
ASEAN GUIDELINE ON STABILITY STUDY OF DRUG...
42
ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT Update revision : 15 May 2013 20 th ACCSQ-PPWG Meeting, Bali, Indonesia, 13 - 17 May 2013
Transcript of ASEAN GUIDELINE ON STABILITY STUDY OF DRUG...

ASEAN GUIDELINE ON STABILITY STUDY OF DRUG PRODUCT
Update revision : 15 May 2013
20th ACCSQ-PPWG Meeting, Bali, Indonesia, 13 - 17 May 2013

LIST OF CONTENTS Page
1. INTRODUCTION 1 2. OBJECTIVES 1 3. SCOPE 1 4. DESIGN 1
4.1. General 1 4.2. Photostability Testing 1 4.3. Selection of Batches 1 4.4. Specification 2 4.5. Testing Parameters 2 4.6. Testing Frequency 6 4.7. Storage Conditions 6
4.7.1. General Case 6 4.7.2. Drug Products Packaged in Impermeable Containers 7 4.7.3. Drug Products Packaged in Semi-Permeable Containers
(Aqueous-Based Products) 8 4.7.4. Drug Products Intended for Storage in a Refrigerator 9 4.7.5. Drug Products Intended for Storage in a Freezer 9 4.7.6. Drug Products Intended for Storage Below -20ºC 9 4.7.7. NCE Drug Products 9 4.7.8. Generic Products 10 4.7.9. Variations (MaV and MiV if appropriate) 10
4.8. In-use Stability 11 4.9. Container Closure System 12 4.10. Evaluation 12 4.10.1. Data Presentation 13 4.10.2. Extrapolation of Data 13 4.10.3. Data Evaluation for Shelf-Life Estimation for Drug Products Intended for
Storage at Room Temperature 14 4.10.4. Data Evaluation for Shelf-Life Estimation for Drug Products Intended for
Storage below Room Temperature 16 4.10.5. General Statistical Approaches 17 4.11. Stability Commitment 18 4.12. Statements/Labeling 19
5. ANNEXES 20 5.1. Protocol of Stability Study (example) 20 5.2. Report Format (example) 24 5.3. Reduced Design (Bracketing and Matrixing) 30 5.4. Examples of Types, Thickness and Permeability Coefficient of Packaging 32 Materials 5.5. Decision Tree for Data Evaluation for Shelf-Life Estimation for Drug Products 34 (excluding Frozen Products) 5.6. Examples of Statistical Approaches to Stability Data Analysis 35
6. GLOSSARY 36 7. REFERENCES 40

1
1. INTRODUCTION 1.1 Stability is an essential factor of quality, safety and efficacy of a drug product.
Insufficient stability of a drug product can result in changes in physical (like hardness, dissolution rate, phase separation, etc.) as well as in chemical characteristics (formation of high risk decomposition substances). Microbiological instability of a sterile drug product could also be hazardous.
1.2 In principle, stability testing should be biased towards more stressful rather than less
stressful conditions so as to provide a margin of error in favour of the patients and to increase the likelihood of identifying substances or formulations that pose particular stability problems.
1.3 The objective of a stability study is to determine the shelf-life, namely the time period of
storage at a specified condition within which the drug product still meets its established specifications.
1.4 The stability study consists of a series of tests in order to obtain an assurance of stability
of a drug product, namely maintenance of the specifications of the drug product packed in its specified packaging material and stored at the established storage condition within the determined time period.
1.5 The general conditions for long term stability testing in the ASEAN region are the Zone
IVb conditions (30oC/75% RH). 2. OBJECTIVES
This guideline is intended to provide recommendations on the core stability study package required for drug products, but leaves sufficient flexibility to encompass the variety of different practical situations that may be encountered due to specific scientific considerations and characteristics of the products being evaluated. This guideline can also be used to propose shelf-life based on the stability data generated from the study package.
3. SCOPE
This guideline addresses the information to be submitted during application for marketing authorization/registration and variations of drug products in ASEAN Member States including examples of a protocol of stability study, a report format, reduced design and extrapolation of data, and examples of types, thickness and permeability coefficient which are covered in Annexes. The drug products covered in this guideline include NCE, Generics and Variations (MaV and MiV) but exclude biologicals and drug products containing vitamin and mineral preparations.
4. DESIGN
4.1. General The design of the stability studies for the product should be based on knowledge of the behavior and properties of the drug substance and dosage form.
4.2. Photostability Testing Photostability testing should be conducted on at least one primary batch of the drug product if appropriate. The standard conditions for photostability testing are described in ICH Q1B.
4.3. Selection of Batches At the time of submission, stability data should be provided for batches of the same formulation and dosage form in the container closure system proposed for marketing.

2
- For NCE stability data should be provided on at least three primary batches of the drug products.
- For Generics and Variations the following will apply : • For conventional dosage forms (e.g., immediate release solid dosage forms,
solutions) and when the drug substances are known to be stable, stability data on at least two pilot scale batches are acceptable.
• For critical dosage forms (e.g., prolonged release forms) or when the drug substances are known to be unstable, stability data on three primary batches are to be provided. Two of the three batches should be at least of a pilot scale; the third batch may be smaller, if justified.
- The manufacturing process used for primary batches should simulate that to be applied to production batches and should provide products of the same quality and meeting the same specification as that intended for marketing.
- Where possible, batches of the drug product should be manufactured by using different batches of the drug substance.
- Stability studies should be performed on each individual strength and container size of the drug product unless bracketing or matrixing is applied.
Other supporting data can be provided.
4.4. Specification i. Specification is a list of tests, reference to analytical procedures, and proposed
acceptance criteria, including the concept of different acceptance criteria for release and shelf-life specifications.
ii. Shelf-life acceptance criteria should be derived from consideration of all available
stability information. It may be appropriate to have justifiable differences between the shelf-life and release acceptance criteria based on the stability evaluation and the changes observed on storage. Any differences between the release and shelf-life acceptance criteria for antimicrobial preservative content should be supported by a validated correlation of chemical content and preservative effectiveness demonstrated during development of the pharmaceutical product with the product in its final formulation (except for preservative concentration) intended for marketing. A single primary stability batch of the drug product should be tested for effectiveness of the antimicrobial preservative (in addition to preservative content) at the proposed shelf-life for verification purposes, regardless of whether there is a difference between the release and shelf-life acceptance criteria for preservative content.
4.5 Testing Parameters
i. Stability studies should include testing of those attributes of the drug product that are susceptible to change during storage and are likely to influence quality, safety and/or efficacy. The testing should cover, as appropriate, the physical, chemical, biological and microbiological attributes, preservative content (e.g. antioxidant, antimicrobial preservative), and functionality tests (e.g., for a dose delivery system). The analytical procedure should be fully validated and stability-indicating according to the ASEAN guideline on Analytical Validation. Whether and to what extent replication should be performed will depend on the results from validation studies.
ii. In general, appearance, assay and degradation products should be evaluated for all
dosage forms. For generic products, degradation products should use current compendia as a minimum requirement. The following list of parameters for each dosage form is presented as a guide for the types of tests to be included in a stability study. The list of tests presented for each dosage form is not intended to be

3
exhaustive, nor is it expected that every listed test be included in the design of a stability protocol for a particular drug product (for example, a test for odour should be performed only when necessary and with consideration for analyst’s safety).
1. Tablets
Tablets should be evaluated for appearance, odour, colour, assay, degradation products, dissolution (or disintegration, if justified), water content, and hardness/friability.
2. Capsules
Hard gelatin capsules should be evaluated for appearance (including brittleness), colour, and odour of content, assay, degradation products, dissolution, water content and microbial limits. Testing of soft gelatin capsules should include appearance, colour, and odour of content, assay, degradation products, dissolution, microbial limits, pH, leakage, and pellicle formation. In addition, the fill medium should be examined for precipitation and cloudiness.
3. Emulsions
Emulsions should be evaluated for appearance (including phase separation), colour, odour, assay, degradation products, pH, viscosity, microbial limits, preservative content, and mean size and distribution of dispersed globules.
4. Oral Solutions and Suspensions
Oral Solutions and Suspensions should be evaluated for appearance (including formation of precipitate, clarity for solutions), colour, odour, assay, degradation products, pH, viscosity, preservative content and microbial limits.
Additionally for suspensions, redispersibility, rheological properties and mean size and distribution of particles should be considered. After storage, sample of suspensions should be prepared for assay according to the recommended labeling (e.g. shake well before using).
5. Oral Powders for Reconstitution
Oral powders should be evaluated for appearance, colour, odour, assay, degradation products, water content, and reconstitution time.
Reconstituted products (solutions and suspensions) should be evaluated as described in Oral Solutions and Suspensions above, after preparation according to the recommended labeling, through the maximum intended use period.
6. Metered-dose Inhalations and Nasal Aerosols
Metered-dose inhalations and nasal aerosols should be evaluated for appearance (including content, container, valve, and its components), colour, taste, assay, degradation products, assay for co-solvent (if applicable), dose content uniformity, labeled number of medication actuations per container meeting dose content uniformity, aerodynamic particle size distribution, microscopic evaluation, water content, leak rate, microbial limits, valve delivery (shot weight) and extractables/leachables from plastic and elastomeric components. Samples should be stored in upright and inverted/on-the-side orientations. For suspension-type aerosols, the appearance of the valve components and container’s contents should be evaluated microscopically for large particles and

