
ATOMIC ENERGY »5Ä L'ENERGIE ATOMIQUE … · cellule partielle cathodique en cours d'oxydation...
39
AECL-8702 ATOMIC ENERGY »"5Ä L'ENERGIE ATOMIQUE OFCANADA LIMITED TiBV DU CANADA LIMITÉE MECHANISMS OF HYDROGEN ABSORPTION BY ZIRCONIUM ALLOYS Mécanismes d'adsorption de l'hydrogène par les alliages de zirconium B. COX Presented at 1984 Fall Meeting of the Materials Research Society, Boston, November 26-30. 1984 Chalk River Nuclear Laboratories Laboratoires nucléaires de Chalk River Chalk River, Ontario January 1985 Janvier
Transcript of ATOMIC ENERGY »5Ä L'ENERGIE ATOMIQUE … · cellule partielle cathodique en cours d'oxydation...

AECL-8702
ATOMIC ENERGY » " 5 Ä L'ENERGIE ATOMIQUE
OFCANADA LIMITED T i B V DU CANADA LIMITÉE
MECHANISMS OF HYDROGEN ABSORPTION BYZIRCONIUM ALLOYS
Mécanismes d'adsorption de l'hydrogène par les alliagesde zirconium
B. COX
Presented at 1984 Fall Meeting of the Materials Research Society,Boston, November 26-30. 1984
Chalk River Nuclear Laboratories Laboratoires nucléaires de Chalk River
Chalk River, Ontario
January 1985 Janvier

ATOMIC ENERGY OF CANADA LIMITED
MECHANISMS OF HYDROGEN ABSORPTION BY ZIRCONIUM ALLOYS
B. Cox
(Presented at 1984 Fall Meeting of the Materials Research Society, Boston,November 26-30, 1984)
AECL-8702
Materials Science BranchChalk River Nuclear Laboratories
Chalk River, Ontario KOJ 1J0 Canada1985 January

L'ENERGIE ATOMIQUE DU CANADA, LIMITEE
Mécanismes d'adsorption de l'hydrogène par les alliages de zirconium
par
B. Cox
(Rapport présenté à la réunion d'automne 1984 de Materials Research Societytenue à Boston du 26 au 30 novembre 1984)
Résumé
Ce rapport résume la façon dont nous comprenons actuellement lestrois mécanismes primaires par lesquels les isotopes d'hydrogène peuventpénétrer dans les alliages de zirconium en service. Voici ces mécanismes:
(i) Réaction avec l'hydrogène et adsorption de cet élément gazeuxprovenant d'atmosphères n'ayant pas suffisamment d'oxydant pour quesoit maintenue la pellicule d'oxyde protectrice.
(ii) Diffusion dans le métal d'une fraction de l'hydrogène dégagé dans lacellule partielle cathodique en cours d'oxydation dans l'eau ou lavapeur.
(iii) Diffusion des isotopes d'hydrogène au travers d'une liaisonmétallurgique comprenant des métaux dissemblables formant un orificetraversant la pellicule d'oxyde normalement protectrice.
Bien que ces trois mécanismes puissent être spécifiés qualitative-ment avec une certaine certitude et bien que des preuves circonstanciéessoient souvent disponibles, on indique dans le rapport que les constantesde vitesse des diverses étapes des réactions sont essentiellement incon-nues.
Département de science des matériauxLaboratoires nucléaires de Chalk RiverChalk River, Ontario, Canada KOJ 1J0
Janvier 1985
AECL-8702

ATOMIC ENERGY OF CANADA LIMITED
MECHANISMS OF HYDROGEN ABSORPTION BY ZIRCONIUM ALLOYS
B. Cox
(Presented at 1984 Fall Meeting of the Materials Research Society, Boston,November 26-30, 1984)
ABSTRACT
This paper summarizes our present understanding of the three primarymechanisms by which hydrogen isotopes can enter zirconium alloys in service.These are:
(i) Reaction with and adsorption of hydrogen gas from atmospheres containinginsufficient oxidant to maintain the protective oxide film.
(ii) Diffusion into the metal of a fraction o.l: the hydrogen released in thecathodic partial cell during oxidation in water or steam.
(iii) Diffusion of hydrogen isotopes through a metallurgical bond with a dissi-milar metal which provides a window through the normally protective oxidefilm.
It is shown that while all three mechanisms can be specified qualita-tively with some certainty, and while supporting circumstantial evidence isoften available, the basic rate constants for the various steps in the reactionsare largely unknown.
AECL-8702
Materials Science BranchChalk River Nuclear Laboratories
Chalk River, Ontario KOJ 1J0 Canada1985 January

-1-
1 . INTRODUCTION
Delayed hydride cracking (DHC) of Zr-2.5 wt% Nb alloy pressure tubes wasfirst observed in CANDU reactors in 1974 (Pickering-3) and 1975 (Pickering-4),and again in 1982 (Bruce-2) [1]. In the first two of these incidents, hydrogenabsorption - as opposed to hydrogen in the tubes as installed - played no partin the process. Nevertheless, some unexpected hydrogen ingress into the tubeends was observed. The sudden failure of a Zircaloy-2 pressure tube inPickering-2 in August 1983 [2], although crack propagation again was essentiallyby delayed hydride cracking until the critical crack length was obtained, wassignificantly influenced by hydrogen absorption after the installation of thetube. Thus, our earlier interests in hydrogen absorption mechanisms, which hadbeen dormant for many years, were revived, not least by the large differences inhydrogen uptake behaviour shown by different zirconium alloys [3].
Zirconium alloys are normally protected against hydrogen ingress by thesurface oxide film, which presents a good barrier both to ingress, and to theegress of hydrogen already in the metal. However, laboratory work has shownthat, under conditions of straining at a notch, this oxide film offers littleprotection, and zirconium alloys are susceptible to rapid cracking (up to10"^ m/s) in hydrogen gas [4,5]. These experiments revealed only smalldifferences in the crack velocities of the same alloys which show surprisinglylarge differences in crack velocity under DHC conditions with only internalhydrogen [6].
As a result of this revived interest in hydrogen absorption, as acritical first step in any hydride cracking processes at elevated temperatures,earlier work on hydrogen absorption (some previously unpublished) has beenreviewed, and new experimental programmes have been started. There are threefundamentally different processes by which hydrogen isotopes can enter afabricated zirconium alloy component. The first, and most trivial mechanistic-ally, is by diffusion into the zirconium alloy via a direct metallurgicalcontact with another metal having a high diffusivlty for hydrogen, and a higherfugacity of hydrogen than the zirconium. The other two ingress routes operatethrough the surface oxide film. The first of these occurs when zirconium isexposed in a hydrogen atmosphere containing insufficient oxidizing species tomaintain the protective nature of the oxide, while the second occurs as part ofthe normal oxidation process in aqueous media. These three processes will bedealt with individually.
2. ABSORPTION OF HYDROGEN GAS
All zirconium components, unless held at high temperature in a goodvacuum, carry a ZrÛ2 film on their surfaces. In air at room temperature, thethickness of this oxide is limited by electron tunneling to 3-5 nm. Thickeningof this oxide can proceed by a variety of processes. By increasing the electricfield across the oxide, further oxygen ion transport through the air-formed filmcan be initiated either at room temperature or above. This same process willproceed by thermal activation if the temperature is raised in the presence of anoxidant. Such an oxide film is normally a very good barrier against reactionwith hydrogen gas and, provided sufficient oxidant is present in the environment,

