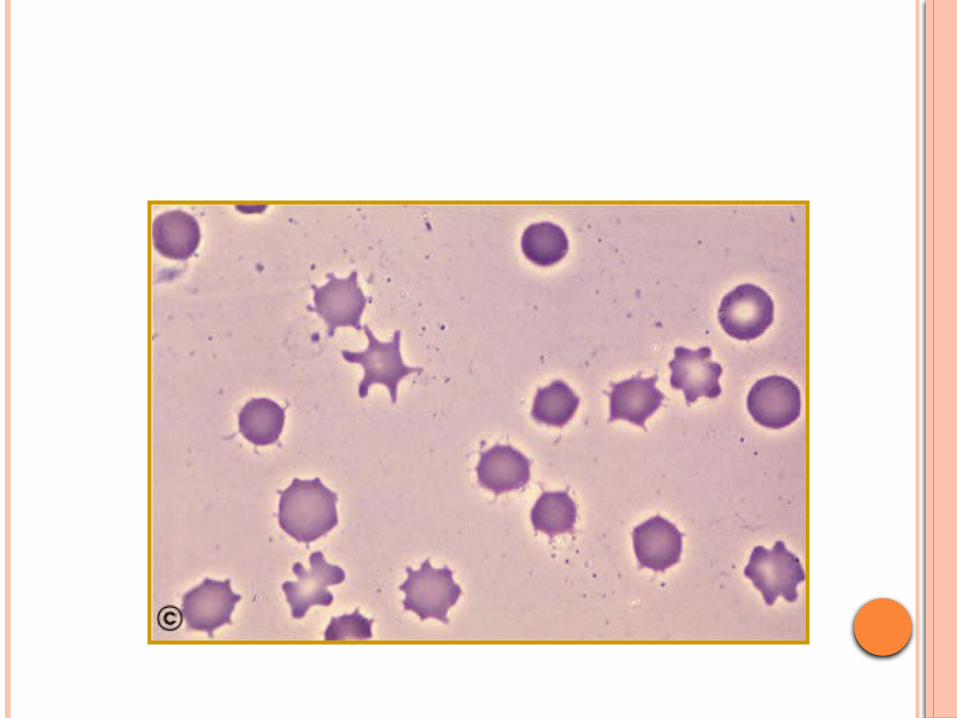
Approach to hemolytic anemia naglaa
61
APPROACH TO HEMOLYTIC ANEMIA Dr NAGLAA MAKRAM CONSULTANT CLINICAL PATHOLOGY
-
Upload
naglaa-makram -
Category
Documents
-
view
286 -
download
4
Transcript of Approach to hemolytic anemia naglaa

APPROACH TO HEMOLYTIC ANEMIA
Dr NAGLAA MAKRAM
CONSULTANT CLINICAL PATHOLOGY

OBJECTIVES
Lab indication of hemolysis
Intravascular v/s extravascular hemolysis
D/D of hemolytic anemia
Diagnose hemo. anemia with peripheral smear &
ancillary lab tests

1ST STEP IN APPROACH TO HAEMOLYTIC ANEMIA
1. Check the reticulocyte count to determine if the anemia is from decreased production (“hypoproliferative”, “reticulocytopenic”) or increased destruction (“hemolytic”)/acute blood loss (“reticulocytosis”)
3. If the the reticulocyte count is increased- Check a direct Coomb’s test
4. Look at the peripheral blood smear to confirm/support the diagnosis
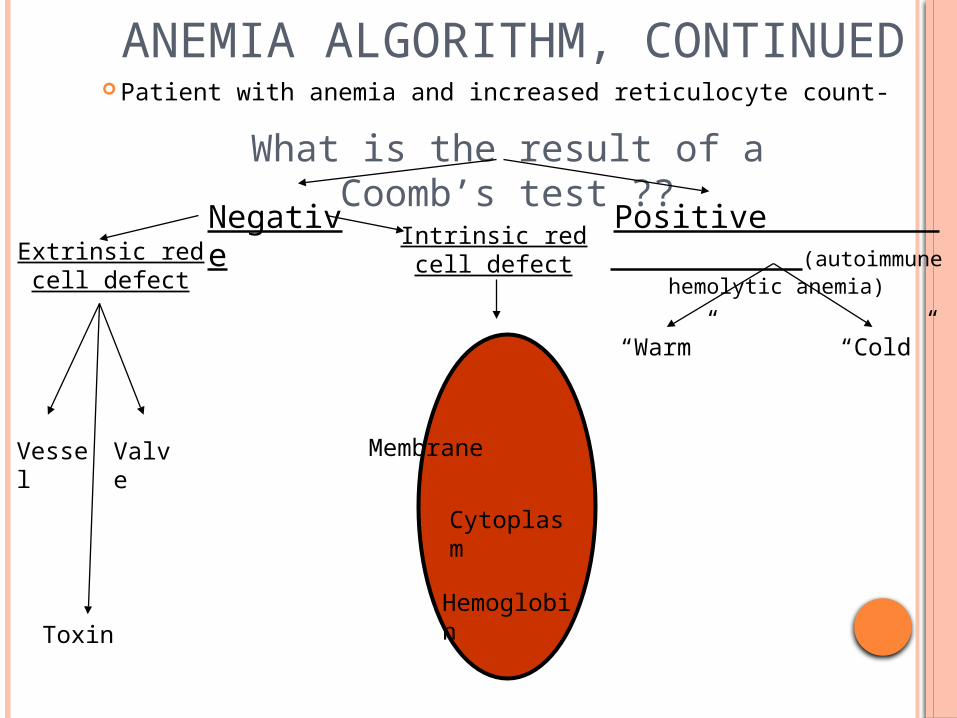
ANEMIA ALGORITHM, CONTINUED Patient with anemia and increased reticulocyte count-
What is the result of a Coomb’s test ??
Extrinsic red cell defect
Vessel Valve
Toxin
Negative Positive (autoimmune hemolytic
anemia)
Intrinsic red cell defect
Membrane
Hemoglobin
Cytoplasm
“Warm” “Cold”

Random hemolysis—:
In addition to the death of aging (senescent) red blood cells (RBCs), there is age-independent RBC destruction (random hemolysis) in normal subjects in range of less than 0.05 to 0.5 percent per day.
When hemolysis occurs, the degree of anemia is minimized by a compensatory increase in the secretion of erythropoietin (EPO), which enhances RBC production .
This response is manifested initially by an increase in the reticulocyte percentage and absolute reticulocyte count, followed by an increase in hemoglobin (Hgb) concentration.

HOW IS HEMOLYTIC ANEMIA DIAGNOSED?
Two main principles
One is to confirm that it is hemolysis
Two is to determine the etiology
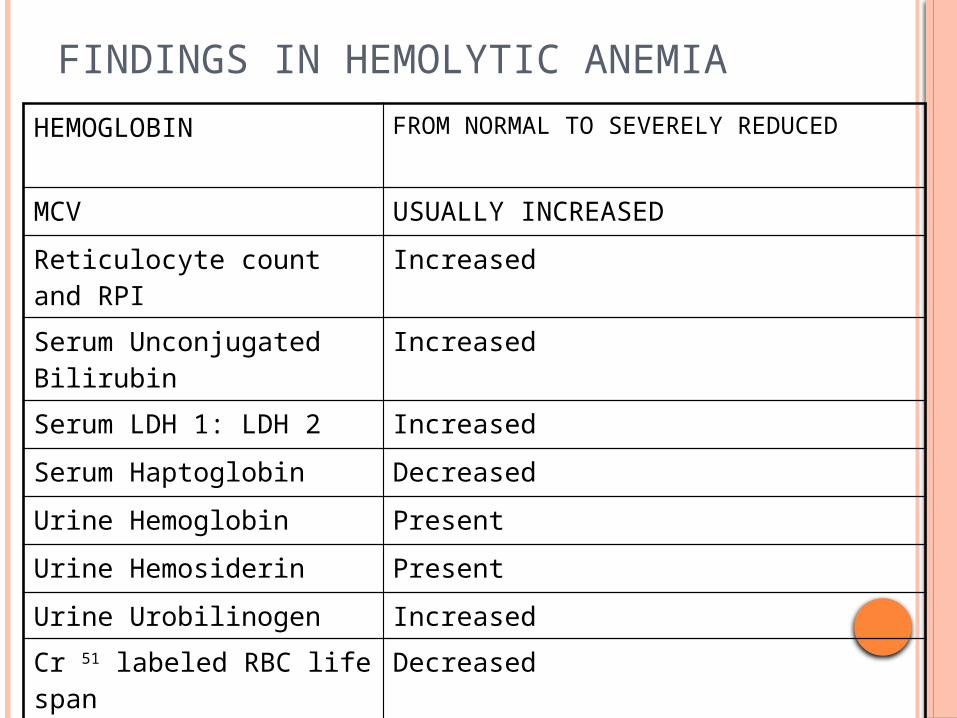
FINDINGS IN HEMOLYTIC ANEMIA
HEMOGLOBIN FROM NORMAL TO SEVERELY REDUCED
MCV USUALLY INCREASED
Reticulocyte count and RPI Increased
Serum Unconjugated Bilirubin Increased
Serum LDH 1: LDH 2 Increased
Serum Haptoglobin Decreased
Urine Hemoglobin Present
Urine Hemosiderin Present
Urine Urobilinogen IncreasedCr 51 labeled RBC life span Decreased
Acid hemolysis test (PNH)