4
changes in morphology of the drug surface particles, extent of agglomerates, crystal growth, as well as foreign particulate matter. These particles lead to clogged valves or non-reproducible delivery of a dose. Corrosion of the inside of the container or deterioration of the gasket may adversely affect the performance of the drug product.
7. Nasal Sprays : Solutions and Suspensions
Nasal solutions and suspensions equipped with a metering pump should be evaluated for appearance, colour, clarity for solution, assay, degradation products, preservative and antioxidant content, microbial limits, pH, particulate matter, unit spray medication content uniformity, number of actuations meeting unit spray content uniformity per container, droplet and/or particle size distribution, weight loss, pump delivery, microscopic evaluation (for suspensions), foreign particulate matter and extractable/bleachable from plastic and elastomeric components of the container, closure and pump.
8. Topical, Ophthalmic and Otic Preparations
Included in this broad category are ointments, creams, lotions, paste, gel, solutions and non-metered aerosols for application to the skin. Topical preparations should be evaluated for appearance, clarity, colour, homogeneity, odour, pH, resuspendability (for lotions), consistency, viscosity, particle size distribution (for suspensions, when feasible), assay, degradation products, preservative and antioxidant content (if present), microbial limits/sterility and weight loss (when appropriate). Ophthalmic or otic products (e.g., creams, ointments, solutions, and suspensions) should be evaluated for the following additional attributes: sterility, particulate matter, and extractable volume. Non-metered topical aerosols should be evaluated for appearance, assay, degradation products, pressure, weight loss, net weight dispensed, delivery rate, microbial limits, spray pattern, water content, and particle size distribution (for suspensions).
9. Suppositories
Suppositories should be evaluated for appearance, colour, assay, degradation products, particle size, softening range, disintegration and dissolution (at 37oC) and microbial limits.
10. Small Volume Parenterals (SVPs) SVPs include a wide range of injection products such as Injection, Powder for Injection, Suspension for Injection, and Emulsion for Injection. Samples should be stored in upright and inverted/on-the-side orientations. Injection products should be evaluated for appearance, clarity, colour, assay, preservative content (if present), degradation products, particulate matter, pH, sterility and pyrogen/endotoxin. Powder for Injection products should be evaluated for appearance, colour, reconstitution time and water content. The stability of Powder for Injection products should also be evaluated after reconstitution according to the recommended labeling. Specific parameters to be examined at appropriate

5
intervals throughout the maximum intended use period of the reconstituted drug product, stored under condition(s) recommended in labeling, should include appearance, clarity, odour, colour, pH, assay (potency), preservative (if present), degradation products/aggregates, sterility, pyrogen/endotoxin and particulate matter. Suspension for Injection products should also be evaluated for particle size distribution, redispersibility and rheological properties in addition to the parameters cited above for Injection and Powder for Injection products. Emulsion for Injection products should be evaluated for in addition to the parameters cited above for Injection, phase separation, viscosity, and mean size and distribution of dispersed phase globules.
11. Large Volume Parenterals (LVPs) LVPs should be evaluated for appearance, colour, assay, preservative content (if present), degradation products, particulate matter, pH, sterility, pyrogen/endotoxin, clarity and volume.
12. Drug Admixture
For any drug product or diluent that is intended for use as an additive to another drug product, the potential for incompatibility exists. In such cases, the drug product labeled to be administered by addition to another drug product (e.g. parenterals, inhalation solutions), should be evaluated for stability and compatibility in admixture with the other drug products or with diluents both in upright and in inverted/on-the side orientations, if warranted. A stability protocol should provide for appropriate tests to be conducted at 0-, 6- to 8- and 24-hour time points, or as appropriate over the intended use period at the recommended storage/use temperature(s). Tests should include appearance, colour, clarity, assay, degradation products, pH, particulate matter, interaction with the container/closure/device and sterility. Appropriate supporting data may be provided in lieu of an evaluation of photo degradation.
13. Transdermal Patches Devices applied directly to the skin for the purpose of continuously infusing a drug substance into the dermis through the epidermis should be evaluated for appearance, assay, degradation products, in-vitro release rates, leakage, microbial limits/sterility, peel and adhesive forces, and the drug release rate.
14. Freeze-dried Products
Freeze-dried products should be evaluated for appearance of both the freeze-dried and its reconstituted product, assay, degradation products, pH, water content and rate of solution.
iii. The microbial quality of multiple-dose sterile and non-sterile dosage forms should be controlled. Challenge tests should be carried out at least at the beginning and at the end of the shelf-life. Such tests would normally be performed as part of the development programme, for example, within primary stability studies. They need not be repeated for subsequent stability studies unless a change has been made which has a potential impact on microbiological status. It is not expected that every test listed be performed at each time point. This applies in particular to sterility testing, which may be conducted for most sterile products at the beginning and at the end of the stability test period. Tests for pyrogens and bacterial endotoxins may be limited to

6
the time of release. Sterile dosage forms containing dry materials (powder filled or lyophilized products) and solutions packaged in sealed glass ampoules may need no additional microbiological testing beyond the initial time point. The level of microbiological contamination in liquids packed in glass containers with flexible seals or in plastic containers should be tested no less than at the beginning and at the end of the stability test period; if the long term data provided to the regulatory authorities for marketing authorization registration do not cover the full shelf-life period, the level of microbial contamination at the last time point should also be provided.
iv. The storage orientation of the product, i.e., upright versus inverted, may need to be
included in a protocol where there has been a change in the container/closure system.
4.6. Testing Frequency For long term studies, frequency of testing should be sufficient to establish the stability profile of the drug product. The frequency of testing at the long term storage condition should normally be every 3 months over the first year, every 6 months over the second year, and annually thereafter through the proposed shelf-life. At the accelerated storage condition, a minimum of three time points, including the initial and final time points (e.g., 0, 3, and 6 months), from a 6-month study is recommended. Where an expectation (based on development experience) exists that results from accelerated studies are likely to approach significant change criteria, increased testing should be conducted either by adding samples at the final time point or by including a fourth time point in the study design. Reduced designs, i.e., matrixing or bracketing, where the testing frequency is reduced or certain factor combinations are not tested at all can be applied, if justified; see Annex 5.3. Storage Condition Products Testing Frequency Long term NCE , Generics,
and Variations (MaV and MiV)
0, 3, 6, 9, 12, 18, 24 months and annually through the proposed shelf-life
Accelerated NCE , Generics, and Variations (MaV and MiV)
0, 3 and 6 months
NCE :New chemical entity; MaV : Major Variation; MiV : Minor Variation 4.7. Storage Conditions
4.7.1. General Case
i. In general, a drug product should be evaluated under storage conditions (with appropriate tolerances) that test its thermal stability and, if applicable, its sensitivity to moisture or potential for solvent loss. The storage conditions and the lengths of studies chosen should be sufficient to cover storage, shipment, and subsequent use (e.g., after reconstitution or dilution as recommended in the labeling).
ii. Stability studies should generally be conducted under the following storage condition:
STUDY/TYPE OF CONTAINER STORAGE CONDITION Long term (for products in primary containers semi-permeable to water vapour)
30oC ± 2oC/75% RH ± 5% RH

7
Long term (for products in primary containers impermeable to water vapour)
30oC ± 2oC /RH not specified
Accelerated 40oC ± 2oC/75% RH ± 5% RH Stress testing* 40oC ± 2oC/75% RH ± 5% RH or at
more stressful conditions * Stress testing is necessary for analytical method validation, pharmaceutical formulation, identifying and monitoring potential degradants during stability testing.
iii. The long term testing will be continued for a sufficient time to cover shelf-life at
appropriate test periods.
iv. Data from the accelerated storage condition can be used to evaluate the effect of short-term excursions outside the label storage conditions (such as might occur during shipping).
v. If submitted data is based on conditions that are less stressful (e.g. 30oC/65% RH,
25oC/60% RH) than those required, the data should be accompanied by appropriate complementary data which will permit conduct of a proper scientific evaluation. Factors to be taken into consideration will include: 1. Whether any instability is seen; 2. Whether data have also been provided under accelerated conditions; 3. Whether more protective packaging is provided/ required. A suitable label recommendation such as “Store below 30oC and protect from moisture” may also be applied.
vi. Additional data accumulated during the assessment period of the registration application
should be submitted to the regulatory authorities if requested.
vii. Other storage conditions are allowable if justified, e.g., under the following circumstances: - Heat sensitive drug products should be stored under an alternative lower temperature
condition which will eventually become the designated long term storage temperature.
* Products containing less stable active ingredients and formulations not suitable for experimental studies on storage at elevated temperature (e.g., suppositories) will need more extensive long term stability studies.
- Special consideration may need to be given to products which change physically or even chemically at lower storage temperature conditions e.g., suspensions or emulsions which may sediment or cream, oils and semi-solid preparations which may show an increased viscosity.
* Where a lower temperature condition is used, the 6 month accelerated testing should be carried out at a temperature at least 15oC above the expected actual storage temperature (together with appropriate relative humidity conditions for that temperature). For example, for a product to be stored long term under refrigerated conditions, accelerated testing should be conducted at 25oC ± 2ºC/ 60% RH ± 5% RH. The designated long term testing conditions will be reflected in the labeling and shelf-life (expiration date).
4.7.2. Drug Products Packaged in Impermeable Containers
i. Generally considered moisture-impermeable containers include glass ampoules,
aluminum/aluminum blisters, High Density Polyethylene (HDPE) or glass bottles fitted with metal or HDPE closures.

8
ii. Sensitivity to moisture or potential for solvent loss is not a concern for drug products
packaged in impermeable containers that provide a permanent barrier to passage of moisture or solvent. Thus stability studies for products stored in impermeable containers can be conducted under any controlled or ambient relative humidity condition.
4.7.3. Drug Products Packaged in Semi-Permeable Containers (Aqueous-Based Products)
i. Aqueous-based products packaged in semi-permeable containers should be evaluated for
potential water loss in addition to physical, chemical, biological and microbiological stability. This evaluation can be carried out under conditions of low relative humidity, as discussed below. Ultimately it should be demonstrated that aqueous-based drug products stored in semi-permeable containers could withstand environments with low relative humidity.
Study Storage Condition Minimum time period
covered by data at submission
Long term 30 °C ± 2 °C/35% RH ± 5% RH 12 months Accelerated 40 °C ± 2 °C/not more than (NMT)
25% RH 6 months
ii. Products meeting either of the long term storage conditions and the accelerated
conditions, as specified in the table above, have demonstrated the integrity of the packaging in semi-permeable containers.
iii. A 5% loss in water from its initial value is considered a significant change for a product packaged in a semi-permeable container after an equivalent of three months’ storage at 40 °C/not more than (NMT) 25% RH. However, for small containers (1 ml or less) or unit-dose products, a water loss of 5% or more after an equivalent of three months’ storage at 40 °C/NMT 25% RH may be appropriate, if justified.
iv. An alternative approach to studies at the low relative humidity as recommended in the table above (for either long term or accelerated testing) is to perform the stability studies under higher relative humidity and deriving the water loss at the low relative humidity through calculation. This can be achieved by experimentally determining the permeation coefficient for the container closure system or, as shown in the example below, using the calculated ratio of water loss rates between the two humidity conditions at the same temperature. The permeation coefficient for a container closure system can be experimentally determined by using the worst-case scenario (e.g. the most diluted of a series of concentrations) for the proposed drug product.
Example of an approach for determining water loss
- For a product in a given container closure system, container size and fill, an appropriate approach for deriving the rate of water loss at the low relative humidity is to multiply the rate of water loss measured at an alternative relative humidity at the same temperature, by a water loss rate ratio shown in the table below. A linear water loss rate at the alternative relative humidity over the storage period should be demonstrated.
- For example, at a given temperature, e.g. 40 °C, the calculated rate of water loss during storage at NMT 25% RH is the rate of water loss measured at 75% RH multiplied by 3.0, the corresponding water loss rate ratio.