-2-
may remain so indefinitely. "Sufficient" in this context was established byShannon [7] to be that required to maintain the normal oxidation rate at thetemperature concerned (Figure 1); the "normal" oxidation rate being that obtainedat environmental pressures sufficiently high for the pressure dependence of theoxidation rate to approach zero [8].
These observations led to the postulate that, for any conditions oftemperature, pressure and oxidant, there should be a critical hydrogen/oxidantratio above which the oxide film would remain protective, and gross reactionwith hydrogen would be prevented [9]. There could still be a slow absorption ofhydrogen by reaction with the oxidant if this were a hydrogen containingmolecule. Several investigators [9,10] have established these critical ratios,for a limited range of environments, and they are generally observed to be inthe range ÎO^-IO^ (Table 1). Thus, very small concentrations of oxidizingspecies in hydrogen gas are sufficient to prevent direct surface reaction withhydrogen at reactor operating temperatures. This effect was borne out inexperiments on cracking in hydrogen gas where small oxygen additions were suffi-cient to inhibit crack growth [5]. However, it is notable (Table 1) that moreoxygen appeared to be necessary to ensure passivation of strained metal at thecrack tip, than appears to be the case for reaction on a smooth surface.
If insufficient oxidant is present to maintain the oxide film in goodrepair, then, after a very variable incubation time required for "breakdown" ofthe surface oxide film [10-13], a rapid direct reaction with hydrogen gas ensues(Figure 2). The progress of the oxide "breakdown" process can be monitored,during the incubation period, by measuring the electrical resistivity (Figure 3)of the oxide film [7]. The resistivity of the oxide declines rapidly with time,as a result of the increasing hypostoichiometry of the oxide, and hydridingcommences once the resistivity declines below a critical level. The hypostoichi-ometry changes because the rate of dissolution of oxygen atoms from the oxideinto the metal now exceeds the rate at which replacement oxygen atoms can beacquired from the environment. In addition to the increasing electronic conduc-tivity associated with this process, an increase in the number of anionvacancies will result.
As a result of these observations, investigators studying the rate ofreaction uf hydrogen with oxidized zirconium surfaces have been tempted topostulate that hydrogen is migrating via these anion vacancies as interstitialH2 [14,15]. From their experiments they have then calculated dif fusivitief;for hydrogen in the Zr(>2 lattice. However, there is no unambiguous evidencefrom these experiments to support an argument that hydrogen is migrating via theZrÛ2 lattice at all. The diffusivities which they calculate are very high forsuch a lattice diffusion process, and lie in a range more typical of surfacedtffusivities. Experiments of this type give permeation rates 10? or moretimes those measured by effusion experiments using tritium [16-18]. From these,a diffusion coefficient for tritium in the surface layer (arbitrarily defined asthe first 5 Mm) of 10~6 cm2/s at 300°C was estimated [17]. This is in good agree-ment with a value of 3 x 10"^ which can be calculated from the autoradiographsin ret.19 for 400-500°C.
There is some circumstantial evidence that hydrogen entering the metalunder these conditions does not pass through the oxide lattice. Tritium

_ r> _
autoradiography [19] has shown that, when a specimen preoxidized in O9 or airexposed to T2 gas, tritium appeared in the metal core but the oxide filmremained essentially free of tritium (Figure 4). Other experiments showed thtritium, built into the oxide fiitn during reaction with T2O, remained fixed othe specimen was cooled and did not migrate into the metal, or exchange withhydrogen in the environment, over periods of years [16] . This again suggeststhat the diffusivity in the oxide is orders of magnitude less than that calcuted from the reaction rate experiments [14,15]. Conversely, tritium absorbedporous oxide films, by exposure to T7O, exchanged rapidly with hydrogen in thenvironment and vanished in periods of, at most, a few days [16]. Thus, at tvery most, any tritium which was present in the oxide on the specimens heatedT'i gas had exchanged with the laboratory environment, and vanished, by thetime the autoradiograph was exposed. This indicates strongly that the T2, whentered the metal in these experiments, did so via pores or cracks in the oxiand not by diffusion through the oxide lattice. Autoradiographs of similarspecimens preoxidized in oxygen and thon exposed to T7O showed tritium both ithe metal and in the oxide (Figr'e 5). This shows the fundamental differencebetween reaction with T2 and T2O.
The experiments of Shannon and others gave no direct clues to the micrcopie nature of the "breakdown process" in the oxide film, other than for theaccompanying changes in conductivity and stoichiometry. The implicit assumptthat these would be homogeneous changes is carried by the deductions subsequemade- Our electron microscope studies [20,21] of the oxide films formed onzirconium, of similar purity to that used in the hydrcgen diffusivity studiesshowed that the oxide films formed in a far from uniform and parallel-sidedmanner. Ridges of thick oxide form preferentially along many grain boundarieand big variations in the oxide thickness occur from grain to grain (Figure 6Many of these features have also been recognized from optical microscopy [22]However, the lines of cracks (Figure 7) which form in the oxide along theseridges, and along grain boundaries separating grains which grow thick and thioxide films [20-23], are less easily seen in the optical microscope. Thesecracks are encouraged by the curvature in the oxide/environment interface intduced by the local variation in oxide thickness and the high Pilling-Bedworthratio of ~1.5. These factors cause the outer surface of the oxide to go inttension, whereas oxide films formed as a planar layer on a smooth surface remin compression at the same point in time.
However, even in regions where cracks form in the outer part of theoxide, the inner layers of the oxide will remain in compression because the nlayers of oxide form at the oxide-metal interface by inward diffusion of oxygThus, there will probably always be a thin layer of protective oxide at thebottom of these cracks. This argument is supported by the observation thatcracks are visible in the oxide surface formed during oxidation, and yet thesoxide films continue to present excellent barriers to the ingress of hydrogenHence, such cracks are not the routes by which hydrogen enters the metal,although they may help to initiate, or become part of such a route during the"breakdown" process. That these ciarks do not penetrate to the oxide/metalinterface is shown in SEM studies of stripped oxide films. Examination of thoxide/metal interfaces [23,24] clearly showed the ridges of thick oxide alonggrain boundaries (Figure 8), and the variations in oxide thickness from graingrain (Figure 9), but did not reveal any yigns of cracks penetrating throughthe interface at these locations. In instances when the stripping process