LAB FINDINGS AND INVESTIGATIONS
Assess Iron Overload via :
• Serum iron level is elevated, with saturation as high as 80%.
• ferritin and transferrin – elevated
• Aim: Serum ferritin <1000ng/ml.
Red cell phenotyping (ideal)–transfusion
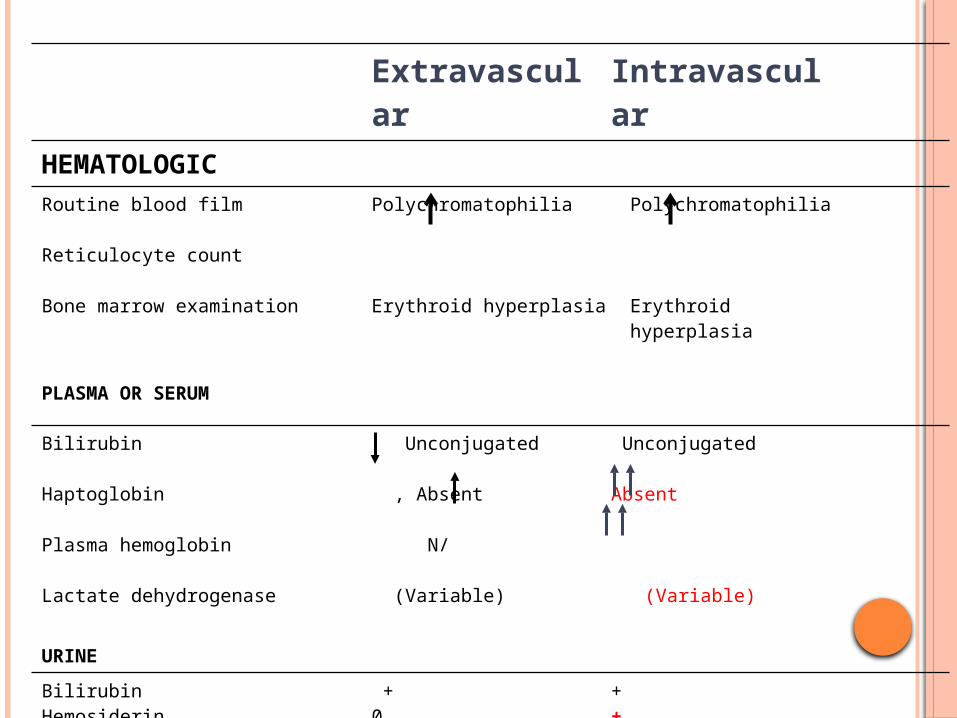
Extravascular Intravascular
HEMATOLOGIC
Routine blood film
Reticulocyte count
Bone marrow examination
Polychromatophilia
Erythroid hyperplasia
Polychromatophilia
Erythroid hyperplasia
PLASMA OR SERUM
Bilirubin
Haptoglobin
Plasma hemoglobin
Lactate dehydrogenase
Unconjugated
, Absent
N/
(Variable)
Unconjugated
Absent
(Variable)
URINE
BilirubinHemosiderinHemoglobin
+00
+++ severe cases
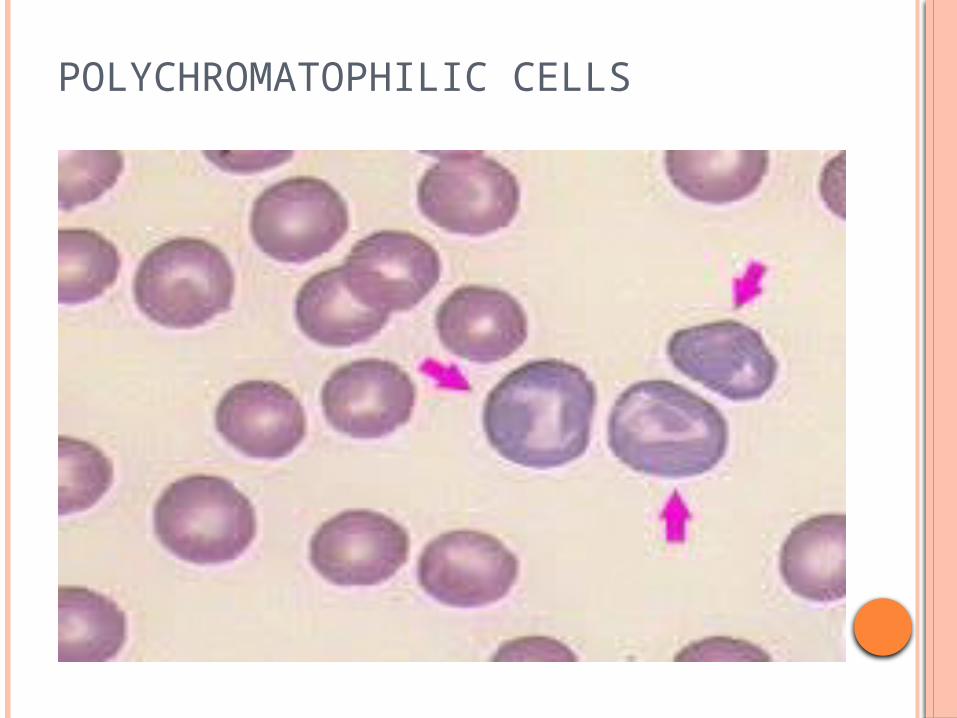
POLYCHROMATOPHILIC CELLS

CORRECTING RETIC COUNT
Retic Index = Retic % x Patient Hct Normal Hct
Absolute Retic = Retic % x RBC/mm3
Retic Production Index = Retic Index Days in circulation

THE KEY TO THE ETIOLOGY OF HEMOLYTIC ANEMIA
The history
The peripheral blood film

CASE 1
3 yr old male child presenting with pallor, jaundice,
Severe pain of long bones, fever CBC-anemia ,reticulocytosis , increased WBC LAB - LDH -600 (normal upto 200) S. bilirubin- 5 mg%
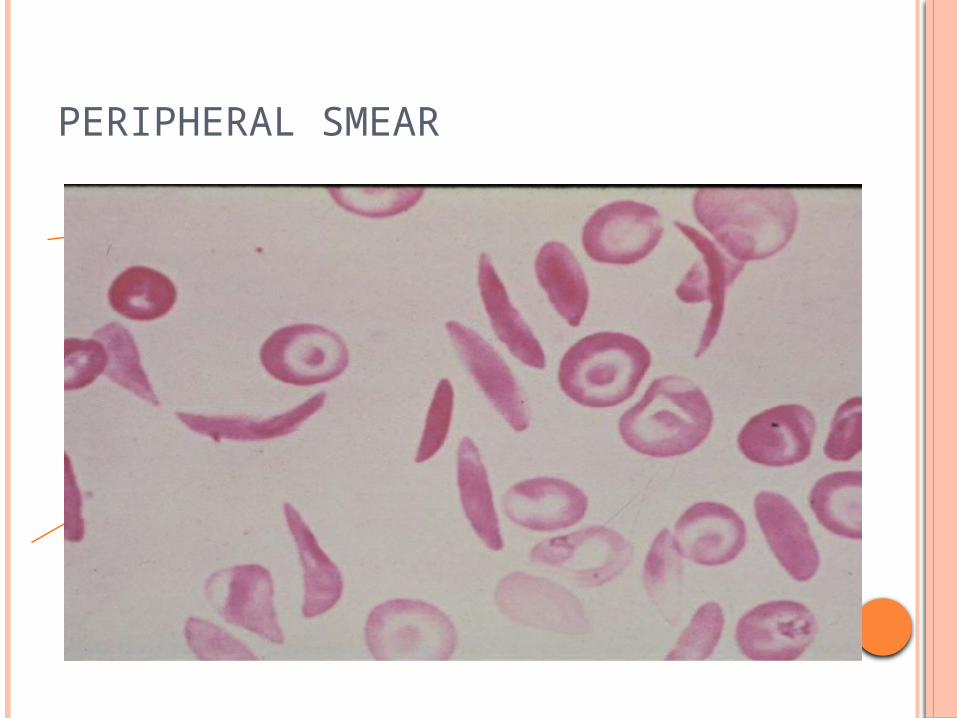
PERIPHERAL SMEAR