9
Val Valid water loss rate ratios at relative humidity conditions other than those shown in the table above can also be used.
v. Other comparable approaches can be developed and reported for non-aqueous, solvent
based products.
4.7.4. Drug Products Intended for Storage in a Refrigerator
Study Storage Condition Minimum Time Period Covered by Data at Submission
Number of Batches
Long term 5oC ± 3oC 12 months
Min. 3
Accelerated 25oC ± 2oC/60% RH ± 5% RH
6 months Min. 3
If the drug product is packed in a semi-permeable container, appropriate information should be provided to assess the extent of water loss. Data from refrigerated storage should be assessed according to the evaluation section of this guideline, except where explicitly noted below.
4.7.5. Drug Products Intended for Storage in a Freezer
Study Storage Condition Minimum Time Period Covered by
Data at Submission Long term -20oC ± 5oC 12 months
For drug products intended for storage in a freezer, the shelf-life should be based on the long term data obtained at the long term storage condition. In the absence of an accelerated storage condition for drug products intended to be stored in a freezer, testing on a single batch at an elevated temperature (e.g. 5oC±3oC or 25oC±2oC) for an appropriate time period should be conducted to address the effect of short term excursions outside the proposed label storage condition.
4.7.6. Drug Products Intended for Storage below -20oC
Drug products intended for storage below -20oC should be treated on a case-by-case basis.
4.7.7. NCE Drug Products
Study Storage Condition Minimum Time
Period Covered by Data at Submission
Number of Batches
Long term 30oC ± 2oC/75% RH ± 5% RH 12 months Min. 3
Accelerated 40oC ± 2oC/75% RH ± 5% RH 6 months Min. 3
Low-humidity testing conditions
Alternative testing condition
Ratio of water loss rates
Calculation
30 °C/35% RH 30 °C/75% RH 2.6 (100-35)/(100-75) 40 °C/NMT 25% RH 40 °C/75% RH 3.0 (100-25)/(100-75)

10
4.7.8. Generic Products
Study Storage Condition Minimum Time Period Covered by Data at Submission
Number of Batches
Long term 30oC ± 2oC/75% RH ± 5% RH
6 months
Min. 2 For conventional dosage form and stable drug substances
12 months
Min.3 For critical dosage form or unstable drug substances
Accelerated 40oC ± 2oC/75% RH ± 5% RH
6 months Min. 2 For conventional dosage form and stable drug substances Min.3 For critical dosage form or unstable drug substances
4.7.9. Variations (MaV and MiV if appropriate)
Once the Drug Product has been registered, additional stability studies are required whenever variations that may affect the stability of the Drug Products are made, refer to ASEAN Variation Guideline Major Variation (MaV)
Study Storage Condition Minimum Time Period Covered by Data at Submission
Number of Batches
Long term 30oC ± 2oC/75% RH ± 5% RH
6 months
Min. 2 For conventional dosage form and stable drug substances Min.3 For critical dosage form or unstable drug substances
Accelerated 40oC ± 2oC/75% RH ± 5% RH
6 months
Min. 2 For conventional dosage form and stable drug substances Min.3 For critical dosage form or unstable drug substances

11
Minor Variaton (MiV)
Study Storage Condition Minimum Time Period Covered by Data at Submission
Number of Batches
Long term 30oC ± 2oC/75% RH ± 5% RH
3 months*
Min. 2 For conventional dosage form and stable drug substances
6 months
Min.3 For critical dosage form or unstable drug substances
Accelerated 40oC ± 2oC/75% RH ± 5% RH
3 months*
Min. 2 For conventional dosage form and stable drug substances
6 months
Min.3 For critical dosage form or unstable drug substances
* Example : replacement of an excipient with a comparable excipient, change in the qualitative and/or quantitative composition of the immediate packaging material, change in the batch size of the finished product, minor change in the manufacture of the finished product, change of colouring system or the flavouring system currently use in the finished product, change in coating weight of tablets or change in weight of capsule shells, and any other minor variation in ASEAN Variation Guideline.
4.8. In-use Stability
i. The purpose of in-use stability testing is to provide information for the labelling on the preparation, storage conditions and utilization period of multidose products after opening, reconstitution or dilution of a solution, e.g. an antibiotic injection supplied as a powder for reconstitution.
ii. As far as possible the test should be designed to simulate the use of the drug
product in practice, taking into consideration the filling volume of the container and any dilution or reconstitution before use. At intervals comparable to those which occur in practice appropriate quantities should be removed by the withdrawal methods normally used and described in the product literature.
iii. The physical, chemical and microbial properties of the drug product susceptible to
change during storage should be determined over the period of the proposed in-use shelf-life. If possible, testing should be performed at intermediate time points and at the end of the proposed in-use shelf-life on the final amount of the drug remaining in the container. Specific parameters, e.g. for liquids and semi-solids, preservatives, per content and effectiveness, need to be studied.
iv. A minimum of two batches, at least pilot-scale batches, should be subjected to the
test. At least one of these batches should be chosen towards the end of its shelf-life. If such results are not available, one batch should be tested at the final point of the submitted stability studies.

12
v. This testing should be performed on the reconstituted or diluted drug product
throughout the proposed in-use period on primary batches as part of the stability studies at the initial and final time points and, if full shelf-life, long term data are not available before submission, at the last time point at which data will be available.
vi. In general this testing need not be repeated on commitment batches.
4. 9. Container Closure System
i. Stability testing should be conducted on the dosage form packaged in the container closure
system proposed for marketing (including, as appropriate, any secondary packaging and container label). Any available studies carried out on the product outside its immediate container or in other packaging materials can form a useful part of the stress testing of the dosage form or can be considered as supporting information, respectively.
ii. Parameters required to classify the packaging materials as semi-permeable or impermeable
depend on the packaging material characteristics such as thickness and permeability coefficient and other relevant parameters. The suitability of the packaging material used for a particular product is determined by its product characteristics. An Example of Types, Thickness and Permeability Coefficient of Packaging Material is provided in Annex 5.4.
iii. When using moisture-permeable containers for packaging, due consideration should be given to the stability of the contents under high humidity conditions.
iv. Moisture may have an undesirable effect on chemical stability (e.g. some antibiotics may
undergo hydrolysis) and physical stability (e.g. dissolution rate may change).
v. The issue of the different permeability of various packaging materials should be addressed. Therefore, it will be necessary to specify parameters, such as the material’s thickness and permeability coefficient. Discussion should be appropriate made under P2 Pharmaceutical Development and P7 Container Closure System of the ACTD.
vi. The effect of high humidity on solid dosage forms packaged in containers permeable to
moisture should be supported by data.
4.10. Evaluation
A systematic approach should be adopted in the presentation and evaluation of the stability information, which should include, as appropriate, results from the physical, chemical and microbiological tests, including particular attributes of the dosage form (for example, dissolution rate for solid oral dosage forms). The purpose of the stability study is to establish, based on testing a minimum of two or three batches of the drug product (refer 4.7 ‘Storage Conditions’), a shelf-life and label storage instructions applicable to all future batches of the drug product manufactured and packaged under similar circumstances. The degree of variability of individual batches affects the confidence that a future production batch will remain within specification throughout its shelf-life.

13
The basic concepts of stability data evaluation are the same for single-versus multi-factor studies and for full versus reduced design studies. Data evaluation from the stability studies and as appropriate, supporting data should be used to determine the critical quality attributes likely to influence the quality and performance of the drug product. Each attribute should be assessed separately and an overall assessment made of the findings for the purpose of proposing a shelf-life. The shelf-life proposed should not exceed that predicted for any single attribute. The decision tree in Annex 5.5 outlines a stepwise approach to stability data evaluation and when and how much extrapolation can be considered for a proposed shelf-life. Annex 5.6 provides (1) information on how to analyze long term data for appropriate quantitative test attributes from a study with a multi-factor, full or reduced design, (2) information on how to use regression analysis for shelf-life estimation, and (3) examples of statistical procedures to determine poolability of data from different batches or other factors. Additional guidance can be found in the references listed. In general, certain quantitative chemical attributes (e.g. assay, degradation products, preservative content) for a drug product can be assumed to follow zero order kinetics during long term storage. Data for these attributes are therefore amenable to linear regression and pool ability testing. Although the kinetics of other quantitative attributes (e.g. pH, dissolution) is generally not known, the same statistical analysis can be applied, if appropriate. Qualitative attributes and microbiological attributes are not amenable to this kind of statistical analysis. The recommendations on statistical approaches in this guideline are not intended to imply that use of statistical evaluation is preferred when it can be justified to be unnecessary. However, statistical analysis can be useful in supporting the extrapolation of shelf lives in certain situations and can be called for to verify the proposed shelf lives in other cases.
4.10.1. Data Presentation
Data for all attributes should be presented in an appropriate format (e.g., tabular, graphical, narrative) and an evaluation of such data should be included in the application. The values of quantitative attributes at all time points should be reported as measured (e.g., assay as percent of label claim). If a statistical analysis is performed, the procedure used and the assumptions underlying the model should be stated and justified. A tabulated summary of the outcome of statistical analysis and/or graphical presentation of long term data should be included.
4.10.2. Extrapolation of Data
Extrapolation is the practice of using a known data set to infer information about future data sets. Limited extrapolation to extend the retest period or shelf-life beyond the observed range of available long term data can be proposed in the application, particularly if no significant change is observed at the accelerated condition. Any extrapolation should take into consideration the possible worst-case situation at the time of batch release. An extrapolation of stability data assumes that the same change pattern will continue to apply beyond the observed range of available long term data. Hence, the use of extrapolation should be justified in terms of, for example, what is known about the mechanisms of degradation, the goodness of fit of any mathematical model, and the existence of relevant supporting data.

