
ASmallMoleculeInhibitsAktthroughDirectBindingtoAkt ... ·...
16
A Small Molecule Inhibits Akt through Direct Binding to Akt and Preventing Akt Membrane Translocation * □ S Received for publication, December 10, 2009, and in revised form, January 11, 2010 Published, JBC Papers in Press, January 12, 2010, DOI 10.1074/jbc.M109.094060 Donghwa Kim ‡1 , Mei Sun ‡1 , Lili He ‡ , Qing-Hua Zhou § , Jun Chen § , Xia-Meng Sun ‡ , Gerold Bepler ¶ , Said M. Sebti , and Jin Q. Cheng ‡2 From the Departments of ‡ Molecular Oncology, ¶ Thoracic Oncology, and Drug Discovery, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612 and the § Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Lung Cancer Institute, Tianjin Medical University General Hospital, Tianjin 300052, China The Akt pathway is frequently hyperactivated in human can- cer and functions as a cardinal nodal point for transducing extracellular and intracellular oncogenic signals and, thus, pre- sents an exciting target for molecular therapeutics. Here we report the identification of a small molecule Akt/protein kinase B inhibitor, API-1. Although API-1 is neither an ATP competi- tor nor substrate mimetic, it binds to pleckstrin homology domain of Akt and blocks Akt membrane translocation. Fur- thermore, API-1 treatment of cancer cells results in inhibition of the kinase activities and phosphorylation levels of the three members of the Akt family. In contrast, API-1 had no effects on the activities of the upstream Akt activators, phosphatidylinosi- tol 3-kinase, phosphatidylinositol-dependent kinase-1, and mTORC2. Notably, the kinase activity and phosphorylation (e.g. Thr(P) 308 and Ser(P) 473 ) levels of constitutively active Akt, including a naturally occurring mutant AKT1-E17K, were inhibited by API-1. API-1 is selective for Akt and does not inhibit the activation of protein kinase C, serum and glucocor- ticoid-inducible kinase, protein kinase A, STAT3, ERK1/2, or JNK. The inhibition of Akt by API-1 resulted in induction of cell growth arrest and apoptosis selectively in human cancer cells that harbor constitutively activated Akt. Furthermore, API-1 inhibited tumor growth in nude mice of human cancer cells in which Akt is elevated but not of those cancer cells in which it is not. These data indicate that API-1 directly inhibits Akt through binding to the Akt pleckstrin homology domain and blocking Akt membrane translocation and that API-1 has anti-tumor activity in vitro and in vivo and could be a potential anti-cancer agent for patients whose tumors express hyperactivated Akt. Akt was first described as the cellular homologue of the prod- uct of the v-akt oncogene (1), and it has three members, Akt1/ PKB, 3 Akt2/PKB, and Akt3/PKB (2–5). Activation of Akt depends on the integrity of the pleckstrin homology (PH) domain, which mediates its membrane translocation, and on the phosphorylation of Thr 308 in the activation loop and Ser 473 (6). Phosphoinositides phosphatidylinositol 3,4-diphosphate and phosphatidylinositol 3,4,5-trisphosphate, produced by PI3K, bind directly to the PH domain of Akt, driving a confor- mational change in the molecule that enables the activation loop of Akt to be phosphorylated by PDK1 at Thr 308 (6). Full activation of Akt is also associated with phosphorylation of Ser 473 within a C-terminal hydrophobic motif (6). Although the role of PDK1 on Thr 308 phosphorylation is well established, the mechanism of Ser 473 phosphorylation is controversial. A num- ber of candidate enzymes responsible for this modification have been put forward, including integrin-linked kinase (7), Akt itself, through autophosphorylation (8) and a DNA-dependent kinase (9). Recent studies indicate that the rictor-mTOR (mTORC2) complex is responsible for phosphorylation of Ser 473 (10, 11). The activity of Akt is negatively regulated by tumor suppressor PTEN, which is frequently mutated or deleted in human malignancy (12). PTEN encodes a dual-spec- ificity protein and lipid phosphatase that reduces intracellular levels of phosphatidylinositol 3,4,5-trisphosphate by convert- ing it to phosphatidylinositol 4,5-diphosphate, thereby inhibit- ing Akt membrane translocation and activation the Akt path- way (13). Akt phosphorylates and/or interacts with a number of mol- ecules to exert its cellular functions, which include roles in cell proliferation, survival, migration, and differentiation (14). Sev- eral lines of evidence demonstrate that Akt is a critical player in tumor development. Hyperactivation of the Akt pathway has been detected in up to 50% all human tumors (15, 16) and is closely associated with chemoresistance (17). Therefore, Akt has been an attracting target for anti-cancer drug discovery (17). A recent study identified a recurring somatic mutation within the PH domain of AKT1 in human breast, colorectal, and ovarian cancers that results in a glutamic acid to lysine * This work was supported, in whole or in part, by National Institutes of Health Grants CA107078, CA137041, and P50 CA119997. This work was also sup- ported by Department of Defense Grant: OC073222, Bankhead-Coley Bridge Grant 09BB-05 (to J. Q. C. and G. B.), National Natural Science Foun- dation of China Grant 30430300, National High Technology Joint Research Program of China Grant 2006DFB32330, and Hi-tech Research and Devel- opment Program of China Grant 2006AA02401 (to Q.-H. Z.). □ S The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1. 1 Both authors contributed equally to the work. 2 To whom correspondence should be addressed, at Molecular Oncology, H. Lee Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL 33612. Tel.: 813-745-6915; Fax: 813-745-3829; E-mail: [email protected]. 3 The abbreviations used are: PKB, PKA, PKC, protein kinase B, C, and A, respec- tively; PI3K, phosphatidylinositol 3-kinase; PH, pleckstrin homology; myr, myristoylation; API-2 (TCN), Akt/PKB signaling inhibitor-2; HA, hemagglu- tinin; GFP, green fluorescent protein; PDK1, phosphatidylinositol-depen- dent kinase-1; GST, glutathione S-transferase; KD, kinase domain; CT, C terminus; EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; WT, wild type; SGK, serum and glu- cocorticoid-inducible kinase. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 285, NO. 11, pp. 8383–8394, March 12, 2010 © 2010 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8383 at Cadmus Professional Communications on September 20, 2016 http://www.jbc.org/ Downloaded from at Cadmus Professional Communications on September 20, 2016 http://www.jbc.org/ Downloaded from at Cadmus Professional Communications on September 20, 2016 http://www.jbc.org/ Downloaded from This article has been withdrawn by the authors. The same data were used to represent different experimental conditions. Specifically, part of the p- PKA immunoblot from Fig. 4D was reused as part of the p-Akt-T308 immunoblot from the same figure. The authors state that this image reuse does not affect the overall conclusions of the study. by guest on June 11, 2018 http://www.jbc.org/ Downloaded from by guest on June 11, 2018 http://www.jbc.org/ Downloaded from by guest on June 11, 2018 http://www.jbc.org/ Downloaded from
Transcript of ASmallMoleculeInhibitsAktthroughDirectBindingtoAkt ... ·...

A Small Molecule Inhibits Akt through Direct Binding to Aktand Preventing Akt Membrane Translocation*□S
Received for publication, December 10, 2009, and in revised form, January 11, 2010 Published, JBC Papers in Press, January 12, 2010, DOI 10.1074/jbc.M109.094060
Donghwa Kim‡1, Mei Sun‡1, Lili He‡, Qing-Hua Zhou§, Jun Chen§, Xia-Meng Sun‡, Gerold Bepler¶, Said M. Sebti�,and Jin Q. Cheng‡2
From the Departments of ‡Molecular Oncology, ¶Thoracic Oncology, and �Drug Discovery, H. Lee Moffitt Cancer Center andResearch Institute, Tampa, Florida 33612 and the §Tianjin Key Laboratory of Lung Cancer Metastasis and TumorMicroenvironment, Lung Cancer Institute, Tianjin Medical University General Hospital, Tianjin 300052, China
The Akt pathway is frequently hyperactivated in human can-cer and functions as a cardinal nodal point for transducingextracellular and intracellular oncogenic signals and, thus, pre-sents an exciting target for molecular therapeutics. Here wereport the identification of a small molecule Akt/protein kinaseB inhibitor, API-1. Although API-1 is neither an ATP competi-tor nor substrate mimetic, it binds to pleckstrin homologydomain of Akt and blocks Akt membrane translocation. Fur-thermore, API-1 treatment of cancer cells results in inhibitionof the kinase activities and phosphorylation levels of the threemembers of the Akt family. In contrast, API-1 had no effects onthe activities of the upstreamAkt activators, phosphatidylinosi-tol 3-kinase, phosphatidylinositol-dependent kinase-1, andmTORC2.Notably, the kinase activity and phosphorylation (e.g.Thr(P)308 and Ser(P)473) levels of constitutively active Akt,including a naturally occurring mutant AKT1-E17K, wereinhibited by API-1. API-1 is selective for Akt and does notinhibit the activation of protein kinase C, serum and glucocor-ticoid-inducible kinase, protein kinase A, STAT3, ERK1/2, orJNK. The inhibition of Akt byAPI-1 resulted in induction of cellgrowth arrest and apoptosis selectively in human cancer cellsthat harbor constitutively activated Akt. Furthermore, API-1inhibited tumor growth in nude mice of human cancer cells inwhich Akt is elevated but not of those cancer cells in which it isnot. These data indicate thatAPI-1directly inhibitsAkt throughbinding to the Akt pleckstrin homology domain and blockingAkt membrane translocation and that API-1 has anti-tumoractivity in vitro and in vivo and could be a potential anti-canceragent for patients whose tumors express hyperactivated Akt.
