
DRAFT GUIDELINES Data Integrity & Computer System Validation

Appendices to the Guidelines for Validation & Qualification, including Change
Control, for Hospital Transfusion Laboratories
This document contains the appendices to the BCSH guidelines on validation and
qualification which is available to download from www.bcshguidelines.com
British Committee for Standards in Haematology
Address for correspondence:
BCSH Secretary
British Society for Haematology
100 White Lion Street
London N1 9PF
e-mail [email protected]
Writing group:
S. Allard1, G Burgess2, B. Cuthbertson3, C Elliott 4, R Haggas5, J Jones6, B Robertson7, D Sadani 8 K Smith2
Disclaimer
While the advice and information in these guidelines is believed to be true and
accurate at the time of going to press, neither the authors, the British Society for
Haematology nor the publishers accept any legal responsibility for the content of
these guidelines.
Date for guideline review
July 2014
1, Barts and the London NHS Trust & NHSBT, Royal London Hospital, 80 Newark Street
London E1 2ES 2 NHSBT, Long Road, Cambridge, CB2 2PT
3 SNBTS, Ellens Glen Rd, Edinburgh
4 James Cook University Hospital, Middlesborough
5 Blood Transfusion Dept, Leeds General Infirmary, Great George Street, Leeds. LS1 3EX
6 WBS, Ely Valley Rd, Cardiff CF72 9WB
7 Guys & St. Thomas‟ Hospital NHS Trust, Westminster Bridge Road, London SE1 7EH
8 Leeds Blood Centre, Bridle Path, Leeds

Contents
1. Information to be included in the Validation Master Plan page 03
2. Example of a VMP currently in use in an NHS Hospital page 07
3. Change Control Request Form page 11
4. Specifications of what could be included in a User Requirement Specification page 14
5. Example of a User Requirement Specification page 17
6. Specifications for inclusion in FDS page 23
7. Example of a Validation Plan page 25
8. Installation Qualification, Operational Qualification and Process Qualification page 30
9. Example of Validation Protocol page 33
10. Example of a qualification Proforma page 35
11. Validation sign off report page 50
12. Acknowledgements and Declarations of interest page 51
Appendix 1: Information to be included in the validation master plan
3
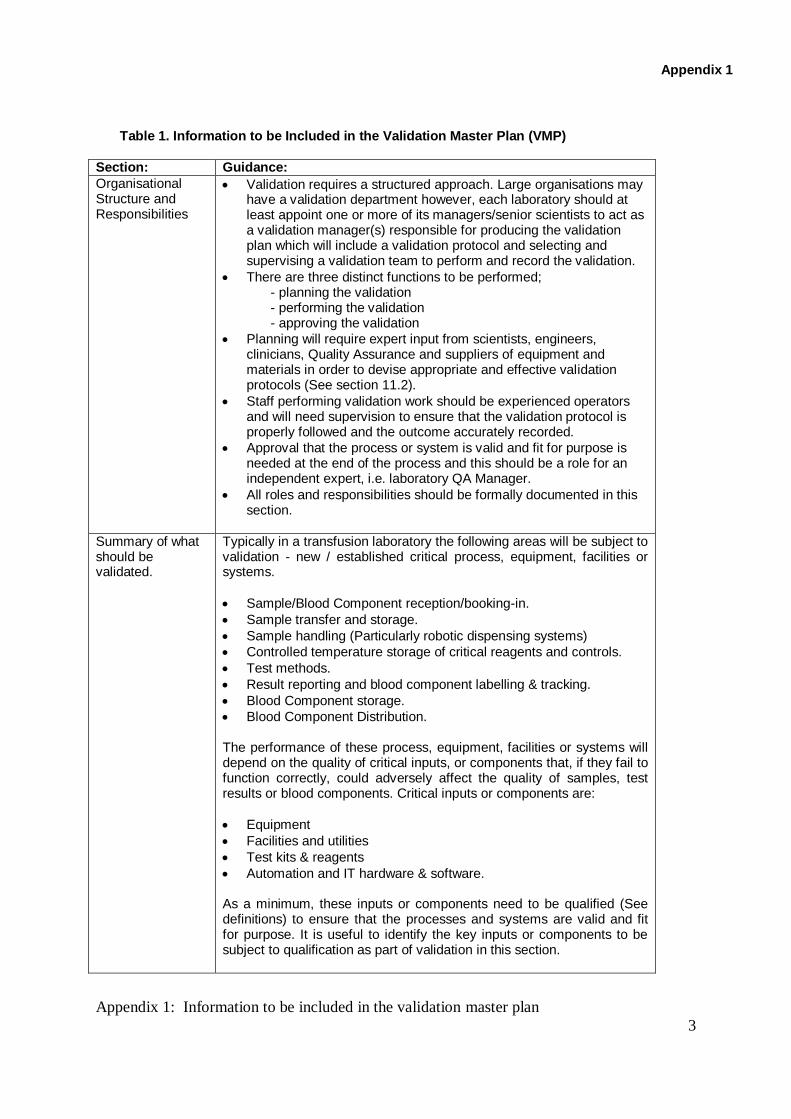
Table 1. Information to be Included in the Validation Master Plan (VMP)
Section: Guidance:
Organisational Structure and Responsibilities
Validation requires a structured approach. Large organisations may have a validation department however, each laboratory should at least appoint one or more of its managers/senior scientists to act as a validation manager(s) responsible for producing the validation plan which will include a validation protocol and selecting and supervising a validation team to perform and record the validation.