14
The correctness of the assumed change pattern is crucial if extrapolation beyond the available long term data is contemplated. For example, when estimating a regression line or curve within the available data, the data themselves provide a check on the correctness of the assumed change pattern, and statistical methods can be applied to test the goodness of fit of the data to the assumed line or curve. No such internal check is available beyond the length of observed data. Thus, shelf-life granted on the basis of extrapolation should always be verified by additional long term stability data as soon as these data become available. Care should be taken to include in the protocol for commitment batches a time point that corresponds to the extrapolated shelf-life. If the long term data are supported by results from accelerated studies, the shelf-life may be extended beyond the end of long term studies. The extrapolated shelf-life may be up to twice, but should not be more than 12 months beyond, the period covered by long term data, depending on the change over time, variability of data observed, proposed storage conditions and extent of statistical analyses performed.
4.10.3 Data Evaluation for Shelf-Life Estimation for Drug Products Intended for Storage
at Room Temperature For drug products intended for storage at room temperature, the assessment should begin with any significant change at the accelerated condition and progress through the trends and variability of the long term data. The circumstances are delineated under which extrapolation of shelf-life beyond the period covered by long term data can be appropriate. A decision tree is provided in Annex 5.5 as an aid.
4.10.3.1 No significant change at accelerated condition
Where no significant change occurs at the accelerated condition, the shelf-life would depend on the nature of the long term and accelerated data.
a. Long term and accelerated data showing little or no change over time and little or no variability Where the long term data and accelerated data for an attribute show little or no change over time and little or no variability, it may be apparent that the drug product will remain well within its acceptance criterion for that attribute during the proposed shelf-life. Under these circumstances, it is normally considered unnecessary to go through a statistical analysis, but justification for the omission should be provided. Justification can include a discussion of the mechanisms of degradation or lack of degradation, relevance of the accelerated data, mass balance, and/or other supporting data.
b. Long term or accelerated data showing change over time and/or variability
If the long term or accelerated data for an attribute show change over time and/or variability within a factor or among factors, statistical analysis of the long term data can be useful in establishing a shelf-life. Where there are differences in stability observed among batches or among other factors (e.g., strength, container size and/or fill) or factor combinations (e.g., strength-by-container size and/or fill) that preclude the combining of data, the proposed shelf-life should not exceed the shortest period supported by any batch, other factor, or factor combination. Alternatively, where the differences are readily attributed to a particular factor (e.g., strength), different shelf-lives can be assigned to different levels within the factor (e.g., different strengths). A discussion should be provided to address the cause for the differences and the overall significance of such differences on the product. Extrapolation beyond the period covered by long term data can be proposed; however, the extent of extrapolation

15
would depend on whether long term data for the attribute are amenable to statistical analysis. • Data not amenable to statistical analysis
Where long term data are not amenable to statistical analysis, but relevant supporting data are provided, the proposed shelf-life can be up to one-and-a-half times, but should not be more than 6 months beyond, the period covered by long term data. Relevant supporting data include satisfactory long term data from development batches that are (1) made with a closely related formulation to, (2) manufactured on a smaller scale than, or (3) packaged in a container closure system similar to, that of the primary stability batches.
• Data amenable to statistical analysis
If long term data are amenable to statistical analysis but no analysis is performed, the extent of extrapolation should be the same as when data are not amenable to statistical analysis. However, if a statistical analysis is performed, it can be appropriate to propose a shelf-life of up to twice, but not more than 12 months beyond, the period covered by long term data, when the proposal is backed by the result of the analysis and relevant supporting data.
4.10.3.2 Significant change at accelerated condition
If a “significant change” occurs between 3 and 6 months’ testing at the accelerated storage condition, the proposed shelf-life should be based on the long term data available at the long term storage condition. Significant Change In general, “significant change” for a drug product is defined as : 1. A 5% change in assay from its initial value, or failure to meet the acceptance
criteria; 2. Any degradation product exceeding the acceptance criterion; 3. Failure to meet the acceptance criteria for appearance, physical attributes, and
functionality tests (e.g. colour, phase separation, resuspendability, caking, hardness, dose delivery per actuation); however, some changes in physical attributes (e.g., softening of suppositories, melting of creams) may be expected under accelerated conditions and as appropriate for the dosage form.
4. Failure to meet the acceptance criteria for pH; 5. Failure to meet the acceptance criteria for dissolution for 12 dosage units (capsule
or tablet).
If the “significant change” occurs within the first 3 months testing at the accelerated storage condition, a discussion should be provided to address the effect of short term excursions outside the label storage condition, e.g., during shipping or handling. This discussion can be supported, if appropriate, by further testing on a single batch of the drug product for a period shorter than 3 months but with more frequent testing than usual. It is considered unnecessary to continue to test a drug product through 6 months when a “significant change” has occurred within the first 3 months. This can be applied to products such as ointments, cream or suppositories that are impossible to test at accelerated condition where only long term testing is required *Note: The following physical changes can be expected to occur at the accelerated condition and would not be considered significant change that calls for long term testing if there is no other significant change:

16
a. softening of a suppository that is designed to melt at 37ºC, if the melting point is clearly demonstrated,
b. failure to meet acceptance criteria for dissolution for 12 units of a gelatin capsule or gel-coated tablet if the failure can be unequivocally attributed to cross-linking.
However, if phase separation of a semi-solid dosage form occurs at the accelerated condition, testing at the long term condition should be performed. Potential interaction effects should also be considered in establishing that there is no other significant change.
4.10.4. Data Evaluation for Shelf-Life Estimation for Drug Products Intended for Storage
below Room Temperature
4.10.4.1. Drug products intended for storage in a refrigerator Data from drug products intended to be stored in a refrigerator should be assessed according to the same principles as described in Section 4.10.3 for drug products intended for room temperature storage, except where explicitly noted in the section below. The decision tree in Appendix 5.5 can be used as an aid.
a. No significant change at accelerated condition
Where no significant change occurs at the accelerated condition, extrapolation of shelf-life beyond the period covered by long term data can be proposed based on the principles outlined in Section 4.10.3, except that the extent of extrapolation should be more limited.
If the long term and accelerated data show little change over time and little variability, the proposed shelf-life can be up to one-and-a-half times, but should not be more than 6 months beyond, the period covered by long term data normally without the support of statistical analysis.
Where the long term or accelerated data show change over time and/or variability, the proposed shelf-life can be up to 3 months beyond the period covered by long term data if (1) the long term data are amenable to statistical analysis but a statistical analysis is not performed, or (2) the long term data are not amenable to statistical analysis but relevant supporting data are provided.
Where the long term or accelerated data show change over time and/or variability, the proposed shelf-life can be up to one-and-a-half times, but should not be more than 6 months beyond, the period covered by long term data if (1) the long term data are amenable to statistical analysis and a statistical analysis is performed, and (2) the proposal is backed by the result of the analysis and relevant supporting data.
b. Significant change at accelerated condition If significant change occurs between 3 and 6 months testing at the accelerated storage condition, the proposed shelf-life should be based on the long term data. Extrapolation is not considered appropriate. In addition, a shelf-life shorter than the period covered by long term data could be called for. If the long term data show variability, verification of the proposed shelf-life by statistical analysis can be appropriate.

17
If significant change occurs within the first 3 months testing at the accelerated storage condition, the proposed shelf-life should be based on long term data. Extrapolation is not considered appropriate. A shelf-life shorter than the period covered by long term data could be called for. If the long term data show variability, verification of the proposed shelf-life by statistical analysis can be appropriate. In addition, a discussion should be provided to address the effect of short-term excursions outside the label storage condition (e.g., during shipping or handling). This discussion can be supported, if appropriate, by further testing on a single batch of the drug product at the accelerated condition for a period shorter than 3 months.
4.10.4.2. Drug products intended for storage in a freezer
For drug products intended for storage in a freezer, the shelf-life should be based on long term data. In the absence of an accelerated storage condition for drug products intended to be stored in a freezer, testing on a single batch at an elevated temperature (e.g., 5°C ± 3°C or 25°C ± 2°C) for an appropriate time period should be conducted to address the effect of short-term excursions outside the proposed label storage condition (e.g., during shipping or handling).
4.10.4.3. Drug products intended for storage below -20°C
For drug products intended for storage below -20°C, the shelf-life should be based on long term data and should be assessed on a case-by-case basis.
4.10.5. General Statistical Approaches Where applicable, an appropriate statistical method should be employed to analyze the long term primary stability data in an original application. The purpose of this analysis is to establish, with a high degree of confidence, a shelf-life during which a quantitative attribute will remain within acceptance criteria for all future batches manufactured, packaged, and stored under similar circumstances. This same method could also be applied to commitment batches to verify or extend the originally approved shelf-life. In cases where a statistical analysis was employed to evaluate long term data due to a change over time and/or variability, the same statistical method should also be used to analyse data from commitment batches to verify or extend the originally approved shelf-life. Regression analysis is considered an appropriate approach to evaluating the stability data for a quantitative attribute and establishing a shelf-life. The nature of the relationship between an attribute and time will determine whether data should be transformed for linear regression analysis. Usually, the relationship can be represented by a linear or non-linear function on an arithmetic or logarithmic scale. Sometimes a non-linear regression can be expected to better reflect the true relationship. An appropriate approach to shelf-life estimation is to analyze a quantitative attribute by determining the earliest time at which the 95 percent confidence limit for the mean around the regression curve intersects the proposed acceptance criterion. For an attribute known to decrease with time, the lower one-sided 95 percent confidence limit should be compared to the acceptance criterion. For an attribute known to increase with time, the upper one-sided 95 percent confidence limit should be compared to the criterion. For an attribute which can either increase or decrease, or whose direction of

18
change is not known, two-sided 95 percent confidence limits should be calculated and compared to the upper and lower acceptance criteria. If analysis shows that the batch-to-batch variability is small, it is advantageous to combine the data into one overall estimate. This can be done by first applying appropriate statistical tests (e.g., p-values for levels of significant of rejection of more than 0.25) to the slopes of the regression lines and zero time intercepts for the individual batches. If it is inappropriate to combine data from several batches, the overall shelf-life should be based on the minimum time a batch can be expected to remain within acceptance criteria. Any evaluation should consider not only the assay, but also the degradation products and other appropriate attributes. Where appropriate, attention should be paid to reviewing the adequacy of the mass balance and different stability and degradation performance. The statistical method used for data analysis should take into account the stability study design to provide a valid statistical inference for the estimated shelf-life. The approach described above can be used to estimate the shelf-life for a single batch or for multiple batches when combined after an appropriate statistical test. Examples of statistical approaches to the analysis of stability data from design study are included in Annex 5.6.
4.11. Stability Commitment 4.11.1. When available long term stability data on primary batches do not cover the
proposed shelf-life granted at the time of approval, a commitment should be made to continue the stability studies post approval in order to firmly establish the shelf-life.
4.11.2. Where the submission includes long term stability data on at least the minimum
number of production batches required covering the proposed shelf-life, a post approval commitment is considered unnecessary. Otherwise, one of the following commitments should be made:
a. If the submission includes data from stability studies on at least the minimum
number of production batches required, a commitment should be made to continue the long term studies through the proposed shelf-life and the accelerated studies for 6 months.
b. If the submission includes data from stability studies on fewer than 3
production batches, a commitment should be made to continue the long term studies through the proposed shelf-life and the accelerated studies for 6 months, and to place additional production batches, to a total of at least the minimum number of production batches required, on long term stability studies through the proposed shelf-life and on accelerated studies for 6 months.
c. If the submission does not include stability data on production batches, a
commitment should be made to place the first 3 production batches on long term stability studies through the proposed shelf-life and on accelerated studies for 6 months.
The stability protocol used for studies on commitment batches should be the same as that for the primary batches, unless otherwise scientifically justified.