Aktwas first described as the cellular homologue of the prod-uct of the v-akt oncogene (1), and it has three members, Akt1/
PKB�,3 Akt2/PKB�, and Akt3/PKB� (2–5). Activation of Aktdepends on the integrity of the pleckstrin homology (PH)domain, which mediates its membrane translocation, and onthe phosphorylation of Thr308 in the activation loop and Ser473(6). Phosphoinositides phosphatidylinositol 3,4-diphosphateand phosphatidylinositol 3,4,5-trisphosphate, produced byPI3K, bind directly to the PH domain of Akt, driving a confor-mational change in the molecule that enables the activationloop of Akt to be phosphorylated by PDK1 at Thr308 (6). Fullactivation of Akt is also associated with phosphorylation ofSer473 within aC-terminal hydrophobicmotif (6). Although therole of PDK1 on Thr308 phosphorylation is well established, themechanism of Ser473 phosphorylation is controversial. A num-ber of candidate enzymes responsible for thismodification havebeen put forward, including integrin-linked kinase (7), Aktitself, through autophosphorylation (8) and a DNA-dependentkinase (9). Recent studies indicate that the rictor-mTOR(mTORC2) complex is responsible for phosphorylation ofSer473 (10, 11). The activity of Akt is negatively regulated bytumor suppressor PTEN, which is frequently mutated ordeleted in humanmalignancy (12). PTEN encodes a dual-spec-ificity protein and lipid phosphatase that reduces intracellularlevels of phosphatidylinositol 3,4,5-trisphosphate by convert-ing it to phosphatidylinositol 4,5-diphosphate, thereby inhibit-ing Akt membrane translocation and activation the Akt path-way (13).Akt phosphorylates and/or interacts with a number of mol-
ecules to exert its cellular functions, which include roles in cellproliferation, survival, migration, and differentiation (14). Sev-eral lines of evidence demonstrate that Akt is a critical player intumor development. Hyperactivation of the Akt pathway hasbeen detected in up to 50% all human tumors (15, 16) and isclosely associated with chemoresistance (17). Therefore, Akthas been an attracting target for anti-cancer drug discovery(17). A recent study identified a recurring somatic mutationwithin the PH domain of AKT1 in human breast, colorectal,and ovarian cancers that results in a glutamic acid to lysine
* This work was supported, in whole or in part, by National Institutes of HealthGrants CA107078, CA137041, and P50 CA119997. This work was also sup-ported by Department of Defense Grant: OC073222, Bankhead-ColeyBridge Grant 09BB-05 (to J. Q. C. and G. B.), National Natural Science Foun-dation of China Grant 30430300, National High Technology Joint ResearchProgram of China Grant 2006DFB32330, and Hi-tech Research and Devel-opment Program of China Grant 2006AA02401 (to Q.-H. Z.).
□S The on-line version of this article (available at http://www.jbc.org) containssupplemental Fig. 1.
1 Both authors contributed equally to the work.2 To whom correspondence should be addressed, at Molecular Oncology, H.
Lee Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL 33612. Tel.:813-745-6915; Fax: 813-745-3829; E-mail: [email protected].
3 The abbreviations used are: PKB, PKA, PKC, protein kinase B, C, and A, respec-tively; PI3K, phosphatidylinositol 3-kinase; PH, pleckstrin homology; myr,myristoylation; API-2 (TCN), Akt/PKB signaling inhibitor-2; HA, hemagglu-tinin; GFP, green fluorescent protein; PDK1, phosphatidylinositol-depen-dent kinase-1; GST, glutathione S-transferase; KD, kinase domain; CT, Cterminus; EGF, epidermal growth factor; ERK, extracellular signal-regulatedkinase; JNK, c-Jun N-terminal kinase; WT, wild type; SGK, serum and glu-cocorticoid-inducible kinase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 285, NO. 11, pp. 8383–8394, March 12, 2010© 2010 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8383
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
at C
admus Professional C
omm
unications on September 20, 2016
http://ww
w.jbc.org/
Dow
nloaded from
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
This article has been withdrawn by the authors. The same data were used to represent different experimental conditions. Specifically, part of the p-
PKA immunoblot from Fig. 4D was reused as part of the p-Akt-T308 immunoblot from the same figure. The authors state that this image
reuse does not affect the overall conclusions of the study.
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018
http://ww
w.jbc.org/
Dow
nloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
substitution at amino acid 17 (E17K) in the lipid binding pocket(18). Lys-17 alters the electrostatic interactions of the pocketand forms newhydrogen bondswith a phosphoinositide ligand.This mutation activates AKT1 through aberrant pathologicallocalization to the plasma membrane, transforms cells, andinduces leukemia in mice (18). Furthermore, the E17K substi-tution reduces the sensitivity to an allosteric Akt kinase inhib-itor (18).In the present report we identified a small molecule Akt
inhibitor, API-1, by screening the compound librariesobtained from NCI/Developmental Therapeutics ProgramOpen Chemical Repository, National Institutes of Health(NCI/DTP) using a cell-based assay. API-1 binds to the AktPH domain and inhibits Akt membrane translocation, whichleads to inhibition of Akt-regulated cell growth and cell sur-vival. In a xenograft nude mouse model, API-1 inhibitsgrowth of tumors with hyperactivated Akt but not in thosewith low levels of phospho-Akt.
EXPERIMENTAL PROCEDURES
Cell Lines, Compounds, and Plasmids—All cell lines used inthis study were either purchased from the ATCC or describedpreviously (19–21). All 2300 compounds were from the NCI/DTP Open Chemical Repository (nci.nih.gov). HA-taggedAkt1, AKT2, and AKT3 expression plasmids have beendescribed previously (21). Wild-type human AKT1 construct
was created by reverse transcription-PCR usingMCF10A RNAas the template. The PCR products were cloned to BamH1-EcoRI sites of pCMV-Myc-Tag2 vector (Stratagene). TheAKT1 primers used for PCR were: forward, 5�-ATGAGC-GACGTGGCTATTGTGAAGG-3�, and reverse, 5�-CTCGC-CCCCGTTGGCGTACTCC-3�. AKT1-E17K plasmid was ob-tained by converting G to A at nucleotide 49 of wild-type AKT1using the QuikChange site-directed mutagenesis kit (Strat-agene). GFP-Akt and GFP-PH domain expression plasmidswere created by ligation of Akt and Akt-PH cDNAs intopEGFP-C1 vector (Clontech).Screening for Inhibition of Akt-transformed Cell Growth—
AKT2-transformed NIH3T3 cells or LXSN vector-transfectedNIH3T3 control cells (19) were plated into a 96-well tissue cul-ture plate. After treatment with 5 �M concentrations of eachcompound, cell growth was detected with a CellTier 96 OneSolution Cell Proliferation kit (Promega). Compounds thatinhibit growth in AKT2-transformed but not LXSN-trans-fected NIH3T3 cells were considered as candidates of Aktinhibitors and subjected to further analysis.In Vitro Protein Kinase andApoptosis Assays—In vitro kinase
was performed as previously described (20, 21). Apoptosis wasdetected with annexin V (BD Biosciences), which was performedaccording to manufacturer’s instruction. Recombinant Akt andPDK1 were purchased fromUpstate Biotechnology, Inc.