induced cracking of the oxide, these cracks generally cut across oxide ridges at
,;rain boundaries (Figure 10), suggesting that these sites were not even regions
of wuakness in the oxide. Only occasionally have cracks running parallel to,
and within, the oxide ridges been seen at the oxide-metal interface. Some other
route, which allows direct access of hydrogen to the oxide/metal interface, must
develop during the incubation period, therefore.
When a specimen, with an oxide film such as those above, is heated in an
environment (vacuum or H2) containing insufficient oxidant to maintain the
normal growth of the oxide, the rate of oxygen dissolution in the metal will
exceed the rate of formation of new oxide. Under these conditions, the oxide
1 ifm will slowly dissolve in the metal. This process has been observed
(.Figure IL) by following the thinning ot interference-coloured oxide films in
the optical microscope [25,26]. On the scale of resolution of this instrument
the dissolution process appears to be uniform, provided that the metal core has
not become saturated in oxygen [26,27]. However, autoradiography ot the oxygen
distribution using the ^ 0 ( p , n ) ^ F reaction [27] shows that preferential diffusion
01 oxygen along zirconium grain boundaries occurs.
When the dissolution of oxide films is studied by electron microscopy,
examination of the oxide/raetal interfaces shows that the thinning of the oxide
is not uniform. Because of the preferential diffusion of oxygen along the grain
boundaries in zirconium, the dissolution of the oxide ridges at these sites occurs
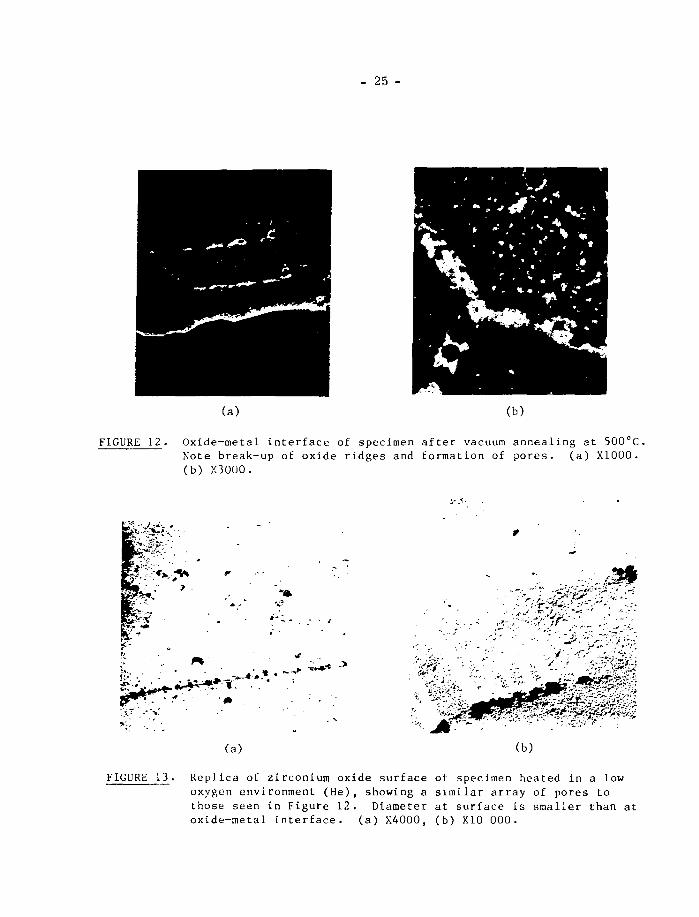
preferentially and inhomogeneously (Figure 12). This irregular dissolution of
the grain boundary ridges leads to the formation of large pores in the oxide.
These pores are thought to pass right through the oxide film because similar
rows ÖL pores have been seen when oxide films on specimens exposed to low oxygen
partial pressure environments were viewed from the outside by a replica technique
(Figure 13). These pores also occur in the oxide away from the prior metal grain
boundaries, perhaps because of enhanced local oxygen diffusion at incoherent
twins, or dislocation pile-ups.
The uniform dissolution of the bulk of the oxide, seen in the interference
colour studies [25,26], is probably the process which leads to the observed drop
in oxide resistivity [7,10]. However, the preferential dissolution of the oxide
at grain boundaries, and other sites, which leads to the formation of arrays of
pores at these locations, is probably the process which terminates the incuba-
tion period, and leads to the rapid direct reaction of hydrogen with the metal.
Since these pores appear to pass right through the oxide, there will be no oxide
diifusion barrier to prevent such a reaction at these sites. Thus, none of the
investigations which purported to measure the diffusivity of hydrogen in ZrO2
by exposing preuxidized specimens in hydrogen gas will have measured such a
quantity, and alternative techniques for measuring the hydrogen diffusion
coel1 ifient in ZrO2 must be sought.
3. HYDROGEN UPTAKE DURING OXIDATION
Direct reaction of gaseous hydrogen with the zirconium at the bottoms of
pores is not a possible route for hydrogen ingress during normal oxidation.
Even when oxide films become porous, in the post-transition region of oxidation
kinetics, the pores which develop will not penetrate up to the oxide/metal

