and regulating the body is a matter of some
debate, but certainly there are plenty of different places where damage to a
"letter" could cause the misspelling of a "word," leading to a loss of infor
mation that could harm the function of the cell. That's a lot of potential
damage that we would expect to need to fix.
The problem is how to do that for so many distinct genes. Any mecha
nism we might use to fix a damaged gene would have to somehow "know"
how to distinguish an intact gene from a damaged one. True, the body al
ready does this, by comparing the damaged DNA to its complementary
strand in the double helix (or sometimes to the homologous chromosome, i.e.
the other copy of the relevant stretch of DNA—remember I just told you that
most genes are present in two copies in each cell), but it's not clear that this
helps us much. It's hard to see how we could improve on our existing, inbuilt
ability to repair DNA, which in humans is already amazingly effective.
We could, in principle, solve this problem if our DNA-repair system
relied on a blueprint independent of the genetic information within the
cell. But this would require a molecular-level tool that would carry in it
self a master copy for each of the many tens of thousands of genes (each
of which is typically a thousand or more DNA letters in length). It's con
ceivable that advanced nanotechnology might somehow be able to pro
vide this level of detail to machines that could then carry out the repairs, 2
but only in the very distant future; we're nowhere near that level of profi
ciency today.
In fact, this summary actually understates the difficulty of the job we
would face if our solution to mutations were to fix them one at a time.
You're probably visualizing that these double-copy gene mutations are
nice, clean breaks cutting across the two strands of the DNA helix—and
sometimes they are. But sometimes, the DNA is either initially hit, or is mis
takenly "fixed" by the body, in a way that leaves two distinct errors on ei
ther strand, separated by a dozen or so DNA letters. This leaves gaps whose
repair, even given a proper blueprint, would entail first fixing the damage

2 7 8 E N D I N G A G I N G
to both sides independently and then realigning the two strands and zip
ping them back together.
This one ready had me stumped back in July 2000, at the conference
where the engineering approach to developing anti-aging biomedicine first
crystallised in my mind. If we can't just throw out damaged DNA, and
there's no clear way to fix all of the possible forms of permanent DNA dam
age that accumulate with age, aren't we stuck with a mechanism of aging
that we can't do anything about—and that will therefore kill us even if we
repair or obviate every other molecular and cellular change that con
tributes to aging? At first I essentially ignored the problem, relying that
other approaches to cancer would suffice, but I became increasingly doubt
ful that they would.
Who 's Afraid of the Big Bad Mutations?
Throughout these chapters, I've always explained how a given kind of molec
ular or cellular change ("damage") probably contributes to aging pathology.
But in most cases, it's not totally clear to what extent a given kind of alter
ation actually adds to the loss of function, increased disease risk, and expo
nential rise in death rates that characterise biological aging. In all the cases
we've discussed so far, however, the answer to this question doesn't ready
matter. Aging damage is, by definition, not a part of a healthy, youthful body,
so removing or obviating that damage certainly won't do us any harm—and,
from what we can ted, will almost certainly do us significant good.
In this case, however, I couldn't see a clear way either to repair or to
render harmless mutations in the nuclear DNA. So I had to take a step back
and ask a more fundamental question: Do we, as a practical matter, actually
have to worry about nuclear DNA damage?
Even to ask such a question sounds a bit crazy to some of my col
leagues. Because the nuclear DNA is so clearly critical to cell structure and
function, it seems beyond debate that mutations are a contributor to aging.
This concept was first formally proposed in the late 1950s, before we ready
even understood what genes were, and it has become almost universally ac
cepted by both scientists and the public at large.
But, however initially intuitive it may sound, this notion has never been
justified with direct (or even good indirect, i.e., correlative) evidence. The
only way to rule out something's involvement with aging is to speed it up
and observe no effect on life span or age-related pathology.3 This has been

N U C L E A R M U T A T I O N S A N D C A N C E R 2 7 9
accomplished for nuclear mutations, but not yet well enough to be decisive.
In mice, deleting a gene that normally fixes up free radical hits to genes be
fore they have a chance to go on to become actual, fixed mutations greatly
increases the steady-state level of such damage in the nuclear DNA. Yet
these animals appear to suffer no pathology as a result, and have normal life
spans. 4 However, we can't read too much into this result because the muta
tion rate is only elevated rather modestly. Similarly, a study was recently
performed using four strains of mutant mice, each with a knockout of a dif
ferent DNA-repair gene. In one such strain, mutations accumulated more
than in normal mice-—and yet, the effects on lifespan were unclear.5
A very good, and equally direct, test for showing that something is a
key contributor to aging is to slow it down or arrest it and observe a direct
anti-aging effect: an increase in the "natural limits" to the organism's life
span and the preservation over time of youthful functionality. Of course, no
one has ever done this with nuclear mutations, or indeed with anything
else. Unfortunately for scientific clarity, all the successful anti-aging inter
ventions that we currently know of in mammals change many things about
the organism, from antioxidant enzymes, to maintenance of proteins, to the
activity of the cell's garbage-disposal system. This prevents us from isolat
ing any one of those changes as the dominant cause of the anti-aging effect,
and thus isolating any particular type of damage as being the dominant
contributor to aging. Calorically restricted animals, for instance, lose a sig
nificant amount of bone mass, but no one thinks that this loss is responsible
for CR's anti-aging effect.
A close approximation of such a test, however, was published a couple
of years ago, in the form of mice that had been given genes allowing them
to produce extra amounts of an antioxidant enzyme (catalase), specifically
targeted to different parts of their bodies. 6 , 7 I mentioned these mice in ear
lier chapters, but the study is worth a recap. Catalase detoxifies an abun
dant oxidizing molecule (hydrogen peroxide), so it can potentially protect
things it's close to from at least one form of damage. Putting catalase into
these animals' mitochondria, which significantly reduced the development
of mitochondrial DNA deletion mutations, reduced their vulnerability to
several age-related diseases, and extended their maximum life span by
about 20 percent—the first unambiguous case of an antioxidant genetic in
tervention with an effect on this key measure of aging in mammals. Yet giv
ing these organisms catalase targeted to the nucleus, which reduced nuclear
mutations, provided no benefit in terms of lifespan.
Another type of evidence that is often raised in support of the idea of

2 8 0 E N D I N G A G I N G
nuclear genetic damage as a contributor to aging is the existence of so-
called "accelerated aging" models whose symptoms arise from accelerated
rates of nuclear mutation accumulation. These are animals either with vari
ous inborn mutations, or subjected to outside assaults (like bombardment
with toxic chemicals or X-ray radiation), that increase the accumulation of
nuclear DNA damage as they age, either by increasing the rate at which it is
formed or by knocking out the machinery that repairs it. This includes such
human genetic diseases as Hutchinson-Gilford syndrome ("progeria") and
Werner's syndrome.
Victims of these diseases, whether they walk on two feet or four, do often
look in many ways like the elderly, suffering pathology that can be eerily par-
allel to that seen as animals get older, from bone diseases and failing hearts to
scruffy fur and cataracts. But the fact that the symptoms of an abnormal
pathology look a lot like the symptoms of "normal" aging doesn't prove that
the mechanisms of one underlie the mechanisms of the other, any more than
a wet lawn proves that the same mechanisms govern rainfall patterns and
sprinkler systems. Almost anything that messes up normal equilibrium in the
body but takes a while to kid you will look like "premature aging;" the ques
tion is what if any relationship a given change bears to aging in the rest of
us. Evolutionary biologist Michael Rose, a geneticist at the University of Cali
fornia who has himself bred remarkably slow-aging, long-lived fruit flies,
puts the point succinctly: "A lot of people can kill things off sooner, by screw
ing around with various mechanisms, but to me that's like killing mice with
hammers: it doesn't show that hammers are related to aging."
Because we don't have this kind of direct evidence, we normally rely on
more indirect, correlative types of evidence of a phenomenon's involvement
with aging. One is to compare the rate of accumulation of some kind of
damage in animals that age at different rates (that is, operationally: whose
"oldest old" finally succumb to "natural" death, after first progressively
losing youthful functionality, at different chronological ages). While all ag
ing animals do accumulate nuclear mutations, the rate at which longer-lived
animals suffer free radical damage to the nuclear DNA doesn't correlate
well with their maximum life spans (unlike the corresponding rate in mito
chondrial DNA, which does).
Frustrating, isn't it? But the good news is that, in my view anyway, the
available evidence allows us to conclude with some confidence that nonspe
cific nuclear DNA damage is not meaningful contributors to aging. I briefly
summarized the argument for this conclusion back in Chapter 4; here's the
full story.

N U C L E A R M U T A T I O N S A N D C A N C E R 281
What gives force to the idea that nuclear mutations are a cause of aging
is the fact that the mutations we inherit from our parents are major risk fac
tors for a wide range of diseases, including age-related diseases from cancer
to heart attacks to Alzheimer's. The same is sometimes true of mutations that
occur very early in development, when there are so few cells in the little ball
that will eventually transform itself into a human body that damage to the
nuclear DNA of just one of them can infect almost our entire being with the
same flaw.
But the situation is quite different after we're born, which is when new
mutations occur and accumulate with the passage of time—and thus, where
we have to think about an effect of nuclear mutations as a form of aging
damage (as opposed to as a source of inborn vulnerability to aging dis
eases). This is because mutations only spread from one cell to another
when the second cell's DNA is actually derived from the DNA of the first,
as happens when (and only when) a cell divides and passes a copy of its
DNA on to its progeny. Thus, whereas inherited mutations infect all of the
cells in our mature bodies (because all of our mature cells derive from a sin
gle, mutated fertilized egg), age-related mutations happen in our mature
bodies one cell at a time, as a result of random events like the radiation that
comes into a plane at high altitude, or toxins produced by invisible mold
spores in your food. Any such mutations can only affect the one cell in
which they occur, and its descendents.
This fact greatly limits the potential of age-related mutations to become
prevalent enough in our tissues to impair function. A lot of our cells—
including the ones in tissues where the impact of aging is most clear, like
the brain and heart—don't divide at all once we mature, so any mutations
in such cells go no further. And even in cells that do divide—skin cells, say,
or the cells lining your gut—the rate of cell division after we mature is bal
anced by the short life of the cells' descendents, so that an individual cell's
mutations are present in only a few cells in the body at any instant and thus
get little chance to "take over" the tissue in which the mutation occurs.
Also, even before we had hard numbers on the subject, we knew that
complete "knockout" mutations would be relatively rare occurrences. For
one thing, most mutations in DNA create errors in the encoded proteins
that, if they affect proteins at all, only make them less effective, rather than
completely dysfunctional. But even in cases where a gene is damaged so
badly that either it can't be used as the basis for protein manufacture at all,
or else the product that it produces will be useless—or even toxic—still
generally no harm will ensue, because the damage will be to a gene that

2 8 2 E N D I N G A G I N G
isn't even being used in that cell! Remember that the DNA of each cell in
cludes not just the genes needed for that cell to do its specialised job, but
the entire instruction manual for producing every cell type in your body.
Individual cells acquire their specialized function—heart cell, liver cell,
kidney cell, skin—by turning off most of their DNA, leaving active only
those genes that they require to perform their particular function. In a typ
ical cell, only about one tenth of a person's full gene complement is active.
So about 90 percent of the genes in a cell could be irreparably damaged
without affecting the cell's function one bit.
It gets better yet. Even though evolution is far too thrifty to allow cells
to go around wasting their resources in producing proteins that aren't im
portant to cell function, the cell can nonetheless suffer the loss of many
proteins and still limp along without harming the body, even contributing
in some degree to its internal economy. Remember, many people are born
with such mutations affecting every single one of their cells, including all
the cells where that protein is normally needed to do their jobs, and these
people still live for decades. The same mutation occurring in only a small
fraction of one's cells might not even be noticed in a normal lifetime, being
compensated for by the fact that the rest of one's cells are fully functional.
Mutations: Few, and Not Far Between
And sure enough, the actual number of mutations that accumulate with ag
ing does indeed appear to be rather low—too low to have any serious effect
on the aging of the tissue in which they appear. We know this thanks in
large part to the work of a team headed by physiology professor Jan Vijg,
now at the Buck Institute for Age Research. Vijg's group came up with a
very ingenious way to obtain a representative sample of both the amount of
mutation that accumulates in different tissues with age, and the general
kind of damage that occurs.
What Vijg's group found was surprising to them and to a lot of other
researchers. They discovered, first, that there were not nearly enough rela
tively minor point mutations ("one-letter" mutations in DNA "words" or
"sentences") to realistically have a meaningful impact on overall tissue
function or on the aging of the organism as a whole.
More important, the number of mutations in a typical cell doesn't even
increase between early and late adulthood in our most critical tissue (the
brain), and the total burden of mutations only goes up by a factor of two to

N U C L E A R M U T A T I O N S A N D C A N C E R 2 8 3
three in any tissue, even in old age. 8 , 9 This may sound like a lot, until you
remember how many genes there are in such a cell and how few mutations
are present in young people. Doubling or tripling a small number of initial
mutations still leaves the cell with only a small number of them; and when
you consider how few of those are occurring in genes actually used by the
cell in which they occur, how few of that subset actually disable (as opposed
to merely inhibit) a critical function of the cell in question, and how little
the loss of a particular activity in a particular cell can really affect the over-
all function of a tissue, an organ, or an entire animal, it becomes clear that
an increase like this is actually more of an inconvenience than a crippling
blow to the aging organism.
But Vijg immediately appreciated that this was not a knockout blow
to the involvement of nuclear mutations in aging. This is because some of
the mutations identified by his group may be much more severe than the
disabling of a single gene. Some of them, for instance, could take the form
of deletions—the total removal of large stretches of DNA, annihilating
many genes at once even though the event is, strictly speaking, only a sin
gle mutational event. Deletions can occur when two widely separated
breaks occur at the same time in the same chromosome, and Vijg's data
showed that a significant proportion of the mutations that accumulate
with age do involve such breaks, which at first glance suggests there are a
significant number of deletions amongst the modest increase in the total
number of mutations that happens with aging. This would have a much
greater impact than would be suggested by a simple tallying-up of muta
tion frequencies.
Luckily, however, it turns out that most of the cases in which a cell suf
fers two distinct breakages of DNA don't actually lead to deletions. 1 0 Fully
half of them are actually cases of fractures happening on two separate chro
mosomes, which are not physically linked to one another and thus do not
lead to a deletion—much as if you cut two separate pieces of string across
their respective middles and switch the pieces' partners, rather than mak
ing two snips in the same length of material and discarding the portion be
tween the two cuts. And even in the remaining cases, where both breaks
occur on the same chromosome, many of the incidents are equally harm
less, leading to mutations called inversions that shuffle chromosomes
around while typically leaving them intact and functional.
For this and other reasons, not many of the events that at first look like
deletions actually seem likely to have much more effect on actual genetic in
tegrity than simple point mutations.

2 8 4 E N D I N G A G I N G
When we look at epimutations, the evidence available so far leads me
to the same conclusion. The most well-studied kind of epimutation is
changes in methylation—a chemical alteration of genes that prevents them
from being expressed. Middle-aged mice 1 1 and humans 1 2 have indeed been
found to have more alterations in their methylation patterns than immature
ones do, but it's not clear that that trend continues further as the organism
actually ages (as would be required of a true, stable form of molecular ag
ing damage). Instead, as they go from adulthood into true old age, the
change in methylation epimutation may stop accelerating and may even
slow down. This could mean, for instance, that the observed methylation
changes occur in the early life of the organism, but that their frequency is
subsequently held to tolerable levels by repair mechanisms or even by the
elimination of unacceptably aberrant cells, rather than accumulating with
age the way that AGE cross-links or mitochondrial mutations do. Just as
important, from our perspective as would-be interveners, there's no evi
dence that the observed methylation changes cause any actual functional
problem over the life span.
Add it all up, and there seems to be remarkably little increase in muta
tions happening in aging cells—and of what there is, again, very few will ac
tually affect the cell's functionality.
The Times, Not the Genes, They Are A-Changin'
Vijg's studies of that period, and those of a few of his forerunners, assessed
the impact of mutations on aging by measuring the actual frequency at
which genes are structurally altered during aging. But there's another kind
of evidence that's often invoked to support the role of mutations in aging:
gene expression studies. Starting in the late 1990s, scientists were able to
use a new technology informally known as "gene chips" to measure the ac
tivity, rather than the structure, of nearly all the genes in the cells of tissues.
This allowed for comparison studies of aging and younger animals—
including humans. 1 3
The results clearly showed that there are pretty substantial changes in
gene expression in the cells of aging animals. This result has often been mis
takenly thought to prove the importance of mutations in aging, because the
gene expression shifts were assumed to be the result of mutations in the genes
themselves. This idea was given some seeming support by evidence that the
changes in gene expression happened alongside markers of "pre-mutagenic"

N U C L E A R M U T A T I O N S A N D C A N C E R 2 8 5
free radical damage to genes—damage that can still be repaired as it stands,
but that can go on to become full-blown mutations if not repaired properly.
But there's a much easier explanation for these shifts, and that is the
very fact that gene expression does change—not just with aging, but all the
time. Your cells are in a state of continuous dynamic adaptation to their en
vironment, changing gene expression in response to new conditions at
every moment. Every time your body needs to respond to its environment,
the expression of genes involved in those responses is altered.
So as cells and tissues acquire aging damage that impairs their normal,
youthful function, the body adapts to the changed circumstances as best it
can. As oxidative stress climbs with the accumulation of cells that have
been taken over by mutant mitochondria, cells ramp up the activity of
genes that produce protective antioxidants and "heat shock" factors that
help to repair some of the ensuing damage to proteins. When the increase
in oxidative stress leads your arteries to become infiltrated with free-
radical-damaged LDL, the surrounding cells produce more inflammatory
factors to attract macrophages to clear the toxic stuff out. When your heart
stiffens as it becomes riddled with cross-linked proteins, the body pro
duces more "remodeling" enzymes that help to degrade the old tissue in
hopes of clearing the way for fresh, undamaged replacement material.
Additionally, changes in the cellular environment imposed by aging
can also interfere with normal gene expression. In some cases, such effects
are easily observed: for example, oxidative stress directly inhibits the ex
pression of some genes. But there are also more subtle effects afoot. For in
stance, you'll recall that free radicals, in addition to being produced as
simple side effects of metabolic forces, are also produced intentionally as
signaling molecules in various systems inside and outside cells. As free rad
ical levels climb in the aging organism's cells, the excessive oxidative stress
distorts these same signalling pathways, introducing "noise" into them.
This can in turn result in inappropriate metabolic and gene-expression
shifts as the cell responds wrongly to misheard, drowned-out, or counter
feit messages from (or superimposed on) the system.
And so on. The point is that the age-related shifts in gene expression
seen in studies of tissues are not the cause of aging, but are an adaptive (and
sometimes maladaptive) response to it. Changes in gene expression in cells
subjected to free radical attack can occur not because that attack has dam
aged the genes whose expression levels change, but because an increase in
oxidative stress requires the cell to change its metabolism to keep going in
face of the challenge.