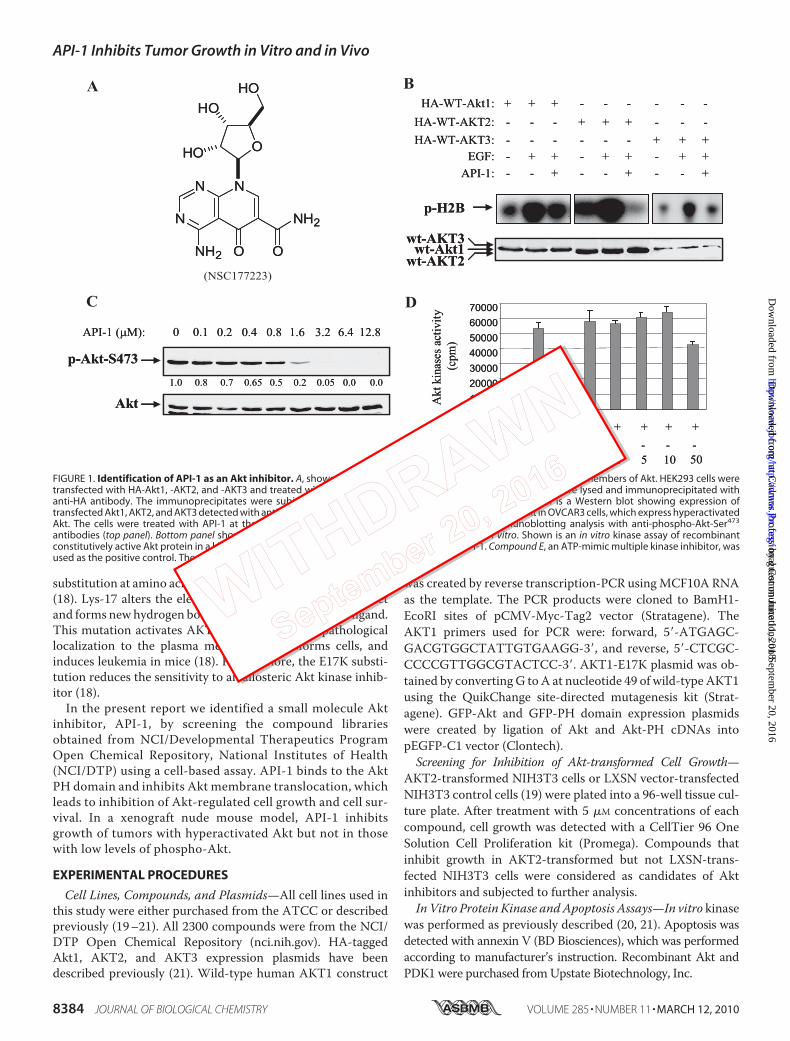
FIGURE 1. Identification of API-1 as an Akt inhibitor. A, shown is the chemical structure of API-1. B, API-1 inhibits three members of Akt. HEK293 cells weretransfected with HA-Akt1, -AKT2, and -AKT3 and treated with API-1 (10 �M) before EGF stimulation, and the cells were lysed and immunoprecipitated withanti-HA antibody. The immunoprecipitates were subjected to an in vitro kinase assay (top). The bottom panel is a Western blot showing expression oftransfected Akt1, AKT2, and AKT3 detected with anti-HA antibody. C, API-1 inhibits phosphorylation levels of Akt in OVCAR3 cells, which express hyperactivatedAkt. The cells were treated with API-1 at the indicated concentrations for 2 h and subjected to immunoblotting analysis with anti-phospho-Akt-Ser473
antibodies (top panel). Bottom panel shows expression of total Akt. D, API-1 does not inhibit Akt in vitro. Shown is an in vitro kinase assay of recombinantconstitutively active Akt protein in a kinase buffer containing the indicated concentrations of API-1. Compound E, an ATP-mimic multiple kinase inhibitor, wasused as the positive control. The experiment was repeated three times.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
8384 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 11 • MARCH 12, 2010
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
API-1 and Akt Protein Binding Assay—The assay for API-1binding to Akt was performed essentially as previouslydescribed for other kinase inhibitors that contain an aminogroup (22–24). API-1was immobilized on Sepharose beads (GEHealthcare) through covalent linkage using its amino group(Fig. 1A). Briefly, NHS-activated Sepharose (1 ml) was equili-brated in DMSO and then incubated with 1 mM API-1 and 100mM triethylamine (the ratio of volumes for coupling solution/Sepharose beads is 0.5:1). The coupling reaction was allowed toproceed on an end-over-end shaker for 16 h. Free NHS groupswere blocked with 0.8 M aminoethanol and then alternatedwashing with two buffers (0.1 M Tris-HCl, pH 8.0, and 0.1 M
acetate, 0.5 M NaCl, pH 4.0) (22, 23). The coupled affinitySepharose beads were incubated with 400 ng of recombinantAkt1 (Upstate Biotechnology) or GST fusion proteins (e.g.GST-PH, GST-KD (kinase domain), or GST-CT (C terminus)
of Akt) overnight at 4 °C in buffer containing 50 mM Tris-HCl,pH 7.5, 50, 100, or 150 mM NaCl, 0.2% Nonidet P-40, 5% glyc-erol, 1.5 mM MgCl2, 25 mM NaF, 1 mM Na3VO4, 1 mM phenyl-methylsulfonyl fluoride, 1 mM dithiothreitol, 2 �g/ml leupep-tin, 2 �g/ml aprotinin. Subsequently, the beads were washedwith the same buffer for 4 times and eluted by heat-denaturingwith the sample buffer. Binding protein was separated by 10%SDS-PAGE and immunoblotted with anti-Akt1 and -GST anti-bodies. NHS-activated Sepharose beads coupling with unre-lated compound (BMS-354825) was used as a negative controland compound E (a pan-kinase inhibitor) as a positive control.Both compounds contain an amino group.Anti-tumor Activity in the Nude Mouse Tumor Xenograft
Model—Tumor cells were harvested, resuspended in phos-phate-buffered saline, and injected subcutaneously into theright and left flanks (2 � 106 cells/flank) of 8-week-old female
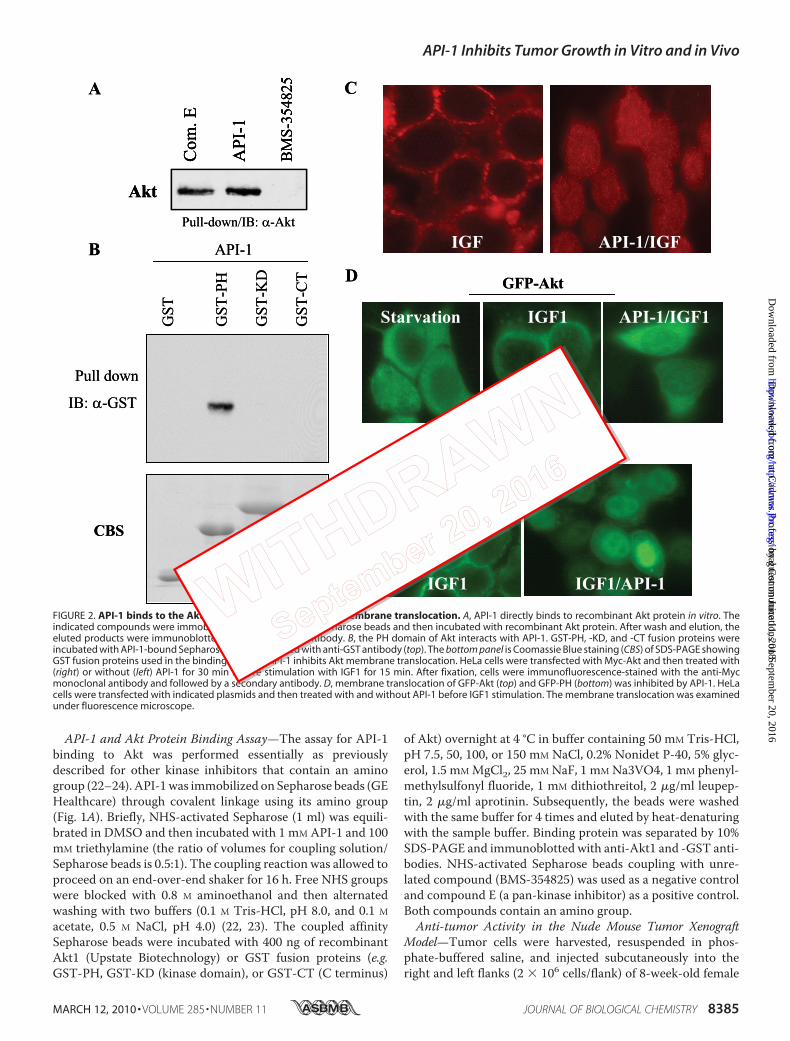
FIGURE 2. API-1 binds to the Akt PH domain and inhibits Akt membrane translocation. A, API-1 directly binds to recombinant Akt protein in vitro. Theindicated compounds were immobilized on NHS-activated Sepharose beads and then incubated with recombinant Akt protein. After wash and elution, theeluted products were immunoblotted (IB) with anti-Akt antibody. B, the PH domain of Akt interacts with API-1. GST-PH, -KD, and -CT fusion proteins wereincubated with API-1-bound Sepharose and then blotted with anti-GST antibody (top). The bottom panel is Coomassie Blue staining (CBS) of SDS-PAGE showingGST fusion proteins used in the binding assay. C, API-1 inhibits Akt membrane translocation. HeLa cells were transfected with Myc-Akt and then treated with(right) or without (left) API-1 for 30 min before stimulation with IGF1 for 15 min. After fixation, cells were immunofluorescence-stained with the anti-Mycmonoclonal antibody and followed by a secondary antibody. D, membrane translocation of GFP-Akt (top) and GFP-PH (bottom) was inhibited by API-1. HeLacells were transfected with indicated plasmids and then treated with and without API-1 before IGF1 stimulation. The membrane translocation was examinedunder fluorescence microscope.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8385
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
nude mice as reported previously (21). When tumors reachedabout 100 mm3, animals were randomized and dosed intraper-itoneal with vehicle or drug daily. Control animals receivedDMSO (20%), and treated animals were injected intraperitone-al with API-1 (10 mg/kg/day) in 20% DMSO.