19
4.11.3. Applicant must submit commitment and protocol on post approval stability study if stability study submitted has been conducted under different storage conditions and it cannot be demonstrated that the drug product will remain within its acceptance criteria stated in this guideline. In such cases, the following options should be considered: (1) a reduced shelf-life, (2) a more protective container closure system, or (3) additional cautionary statements in the labeling.
4.11.4. Post approval stability can be conducted in any ASEAN member country,
country of origin, or any country that can meet the required storage condition.
4.12. Statements/Labeling
A storage statement should be established for the labeling in accordance with relevant national/regional requirements. The statement should be based on the stability evaluation of the drug product. Where applicable, specific instructions should be provided, particularly for drug products that cannot tolerate freezing. Terms such as “ambient conditions” or “room temperature” should be avoided. There should be a direct link between the label statement and the demonstrated stability characteristics of the drug product. The storage conditions (temperature, light, humidity) indicated should refer to the relevant national/regional requirements or following the recommendations below. The range should be based on the stability evaluation of the drug product.
Table 1 Recommended labelling statements for Drug Products
a During storage, shipment and distribution of the Drug Products, the current good distribution practices (GDP) for pharmaceutical products are to be observed.
If testing conditions different from above table, the recommended labeling statement should justified with supported stability studies. In principle, Drug Products should be packed in containers that ensure stability and protect the Drug Product from deterioration. A storage statement should not be used to compensate for inadequate or inferior packaging. Additional labeling statements that could be used in cases where the result of the stability testing demonstrates limiting factors are listed in Table 2 below.
Testing condition under which the stability of the drug product has been demonstrated
Recommended labeling statement a
30 °C/75% RH (long term) 40 °C/75% RH (accelerated)
“Do not store above 30 °C”
5 °C ± 3 °C ”Store in a refrigerator (2 °C to 8 °C)” -20 °C ± 5 °C “Store in a freezer”

20
Table 2 Additional labeling statements for use where the result of the stability testing demonstrates limiting factors
Limiting factors Additional labeling statement,
where relevant Drug Products that cannot tolerate refrigeration
“Do not refrigerate or freeze”a
Drug Products that cannot tolerate freezing “Do not freeze”a Light-sensitive Drug Products “Protect from light” Drug Products that cannot tolerate excessive heat, e.g. suppositories
“Store and transport not above 30 °C”
Hygroscopic Drug Products “Store in dry conditions”
a Depending on the pharmaceutical form and the properties of the Drug Product, there may be a risk of deterioration due to physical changes if subjected to low temperatures, e.g. liquids and semi-solids. Low temperatures may also have an effect on the packaging in certain cases. An additional statement may be necessary to take account of this possibility.
1. The use of terms such as “ambient conditions” or “room temperature” is unacceptable.
2. If applicable, recommendations should also be made as to the utilization period and storage conditions after opening and dilution or reconstitution of a solution, e.g., an antibiotic injection or suspension supplied as a powder for reconstitution.
5. ANNEXES
5.1 Protocol of Stability Study (example)
5.1.1 PARACETAMOL TABLET 500 MG PACKED IN PVC BLISTER OF 10 TABLETS
1. Purpose
To evaluate stability of product due to the scaling up from the Research and Development to the Manufacturing Site.
2. Test Design
The product is packed in PVC blister and will be stored according to the storage condition mentioned in the manufacturing instruction 2.1 Test Material
- Push-through foil Alufoil of 20 micron thickness, heat-seal lacquered, PVC layered (8 g/m2),
hard temper, bright side finish silver-tinted. Forming foil
PVC foil of 250 micron thickness.

21
Batch No. Packaging type Storage Condition/Period
001 PVC Blister Long term (60 months); Accelerated (6 months)002 PVC Blister Long term (60 months); Accelerated (6 months)003 PVC Blister Long term (60 months); Accelerated (6 months)
2.2 Testing Plan
2.2.1 Storage condition and sampling intervals
Paracetamol tablet is filled and sealed in PVC blister, 10 blisters are packed in carton folding box and stored at the following storage condition:
Storage Condition Sampling Intervals
Long term 30oC/75% RH 0, 3, 6, 9, 12, 18, 24, 36, 48, 60 months Accelerated 40oC/75% RH 0, 1, 3, 6 months
The detailed schedule is attached.
2.2.2 Testing and Test Criteria
QA/QC Dept. is responsible for storing and testing the sample in accordance with the storage condition and the valid test method. The samples are taken out of the storage prior to the planned testing date, and kept at 5oC until the time for analysis. The analytical work should be concluded not later than 4 weeks after the samples have been out of storage. The testing procedure is: No. XXXX and the parameters to be tested are as follows: a. Physical test
- appearance - average weight - dissolution - disintegration time - hardness - friability - water content
b. Content : Paracetamol c. Degradation Product : p-aminophenol
3. Number of Samples (of one batch / storage condition)
Accelerated Test
- Appearance : 0* tablets - water content : 10 tablets - disintegration : 6 tablets - dissolution : 6 tablets - content & impurity : 10 tablets - hardness : 10 tablets - friability : 50 tablets = 92 tablets ~ rounded to 100 tablets

22
Number of testing : 4 times Quality needed = 4 x 100 tablets = 400 tablets = 40 blisters of 10 tablets = 4 boxes
Long Term Stability Study
- Appearance : 0* tablets - water content : 10 tablets - disintegration : 6 tablets - dissolution : 6 tablets - content & impurity : 10 tablets- hardness : 10 tablets - friability : 50 tablets = 92 tablets ~ rounded to100 tablets
* = observation made on tablets allocated for other tests Number of testing: 9 times Quality needed = 9 x 100 tablets = 900 tablets = 90 blisters 0f 10 tablets = 9 boxes
Total for long term and accelerated stability studies = 4 boxes + 9 boxes = 13 boxes of 10 blisters
4. Report Content :
1. Responsibility 2. Summary 3. Objective 4. Test Material 5. Composition 6. Packaging 7. Storage condition and testing materials (Schedule) 8. Analytical Procedures 9. Reference Standard 10. Results
10.1. Physical Stability 10.2. Chemical Stability
10.2.1. Stability under long term storage condition 10.2.2. Stability under accelerated storage condition
11. Discussion/Conclusion 12. Test result in tabular form
Approved by : Checked by: Prepared by :

23
5.1.2. Schedule for Stability Study Paracetamol Tablet 500 mg
Dated: 02.07.1997
Storage Schedule
Batch No. Batch No. Batch No.
Period Condition 001 002 003
Initial Accelerated July 02, 1997 July 09, 1997 July 16, 1997
Long term July 04, 1997 July 12, 1997 July 18, 1997
1 Month Accelerated Aug 02, 1997 Aug 09, 1997 Aug 16, 1997
3 Months Accelerated Oct 02, 1997 Oct 09, 1997 Oct 16, 1997
Long term Oct 04, 1997 Oct 12, 1997 Oct 18, 1997
6 Months Accelerated Jan 02, 1998 Jan 09, 1998 Jan 16, 1998
Long term Jan 04, 1998 Jan 12, 1998 Jan 18, 1998
9 Months Long term Apr 04, 1998 Apr 12, 1998 Apr 18, 1998
12 Months Long term Jul 04, 1998 Jul 12, 1998 Jul 18, 1998
18 Months Long term Jan 02, 1999 Jan 12, 1999 Jan 18, 1999
24 Months Long term Jul 04, 1999 Jul 12, 1999 Jul 18, 1999
36 Months Long term Jul 04, 2000 Jul 12, 2000 Jul 18, 2000
48 Months Long term Jul 04, 2001 Jul 12, 2001 Jul18, 2001
60 Months Long term Jul 04, 2002 Jul 12, 2002 Jul 18, 2002 Remarks :
Accelerated : 40oC + 2C/75% RH + 5% RH Long term: 30C + 2C/75% RH + 5% RH
Approved by: Checked by: Prepared by:

24
5.2. Report Format (example) DRUG PRODUCT: PARACETAMOL TABLET
STRENGTH: 500 mg Date: 23/07/02 Doc. No.: XXXX. Page 1 of 20 Study Type: Pre- and post-market Stability Objective: Stability profile of the drug product for storage under long
term and accelerated conditions Period of Investigation: 60 Months Packaging: PVC Blister Originating Site : MMM Ltd Jakarta – Indonesia Stability Study Unit : R&D Dept.
John Doe
Quality Assurance : Tom Smith

25
1. RESPONSIBILITY
Persons in Charge Site / Department Responsibility
John Doe R&D Physical and chemical tests
John Doe R&D Microbiological tests
2. SUMMARY
This report presents the stability data on Paracetamol tablet 500 mg stored up to 60 months in the primary packaging used for marketing. Any storage-related changes occuring in the finished product were monitored by means of stability-specified control tests. The test design was based on the stability profile of the drug substance paracetamol and on the specific requirements of the dosage form.
Shelf-life: The product has a shelf-life of five years Storage Directions: The finished product is not labelled with any storage directions.
3. OBJECTIVE
The objective of the present study on Paracetamol tablet 500 mg is the assessment of the stability profile for storage under long term and accelerated conditions. The samples were in inverted position to ensure contact with the container closure system.
4. TEST MATERIAL
The batches under stability testing are listed in the following table with further details: 4.1. Starting Material
MATERIAL PRODUCT BATCH NO SOURCE #01 #02 # 03
Paracetamol Note: Batch API ...................... ................... .................. Lactose 1H2O ............................ ...................... .................... .................. Maize Starch ............................ ...................... .................... .................. Pregelatinized Maize Starch
............................ ...................... ................... ..................
Talc ........................... ...................... .................... .................. Colloidal Anhydrous Silica (Aerosil 200)
........................... ...................... .................... ..................
Magnesium Stearate ........................... ...................... .................... ..................