DIAGNOSIS – OTHER TESTSSICKLING TEST :
It should not be performed on infants until they are at least 6 month old because of the presence of hemoglobin F as the predominant hemoglobin at birth , therefore, this test may give a false-negative result if performed too early (if hemoglobin S is <10%).
Recent blood transfusions, typically within the last three months of the date of testing, may cause a false-negative test result .
False positive results may be due to polycythaemic , high proteins and a variety of abnormal haemoglobins including Hb’s I, Bart’s, C-Georgetown, Alexandra and C-Harlem.

Hemoglobin electrophoresis
Hemoglobin fractionation by HPLC, the most frequently used method to screen for hemoglobin variants, including Hb S .
Isoelectric focusing, a highly sensitive method that is often used at large reference laboratories .
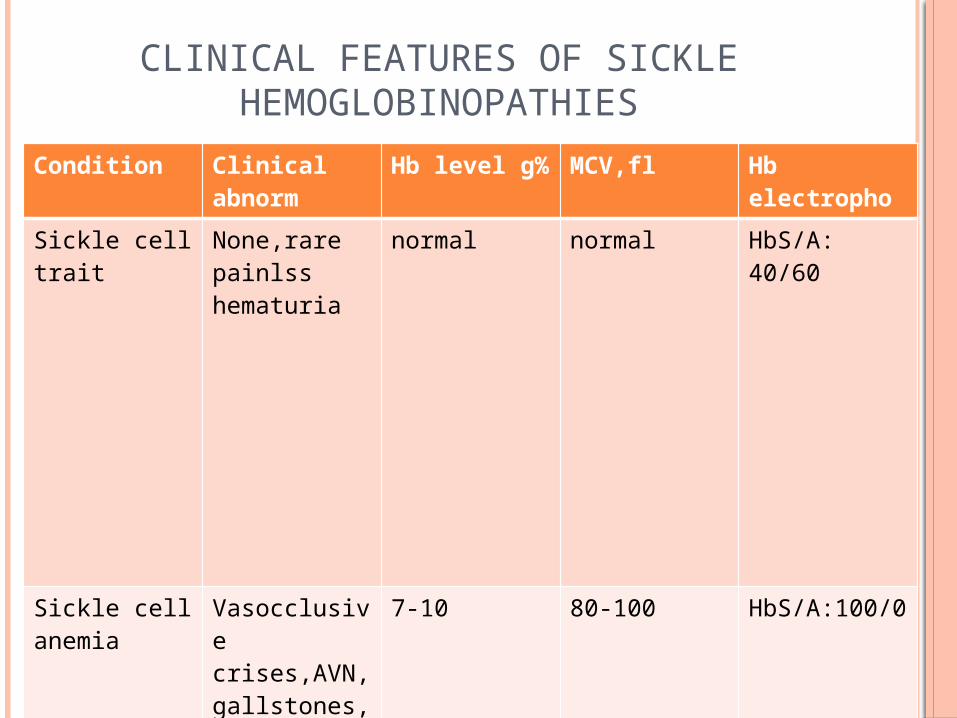
CLINICAL FEATURES OF SICKLE HEMOGLOBINOPATHIES
Condition Clinical abnorm
Hb level g% MCV,fl Hb electropho
Sickle cell trait
None,rare painlss hematuria
normal normal HbS/A: 40/60
Sickle cell anemia
Vasocclusive crises,AVN,gallstones, priapism
7-10 80-100 HbS/A:100/0
S/beta0thalasssemia
VasoocclusiveCrises,AVN
7-10 60-80 HbS/A-100/0HbF; 1-10%
S/beta+ thalassemia
Rare crises,AVN
10-14 70-80 HbS/A:60/40
HbSC --do--, retinopathy
10-14 80-100 HbS/A;50/0HbC;50%

DNA analysis :
This test is used to investigate alterations and mutations in the genes that produce hemoglobin components.
It may be performed to determine whether someone has one or two copies of the Hb S mutation or has two different mutations in hemoglobin genes (e.g., Hb S and Hb C).
Genetic counseling :used for prenatal testing: amniotic fluid may be tested at 14 to 16 weeks to provide a definitive answer. It can also be performed earlier with chorionic villus sampling.
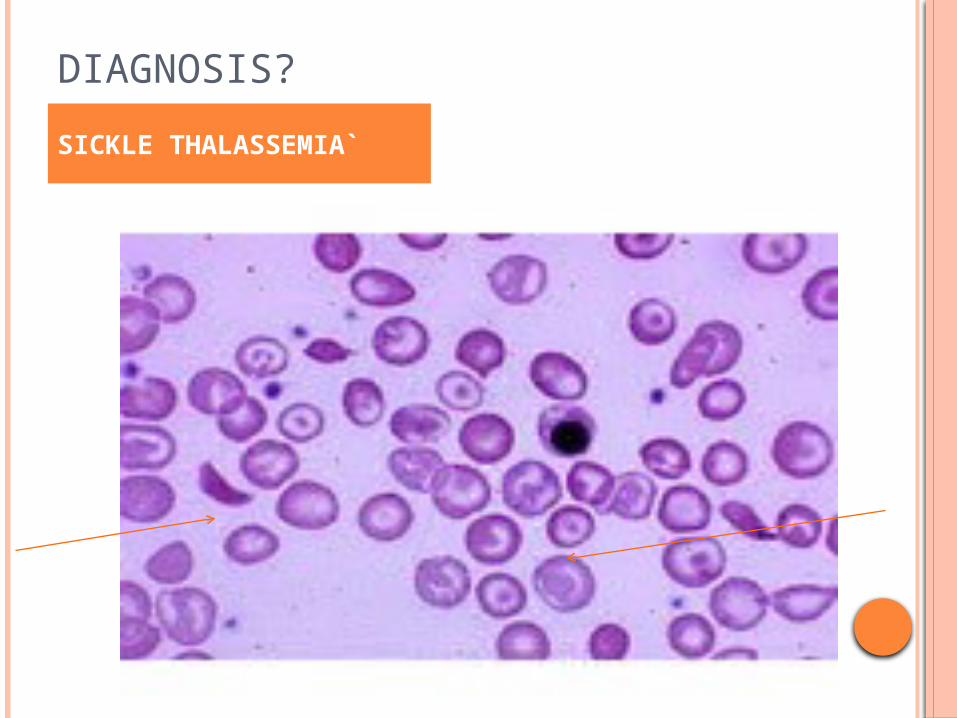
DIAGNOSIS?
SICKLE THALASSEMIA`