-5-
interlace, .'. 1 thuu ii L lit? y may approach quiLe close to it, because there willalways bo c-nougli oxi.dant available to maintain at least a thin barrier layer atthe bottom of the pore.
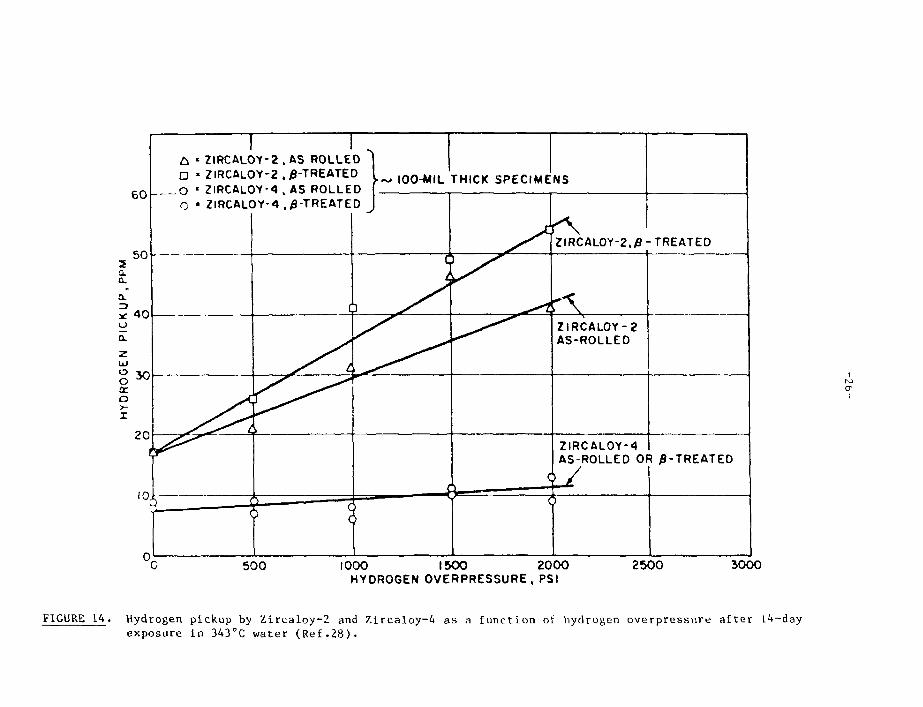
Oxid.ition studies using T2/H2O mixtures [16] have shown that, duringnormal oxidation, no 'Ij enters the metal (Table 2) until the thermally-inducedexchange reaction has progressed to the point where a measurable fraction ofT2O has been formed. Thus, the hydrogen isotopes which enter the metal do soas an integral part of the reaction of the zirconium with water molecules, andnot by reaction with any dissolved hydrogen in the water- Studies have shownthat this situation persists (Figure 14) until hydrogen overpressures in thesystem o(" tens of MPa are present [28].
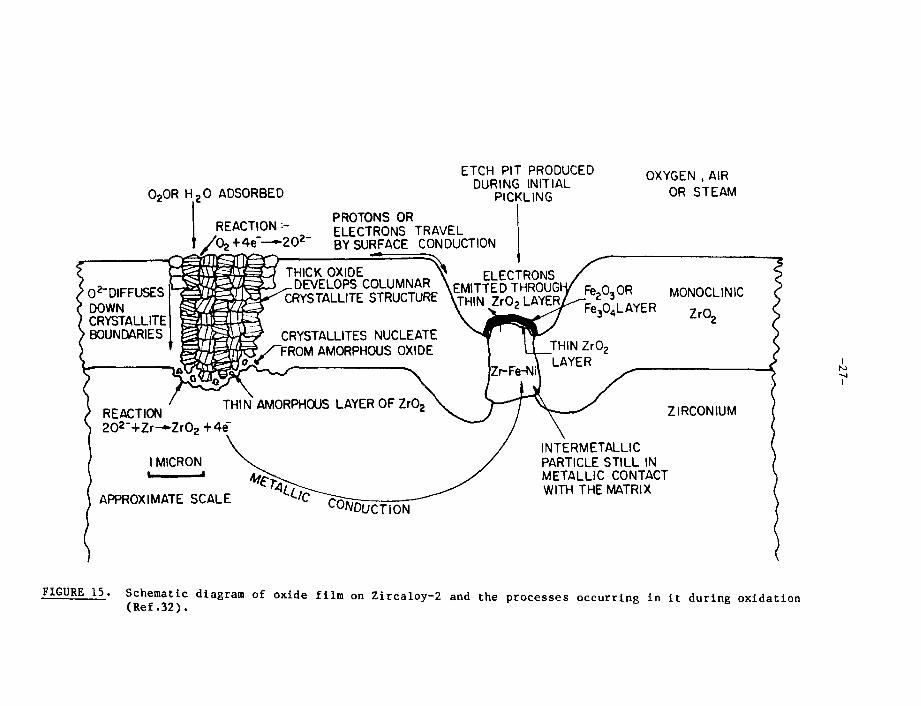
During the reaction cf zirconium alloys with water the hydrogen isliberated, initially, as an adsorbed hydrogen atom on the oxide surface, when aproton is discharged by an e lee. L run ei;u--rt; i. :,;, through the oxide film. The siteat which this occurs depends on the morphology of the oxide film (Figure 15).Thus, for alloys of the Zircaloy type, which contain additions of Fe, Cr and Ni(to a total of~0.3 wt%) in the form of a distribution of second-phase particles,the elecuron current flows primarily at sites where intermetallics partially, orcompletely, short-circuit the oxide [29]. In alloys containing no, or few, suchintermetallic particles of the transition metals (e.g. Zr-2.5 wt% Nb) the elec-tron current flows homogeneously through the bulk of the oxide [29].
The hydrogen atoms released by the discharge of the protons (eitherlocally at intermetallics, or uniformly over the surface) then either recombine,and are evolved as hydrogen gas, or diffuse into the metal. It is the competi-tion between the rates of these two processes which determines the proportion ofthe corrosion-hydrogen which enters the metal. Any local situation whichaffects the relative probabilities of recombination or ingress into the metalwill, therefore, affect the hydrogen uptake percentage. Despite the importanceof this step in the reaction no measurements of hydrogen recombination rates oneither pure or doped zirconia surfaces are known.
The presence of alloying additions, such as nickel, which are thought todecrease the rate of the recombination reaction, or even, under some conditions,to directly dissociate molecular hydrogen dissolved in the water [28], couldthen lead to high percentage uptakes for the corrosion hydrogen by allowing agreater opportunity for the hydrogen atoms to diffuse into the metal. It wasfor this reason that nickel was eliminated from the alloy in the development ofZircaloy-4 from Zircaloy-2 [30]. It is also notable that the hydrogen uptakepercentage shown by Zircaloy-4 in water is virtually independent of the hydrogenoverpressure (Figure 14), unlike that of Zircaloy-2 [28], thus adding furtherweight to the argument that the presence of nickel can lead to the direct disso-ciation and ingress of hydrogen dissolved in the water, when this is present toexcess.
The presence of dissolved oxygen, or other oxidizing species (e.g. nitricacid) is able to speed up the removal of the discharged protons from the Zr02surface and hence reduce the percentage hydrogen uptake (Figure 16). The precisemechanism by which this is achieved, whether by direct reaction of oxygendissolved in the water with hydrogen atoms on the surface, or by the substitution

-6-
of the reduction of oxygen by the electrons for the reduction of protons (as thecathodic reaction), has not been identified. The presence of dissolved hydrogen,as we have seen above, has virtually no effect on alloys without nickelcontaining precipitates, and only a small effect on Zircaloy-2, until very largeconcentrations are present. Once the oxide film becomes porous, in the post-transition oxidation region, the hydrogen apparently finds it more difficult toescape from the oxide, for there is a general tendency for percentage uptakes toincrease [31]. However, these effects are smaller at temperatures typical ofreactor operation than at higher temperatures (Figure 17).
The strong correlation (Figure 18) of hydrogen uptake percentages withthe nature of the elements in the intermetallic precipitates [32], coupled withthe observations of localized electron conduction at these sites, leads to theinference that the hydrogen entering the metal does so via some specific propertyof the oxide formed over these intermetallics. We are only now elucidating thenature of these oxide films [33], so the understanding of this aspect of theuptake mechanism may change rapidly in the near future. However, preliminaryresults suggest that elements like Fe and Ni may remain in the zero oxidationstate in the oxide on an intermetallic. The state of agglomeration of theseatoms has not yet been established, but the possibility of metallic stringerswithin these oxides cannot yet be ruled out. Such stringers could account forboth the high local electrical conductivity and the easy ingress of hydrogen atthese sites.
Although it is thought that, in alloys containing large numbe's of inter-metallic paticles large enough to partially, or completely short-c i_rcuit theoxide, the hydrogen from the corrosion reaction enters the metal at these sites,it is known from tritium autoradiography that the uniform oxide between theintermetallics contains a significant (Figure 19), but unknown, concentration ofhydrogen [19]. That the quantity of tritium in such oxides appears to be roughlyproportional to oxide thickness (Figure 20), for interference-coloured oxides,suggests a relatively constant concentration of tritium in these oxides, withlittle concentration gradient thrcigh them. Attempts .-, measure this concentra-tion using early infra-red spectroscopy techniques [34] were unsuccessful, butindicated that the concentration of OH~ in Zr02 films was probably ~ 5%. Noapplication of modern infra-red techniques to this problem has yet been made.
Despite the known presence of hydrogen in the uniform oxide film there isno knowledge of its mobility. The long-term stability of autoradiographicspecimens against exchange of the tritium with laboratory water vapour suggeststhat the mobility is low, but does not permit the calculation of other than anupper bound for the mobility. If, as observed, the tritium content of an 0.5 fimoxide film remains apparently unchanged after one year at room temperature, thepermeability of tritium through these films must have been less than~ 1 0 ~ ^ mole cm~2.s~l at room temperature. This is very much the value extrapo-lated from the elevated temperature tests [14,15], which we have already arguedrepresents surface diffusion down pores (Figure 21).
Work presently in progress by Fourier Transform NMR on ZrO2 filmsformed in 500°C steam, suggests that the hydrogen in the ZrO2 is tightly bondedand relatively immobile. However, we have not yet been able to extract a valuefor the diffusivity from these experiments.

