Application and implementation of probabilistic profile profile comparison methods for protein fold...
23
Application and implementation of probabilistic profile-profile comparison methods for protein fold recognition mgr inż. Jakub Paś Dissertation supervisor: dr hab. Marcin Hoffmann Auxiliary supervisor: dr Krystian Eitner
-
Upload
jakub-pas -
Category
Health & Medicine
-
view
40 -
download
0
Transcript of Application and implementation of probabilistic profile profile comparison methods for protein fold...

Application and implementation of probabilistic
profile-profile comparison methods
for protein fold recognition
mgr inż. Jakub Paś
Dissertation supervisor: dr hab. Marcin Hoffmann
Auxiliary supervisor: dr Krystian Eitner

Introduction
● The purpose of Phd thesis is to show that profile - profile methods may
outperform other fold recognition approaches in comparison and analysis
of distantly related proteins.
● The work presents advantages of usage probabilistic profile-profile
methods over comparable fold recognition techniques in research
performed by author.
● The thesis is based on several author’s publications in area of gene
identification, detection of distant homologous, domain boundaries
detection, protein modeling, evolutionary analysis and protein - ligand
interaction
● The work shows both applications and implementations of such methods in
molecular biology software.

What is fold Recognition
● Fold recognition are the methods of fold detecting and protein tertiary
structure prediction applied for proteins lacking homologues sequences of
known fold and structure deposited in the Protein Data Bank.
● They are based on assumption that there is strictly limited number of
different protein folds in nature, mostly as a result of evolution and due to
basic physical and chemical constraints of polypeptide chains.
● Fold recognition methods are useful for protein structure prediction,
evolutionary analysis, metabolic pathways analysis, enzymatic efficiency
prediction, molecular docking and drug design.

Sequence comparison methods
used for fold recognition
Sequence based
comparison methods:
● Smith Waterman
● BLAST
Sequence - Profile methods:
● PSI - BLAST
● RPS-BLAST
Profile - profile methods.
FFAS:
● Meta-BASIC
● ORFEUS
Other (non sequence based) fold
recognition methods:
● Threading (HHpred, Raptor)
● Ab initio (Rosetta)

What are probabilistic profile -
profile comparison methods?
● A profile or PSSM simply is a set of vectors, where each vector contains
the frequency of each type of amino acid in a particular position of the
multiple sequence alignment.
● In profile - profile alignments, we have to compare two frequency vectors
and this can be done in several different ways, including calculating the
sum of pairs, the dot-product, or a correlation coefficient between the two
vectors
● The performance of a profile - profile methods depends on: calculation of
the score between two profile positions, alignment methodology and
evolutionary distance between two sequences in study.
● Because profiles contain more information they are more sensitive and
provide better alignments than sequence - sequence methods.

Improvement and assessment
of fold recognition methods
Benchmarking of fold recognition methods:
● CASP(Critical Assessment of protein Structure Prediction)
● CAFASP(Critical Assessment of Fully Automated Structure Prediction)
● LIVEBENCH (Live Benchmark)
● EVA

Application vs implementation
Implementation - realization of an application, or execution of
a plan, idea, model, design, specification, standard, algorithm,
or policy.
Eg. Implementation of profile - profile comparison methods to
create “tools” (molecular biology software)
Application - to put something into effect or into use after it
was implemented.
Eg. Usage of such software in for science discovery
(operating “tools”)

Implementations of profile-profile
comparison methods
GRDB
J Pas, P Stepniak, L Rychlewski, GRDB – Gene
Relational DataBase. Bioinfobank Library Acta, 2011.
2659.
ELM
CM Gould, F Diella, A Via, P Puntervoll, C Gemünd, S
... J Pas, S … ELM: the status of the 2010 eukaryotic
linear motif resource
Nucleic acids research 38 D167-D180 137 2010

Implementations of profile-profile
comparison methods
ORFeus
K Ginalski, J Pas, LS Wyrwicz, M Von Grotthuss, JM
Bujnicki, L Rychlewski ORFeus: Detection of distant
homology using sequence profiles and predicted
secondary structure.
Nucleic Acids Res, 2003. 31(13): p. 3804-3807
PDB-Preview
Fischer, D., J. Pas, and L. Rychlewski, The PDB-
Preview database: a repository of in-silico models
of 'on-hold' PDB entries.
Bioinformatics, 2004. 20(15): p. 2482-4.004. 20(15)

Applications of profile-profile
comparison methods
ELM: the status of the 2010 eukaryotic linear motif resource
CM Gould, F Diella, A Via, P Puntervoll, C Gemünd, S ... J Pas at al.
Nucleic acids research 38 (suppl 1), D167-D180 137 2010
Predicting protein structures accurately
M von Grotthuss, LS Wyrwicz, J Pas, L Rychlewski
Science 304 (5677), 1597-1599 5 2004
How unique is the rice transcriptome?
LS Wyrwicz, M von Grotthuss, J Pas, L Rychlewski
Science (New York, NY) 303 (5655), 168; author reply 168 6 2004
Structure prediction, evolution and ligand interaction of CHASE
domain.
J Pas., LS Wyrwicz, L Rychlewski, J Barciszewski
FEBS Lett, 2004. 576(3): p. 287-90.
Linear motif identification in
distant proteins
Protein structure prediction
Gene identification
Protein topology Detection