CASE 2
6 yr old child presenting with severe pallor , jaundice growth delay
Abnormal facies , hepatosplenomegaly+ h/o recurrent blood transfusions CBC-Hb -3gm%, MCV-58FL, -MCH- 19pg (nl-28-33) P.S- MICROCYTIC,HYPOCHROMIA with target cells +
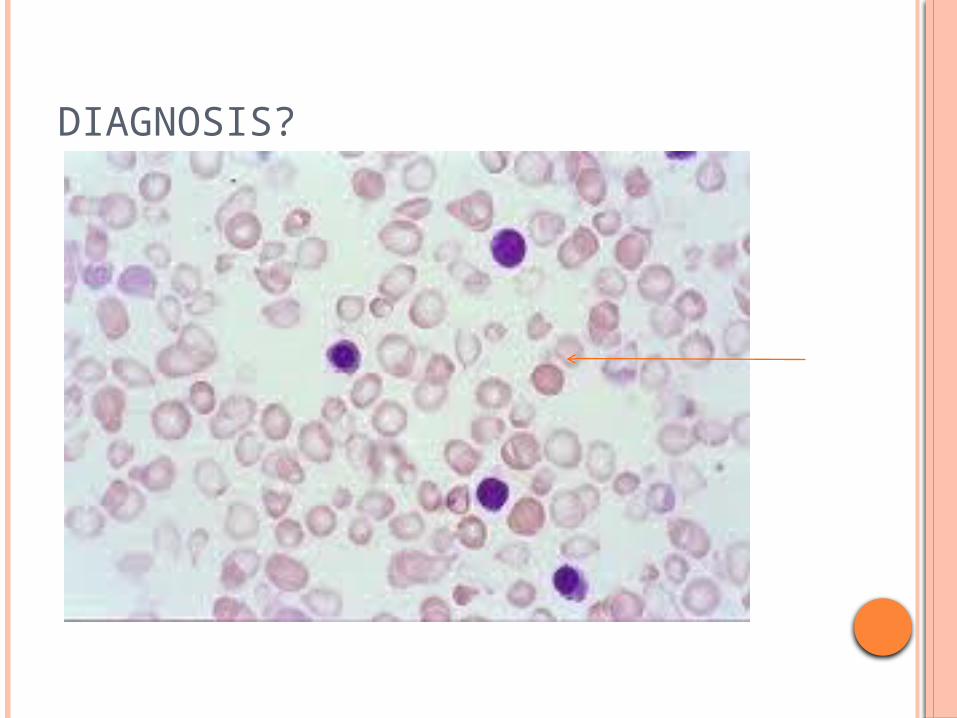
DIAGNOSIS?
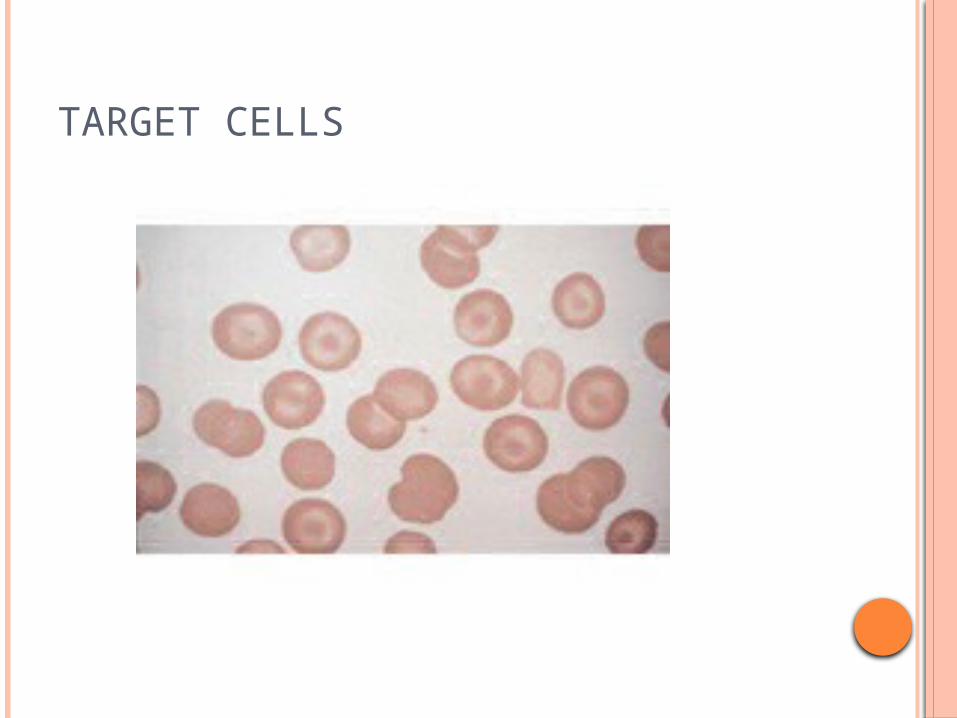
TARGET CELLS
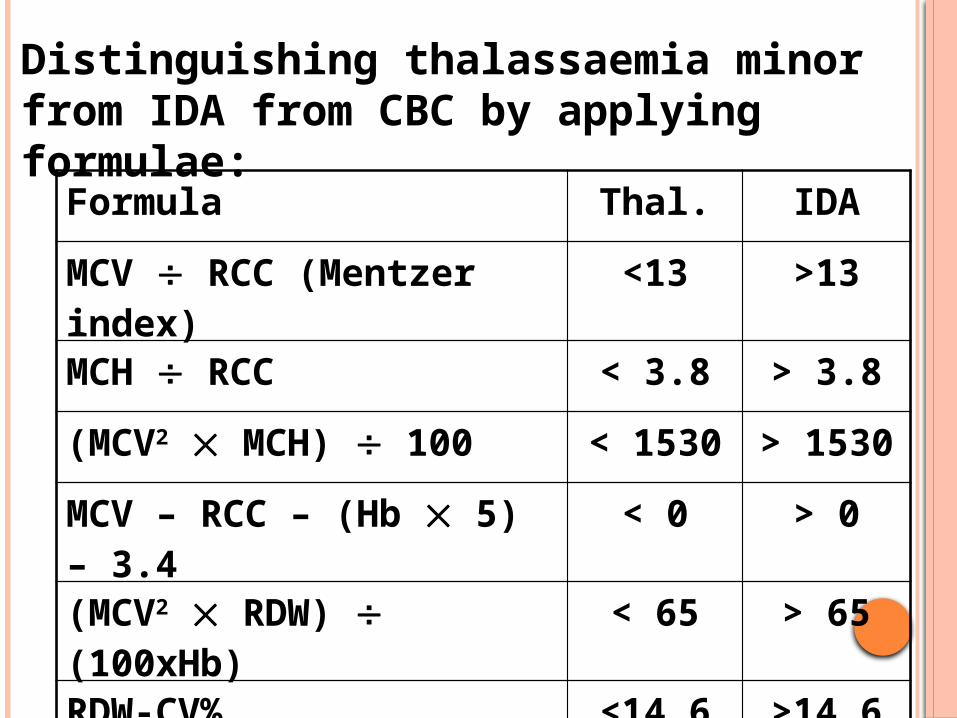
Distinguishing thalassaemia minor from IDA from CBC by applying formulae:
Formula Thal. IDA
MCV RCC (Mentzer index) <13 >13
MCH RCC < 3.8 > 3.8
(MCV2 MCH) 100 < 1530 > 1530
MCV – RCC – (Hb 5) – 3.4 < 0 > 0
(MCV2 RDW) (100xHb) < 65 > 65
RDW-CV% <14.6 >14.6
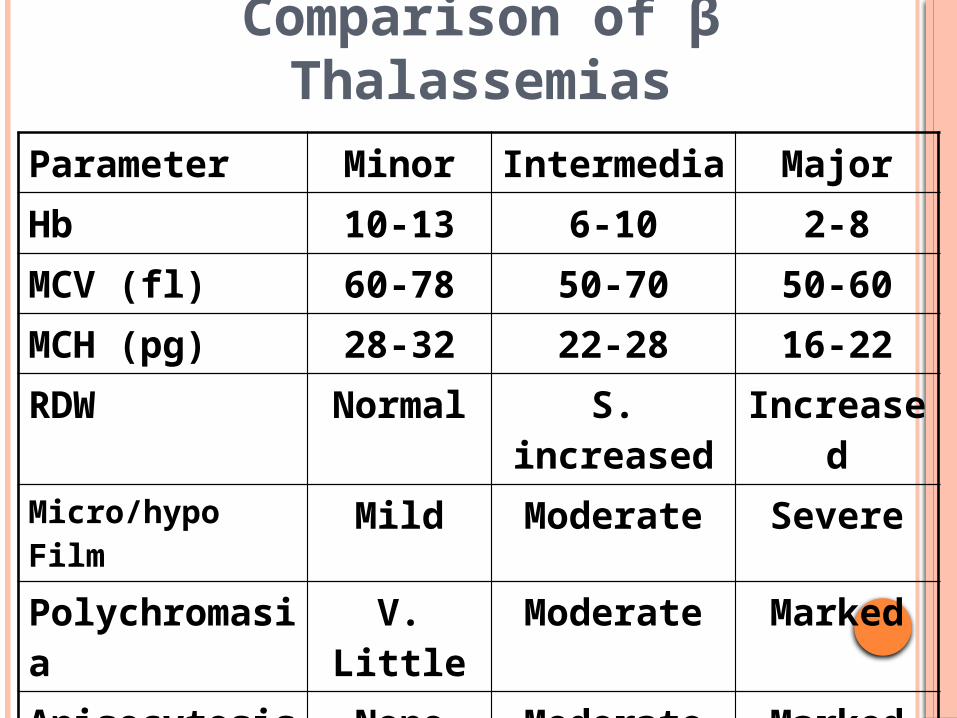
Comparison of β Thalassemias
Parameter Minor Intermedia MajorHb 10-13 6-10 2-8MCV (fl) 60-78 50-70 50-60MCH (pg) 28-32 22-28 16-22RDW Normal S. increased IncreasedMicro/hypo Film Mild Moderate SeverePolychromasia V. Little Moderate MarkedAnisocytosis None Moderate MarkedPoikilocytosis None Moderate MarkedTargetting Present Present Present