RESULTS AND DISCUSSIONS
Identification of a Small Molecule Akt/PKB Inhibitor-1(API-1)—The fact that aberrant activation of the Akt pathwayoccurs in almost 50% all the human malignancy (15, 16) andinhibition of Akt induces cell growth arrest and apoptosisprompted industry and academia to develop Akt inhibitors asanti-cancer drugs (25, 26). Although several Akt inhibitors havebeen reported, many lack anti-tumor activity in vivo. A lipid-based non-selective Akt inhibitor, perifosine, has been evalu-
ated in phase I and II studies (27, 28). However, in neither studywas modulation of Akt assessed. A recent phase II study ofperifosine in pancreatic cancer was terminated as a result ofunacceptable adverse events during the first stage (29).Therefore, there is an unmet need to develop potent andselective Akt inhibitors that are void of inhibiting otherkinase activities with minimal adverse effect. To identifysmall a molecule inhibitor(s) of Akt, we have evaluated 2300compounds from the NCI/DTP Open Chemical Repositoryfor agents capable of inhibition of growth of AKT2-trans-formed but not empty vector LXSN-transfected NIH3T3cells as described under “Experimental Procedures.” Tripleexperiments showed that 32 compounds inhibited growthonly in AKT2-transformed cells. We previously character-ized one of them, named API-2/triciribine, which is a pan-
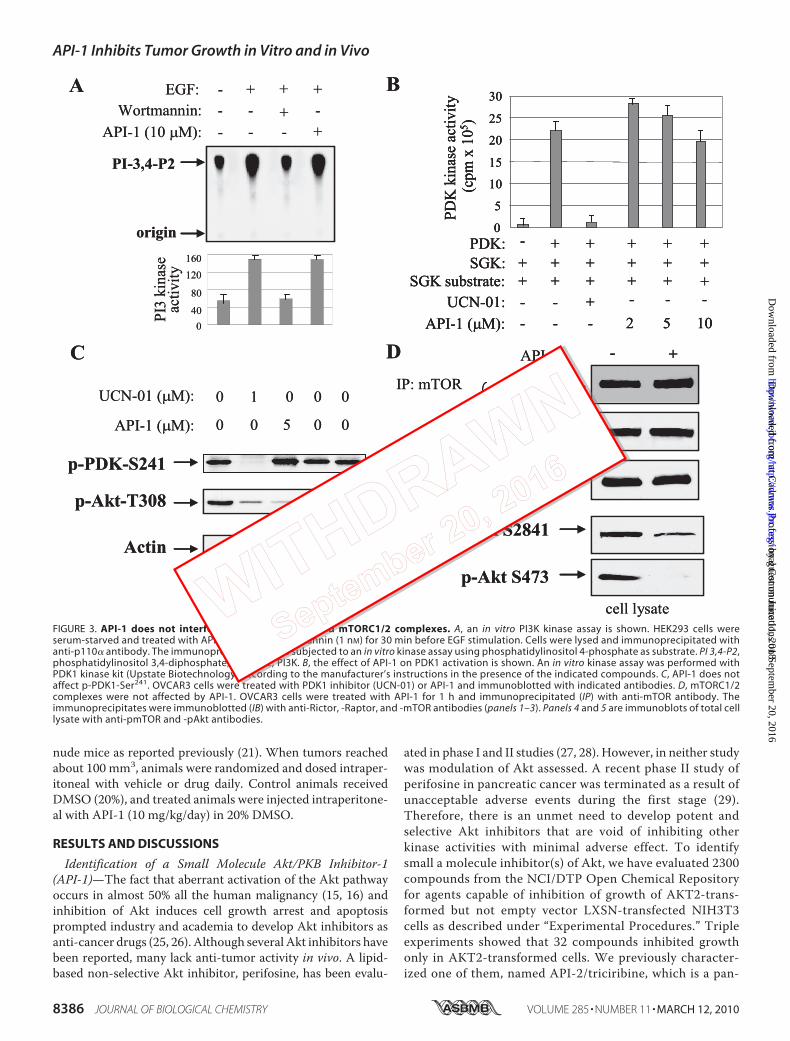
FIGURE 3. API-1 does not interfere with PI3K, PDK1, and mTORC1/2 complexes. A, an in vitro PI3K kinase assay is shown. HEK293 cells wereserum-starved and treated with API-1 (10 �M) or wortmannin (1 nM) for 30 min before EGF stimulation. Cells were lysed and immunoprecipitated withanti-p110� antibody. The immunoprecipitates were subjected to an in vitro kinase assay using phosphatidylinositol 4-phosphate as substrate. PI 3,4-P2,phosphatidylinositol 3,4-diphosphate; PI kinase, PI3K. B, the effect of API-1 on PDK1 activation is shown. An in vitro kinase assay was performed withPDK1 kinase kit (Upstate Biotechnology) according to the manufacturer’s instructions in the presence of the indicated compounds. C, API-1 does notaffect p-PDK1-Ser241. OVCAR3 cells were treated with PDK1 inhibitor (UCN-01) or API-1 and immunoblotted with indicated antibodies. D, mTORC1/2complexes were not affected by API-1. OVCAR3 cells were treated with API-1 for 1 h and immunoprecipitated (IP) with anti-mTOR antibody. Theimmunoprecipitates were immunoblotted (IB) with anti-Rictor, -Raptor, and -mTOR antibodies (panels 1–3). Panels 4 and 5 are immunoblots of total celllysate with anti-pmTOR and -pAkt antibodies.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
8386 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 11 • MARCH 12, 2010
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
Akt inhibitor, with anti-tumor activity in vitro and in vivoand currently in phase I clinic trial (21).In the present study we characterized a secondAkt inhibitor,
API-1. API-1 specifically inhibits the kinase activity and phos-phorylation (Thr(P)308 and Ser(P)473) levels of Akt in livingcells. Fig. 1A shows the chemical structure of API-1 (CancerChemotherapy National Service Center (NSC) 177223;pyrido[2,3-d]pyrimidines), which is structurally related to theantibiotic sangivamycin (30). Although the sangivamycin hasbeen shown to have anti-tumor activity (31–33), NSC 177223/API-1 has not been tested in cancer cells including NCI 60 celllines (nih.gov). Because API-1 inhibited AKT2-transformedcells over untransformed parental cells, we first examinedwhether API-1 is an inhibitor of AKT2 kinase and whether italso inhibits the other two members of Akt family. HEK293cells, which are commonly used to robustly express the proteinof interest, were transfected with HA-tagged wild-type Akt1,AKT2, and AKT3. After serum starvation overnight, cells weretreated with API-1 for 60 min before EGF stimulation andimmunoprecipitated with anti-HA antibody. The immunopre-cipitates were subjected to in vitro kinase assay. Fig. 1B showsthat API-1 inhibited EGF-induced kinase activity of Akt1,AKT2, and AKT3.We next examined if API-1 decreases phospho-Akt levels in
living cells. OVCAR3 cells, which express elevated levels of
phospho-Akt, were treatedwith different doses ofAPI-1 for 3 h.Immunoblotting analysis with anti-phospho-Akt-S473 anti-body showed that API-1 efficiently reduced the phosphoryla-tion levels of Akt with an IC50 of �0.8 �M. However, total Aktlevels were not changed (Fig. 1C). Furthermore, we examined ifAPI-1 directly inhibitsAkt kinase activity in vitro. Recombinantconstitutively active Akt protein was incubated with Akt/SGKsubstrate peptide (Upstate Biotechnology) in a kinase buffercontaining different amounts of API-1 and compound E, a pan-kinase ATP-competitive inhibitor as positive control. Tripleexperiments showed that API-1 did not reduce in vitro Aktkinase activity at concentrations that inhibit pAkt in cell cul-ture, whereas a high dose (e.g. 50 �M) of API-1 decreased Aktactivity about 20% (Fig. 1D), suggesting that API-1 functionsneither as ATP nor substrate competitor.API-1 Directly Binds to Akt Protein and Inhibits Akt Mem-
brane Translocation—To explore the mechanism by whichAPI-1 inhibits Akt, we performed a protein kinase-compoundbinding assay because API-1 contains an amino group that hasbeen shown to bind toNHS-activated Sepharose (22–24). Afterimmobilization, compound-bound Sepharose beadswere incu-bated with recombinant Akt protein. After washing and elu-tion, the products were immunoblotted with an anti-Akt anti-body. Fig. 2A shows that API-1 and compound E (positivecontrol), but not BMS-354825 (negative control), pulled down
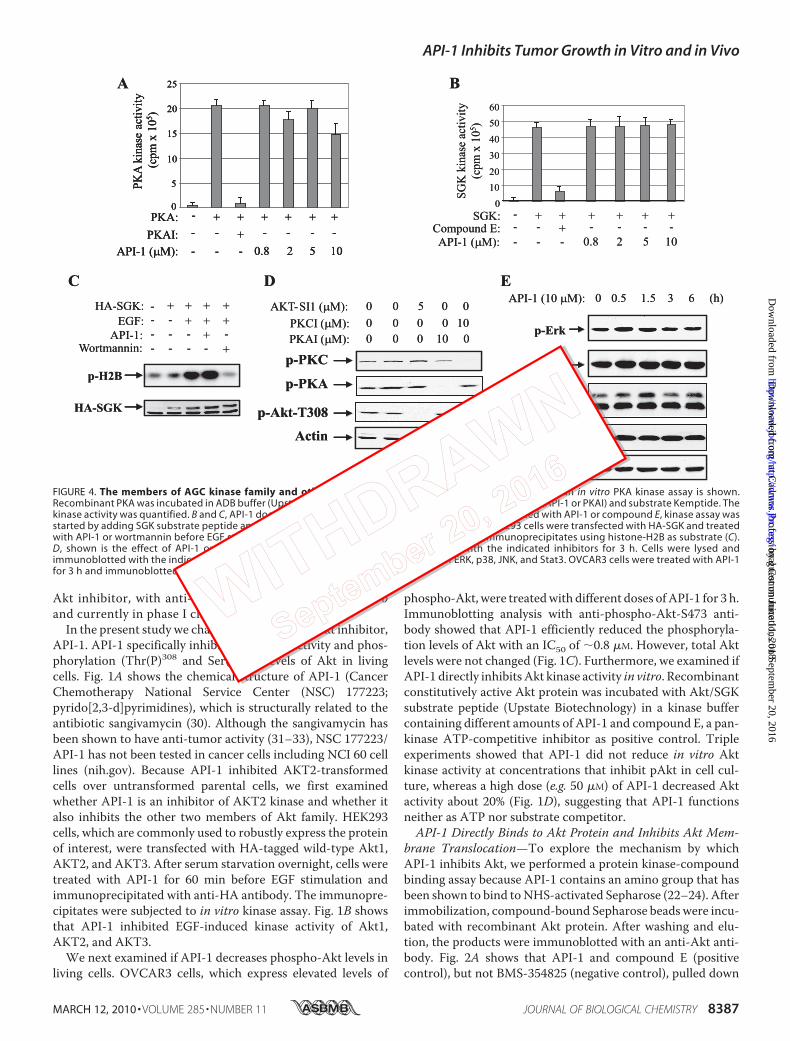
FIGURE 4. The members of AGC kinase family and other signal molecules are not affected by API-1. A, an in vitro PKA kinase assay is shown.Recombinant PKA was incubated in ADB buffer (Upstate Biotechnology) containing the indicated inhibitors (API-1 or PKAI) and substrate Kemptide. Thekinase activity was quantified. B and C, API-1 does not inhibit SGK. Recombinant SGK protein was incubated with API-1 or compound E, kinase assay wasstarted by adding SGK substrate peptide and [�-32P]ATP. The kinase activity was quantified (B). HEK293 cells were transfected with HA-SGK and treatedwith API-1 or wortmannin before EGF stimulation. In vitro kinase was performed with HA-SGK immunoprecipitates using histone-H2B as substrate (C).D, shown is the effect of API-1 on pPKC and pPKA levels. OVCAR3 cells were treated with the indicated inhibitors for 3 h. Cells were lysed andimmunoblotted with the indicated antibodies. E, effects of API-1 on phosphorylation of ERK, p38, JNK, and Stat3. OVCAR3 cells were treated with API-1for 3 h and immunoblotted with the indicated antibodies.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8387
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
API-1 Inhibits Tumor Growth in Vitro and in Vivo
8388 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 11 • MARCH 12, 2010
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Akt. To define the domain(s) of Akt that interacts with API-1,we generated GST-PH, -KD, and -CT fusion proteins and incu-bated them with API-1. Immunoblotting analysis of the elutedproducts revealed that the PHdomain of Akt bound toAPI-1 ina buffer containing 50, 100, or 150 mM NaCl (Fig. 2B).