-7-
Thus, we arrive at a picture whereby hydrogen uptake in alloys containing
intermetalLics of Fe, Cr and Ni is controlled by reLease of the hydrogen atoms
at the sites of these particles, and migration of them via the same intermetal-
llcs, perhaps by way of stringers of metallic Fe or Ni in the oxide, into V. he
•netal. [n alloys containing few sucli particles (e.g. Zr-2.5 wt% Mb) the hydrogen
must migrate nore uniformly through the oxide, which has a low diffusivity for
hydrogen. This difference in mechanism can account for the large differences in
the observed deuterium uptake rates in Zircaloy-2 and Zr-2.5% wt Mb alloy pressure
tubes [3]. If we assume that the very low rates of uptake by Zr-2.5 wt% Nb are
entirely controlled by diffusion through the oxide film, then we can use these
rates to calculate a lower bound for the diffusivity in ZrO2• This gives us a
value of ~ 1 0 ~ ^ moles.cm~^.s~^ for the permeability at 290°C. This is in the
same range as rhe results from effusion into steam [16], especially if the
actual barrier layer oxide thickness is quite thin (i.e. If the oxide is post-
transition)- Without a knowledge of the hydrogen concentrations in these
oxides, it is not possible to estimate a true dif tus ion coefficient, but the
permeabilities estimated above are not inconsistent with the very low diffusion
coefficients in the surface layer measured by the workers at North Carolina
State University [17,18].
4. HYDROGEN ABSORPTION VIA METALLIC CONTACTS
Although hydrogen absorption through a metallic contact may be the most
trivial of the three mechanisms mechanistically, it nevertheless has important
practical aspects as it may be one route for ingress of excess deuterium into
the ends of pressure tubes (Figure 22) from the 403 SS end fitting [1]. The
zirconium alloy pressure tube is in good metallic contact with the end-fitting
steel in the three grooves of the rolled joint. The factors which control
ingress by such a route should, therefore, be considered.
When a galvanic couple between a zirconium alloy and a nickel or iron
alloy is exposed to high temperature water the zirconium alloy becomes anodic and
the transition metal alloy cathodic. The cathodic part of the oxidation process
then occurs on the transition metal and protons are discharged there and may be
absorbed by the transition metal. Thj effective surface areas of the two parts
ot the couple will be determined by the conductivity of the water, and in high
purity water only small areas adjacent to the point of contact should be effect-
ive, although under irradiation this limitation may be relaxed by the effect of
irradiation on the conductivity of the electrolyte [36]. The distance from the
points of closest approacli of the two metals in the water and the point of
metallurgical contact will be important for the hydrogen migration, but not lor
the oxidation. This is evident in the instance of localized oxidation opposite
a platinum implant [36], where the electrical contact was distant from the point
of closest approach of the platinum implant and the facing zirconium surface. In
this instance, accelerated oxidation but no excess hydriding occurred, whereas
adjacent to the platinum implant both accelerated oxidation and enhanced
hydrogen contents were observed.
In the case of the Inconel to Zircaloy bonds [35,37], where the interface
between the Inconel and the Zircaloy-2 was exposed to the water, and diffusion
distances for both hydrogen and electrons were short, both excess oxidation and

-8-
excess hydriding were observed adjacent to the bond. No enhanced hydrogen uptakewas observed in Zircaloy/Zircaloy crevices [36,37], but enhanced oxidation,perhaps from enhanced radiolysis, was seen. Although it might be expected thatthe diffusivity of hydrogen in the transition metal would affect the rate ofmigration into the zirconium alloy via the metallurgical bond there is noevidence for comparison (say) between Inconel and stainless steel. However,where high temperature water is in contact with zirconium alloy/transition metalbonds close to or at the interface between them the rate of the cathodic reac-tion on the transition metal may be more important than the hydrogen diffusivityin it. In this circumstance, the higher nickel content: of Inconel than ofstainless steel and the specific effects of nickel with regard to the hydrogenrecombination reaction may mean higher excess hydrogen absorption for zirconiumalloy/lnconel bonds than for zirconium alloy/stainless steel bonds. This seemsto be borne out by the results of Urbanic's [38] experiments on Zr-2.5% Nb tubeswith plugs of various metals inserted in them. If these results are comparedwith some properties of the various metals (Table 3) it would appear that theexchange current density for the hydrogen evolution reaction is more importantthan the hydrogen permeability in determining the uptake by zirconium alloys.However, there is insufficient evidence at present to reach a firm conclusionabout the important factors determining the rate of excess hydrogen uptake inzirconlum alloys metallurgically bonded to transition metals; tue indicationsare that we should look at the efficiency of the coupled metal as a cathode,rather than its diffusion properties.
5. CONCLUSIONS
1 have summarized, above, our present understanding of the three princi-pal mechanisms by which hydrogen isotopes may enter zirconium alloy componentsin service. It will be seen that many of the basic physical rate constantswhich would be useful in confirming these uiechanisms have not been measured. Thediffusion coefficients for hydrogen isotopes are not known to within many ordersof magnitude, and other rate constants such as the recombination rate of hydro-gen atoms on zirconia surfaces (doped with realistic alloying additions) are notknown at all. The radiation chemistry within porous ZrO2 films is, likewise,an unknown quantity with regard to any differences between it and that in bulkwater. There is much scope, therefore, for further work in t'.ls area.
REFERENCES
E.C.W. Ferryman, "Pickering Pressure Tube Cracking Experience", Nucl.En., 1978, 1_7_, 95.
G.J. Field, J.T. Dunn and B.A. Cheadle, "Analysis of the Pressure TubeFailure in Pickering NGS 'A1 Unit 2", Presented at Can. Nucl. Ass. Ann.Meeting, Saskatoon, Jun.- 1984; Atomic Energy of Canada Ltd., Report AECL-8335 (June 1984).