2 8 6 E N D I N G A G I N G
Now, you may be thinking that I've jumped the gun here. So far, I've
explained how gene expression changes can be a compensatory response to
aging rather than a cause of aging—but surely I have not shown that those
changes are, overwhelmingly, responses and not causes. But it's easy to see
that they must be responses, because they're coordinated. The only reason
the studies I'm referring to were able to detect a change in expression of a
given gene was because it was occurring in the bulk of the cells within the
tissue being analyzed. Mutations and epimutations, by contrast, would af
fect one gene in one cell and a different gene in the next cell.
This coordinated change of gene expression in response to aging is
shown most clearly in gene-chip studies that compare normally aging animals
to those undergoing calorie restriction ( C R ) , 1 4 , 1 5 , 1 6 which is, again, the only
intervention now at our disposal that slows down aging in mammals, short of
tinkering with their genes. These studies show that the greatest number of
changes in gene expression that happen with aging occur in the blueprints
for antioxidant, inflammatory, remodelling, and heat shock proteins—
exactly the ones needed to respond to the damage inflicted by aging. They
also show that CR not only slows down, but reverses, many of these changes
when it's implemented late in life. This shows that the changes in gene ex
pression can't be the result of mutations, because while CR can slow down
the accumulation of mutations and other aging damage, it cannot undo dam
age that has already been done. Rather, it reduces the sources of damage that
are leading to the changes in gene expression, cutting down free radical pro
duction in the mitochondria, reducing the accumulation of mitochondrially
mutant cells, lowering blood glucose to prevent glycation cross-links, and so
forth. Change the cells' pro-aging environment, and they scale back their
gene-expression adaptations to that environment.
This sort of logic has led some, among whom Vijg's team are again
prominent, to conduct much trickier studies assessing the variation of gene
expression from one cell to the next in old animals as compared with young
ones It was found that there is a dramatic increase with age in that varia
tion. 1 7 On the face of it, this increased variability doesn't seem likely to be
the result of the kinds of adaptation that are responsible for the dominant
pattern of age-related gene expression shifts that we were talking about a
moment ago, because the conditions that create them—and the adaptive re
sponses needed to keep going under their influence—are more or less the
same from one cell to the next in a given tissue.
But there are a lot of alternatives to explore before we jump to the con
clusion that this increase in the amount of difference in gene expression be-

N U C L E A R M U T A T I O N S A N D C A N C E R 2 8 7
tween a given aging cell and its neighbors is the result of mutations (or
epimutations). It may be due to some other age-related stressor, for in
stance. Vijg's team found that they could reproduce the increase in variabil
ity by exposing cells in culture to oxidative stress—and again, oxidative
stress disrupts the ability of cells to relay signals within themselves. Such
disruptions could lead to a failure to respond to signals coming in from the
cell's environment—either local signaling molecules from its neighbors, or
more "broadcast" signals like hormones and other factors. The effect of
this would naturally vary from one cell to the next, because it results from
noise in intra- and intercellular signaling, but if the oxidative stress were
then removed, the variability might return to its original level.
Variability from one cell to the next can also result from other kinds of
damage that have accumulated, randomly, to different degrees from one
cell to another: telomere loss in one cell, high levels of AGEs in another,
and the presence of mitochondrial mutations (without nuclear mutations)
in still a third. Intuitively, the gene expression shifts required to increase
the degradation of AGE-ridden proteins on the surface of heart muscle
cells are quite distinct from those that occur in response to accumulated
junk within them. When neighboring cells suffer different levels of differ
ent kinds of molecular damage, they mount different adaptive gene expres
sion responses, both to counterbalance the effect of the damage on their
ongoing metabolic processes and in many cases to attempt to repair it.
Such effects have been observed at the cellular level in roundworms, 1 8 and
preliminary studies suggest that similar factors could be at play in increases
in gene expression variability in rats and humans, too. 1 9
Also, such damage can send shock waves outside the cell in which it oc
curs, which will elicit changes in gene expression in nearby cells trying to
cope with its impact. Consider cell senescence, which I discussed in Chap
ter 10. The activation of the senescence program will certainly change the
expression of genes in the senescent cell as compared to its neighbors, but
it will also change the expression profile of those neighbors, as they re
spond to the flood of growth factors, inflammatory signals, and remodeling
proteins that it produces. Cells closer to the senescent one will be more af
fected than cells further away.
Importantly, these changes would distinguish the neighbors of a senes
cent cell both from the original culprit and from other cells not directly af
fected by its output. To pick the most obvious example: growth factors
secreted by a senescent cell will trigger cell-replication programs in nearby
cells. These programs, executed by gene expression changes, are ones that

2 8 8 E N D I N G A G I N G
have been permanently shut off in the senescent cell itself, and that are qui
escent in cells far enough removed from the senescent cell to evade its in
fluence.
The same would be true of cells that had suffered any number of prob
lems. And the variation can flow in the opposite direction, too. That is, in
addition to damaged cells playing havoc with their neighbors by exporting
their own internal woes, healthy cells can communicate with damaged ones
to keep them functioning more normally. The best-researched case of this is
cancer. 2 0 , 2 1 A single cell can harbor mutations that would by default lead to
malignancy, but be held in check by its neighbors, which can do everything
from inhibiting its proliferation (so-called contact inhibition) to inducing
apoptosis. As with all other adaptive changes, the output required to impose
this control requires shifts in gene expression that will distinguish cells work
ing to keep their antisocial neighbor under control from the patterns of both
the would-be cancer cell and noncancerous cells elsewhere in the tissue.
Hence, even the fact that there are more pronounced differences in the
activity of genes from one cell to the next in the tissue of old animals (as
compared with young) doesn't necessarily pin the blame on nuclear muta
tions. Moreover, much of the variability observed is a good thing—the dis
tinct efforts, futile though they may ultimately be, of each cell to preserve its
integrity under the unique burden of aging damage that each of them faces.
If, then, age-related gene expression changes don't flow out of a signif
icant increase in nuclear mutations, but are instead the result of other kinds
of damage (and cells' attempts, successful and otherwise, to carry on in the
face of it), the proper engineering response is to leave nuclear mutations
alone, and focus our attention on the real culprit in age-related gene dys-
regulation: the aging damage that forces our cells to flail about in increas
ingly desperate, disorderly, and panicked attempts to keep their heads
above the waters of the aging process. Once our cells are no longer suffer
ing under the onslaught of problems ranging from mitochondrial mutations
to AGE cross-linking to visceral fat accumulation, their gene expression
profiles can be expected to normalize, because both they and their environ
ment will be normalized—returned to a youthful state of functionality.
Ironically, it may be precisely after we have cleaned up all of these
sources of damage that nuclear DNA mutations may, eventually, begin to
contribute to age-related death. The evidence shows only that cells accumu
late relatively few nuclear DNA mutations, and those mutations do relatively
little to impede cell function within a normal lifetime. But what happens
when that life span is extended to centuries, with the same amount of meta-

N U C L E A R M U T A T I O N S A N D C A N C E R 2 8 9
bolic damage to our DNA occurring, and with our DNA-replicating and
DNA-repairing machinery no more perfect than it is today? It may wed be
that effects that are too subtle ever to reach a pathological stage within nine
decades or so could build up into a real threat over the longer term.
However, we don't need to worry about that now. As I explained in re
spect of AGE cross-links in Chapter 9, one cornerstone of the engineering
approach to developing anti-aging biotechnology is that we don't need to
fix all possible forms of damage at once—we only need to do a good
enough job of cleaning up those insults that meaningfully contribute to
age-related frailty within today's life expectancies. Once this is accom
plished, our bodies will remain youthful during the years in which they are
now undergoing a slow descent into decrepitude. Some forms of molecular
damage that aren't causing us problems now will then begin to reach the
threshold beyond which we begin to meaningfully, functionally decay.
At that point, the next generation of SENS therapies will need to be
brought in, to repair that newly pathological (and in some cases even newly
identified) damage to the same standard. Fortunately, our extended lives
will buy us the time to observe such damage accumulating in our bodies,
and in those of shorter-lived animals whose lives have been extended in the
lab, and whose youths renewed, using the same treatment—and as time
goes by, we'd build better and better tools for identifying that new damage
and for developing weapons to fight it. The key is to get past the barriers
that hold us back today, and then be ready to break new ones as they ap
proach. From all available evidence, nuclear mutations aren't yet such a
limit—but they may well be one in the future.
The Exception That Creates the Rule
So far in this chapter, I've explained that one of the reasons not to worry
too much about nuclear DNA mutations is that even the ones that are actually harmful to the cell are usually minor and, even when they are major, the
damage that they inflict is mostly constrained to one or a few cells, so that
any slack that their dysfunction creates can be picked up by other cells in
the tissue. But there is, of course, one screamingly important exception to
this rule: cancer. Cancer is famously a disease of nuclear DNA mutations
(though, as we've seen, it usually takes more than just mutations to turn an
aberrant cell into full-blown cancer). And the incidence of cancer clearly
goes up with age.

2 9 0 E N D I N G A G I N G
Shortly, I'll discuss how I think we can really defeat cancer. But first,
I'm going to explain why cancer is the reason for our apparently "unneces
sarily" good defenses against the accumulation of nuclear mutations.
First, let's recall that one of the reasons cancer is so formidable an ad
versary is that any number of different mutations can contribute to the can
cer process. A breakdown in any one of the many systems that protect
against cancer—mutations that lead to the formation of a defective senes
cence or apoptosis "tumor suppressor" protein, or to the excessive produc
tion of a cell receptor for growth signals, or to the reactivation of a
suppressed telomerase-coding gene—can be a key step along the broad
and winding road that leads a healthy cell to become a renegade.
As with all other causes of age-related death, the evolved level of pro
tection against potentially cancer-causing mutations is limited by the cost of
creating and maintaining the machinery to achieve that protection. On the
one hand, there's no sense in putting all of an organism's eggs into the bas
ket of being so resistant to aging processes that it can stay young and
healthy for two hundred years, if there are strong odds that it will freeze,
starve, sicken, or become something's lunch in its third decade; those re
sources would be better spent on warmer fur, sharper claws, or simply a
shorter gestational period. But on the other hand, the animal does need to
remain internally intact for as long as it can reasonably be expected to sur
vive those threats from the external environment, because every year of
youth is another opportunity to spread one's genes around. If you're unsure
about this, go back and reread Chapter 3.
Given those opposing priorities, natural selection will push hard to
create machinery that protects against potentially cancerous mutations rig
orously enough to keep cancer at bay for at least as long as the organism
would likely get through winters, wars, and attacks from predators. But,
precisely because of the nature of the cancer threat—that so many alterna
tive mutations can contribute to the cancer—the body can't afford to
cherry-pick the genes that it's going to protect. The only effective defense
against cancer is to safeguard the integrity of every single gene we possess.
So this is how it comes to be that new, age-related nuclear mutations
are so rare—why we possess such an elaborate system of oversight of DNA
synthesis repair guarding our every gene, despite the fact that the great ma
jority of mutations have negligible effect on the overall economy of the
body. To protect the organism adequately against cancer, evolution gets the
best bang for its buck from a system in which every gene gets the kind of

N U C L E A R M U T A T I O N S A N D C A N C E R 2 9 1
gold-plated antimutation defense that you might think it would reserve for
a privileged few, mission-critical, cancer-avoidance genes . 2 2 , 2 3
A 2015 Challenge
This analysis shows that we don't need to address all mutations in order to
develop a panel of interventions comprehensive enough to result in the first
dramatic extensions of human life span. My realization of this was crucial
to the nascent development of the SENS platform back in 2000, because it
showed me that the sheer scope of the nuclear mutation problem was in
one sense much smaller than I had at first feared. It had become clear to me
that, for the most part, age-related accumulation of nuclear mutations are
basically harmless over the course of a currently normal lifespan: the rate of
their accumulation is quite insufficient to contribute significantly to age-
related decline. But we emphatically do need to confront the one enormous
exception to that rule: cancer. In principle, we can simply ignore nuclear
mutations, if and only if we can find a truly effective way to protect our
selves against this one fatal disease.
The stakes here are high. Cancer is a deal-breaker for building an age
less organism. We can shatter the cellular manacles of AGE, free our brains
and hearts of the webs of amyloid, clean out the dirty depths of our lyso
somes, and all the rest—but if we fad to make a breakthrough against this
one disease, we can still expect to be dead in our mid-eighties.
If you've been listening to the popular press accounts of progress in the
War on Cancer, you may now be feeling much less alarmed than you
should. The media, and also the scientists and bureaucrats on whom they
report, love to trumpet every advance (indeed, every hint of an advance) in
the treatment of cancer. There are so many reports of potential new cancer
treatments that one might well think we were already far down the path to
the day when we will have cancer mastered. This is especially so in light of
the targeted cancer therapies that I reviewed in Chapter 10—therapies that
generally are, or can be predicted to be, far safer and more effective than
the knives, poisons, and radiation that have been the staples of cancer man
agement for decades.
And you wouldn't be alone, nor even outside the scientific mainstream,
in thinking this. In 2003, none other than Dr. Andrew von Eschenbach, the
Director of the National Cancer Institute, famously put forward an ambitious

2 9 2 E N D I N G A G I N G
but (so he claimed) realistic vision for his organization: to eliminate suffering
and death from cancer by the year 2015. Dr. von Eschenbach wasn't just
whimsically putting his dreams into words: he was putting forward his sober
assessment of what the world scientific community, spearheaded by the NCI,
could achieve in little over a decade. It became the formal agenda of the In
stitute: "Challenge Goal 2015." The timetable is now so embedded in the or
ganization that it is routinely alluded to as simply "2015," with no further
elaboration necessary—in the same way that we once talked about "Y2K." 2 4
I think that this goal is utterly unrealistic—and that it only arose be
cause of a failure to appreciate the flaws in the assumptions that are built
into it. First, and quite explicitly, "it does not mean 'curing' cancer but,
rather, it means that we will eliminate many cancers and control the others,
so that people can live with—not die from—cancer."2 5 If feasible, this is a
perfectly legitimate medical goal: to have cancer under the same level of
control that we today have over adult-onset diabetes or AIDS—in which
the disease is still present but is so well managed that patients can lead
nearly normal lives—would represent an enormous alleviation of human
suffering and death from a terrible disease.
But cancer is fundamentally different from these diseases in a way that
precludes its chronic "management." Diabetes and hypertension can be
held at a safe, manageable level precisely because they are essentially stable
diseases. By contrast, what makes cancer so fearsome a foe is that it's a con
stantly evolving disease, a hive of genetic inventiveness that continuously
finds new and better ways to outwit our attempts to control it. Relegating
cancer to the level of a chronic disease is an idea that could only ever be en
tertained by completely ignoring the basics of natural selection.
Cancer cells are characterized by an immense genetic instability, which
results in large part from the fact that they nearly all get started from a mu
tation in one or more of the "guardians of the genome"—the genes that po
lice mutations and direct either repair of DNA damage or the activation of
senescence and apoptosis programs. Without this constant surveillance and
maintenance, the random damage that cells suffer every day is allowed to
develop into full-blown mutations, and the process feeds on itself as more
regulatory genes are lost.
Many of these mutations are fatal to the cancer cell, but a few of them
result in viable progeny that are just different from their parents and half-
siblings. And that's where natural selection comes into play. Cancer cells,
by definition, are reproducing themselves at an astonishing pace. They
throw their bastard children out into the world and let survival of the fittest

N U C L E A R M U T A T I O N S A N D C A N C E R 2 9 3
reign. The immune system or oncologists soon take their best shots at the
tumor, exploiting the soft spots in the cancer cells' metabolism: their re
liance on particular growth factors, for example, or their need for a reacti
vated telomerase gene, or their hunger for folic acid. But within a single
tumor exists such an astonishingly varied population of cells, each with its
own combination of normal and abnormal genes, that at least some of those
cells nearly always have a way to survive any particular attack: a greater abil
ity to detoxify a particular toxin, or an alternative way to keep their growth
fueled even when a particular signal transduction pathway is shut down.
As a result, it ultimately just doesn't matter if a given therapy kids 99
percent of the cells in a tumor. Somewhere within its heart lurks the dark fa
ther of a "strain" of the cancer with a novel mutation that allows it to survive
the drug that destroyed its cousins. This founding cell's furious growth con
tinues even as we decimate its cousins, or resumes when the patient can no
longer tolerate the stresses of the treatment. Its descendents remain standing
after the assault, and are thereby selected for survival by the very thing that
killed their cousins. When the ensuing tumor becomes large enough for us to
detect, we attack what seems to be the same cancer in the same patient using
the same treatment, but this time, the old tricks don't work. There really is
plenty of truth in the saying that you can't outsmart evolution.
As I sat reflecting on all of this in the wake of my original SENS "Eu
reka! " moment at the turn of the millennium, a grim formulation of the prob
lem crystallized in my mind. It is not my goal, I thought, to buy time for cancer.
Making Cancer Wilt Away
Soberly, I contemplated my own cancer challenge: to develop a cancer
treatment that would keep us free of clinical cancer for just as long as the
other SENS therapies would keep us free of other age-related maladies.
The reasoning outlined above immediately ruled out all existing ap
proaches, which leave us fighting one battle after another against an enemy
with the implacable force of evolution on its side—a war in which we can
win individual campaigns but must ultimately be defeated.
The answer came to me in March 2002 while nursing a beer in a cafe in
Italy. As my mind had done late that night in California in 2000, it now leapt
upon an insight that was in some ways completely obvious, and yet led inex
orably to revolutionary conclusions. To defeat cancer, I saw, we would need
a therapy that does not depend on anything that a cancer could escape