Applications of profile-profile
comparison methods
Two sequences encoding chalcone synthase in yellow lupin
(Lupinus luteus l.) may have evolved by gene duplication
D Narożna, J Pas, J Schneider, CJ Mądrzak
Cellular & molecular biology letters 9 (1), 95-105 5 2004
Molecular phylogenetics of the RrmJ/fibrillarin superfamily of
ribose 2'-O-methyltransferases
M Feder, J Pas, LS Wyrwicz, JM Bujnicki
Gene 302 (1-2), 129-138 66 2003
Application of 3D-Jury, GRDB, and Verify3D in fold recognition.
Proteins
M Grotthuss, J Pas, L Wyrwicz, K Ginalski, L Rychlewski
Proteins, 2003. 53 Suppl 6: p. 418-23.
Predicting protein structures accurately
LS Wyrwicz, M Von Grotthuss, J Pas, L Rychlewski
Science 304 (5677), 1597
Gene duplication detection
Molecular phylogeny
Fold Recognition
Distant homologues detection

Gene identification and detection of
distinct homologuesApplications of profile-profile comparison methods
D Narożna, J Pas, J Schneider, CJ Mądrzak, Two sequences encoding
chalcone synthase in yellow lupin (Lupinus luteus l.) may have evolved by gene
duplication. Cell Mol Biol Lett, 2004. 9(1): p. 95-105.
● In the publication about detection of chalcone synthase (CHS)
sequences encoded in yellow lupin profile-profile fold recognition
methods were used to detect two full copies of CHS.
● Using the molecular clock calibration the duplication of genes was
estimated to happened about 16 millions years ago.
● Initial multiple alignment of distant homologues from 52 plant
species was created using multiple profile-profile comparison
methods.

Gene identification and detection of
distinct homologuesApplications of profile-profile comparison methods
D Narożna, J Pas, J Schneider, CJ Mądrzak, Two sequences encoding
chalcone synthase in yellow lupin (Lupinus luteus l.) may have evolved by gene
duplication. Cell Mol Biol Lett, 2004. 9(1): p. 95-105.
● Amino-acids involved in ligand binding has been
detected.
● Catalytic, evolutionary conserved amino acids
helped to produce full multiple sequence alignment
of known sequences of whole CHS family.

Detection of domain boundaries and
modeling of complex proteinsApplications of profile-profile comparison methods
● Domain boundaries and the homologs of the Tn-C domains were
identified using Gene Relate Sequence Database (GRDB)
● The characteristic profiles were computed for every domain using
protein families collected from Pfam, COG and from other genomic
sources.
● The comparison of the target families with about 100,000 other
families was performed using Meta-BASIC program
● The models of Tenascin-C domains were performed
J Pas., et al., Analysis of structure and function of tenascin-C. Int J Biochem
Cell Biol, 2006. 38(9): p. 1594-602.

Detection of domain boundaries and
modeling of complex proteinsApplications of profile-profile comparison methods
● Usage of sensitive profile - profile sequence comparison analysis allowed to detect the order of
functional elements in large multidomain tenascin-C protein, all variable part of a molecule as
well as all isoforms.
● The number of putative fibronectin repeats was corrected. Also previously not identified HSP33
domain with was described.
J Pas., et al., Analysis of structure and function of tenascin-C. Int J Biochem
Cell Biol, 2006. 38(9): p. 1594-602.
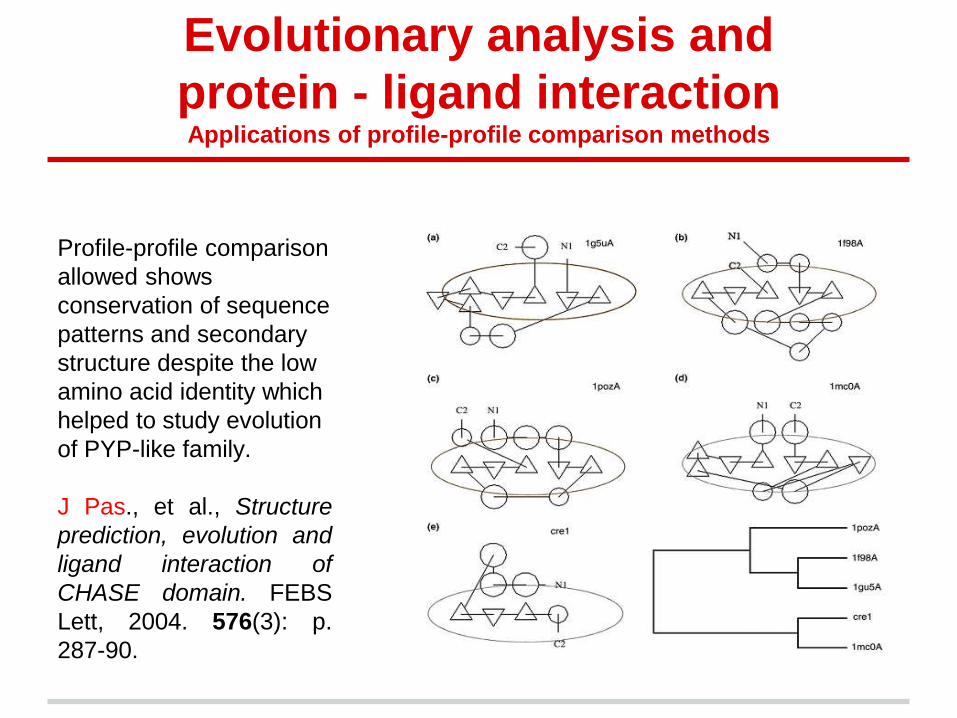
Profile-profile comparison
allowed shows
conservation of sequence
patterns and secondary
structure despite the low
amino acid identity which
helped to study evolution
of PYP-like family.
J Pas., et al., Structure
prediction, evolution and
ligand interaction of
CHASE domain. FEBS
Lett, 2004. 576(3): p.
287-90.
Evolutionary analysis and
protein - ligand interactionApplications of profile-profile comparison methods