25
COMPARISON OF BETA THALASSEMIAS
GENOTYPE HGB A HGB A2 HGB F
NORMAL Normal Normal Normal
MINOR 80-95%, 3.5-7.5% 1-5%
INTERMEDIA
0-30% 3.5%-5.5%
20-70%
MAJOR 0 % 3.5 %- 5.5%
90-96 %
DNA analysis for mutations
Globin Chain Testing - determines ratio of globin chains being produced.

ALPHA THALASSEMIAS
disease Hb A %
HbH %
Hb , % MCV,fl
Normal α,α/α,α 97 0 15 90
Thalassemia traits(genotype α,-/α,α)
90-95 rare 12-13 70-80
HbH (b4) (genotype α,-/-,-) 70-95 5-30 6-10 60-70
Hb Bart (genotype -,-/-,-)(hydrops fetalis)
0 5-10 Fatal inutero or at birth
α+ thalassaemia homozygous (genotype α,-/α,-)αo thalassaemia heterozygous (genotype α,α/,--)

HB-BART’S
Is only detected at birth. But then disappears (WHY???). So diagnosis of alpha thalassemia could be established at birth directly in comparison of beta thalassemia.
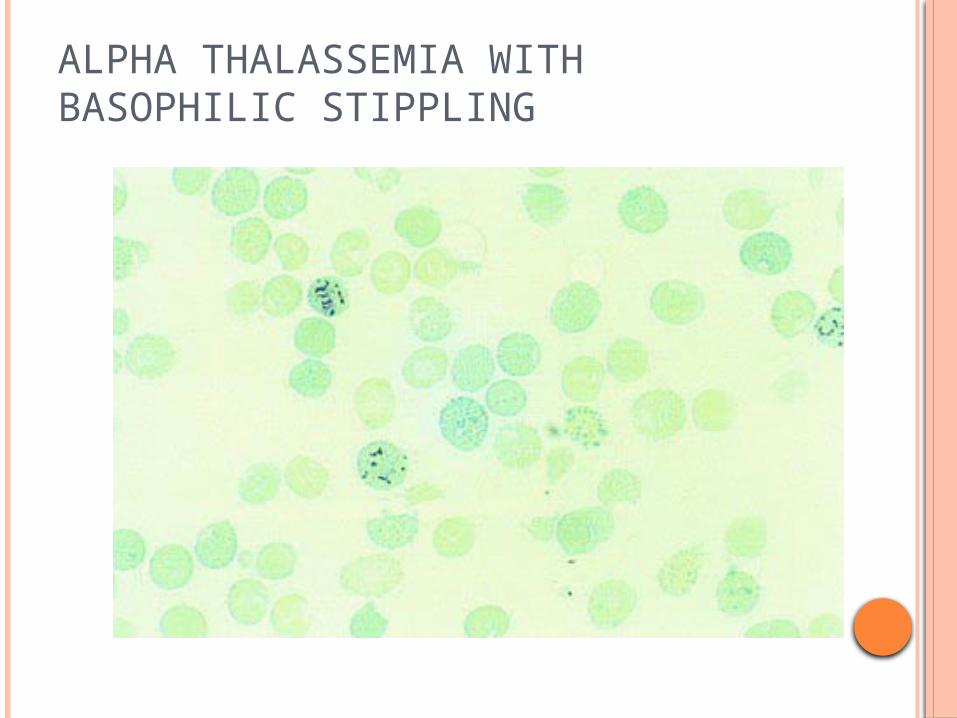
ALPHA THALASSEMIA WITH BASOPHILIC STIPPLING

HB-H PREPARATION
Same preparation as Retic count stain, but with extended time of incubation, instead of 15 minutes, 2 hours incubation is required.
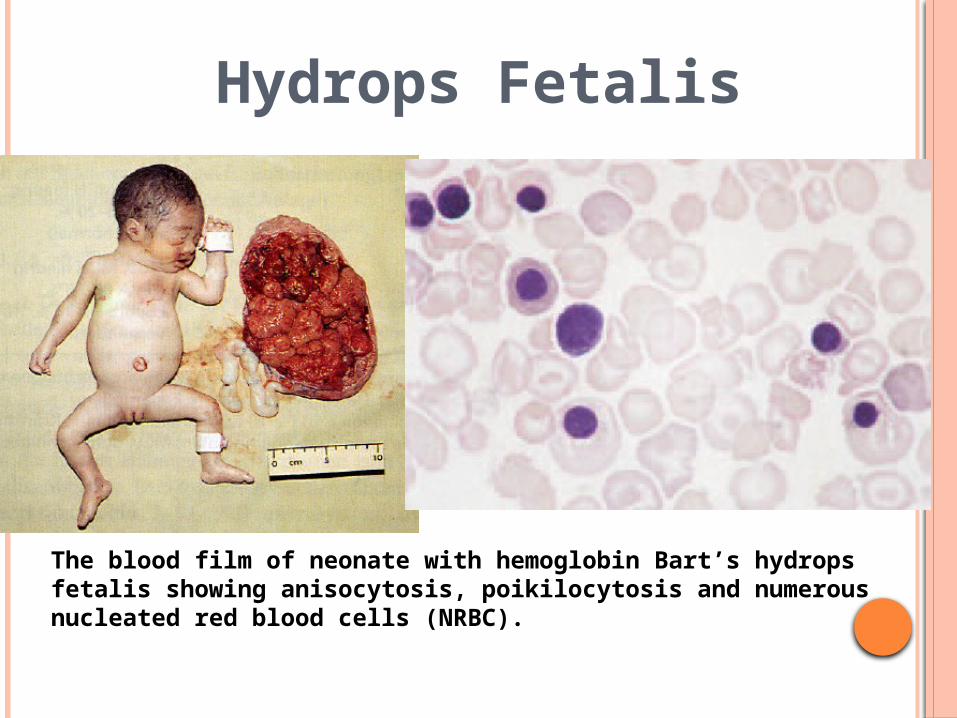
Hydrops Fetalis
The blood film of neonate with hemoglobin Bart’s hydrops fetalis showing anisocytosis, poikilocytosis and numerous nucleated red blood cells (NRBC).