There are three distinct functions to be performed; - planning the validation - performing the validation - approving the validation
Planning will require expert input from scientists, engineers, clinicians, Quality Assurance and suppliers of equipment and materials in order to devise appropriate and effective validation protocols (See section 11.2).
Staff performing validation work should be experienced operators and will need supervision to ensure that the validation protocol is properly followed and the outcome accurately recorded.
Approval that the process or system is valid and fit for purpose is needed at the end of the process and this should be a role for an independent expert, i.e. laboratory QA Manager.
All roles and responsibilities should be formally documented in this section.
Summary of what should be validated.
Typically in a transfusion laboratory the following areas will be subject to validation - new / established critical process, equipment, facilities or systems.
Sample/Blood Component reception/booking-in.
Sample transfer and storage.
Sample handling (Particularly robotic dispensing systems)
Controlled temperature storage of critical reagents and controls.
Test methods.
Result reporting and blood component labelling & tracking.
Blood Component storage.
Blood Component Distribution. The performance of these process, equipment, facilities or systems will depend on the quality of critical inputs, or components that, if they fail to function correctly, could adversely affect the quality of samples, test results or blood components. Critical inputs or components are:
Equipment
Facilities and utilities
Test kits & reagents
Automation and IT hardware & software. As a minimum, these inputs or components need to be qualified (See definitions) to ensure that the processes and systems are valid and fit for purpose. It is useful to identify the key inputs or components to be subject to qualification as part of validation in this section.
Appendix 1
Appendix 1: Information to be included in the validation master plan
4
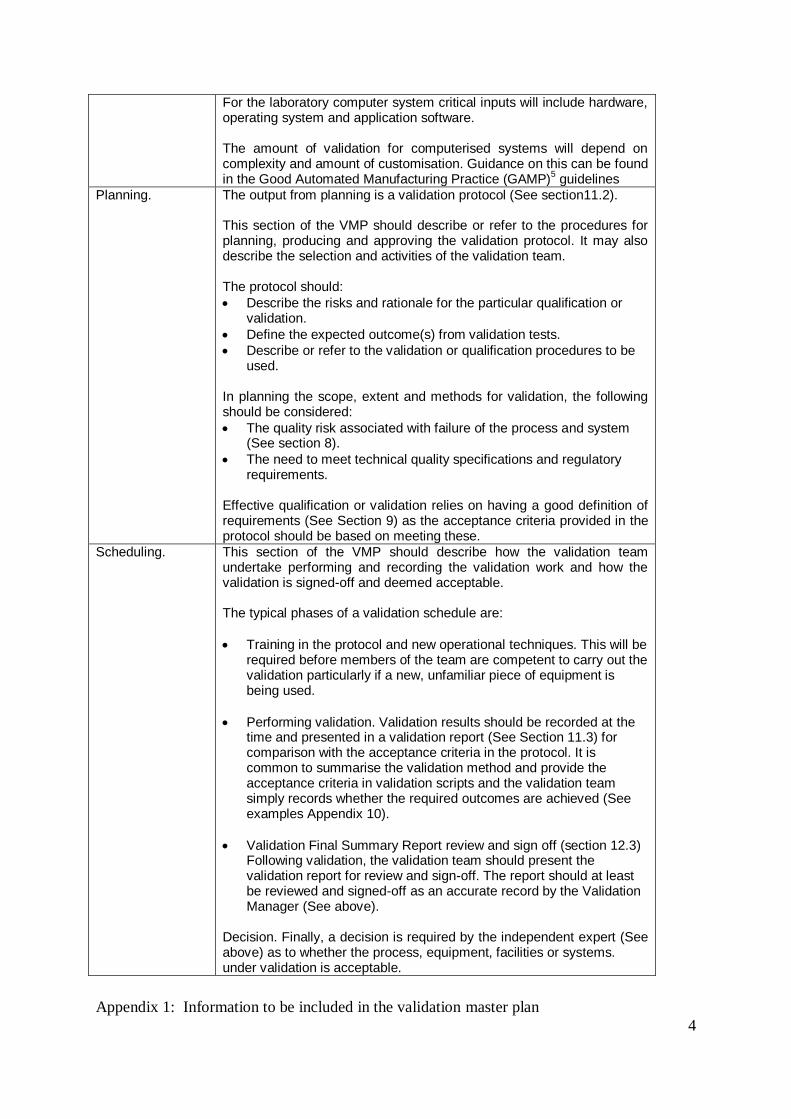
For the laboratory computer system critical inputs will include hardware, operating system and application software. The amount of validation for computerised systems will depend on complexity and amount of customisation. Guidance on this can be found in the Good Automated Manufacturing Practice (GAMP)
5 guidelines
Planning. The output from planning is a validation protocol (See section11.2). This section of the VMP should describe or refer to the procedures for planning, producing and approving the validation protocol. It may also describe the selection and activities of the validation team. The protocol should:
Describe the risks and rationale for the particular qualification or validation.