2 9 4 E N D I N G A G I N G
through a mutation-driven change in gene expression. So any solution
would have to have three key characteristics to be viable. First, it would nec
essarily involve denying cancerous cells access to some tool that is absolutely
indispensable to any cancer's survival, so that they couldn't just make up for
its loss by tweaking some other gene expression pathway through mutating
its other genes. Second, we would have to take away that tool in such a way
that no mutation could restore it, either. And third, this tool would have to
be one that our normal, noncancer tissues could do without.
I quickly saw the tool that I wanted to lock up: telomerase. I mentioned
this enzyme back in Chapter 10, when discussing senescent cells; now's the
time to explore it in more detail. Our DNA comes equipped with a stretch
of nonsense or "noise" DNA called the telomere. Telomeres are to our
genes as the brief, silent stretch of "leader tape" at the beginning of a music
cassette is to the songs on the tape: they give the "cassette player" (the
DNA-replicating machinery) something to hold on to and advance over, so
that it won't skip over the essential information at the beginning of the very
first "song" (gene) on the tape.
One key difference between telomeres and cassette leaders is that lead
ers stay intact as long as the tape does, whereas telomeres become ever-so-
slightly shorter every time the cell replicates itself or is hit by damaging
agents like free radicals. If it weren't for telomerase, this gradual shortening
would eventually lead to the complete loss of the telomeres in cells that
replicate frequently during the lifespan, and thus the gradual erosion of the
genes themselves. Telomerase periodically relengthens the telomere before
it becomes critically short.
As with all of our other genes, the DNA that encodes the telomerase
enzyme is present in all of our cells—but, because it's only needed after
quite a few cell divisions have occurred, it's not needed in most cells for
most or all of the time, so it's turned off. This widespread lack of the need
for telomerase is used by evolution as a key component of our defense
against cancer, because having a limit to the size and renewal of our telom
eres prevents our cells from replicating themselves indefinitely—the crucial
hallmark of cancer.
To become a full-blown cancer (as opposed to a cell with a single, po
tentially threatening mutation—a genetic risk factor for becoming a cancer)
requires the accumulation of five to ten mutations, and statistically that re
quires multiple rounds of cell division and selection. The arithmetic is com
plex, but the consensus is that, to pose a health threat, cancers have to
replicate at least two to three hundred times, even though a clinically rele-

N U C L E A R M U T A T I O N S A N D C A N C E R 2 9 5
vant tumor contains "only" a million million (a " 1 " with twelve zeroes after
it) cells, which could be achieved by "only" forty or so divisions if the orig
inating cell had all the necessary mutations from the outset. And to be gen
uinely malignant (i.e., to be the founder of a colony of cancer cells that
spreads its way throughout the body, as opposed to a localised tumor that
could be simply removed with surgery and forgotten about), cancers must
then be able to keep up the feverish pace of their replication even longer.
The frenzied reproduction of cancer cells is also a key part of their ability to
evade our assaults, because it is essential to their capacity to evolve new so
lutions to the challenges that we throw up against them.
It's no surprise, then, that mutations that unleash telomerase from the re
pressive strictures imposed on it in normal cells are found in over 90 percent
of cancers. The remaining 10 percent also have a way to renew their
telomeres—a little-understood mechanism called the "Alternative Lengthen
ing of Telomeres" (ALT) pathway, which I'd discuss a little later on. Either
way, without a way to renew their telomeres, the single-minded multiplication
of potential cancer cells rapidly grinds to a halt as it reaches the end of its
telomere "rope," and we wind up with a tiny (and generally short-lived) lump
in our bodies instead of a life-threatening, malignant disease.
If, then, we could snatch this one tool out of the hands of cancers, we
would cause any and all the aspiring cancers we developed to fizzle out be
fore they became life-threatening—indeed, before many of them even be
came actual cancers, because they wouldn't get the opportunity to undergo
the full spectrum of mutational events needed to give rise to the kind of
renegade cell that can truly pose a threat to the body.
Of course, I was hardly the first to think the problem this far through.
Several biotech companies—most prominently Geron, which first made a
name for itself in telomere research—are working to develop anticancer
drugs that would work by deactivating telomerase. But these pharmaceuti
cals suffer the same problem as all other approaches based on drugs that af
fect gene expression: they act as a force of natural selection against a
disease with evolution at its disposal. A telomerase inhibitor would kill off
those cancer cells in which it effectively turns off the enzyme (and in which
ALT didn't take telomerase's place), but it would leave behind any cells that
harbored mutations allowing them to keep on renewing their telomeres in
the face of it. Different cancer cells might bear any number of variations
that let them escape the drug's effects. Some would simply crank their
telomerase activity up even further; some would enhance the activity of
drug-metabolizing enzymes that degrade the inhibitor; still others would

2 9 6 E N D I N G A G I N G
change their cell surface proteins in ways that would make it harder for the
drug to penetrate into the cell. Whatever the mechanism, if even one can
cer cell can evade the effects of such a drug, it can act as the seed for the tu
mor's renewed blossoming in a dark spring.
So, again, there was no sense in doing the job only halfway. If we're go
ing to snatch telomerase out of the hands of cancer cells, I thought, we must
really take it away. And there was only one way that I could think to do that
reliably: by deleting the gene that encodes it.
Evolution can, of course, create whole new genes—but it takes a very,
very long time to do it. Indeed, very little evolutionary change actually in
volves the creation of new genes, or even the removal of old ones, precisely
because it's so hard to do: instead, evolution finds new ways to regulate old
genes, or new functions for gene products other than the ones for which
they originally evolved. 2 6 Thus, for example, the lens of the eye is made up
of clear, flexible proteins called crystallins, which would seem to have no
purpose other than to be used to focus light. Yet, there it is in the nervous
system of the sea squirt, where it is part of an organ that keeps track of
"down" by sensing gravity. The gene is of course present in every cell in its
body, and a mutation in a proto-eye cell of one of our ancestors that carried
this gene may have caused it to be expressed there, where previously it
would have been turned off, making the protein available to let the light
shine on in. And our genome contains no genes similar to the ones for
telomerase, ready to mutate to replace it on cancer's demand.
So deleting the telomerase gene, unlike trying to inhibit it somehow,
would be an almost sure-fire way to shut down cancer cells permanently.
(Again: this logic ignores Alternative Lengthening of Telomeres but fear
not—I will get to ALT shortly.) I had hoped back in 2000, and even specu
lated in the paper arising from the first SENS workshop, 2 7 that there might
be some way that we could do this only in cancer cells. I became increas
ingly aware, however, that while it might wed be possible to do this for most
cancer cells, we'd never be able to do it for all, for the same evolutionary
reasons that I keep hammering on: any mechanism that targeted cancer
cells exclusively would have to have some mechanism for selecting a differ
ence between them and normal cells—and of course, those differences
would have a genetic basis, leaving the flap of the "tent" open for a mutant
subpopulation of cancer cells to stick its evolutionary "nose" under.
Finally, fully eighteen months later in that Italian cafe, I stopped trying
to run from this conclusion. The only way to be sure that we were denying
telomerase to cancer cells would be to deny it to all cells. What we needed,

N U C L E A R M U T A T I O N S A N D C A N C E R 2 9 7
I realized, was to take the telomerase gene out of every cell in the body,
along with the ALT mechanism whereby a small minority of cancer cells
manage to lengthen their telomeres without relying on telomerase itself. I
would soon term this therapeutic target the "Whole-body Interdiction of
Lengthening of Telomeres" (WILT).
Removing telomerase from every cell in our body would preempt can
cer before it got a chance to get started. But you can surely see why I took
so long to explore this option—and why no one else had explored it before
me. Deleting telomere elongation capacity throughout the body would also
be life-threatening, because it would mean that our regular, proliferating
cells (like those in the skin or the lining of the gut) would suddenly have
iron limits on their ability to reproduce themselves and thus replenish tis
sue. From the moment that we denuded our cells of telomerase, a clock
would be ticking. With each division the telomere would shorten by a
notch from whatever it had been when we took telomerase out. We would
be under the specter of a rather horrible death, as our stem cells went off
line one by one under replicative senescence (see Chapter 10): with each
failure of a stem cell responsible for supplying key functions, the tissue
would fail to be renewed and would slowly degenerate.
So, the effect of telomerase deletion on frequently dividing cells would
indeed be very serious indeed—fatal, in fact, in what I calculated to be
around a decade from the point when telomerase was deleted.
But hang on, I immediately thought, SENS already has a proposed solu
tion to "normal," age-related cell loss: stem cells. So we might just be able to
deal with cell loss if we had a sufficiently sophisticated program of stem-
cell replenishment—using cells engineered to lack the one linchpin func
tion for cancer, namely telomere elongation.
Of course, these stem cells would eventually peter out, too, as their
telomeres were worn down—but this is just the same situation that we face
with all aging damage. The engineer knows that we don't have to root every
last trace of cellular and molecular injury out of our systems in order to
build a body that will not suffer age-related degeneration and death. The
tissues of a twenty-year-old are already riddled with aging damage, and the
level climbs every day, but you'd be hard-pressed to find much of a health
difference between a basically clean-living person at twenty-five and the
same person at thirty-five, because their level of damage at thirty-five is still
beneath the threshold at which it causes functional deficits. As long as we
keep it there, we will remain biologically young.
So if we introduced stem cells with nice, long telomeres in the first

2 9 8 E N D I N G A G I N G
place, we could let them wind down and eventually be lost to apoptosis,
senescence, or other sources of damage—and just top our tissues up with
more stem cells before enough of those cells were lost to begin to impair tis
sue function. The need for regular treatments in this case would, ultimately,
be no different from the need for regular rounds of AGE-breakers or of
purges of anergic T-cell clones. Neglect your medicine and you will eventu
ally suffer the consequences; keep up with your schedule, and stay young
and healthy into a boundless future.
In this case, the engineer's logic is even stronger, because the same
"damage" that might eventually kill us (in this case, the running down of
our telomeres) is simultaneously the very thing that we need to ensure does
happen, or else we will be killed by another means (the unchecked cell di
vision at the heart of cancer). Putting an expiry date on all of our cells, but
ensuring that they are regularly replenished with new ones, erases both
problems at once.
In fact, the case for deleting telomerase was even stronger than this, be
cause placing an absolute limit on the number of cell divisions that our stem
cells (and the mature cells derived from them) could undergo would actually
bring us an additional anticancer and anti-aging benefit. While we often are
given the impression that most age-related nuclear DNA mutations are the
result of damaging agents like free radicals, radiation, and mutagenic chem
icals, the reality is that most nuclear mutations are the result of errors made
in copying the DNA during cell division. And while it's almost never men
tioned in the popular press, most cancers arise not in the mature cells of our
bodies, but in our stem cells, where regular cell division and an active telom
erase gene makes it relatively easy to take the brakes off cell growth. 2 8 (It
was, in fact, precisely my increasing appreciation of this fact during 2001
that forced me along the line of thinking that led to the WILT concept.) By
cutting down on the number of divisions that our stem cells can undergo be
fore they die, we would simultaneously reduce the number of mutations that
they would ever accumulate—and thus, the risk that they might suffer the
combination of mutations that would turn them cancerous.
At that point, we'd have cancer licked. No cancer could reach a clini
cally significant stage. At worst, we would end up with a few little pebble-
tumors, small bads of abnormal cells that have exhausted their ability to
grow, no more life-threatening than a mole or a small cyst. And our normal
tissues would be preserved intact, provided that we underwent regular
rounds of replacement of stem cells. See Figure 1.

N U C L E A R M U T A T I O N S A N D C A N C E R 2 9 9
Figure 1. The effects of traditional (late-acting) cancer therapies (b), telomerase deletion (c), and telomerase deletion plus stem cell reseeding (d) on the prognosis for cancer (a).
- * P So Crazy, I t Just Might Work
I will not conceal that, when they first hear about it, virtually all my col
leagues think the WILT proposal is utterly mad. Indeed, I myself, while not
doubting my own sanity, initially worried that I must surely be missing
some life-threatening side effect of the two-in-one intervention that was be
ginning to firm itself up in my mind. So I began consulting experts in all of
the relevant fields—telomere biology, mutant humans, mice lacking func
tional telomerase genes, the ALT mechanism, stem cells, bone marrow
transplantation, and of course cancer as a disease above and beyond its
characteristic preservation of its telomeres—to confirm that I had my facts
straight and hadn't neglected any actual showstoppers, and to ask them

300 E N D I N G A G I N G
what they thought of the technical challenges facing the development of
each of the biotechnologies that would be required to implement it.
Their reaction was interesting, and typical of my experience in net
working with experts from different fields on interdisciplinary projects.
Presented with the whole scheme, each of these experts thought the project
as a whole was audacious at best, and something straight out of "soft" sci
ence fiction at worst. But to my moderate surprise, when I asked each of
them to assess the feasibility and timescales for the development of the in
dividual components that WILT would take from their own discipline,
each and every one thought them achievable (albeit ambitious), and felt
that nothing in the subfield of biomedicine in which they worked every day
would pose an insurmountable challenge. It was in the areas that they
didn't have intimate, working familiarity with the science or the biotechnol
ogy involved that they made assumptions of intractability.
Encouraged by these discussions, I held the third SENS workshop
specifically on WILT, inviting many of the experts I'd already been con
sulting about the field. As with the 2000 meeting, my purpose was to put
the participants in a room together and simultaneously put them to work
fleshing out the way forward (or else the proof that none existed) to the
complete, integrated intervention, thus showing them (I hoped) that the
plan was sound in its elements and as a whole.
To my enormous satisfaction, it achieved both ends, and we published
the results together in the Annals of the New York Academy of Sc iences .
And of all the experts that were involved in the roundtable, only one scien
tist objected to being given the credit (and blame) for authorship of the
paper—and her reasons were extremely revealing, as the Acknowledge
ments section of the paper, written with her approval, indicates. Dr. Nicola
Royle, senior lecturer at the University of Leicester's Department of Gene
tics and an expert on telomeres (and especially the ALT mechanism), in
sisted that her name be withdrawn from its author list, not because she
didn't think that WILT would work, but because she very much feared that
it would. Her concern was that, as far as she could see, there was nothing
stopping us from developing WILT as a final cure for cancer, and especially
as part of a complete panel of SENS interventions that would finally free
humanity from age-related degeneration, leading to indefinite youthful,
healthy lifespans. But she was not ready to embrace that future: like so
many others, her principled (but, I of course maintain, misplaced) fear
about the potential drawbacks of unbounded human lifespans on the envi
ronment and on existing social structures was so great that she wanted no

N U C L E A R M U T A T I O N S A N D C A N C E R 3 0 1
further part in promoting our progress toward any part of the SENS
agenda. 3 0
Let's look at some of the technical challenges and concerns that were
discussed at this SENS roundtable—and, of course, in follow-ups with
these scientists and other colleagues at other venues ever since. 3 1
What Happens When We Take Telomerase Away?
This is an obvious one. We're talking about taking away a gene that is at
least present in all of our cells, though it's permanently turned off in tissues
where the cells never have to divide, like the muscles, the heart, and the
brain. At least on the face of it, we wouldn't think that taking the gene (ei
ther or both of two genes in fact, as telomerase has two subunits) right out
of the cell would cause these cells any problems. On the other hand, the en
zyme is expressed and routinely used (under strict controls) by cells that
need to undergo regular division—most notably stem cells. What would be
the effects of taking it away from every last one of them?
Fortunately, we have some pretty reliable evidence on this point,
thanks to two models: mice that have been genetically engineered to lack
one or other of the two subcomponents of the telomerase enzyme, and a
human inherited disease called dyskeratosis congenita (DKC). The picture
here, overall, is a pretty optimistic one.
Unlike humans, mice are born with telomeres long enough to last
them their whole lives without telomerase. (This makes mice a rather
tricky species to use as models for human cancer, in fact.) Therefore, mice
with their telomerase genes deleted have to pass their progressively short
ening telomeres on through several successive generations before much of
anything bad occurs. At that point, they develop the symptoms that you'd
expect from a lack of stem cells, which appear first in the tissues that mul
tiply the most. They become sterile as their sperm-forming cells run out of
steam, and their guts and skin start to become depleted of cells and fragile.
They also, ironically, start developing high levels of cancer, which
might at first seem to fly in the face of the entire program—but this is just
one of the ways in which mice are a fraught model for human cancer. First,
even though these mice telomeres are short enough to mess up stem cell
proliferation, they're still long enough to let cancers grow to a size that's
dangerous for a mouse, simply because mice's small size allows tumors
that would be harmlessly small in a human to impede organ function and

3 0 2 E N D I N G A G I N G
siphon off a fatally high percentage of their tiny bodies' resources. Second,
mouse cells find it relatively easy to activate ALT, so the fact that these
mice lack functional telomerase doesn't guarantee that they can't lengthen
those telomeres anyway. Clever combinations of telomerase deletion with
other mutations can largely sidestep these differences between mice and
humans, though, and when this has been done, 3 2 , 3 3 cancer risk went down
dramatically—in one case, to such low levels that none of the telomerase-
lacking mice had died of the disease at a point when all of the animals
from the same strain but with functional telomerase had been consumed.
DKC patients also give us reason for hope in the midst of their despair.
They have a variety of mutations that prevent the effective functioning of
their telomerase enzymes—either in telomerase itself, or in genes encoding
proteins needed for its normal working. Patients do hold on to some lim
ited telomerase activity, however. The likely reason we never see people
completely lacking in the gene is that humans have to undergo a lot more
cell division in the womb than mice do (and have shorter telomeres than
mice at conception), so fetuses with a more severe telomerase mutation are
probably aborted.
But telomerase activity in DKC patients is certainly very low. As a re
sult, their telomeres are shorter than normal folks', and they develop pre
dictable symptoms similar to those suffered by telomeraseless mice:
mottled or web-patterned skin; patches of abnormal, thickened white cells
in the mucous membranes, similar to those often seen in lifetime smokers;
weak, thin nails with ridges and fissures; hair loss and lung problems; and
bone marrow failure, causing problems with immunity, blood clotting, and
delivery of oxygen and iron to their tissues. 3 4
It used to be thought that the worst symptoms of DKC usually occur in
people in their teens and twenties, but we now know that it isn't quite that
simple—and the reason why that's so turns out to be very important. Dr.
Inderjeet Dokal, who works extensively with DKC patients at the Depart
ment of Haematology at Imperial College in London, told me and the other
participants back in 2002 at the WILT summit that he had noted that first-
generation DKC patients of a particular type, the ones that have a mutation
in one copy of a telomerase gene, don't develop symptoms until they're in
their forties—but that, as they pass on their preshortened telomeres to their
children and grandchildren, those that inherit the disease develop symp
toms earlier and earlier. He and colleagues later confirmed this preliminary
observation in rigorous studies of several families. 3 5 , 3 6
This fact reinforces the principle that it is not the lack of telomerase per

N U C L E A R M U T A T I O N S A N D C A N C E R 3 0 3
se, but the reaching of a threshold length by the telomeres themselves, that
causes symptoms. This is an optimistic finding, because it implies exactly
what we would expect (and hope) would be the case: that if we can period-
ically replenish the bone marrow and other stem cell pools with new stem
cells whose telomeres are wed above the critical length, we should be able
both to cure DKC and also to avoid all the problems of the disease in peo
ple with intentionally extinguished telomerase. Indeed, the best treatment
for DKC today is a bone marrow transplant, introducing new stem cells
taken from people without DKC to replace the ones that are being de
pleted.
What About This Periodic Stem Cell Replenishment?
T h e B o n e M a r r o w
Bone marrow transplants are, of course, already a common and nearly rou
tine procedure, not only for DKC sufferers, but also for patients with a
range of blood disorders, cancer patients who have lost their bone marrow
to radiation therapy, and many others. Still, there are many complications
in recipients today, and many technologies that we will absolutely have to
master if we are to use bone marrow transplants for WILT.
One reason why bone marrow transplants often don't "take" is that
they are not robustly incorporated into their niche in the bone while the
original stem cells are still there. For the first round of WILT bone marrow
replacement, we may have to perform chemotherapy to wipe out the native
cells—but we'd want to do this anyway to minimize the cancer risk of hav
ing those old, telomerase-competent cells left behind. In subsequent rounds,
the process will be easier, because we will intentionally wait to replace the
cells from the first round of transplantation until the cells introduced in the
previous round are beginning to die as their nonrenewing telomeres wear
down.
How often will this stem cell replenishment have to be performed? It's
looking good. People have done clever experiments to measure the average
time between divisions of blood stem cells, and it's at least a couple of
months in humans. Because it takes around fifty divisions before human
cells not expressing telomerase start to feel the shortness of their telomeres,
this rate of division should be slow enough to enable us to function just fine
for about a decade between successive rounds of bone marrow replacement.