26
4.2 Drug Product
Dosage Batch
No.
Manufacturing Scale
Batch Size
(Unit) Date Site
500 mg/tab 001 July 02, 1997 Jakarta Production 280000
500 mg/tab 002 July 09, 1997 Jakarta Production 280000
500 mg/tab 003 July 16, 1997 Jakarta Production 280000
5. COMPOSITION
1 tablet of Paracetamol contains :
Composition Weight [mg] Source (API produsen)
Paracetamol 500.00 .................................. Lactose 1H2O 79.00 .................................. Maize Starch 65.50 .................................. Pregelatinized Maize Starch 5.00 ................................... Talc 3.00 ................................... Colloidal Anhydrous Silica (Aerosil 200)
2.00 ...................................
Magnesium Stearate 0.50 ................................... Total 655.00
6. PACKAGING
The stability tests on the batches listed above are performed in the following primary packaging: The product is packed in PVC blister consisting of:
Push-through foil : Alufoil of 20 micron thickness, heat-seal lacquered, PVC layered (8
g/m2), hard temper, bright side finish silver-tinted. Forming foil : PVC foil of 250 micron thickness.

27
7. STORAGE CONDITIONS AND TESTING INTERVALS
The various samples of the packaged drug product have been / will be tested according to the following schedule:
Storage Condition Months
0 1 3 6 9 12 18 24 36 48 60
30C + 2C/75% RH
+ 5% RH X - X X X X X X X X X
40C + 2C/75% RH
+ 5% RH X X X X - - - - - - -
8. ANALYTICAL PROCEDURES
The stability tests on Paracetamol were performed according to the control tests of USP. In the course of the stability testing the main emphasis was put on the stability-relevant test items as listed below:
Test Item Control Test No. Specification
Hardness USP > 70 N
Friability USP < 2%
Degradation Product USP
p-aminophenol < 0.005%
Microbial Contamination USP Total count < 102 CFU E.coli : absent
Content (LC) USP 95.0 – 105.0 % Note: As mentioned in 2.1.2, 3.1 and 3.2, Disintegration Time and Dissolution should be
added.
9. REFERENCE STANDARD
Standard Paracetamol USP, 99.5%, was used.
10. RESULTS
The test results of the study are presented in the tables attached. Physical Stability The physical stability of Paracetamol tablet 500 mg proved to be unchanged after storage up to 60 months at 30oC/75% RH and after 6 months under accelerated conditions at 40oC/75% RH.

28
The result obtained for the test item’s "appearance" was not changed significantly.
Chemical Stability Stability under Long term Conditions Storage for up to 60 months at 30oC/75% RH had no significant effect on the chemical stability of the drug product. With regard to test item "Organic Impurity" only slight changes were observed. The p-aminophenol concentration was below 0.005%. The content of Paracetamol did not change significantly after storage under long term conditions compared to initial assay of the batches.
Stability under Accelerated Conditions
Storage under accelerated conditions for 6 months did not affect the chemical stability. The content of paracetamol was not significantly changed compared to the initial value of the batches.
11. DISCUSSION / CONCLUSIONS Storage under long term testing conditions causes insignificant change of assay results of paracetamol. Significant changes in physical and chemical stabilities were not observed. Since the long-term data and accelerated data show little or no change over time and little variability, a statistical analysis is considered unnecessary.
Shelf-life: Based on the resulting data the shelf-life has been established for five years.
Storage Directions: The product can be labelled with ”Store below 30oC”
29
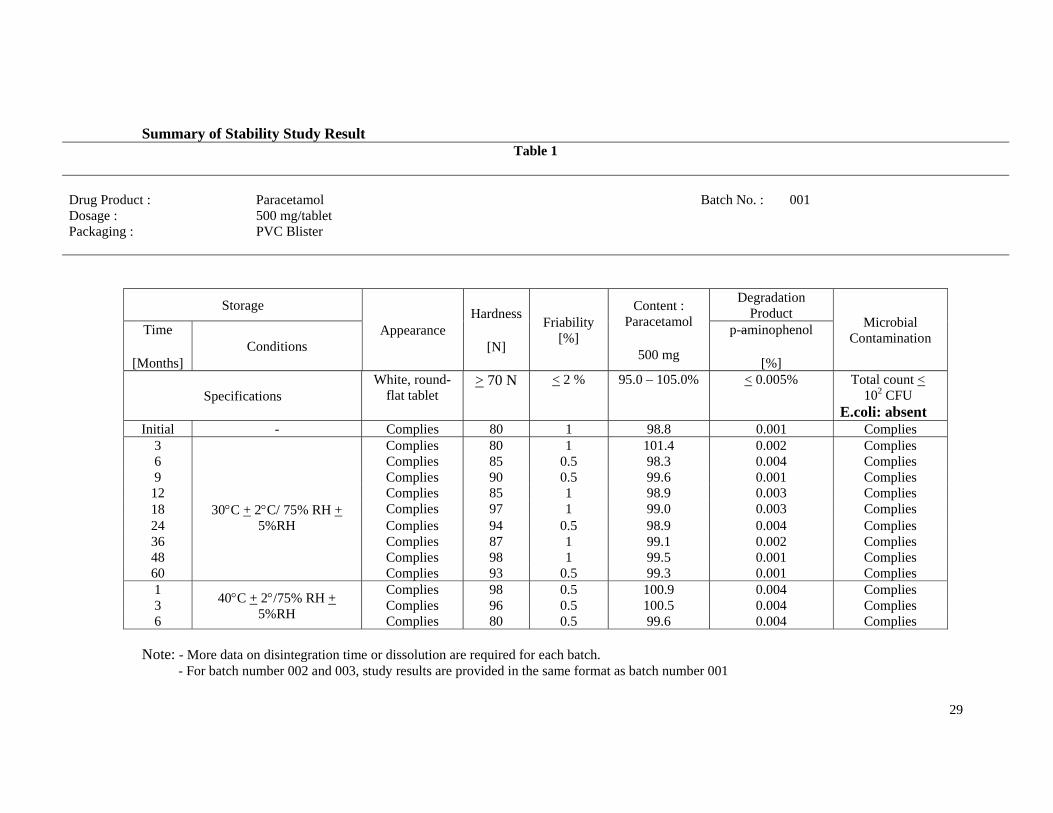
Summary of Stability Study Result Table 1
Drug Product : Paracetamol Batch No. : 001 Dosage : 500 mg/tablet Packaging : PVC Blister
Storage
Appearance Hardness
[N]
Friability [%]
Content : Paracetamol
500 mg
Degradation Product
Microbial Contamination
Time
[Months] Conditions
p-aminophenol
[%]
Specifications White, round-
flat tablet > 70 N < 2 % 95.0 – 105.0% < 0.005% Total count <
102 CFU E.coli: absent
Initial - Complies 80 1 98.8 0.001 Complies 3 Complies 80 1 101.4 0.002 Complies 6 Complies 85 0.5 98.3 0.004 Complies 9 Complies 90 0.5 99.6 0.001 Complies12 Complies 85 1 98.9 0.003 Complies 18 30C + 2C/ 75% RH + Complies 97 1 99.0 0.003 Complies 24 5%RH Complies 94 0.5 98.9 0.004 Complies 36 Complies 87 1 99.1 0.002 Complies 48 Complies 98 1 99.5 0.001 Complies 60 Complies 93 0.5 99.3 0.001 Complies 1
40C + 2/75% RH + 5%RH
Complies 98 0.5 100.9 0.004 Complies 3 Complies 96 0.5 100.5 0.004 Complies 6 Complies 80 0.5 99.6 0.004 Complies
Note: - More data on disintegration time or dissolution are required for each batch.
- For batch number 002 and 003, study results are provided in the same format as batch number 001

30
5.3 Reduced Design (Bracketing and Matrixing)
A full study design is one in which samples for every combination of all design factors are tested at all time points. A reduced design is one in which samples for every factor combination is not all tested at all time points. A reduced design can be a suitable alternative to a full design when multiple design factors are involved. Any reduced design should have the ability to adequately predict the shelf-life. Before a reduced design is considered, certain assumptions should be assessed and justified. The potential risk should be considered of establishing a shorter shelf-life than could be derived from a full design due to the reduced amount of data collected. During the course of a reduced design study, a change to full testing or to a less reduced design can be considered if a justification is provided and the principles of full designs and reduced designs are followed. However, proper adjustments should be made to the statistical analysis, where applicable, to account for the increase in sample size as a result of the change. Once the design is changed, full testing or less reduced testing should be carried out through the remaining time points of the stability study. Applicability of Reduced Designs Reduced designs can be applied to the stability study of most types of drug products, although additional justification should be provided for certain complex drug delivery systems where there are a large number of potential drug-device interactions. Bracketing Bracketing is the design of a stability schedule such that only samples on the extremes of certain design factors (e.g., strength, container size and/or fill) are tested at all time points as in a full design. The design assumes that the stability of any intermediate levels is represented by the stability of the extremes tested. Design Example An example of a bracketing design is given in Table 1. This example is based on a product available in three strengths and three container sizes (P1, P2 and P3). In this example, it should be demonstrated that the 15 ml (P1) and 500 ml (P3) high-density polyethylene container sizes truly represent the extremes. The batches for each selected combination should be tested at each time point as in a full design.
Table 1: Example of a Bracketing Design
Strength 50 mg 75 mg 100 mg Batch 1 2 3 1 2 3 1 2 3Container size
15 ml T T T T T T 100 ml 500 ml T T T T T T
Key: T = Sample tested The bracketing design assumes that the stability of the intermediate strengths or sizes is represented by the stability at the extremes. If the statistical analysis indicates that the stability of the extreme strengths or sizes is different, the intermediate strengths or sizes should be considered no more stable than the least stable extreme. For example, if P1 from the above bracketing design is found to be less stable than P3, the shelf-life for P2 should not exceed that for P1. No interpolation between P1 and P3 should be considered.