Because Akt activation is initiated by PH domain binding tophosphatidylinositol 3,4,5-trisphosphate in the cell membrane,we reasoned that API-1 inhibits Akt through blockage of itsmembrane translocation. To this end, HeLa cells, which arecommonly used for immunofluorescence staining, were trans-fectedwithMyc-Akt and then treatedwith orwithoutAPI-1 for30 min before stimulation with IGF1. Immunofluorescencestaining revealed that Akt membrane translocation induced byIGF1was abrogated by API-1 (Fig. 2C). To further demonstrateAPI-1 inhibition of Akt through targeting the PH domain, wetransfected HeLa cells with the GFP-Akt and GFP-PH domainof Akt. Before stimulation with IGF1, cells were treated withand without API-1 for 1 h and examined under a fluorescencemicroscope. Fig. 2D shows that IGF1-induced GFP-Akt, andGFP-PH membrane translocation was largely attenuated byAPI-1. Moreover, endogenous AKT2 membrane translocationinduced by EGF was also inhibited by API-1 (supplemental Fig.S1). Collectively, these findings indicateAPI-1 inhibition of Aktthrough binding to theAkt-PHdomain and blockingAktmem-brane translocation.API-1 Does Not Inhibit Upstream Activators of Akt—Akt is
activated by extracellular stimuli and intracellular signal mole-cules through a PI3K-dependent manner. Activation of PI3Kwill activate PDK1 leading to induction of Akt kinase activity.Therefore, API-1 inhibition of Akt could result from targetingupstream regulators of Akt, such as PI3K and PDK1. To thisend, we examined if API-1 inhibits PI3K and/or PDK1.HEK293cells were serum-starved and then treated with API-1 or PI3Kinhibitor, wortmannin, for 1 h before EGF stimulation. PI3Kwas immunoprecipitated with anti-p110� antibody. The im-munoprecipitates were subjected to in vitro PI3K kinase assayusing phosphatidylinositol 4-phosphate as a substrate. Asshown in Fig. 3A, the EGF-induced PI3K activity was inhibitedby wortmannin but not by API-1.To evaluate the effect of API-1 on PDK1, we performed an in
vitro PDK1 kinase assay using SGK as a readout (Upstate Bio-technology). Unlike PDK1 inhibitor UCN-01 (34), API-1 hadno effect on in vitro PDK1 kinase activity (Fig. 3B). To furtherevaluate the effect of API-1 on PDK1 activation in living cells,we examined the phosphorylation level of PDK1-Ser241, a resi-due that is autophosphorylated and is critical for its activation
(35). After API-1 treatment of OVCAR3 cells, immunoblottinganalysis shows that phosphorylation levels of PDK1were inhib-ited by UCN-01 but not API-1 (Fig. 3C).In addition to PDK1, Rictor-mTOR (mTORC2) complex
activates Akt by phosphorylation of Ser473, whereas Raptor-mTOR (mTORC1) negatively regulates Akt by phosphoryla-tion of IRS1 (10, 37). Because API-1 did not inhibit PDK1, wenext examined whether mTORC1 and mTORC2 complexesare affected by API-1. Co-immunoprecipitation and immuno-blotting experiments show that API-1 had no effects on theinteraction of Rictor-mTOR and Raptor-mTOR, whereas itinhibits phospho-mTOR-S2841 (Fig. 3D), which is resultedfrom inhibition of Akt-TSC2-Rheb-mTOR cascade (11). Thesefinding further suggest that API-1 directly inhibits Akt.API-1 Is Selective for the Akt over the AGC Kinase Members
PKA, PKC, and SGK and Other Signaling Molecules ERK, JNK,p38, and STAT3—In addition to Akt, the AGC (PKA/PKG/PKC) kinase family also includes PKA, PKC, SGK, p90 ribosomalS6 kinase, p70S6K,mitogen- and stress-activated protein kinase,and PKC-related kinase. The protein structures of PKA, PKC,and SGK are much closer to Akt kinase than other members.Therefore, we next examined the effects of API-1 on the enzy-matic activities of these three kinases. In vitro PKA and SGKkinase assays were performed by preincubation of increasingdoses of API-1 or the indicated PKA and SGK inhibitors withrecombinant PKA or SGK protein for 30 min before addingKemptide (Leu-Arg-Arg-Ala-Ser-Leu-Gly) or SGK substratepeptide and [�-32P]ATP. Fig. 4, A and B, show that the kinaseactivities of PKA and SGK were inhibited by PKAI and com-pound E, respectively, but not by API-1. In addition, we carriedout an in vitro SGK kinase assay in HEK293 cells, which weretransfected with HA-tagged SGK, which showed that EGF-in-duced SGK kinase activity was attenuated by wortmannin butnot API-1 (Fig. 4C). To evaluate the effect of API-1 on the PKCand PKA activation in living cells, OVCAR3 cells were treatedwith the indicated doses of API-1 or a specific inhibitor of PKCand PKA, and immunoblotting analysis revealed that phospho-rylation levels of PKC and PKA were not inhibited by API-1(Fig. 4D).To determine whether API-1 has effects on other oncogenic
survival pathways, OVCAR3 cells were treated with API-1 (10�M) for different times and immunoblotted with commerciallyavailable anti-phospho-antibodies. We did not observe detect-able changes of phosphorylation levels of Stat3, JNK, p38, andERK1/2 after API-1 treatment (Fig. 4E). These data suggest thatAPI-1 is a specific Akt inhibitor.
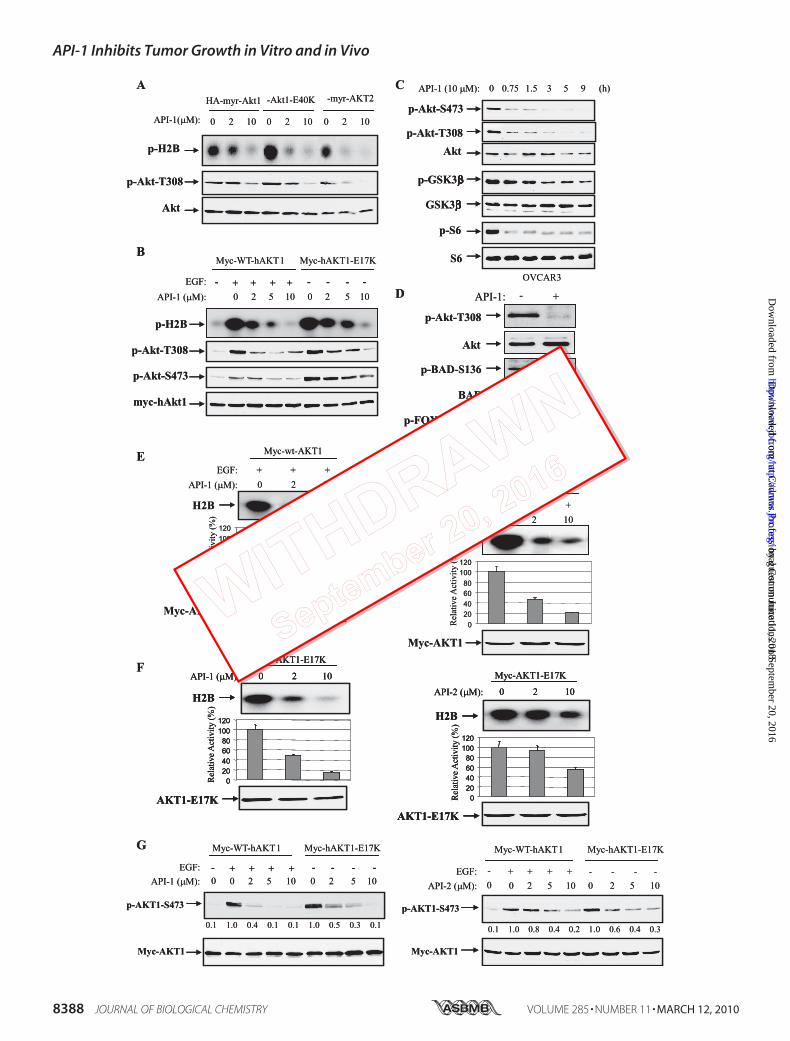
FIGURE 5. API-1 inhibits constitutively active Akt and its downstream targets. A, API-1 inhibited constitutively active Akt. HEK293 cells were transfectedwith HA-myr-Akt1, HA-Akt1-E40K, and HA-myr-AKT2. After treatment with API-1 for 1 h, cells were lysed and immunoprecipitated with anti-HA antibody. Theimmunoprecipitates were subjected to in vitro kinase assay (top) and immunoblotting with the indicated antibodies (middle and bottom panels). Note that thesecond lane from the left (lane 2) shows no significant change of pAkt as compared with lane 1 (middle panel), which is due to higher expression of myr-Akt1 inthe transfection (bottom panel). B, kinase activity and phospho-Thr308 and -Ser473 of AKT1-E17K were inhibited by API-1. Myc-tagged human WT AKT1 andAKT1-E17K were introduced into HEK293 cells. After serum starvation overnight, the cells were treated with API-1 at indicated concentration for 1 h. After EGFstimulation of WT-AKT1-transfected cells for 30 min, the cells were subjected in vitro kinase and immunoblotting analysis as described in panel A. C and D, API-1inhibited downstream targets of Akt. OVCAR3 and H661 cells were treated with API-1 (10 �M) and immunoblotted with the indicated antibodies. GSK3�,glycogen synthase kinase 3. E–G, API-1 is more potent than API-2. HEK293 cells were transfected with wild-type Myc-AKT1 (E) and constitutively activemyc-AKT1-E17K (F). After 36 h incubation, cells were serum-starved overnight. WT-AKT1-transfected cells were treated with API-1 (left) or API-2 (right) for 30 minand subsequently stimulated with EGF for 15 min. Immunoprecipitation was carried out with anti-myc antibody, and the immunoprecipitates were subjectedto in vitro kinase assay (top). Akt kinase activity was quantified (middle). The bottom panels show expression of transfected plasmids. The experiments wererepeated three times. Shown is a Western blot analysis of pAkt (G) in HEK293 cells that were transfected with Myc-AKT1 and Myc-AKT1-E17K and treated withor without API-1 or API-2 at the indicated doses for 1 h.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8389
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