CASE 3
45 yr old male came to OPD in a remote PHC with burning micturition
Urine R/M shows numerous pus cells++++ UTI diagnosed & medical officer gave cotrimoxazole 2
bd X 5days 1 wk later,pt developed severe
pallor,palpitation,jaundice
Lab- increased LDH, S.BILIRUBIN, RETIC COUNT P.S- shows irreg cells like
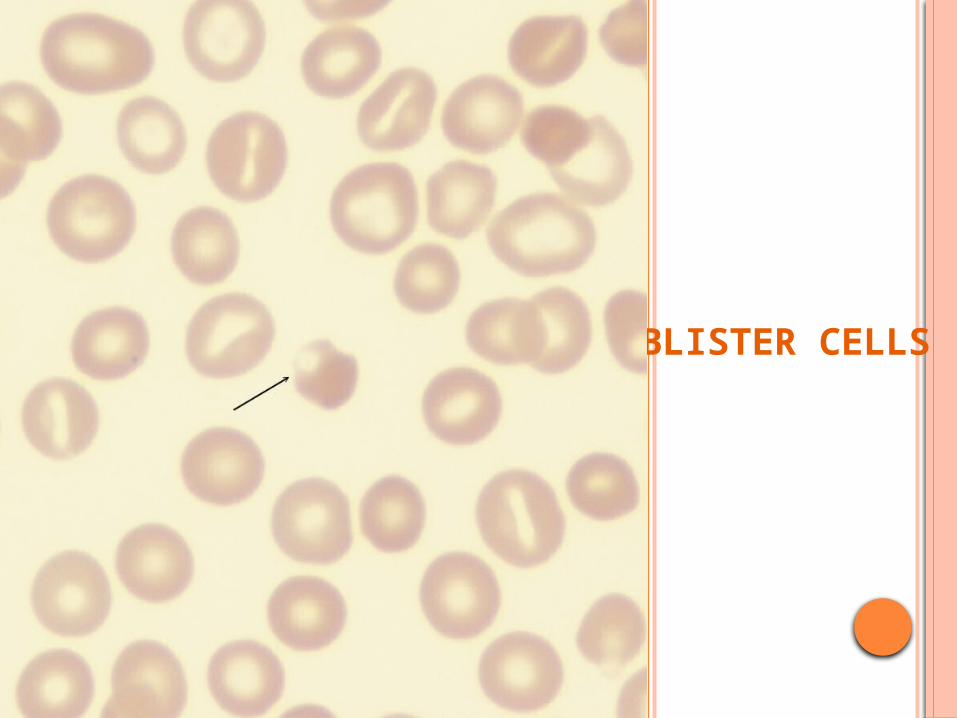
BLISTER CELLS
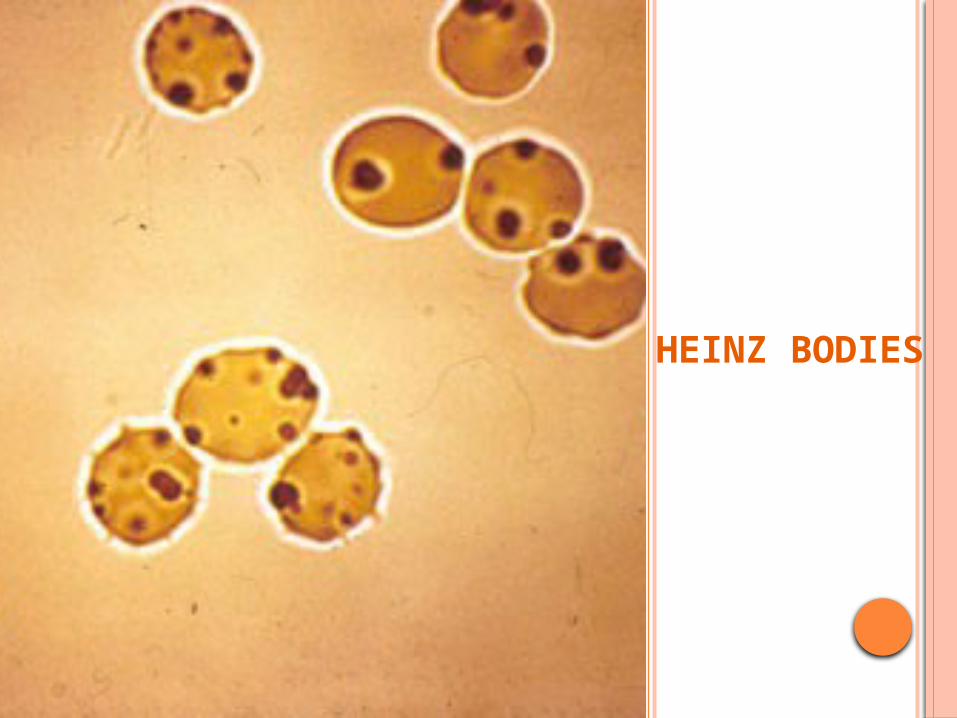
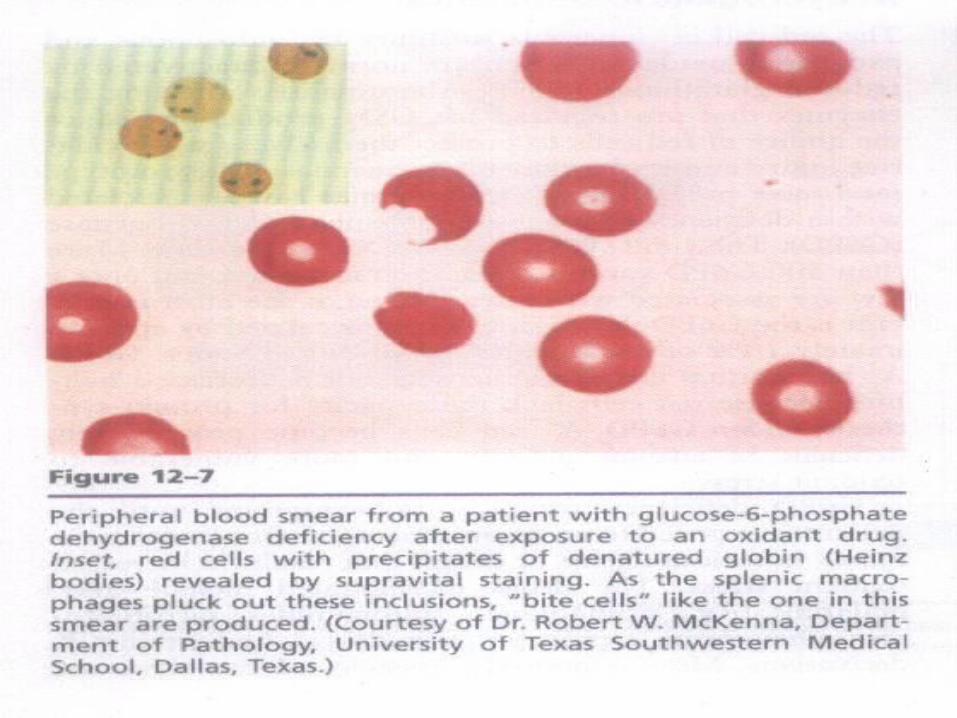
HEINZ BODIES

DIAGNOSIS?
G-6PD DEFICIENCY
INVESTIGATION-• Peripheral smear: bite cells , heinz bodies -
polychromasia
• G-6PD LEVEL
Quantitative , qualitative , PCR

Patients with acute hemolysis, testing for G6PD deficiency may be falsely negative because older erythrocytes with a higher enzyme deficiency have been hemolyzed.
Young erythrocytes and reticulocytes have normal or near-normal enzyme activity.
Female heterozygotes may be hard to diagnose because of X-chromosome mosaicism leading to a partial deficiency that will not be detected reliably with screening tests.
G6PD deficiency should be considered in neonates who develop hyperbilirubinemia