31
Matrixing Matrixing is the design of a stability schedule such that a selected subset of the total number of possible samples for all factor combinations would be tested at a specified time point. At a subsequent time point, another subset of samples for all factor combinations would be tested. The design assumes that the stability of each subset of samples tested represents the stability of all samples at a given time point. The differences in the samples for the same drug product should be identified as, for example, covering different batches, different strengths, different sizes of the same container closure system, and possibly, in some cases, different container closure systems. When a secondary packaging system contributes to the stability of the drug product, matrixing can be performed across the packaging systems. Each storage condition should be treated separately under its own matrixing design. Matrixing should not be performed across test attributes. However, alternative matrixing designs for different test attributes can be applied if justified. Design Examples Examples of matrixing designs on time points for a product with two strengths (S1 and S2) are shown in Table 2. The terms “one-half reduction” and “one-third reduction” refer to the reduction strategy initially applied to the full study design. For example, a “one-half reduction” initially eliminates one in every two time points from the full study design and a “one-third reduction” initially removes one in every three. In the examples shown in Table 2, the reductions are less than one-half and one-third due to the inclusion of full testing of all factor combinations at some time points. These examples include full testing at the initial, final, and 12- month time points. The ultimate reduction is therefore less than one-half (24/48) or one-third (16/48), and is actually 15/48 or 10/48, respectively. Table 2: Examples of Matrixing Designs on Time Points for a Product with Two Strengths
“One-Half Reduction”
Time point (months) 0 3 6 9 12 18 24 36 STRENGTH
S1 Batch 1 T T T T T T Batch 2 T T T T T T Batch 3 T T T T T
S2 Batch 1 T T T T TBatch 2 T T T T T T Batch 3 T T T T T
Key: T = Sample tested
“One-Third Reduction” Time point (months) 0 3 6 9 12 18 24 36
STRENGTH
S1 Batch 1 T T T T T T Batch 2 T T T T T T Batch 3 T T T T T T T
S2 Batch 1 T T T T T T T Batch 2 T T T T T T Batch 3 T T T T T T
Key: T = Sample tested
More details are described in ICH Q1D.

32
5.4. Example of Types, Thickness and Permeability Coefficient of Packaging Materials can be seen in Table-1 and Permeability to Vapour of Various Packaging Materials can be seen in Figure-1.
Table-1: Example of Types, Thickness and Permeability Coefficient of Packaging Materials
No. Material Thickness
Thickness Commonly
Used (µm)
SPECIFICATION PERMEABLITY
Thermo- formability
At 23°C / 85%RH (g/m².d)
At 38°C / 90%RH (g/m².d)
1 PVC 250 µm 200 - 250 µm 1,6 - 1,8 3,0 - 3,2 Good (Polyvinyl Chloride) 2 Duplex (PVC + PVDC) 270 µm Good /
Excellent PVC (Polyvinyl Chloride) 200 – 250 µm PVDC 5 µm for spread of 10 g/m² 40g/m² 0,15 0,6 (Polyvinylidene Chloride) (40 - 60 - 80 g/m²) 60g/m² 0,1 0,4 80g/m² 0,05 0,3 3 Triplex (PVC + PE + PVDC) 300 µm Good/Excellent
(according to thickness)
PVC (Polyvinyl Chloride) 200 – 250 µm PE (Polyethylene) 25 µm PVDC 5 µm for spread of 10 g/m² 40g/m² 0,12 0,55 (Polyvinylidene Chloride) (40 - 60 - 90 g/m²) 60g/m² 0,06 0,35 90g/m² 0,02 0,2 4 Starflex (PVC + TE + PVDC) Max. 300 µm Good/Excellent
(according to thickness)
PVC (Polyvinyl Chloride) 200 - 250 µm TE (Thermolast) Spreading TE (coating) 5 g/m² PVDC (Polyvinylidene 5 µm for spread of 10 g/m² 60g/m² 0,06 0,35 Chloride) (60 - 90 - 120 g/m²) 90g/m² 0,03 0,2 120g/m² 0,01 0,15 5 PVC + ACLAR 270 µm Excellent PVC (Polyvinyl Chloride) 200 - 250 µm
ACLAR (Polyfluor Carbonat) 15-23-51 µm 15g/m² - 0,39 23g/m² - 0,22 51g/m² - 0,11 6 PVC/PE/ACLAR 280 µm Excellent PVC (Polyvinyl Chloride) 200- 250 µm PE (Polyester) 25 µm ACLAR (pfc) 15 - 51 µm 15 µm - < 0.32 51µm - < 0.11 7 Aluminum Cold Forming 130 µm - 0 Excellent Aluminum 40 µm - 45 µm - - PVC rigid 60 µm - - OPA 25 µm - - 8 Aluminum Foil Hard Temper (Lidding Foil) 20 µm - - Alublister for PVC Foil - - - Aluminum 20 µm - - - PVC min. 7 g/m² - - Alublister for PVC - PVDC Foil 30 µm - - - Aluminum 20 µm - - - PVDC 15 g/m² - - 9 Aluminum Foil for Soft Temper 40 µm - - - Aluminum 30 µm - PVDC 15 g/m²

33
Figure 1 Permeability to Vapour of Various Packaging Materials (Method ASTM F1249, 38C/90%RH)
4,00
3,00
0,750,55 0,55 0,50
0,39 0,35 0,35 0,35 0,28 0,25 0,22 0,20 0,13 0,110,00
0,00
0,50
1,00
1,50
2,00
2,50
3,00
3,50
4,00
4,50
PET 200
PVC 250
PVDC 250/4
0gr
PVDC 250/6
0gr
TRIPLEX 25
0/25/4
0gr
PP 300
ACLAR 25
4/15
COC/PVC 35/19
0/35
TRIPLEX 25
0/25/6
0gr
STARFLEX 25
0/5gr/
60gR
COC/PVC 35/24
0/35
TRIPLEX 25
0/25/9
0gr
ACLAR 25
4/23
STARFLEX 25
0/5gr/
90gr
COC/PVDC 60/24
0/90g
rACLA
R 254/5
1
ALU C
OLD FORMIN
G
g/m
2 .d
Per
mea
bilit
y to
vap
our
Type of Various Packaging Materials Permeable Impermeable

34
5.5. Decision Tree for Data Evaluation for Shelf Life Estimation for Drug Products (excluding Frozen Products)
Yes
Yes
Yes
No
No to (1) Or (2)
No to (1) or (2) or both
Yes to both
No to (1) Or (2)
Yes to both
Yes
Yes
Yes
Yes
No
No to (1) Or (2)
No to (1) or (2) or both
Yes to both
No to (1) Or (2)
Yes to both
Yes
Yes
Tabulate and/or plot stability data on all
attributes at all storage condition and evaluate
each attribute separately
Significant change at
accelerated condition within
6 months?
Significant change at
accelerated condition within
3 months?
Long term data show: (1) little or no change over time and (2) little or no variability?
Accelerated data show: (1) little or no change over time and (2) little or no variability?
No Extrapolation; shorter shelf life and data covering
excursions can be called for; statistical analysis if real time
data show variability
No Extrapolation; shorter shelf life can be called for;
statistical analysis if real time data show variability
Intended to be stored in a
refrigerator?
(1) Long term data amenable to statistical analysis and (2) statistical
analysis perfomed?
No
No to (1) or (2) or both
No
(1) Long term data amenable to
statistical analysis and (2) statistical analysis
perfomed?
If backed by relevant supporting data:
Y = up to X + 3 months
If backed by statistical analysis and relevant
supporting data: Y = up to 1.5X, but not
exceeding X + 6 months
Yes
Statistical analysis is normally unnecessary
Y= up to 2X, but not exceeding X + 12 months;
or if refrigerated, Y= up to 1.5X, but not exceeding X + 6 month
If backed by statistical analysis and relevant
supporting data: Y = up to 2X, but not exceeding X +
12 months: or if refrigerated Y = up to 1.5 X, but not exceeding X + 6 months
If backed by relevant supporting data: Y = up
to 1.5X, but not exceeding X + 6 months; or if refrigerated, Y = up
to X + 3 months
Y = Proposed shelf life X = Period covered by real time data
Yes to both
Yes to both
No
No to (1) or (2)
No to (1) or (2) or both
Yes to both
No to (1) or (2)
Yes to both
Yes
Yes

35
5.6. Examples of Statistical Approaches to Stability Data Analysis
Linear regression, poolability tests, and statistical modeling, described below, are examples of statistical methods and procedures that can be used in the analysis of stability data that are amenable to statistical analysis for a quantitative attribute for which there is a proposed acceptance criterion. Data Analysis for a Single Batch In general, the relationship between certain quantitative attributes and time is assumed to be
linear1
. Figure 1 shows the regression line for assay of a drug product with upper and lower acceptance criteria of 105 percent and 95 percent of label claim, respectively, with 12 months of long term data and a proposed shelf-life of 24 months. In this example, two-sided 95 percent confidence limits for the mean are applied because it is not known ahead of time whether the assay would increase or decrease with time (e.g., in the case of an aqueous-based product packaged in a semi-permeable container). The lower confidence limit intersects the lower acceptance criterion at 30 months, while the upper confidence limit does not intersect with the upper acceptance criterion until later. Therefore, the proposed shelf-life of 24 months can be supported by the statistical analysis of the assay, provided the recommendations in Sections 4.10.1 and 4.10.2 are followed.
When data for an attribute with only an upper or a lower acceptance criterion are analyzed, the corresponding one-sided 95 percent confidence limit for the mean is recommended. Figure 2 shows the regression line for a degradation product in a drug product with 12 months of long term data and a proposed shelf-life of 24 months, where the acceptance criterion is not more than 1.4 percent. The upper one-sided 95 percent confidence limit for the mean intersects the acceptance criterion at 31 months. Therefore, the proposed shelf-life of 24 months can be supported by statistical analysis of the degradation product data, provided the recommendations in Sections 4.10.1 and 4.10.2 are followed.