3 0 4 E N D I N G A G I N G
T h e S k i n
Because of the pressure to supply skin grafts for burn patients, disfigured
children, and cosmetic surgery, we've made remarkably fast progress in
mastering the art of making new skin from stem cells. In mice, we can now
peel the skin right down to the dermis (the layer of tissue beneath what we
normally think of as "skin," which houses hair follicles, sweat and skin oil-
secreting glands, and blood vessels), and fully reconstitute the old skin that
we've removed. The cells of the dermis do not divide regularly, so we don't
need to worry about their telomeres running out. Remarkably, the epider
mis (the layer on top of the dermis, which is where we do need to replace
stem cells) can be replaced using stem cells derived even from such differ-
ent locations as the cornea of the eye, and the dermis orchestrates their
rapid transformation into hair follicle stem cells that then expand outward,
renewing the tissue.
Again based on how often skin stem cells divide under the status quo,
a round of skin stem cell replacement should last us about ten years. Be
cause the skin is so easy to access, it should be among the easiest and least
invasive of WILT stem cell replacement routines. *
T h e L u n g s
The innermost layer of the lung, like the skin and the gut, is continuously
sloughed off and is thus in continuous need of renewal. Unsurprisingly,
lung complications are a major cause of death in DKC patients. Because
the lung is in important ways similar to the skin, and is relatively easy to ac
cess, there is no reason to think that we won't make quick and relatively
painless progress on this front once we put our mind to it. Indeed, some sci
entists are doing this already, mostly in hopes of treating cystic fibrosis pa
tients. Better yet, the latest estimates are that lung stem cells divide
considerably less often than even those in the skin.
Work to date has gone forward using similar approaches to those used
in skin, though not yet from stem cells. Even so, progress is being made. In
two different models of immunodeficient mice, scientists have purged the
lung of its "skin" (epithelium), "scraping" it down to the underlying base
ment membrane, and then replaced the lost tissue using restructured cells
taken from the innermost layer of the human lung. The next step will be to
do it with stem cells.

N U C L E A R M U T A T I O N S A N D C A N C E R 305
T h e G u t
So far, there are still significant challenges facing the replacement of stem
cells in the gastrointestinal tract in humans. Several years ago, Dr. F.
Charles Campbell, now Professor of Surgery at the Queen's University
Belfast's School of Medicine, made the first major advance. His team ex
tracted stem cells from mouse intestinal tissue, wiped out the cells from
small stretches of the colon, and then repopulated the tissue with stem
cells, which differentiated into all the appropriate cell types and made fully
functional new tissue. Progress has not been rapid since then, however. At
the WILT summit Campbell explained that, in studies he has never re
ported in print, his team tried the same approach in pigs, but the result was
a mass of dysfunctional scar tissue. Since then, however, considerable
progress has been made by another group working in dogs, 3 7 and the same
group has further advanced the technique in rats and mice.
Much work remains to be done here: not least, the opening-up of sec
tions of the gut to remove the existing cells would be far too invasive for
human use in WILT. Endoscopy, similar to what's currently in use to re
move potentially cancerous colon polyps, may provide us with a more tol
erable solution, and should be much more advanced in a few decades,
when we'll actually need it.
A further question is how often we will need to replace stem cells in the
gut. Previous estimates suggested that it would be much more frequently
than the decade or so needed in these other tissues—more like a few times
a year. Luckily, however, there is an easy way to see that those numbers
must be wrong. If gut stem cells divided so much faster than those in other
tissues, DKC sufferers and telomerase-lacking mice would suffer gut fail
ure at a younger age than they suffer failure of the bone marrow or skin—
but they generally don't. Rather, all these tissues fail at about the same age
in any given patient; if anything, the blood is most often the first to go in
humans, which is why bone marrow transplants are temporarily helpful.
But still, the replacement of the stem cells of the gut seems likely to be a
more invasive procedure than that in bone.
T h e R e s t o f U s
The rest of our bodies is, in the relevant respect, like the dermis: composed
of cells that don't divide regularly. Some of these cell types (including the
dermal cells, which are called fibroblasts) are quiescent: they can divide but

3 0 6 E N D I N G A G I N G
they only do so when called upon to do so, such as to close a wound. Oth
ers are postmitotic: totally unable to divide, and instead renewed by incom
ing progenitor cells, if at all.
Postmitotic cell types are obviously no problem in cancer terms, but
quiescent cells require a little more thought. The fact that they are not sig
nificantly affected in DKC or telomerase-knockout mice, and the relative
rarity of cancers derived from them, gives us a bit more time to work on
the job—and, once we have mastered it, probably a lot more time between
each round of stem cell reseeding (as opposed to the more targeted re
placement of "normal" and pathological cell loss to aging damage, which
we will be tackling earlier on using ESCs, as outlined in Chapter 11).
But, precisely because they don't divide, these cells aren't going any
where for a while either—and they do become cancerous occasionally.
Thus, we do want to protect ourselves from cancer in them as much as pos
sible. Removing senescent cells will reduce the already-low risk in these tis
sues considerably, but we will still want to cut it down much further.
The likely solution to this problem is targeted gene deletion. This is
again a major challenge, because even in mice (where gene therapy is now
routine) it's very difficult to target genes for insertion or deletion in cells
that are already inside the organism (as opposed to putting genes into
sperm or egg, or embryos, or into cells that have been taken out of the ani
mal, modified, and replaced). We are, however, getting better at this.
Hopes are high for using this technique for gene therapy generally, and it
could potentially be used to remove telomere-renewing potential from tis
sues that aren't maintained by stem cells.
Over time, however, between replacing age-related cell loss using stem
cells engineered to lack telomerase, and eventually replacing the original
stem cells for these tissues with new ones engineered the same way, we will
slowly "WILT-ify" these tissues a few cells at a time, progressively lowering
the cancer risk they may pose.
Does WILT Underestimate Cancer 's Evolutionary
Ingenuity?
WILT is a highly complex, multifaceted proposal—but it rests, as I've ex
plained, on one absolutely essential assumption. WILT will fail if cancers
can figure out a way to grow indefinitely without the genes that we delete.
Let's look at that eventuality in detail.

N U C L E A R M U T A T I O N S A N D C A N C E R 307
One formal possibility is that cells could develop a way to replicate their
chromosomes without telomere shortening, and thereby avoid the need for
any enzyme to reverse that shortening. It's been hypothesized that some stem
cells effectively do just this. When stem cells divide, they typically produce
one daughter that's still a stem cell and one "amplifying" cell that is set on the
path to differentiation into its required function. (Indeed, the ability to do
this is an essential feature of most people's definition of a stem cell.) It's pos
sible that, by a clever system of controlling which DNA strands stay in the
daughter stem cell and which ones go into the amplifying cell, stem cells
could stop their DNA from shortening. But we don't need to worry about
this possibility in respect of cancer, because it can only confer linear growth
rates, not the exponential growth that is seen in cancers. With linear growth,
a cancer would take thousands of years to grow large enough to kid us.
Another way to avoid telomere shortening is the way that bacteria, and
indeed our own mitochondria, do it: not to have any telomeres! Bacterial
and mitochondrial DNA are circles, so there's no end-replication problem.
But it seems that there's no meaningful risk of this happening in humans.
When telomeres get really short, the cell's DNA-repair machinery mistakes
the raw ends of the exposed chromosomes for the result of a chromosome
break. That causes the cell to make a misguided attempt at repair, joining
the chromosomes together, end to end—which is rapidly fatal for a dividing
cell, because the cell division machinery snaps the double chromosome
apart again, not at the original join but at some random place, scrambling
the genes into a dysfunctional mess.
The final possibility is altogether more real, unfortunately. I've men
tioned it a few times in passing during this chapter: it is that cancers can oc
casionally relengthen their telomeres using enzymes other than telomerase.
These enzymes have not yet been identified, so the system is given the un-
informative name ALT, or Alternative Lengthening of Telomeres. The good
news here is that ALT is quite rare—in some tissues it's almost never seen,
and in those few tissues where it does appear it occurs in no more than
about half of all tumors. This means that at least as much mutation is
needed to activate it as to activate telomerase—and that teds us that ALT is
almost certainly dependent on the turning-on of a gene that's usually very
firmly off, rather than only the loss of activity of genes that are normally on.
This is great news for WILT, because, if ALT is indeed dependent on the
activation of some gene or other, rather than just on the inactivation of
genes, we will be able to do the same for that gene (once we identify it) as
WILT proposes to do with telomerase. There may be side effects, just as

308 E N D I N G A G I N G
there are with deleting telomerase, but they should be addressable with
stem cell or other regenerative therapies, just as telomerase deletion seems
likely to be.
A S IDE E F F E C T : WILTING F E R T I L I T Y ?
One potential side effect of the loss of telomerase from all of
our cells might be eventual sterility for men. If having children is still
a priority in a post-rejuvenation world, then men may choose to
freeze their sperm in advance, as is presently done for sperm
donors, for IVF. The sole sexual issue will be the actual making of
babies, of course: nothing else will wilt from WILT.

Part Three


13
This book has presented, in as simple terms as possible, the biolog
ical details of what human aging is and how we can realistically set about de
feating it. However, from what I've presented so far, you would be entirely
justified in concluding that I've only made a case for the eventual develop
ment of therapies that could modestly delay aging. You might think that a re
alistic time frame for getting encouraging results in mice might indeed be a
decade or so, but you're probably already thinking about the problems that
there would be in negotiating FDA obstacles and such like in translating
these therapies to humans. And you may be concluding that the sort of time
frame I've been predicting for the arrival of widely-available therapies—a
few decades with 50 percent probability—is too short by a factor of at least
three. You're probably also pretty skeptical about the degree of life exten
sion that the techniques described in the last seven chapters could practi
cally achieve, even in that extended timeframe: sure, they might be truly
comprehensive if they worked absolutely perfectly, eliminating every scrap
of their respective type of target damage, but we all know that no therapy is
that perfect, and certainly not in its early versions. Thus, you're probably
thinking that I have fallen drastically short of making my well-publicized
case that a lot of people alive today may wed live to be one thousand—and
G e t t i n g f r o m H e r e t o T h e r e
T h e War o n A g i n g

312 E N D I N G A G I N G
you'd be absolutely right. Thus, in this chapter and the next I'm going to put
the scientific details to one side and directly address these two important
and legitimate concerns. I'm going to start with the time frame for wide
spread availability of the panel of therapies described in this book.
I use the phrase "the War on Aging" to describe a specific phase in the
process leading to the defeat of aging. I define it as the period beginning
with the destruction of the pro-aging trance and ending with the wide
spread availability of therapies that can add a few decades to the life span
of people who are already middle-aged. First I'll elaborate on this defini
tion, then I'll explain why I call it the war on aging, and finally I'll explain
why it has a good chance of lasting only fifteen to twenty years.
The pro-aging trance—the "rational irrationality" about aging that I
described and critiqued in Chapter 2—will end only when its claim to ra
tionality becomes unable to withstand even simple assaults, the sort that
most people can understand. I believe that this will truly occur only when
scientists obtain results in the laboratory—mainly with mice, I expect—
that are so impressive that the majority of professional biogerontologists
are finally prepared to say publicly that it's only a matter of time before we
can postpone aging by at least a few decades in humans. Science is in a very
real sense the new religion: what individual scientists say can be doubted,
but the public scientific consensus is gospel. The result that I think is
needed is something that I've called "robust mouse rejuvenation" or RMR.
RMR is a mouse life-extension result, and it's a rather precisely defined
one. That's what's needed if we want scientists to set aside their paranoia
about making predictions about what might happen in the future—we need
to close all the major loopholes. By my definition, RMR will be achieved
when:
at least twenty mice of the species Mus musculus,
from a strain whose natural average life span is at least three years,
receive treatments starting only when they are at least two years old,
and l i v e to an average of five years of age, with all the extra time be
ing healthy.
I thought about this definition pretty carefully before I publicized it,
and it seems to be standing the test of time: no one has pointed out any way
in which it could be achieved by "uninteresting" means, i.e., by means that
would not convince knowledgeable scientists that a massive breakthrough
had been made that was likely to be relevant to humans. The requirement

G E T T I N G F R O M H E R E T O - T H E R E 313
to use at least twenty mice is so that we can be sure the age wasn't a fluke or
a bookkeeping error. The requirement to use Mus musculus is because
other mouse species already live longer than Mus musculus but are less wed
characterised by scientists. The requirement to use a strain of that species
that naturally lives to three, which is unusually long for that species, is to
avoid any possibility that the mice have some specific congenital problem,
something that normally kills them rather young, and that the treatment
merely alleviates that defect rather than comprehensively affecting aging.
And of course the requirement that the treatment begin only when the mice
are already two-thirds of the way through their natural life span is to ensure
that it has potential relevance to people who are already alive, read the
newspapers, pay taxes—and vote.
My reason for calling the period that begins with the achievement of this
milestone the War on Aging arises from the initial, essentially immediate re
action that I expect society to have to it. In order to describe this reaction, I
must first describe a side effect of the pro-aging trance that determines soci
ety's current reluctance to take aging seriously. I have a name for this, too: it's
the triangular logjam. See Figure 1.
Experimental biology, like any other area of science, costs money—ready
quite a lot of money. Most biology isn't nearly as expensive as high-energy
physics or astronomy, but it's expensive enough that professors have to spend
Figure 1. The triangular logjam impeding funding, and how philanthropy can unlock it.

314 E N D I N G A G I N G
a hideous amount of their time fundraising. The overwhelming majority of
the funds that support experimental biology come from the public purse.
Biogerontology is typical in the above regard, but it's extremely un
usual in one way: the public are absolutely fascinated by it, so biogerontol
ogists get on the television all the time. I mean really, all the time. The
difference in this regard between biogerontology and other biological
fields—even really high-profile medical fields—cannot be overstated: even
quite junior biogerontologists get more press attention than the world's
leaders in other areas. And of course, when given that chance, biogerontol
ogists are just as keen as any other scientist would be to talk about their
own research—which, necessarily, is the research that they were able to ob
tain the funding to perform.
Consider, for a moment, what else a biogerontologist might choose to
talk about to the media. In particular, consider the possibility of talking about
research avenues that the public consider distinctly suspect: defeating aging,
for example. What are the attractions of discussing such topics? Wed, your
name might get quite widely known to the general public, and you might get
more media exposure. But hang on: What is the media exposure for? Scien
tists are intensely preoccupied, as I just mentioned, with the miserable busi
ness of maintaining a funding stream for their laboratories. How, exactly,
would a high media profile achieve this—or, conversely, make it harder?
In order to explain the answer to that question, I must make sure you
are aware of a key feature of the way in which public funding for science is
allocated. When scientists want to do a particular series of experiments,
they write a detailed description of what they want to do, how long they
think it'll take and how much it'll cost, and they send it to the appropriate
government agency. But the government agency doesn't then decide on its
own whether the scientist can have the money. No: even though such agen
cies employ highly experienced ex-scientists as administrators of grant
funds, those ex-scientists don't have anywhere near broad enough expert
ise to be able to tell the difference between a good idea and a poor one
across the whole range of scientific disciplines that they're responsible for.
So instead, they seek specialist advice from other scientists. This is called
"peer review" and it's an absolutely universal component of the process of
evaluating applications for government grants to do science.
Selection to evaluate your colleagues' ideas for experiments is an im
mense privilege and responsibility. It's not something that junior scientists
get to do very often; generally the most senior scientists are the ones who
do it most.