Pyruvate Kinase Deficiency
Deficient ATP production, Chronic hemolytic anemia
Inv;P. Smear: PRICKLE CELLS ( Contracted rbc with
spicules)
Decreased enzyme activity
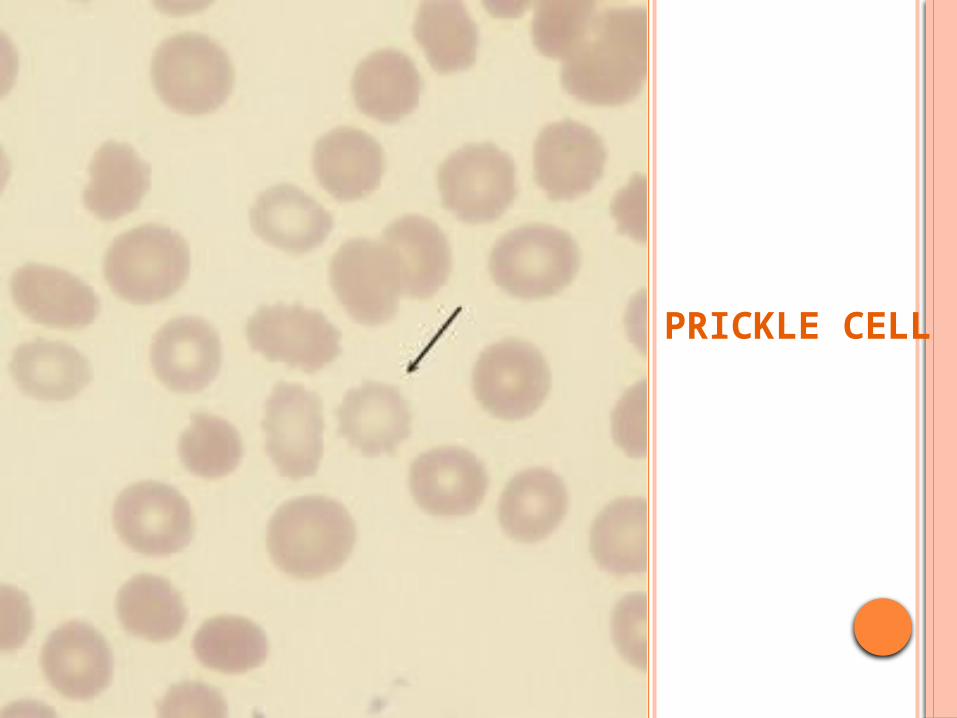
PRICKLE CELL

CASE 4
14 YR old female present with anemia, jaundice Rt hypochondrial pain o/e- vitals stable.pallor+,icterus+,splenomegaly
+ Usg- cholilithiasis
Lab; elevated LDH, S.Bilirubin , reticulocyte count MCHC, , RDW decreased Hb, MCV.
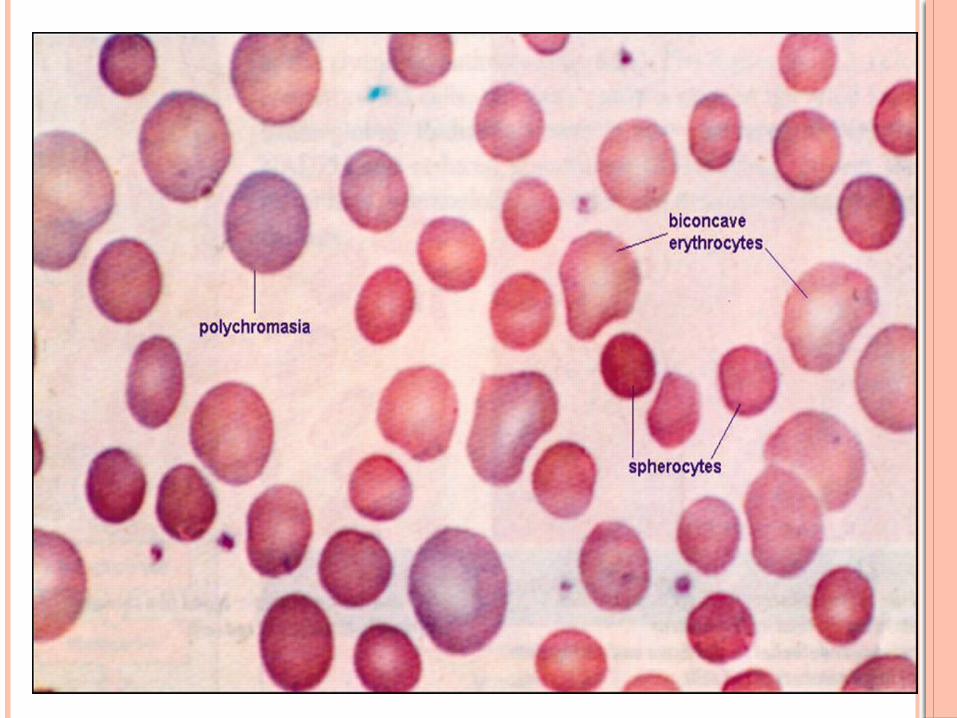
Peripheral smear shows- hyperdense cells

DIFFERENTIAL DIAGNOSIS
Hereditary spherocytosis
Autoimmune hemolytic anemia
Other diagnostic tests- osmotic fragility - coombs test
The acid glycerol lysis test Screen family members
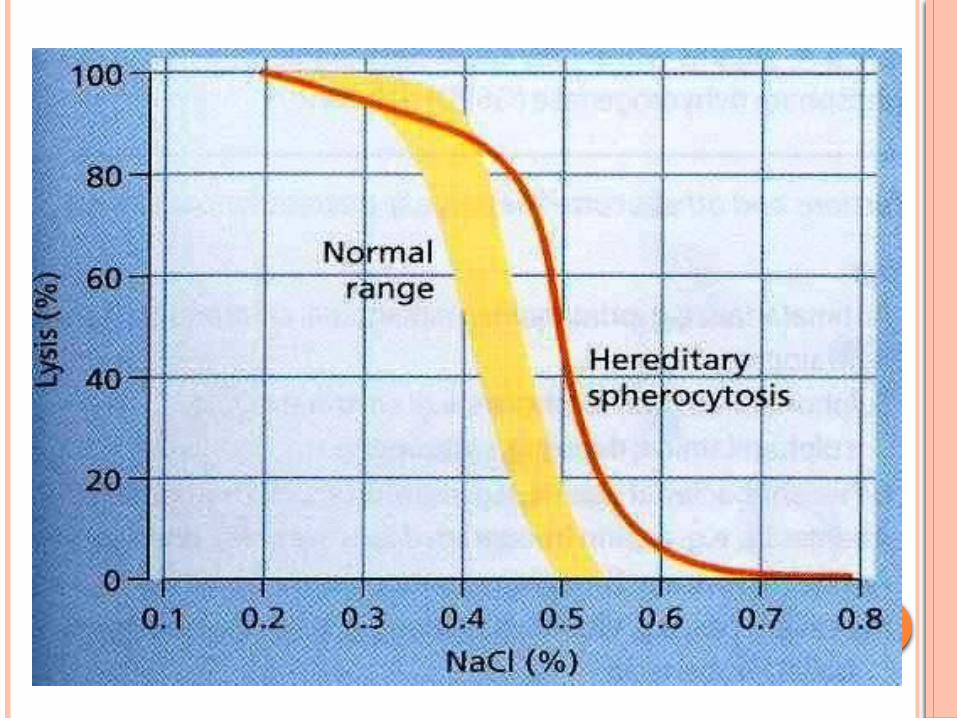

The red cell lipid composition regulates the membrane permeability to glycerol.
The added glycerol slows down the rate of water entry into the red cells.
Measure the time taken for absorbance of red cell suspension at 625 nm in glycerol to fall to half of its original value before glycerol addition (AGLT50)
Also detects autoimmune haemolytic anaemia, hereditary persistence of fetal haemoglobin, pyruvate kinase deficiency, severe glucose- 6-phosphate dehydrogenase deficiency, pregnant women (one-third), chronic renal failure on dialysis
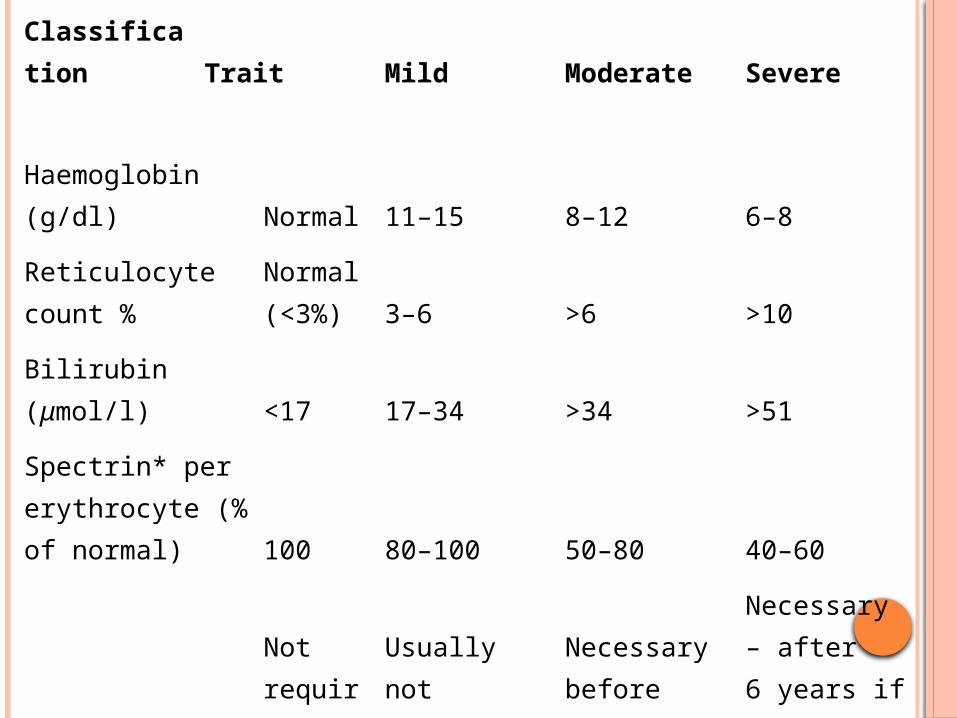
Classification Trait Mild Moderate Severe
Haemoglobin (g/dl) Normal 11–15 8–12 6–8
Reticulocyte count %Normal (<3%) 3–6 >6 >10
Bilirubin (μmol/l) <17 17–34 >34 >51
Spectrin* per erythrocyte (% of normal) 100 80–100 50–80 40–60
SplenectomyNot required
Usually not necessary
Necessary before puberty
Necessary – after 6 years if possible