API-1 Inhibits Tumor Growth in Vitro and in Vivo
8390 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 11 • MARCH 12, 2010
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

API-1 Inhibits Constitutively Active Akt, Including NaturallyOccurring Mutant AKT1-E17K and Its Downstream Targets—BecauseAPI-1 abrogates kinase activity and phosphorylation ofAkt in living cells and binds to the PH domain of Akt, weassumed API-1 may inhibit Akt1E40K and Akt1E17K but notMyr-Akt as Glu40 and Glu17 locate in the PH domain, whereasmyristoylation (myr) could directly bind to membrane. To thisend we transfected HEK293 cells with HA-tagged myr-Akt1,myr-AKT2, and Akt1E40K and myc-tagged AKT1E17K. Afterserum starvation overnight, cells were treated with or withoutAPI-1. Akts were immunoprecipitated with anti-HA or anti-myc antibody. The immunoprecipitates were subjected to invitro kinase assay and immunoblotting analysis with anti-phos-pho-Akt-Thr308 antibody. Unexpectedly, API-1 inhibited allfour forms of constitutively activeAkt (Fig. 5,A andB), suggest-ing that API-1 also interferes with myristoylation signal bybinding to Akt. Nevertheless, API-1 inhibition of AKT1-E17Kis clinically significant because the E17K mutation, which wasdetected in human tumors, leads to constitutively activation ofAKT1 through aberrant pathological localization to the plasmamembrane (18). An allosteric Akt kinase inhibitor AKT1/2inhibitor VIII (22) could not efficiently inhibit AKT1-E17K(18).Akt exerts its cellular function through phosphorylation of a
number of proteins (6, 14). Thus, we next examined whetherAPI-1 inhibits downstream targets of Akt. Because glycogensynthase kinase 3�, mTOR/p70S6K, Bad, and FOXO3a aremajor Akt targets, we treated OVCAR3 and H661 cells andevaluated the effects of API-1 on phosphorylation levels ofthese targets. After treatment with API-1, immunoblottinganalysis revealed that API-1 inhibited their phosphorylation(Fig. 5, C and D).We previously reported an Akt inhibitor, API-2/triciribine
(21), which is currently in clinical trial (38). We next comparedthe potency of API-1 and API-2 in inhibition of Akt. HEK293cells were transfected with wild-type (WT)-AKT1 and consti-tutively active AKT-E17K. After treatment with or withoutAPI-1 or API-2 and EGF, in vitro kinase and immunoblottinganalyses revealed that API-1 is more potent than API-2 in inhi-bition of both growth factor-induced Akt and constitutivelyactive Akt (Fig. 5, E–G).API-1 Suppresses Cell Growth and Induces Apoptosis Selec-
tively in Akt-overexpressing/activating Human Cancer CellLines—Accumulated studies have shown that cancer cells withan elevated Akt exhibit more resistance to chemotherapeuticdrug-induced apoptosis, whereas knockdown of Akt sensitizescells to the programmed cell death (39–41). The ability ofAPI-1 to selectively inhibit the Akt suggests that it shouldinhibit cell survival and growth preferentially in those tumorcells with aberrant expression/activation of Akt. To test this,API-1 was used to treat the cells that express constitutively
active Akt, caused by overexpression of Akt (OVCAR3,OVCAR8, MCF7, and PANC1) or mutations of the PTEN gene(MDA-MB-468) and cells that do not contain hyperactivatedAkt (OVCAR5,MDA-MB-435s, and COLO357). Immunoblot-ting analysis shows that phosphorylation levels of Akt were sig-nificantly inhibited by API-1 in the cells expressing elevatedAkt,whereas phospho-Aktwas also reduced byAPI-1 in the celllines exhibiting low levels of Akt (Fig. 6A and data not shown).However, API-1 induces poly(ADP-ribose) polymerase (PARP)cleavage and inhibits cell growth to a much higher degree incells with hyperactivated Akt as compared with those with lowlevels of activatedAkt (Fig. 6,A andB). API-1 (10�M) treatmentinhibited cell proliferation by approximate 50–70% in Akt-overexpressing/activating cell lines, OVCAR3, OVCAR8,MDA-MB-468, and MCF7 but only by about 10–30% inOVCAR5 and MDA-MB-435s cells (Fig. 6B). Moreover, API-1induces apoptosis by 9- and 4.3-fold in OVACAR3 and MDA-MB-468, respectively, whereas much less apoptosis wasobserved in API-1-treated OVCAR5 and MDA-MB-435s cells(Fig. 6C). In addition, we have introduced wild-type Akt1 andactive mutant AKT1-E17K into Akt1-knock out mouse embry-onic fibroblasts. After treatment with API-1, the AKT1-E17Kcells underwent apoptosis more than the mouse embryonicfibroblasts transfected with WT-Akt1 (Fig. 6D). These resultsindicate that API-1 inhibits cell growth and induces apoptosispreferentially in the cells that express aberrant Akt.API-1 Inhibits the Growth in Nude Mice of Tumors with
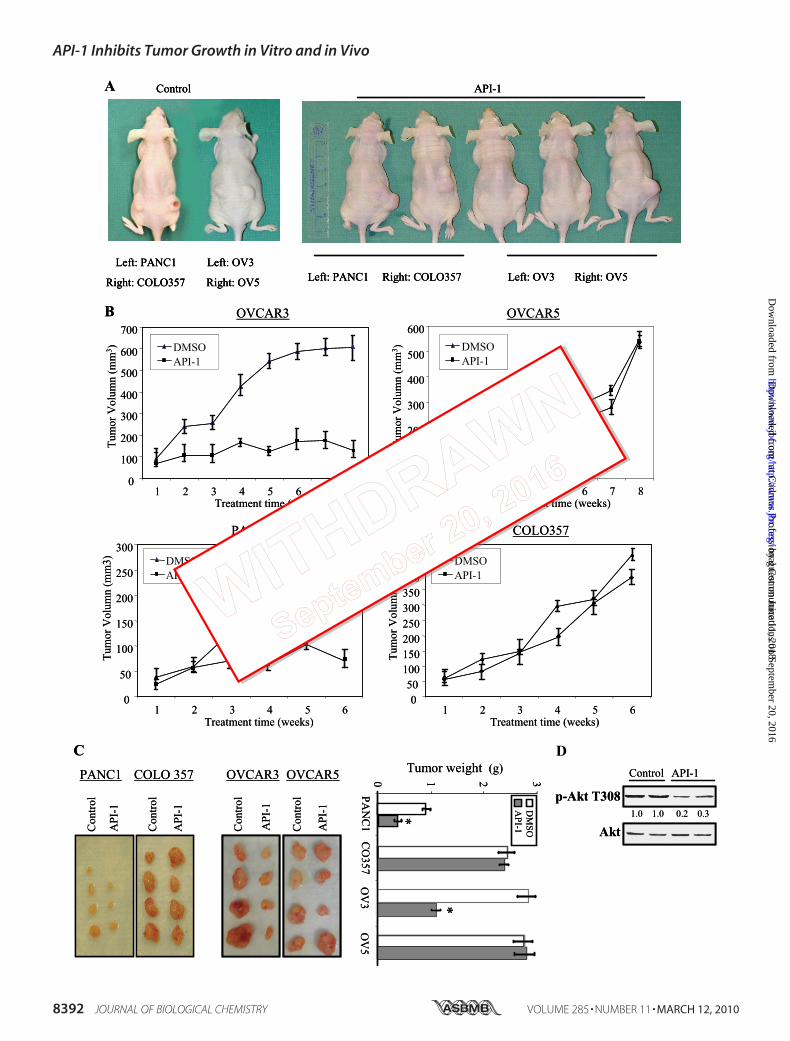
Hyperactivated Akt—We and others have previously shownthat aberrant activation and overexpression of Akt are fre-quently detected in human cancer (2, 15, 16) and that anti-sense of Akt significantly inhibits tumor cell growth (42).Furthermore, inhibition of Akt pathway by inhibitors ofPI3K, HSP70, Src, and farnesyltransferase resulted in cellgrowth arrest and induction of apoptosis (20, 39, 40).Because API-1 inhibits Akt signaling and induces apoptosisand cell growth arrest in cancer cells with elevated levels ofAkt (Fig. 6), we reasoned that the growth of tumors withelevated levels of Akt should be more sensitive to API-1 thanthat of tumors with low levels of Akt in nude mice. To thisend, we subcutaneously implanted tumors with hyperacti-vated Akt (OVCAR3 and PANC-1) into the left flank andthose tumors that express low levels of activated Akt(OVCAR5 and COLO357) into the right flank of mice. Whenthe tumors reached an average size of about 100 mm3, theanimals were randomized and treated intraperitoneal witheither vehicle or API-1 (10 mg/kg/day). As illustrated in Fig.7, A–C, OVCAR3 and PANC1 tumors treated with vehiclecontrol continued to grow. API-1 inhibited OVCAR3 andPANC1 tumor growth by 70 and 50%, respectively (Fig. 7, Band C). In contrast, API-1 had little effect on the growth ofOVCAR5 and COLO357 cells in nude mice (Fig. 7, A–C). At
FIGURE 6. API-1 inhibits Akt activity and cell growth and induces apoptosis in the cells with hyperactivated Akt. A, a Western blot is shown. Aftertreatment with API-1, phosphorylation levels of Akt and poly(ADP-ribose) polymerase (PARP) cleavage were detected with anti-pAkt-Thr308 and cleaved PARPantibodies, respectively, in the indicated human cancer cell lines (top and middle panels). The blots were reprobed with anti-actin antibody (bottom panel).B, a cell proliferation assay is shown. The indicated cell lines were treated with different doses of API-1 for 24 h and then analyzed with CellTier96 One SolutionCell Proliferation kit (Promega). C, shown is an apoptosis analysis. Cells were treated with API-1 and stained with annexin V and propidium iodide and analyzedby FACScan. D, Akt1-knock-out mouse embryonic fibroblasts were transfected with wild-type Akt1, constitutively active AKT1-E17K, or vector alone (left). Aftertreatment with API-1 for 24 h, cells were assayed with annexin V labeling and FACScan (middle). The right panel shows inhibition of AKT1-E17K by API-1.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8391
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from