G E T T I N G F R O M H E R E T O T H E R E 315
Do you see the problem yet?
Science is about the testing and refinement of hypotheses and theories.
In principle, the most important quality of a scientist should be their ability
to accept, with an open mind, evidence that challenges theories that they
had believed for many years. But scientists are human, and moreover they
know that the scientists that produced the new evidence are also human. In
particular, they know that when a result is reported that contradicts estab
lished conventional thinking, the new evidence is often found later on to
have been the result of experimental error. Thus, it is generally pretty hard
to get senior scientists to change their mind about things, even if your evi
dence is really strong. The legendary physicist Max Planck famously re
marked in the 1920s that "Science advances funeral by funeral" and this is
only barely an exaggeration: It can take wed over a decade for really funda
mental changes of understanding of aspects of science to become generally
accepted. A famous example in biology is the mechanism of action of mito
chondria, cellular components that we've heard a lot about in this book. 1
And inevitably, this resistance to new ideas carries over heavily into how se
nior scientists evaluate grant applications.
So far, so unproblematic. After all, a modest degree of resistance to
new ideas that you may not yet fully grasp is a good thing in some ways—
we wouldn't want the scientific consensus to flit from one new idea to an
other too easily either, because (as I just mentioned) new ideas are often
wrong. But the inertia that exists in scientific thinking is generally greater
than this happy medium. And unfortunately, it's not just inertia of ideas, it's
inertia of reputations. Senior scientists who have been appointed by the
government to evaluate their colleagues' grant applications are, essentially
by definition, members of the establishment. If they receive two applica
tions of equal scientific merit, only one of which they have the resources to
fund, and one is from a scientist who has a history of radical views about
what science may shortly achieve, while the other is from a scientist who
has never said anything outrageous in his life, you can bet that it's the latter
who will get the money.
And that's not all. Reviewers of grant applications are, of course, given
guidelines telling them what aspects of the applications' scientific merit or
demerit they should consider particularly important. One aspect that is in
variably high on this list, if not at the very top, is feasibility: perceived likeli-
hood that the investigator will complete the proposed experimental program
within the time and budget requested and obtain results that will merit pub
lication in a reputable scientific journal. Sounds pretty uncontroversial,

316 E N D I N G A G I N G
doesn't it?—but in fact this policy is a huge problem for science, because it is
not (in practice) weighted by scientific significance. That's to say: A proposal
for a study that will almost certainly tell us something whatever its outcome
will fare much better in peer review than a study that may wed ted us noth
ing, even if what the second study might ted us is far more important than
what any outcome of the first study would ted us. This bias in favor of low-
risk, low-gain research at the expense of high-risk, high-gain research per
vades the whole of science and is extremely strong in biogerontology.
Well, I've spent quite a while in this chapter denouncing the stubborn
closed-mindedness of senior scientists, but I hope you've taken on board
that in the past couple of paragraphs I've explained that it's not entirely their
fault: it's really the fault of their paymasters, the public funding agencies,
a.k.a. the government. Grant reviewers are also grant recipients, by and
large (though they don't review their own applications, of course). Thus, if
the funding agency makes it known—either explicitly via written guidelines,
or implicitly by their actions and off-the-record remarks—that they would
prefer to fund middle-of-the road people to do dependable work than to
fund troublemakers to do controversial work, the reviewers are hardly likely
to dissent. It would be a very good thing if more such scientists did resist
this sort of instruction, but realistically that's too much to ask.
But actually . . . it's not the government's fault either. The real culprit is
you, the public.
This should not be a surprise to you. It's hardly a secret that govern
ments in all democracies ultimately act to achieve one thing above all oth
ers: their own re-election. That will not be aided if the government spends,
and is seen to spend, appreciable amounts of taxpayers' money on what the
public regards as blue-sky pipe dreams: research that will probably lead
nowhere. If the public were scientifically mature enough to appreciate that,
in the long run, the rate of scientific progress is slowed by this overcautious
approach, their elected representatives would be able to exercise similar vi
sion and to instruct grant reviewers accordingly. But the public do not ade
quately understand how science works, so this doesn't happen. (A similar
and even worse problem exists when it comes to medicine; I'll explore that
topic later in this chapter.) This is basically because the judgment of how
likely a hypothesis is to be true, or of how likely an experimental result is to
lead to further knowledge, is actually one of the most sophisticated and un-
teachable aspects of doing science.
In biogerontology, however, there is potentially a way out—and this
brings me back full circle (or triangle!) to the thing that distinguishes

G E T T I N G F R O M H E R E T O T H E R E 317
biogerontology from all other scientific fields in terms of its interaction
with the public: the sheer extent of that interaction. Though there is no
hope of turning the public into scientists sophisticated enough to under
stand the merits of highly ambitious experiments, there is ample chance
simply to ted them that such-and-such an experimental approach is well
worth pursuing. There's probably not enough chance of this for most sci
entific fields, but in the case of biogerontology it's abundantly possible. So,
why don't biogerontologists do just that whenever they find themselves in
front of a camera? I already told you: They don't want to gain a reputation
for irresponsibility among their peers, because to do so would be to jeopar
dise their own chances of being funded even for unambitious work.
So there you have it—the triangular logjam. Biogerontologists are cau
tious in what they say to the public, in order to protect their funding, which
is provided by the government, which are cautious in what and whom they
fund, in order to protect their votes, which are provided by the public, who
are fatalistic about what's even worth trying to achieve, because they see the
biogerontologists saying only cautious things on the TV.
In order to defeat aging any time soon, I believe that an essential first
step must be to break the triangular logjam. How can this be done?
Since I entered biogerontology, I've been chipping away primarily at
one corner of the triangle: my fellow biogerontologists, especially the se
nior ones. Scientists are very politically aware, as described above, but
they're also honest and sincere people. Moreover—and this is a key point—
hardly any biogerontologists suffer from the pro-aging trance themselves.
They know full well how horrific aging is, and with very few exceptions
they want an end to it just as much as I do. Thirdly, there aren't very many
of them, so personal contact is easy: I've known essentially everyone in the
field personally for several years now. And finally, they're all smart enough
to have earned doctorates. All in all, if I have a good strong case that we
may be much closer to fixing aging than people have hitherto realized,
shouldn't I be able to convince them—and even convince them to say so
publicly?
Wed, not quite—but nearly. As in any walk of life, what people say is
important but what they don't say is also important. Those of my col
leagues to whom I have presented the SENS panel in detail have mostly
concluded that it is not fantasy, even though it's certainly very ambitious—
but that hasn't translated into explicit public calls for SENS to be funded.
What it has led to, however, is a variety of demonstrations of tacit support.
It started with coauthorship by five senior colleagues of the first paper on

318 E N D I N G A G I N G
SENS, which arose from a workshop in 2000 (see Chapter 4); it's continued
with refusal of several eminent colleagues to coauthor a denunciation of
SENS published in the respected journal EMBO Reports and orchestrated
by some of the less open-minded members of the community;2 and most re
cently it's included the remarkable development that some people who did
sign that denunciation have taken the initiative to divorce themselves from
it by publishing constructive responses to the SENS agenda, 3 , 4 something
that the EMBO Reports tirade specifically counselled against. While this
may seem pretty tame as seen from the outside, I can assure you that it's an
about-face as thorough as one ever sees in science.
It's obviously not enough, though. But it's all I'm likely to get from my
senior colleagues in biogerontology for the time being, i.e., until I can piece
together enough funding to give SENS research serious momentum despite
its radical nature. Thus, in the (hopefully brief!) meantime I must address
the other points of the triangle, too.
It's just conceivable that the government could be influenced directly.
There are visionaries in government, and just occasionally they find a way
to realize those visions. But in order to have any real chance on Capitol Hill
or its counterparts in other countries, you really have to know the minds of
the major players well—and that is something you don't pick up overnight.
Thus, I've continued to leave that strategy to others—and I'm delighted to
say that the ball seems to be slowly being picked up, most notably with the
splendid "Longevity Dividend" initiative, a new effort spearheaded by the
veteran lobbyist Dan Perry of the Alliance for Aging Research in collabora-
tion with three gerontologists.5 Whether they have much chance of success
remains to be seen, but I emphatically wish them luck.
As I've become more prominent, however, I have been able to start ad
dressing the third corner of the logjam: the public. You may recall that I
started this book with a somewhat cantankerous complaint to the effect
that if it weren't for the pro-aging trance I would be able to get on with the
actual science and technology of defeating aging. Well, that's certainly
true—but once given the chance, I have thrown as much energy into my
advocacy and outreach efforts as I was already throwing into the science.
Apart from anything else, I'm aware that the public are a source of funds in
their own right, as well as a source of pressure on governments to alter their
funding priorities.
The pro-aging trance dominates the nature of my interaction with the
media, and via them with the public. The overwhelming majority of my
time in interviews is occupied in discussions of the desirability of defeating

G E T T I N G F R O M H E R E T O T H E R E 319
aging, rather than the feasibility. But the good news, which I encounter
mercifully often, is that it generally takes only a little bit of probing to re
veal that the ultimate basis of my interlocutor's concern is their reluctance
to accept the feasibility. It is this that convinces me so thoroughly that the
achievement of robust mouse rejuvenation will consign the pro-aging
trance to history in the twinkling of an eye.
The Intensity of the War on Aging, and Its Consequent
Likely Duration
In order to give you a sense of what the world is likely to be like after RMR
is achieved, I'm going to review some basic epidemiological and biomedical
facts about three well-known viruses—HIV, CMV, and avian flu—and then
examine an interesting scenario.
HIV has become one of the world's major killers. Belatedly, drugs that
can suppress it and prevent it from progressing to full-blown AIDS are
gradually becoming available in the developing world—still nowhere near
in the quantities needed, but maybe soon even in those quantities. In the
developed world, however, HIV is in a meaningful sense under control. It's
possible to live with HIV for decades without any symptoms whatsoever,
by the regular administration of expensive but (in the West) affordable
drugs. What we still don't have, of course, are two key things:
• a drug to eliminate HIV from the body;
• a vaccine to stop it from infecting people.
CMV, cytomegalovirus, is not one of the world's major killers. Wed, not
obviously. In people with a normal immune system, it is completely sup
pressed and causes no symptoms at all. (My "not obviously" qualification
arises from the fact that this suppression gradually wears down the immune
system during aging, so that eventually people become more susceptible to
more aggressive infections such as pneumonia; in this sense CMV is indi
rectly life-threatening. For more details on this and what we need to do
about it, see Chapter 10.) But it is incredibly widespread: most Westerners
are infected with it.
Avian flu is big news as I write these words (mid-2007), because for the
past few years we have been somberly informed that it could soon mutate
into a form that would cause a pandemic and potentially kid tens of millions

320 E N D I N G A G I N G
or even hundreds of millions of people. Ad that needs to happen is for the
avian flu virus to acquire genetic changes so that it can easily be passed from
humans to other humans, the same as more familiar (and much less deadly)
flu viruses can do. Such mutations are rare, but not astronomically rare; this
could happen any time. Vaccines for avian flu are under development, but
how well they will work depends on what mutations the virus picks up in the
process of becoming human-to-human transmissible, and anyway vaccines
often don't work so well on the elderly, who will be most at risk. Hence the
possible death rates.
I've summarized these viruses as background for a scenario that I now
want to explore in some detail, by way of an analogy with the situation after
RMR has been achieved. I needed to lay out this background so that you
appreciate that the scenario is reasonably realistic; I don't think I'll have
any trouble convincing you that it's a valid analogy.
Let's suppose that HIV mutated to become transmissible by air, just
like flu.
That's it. That's the situation I want to explore. Nothing else changes:
the drugs to suppress HIV still work, they're still pretty expensive, and vac
cines for HIV are still far away from development.
In this scenario, it's essentially certain that almost everyone in the
world would have HIV within a couple of years. Not everyone gets flu in a
pandemic, of course, but the difference is that once you get flu, if you don't
die you mount an immune response that actually works, i.e., that eliminates
the virus from your body. Thus, any given individual is infectious only for a
rather short period (and after they've recovered, they can't be infected
again, either). In the scenario we're looking at, once you get it you have it
forever, and you're infectious forever. There will be no hiding place.
Pretty apocalyptic, isn't it? (Lucidly, virologists think that in actual fact
this scenario is vastly more unlikely than the corresponding mutation for
avian flu.)
Well, hang on—is it so apocalyptic? We do have these drugs . . .
Let's look at a few round numbers. In the United States, roughly one
person in every 250 has HIV, according to the Populations Reference
Bureau—that's about one million people. The drug treatment to keep HIV
under control costs about $30,000 per year, so that adds up to about $30
billion per year. Thus, if everyone in the United States had HIV, we'd be
talking about $30 trillion per year. But the actual cost of production of
these drugs is far, far lower: generic forms of them are being synthesized in

G E T T I N G F R O M H E R E T O T H E R E 321
India and sold (still at a profit, mind) for only $300 per year, and even lower
prices are in the offing. So actually we're looking at only $300 billion per
year—$1 billion per day—to keep everyone in the United States healthy
even if they all have HIV.
Now, a couple of points. First, you might think that "only $300 bil
lion per year" is a pretty curious use of the word "only." Well, think
again, because that's almost exactly what the United States is currently
spending on the war in Iraq. (I'm not commenting here on the relative
merits of these expenditures, you understand—I'm just pointing out that
we have a precedent of an unexpected expense of the same size that is not
bankrupting the nation.) Second, you might be against the infringing of
patents, so you might object to my slashing the cost by a factor of a hun
dred. But is your belief in the patent system stronger than your belief in
stopping your neighbors—or yourself, or your family—from coming
down with AIDS and dying horribly? Ask yourself honestly: If this sce
nario actually happened, and one major party campaigned on a manifesto
to raise taxes by $300 per year for the average person and to spend that
money on generic drugs to prevent AIDS, and the other party cam
paigned on a commitment either to raise taxes by $30,000 per year for the
average person or not to provide the drugs at all, who do you seriously
think would get elected?
I hope I've convinced you what would happen in the above scenario:
we would find the resources to treat everyone. We'd probably find the re
sources to treat everyone in the developing world, too, just as we're now
stirring ourselves to treat everyone who needs such drugs in the developing
world today.
Now, let's look at society's post-RMR view of aging in the same way. I
suspect you can quickly see the similarities. Everyone has aging. The ther
apies we'll be looking to make available will be suppression therapies,
which we will have to take for as long as we live (though much less fre
quently than those with HIV need to take their drugs). Within that limita
tion, however, the therapies will work: people's aging won't progress. But
the therapy will be very expensive. (In the first instance that expense will
be mainly for funding of research, training greatly increased numbers of
medical personnel, building additional drug synthesis facilities and such
like. The same figure I discussed above, $1 billion per day, is as good an es
timate as any.)
So let's ask the opposite question: what are the differences between a

322 E N D I N G A G I N G
post-RMR world and a universal-HIV world? I would say that there are re
ally only two:
• the therapies won't yet exist at the time RMR is achieved;
• our acceptance that human aging can probably be defeated fairly
soon will be new, while the universality of aging is age-old: this is
the reverse of the situation with HIV in the above scenario.
I would argue that neither of these differences has any real chance of
causing society to behave any differently in the aftermath of RMR than in
the universal-HIV scenario. The nonexistence of the therapies is really no
different than the nonexistence of enough antiretroviral drugs, which
would certainly be the initial situation in the scenario I've described: we
will work to develop those therapies as fast as possible, just as we would
work to scale up production of antiretrovirals as fast as possible. The idea
that the order of events could matter seems equally far-fetched; if everyone
has a life-threatening health condition and we have a shot at making it no
longer life-threatening, we'd clearly strive to do so.
Why "the War on Aging"?
I think you can probably see by now my reasons for calling this period "the
War on Aging." In the early 1970s, there was an initiative called "the War
on Cancer" that involved a sharp and sustained hike in the funding for can
cer research fueled by the hope that cancer could be cured within as few as
five years. 6 The war on cancer was not as abject a failure as some people
tend to suggest—without that funding we would not have advanced nearly
as fast as we have in our understanding of cancer, so there's little doubt that
that initiative will have brought forward the true defeat of cancer quite
substantially—but it was a complete misnomer, for one simple reason: the
amount of money involved was really quite small, imperceptible to the U.S.
taxpayer. As summarized above, the war on aging will be extremely
expensive—not imperceptible at all. And yet, it is clear that the public will
embrace the necessary tax rises: it will be quite obviously impossible to get
elected except on a manifesto commitment to attack aging with all avail
able resources. This is a mind-set that has previously been seen only at one
type of stage in a wealthy nation's history: wartime.

G E T T I N G F R O M H E R E T O T H E R E 323
Hippocrates and Gelsinger
In closing this chapter, I want to touch on one final aspect of the war on
aging—one which completes my case that it may well last only 15-20 years.
In 1999, a teenager named Jesse Gelsinger died of anaphylactic shock in
a trial of a gene therapy procedure at a hospital in Philadelphia. 7 This was the
first such incident of its kind, and it sent the gene therapy world into its own
form of anaphylactic shock. The bottom l i n e was that essentially all gene
therapy trials worldwide were suspended for about a year. We don't know
how much delay that will eventually turn out to translate into in terms of the
development of safe and effective gene therapy, but the chances are pretty
good that it'll be a few weeks at least, and given the enormous breadth of ap
plicability of gene therapy, that could mean thousands of lives lost—maybe
even hundreds of thousands if it delays the defeat of aging. Bearing this in
mind, was the suspension of trials for so long a proportionate response?
The U.S. Food and Drug Administration (FDA) would answer in the af
firmative, as would their counterparts around the world. Regulation of exper
imental drugs and therapies, whether it be in terms of what results are
needed or how they are obtained, is based on one abiding principle above all
others: the minimization of risk that the therapy might make the patient
worse. Specifically, this minimization of risk explicitly counts for more, much
more, than maximizing benefit. In this way, the FDA is following a principle
that has existed since medicine's earliest days: the famous edict of Hip
pocrates, primum non nocere, or "first do no harm." (Note that this phrase is
actually not part of the Hippocratic Oath, the set of principles by which med
ical professionals swear to abide as part of their qualification process.)
I take the view, quite simply, that Hippocrates has had his day. The
avoidance of harm was a rational strategy to adopt during the early days of
medicine, when people very often recovered spontaneously from what their
doctors thought were fatal conditions simply because the doctors had inad
equate diagnostic tools. In such a situation, the psychological effect of pos
sibly causing harm, whether it be the effect on the doctor or on the patient's
loved ones, legitimately skews the objective cost-benefit analysis of a given
treatment. In the modern world, however, such recoveries are relatively
very rare. I therefore believe that the 10:1 (at least) ratio of lives lost
through slow approval of safe drugs to lives lost through hasty approval of
unsafe drugs 8 is no longer acceptable.
Furthermore, I believe that in the turbulence of the War on Aging, the
general public will also come to the view that it is unacceptable. This will

324 E N D I N G A G I N G
lead, in a matter of months from the achievement of RMR, to a root-and-
branch revision of the laws and regulations governing clinical trials and ap
proval of drugs and therapies. A fair guess is that drugs will be approved
for universal use (via prescription) after a degree of testing that approxi
mates today's Phase 2. People will die as a result; the 10:1 ratio mentioned
above will probably reduce to 2:1. And people will be happy about this
change, because they'll know that it's wartime, and the first priority—even
justifying considerable loss of life in the short term—is to end the slaughter
as soon as humanly possible. I am the first to acknowledge that, without
such a change of priorities, my prediction that the war on aging may well
last only fifteen years would be totally absurd. But with that change, only
the pace of research will be limiting.

14
B o o t s t r a p p i n g O u r W a y t o
a n A g e l e s s F u t u r e
I have a confession to make. In Chapters 5 through 12, where I
explained the details of SENS, I elided one rather important fact—a fact
that the biologists among my audience will very probably have spotted. I'm
going to address that omission in this chapter, budding on a line of reason
ing that I introduced in an ostensibly quite circumscribed context toward
the end of Chapter 9.
It is this: The therapies that we develop in a decade or so in mice, and
those that may come only a decade or two later for humans, will not be per
fect. Other things being equal, there will be a residual accumulation of
damage within our bodies, however frequently and thoroughly we apply
these therapies, and we will eventually experience age-related decline and
death just as now, only at a greater age. Probably not all that much greater
either—probably only thirty to fifty years older than today.
But other things won't be equal. In this chapter, I'm going to explain
why not—and why, as you may already know from other sources, I expect
many people alive today to live to one thousand years of age and to avoid
age-related health problems even at that age.
I'll start by describing why it's unrealistic to expect these therapies to
be perfect.