Symptoms of HS may appear in the perinatal period: jaundice is common in the first 2 d of life.
Some neonates with HS may be transfusion-dependent(unusual) due to their inability to mount an adequate erythropoietic response in the first year of life .
Recent evidence suggests that erythropoietin may be of benefit in reducing or avoiding transfusion, and can usually be stopped by the age of 9 months .
Children who require one or two transfusions early in life frequently become transfusion independent

An artifact showing ‘macrospherocytosis’ on a blood film can be produced as a result of cold storage of blood samples from patients with cryohydrocytosis, which is a variant form of hereditary stomatocytosis (‘atypical HS’)
It is particularly important to rule out stomatocytosis where splenectomy is contraindicated because of the thrombotic risk.
Atypical cases may require measurement of erythrocyte membrane proteins to clarify the nature of the membrane disorder and in the absence of a family history.
Occasionally molecular genetic analysis will help to determine whether inheritance is recessive or non-dominant.

Iron, folate or vitamin B12 deficiencies can mask the laboratory features.
Obstructive jaundice alters the lipid composition of the red cell membrane, masking the film appearances andreducing the haemolysis.
Co-inheritance of other haematological disorders, such as beta thalassaemia trait or SC disease, can lead to confusion in the diagnosis and variable clinical effects
.

AUTOIMMUNE HAEMOLYTIC ANAEMIA
hemolysis, MCV decreased
P Smear: microspherocytosis,
Confirmation: Warm Direct Coomb’s Test / Antiglobulin test
ColdDAT positive with polyspecific and anticompliment antisera

DAT
•Sensitivity/Specificity: >99% patients with warm agglutinin AIHA will exhibit a positive result; <1% normal population have + result.

In some cases the density of attached autoantibody may be too low for detection by DAT. This may be resolved by flow cytometric assessment of red cell Ig density .
Polyspecific DAT reagents may also fail to detect some autoantibodies, particularly IgA, as anti-IgA is not usually incorporated into the reagent.
Application of specific anti G, A, M and complement DAT reagents may resolve some cases
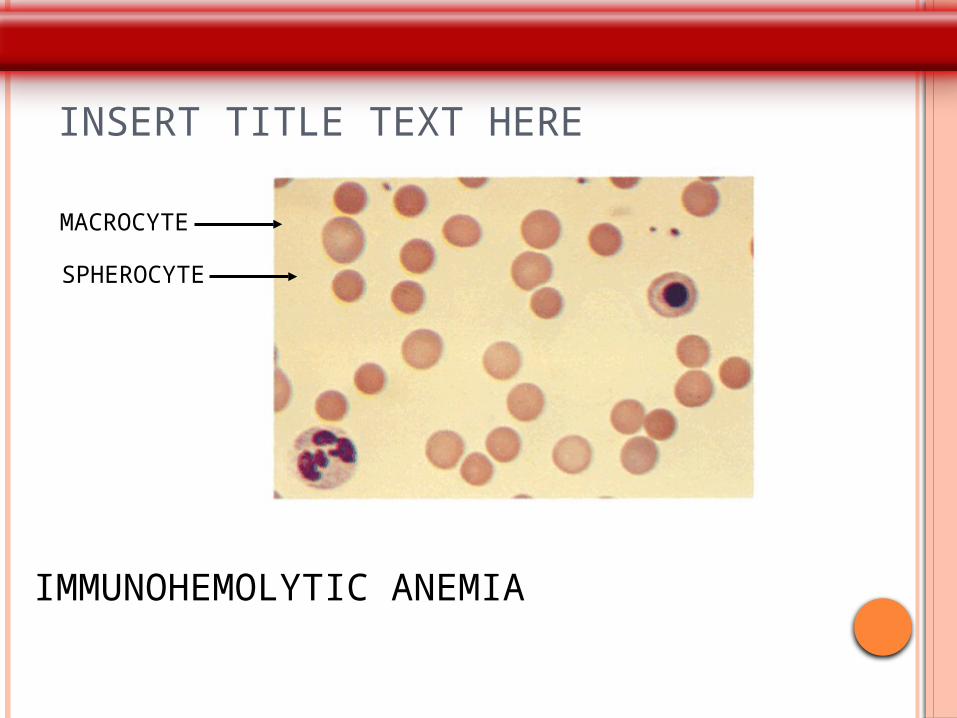
INSERT TITLE TEXT HERE
MACROCYTE
SPHEROCYTE
IMMUNOHEMOLYTIC ANEMIA

CASE 5
32 yr old presented 4 days history of distention of abdomen and rt hypochondrial pain and has h/o passage of dark colored urine at night for weeks
On USG- hepatomegaly,gross ascites,hepatic vein
thrombosis Lab : Hb – 7gm%. WBC- 2200, PLC- 80,000 LDH- 600, S.BR- 4 mg% urine bile pigment +,heme dip
stick++
What is the diagnosis?

PAROXYSMAL NOCTURNAL HEMOGLOBINURIA
Acquired chronic H.A
Persistent intra vascular hemolysis Pancytopenia Lab :HBuria,hemosiderinuria,increased LDH,bilirubin Risk of venous thrombosis C/F – hemoglobinuria during night P.S – polychromatophilia, normoblasts B.M – normoblastic hyperplasia Def.diagnosis-flow cytometry CD59-,CD55- RBC,WBC Acid haemolysis test Acid hemolysis test (PNH)A

Caused from a defect in the production of GPI protein anchors on the surface of all blood cell
Protect red cells from the activity of the complement system. but 2 are important in protecting red cells from destruction: CD55 (DAF) and CD59
Defect makes the red cells in particular susceptible to destruction by the complement system. Intravascular hemolytic anemia.

“FOOT STRIKE HEMOLYSIS”
Caused by RBC destruction from repeated trauma
• Elevated temperature in muscle, turbulence and acidosis may also be involved
Bilirubin Haptoglobin Schistocytes Slight MCV & Reticulocytes
Preferential breakdown of older rbcs Hemoglobinuria Anemia resolves w/ d/c exercise

THANK YOU

BETA THALASSEMIA
Mutn. Beta globin expression
M.C- derange splicing of m-RNA
HYPOCHROMIA ,MICROCYTIC anemia

AUTOIMMUNE HEMOLYTIC ANEMIA
Result from RBC destruction due to RBC autoantibodies: Ig G, M, E, A
Most commonly-idiopathic Classification
Warm AI hemolysis:Ab binds at 37 C Cold AI Hemolysis: Ab binds at 4 C

1.Warm AI Hemolysis: Can occurs at all age groups F > M Causes:
50% IdiopathicRest - secondary causes:
1.Lymphoid neoplasm: CLL, Lymphoma, Myeloma
2.Solid Tumors: Lung, Colon, Kidney, Ovary, Thymoma
3.CTD: SLE,RA4.Drugs: Alpha methyl DOPA, Penicillin , Quinine, Chloroquine
5. UC, HIV