36
If the above approach is used, the mean value of the quantitative attribute (e.g., assay, degradation products) can be expected to remain within the acceptance criteria through the end of the shelf-life at a confidence level of 95 percent.
6. GLOSSARY
Accelerated Testing Studies designed to increase the rate of chemical degradation or physical change of a drug substance or drug product by using exaggerated storage conditions as part of the formal stability studies. (Data from these studies, in addition to long term stability studies, can be used to assess longer term chemical effects at non-accelerated condition and to evaluate the effect of short term excursions outside the label storage conditions such as might occur during shipping. Results from accelerated testing studies are not always predictive of physical changes; see also Stability and related terms) Batch A defined quantity of starting material, packaging material or Drug Product processed in a single process or series of processes so that it is expected to be homogeneous. It may sometimes be necessary to divide a batch into a number of sub-batches, which are later brought together to form a final homogeneous batch. In the case of terminal sterilization, the batch size is determined by the capacity of the autoclave. In continuous manufacture, the batch must correspond to a defined fraction of the production, characterized by its intended homogeneity. The batch size can be defined either as a fixed quantity or as the amount produced in a fixed time interval. Bracketing The design of a stability schedule such that only samples on the extremes of certain design factors, e.g., strength, package size, are tested at all time points as in a full design. (The design assumes that the stability of any intermediate level is represented by the stability of the extremes tested. Where a range of strengths is to be tested, bracketing is applicable if the strengths are identical or very closely related in composition [e.g., for a tablet range made with different compression weights of a similar basic granulation, or a capsule range made by filling different plug fill weight of the same basic composition into different size capsule shell]. Bracketing can be applied to different container sizes or different fills in the same container closure system).

37
Climatic Zones Climatic Zone
Definition
Long-term testing conditions
I Temperate climate
21 °C / 45% RH
II Subtropical and Mediterranean climate
25 °C / 60% RH
III Hot and dry climate
30 °C / 35% RH
IVA Hot and humid climate
30 °C / 65% RH
IVB Hot and very humid climate
30 °C / 75% RH
Commitment batches Production batches of a drug substance or drug product for which the stability studies are initiated or completed post approval through a commitment made in the registration application. Container Closure System The sum of packaging components that together contain and protect the dosage form. This includes primary packaging components and secondary packaging components, if latter are intended to provide additional protection to the drug product. A packaging system is equivalent to a container closure system. Dosage Form A pharmaceutical product type (e.g., tablet, capsule, solution, cream) that contains a drug substance generally, but not necessarily, in association with excipients. Drug Product/Pharmaceutical Product Any preparation for human use that is intended to modify or explore physiological systems or pathological states for the benefit of the recipient. Drug Substance The unformulated drug substance that may subsequently be formulated with excipients to produce the dosage form.(See also Active Pharmaceutical Ingredient in the Glossary of Terms of ACTD Quality) Excipient An ingredient, added intentionally to the drug substance, which should not have pharmacological properties in the quantity used. Expiry Date The date placed on the container label of a drug product designating the time prior to which a batch of the product is expected to remain within the approved shelf-life specification if stored under defined conditions. (After the expiry date, there is no guarantee that the product will remain within the approved specifications and, therefore, it may be unsuitable for use and should not be used). Formal Stability Studies Long term and accelerated studies undertaken on primary and/or commitment batches according to a prescribed stability protocol to establish or confirm the shelf-life of a drug product. Impermeable Containers Containers that provide a permanent barrier to the passage of gases or solvents, e.g., sealed aluminum tubes for semi-solids, sealed glass ampoules for solutions and aluminium/aluminium blisters for solid dosage forms.

38
Long Term Testing Stability studies under the recommended storage condition for the re-test period or shelf life proposed (or approved) for labeling.
Major Variation (MaV) Variation to authorized pharmaceutical product affecting one or more of the following aspects : • route of administration • strength, posology • indication, or • or that does not fall within the definition of minor variation (Applications for major variations usually require the submission of data necessary to establish quality, safety and efficacy of the new formulation resulting from the variation).
Mass Balance The process of adding together the assay value and levels of degradation products to see how closely these add up to 100% of the initial value, with due consideration of the margin of analytical error. Matrixing The design of a stability schedule such that a selected subset of the total number of possible samples for all factors combinations is tested at a specified time point. (At a subsequent time point, another subset of samples for all factor combinations is tested. The design assumes that the stability of each subset of samples tested represents the stability of all samples at a given time point; the differences in the samples for the same drug product should be identified as, for example, covering different batches, different strengths, different sizes of the same container closure system, and, possibly in some cases, different container closure systems).
Minor Variation (MiV) Variation to authorized pharmaceutical product not affecting one or more of the following aspects :
• route of administration • strength, posology • indications, and • active ingredient(s)
(Applications for minor variations usually require the submission of data necessary to establish quality of the new formulation resulting from the variations). Pilot Scale Batch A batch of a drug product manufactured by a procedure fully representative of and simulating that to be applied to a full production scale batch. (For solid oral dosage forms, a pilot scale is generally, at a minimum, one-tenth that of a full production scale or 100,000 tablets or capsules, whichever is the larger unless otherwise justified). Primary Batch A batch of a drug product used in a stability study, from which stability data are submitted in a registration application for the purpose of establishing a re-test period or shelf-life, respectively. For a drug product, two of the three batches should be at least pilot scale batch, and the third batch can be smaller if it is representative with regard to the critical manufacturing steps. However, a primary batch may be a production batch). Production Batch A batch of a drug product manufactured at production scale by using production equipment in a production facility as specified in the application.

39
Semi-Permeable Containers Containers that allow the passage of solvent, usually water, while preventing solute loss. The mechanism for solvent transport occurs by absorption into one container surface, diffusion through the bulk of the container material, and desorption from the other surface. Transport is driven by a partial-pressure gradient. Examples of semi-permeable containers include plastic bags and semi-rigid, low-density polyethylene (LDPE) pouches for large volume parenterals (LVPs), and LDPE ampoules, bottles and vials. Shelf-life (also referred to as expiration dating period) The time period during which a drug product is expected to remain within the approved shelf life specification, provided that it is stored under the condition defined on the container label. Specification A list of tests, references to analytical procedures, and appropriate acceptance criteria which are numerical limits, ranges, or other criteria for the tests described. (It establishes the set of criteria to which a drug substance, drug product or material at other stages of its manufacture should conform to be considered acceptable for its intended use. "Conformance to specification" means that the drug substance and drug product, when tested according to the listed analytical procedures, will meet the acceptance criteria. Specifications are critical quality standards that are proposed and justified by the manufacturer and approved by regulatory authorities as conditions of approval). Specifications – Release The specifications that determine the suitability of a drug product at the time of its release. (See also Specification) Specifications – Shelf-life The specifications that determine the suitability of a drug substance throughout its re-test period, or that a drug product should meet throughout its shelf-life. Stability The ability of an active ingredient or a drug product to retain its properties within specified limits throughout its shelf-life. (The chemical, physical, microbiological and biopharmaceutical aspects of stability must be considered). Stability Studies Long term and accelerated (and intermediate) studies undertaken on primary and/or commitment batches according to a prescribed stability protocol to establish or confirm the re-test period of a drug substance or shelf-life of a drug product. Storage Condition Tolerances The acceptable variations in temperature and relative humidity of storage facilities for formal stability studies. (The equipment should be capable of controlling the storage condition within the ranges defined in the current relevant guidelines. The actual temperature and humidity - when controlled - should be monitored during stability storage. Short-term spikes due to opening of doors of the storage facility are accepted as unavoidable. The effect of excursions due to equipment failure should be addressed, and reported if judged to affect stability results. Excursions that exceed the defined tolerances for more than 24 hours should be described in the study report and their effect assessed). Stress Testing (Drug Product) Studies undertaken to assess the effect of severe condition on the drug product. (Such studies include photo-stability testing; - see ICH Q1B - and specific testing on certain products, e.g., metered dose inhalers, creams, emulsions, refrigerated aqueous liquid products).

40
Supporting Data Data, other than those from formal stability studies, that support the analytical procedures, the proposed re-test period or shelf-life, and the label storage statements. (Such data include (1) stability data on early synthetic route batches of drug substance, small scale batches of materials, investigational formulations not proposed for marketing, related formulations, and product presented in containers and closures other than those proposed for marketing; (2) information regarding test results on containers; and (3) other scientific rationales).
REFERENCES
1. Note for Guidance on Stability Testing of Existing Active Substance and Related Finished Product (Draft), February 2002, The European Agency for The Evaluation of Medicinal Product (EMEA)
2. ICH Q1A (R2) Guideline on Stability Testing of New Drug Substances and Product, February 2003 and its annexes (Q1B Photostability Testing of New Drug Substances and Producsts, Q1C Stability Testing : Requirements for New Dosage Forms, Q1D Bracetting and Matrixing Designs for Stability Testing of New Drug Substances and Products, Q1E Evaluation for Stability Data, Q1F Stability Data Package for Registration Applications in Climatic Zones III and IV).
3. Guidelines for Stability Testing of Pharmaceutical Products Containing Well Established Drug Substances in Conventional Dosage Form, WHO Technical Report Series No. 863, 1996.
5. Carstensen, J.T., “Stability and Dating of Solid Dosage Forms,” Pharmaceutics of Solids and Solid Dosage Forms, Wiley-Interscience, 182-185, 1977.
6. Ruberg, S.J. and Stegeman, J.W., “Pooling Data for Stability Studies: Testing the Equality of Batch Degradation Slopes,” Biometrics, 47:1059-1069, 1991.
7. Ruberg, S.J. and Hsu, J.C, “Multiple Comparison Procedures for Pooling Batches in Stability Studies,” Technometrics, 34:465-472, 1992.
8. Shao, J. and Chow, S.C., “Statistical Inference in Stability Analysis,” Biometrics, 50:753-763, 1994.
9. Murphy, J.R. and Weisman, D., “Using Random Slopes for Estimating Shelf-life,” Proceedings of American Statistical Association of the Biopharmaceutical Section, 196-200, 1990.
10. Yoshioka, S., Aso, Y, and Kojima, S., “Assessment of Shelf-life Equivalence of Pharmaceutical Products,” Chem. Pharm. Bull., 49:1482-1484, 1997.
11. Chen, J.J., Ahn, H., and Tsong, Y., “Shelf-life Estimation for Multifactor Stability Studies,” Drug Inf. Journal, 31:573-587, 1997.
12. Fairweather, W., Lin, T.D., and Kelly, R., “Regulatory, Design, and Analysis Aspects of Complex Stability Studies,” J. Pharm. Sci., 84 (11): 1322 – 1326, 1995.
13. WHO Expert Committe on Specifications for Pharmaceutical Preparation, Annex 2 : Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products, WHO Technical Report Series No. 953, 2009.
14. WHO Expert Committee on Specifications forPharmaceutical Preparations, Annex 6: Guidance on Variations to a Prequalified Product Dossier, WHO Technical Report Series No. 943, 2007.


