Evolutionary analysis and
protein - ligand interactionApplications of profile-profile comparison methods
● Molecular model of CRE1
receptor from A. thaliana
docked with (a)trans-zeatin, (b)
kinetin confirms that ligands are
entirely buried
● The visible side chain belongs
to threonine 278 whose
mutation is responsible for loss
of function.
● Molecular modeling and
docking confirmed that ligand
was entirely buried.
● Importance of threonine 278 for
the catalytics activity of the
enzyme was confirmed.
J Pas., et al., Structure prediction, evolution and ligand interaction of CHASE
domain. FEBS Lett, 2004. 576(3): p. 287-90.

Implementation: PDB PreviewImplementations of profile-profile comparison methods
● Not all the entries in the Protein Data Bank (PDB) are publicly available.
● A new structure can be deposited as an “on-hold” entry, non-accessible for
public before final release.
● To access 3D structure before release it is possible in most cases to
generate relatively accurate automatically created computational models.
● The PDB-Preview provide biologists with relatively accurate 3D models for
not yet released PDB shortly after they are deposited in the PDB, and well
before the experimental structure is released.
● Additionally the resulting PDB-CAFASP analysis provides computational
biologists with a continuous blind evaluation of their methods.
D Fisher., J Pas, and L Rychlewski, The PDB-Preview database: a
repository of in-silico models of 'on-hold' PDB entries. Bioinformatics,
2004. 20(15): p. 2482-4.

Implementation: Gene Relational
DataBase (GRDB)Implementations of profile-profile comparison methods
● GRDB is the web service dedicated for searching for distant homologues
of protein sequences which may not be detected using different
approaches such as direct sequence search.
● It performs the comparison of the target family with 100,000 other families,
using Meta BASIC profile-profile comparison methods. (SCOP, CATH,
COG)
● In contrast other methods it allows to use manually build profile as input
and perform comparison between whole protein families.
● GRDB was successfully used for comprehensive classification of proteins
folds and identification of novel families and their representatives in human
(Kuchta, et al., 2009).
J Pas., et al., GRDB – Gene Relational DataBase. Bioinfobank Library Acta,
2011. 2659.

Conclusions
● Profile-profile based sequence comparison methods are usually superior to
sequence methods and may have more possible applications in molecular
biology.
● PDB statistics shows that in recent years, only a limited number of
completely new protein folds appear although several thousand new
structures are deposited to the PDB. Most of the single-domain proteins
can be aligned to a protein already deposited in the PDB.
● The output of experimentally determined protein structures from X-ray
crystallography and NMR spectroscopy are still expensive and time
consuming despite the efforts in structural genomics.
● As more and more novel sequences are produce from the genome projects,
the profile-based methods can be expected to become even more sensitive
(new alignment, scoring methods etc.)

Acknowledgements
Dissertation supervisor: dr hab. Marcin Hoffmann
Auxiliary supervisor: dr Krystian Eitner
My wife: Agnieszka Paś

Recenzja: Prof. Tadeusz Kuliński
● Współczynnik impact factor oraz cytowalność. Autor niestety w żaden
sposób nie wyróżnia tych prac, które stanowią spójny tematyczne zbiór
artykułów i faktycznie wchodzą w skład dysertacji.
● Literówki i błedy językowe
● Błąd merytoryczny. W publikacji:Two sequences encoding chalcone synthase in yellow lupin (Lupinus luteus l.) may have
evolved by gene duplication
Narożna, J Pas, J Schneider, CJ Mądrzak
Cellular & molecular biology letters 9 (1), 95-105 5 2004
istotnie chodziło o 52 sekwencje roślinnej suntazy chalkonowej
w tym 2 z L. Luteus

Recenzja: Prof. Andrzej Koliński
● Uwagi formalne - brak oświadczeń - zostały dosłane
● Uwagi dotyczace polskich odpowiedników wyrazów angielskich w polskim
streszczeniu.
● ab inition - de novo