326 E N D I N G A G I N G
Evolution Didn' t Leave Notes
I emphasized in Chapter 3 that the body is a machine, and that that's both
why it ages and why in principle it can be maintained. I made a comparison
with vintage cars, which are kept fully functional even one hundred years
after they were built, using the same maintenance technologies that kept
them going fifty years ago when they were already far older than they were
ever designed to be. More complex machines can also be kept going indef
initely, though the expense and expertise involved may mean that this never
happens in practice because replacing the machine is a reasonable alterna
tive. This sounds very much like a reason to suppose that the therapies we
develop to stave off aging for a few decades will indeed be enough to stave
it off indefinitely.
But actually that's overoptimistic. All we can reliably infer from a com
parison with man-made machines is that a truly comprehensive panel of
therapies, which truly repairs everything that goes wrong with us as a result
of aging, is possible in principle—not that it is foreseeable. And in fact, if
we look back at the therapies I've described in this book, we can see that
actually one thing about them is very unlike maintenance of a man-made
machine: these therapies strive to minimally alter metabolism itself, and tar
get only the initially inert side effects of metabolism, whereas machine
maintenance may involve adding extra things to the machinery itself (to
the fuel or to the oil of a car, for example). We can get away with this sort
of invasive maintenance of man-made machines because we (well, some of
us!) know how they work right down to the last detail, so we can be ade
quately sure that our intervention won't have unforeseen side effects.
With the body—even the body of a mouse—we are still profoundly igno
rant of the details, so we have to sidestep our ignorance by interfering as
little as possible.
What that means for efficacy of therapies is that, as we fix more and
more aspects of aging, you can bet that new aspects will be unmasked.
These new things—eighth and subsequent items to add to the "seven
deadly things" listed in this book—will not be fatal at a currently normal
age, because if they were, we'd know about them already. But they'll be fa
tal eventually, unless we work out how to fix them, too.
It's not just "eighth things" we have to worry about, either. Within each
of the seven existing categories, there are some subcategories that will be
easier to fix than others. For example, there are lots of chemically distinct
cross-links responsible for stiffening our arteries; some of them may be

B O O T S T R A P P I N G O U R W A Y 327
broken with alagebrium and related molecules, but others will surely need
more sophisticated agents that have not yet been developed. Another ex
ample: obviating mitochondrial DNA by putting modified copies of it into
the cell's chromosomes requires gene therapy, and thus far we have no gene
therapy delivery system ("vector") that can safely get into all cells, so for the
foreseeable future we'd probably only be able to protect a subset of cells
from mtDNA mutations. Much better vectors will be needed if we are to
reach all cells.
In practice, therefore, therapies that rejuvenate sixty-year-olds by
twenty years will not work so well the second time around. When the ther
apies are applied for the first time, the people receiving them will have sixty
years of "easy" damage (the types that the therapies can remove) and also
sixty years of "difficult" damage. But by the time beneficiaries of these
therapies have returned to biologically sixty (which, let's presume, will hap
pen when they're chronologically about eighty), the damage their bodies
contain will consist of twenty years of "easy" damage and eighty years of
"difficult" damage. Thus, the therapies will only rejuvenate them by a
much smaller amount, say ten years. So they'd have to come back sooner
for the third treatment, but that will benefit them even less . . . and very
soon, just like Achilles catching up with the tortoise in Zeno's paradox, ag
ing will get the better of them. See Figure 1.
Back in Chapters 3 and 4, I explained that, contrary to one's intuition,
rejuvenation may actually be easier than retardation. Now it's time to intro
duce an even more counterintuitive fact: that, even though it will be much
harder to double a middle-aged human's remaining life span than a middle-
aged mouse's, multiplying that remaining life span by much larger factors—
ten or thirty, say—will be much easier in humans than in mice.
The Two-Speed Pace of Technology
I'm now going to switch briefly from science to the history of science, or
more precisely the history of technology.
It was well before recorded history that people began to take an inter
est in the possibility of flying: indeed, this may be a desire almost as ancient
as the desire to live forever. Yet, with the notable but sadly unreproduced
exception of Daedalus and Icarus, no success in this area was achieved
until about a century ago. (If we count balloons then we must double that,
but really only airships—balloons that can control their direction of travel

3 2 8 E N D I N G A G I N G
Figure 1. The diminishing returns delivered by repeated application of a rejuvenation regime.
reasonably well—should be counted, and they only emerged at around the
same time as the airplane.) Throughout the previous few centuries, engi
neers from Leonardo on devised ways to achieve controlled powered flight,
and we must presume that they believed their designs to be only a few de
cades (at most) from realization. But they were wrong.
Ever since the Wright brothers dew at Kitty Hawk, however, things
have been curiously different. Having mastered the basics, aviation engi
neers seem to have progressed to ever greater heights (literally as well as
metaphorically!) at an almost serenely smooth pace. To pick a representa
tive selection of milestones: Lindbergh dew the Atlantic twenty-four years
after the first powered flight occurred, the first commercial jetliner (the
Comet) debuted twenty-two years after that, and the first supersonic air
liner (Concorde) followed after a further twenty years.
This stark contrast between fundamental breakthroughs and incre
mental refinements of those breakthroughs is, I would contend, typical of
the history of technological fields. Further, I would argue that it's not sur
prising: both psychologically and scientifically, the difficulty of bigger ad
vances is harder to estimate.
I mention all this, of course, because of what it tells us about the likely
future progress of life extension therapies. Just as people were wrong for
centuries about how hard it was to fly but eventually cracked it, we've been
wrong since time immemorial about how hard aging is to combat but we'll

B O O T S T R A P P I N G O U R W A Y 329
eventually crack it, too. But just as people have been pretty reliably correct
about how to make better and better aircraft once they had the first one, we
can expect to be pretty reliably correct about how to repair the damage of
aging more and more comprehensively once we can do it a little.
That's not to say it'll be easy, though. It'll take time, just as it took time
to get from the Wright Flyer to Concorde. And that is why, if you want to
live to one thousand, you can count yourself lucky that you're a human and
not a mouse. Let me take you through the scenario, step by step.
Suppose we develop Robust Mouse Rejuvenation in 2016, and we take a
few dozen two-year-old mice and duly treble their one-year remaining life
spans. That will mean that, rather than dying in 2017 as they otherwise
would, they'd die in 2019. Well, maybe not—in particular, not if we can de
velop better therapies by 2018 that re-treble their remaining life span (which
will by now be down to one year again). But remember, they'd be harder to
repair the second time: their overall damage level may be the same as before
they received the first therapies, but a higher proportion of that damage will
be of types that those first therapies can't fix. So we'd only be able to achieve
that re-trebling if the therapies we have available by 2018 are considerably
more powerful than those that we had in 2016. And to be honest, the chance
that we'd improve the relevant therapies that much in only two years is ready
pretty slim. In fact, the likely amount of progress in just two years is so small
that it might as well be considered zero. Thus, our murine heroes will indeed
die in 2019 (or 2020 at best), despite our best efforts.
But now, suppose we develop Robust Human Rejuvenation in 2031,
and we take a few dozen sixty-year-old humans and duly double their
thirty-year remaining life spans. By the time they come back in, say, 2051,
biologically sixty again but chronologically eighty, they'd need better thera
pies, just as the mice did in 2018. But luckily for them, we'll have had not
two but twenty years to improve the therapies. And twenty years is a very
respectable period of time in technology—long enough, in fact, that we will
with very high probability have succeeded in developing sufficient im
provements to the 2031 therapies so that those eighty-year-olds can indeed
be restored from biologically sixty to biologically forty, or even a little
younger, despite their enrichment (relative to 2031) in harder-to-repair
types of damage. So unlike the mice, these humans will have just as many
years (twenty or more) of youth before they need third-generation treat
ments as they did before the second.
And so on . . . see Figure 2.

330 E N D I N G A G I N G
Figure 2. How the diminishing returns depicted in Figure 1 are avoided by repeated application of a rejuvenation regime that is sufficiently more effective each time than the previous time.
Longevity Escape Velocity
The key conclusion of the logic I've set out above is that there is a threshold
rate of biomedical progress that will allow us to stave off aging indefinitely,
and that that rate is implausible for mice but entirely plausible for humans.
If we can make rejuvenation therapies work well enough to give us time to
make them work better, that will give us enough additional time to make
them work better still, which wi l l . . . you get the idea. This will allow us to
escape age-related decline indefinitely, however old we become in purely
chronological terms. I think the term "longevity escape velocity" (LEV)
sums that up pretty well.1
One feature of LEV that's worth pointing out is that we can accumu
late lead time. What I mean is that if we have a period in which we improve
the therapies faster than we need to, that will allow us to have a subsequent
period in which we don't improve them so fast. It's only the average rate of
improvement, starting from the arrival of the first therapies that give us just
twenty or thirty extra years, that needs to stay above the LEV threshold.
In case you're having trouble assimilating all this, let me describe it in
terms of the physical state of the body. Throughout this book, I've been dis
cussing aging as the accumulation of molecular and cellular "damage" of

B O O T S T R A P P I N G O U R W A Y 331
various types, and I've highlighted the fact that a modest quantity of dam
age is no problem—metabolism just works around it, in the same way that
a household only needs to put out the garbage once a week, not every hour.
In those terms, the attainment and maintenance of longevity escape veloc
ity simply means that our best therapies must improve fast enough to out
pace the progressive shift in the composition of our aging damage to more
repair-resistant forms, as the forms that are easier to repair are progres
sively eliminated by our therapies. If we can do this, the total amount of
damage in each category can be kept permanently below the level that ini
tiates functional decline.
Another, perhaps simpler, way of looking at this is to consider the anal
ogy with literal escape velocity, i.e. the overcoming of gravity. Suppose
you're at the top of a cliff and you jump off. Your remaining life expectancy
is short—and it gets shorter as you descend to the rocks below. This is ex
actly the same as with aging: The older you get, the less remaining time you
can expect to live. The situation with the periodic arrival of ever better re
juvenation therapies is then a bit like jumping off a cliff with a jet pack on
your back. Initially the jet pack is turned off, but as you fall, you turn it on
and it gives you a boost, slowing your fall. As you fad farther, you turn up
the power on the jet pack, and eventually you start to pull out of the dive
and even start shooting upward. And the farther up you go, the easier it is
to go even further.
The Political and Social Significance of Discussing LEV
I've had a fairly difficult time convincing my colleagues in biogerontology
of the feasibility of the various SENS components, but in general I've been
successful once I've been given enough time to go through the details.
When it comes to LEV, on the other hand, the reception to my proposals
can best be described as blank incomprehension. This is not too surprising,
in hindsight, because the LEV concept is even further distant from the sort
of scientific thinking that my colleagues normally do than my other ideas
are: it's not only an area of science that's distant from mainstream gerontol
ogy, it's not even science at all in the strict sense, but rather the history of
technology. But I regard that as no excuse. The fact is, the history of tech
nology is evidence, just like any other evidence, and scientists have no right
to ignore it.
Another big reason for my colleagues' resistance to the LEV concept is,

332 E N D I N G A G I N G
of course, that if I'm seen to be right that achievement of LEV is foresee
able, they can no longer go around saying that they're working on postpon
ing aging by a decade or two but no more. As I outlined in Chapter 13,
there is an intense fear within the senior gerontology community of being
seen as having anything to do with radical life extension, with all the un
certainties that it will surely herald. They want no part of such talk.
You might think that my reaction to this would be to focus on the short
term: to avoid antagonising my colleagues with the LEV concept and its
implications of four-digit life spans, in favor of increased emphasis on the
fine details of getting the SENS strands to work in a first-generation form.
But this is not an option for me, for one very simple and incontrovertible
reason: I'm in this business to save lives. In order to maximize the number
of lives saved—healthy years added to people's lives, if you'd prefer a more
precise measure—I need to address the whole picture. And that means en
suring that you, dear reader—the general public—appreciate the impor
tance of this work enough to motivate its funding.
Now, your first thought may be: Hang on, if indefinite life extension is
so unpalatable, wouldn't funding be attracted more easily by keeping quiet
about it? Well, no—and for a pretty good reason.
The world's richest man, Bid Gates, set up a foundation a few years ago
whose primary mission is to address health issues in the developing world. 2
This is a massively valuable humanitarian effort, which I wholeheartedly
support, even though it doesn't directly help SENS at all. I'm not the only
person who supports it, either: In 2006 the world's second richest man,
Warren Buffett, committed a large proportion of his fortune to be donated
in annual increments to the Gates Foundation.3
The eagerness of extremely wealthy individuals to contribute to world
health is, in more general terms, an enormous boost for SENS. This is
mainly because a rising tide raises all boats: once it has become acceptable
(even meritorious) among that community to be seen as a large-scale health
philanthropist, those with "only" a billion or two to their name will be
keener to join the trend than if it is seen as a crazy way to spend your hard-
earned money.
But there's a catch. That logic only works if the moral status of SENS is
seen to compare with that of the efforts that are now being funded so well.
And that's where LEV makes all the difference.
SENS therapies will be expensive to develop and expensive to admin
ister, at least at first. Let's consider how the prospect of spending all that
money might be received if the ultimate benefit would be only to add a cou-

B O O T S T R A P P I N G O U R W A Y 333
ple of decades to the lives of people who are already living longer than most
in the developing world, after which those people would suffer the same
duration of functional decline that they do now.
It's not exactly the world's most morally imperative action, is it?
Indeed, I would go so far as to say that, if I were in control of a few bil
lion dollars, I would be quite hesitant to spend it on such a marginal im
provement in the overall quality and quantity of life of those who are
already doing better in that respect than most, when the alternative exists
of making a similar or greater improvement to the quality and quantity of
life of the world's less fortunate inhabitants.
The LEV concept doesn't make much difference in the short term to
who would benefit from these therapies, of course: it will necessarily be
those who currently die of aging, so in the first instance it will predomi
nantly be those in wealthy nations. But there is a very widespread appreci
ation in the industrialised world—an appreciation that, I feel, extends to
the wealthy sectors of society—that progress in the long term relies on aim
ing high, and in particular that the moral imperative to help those at the
rear of the field to catch up is balanced by the moral imperative to maxi
mize the average rate of progress across the whole population, which ini
tially means helping those who are already ahead. The fact that SENS is
likely to lead to LEV means that developing SENS gives a huge boost to the
quality and quantity of life of whomever receives it: so huge, in fact, that
there is no problem justifying it in comparison to the alternative uses to
which a similar sum of money might be put. The fact that life span is ex
tended indefinitely rather than by only a couple of decades is only part of
the difference that LEV makes, of course: arguably an even more important
difference in terms of the benefit that SENS gives is that the whole of that
life will be youthful, right up until a beneficiary mistimes the speed of an
oncoming truck. The average quality of life, therefore, will rise much more
than if all that was in prospect were a shift from, say, 7:1 to 9:1 in the ratio
of healthy life to frail life.
Quantifying Longevity Escape Velocity More Precisely
This chapter has, I hope, closed down the remaining escape routes that
might still have remained for those still seeking ways to defend a rejection of
the SENS agenda. I have shown that SENS can be functionally equivalent to
a way to eliminate aging completely, even though in actual therapeutic terms

334 E N D I N G A G I N G
it will only be able to postpone aging by a finite amount at any given mo
ment in time. I've also shown that this makes it morally just as desirable—
imperative, even—as the many efforts into which a large amount of private
philanthropic funding is already being injected.
I'm not complacent though: I know that people are quite ingenious
when it comes to finding ways to avoid combating aging. Thus, in order to
keep a few steps ahead, I have recently embarked on a collaboration with a
stupendous programmer and futurist named Chris Phoenix, in which we are
determining the precise degree of healthy life extension that one can expect
from a given rate of progress in improving the SENS therapies. This is lead
ing to a series of publications highlighting a variety of scenarios, but the short
answer is that no wool has been pulled over your eyes above: the rate of
progress we need to achieve starts out at roughly a doubling of the efficacy of
the SENS therapies every forty years and actually declines thereafter. By
"doubling of efficacy" I mean a halving of the amount of damage that still
cannot be repaired.
So there you have it. We will almost certainly take centuries to reach
the level of control over aging that we have over the aging of vintage cars—
totally comprehensive, indefinite maintenance of full function—but be
cause longevity escape velocity is not very fast, we will probably achieve
something functionally equivalent within only a few decades from now, at
the point where we have therapies giving middle-aged people thirty extra
years of youthful life.
I think we can cad that the fountain of youth, don't you?

15
War B o n d s fo r t h e
C a m p a i g n A g a i n s t A g i n g
It is, as you now know, my bold but solidly grounded contention
that there is a strong chance that you—the reader of this book—will live to
experience the rejuvenation of your body into a flesh that is years or even
decades younger, biologically, than your chronological age, leading ulti
mately to an endless summer of literally perpetual youth. But that is a pre
diction about what could be—not about what must be. Make no mistake:
Once the War on Aging begins, it must end in victory, and the future of in
definite health will be ours. But whether that process begins in time to save
our parents, or only ourselves, or only our children, or even their children
depends entirely on when the first bomb of that war—the achievement of
robust mouse rejuvenation (RMR)—is finally dropped. So with that under
standing, the key question for each of us is, What am I going to do about it?
Granted that the War on Aging could begin in as little as a decade, it
makes sense to take care of yourself and your family, so that you don't miss
out on the arrival of robust human rejuvenation by just a few years. There is
plenty of well-established science on this front: eat more fruits and vegetables,
get your essential fatty acids, exercise, and maintain a healthy weight. But a
much more effective way to increase your personal odds of seeing your body
rejuvenated—and one that has the decided advantage that it also increases

336 E N D I N G A G I N G
the odds of survival for those close to you, and for untold thousands that you
have never even met—is to hasten the day when the battle is well and truly
joined. Fortunately, there are some very powerful things that you can do, to
day, to help ensure the saving of lives, again of tens of thousands of lives a day,
possibly including your own or your most dearly beloved. The most immedi
ately obvious actions would be to lobby for more funding for rejuvenation re
search, and for the crucial lifting of restrictions on federal funding to
embryonic stem cell research in the United States, by writing letters to your
political representatives, demanding change.
But an even more powerful thing you can do is to donate to the
Methuselah Foundation.
Let's step back a moment to remind ourselves of where we are today,
and see how we can affect our future.
Remember the logjam that I outlined in Chapter 13? The reason why
the investments necessary to bring forward robust mouse rejuvenation—
the first and critical benchmark for the instigation of a total life-and-death
struggle against biological aging—are so hard to obtain lies in a mutually
reinforcing ring of politically directed funding restrictions, scientific over-
caution in public statements and grant requests, and public opinion. The
fastest way out of this vicious circle is to create an independent source of
funding, pouring the needed billion or so dollars directly into work de
signed to achieve RMR. Unfortunately, it would be very difficult to raise the
funds required to do this in a short time frame, precisely because of the
widespread pessimism engendered in the public by scientists afraid for
their careers in a world where the logjam exists.
There are two plausible ways to change that, and the Methuselah Foun
dation is the worldwide spearhead for both of them.
The first is to support SENS research directly. You can do that by do
nating to the Methuselah Foundation, because we are a standard science
funding organization. Just like the National Institutes of Health or the Na
tional Science Foundation, our scientific team, headed by me as Chief Sci
ence Officer, evaluate scientific publications and research findings and
provide funds to professors around the world. The difference between us
and other agencies, of course, is that we are focused on a particular goal
and we are not afraid to fund projects that may take a long time to succeed.
Or . . . that may not succeed at all. And that's why, by way of hedging
our bets, we have the second strategy.
I believe that SENS is far and away the most promising way to achieve
RMR soon and corresponding human therapies thereafter—but, like any

W A R B O N D S F O R T H E C A M P A I G N 337
scientist, I could be wrong. Really hard technological goals, whether in
medicine or elsewhere, vary a lot in terms of how confident the experts in
the relevant field are that their preferred approach will work. At one ex
treme, in some cases there is almost no doubt about how to proceed—all
that holds the project back is availability of resources. The Apollo project
was a fine example of this: once national pride freed up the necessary cash,
the project went from start to finish faster than Boston's Big Dig. But at the
other extreme—powered flight before 1900, for example, or essentially all
medicine before 1800—people have ideas about what might work that are
still extremely speculative, and attempt after attempt fails or never even
gets tested. Testing everything that might work just costs too much for any
organization, even a highly motivated government, to afford when there are
so many comparably plausible possibilities.
Luckily, there is one time-tested strategy that has been successfully
used, again and again, to solve engineering challenges of this sort without
having to raise even a small fraction of the full sum required to complete
the project. That strategy is the research prize.
Research prizes with a specific benchmark have a long and illustrious
track record of producing the development of effective prototype tech
nologies with only a small investment of funds. Charles Lindbergh's famous
transatlantic flight in 1927; John Harrison's invention of a way to fix the
longitude location of a vessel at sea (crucial to successful navigation away
from a coastline); and the dramatic photo-finish race for the first privately
funded suborbital flight with human cargo that was driven by the Ansari
X-Prize, are all examples of the power of such prizes to spur daring tech
nological innovation.
What is so powerful about research prizes is that they only reward suc
cess. Not a single dollar of the prize goes out until someone achieves the
goal laid out by its creators. As a result, the existence of a single prize moti
vates many teams of independent scientists and engineers to go after it—
each of them using a different approach, and each of them investing their
own dollars independently. As a result, the money that is mistakenly in
vested in approaches that are ultimately shown to be unsuccessful does not
deplete the prize jackpot by a single penny. In the end, about ten to twenty
dollars is ultimately laid out by the contestants for every dollar raised to put
in the jackpot of a research prize—even though each contestant usually
gambles less private money than is sitting in the pot—and the goal of the
prize is achieved at a cost to the prize's principals that is far less than would
be required for a monolithic "Apollo Project" approach.