API-1 Inhibits Tumor Growth in Vitro and in Vivo
8392 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 11 • MARCH 12, 2010
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

a dose of 10 mg/kg/day, API-1 had no effects on blood glu-cose level, body weight, activity, and food intake of mice(data not shown). In treated tumor samples, phosphoryla-tion levels of Akt were reduced by API-1 about 70% withouta change of total Akt content (Fig. 7D). Taken together, theseresults indicate that API-1 selectively inhibits the growth oftumors with hyperactivated Akt.In the last several years, through combinatorial chemistry,
high throughput and virtual screening, and traditional medici-nal chemistry, several inhibitors of the Akt pathway have beenidentified (25, 26). Lipid-based inhibitors ofAktwere the first tobe developed, including perifosine (43), PX-316 (44), and phos-phatidylinositol ether lipid analogues (45), which weredesigned to interact with the PH domain of Akt. In addition,several Akt antagonists have been identified using highthroughput screening of chemical libraries and rational design.These inhibitors include 9-methoxy-2-methylellipticiniumacetate (46), the indazole-pyridineA-443654 (47), isoform-spe-cific allosteric kinase inhibitors (48), and Akt/PKB signalinginhibitor-2 (API-2), also called triciribine/TCN (21). TCN is atricyclic nucleoside that has previously been evaluated clinicallyand showed antitumor activity in some patients, but toxicitiesprecluded further development (36, 49). These clinical studieswere performed before the TCN inhibition of Akt activity wasdiscovered. Our recent discovery of the TCN ability to inhibitAkt activities (21) prompted new interest in studying this drugand raises the possibility that lower doses that inhibit Akt mayresult in a clinical response with less toxicity in those patientswhose tumors have hyperactivatedAkt (21, 26). Presently, TCNis undergoing phase I trials in patients with solid tumors andleukemia. Unlike API-2/TCN, API-1 is a new small moleculeinhibitor of Akt and has not been studied previously. It inhibitsnot only activated wild-type Akt, which results from alterationsof upstream regulators such as PTEN mutations, but alsoconstitutively active Akt mutants including myr-Akt1, myr-AKT2, and E40K-Akt1 as well as naturally occurring mutantAKT1-E17K.In summary, we have identified an Akt inhibitor, API-1, that
inhibits Akt by binding to the PH domain of Akt and blockingAkt membrane translocation. As a result, API-1 selectivelyinduces apoptosis and inhibits cell growth. The ability of API-1to inhibit growth of human tumor xenografts in nude miceprovides validation for the development of drugs targeting Aktto treat cancers displaying hyperactivated Akt. Further investi-gation is needed to evaluate whether API-1 is clinically useful inthis setting. In addition, API-1 could be further chemicallymodified and optimized to develop it into a more effectivelytherapeutic agent.
Acknowledgments—We thank Drs. Edward Sausville, Jill Johnson,George Johnson, Daniel Zaharevitz, and Robert Schultz from the NCIDevelopmental Therapeutics Program for providing us with the com-pounds. We also thank Drs. Andrew Hamilton and Nicholas Law-rence for their thoughtful input as well as the Core Facilities of HighThroughput Screening, Mouse Models, Molecular Biology, and FlowCytometry at H. Lee Moffitt Cancer Center.
REFERENCES1. Bellacosa, A., Testa, J. R., Staal, S. P., and Tsichlis, P. N. (1991) Science 254,
274 –2772. Cheng, J. Q., Godwin, A. K., Bellacosa, A., Taguchi, T., Franke, T. F.,
Hamilton, T. C., Tsichlis, P. N., and Testa, J. R. (1992) Proc. Natl. Acad. Sci.U.S.A. 89, 9267–9271
3. Jones, P. F., Jakubowicz, T., Pitossi, F. J., Maurer, F., and Hemmings, B. A.(1991) Proc. Natl. Acad. Sci. U.S.A. 88, 4171– 4175
4. Jones, P. F., Jakubowicz, T., and Hemmings, B. A. (1991) Cell Regul. 2,1001–1009
5. Konishi, H., Kuroda, S., Tanaka, M., Matsuzaki, H., Ono, Y., Kameyama,K., Haga, T., and Kikkawa U. (1995) Biochem. Biophys. Res. Commun. 216,526 –534
6. Datta, S. R., Brunet, A., and Greenberg, M. E. (1999) Genes Dev. 13,2905–2927
7. Persad, S., Attwell, S., Gray, V., Mawji, N., Deng, J. T., Leung, D., Yan, J.,Sanghera, J., Walsh, M. P., and Dedhar, S. (2001) J. Biol. Chem. 276,27462–27469
8. Toker, A., and Newton, A. C. (2000) J. Biol. Chem. 275, 8271– 82749. Feng, J., Park, J., Cron, P., Hess, D., and Hemmings, B. A. (2004) J. Biol.
Chem. 279, 41189 – 4119610. Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005)
Science 307, 1098 –110111. Huang, J., and Manning, B. D. (2009) Biochem. Soc. Trans. 37, 217–22212. Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S. I., Puc, J.,
Miliaresis, C., Rodgers, L., McCombie, R., Bigner, S. H., Giovanella, B. C.,Ittmann, M., Tycko, B., Hibshoosh, H., Wigler, M. H., and Parsons, R.(1997) Science 275, 1943–1947
13. Stambolic, V., Suzuki, A., de la Pompa, J. L., Brothers, G. M., Mirtsos, C.,Sasaki, T., Ruland, J., Penninger, J. M., Siderovski, D. P., and Mak, T. W.(1998) Cell 95, 29 –39
14. Cheng, J. Q., and Nicosia, S. V. (2001) Cancer, pp. 35–37, Springer-VerlagNew York Inc.
15. Altomare, D. A., and Testa, J. R. (2005) Oncogene 24, 7455–776416. Sun, M., Wang, G., Paciga, J. E., Feldman, R. I., Yuan, Z. Q., Ma, X. L.,
Shelley, S. A., Jove, R., Tsichlis, P. N., Nicosia, S. V., and Cheng, J. Q. (2001)Am. J. Pathol. 159, 431– 437
17. West, K. A., Castillo, S. S., and Dennis, P. A. (2002) Drug Resist. Updat. 5,234 –248
18. Carpten, J. D., Faber, A. L., Horn, C., Donoho, G. P., Briggs, S. L., Robbins,C. M., Hostetter, G., Boguslawski, S., Moses, T. Y., Savage, S., Uhlik, M.,Lin, A., Du, J., Qian, Y. W., Zeckner, D. J., Tucker-Kellogg, G., Touchman,J., Patel, K., Mousses, S., Bittner, M., Schevitz, R., Lai, M. H., Blanchard,K. L., and Thomas, J. E. (2007) Nature 448, 439 – 444
19. Cheng, J. Q., Altomare, D. A., Klein, M. A., Lee, W. C., Kruh, G. D., Lissy,N. A., and Testa, J. R. (1997) Oncogene 14, 2793–2801
20. Jiang, K., Coppola, D., Crespo, N. C., Nicosia, S. V., Hamilton, A. D., Sebti,
FIGURE 7. API-1 exhibits anti-tumor activity in cancer cell lines with hyperactivated Akt in mouse xenografts. A and B, API-1 inhibits tumor growth. Tumorcells were subcutaneously injected into nude mice with tumors presenting a low level of pAkt on the right flank and tumors with elevated level of pAkt on theleft flank. When the tumors reached an average size of about 100 mm3, animals were treated with either vehicle or 10 mg/kg/day API-1 as described under“Experimental Procedures.” shown is a representation of the mice with PANC1/OVCAR3 (left flank), which express elevated levels of pAkt, and COLO357/OVCAR5 (right flank), which exhibit low levels of pAkt, xenografts treated with API-1 or vehicle (A). Panel B shows tumor growth curve with 10 mice/group.C, shown are examples of tumor size (left) and weight (right) at the end of experiment. API-1 significantly reduced tumor weight in PANC1 and OVCAR3xenografts (*, p � 0.02) compared with vehicle control. D, API-1 inhibits Akt phosphorylation in vivo. API-1-treated and untreated tumor specimens were lysedand immunoblotted with the indicated antibodies.