-9-
[3] V.F. Urbanic and B. Cox, "Long-Term Corrosion and Deuteriding Behaviourof Zircaloy-2 Under Irradiation", Proc. 1984 C.I.M. Conf., Quebec City,Aug. Can. Met. Quart, (to be published).
[4] H.G. Nelson and H.F. Wachob, "Stress Corrosion Cracking of Zircaloy" inElectric Power Research Institute Report EPRI-NP-717, Palo Alto, CA,March 1978, Section 6.
[5] C.E. Coleman and B. Cox, "Cracking of Zirconium Alloys in Hydrogen",Proc. of 6th Int. Conf. on Zirconium in the Nuclear Industry, Vancouver,1982, ASTM-STP-824, p.675.
[6] C E . Coleman and J.F.R. Ambler, "Delayed Hydrogen Cracking in Zr-2.5 wt%Nb Alloy", Revs, of Coatings & Corrosion, 1979, 3^ 105, Freund, Tel Aviv.
[7] D.W. Shannon, "Role of Oxidation Rate on the Hydriding of ZirconiumAlloys in Gas Atmospheres Containing Hydrogen", Corrosion, 1963, 19,414t, and U.S. Report HW-76562 REV. (Feb.1963).
[8] B. Cox, "Some Effects of Pressure on the Oxidation of Zircaloy-2 in Steamand Oxygen", J. Less Common Met., 1963, _5_, 325.
[9] R.F. Boyle and T.J. Kisiel, "Hydrogen Permeation of Zircaloy-2 CorrosionFilms", U.S. Report, Bettis Tech. REv., WAPD-BT-10 (1958) p.31.
[10] Katsumi Une, "Kinetics of Reaction of Zirconium Alloy with Hydrogen",J. Less. Common Met., 1978, _57_, 93.
[11] S. Aronson, "Some Experiments on the Permeation of Hydrogen Through OxideFilms on Zirconium", U.S. Report, Bettis Technical Rev., WAPD-BT-19(1960) p.75.
[12] E.A. Gulbransen and K.F. Andrew, "Mechanism of the Reaction of Hydrogenwith Zircaloy I. Role of Oxide Films, Pretreatments and Occluded Gases",J. Electrochem. Soc, 1954, 101, 348.
[13] Idem, "Reaction of Hydrogen with Preoxidized Zircaloy-2 at 300° to400°C", ibid., 1957, 104, 709.
[14] T. Smith, "Mechanism of Hydrogen Permeation of Oxide Films on Zirconium",J. Electrochem. Soc., 1965, 112, 560.
[15] Idem, "Kinetics and Mechanisms of Hydrogen Permeation of Oxide Films onZirconium", J. Nucl. Mat., 1966, 1£, 323; and U.S. Report, NAA-SR-6267.
[16] B. Cox and C. Roy, "The Use of Tritium as a Tracer in Studies of HydrogenUptake by Zirconium Alloys", Atomic Energy of Canada Ltd., Report AECL-2519 (Dec.1965).
[17J T.S. Elleman and K. Verghese, "Surface Effects on Tritium Diffusion inNiobium, Zirconium and Stainless Steel", J. Nucl. Mat., 1974, 53, 299.

-10-
[18J J.H. Austin, T.S. Ellenan and K. Verghese, "Tritium Diffusion inZircaloy-2 in the Temperature Range -78 to 204°C", J. Nucl. Mat., 1974,2l_, 321 .
[19] C. Roy, "Hydrogen Distribution in Oxidized Zirconium Alloys by Autoradio-graphy", Atomic Energy of Canada Ltd., Report AECL-2085 (Sept.1964).
[20] B. Cox, "Oxide Breakdown on Arc-Melted Sponge Zirconium", Corrosion,1960, lb_, 306; and U.K. Report AERE-2874 (1959).
[21] B. Cox and A.B. Mclntosh, "The Oxide Topography on Crystal-Bar andReactor Grade Sponge Zirconium", Atomic Energy of Canada Ltd., ReportAECL-3223 (Nov.1968).
[22] J.K. Wanklyn, C F . Britton, D.R. Silvester and N.J.M. Wilkins, "TheCorrosion of Zirconium and its Alloys. Part III. The Influence of theEnvironment on Oxidation", J. Electrochem. Soc., 1963, 110, 856; and U.K.Report AERE-R4130 (August 1962).
[23] B. Cox, "Examination of Oxidized Zirconium Alloys with the Photo-Emissionand Scanning Electron Microscopes", Atomic Energy of Canada Ltd., Prog.Rep. PR-CMa-9 (1969) p.76.
[24] B. Cox, "The Zirconium-Zirconia Interf?r" ", J. Aust. Inst. Met., 1969,
[25] J.P. Pemsler, "Diffusion of Oxygen in Zirconium and its Relation toOxidation and Corrosion", J. Electrochem. Soc, 1958, 105, 315.
[26] J.P. Pemsler, "Studies of Oxygen Gradients in Corroding ZirconiumAlloys", J. Nucl. Mat., 1962, ]_, 16.
[27] W.W. Doertfler, "A Contribution to the Mechanism of Dissolution andDiffusion of Oxygen in Zirconium", Swiss Report, EIR-82 (Sept.1965).
[28] E. Hillner, "Hydrogen Absorption in Zircaloy During Aqueous Corrosion.Effect of Environment", U.S. Report, WAPD-TM-411 (1964).
[29] N. Ramasubramanian, "Localized Electron Transport in Corroding ZirconiumAlloys", Proc. 1975 NACE Annual Meeting, Toronto, Preprint 160; andJ. Nucl. Mat., 1975, 55_, 134.
[30] S. Kass and W.W. Kirk, "Corrosion and Hydrogen Absorption Properties ofNickel-Free Zircaloy-2 and Zircaloy-4" , ASM Trans. Quart., 1962, _56_, 77.
[31] B. Cox, "Some Factors which Affect the Rate of Oxidation and HydrogenAbsorption of Zircaloy-2 in Steam", U.K. Report AERE-R4348 (July 1963).
[32] B. Cox, "Oxidation of Zirconium and its Alloys", Adv. in Corr. Sei. andTech., Eds. Fontana and Staehle, Plenum, N.Y., Vol.5 (1976) p.173.

-11-
[33] R.A. Ploc and R.D. Davidson, "Auger Electron Analysis of Oxides Grown onDilute Zirconium Alloys", Proc. of Int. Metall. Soc. Ann. Conf.,Philadelphia, July 16-19, 1984.
[34] T. Maekawa and M. Terada, "Infra-Red Spectra Study on the Oxidation Filmof Zirconium", Trans. Jap. Inst. Met., 1963, 4L, 47.
[35] A.B. Johnson, Jr., "Aqueous Corrosion and Hydriding of Zirconium Alloysin Nuclear Reactor Environments", Proc. 4th Int. Cong, on Met. Corr.,Amsterdam, Sept. 1969, NACE, p.168.
[36] A.B. Johnson Jr., J.E. LeSurf and R.A. Proebstle, "Study of ZirconiumAlloy Corrosion Parameters in the Advanced Test Reactor", Proc. of Symp.on Zirc. in Nucl. Appl., Portland, Aug. 1973, ASTM-STP-551, p.495.
[37] A.B. Johnson, Jr., "A Review of Corrosion Phenomena on Zirconium Alloys,Niobium, Titanium, Inconel, Stainless Steel and Nickel Plate under Irra-diation", Revs, on Coatings and Corro., Freund, Tel Aviv, 1975, 4_, 299.
[38] V.F. Urbanic, "Observations of Accelerated Hydriding in ZirconiumAlloys", Proc. 6th Int. Symp. on "Zirconium in the Nuclear Industry",Vancouver, 1982, ASTM-STP-824, p.554.