338 E N D I N G A G I N G
You can see where this is going.
The Methuselah Mouse Prize (or Mprize, with a tip of the hat to the re
cent success of the X Prize) is a project that was dreamed up independently
around the turn of the century by a few biogerontologists (starting with
Gregory Stock's concept of the Prometheus Prize) and by long-time hu
manitarian visionary David Gobel. When David and I discovered each
other, our complementary talents allowed us very rapidly to bring the prize
to fruition. The purpose of the project is to break the logjam that we've
been discussing using a research prize structured similarly to the X Prize
model. Enjoying the support of X Prize Chair and CEO Peter Diamandis as
a chief advisor, the Mprize's chief project is the Rejuvenation Prize for the
greatest extension of life span in mice that are already elderly—in other
words, for progress toward fully-fledged robust mouse rejuvenation.
The Mprize has the potential to remove the stumbling blocks that cur
rently restrict scientists in government and industry from taking on the ag
ing process as a curable disease. For the scientists in academia, it creates an
incentive to write the right grant proposals, in hopes of obtaining more
funding directly and greater prestige for their institutions by winning the
prize—prestige that itself tends to attract more funding from outside
sources.
But not only that: it's a popular win, too. The prize concept, by its na
ture, captures public imagination and provides a dramatic way to inform
the public and the media that scientists are working on extending healthy
life spans in mammals. This increases the credibility of any similar rep
utable efforts, and wins acceptance for the idea that it can be done in hu
mans. In turn, changes in public opinion ease political constraints on
awarding public funding for such projects, and may even generate active
pressure for such awards to be made before RMR has occurred.
And the Mprize also reorients the incentives for industry. Right now,
there is no specific motivation for private researchers to perform life span
studies in mice: at most, they are a stepping stone toward long, expensive,
human trials. When a significant financial reward—and the promise of sub
stantial publicity—is on the table, however, a business case is created for
spending a few years rather than a few months in testing a compound in
mice. Should a startup company succeed in rejuvenating mice, you can bet
that Big Pharma will be beating down its door for the rights to translate
their intervention to humans.
Thus, the Mprize can bypass the vicious circle that has put the chid on
serious biomedical gerontology in academic research. More than this: it can

W A R B O N D S F O R T H E C A M P A I G N 339
reverse its course, putting its converging, self-reinforcing mechanisms to
work in a new, virtuous circle. Scientific results will drive public optimism,
in turn driving political acceptability, leading to more public and private
funding. Those investments will eventually—maybe very rapidly—lead to
robust mouse rejuvenation even if the SENS approach were to falter; and
then, the War on Aging will begin in earnest.
Eat well, exercise, and support the Methuselah Foundation, and I shad
look forward to shaking your hand in a future where engineered negligible
senescence is a reality: where we can enjoy dramatically extended lives in a
new summer of vigor and health, the dark specter of the age plague driven
away by the sunshine of perpetual youth.

Glossary
2-deoxyglucose (2-DG): a compound that closely resembles blood sugar, but
unlike sugar cannot be metabolized into energy in the mitochondria. For a
while, it looked as if 2-DG were a calorie restriction mimetic.
Active vaccination: giving a person the antigen against which doctors want to
provide immune protection, to trigger the immune system to produce anti
bodies that will attack it.
Adaptive immune system: the branch of the immune system that learns about
and targets specific foreign and "hijacked" cells though their antigens. The
existence of the adaptive immune system provides the basis of vaccines.
Adenosine triphosphate: see ATP.
Adult stem cell: stem cells found in mature bodies, that can give rise to only a
relatively narrow range of specialized, mature cells. Compare embryonic
stem cell.
Advanced glycation endproducts (AGE): highly chemically stable compound
that results as the "end product" of a series of chemical reactions in glyca
tion chemistry.
Advanced lipoxidation endproducts (ALEs): stable chemical damage to pro
teins caused by their interactions with fats; similar to advanced glycation
endproducts.
Aging damage: from the practical viewpoint of the engineer's school of anti-aging
medicine, changes at the molecular level to the structure of the organism

364 G L O S S A R Y
that distinguish an organism that has been around for a long time from one
that has not. On an abstract, theoretical level, we would only want to in
clude those changes that actually contribute to biological aging—i.e., those
that contribute to the increasing risk of disease, dysfunction, and death as
we go on living. From the point of view of anti-aging medicine, however, this
distinction becomes problematic, because it would require us to first deter
mine which changes genuinely contribute to biological aging and which do
not—which would require many more decades of research. The engineer's
school of anti-aging medicine sidesteps this ignorance by classifying all ag
ing changes that might contribute to biological aging as "damage," and
then intervening to repair or obviate it.
Aging: the word is used in many senses, but the one that's important for our
purpose is biological aging.
Alagebrium: a drug that appears to break advanced glycation end-product cross
links. Chemically, 4,5-dimethyl-3-(2-oxo-2-phenylethyl)-thiazolium chlo
ride. Originally code-named ALT-711.
Allotopic expression (AE): Expression of proteins from a different (Greek allo-)
place (topos). The creation of "backup copies" in the nucleus of the
protein-coding genes now housed in mitochondrial DNA.
Alpha-diketone: an advanced glycation end-product cross-link. Hypothetically,
the AGE broken by alagebrium.
Alpha-secretase: an enzyme required for the normal processing of Amyloid
Precursor Protein. APP does not form beta-amyloid protein when it is pro
cessed by this enzyme.
ALT: see Alternative Lengthening of Telomeres pathway.
ALT-711: the original code-name of alagebrium.
Alternative Lengthening of Telomeres (ALT): a poorly understood mecha
nism whereby some cancer cells relengthen their telomeres without using
telomerase.
Amadori product: a somewhat stable intermediate compound of glycation
chemistry, linking Schiff bases to advanced glycation endproducts (AGE).
Glycated hemoglobin or HbA1c is an Amadori product.
Aminoguanidine: drug that inhibits the formation of advanced glycation end
products, first inhibiting the initial glycation reaction, and then reducing
glycoxidation through antioxidant and metal-chelating effects, and above
all through its ability to mop up oxoaldehydes. Trade name Pimagidine.
Amyloid beta: see beta-amyloid protein-:
Amyloid precursor protein (APP): a protein produced in the brain that, when
damaged, can form beta-amyloid protein. Has some essential function in
our bodies, possibly including being necessary to allow neurons to rewire
themselves in response to new learning and to grow out neurites.
Amyloid protein A amyloidosis (AA): an amyloidosis caused by the excessive

G L O S S A R Y 365
production of amyloid A protein, a protein involved in the inflammatory
response, usually resulting from chronic inflammatory conditions. The
most common amyloid disorder outside the United States. Also termed
"inflammatory" or "secondary" amyloidosis.
Amyloid: any one of a range of cell-snaring chains of molecules that are created
by damage to healthy proteins naturally present in the body, causing them to
become twisted out of their proper configuration in ways that cause them to
undergo toxic interactions with each other, or with other constituents of the
cell. Typically form chemically "sticky" "webs" around cell structures that in
hibit their functions. See for example beta-amyloid protein.
Amyloidoses: Diseases caused by the accumulation of amyloids.
Anergic T cells: T cells that can no longer carry out their duties.
Angiogenesis: the growth of new blood vessels.
Anti-aging medicine: Biomedical attempts to intervene in the pathological ef
fects of biological aging. See geriatrician's school, gerontologists' school, and
engineer's school of anti-aging medicine. Most of what is called "anti-aging
medicine" in the marketplace today is either sheer hokum, or is a mixture
of (a) crude and unproven attempts at the gerontologist's school of anti-
aging medicine (hormone shots, antioxidant vitamins), combined with ba
sic (though unfortunately rarely practiced) preventive medicine (improved
diet and exercise) and the geriatrician's school of anti-aging medicine.
Antibodies: Proteins that specifically recognize and bind to antigens of the or
ganisms they're intended to fight, either by flagging the invader for attack
by other components of the immune system, or by blocking receptors and
other proteins that are needed for the pathogen's survival.
Antigen: a protein recognized by the immune system.
Antigen-presenting cells (APCs): the immune system's reconnaissance teams,
which identify enemy combatants' antigens through direct encounters
with them or by digging through the rubble of old battlegrounds (the re
mains of cells ravaged by them)and then alert T cells specialized in waging
war against the specific invaders that carry them.
Antisense mRNA: genetic material that binds to a pre-defined transcribed
gene as it emerges, preventing it from being used to make the protein that
it encodes.
Apoptosis: often referred to as "programmed cell death" or "cellular suicide."
A carefully orchestrated programmed process of self-destruction carried
out in cells that have been hijacked by "enemy forces" (viruses or cancer,
for example) to prevent them from threatening the organism as a whole.
Also used extensively during development to cull unnecessary cells. Apop
tosis is carefully sequenced to prevent damage to surrounding cells (see
necrosis).
ASP-2: see beta-secretase.

366 G L O S S A R Y
ATP: adenosine triphosphate. The cellular "energy currency." Just as you can
use many different fuels (enriched uranium, coal, solar energy) and turn it
into useable "universal energy" (electricity) to fuel a wide range of devices
(DVD players, food processors, washing machines), so the body uses the
chemical energy stored in various fuels (glucose, amino acids, fatty acids,
etc) to make ATP to drive many of its metabolic processes.
ATPase, vacuolar: an energy- (that is, ATP-) consuming pump located on the
membrane of the lysosome that drives extra protons into the lysosome
from the main body of the cell, increasing its acidity.
B cells: cells of the immune system that recognise specific markers (antigens)
on the surfaces of invaders that mark them as foreign, and churn out anti
bodies to them. Mostly responsible for defending us against pathogens like
bacteria and parasites that are purely foreign to the body, and that can
therefore be targeted directly for destruction.
BACE: see beta-secretase.
Basement membrane: the biological filter material of the kidney.
Beta-amyloid protein (also called "amyloid beta"): a peptide formed from amy
loid precursor protein that accumulates as the waxy "senile plaques" that
cluster around the brain cells of people with Alzheimer's disease, choking
off the neurons' nourishment and preventing their normal functioning.
Beta-secretase: an enzyme with an uncertain function that sometimes mistak
enly processes Amyloid Precursor Protein, contributing to the formation of
beta-amyloid protein.
Biogerontology: The study of biological aging aimed at understanding it better.
Biological aging: the universal, progressive, and deleterious process of escalat
ing loss of molecular fidelity with age, resulting stochastically from the in
trinsic metabolic processes of the organism, that degrades its ability to
maintain homeostasis in the face of environmental stressors, leading to in
creased intrinsic vulnerability to pathology and mortality.
Biomedical gerontology: The study of biological aging aimed at combating it
in humans.
Blastocyst: the very primitive ball of cells that is formed within just a few days
after sperm meets egg. The embryo only remains in this stage of develop
ment very briefly; it has developed much further by the time the embryo is
implanted in the womb.
Blood-brain barrier: the protective layer of cells surrounding the blood vessels
that feed the brain, that denies many molecules in the circulation free ac
cess to the brain.
C. elegans: see Caenorhabditis elegans.
Caenorhabditis elegans (C. elegans): a nematode (roundworm) that is now
widely used to study genetic pathways involved in the rate of aging.
Calorie restriction (CR): Reducing the amount of food energy (calories) in

G L O S S A R Y 367
the diet while maintaining adequate levels of essential vitamins, minerals,
fats, and protein. In laboratory rodents and many other species, calorie
restriction slows down aging—extending life beyond its 'natural' limits
while preserving youthful functionality and protecting against nearly all
age-related diseases and degenerative processes—in direct proportion to
the level of restriction: fewer calories lead to more healthy, youthful life
span. The first and best-studied way to slow down aging in mammals. Also
known as "dietary restriction," "energy restriction," or "food restriction."
Calorie restriction mimetic (CR mimetic): A substance that would induce
metabolic changes that would reproduce the essential anti-aging features
of calorie restriction.
Carbonyl: an organic molecule with a carbon atom double-bonded to an oxy
gen atom. This structure makes many carbonyls highly biologically active,
and many virulent precursors of advanced glycation end products are car
bonyls such as oxoaldehydes.
Carboxymethyllysine (CML): a common advanced glycation endproduct de
rived from glycoxidation. Can also be an advanced lipoxidation endproduct.
Catalase: an enzyme produced by the body that detoxifies hydrogen peroxide
by breaking it down into water and oxygen.
CD28: a receptor on the surface of T cells that allows antigen-presenting cells to
identify the CD8 cells that target the antigen found by the APCs.
CD4 cells: cells of the immune systems that help other immune cells to ramp
up their counteroffensive when pathogens first invade. Also known as
"T-helper" cells.
CD8 cells: see cytotoxic T cells.
Cerebral amyloid angiopathy (CAA): a disease caused when beta-amyloid
binds up the interior surfaces of the brain's blood vessels, crusting them
up, weakening them, and reducing their ability to flex in response to the
surging flow of the pulse. This leaves them vulnerable to bursting open in
a bleeding stroke.
Cerebrospinal fluid (CSF): the fluids bathing the brain and spinal cord.
Ceroid: substances that share many of lipofuscin's properties (and are therefore
often confused for it) but are much easier for the cell to break down and
do not accumulate in "normal" biological aging.
Chelate: to tie up a metal in unreactive form.
Clonal expansion: the process whereby a single cell (such as a memory cyto
toxic T cell) reproduces itself in large numbers, creating a "clone" of iden
tical cells.
CML: see carboxymethyllysine.
CMV: see cytomegalovirus.
Code disparity: differences in the "languages" made from the DNA "letters" of
genes in the mitochondria and cell nucleus. Code disparity makes some of

368 G L O S S A R Y
the genes in the mitochondria unreadable by the expression machinery of the
nucleus (and vice versa).
Complex V: the mitochondrial "turbine" that uses the flow of protons to fuel
the storage of energy as ATP.
CR: see Calorie restriction.
Creatinine: a waste product of protein breakdown that healthy kidneys effi
ciently remove, and is therefore used as a blood test of kidney function.
Crescentic glomerulonephritis: a form of highly inflammatory kidney disease
named for the crescent-shaped abnormalities that are seen in biopsies of
victims' kidneys. Once it develops, crescentic glomerulonephritis leads to
very rapid loss of kidney function.
CR mimetic: see Calorie restriction mimetic.
Cross-link: a molecular "handcuff" between adjacent proteins.
Crystallin: the clear, flexible proteins that make up the structure of the lens of
the eye.
Cytochrome c oxidase: one of the pumping complexes of the electron transport
chain.
Cytokine: an inflammatory signalling molecule of the immune system.
Cytomegalovirus (CMV): a persistent virus in the herpes family.
Cytotoxic T cells: T cells responsible for rooting out cells that are native to the
body but that have now been turned against it, such as cancer cells or cells
hijacked by viruses. Also called CD8 cells because of the characteristic re
ceptor they bear.
Dauer pathway: a hibernation-like state in roundworms like C. elegans. In the
dauer pathway, a larva suspends its development for a period than can last
much longer than the entire lifetime of a nematode that follows the nor
mal, nondauer trajectory.
Deletion: mutation that involves the total removal of large stretches of DNA,
annihilating many genes at once even though it is, strictly speaking, only a
single mutation.
Dendrimer: a tiny nanotechnology structure with exquisitely complex branch
ing structures that extend outward like bushes, forming a spherical shape.
Dendrimers' branches are engineered in a way that allows us to bind a
wide range of molecules to them.
Diastolic heart failure (DHF): heart failure when the pumping chamber of the
heart can't expand sufficiently well to take in the required volume of blood.
Diastolic pressure: the second number that you get from a blood pressure cuff,
like the "80" in "110 over 80." The baseline pressure in the arteries at rest.
DNA polymerase: the enzyme responsible for making a new copy of a cell's
DNA.
EGFR and EGFRvIII: see epidermal growth factor receptor.
Electron transport chain (ETC): the series of "pumps" that use food energy in