API-1 Inhibits Tumor Growth in Vitro and in Vivo
MARCH 12, 2010 • VOLUME 285 • NUMBER 11 JOURNAL OF BIOLOGICAL CHEMISTRY 8393
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

S. M., and Cheng, J. Q. (2000) Mol. Cell. Biol. 20, 139 –14821. Yang, L., Dan, H. C., Sun, M., Liu, Q., Sun, X. M., Feldman, R. I., Hamilton,
A. D., Polokoff, M., Nicosia, S. V., Herlyn, M., Sebti, S. M., and Cheng, J. Q.(2004) Cancer Res. 64, 4394 – 4399
22. Bantscheff, M., Eberhard, D., Abraham, Y., Bastuck, S., Boesche, M., Hob-son, S., Mathieson, T., Perrin, J., Raida, M., Rau, C., Reader, V., Sweetman,G., Bauer, A., Bouwmeester, T., Hopf, C., Kruse, U., Neubauer, G., Rams-den, N., Rick, J., Kuster, B., and Drewes, G. (2007) Nat. Biotechnol. 25,1035–1044
23. Rix, U., Hantschel, O., Durnberger, G., Remsing Rix, L. L., Planyavsky, M.,Fernbach, N. V., Kaupe, I., Bennett, K. L., Valent, P., Colinge, J., Kocher, T.,and Superti-Furga, G. (2007) Blood 110, 4055– 4063
24. Oppermann, F. S., Gnad, F., Olsen, J. V., Hornberger, R., Greff, Z., Keri, G.,Mann, M., and Daub, H. (2009) Mol. Cell. Proteomics 8, 1751–1764
25. Cheng, J. Q., Lindsley, C. W., Cheng, G. Z., Yang, H., and Nicosia, S. V.(2005) Oncogene 24, 7482–7492
26. Granville, C. A., Memmott, R. M., Gills, J. J., and Dennis, P. A. (2006) Clin.Cancer Res. 12, 679 – 689
27. Van Ummersen, L., Binger, K., Volkman, J., Marnocha, R., Tutsch, K.,Kolesar, J., Arzoomanian, R., Alberti, D., and Wilding, G. (2004) Clin.Cancer Res. 10, 7450 –7456
28. Bailey, H. H., Mahoney, M. R., Ettinger, D. S., Maples, W. J., Fracasso,P. M., Traynor, A. M., Erlichman, C., and Okuno, S. H. (2006) Cancer 107,2462–2467
29. Marsh Rde, W., Rocha Lima, C. M., Levy, D. E., Mitchell, E. P., Rowland,K. M., Jr., and Benson, A. B., 3rd (2007) Am. J. Clin. Oncol. 30, 26 –31
30. Rizkalla, B. H., and Broom, A. D. (1972) J. Org. Chem. 37, 3980 –398531. Ritch, P. S., Glazer, R. I., Cunningham, R. E., and Shackney, S. E. (1981)
Cancer Res. 41, 1784 –178832. Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003) Cancer
Cell 3, 285–29633. Lee, S. A., and Jung, M. (2007) J. Biol. Chem. 282, 15271–1528334. Sato, S., Fujita, N., and Tsuruo, T. (2002) Oncogene 21, 1727–173835. Casamayor, A., Morrice, N. A., and Alessi, D. R. (1999) Biochem. J. 342,
287–29236. Hoffman, K., Holmes, F. A., Fraschini, G., Esparza, L., Frye, D., Raber,
M. N., Newman, R. A., and Hortobagyi, G. N. (1996) Cancer Chemother.Pharmacol. 37, 254 –258
37. Harrington, L. S., Findlay, G. M., Gray, A., Tolkacheva, T., Wigfield, S.,Rebholz, H., Barnett, J., Leslie, N. R., Cheng, S., Shepherd, P. R., Gout, I.,Downes, C. P., and Lamb, R. F. (2004) J. Cell Biol. 166, 213–223
38. Ravandi, F., Lancet, J., Giles, F., Plunkett, W., Williams, B., Burton, M.,Faderl, S., Estrov, Z., Borthakur, G., Akinsanmi, L., and Kantarjian, H.(2007) Blood 110, 913 (The American Society of Hemotology 49th AnnualMeeting, Atlanta, Georgia, December 8 –11, 2007)
39. Solit, D. B., Basso, A. D., Olshen, A. B., Scher, H. I., and Rosen, N. (2003)Cancer Res. 63, 2139 –2144
40. Xu, W., Yuan, X., Jung, Y. J., Yang, Y., Basso, A., Rosen, N., Chung, E. J.,Trepel, J., and Neckers, L. (2003) Cancer Res. 63, 7777–7784
41. Jetzt, A., Howe, J. A., Horn, M. T., Maxwell, E., Yin, Z., Johnson, D., andKumar, C. C. (2003) Cancer Res. 63, 6697– 6706
42. Cheng, J. Q., Ruggeri, B., Klein, W. M., Sonoda, G., Altomare, D. A.,Watson, D. K., and Testa, J. R. (1996) Proc. Natl. Acad. Sci. U.S.A. 93,3636 –3641
43. Kondapaka, S. B., Singh, S. S., Dasmahapatra, G. P., Sausville, E. A., andRoy, K. K. (2003) Mol. Cancer Ther. 2, 1093–1103
44. Meuillet, E. J., Ihle, N., Baker, A. F., Gard, J. M., Stamper, C., Williams, R.,Coon, A., Mahadevan, D., George, B. L., Kirkpatrick, L., and Powis, G.(2004) Oncol. Res. 14, 513–527
45. Castillo, S. S., Brognard, J., Petukhov, P. A., Zhang, C., Tsurutani, J., Gran-ville, C. A., Li, M., Jung, M., West, K. A., Gills, J. G., Kozikowski, A. P., andDennis, P. A. (2004) Cancer Res. 64, 2782–2792
46. Jin, X., Gossett, D. R., Wang, S., Yang, D., Cao, Y., Chen, J., Guo, R.,Reynolds, R. K., and Lin, J. (2004) Br. J. Cancer 91, 1808 –1812
47. Luo, Y., Shoemaker, A. R., Liu, X., Woods, K. W., Thomas, S. A., de Jong,R., Han, E. K., Li, T., Stoll, V. S., Powlas, J. A., Oleksijew, A., Mitten, M. J.,Shi, Y., Guan, R., McGonigal, T. P., Klinghofer, V., Johnson, E. F., Lever-son, J. D., Bouska, J. J., Mamo, M., Smith, R. A., Gramling-Evans, E. E.,Zinker, B. A., Mika, A. K., Nguyen, P. T., Oltersdorf, T., Rosenberg, S. H.,Li, Q., and Giranda, V. L. (2005) Mol. Cancer Ther. 4, 977–986
48. Lindsley, C. W., Zhao, Z., Leister, W. H., Robinson, R. G., Barnett, S. F.,Defeo-Jones, D., Jones, R. E., Hartman, G. D., Huff, J. R., Huber, H. E., andDuggan, M. E. (2005) Bioorg. Med. Chem. Lett. 15, 761–764
49. Feun, L. G., Blessing, J. A., Barrett, R. J., and Hanjani, P. (1993) Am. J. Clin.Oncol. 16, 506 –508
API-1 Inhibits Tumor Growth in Vitro and in Vivo
8394 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 285 • NUMBER 11 • MARCH 12, 2010
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

α-AKT2/OVCAR3
EGF API-1/EGFStarvation
Fig. S1
by guest on June 11, 2018 http://www.jbc.org/ Downloaded from

Bepler, Said M. Sebti and Jin Q. ChengDonghwa Kim, Mei Sun, Lili He, Qing-Hua Zhou, Jun Chen, Xia-Meng Sun, Gerold
Membrane TranslocationA Small Molecule Inhibits Akt through Direct Binding to Akt and Preventing Akt
doi: 10.1074/jbc.M109.094060 originally published online January 12, 20102010, 285:8383-8394.J. Biol. Chem.
10.1074/jbc.M109.094060Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2010/01/26/M109.094060.DC1.html
http://www.jbc.org/content/285/11/8383.full.html#ref-list-1
This article cites 48 references, 27 of which can be accessed free at
at Cadm
us Professional Com
munications on Septem
ber 20, 2016http://w
ww
.jbc.org/D
ownloaded from
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

Bepler, Said M. Sebti and Jin Q. ChengDonghwa Kim, Mei Sun, Lili He, Qing-Hua Zhou, Jun Chen, Xia-Meng Sun, Gerold
Membrane TranslocationA Small Molecule Inhibits Akt through Direct Binding to Akt and Preventing Akt
doi: 10.1074/jbc.M109.094060 originally published online January 12, 20102010, 285:8383-8394.J. Biol. Chem.
10.1074/jbc.M109.094060Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
Supplemental material:
http://www.jbc.org/content/suppl/2010/01/26/M109.094060.DC1
http://www.jbc.org/content/285/11/8383.full.html#ref-list-1
This article cites 48 references, 27 of which can be accessed free at
by guest on June 11, 2018http://w
ww
.jbc.org/D
ownloaded from

VOLUME 285 (2010) PAGES 8383– 8394DOI 10.1074/jbc.A109.094060
A small molecule inhibits Akt through direct binding toAkt and preventing Akt membrane translocation.Donghwa Kim, Mei Sun, Lili He, Qing-Hua Zhou, Jun Chen, Xia-Meng Sun,Gerold Bepler, Said M. Sebti, and Jin Q. Cheng
This article has been withdrawn by the authors. The same data wereused to represent different experimental conditions. Specifically, part ofthe p-PKA immunoblot from Fig. 4D was reused as part of the p-Akt-T308 immunoblot from the same figure. The authors state that thisimage reuse does not affect the overall conclusions of the study.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 291, NO. 43, p. 22856, October 21, 2016© 2016 by The American Society for Biochemistry and Molecular Biology, Inc. Published in the U.S.A.
22856 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 291 • NUMBER 43 • OCTOBER 21, 2016
ADDITIONS AND CORRECTIONS
Authors are urged to introduce these corrections into any reprints they distribute. Secondary (abstract) services are urged to carry notice ofthese corrections as prominently as they carried the original abstracts.


