-12-
TABLE 1
Critical H2/0xidant Ratios for Direct Hydriding
Temperature H. Pressure Critical(*C) (atm) RatioMaterial (*C) (atm) Ratio Author Ref.
Zircaloy-2 343 6.5 x 10"2-1.0 106-108 Boyle & Kisiel 9
400 1.3 x 10 102 Shannon 7
320 1.0 105 Gibby BNWL-150 (1965)
300 .-v .„ , „ ..
Une 10400 1.0 10Z
25 0.1 102-103 .„ ._ . / Coleman & Cox 5
Zr-2.5% Nb 25 0.1 10-10

-13-
TABLE 2
Oxidation in T2/H2O Mixtures
Hydrogen Absorption
SampleTemp. Time Wt. Gain(°C) (h) (mg/dm2) ppm % Theor. Observations
GRAO Zr-2.5 wt% Nb (1) 400 66.0 15.5 14 21.2 No significanttritium uptake.
GR41 Zr-2.5 wt% Nb (2) 400 113.0 14.5 13 23.4 Small amount oftritium in oxideand metal.
GR42 Zircaloy-2 (3) 500 24.5 24.5 39 23.0 No tritiumdetected in oxideor metal.
(1) Heated at 1000°C in vac. for 1 h and quenched.
(2) Heated at 88CCC in vac. for 72 h and quenched, reheated at 500°C for 6 h.The large amount of isothermal a-phase present in this sampleprobably results from oxygen absorption during the long anneal at 880°C.
(3) Preoxidized in 0 2 at 500°C for 24 h; Aw = 28.2 mg/dm2.

TABLE 3
Comparison of H Uptake by Zirconium Alloy/Metal Couples with Physical Properties of the Coupled Metal
Metal
Cu
Fe
Inconel
30A S/S
A03 S/S
Ni
Pt
Exchange CurrentDensity for H, (1)
dog io>2
-6.70
-5.93
-5.46
-5.37
-
-5.17
-3.73
Diffusion Coeff.of B2 at 400*C
9.0 x 10"6 (4)
1.5 x 10"A (4)
8.0 x 10"7 (7)
3.0 x 10"7 (8)
5.8 x 10"5 (9)
5.0 x 10~6 (4)
5.1 x 10"3 (4)
Enthalpy ofSolution of
H2
+0.47 (A)
+0.28 (A)
-
-
-
+0.16 (A)
+0.75 (A)
Permeabilityto B, at 400*C
1 J -1 -2 -I( « (STP)aawb .cm at» '}
3.6 x 10"4 (5)
1.5 x lO"1 (6)
5.4 x 10"3 (7)
3.5 x 10~3 (6)
4.6 x 10~2 (9)
3.0 x 10"2 (6)
1.1 x 10~3 (6)
Hydrogen Uptake
Ref.37
-
-
152
-
-
-
354
Ref.38(3)
13
-
-
380
1380
3730
>10000(9)
(1) Comprehensive Treatise of Electrochemistry, Vol.2, Eds. Bockris, Conway, Yeager & White, Plenum, NY, 1981.(2) mg/kg H2 in Zircaloy-2 after 60 d coupled to metal in 300°C water under irradiation.(3) Peak H2 concentration at centre of Zr-2.5 wtZ Nb tube after 10 days in pHIO LiOH with 1.1 MPa H2 overpressure.(4) J. Vblkl and G. Alefeld, Chapter 5, Diffusion in Solids, Recent Developments, Eds. Nowick and Burton,
Ac. Press, New York, 1975.(5) G.R. Caskey et al., Corrosion, 1976, 32. (9).(6) R.W. Webb, Permeation of H through Metals, NAA-SR-10462 (1965).(7) W.M. Robertson, Met. Trans. A, 1977, 8A.(8) M.R. Louthan and R.G. Derrick, Corr. Sei., 1975, JJ>,(9) V.F. Urbanic and A.F. Coles, unpublished results, CRNL.

- 1 5 -
100
CNi
3
I v - Samples that Picked Up More than 100%of Corrosion Product H2
o - Samples that Picked Up Less than 100%of Corrosion Product H2
• - Weight Gain in Pure ^ O Vapor
<7 1 1 I I I 1 l 1 1
10 50
Time (h)
FIGURE 1. Plot showing influence of oxidation rate during exposure to H2/H2Omixtures at 400°C on hydrogen uptake (Ref.7)

320
2*0
Û CURVE I , UNOXlOlZED PLATESQ CURVES 2 , OXIOiZEO PLATESO CURVES 3 , OXIDIZED PLATES
• CURVE 4 , OXIDIZED PLATES, TREATED IN VACUO
• CURVE S, OXIDIZED PLATES, TREATED IN VACUO
}} INDICATE!. RUN INTERRUPTED AT END OF WORKING DAY
(SEE TABLE I )
6 7TIME IN HOURS
10 12
FIGURE 2. Incubation times for absorption of hydrogen gas by zirconium at 400°C (Ref.11).

-17-
500 K —
Time (days)
FIGURE 3. Plot of electrical resistance for Zircaloy-2 exposed to gaseousenvironments at 45O°C (Ref.7).

Oxide
OxygenDi ffus ionZone
aZr+Zriu
X500 Autoradiograph X500
03I
Autoradiograph in-situ
FIGURE 4. Autoradiograph showing lack of tritium absorption by a thick oxide film formed in O2 when subsequentlyexposed to T2• (Oxidized in O2 at 6OOCC for 66.5 h to weight gain of 6.84 mg/dm2; Exposed to T2 gas at800°C for 16.5 h; Réf.19).

ox ido
diffusion zone
aZr+ZrHx
^ % V ^
X500 Autoradiograph X500 Micrograph
FIGURE 5. Autoradiograph showing tritium incorporated in a thick oxide film formed in O2 when subsequentlyexposed to T2O. (Oxidized in O2 at 600°C for 66.5 h to weight gain of 727 mg/dm2; Exposed in T2Ovapour at 400°C for 72 h, additional weight gain 5 mg/dm2; Ref.19).

,*^ Î^V y*» *
*«•**•"
FIGURE 6. Scanning electron micrograph showing regions of thick and thin oxideformed on different zirconium grains together with ridges of thickoxide along grain boundaries and micro-twins, X2000 (Ref.21).

-21-
FIGURE 7• Higher magnification micrograph of junction between thick and thinoxide areas and oxide ridge, X6000 (Ref.21).

FIGURE 8. View of oxide-metal interface showing appearance of oxide ridges at grain boundaries, X3000 (Ref.24)

-23-
.'. " ~ "'''^.. '.:....'••- :.i-^..- -' • • - - - - - - J
FIGURE 9. View of oxide-metal interface showing areas of thick and thin oxidecomparable to those seen in Figure 6, X1000. Note oxide has notfractured along grain boundaries.
crack crack
(a)
crack
(b)
FIGURE 10. Oxide-metal interface of stripped oxide films showing cracks inducedby the stripping, X3000. Note these cracks do not follow oxideridges .