G L O S S A R Y 369
the form of electrons provided by NADH to fill up a "reservoir" of protons
held back by the mitochondrial inner membrane (the mitochondrial "dam"),
to drive the production of energy in the form of ATP by complex V via ox
idative phosphorylation.
Electron: a charged particle in an atom. The flow of electrons is the basis for
electricity and of oxidative phosphorylation.
Embryonic stem cell: the primordial "master cells" from which our mature
cells spring. Stem cells apparently found only in embryos, that have the
ability to become any of the many different mature cells of the body. Com
pare adult stem cell.
Engineer's school of anti-aging medicine: direct intervention in the molecular
damage of aging. Leaving metabolism alone to wreak damage on our mo
lecular structures, but preventing the ensuing damage from leading to
pathology, either by cleaning up the damage (keeping it (or restoring it to)
below the threshold at which it becomes pathological), or by devising ways
of rendering the damage itself harmless.
Enzyme: a biological catalyst that facilitates a chemical reaction in the body.
Epidermal growth factor receptor (EGFR): a receptor that stimulates cell
growth. Excessive production of EGFR, or production of a mutated form
called EGFRvIII, is implicated in many cancers.
Epigenetic structures: the "scaffolding" that is anchored to the DNA in our
chromosomes. Epigenetic structures help determine which genes are
turned on in a cell and which are turned off, allowing the same overall
DNA to be used to create cells as diverse as liver, heart, and kidney cells.
Epimutation: a permanent, unprogrammed structural alteration in the "super
structure" or "scaffolding" that controls the expression of genes. Because
changes in this "scaffolding" induce changes in the regulation of gene ex
pression (turning genes on or off), epimutations are functionally the same
as mutations and are treated as such by SENS.
Expression: the process by which the "blueprints" present in genes are exe
cuted in the creation of proteins.
Fenton reaction: chemical reaction in which transition metals make preexist
ing but relatively harmless, free radicals become more virulent.
F0/F1 ATP synthase: see complex V.
Free radicals: Usually defined as an electrically neutral atom or molecule con
taining an electron that is missing its twinned "pair." This deficiency
makes free radicals unstable and highly chemically reactive. Many free rad
icals "steal" electrons from other molecules to stabilize themselves, in the
process damaging the molecule from which the electrons are "stolen" and
often turning that molecule into a free radical itself. An excess of free rad
icals in the cell can cause oxidative stress, leading to dysfunctional meta
bolic imbalances and direct damage to key structural components such as

370 G L O S S A R Y
proteins, DNA, and fatty membranes. Many substances behave like "free
radicals" and are often called free radicals but don't, strictly, meet this def
inition (e.g., reduced iron ions (Fe 2 +); equally, some molecules that strictly
are free radicals are not harmful at all.
Gamma-secretase: an enzyme required for the normal processing of Amyloid
Precursor Protein but that can also play a role in its abnormal processing
into beta-amyloid protein, particularly if the enzyme is mutated and thus
produces an abnormal form of the enzyme.
Geriatrician's school of anti-aging medicine: attempting to interfere with the
pathological consequences of aging as they come up, by treating them med-
ically on a one-by-one basis.
Gerontologists' school of anti-aging medicine: attempting to interfere with the
mechanisms of aging, by cleaning up the biochemically "messy" processes
of metabolism or by neutralizing the reactive by-products of metabolism
before they cause damage to our molecular structures.
Gerontology: the study of "aging," in any of its aspects or meanings: the psy
chology of the old in our society; the prejudices against old people; the so
cial structures that support or restrict the access of the frail elderly to
society; and the biology of aging (biogeronto logy) .
Glial cell: part of the caretaking support staff of neurons.
Glioblastoma multiforme: a rare, extremely aggressive, and hard-to-treat brain
cancer.
Glucosepane: a complex advanced glycation end-product that is the single most
important contributor to the body's AGE burden known to date, tying up
as much as one out of every five molecules of the key structural protein col
lagen in old, nondiabetic humans.
Glycated hemoglobin (HbA1c): an Amadori product that forms on red blood
cells. Because HbA1c persists for 2-3 months, it reflects average blood
sugar levels for that time period, and so is used as a lab test to measure
overall blood sugar control.
Glycation: the spontaneous chemical bonding of sugar molecules to proteins.
Glycoxidation: the accelerated conversion of some precursors of advanced gly
cation end-products into those end-products by free radicals.
HbA1c: see glycated hemoglobin.
Herceptin: a monoclonal antibody used as a targeted cancer therapy. Herceptin
targets a receptor called HER-2 that stimulates cell growth. By tying up
HER-2, herceptin prevents the excessive growth of cancer cells dependent
on overstimulation from HER-2 to keep up their very rapid rate of repro
duction.
Homeostasis: the ability of the cell or organism to maintain a defined meta
bolic equilibrium.

G L O S S A R Y 371
Hydrogen peroxide: a molecule that acts like a free radical. Produced from the
breakdown of superoxide radicals.
Hydrolase: an enzyme that breaks down a compound by combining it with wa
ter molecules. Most of the enzymes of the lysosome are hydrolases.
Innate immune system: the branch of the immune system that doesn't have to
"learn" to identify a specific enemy. Its job is similar to that of regular sol
diers on patrol in a demilitarized zone, trying to maintain order but unsure
of who might be the enemy, ready to confront anything suspicious-looking
that it happens upon.
Insulin: the hormone whose job it is to move carbohydrates and amino acids
into fat and muscle cells.
Intein: sequences that are inserted temporarily into some proteins when they
are first synthesised, possibly to help the protein mature into its final form
properly, and are then snipped out once they've served their purpose.
Isolated systolic hypertension (ISH): the kind of high blood pressure in which
a person's systolic reading (the first of the two numbers that you get from a
blood pressure cuff, like the "110" in "110 over 80") is high, even though
their diastolic pressure (the second number) is fine
K2P: a major advanced glycation endproduct in the lenses of our eyes and possi
bly other tissues.
KLRG1: a receptor that prevents cytotoxic T cells from proliferating when no
infection is present. Healthy cells bearing KLRG1 are able to reproduce
when a threat is actually present; anergic CD8 cells cannot, because of an
other receptor called CD57.
LEV: see Longevity Escape Velocity.
Lipofuscin (lip-oh-FEW-sin): a catch-all term for the mixture of stubborn
waste products that builds up in the lysosomes of long-lived cells like those
of the heart and the brain as we age. A chemical hodgepodge of fatty and
proteinaceous materials derived from membranes, reactive metals like iron
and copper, and a variety of other organic molecules that the normal com
plement of enzymes in the lysosome doesn't know how to deal with and so
refuse to be broken down after being sent there. Glows red when exposed
to light of a particular wavelength. Called popularly, "age pigment."
Longevity escape velocity (LEV): a threshold rate of biomedical progress that
will allow us to stave off aging indefinitely. The point at which each suc
cessive round of refinements to the SENS age-reversing toolkit is buying
us more time than we need to develop the next round of refinements, until
we can eventually escape age-related decline indefinitely, however old we
become in purely chronological terms.
Lysosomal storage diseases (LSDs): a range of genetic disorders caused by
failure, by one mechanism or another, of the lysosomes. Many sufferers

372 G L O S S A R Y
completely lack the gene for a lysosomal enzyme, or bear a mutated copy of
it, resulting in a misshapen and ineffective version of the protein. In other
cases, the problem is that one of the specialized transport proteins on the
surface of the lysosomal membrane is missing or defective, so that the lyso
some can't bring the junk into itself to break it down. LSDs all result in
deadly degenerative diseases, with specific symptoms varying based on
which organs a given mutation affects, and how severely.
Lysosome: An acidic organelle that uses enzymes to break damaged cellular
components down at the molecular level into more basic constituents that
can be used as raw materials for the biosynthesis of new cellular mem
branes, enzymes, and other important components of the cellular machin
ery. The biological "garbage incinerator" or "recycling center." The extra
protons that create the lysosome's acidity are actively pumped out of the
main chamber of the cell and into the lysosome by an energy- (that is,
ATP-) consuming pump located on its membrane (the vacuolar ATPase).
Maillard reaction: a major form of glycation chemistry, in which a molecule of
sugar opens its structure and glues onto a protein molecule, forming a
Schiff base. This structure is relatively unstable, so the Schiff base will of
ten spontaneously fall apart. Sometimes, however, it will collapse into a
more stable Amadori product.
Matrix metalloproteinases (MMPs): protein-digesting enzymes that act as the
"demolition teams" of tissue remodeling, clearing away the old, damaged
"scaffolding" in which cells are embedded in a tissue, making space for
new growth.
Maximum life span: see species maximum life span.
Memapsin: see beta-secretase.
Memory cytotoxic T cells: specialized to attack hijacked cells bearing antigens
encountered in previous immunological battles.
Meningoencephalitis: life-threatening swelling of the brain, apparently as a re
sult of an overreaction of the immune system inside the brain.
Metabolism: the sum of the physical and chemical processes that occur in the
living body, including the breakdown and buildup of body structures and
proteins, and the uptake, distribution within the body, chemical and phys
ical transformation, and ultimate elimination of food, air, and other com
pounds taken in from the environment.
Metastasis: the process whereby cancerous cells escape from the restraints of
the tissue in which they were originally embedded, and begin a new colony
in tissues far removed from the original cancer site.
Methylglyoxal: a major oxoaldehyde precursor of advanced glycation end prod
ucts, up to 40,000 times more reactive than blood sugar.
Microglia: the immune cells of the brain.
Mitochondria: the cellular "power plants" that take the body's raw fuels (glu-

G L O S S A R Y 373
cose, amino acids, fatty acids, etc) and turn them into useable cellular en
ergy (adenosine triphosphate—ATP).
Mitochondrial inner membrane: mitochondrial "dam" that holds back the
"reservoir" of protons used to drive the production of ATP by Complex V
(the "turbine" of the mitochondrial "hydroelectrical dam").
Mitochondriopathies: a class of diseases caused by defects in the inherited mi
tochondrial DNA (or, more rarely, by mutations acquired through causes
independent of the aging process). These mutations lead to a failure of en
ergy production that causes a spectrum of dysfunctions in various organs,
depending on the exact mutation involved.
MMPs: see matrix metalloproteinases.
Monoclonal antibody: an antibody produced on a mass scale in the laboratory
to target a specific antigen.
Monomer, beta-amyloid: an individual fragment of beta-amyloid protein that
initially floats free in the brain.
mRNA: the cell's transcripts of the DNA instructions.
Mutation: a permanent, unprogrammed structural alteration in the DNA. As
used in Chapter 12, includes epimutations.
Myelin sheath: insulating material that surrounds nerves.
Myeloperoxidase: an enzyme used by macrophages to kill bacteria by generat
ing toxic hypochlorous acid.
NAD+/NADH: A biological "carrier molecule" that shuttles electrons from
food into the mitochondria.
Naive cytotoxic T cells: a reserve of as-yet-unspecialized cytotoxic T cells that
are ready to identify new threats, "learn" about their key antigens, and
then mount an attack.
Nanotechnology: engineering performed at the molecular level.
Necrosis: traumatic, uncontrolled death of a cell. Necrosis usually causes the
cell to swell and break open, harmfully releasing its contents and damag
ing neighboring cells.
Neurite: the branching "fiber optic cables" that allow neurons to talk with one
another.
Neuron: a main type of cell of the brain and nervous system, specialized to re
ceive and transmit information from the body and from other neurons.
Neuropathy, diabetic: the debilitating damage to the nerves that is suffered
by so many diabetics, resulting from the degeneration of the insulating
myelin sheaths, complicated by the slow shrinking away of the "electric ca
bles" (dendrites and axons) through which nerves communicate with one
another.
Notch receptor 1 (NOTCH1), a protein required for the activation of stem
cells, the growth of new blood vessels, and the maturation of some kinds of
immune cells.

374 G L O S S A R Y
N-phenacylthiazolium bromide (PTB): a thiazolium breaker of advanced glyca
tion end products. Chemical cousin and predecessor of alagebrium.
Nucleus: the part of the cell that houses its central DNA genetic instructions.
Oligomer, beta-amyloid: a short chain of monomers of beta-amyloid protein
that can still float free in the brain.
Organelle: a self-contained cellular "factory" that exists outside the nucleus
and carries out specific metabolic functions for the cell as a whole. Exam
ples include mitochondria and lysosomes.
Oxidant: substances that chemically "need" electrons. Includes many free radi
cals, but also normal biochemical intermediates that are sometimes re
quired to be in this state.
Oxidating agent: see oxidant.
Oxidative phosphorylation (OXPHOS): The "charging up" of the "battery"
of ATP by complex V, achieved by adding phosphorus to its precursor mol
ecule. Consumes oxygen and produces carbon dioxide and water, and thus
called cellular respiration.
Oxidative stress: the imbalance of those substances in the body that tend to
chemically "need" electrons (oxidants including free radicals, but also nor
mal biochemical intermediates that are sometimes required to be in this
state) relative to substances that chemically "want" to donate them (reduc-
tants or reducing agents). Oxidative stress increases the risk that free radi
cals will damage key cellular components instead of being detoxified, and
can also cause dysfunctional imbalances in metabolic processes by shifting
the electrical homeostasis of the cell.
Oxoaldehyde: a highly reactive carbonyl intermediate product of glycation
chemistry.
OXPHOS: see oxidative phosphorylation.
Passive vaccination: directly providing antibodies against an antigen that we
want to target for immune attack, bringing out the immune response that
the same antibodies elicit when they are produced by the body.
Pentosidine: one of the more easily measured advanced glycation end products.
Pimagidine: see aminoguanidine.
Point mutations: mutations that change only one, or a few, of the "letters" in one
"word" in the "sentence" of instructions comprising an individual gene.
Prodrug: a substances that is inactive until metabolized in some way, where
upon it is chemically transformed into a pharmacologically active product.
Proton: an electrically charged particle present in the atom. The flow of pro
tons across the mitochondrial inner membrane through complex V provides
the power to drive the storage of cellular energy as ATP.
Receptor: a molecular "lock" on the surface of the cell that responds to the
correct molecular "key" by performing functions like opening the cell up
to a needed nutrient or inducing a signaling cascade.

G L O S S A R Y 375
Reductant: a substance that characteristically "wants" to give electrons to
other compounds.
Retinopathy, diabetic: vision loss in diabetics linked to damage to the fine
blood vessels feeding the light-absorbing tissues at the back of the eyeball.
RMR: see robust mouse rejuvenation.
Robust mouse rejuvenation (RMR): the irrefutable reversal of aging in mice.
RMR will be achieved when we can take at least twenty mice of the
species Mus musculus (the common house or laboratory mouse), of a
healthy strain (one with a normal average life span of at least three years),
and administer anti-aging treatments starting only when they are at least
two years old that lead them to be able to live in good health to an average
of five years.
SA-beta-gal: see Senescence-associated beta-galactosidase.
Schiff base: a compound that can result from various glycation reactions that
contains a double bond between carbon and nitrogen atom, with specific
classes of compounds connected to it in turn through the nitrogen atom.
The Schiff bases that result from glycation are unstable and tend to go on
to form Amadori products.
SCNT: see somatic cell nuclear transfer.
Senescence-associated beta-galactosidase (SA-beta-gal): an enzyme whose ac
tivity identifies senescent cells.
Senescent cells: cells that have lost the ability to divide as a result of aging dam
age (and, usually, the body's response to that damage).
SENS: Strategies for Engineered Negligible Senescence. The scientific plat
form for anti-aging medicine based on the heuristic of the engineer's school
of anti-aging medicine.
Serotonin: a chemical messenger in neurons (and elsewhere) involved in mood,
appetite, thought, and sensory perception. Serotonin is the substance whose
metabolism is modulated by drugs like Prozac.
Somatic cell nuclear transfer (SCNT): The process of making new, perfectly-
matched embryonic stem cells out of a specialized mature cell (a "somatic
cell") from a patient by fusing it with an egg cell, provided by a prospec
tive stem cell recipient, whose cell nucleus is removed to make way for
the one from the patient's cell. When the fused cell begins dividing, it
creates ESCs with the donor's genetic code, and so with absolutely no
risk of rejection.
Species maximum life span: how long the oldest old of the species (and not just
of a particular strain of them, or of the cohort of animals in which the in
tervention was tested) can live under the best possible conditions.
Stem cell: an early, unspecialized cell that can renew itself indefinitely and de
velop into one or more of the highly specialized, mature cells of each tissue
in the body. Includes embryonic stem cells and adult stem cells.

376 G L O S S A R Y
Subcutaneous fat: the fat that lies under your skin all over the body, producing
a "pear shape" when present in excess. Compare visceral fat.
Superoxide radical: a free radical produced by the "fumbling" of electrons by
the electron transport chain in the mitochondria.
Systemic AL amyloidosis: the most common form of amyloidosis in the United
States and some other industrialised countries, caused by overproduction by
a kind of blood cell of a component of a class of antibodies called "im
munoglobulin light chain" (L—thus "AL," for "Amyloidosis Light-chain").
Systolic blood pressure: the first of the two numbers that you get from a blood
pressure cuff, like the "110" in "110 over 80." A measure of how much
pressure is applied to the artery wall by the surge of blood into the vessel
as the heart contracts.
T cells: immune cells that mature in the thymus. Includes cytotoxic T cells and
helper T cells.
Targeted cancer therapy: a selective cancer treatment that "targets" cancer
cells selectively. Usually refers specifically to drugs that interfere with spe
cific receptors or signaling processes upon which a particular cancer relies
strongly.
Targeting sequence: a special string of amino acids that when appended to the
"nose" of a protein produced in the main body of the cell, directs it into a
particular location such as the mitochondria.
Telomerase: the enzyme that relengthens shortened telomeres.
Telomeres: long stretches of DNA present at the ends of all our chromosomes
that contain no protein blueprints. Telomeres are worn down with each
round of DNA replication.
T-helper cells: see CD4 cells.
Therapeutic cloning: see somatic cell nuclear transfer.
Thiazolium: member of a class of compounds with a chemical structure related
to thiamin (vitamin B1).
Thymidine kinase (TK): an enzyme that is required for the synthesis of DNA.
TIM/TOM complex: short for "Translocase of the Inner Mitochondrial mem
brane" (TIM) and the "Translocase of the Outer Mitochondrial membrane"
(TOM). The elaborate machinery that specifically moves ("translocates")
proteins through the mitochondrial membranes.
TK: see thymidine kinase.
Transition metals: elements like iron and copper that can intensify oxidative
stress through their role in the Fenton reaction.
Translocase of the Inner Mitochondrial membrane (TIM): see TIM/TOM
Complex.
Translocase of the Outer Mitochondrial membrane (TOM): see TIM/TOM
Complex.
Triglycerides: fats; especially fats circulating in the blood.

G L O S S A R Y 377
Vacuolar ATPase: see ATPase, vacuolar.
Vascular endothelial growth factor (VEGF): a chemical messenger that stimu
lates new blood vessel growth.
VEGF: see vascular endothelial growth factor.
Visceral fat: fat tissue that surrounds your internal organs, as opposed to the
subcutaneous fat that lies under your skin all over the body. Too much vis
ceral fat is responsible for an "apple-shaped" or "beer-bellied" overweight
appearance. Visceral fat appears to be implicated in much of the metabolic
derangement of diabetes and of aging.


