-24-
490
420 -
e& 3504
i 280 -ur.•-
6 2'°ç
S 140D
370
1 1 1 1 1 1 1 1 1 1
1 1
1 \
1
....I_J
1 -/l 1 1 1 1 1 1 1 1
a
i i i
2tfcotoy
* * •
t i
1 I
/
î * Siol.c
3A Or«*»
t |
9 ' •
• •
i i
i
Vacuum
1.0 2JO
Tim«, (days)
4.0
FIGURE 11. Decrease in thickness of interference-colour oxide films as afunction of annealing time in vacuo at 400°C (Ref.25).
- 25 -
(a) (b)
FIGURE 12. Oxide-metal interface of specimen after vacuum annealing at 500°C.Note break-up of oxide ridges and formation of pores. (a) X1000.(b) X3000.
(a) (b)
FIGURE 13 . Replica of zirconium oxide surface of specimen heated in a lowoxygen environment (He), showing a similar array of pores tothose seen in Figure 12. Diameter at surface is smaller than atoxide-metal Interface. (a) X4000, (b) X10 000.
60
50
A * ZlRCALOY-2,AS ROLLEDD * 2IRCALOY-2 ,/9-TREATED
_O ' ZIRCALOY-4 , AS ROLLEDO • ZIRCAL0Y-4, ^-TREATED
•~ I00-WIL THICK SPECIMENS
ZIRCALOY-2,/3-TREATED
ZIRCALOY-2AS-ROLLED
ZIRCALOY-4AS-ROLLED OR 0-TREATED
500 1000 1500 2000HYDROGEN OVERPRESSURE, PS!
2500 3000
FIGURE 14. Hydrogen pickup by Zircaloy-2 and Zircaloy-4 as a function of hydrogen overpressure a f te r 14-dayexposure in 343°C water (Ref .28) .
0 2 0R H 2 0 ADSORBED
REACTION :-—2O 2~
0 2" DIFFUSESDOWNCRYSTALLITEBOUNDARIES
ETCH PIT PRODUCEDDURING INITIAL
PICKLING
PROTONS ORELECTRONS TRAVELBY SURFACE CONDUCTION
OXYGEN , AIROR STEAM
THICK OXIDE NJ ELECTRONS^DEVELOPS COLUMNAR \ E M 1 T T E D THR0UC4CRYSTALLITE STRUCTURE \ T H , N | r O 2 LAYER,
CRYSTALLITES NUCLEATEAMORPHOUS OXIDE
Fe2O3ORFe3O4LAYER
...„,„, THIN AMORPHOUS LAYER OF ZrO2
REACTION2O2~+Zr-»ZrO2+4e
I MICRON
APPROXIMATE SCALE
MONOCLINIC
ZIRCONIUM
INTERMETALLICPARTICLE STILL INMETALLIC CONTACTWITH THE MATRIX
FIGURE 15. Schematic diagram of oxide film on Zircaloy-2 and the processes occurring in i t during oxidation(Ref.32).

ZIRCALOY - 2680*F DEGASSED H2O
NICKEL-FREE ZIRCALOY-2680'F DEGASSEO H,0
>^680*F HzO + IOOO PSIALL MATERIALS
THEORETICAL (INSTANTANEOUS)
ù, • ZIRCALOY-2 • AÏ«... I S SURVEY EXPREfEATJ O « Ni "FREE ZIRCALOY-2 • »J
D • ZIRCALOY-4
IcoI
SO 73 100 125OXYGEN WEIGHT GAIN, MG/DM 2
ISO 175 2 0 0
FIGURE 16. Effect of oxygen content of water on hydrogen uptake by Zircaloys in 360°C water (Ref.28)

-29-
O6
O-4
O2
O
V
/
/ STEAM' latm
3OO°C
% a.
ZIRCALOY-2O-OOS'thcct
•0 2 4
g.
oo.
e
o
z
IO SO 6O 7O 8OOxygen uptake
FIGURE 17. Change in hydrogen uptake ra te with oxide film thickness forZircaloy-2 (Ref .31) .

»OR
BED
.
CD
HVDR
OG
EhCA
L 1
100
9 0
80
7O
60
5 0
30
20
10
BASED ON ALLOYS CONTAINING1-5 7© ADDITIONS
1. THIS WORK.2. WE. BERRY3. J. N. WANKLYN (UNPUBLISHED DATA)
iu>oi
INl
IZn
IGo
TGeTi V Cr Mn Fe Co NL Cu
FIGURE 18. Effect of alloying elements in intermetallic form on hydrogen uptake percentage (Ref.32)

X1000 Autoradiograph X1000 Micrograph
FIGURE 19. Autoradiograph showing uniform tritium content in thin zirconia film formed in T2O for long enough tosaturate oxide (oxidized in O2 at 400°C for 66.5 h to weight gain of 12.7 mg/dm2, oxidation conti-nued in T2O at 400°C for 48 h to total weight gain of 114.4 mg/dm2). Compare with non-uniformdistribution in Figure 5 (Ref.19).

X200 Autoradiograph X200 Micrograph
FIGURE 20. Surface autoradiograph shows tritium content proportional to oxide thickness. Note that the darknessof the interference colour oxides in the micrograph is not proportional to thickness because ofcolour response of black and white film. (Oxidized in T2O at 400°C to weight gain of 1.9 mg/dm2,Ref .19).

FIGURE 21. Comparison of permeability data of T. Smith with other estimates ofhydrogen in ZrC>2 films.

FIGURE 22. Increase in deuterium concentration at the inlet ends of Zr-2.5 wt% t?b alloy pressure tubes in thePickering reactors (Ref.l).

ISSN 0067 0367
To identify individual documents in the serieswe have assigned an AECL number to each.
Please refer to the AECL numberwhen re-questing additional copies of this document
from
Scientific Document Distribution OfficeAtomic Energy of Canada Limited
Chalk River, Ontario, CanadaKOJ 1J0
ISSN 0067-0367
Pour identifier les rapports individuels fai: 3partie de cette série nous avons assignéun numéro AECL- àchacun.
Veuillez faire mention du numéro AECL- sivous demandez d'autres exemplaires de cerapport
Servicede Distribution des Documents OfficeL'Energie Atomique du Canada Limitée
Chalk River, Ontario, CanadaK0J1J0
Price $3 00 per copy Prix $3.00 par exemplaire
ATOMIC ENERGY OF CANADA LIMITED, 1985
0285-85













![9RGLþ ]D - UNESCO · 5 6$'5ä$- 8nudwnr ± ]d nrjd mh rydm 3uluxþqln"7 'ydghvhw vdymhwd ]d suh]dsrvohqh qrylqduh 10 , 3udyr qd sulvwxs lqirupdflmdpd l qrylqduvnr lvwudålydqmh 15](https://static.fdocuments.us/doc/165x107/5ec3bd8d11af562f7609d22e/9rgl-d-5-65-8nudwnr-d-nrjd-mh-rydm-3uluxqln7-ydghvhw-vdymhwd.jpg)