Define the expected outcome(s) from validation tests.
Describe or refer to the validation or qualification procedures to be used.
In planning the scope, extent and methods for validation, the following should be considered:
The quality risk associated with failure of the process and system (See section 8).
The need to meet technical quality specifications and regulatory requirements.
Effective qualification or validation relies on having a good definition of requirements (See Section 9) as the acceptance criteria provided in the protocol should be based on meeting these.
Scheduling. This section of the VMP should describe how the validation team undertake performing and recording the validation work and how the validation is signed-off and deemed acceptable. The typical phases of a validation schedule are:
Training in the protocol and new operational techniques. This will be required before members of the team are competent to carry out the validation particularly if a new, unfamiliar piece of equipment is being used.
Performing validation. Validation results should be recorded at the time and presented in a validation report (See Section 11.3) for comparison with the acceptance criteria in the protocol. It is common to summarise the validation method and provide the acceptance criteria in validation scripts and the validation team simply records whether the required outcomes are achieved (See examples Appendix 10).
Validation Final Summary Report review and sign off (section 12.3) Following validation, the validation team should present the validation report for review and sign-off. The report should at least be reviewed and signed-off as an accurate record by the Validation Manager (See above).
Decision. Finally, a decision is required by the independent expert (See above) as to whether the process, equipment, facilities or systems. under validation is acceptable.
Appendix 1: Information to be included in the validation master plan
5
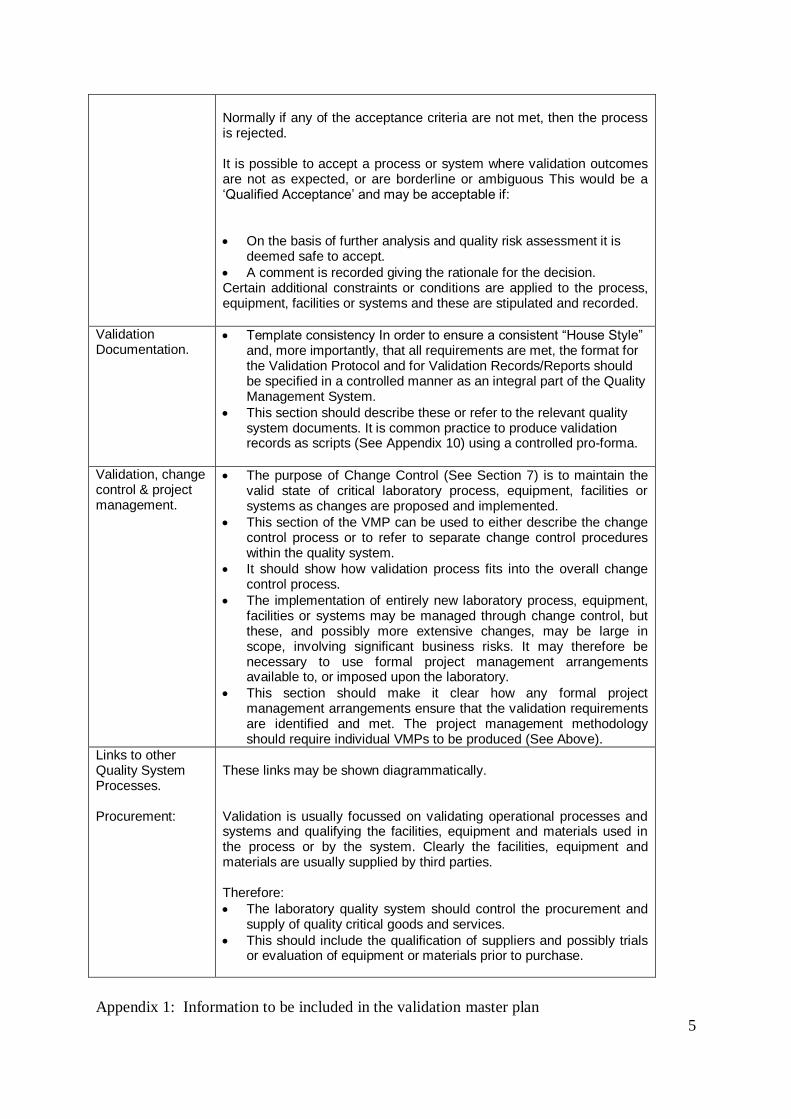
Normally if any of the acceptance criteria are not met, then the process is rejected. It is possible to accept a process or system where validation outcomes are not as expected, or are borderline or ambiguous This would be a „Qualified Acceptance‟ and may be acceptable if:
On the basis of further analysis and quality risk assessment it is deemed safe to accept.
A comment is recorded giving the rationale for the decision. Certain additional constraints or conditions are applied to the process, equipment, facilities or systems and these are stipulated and recorded.
Validation Documentation.
Template consistency In order to ensure a consistent “House Style” and, more importantly, that all requirements are met, the format for the Validation Protocol and for Validation Records/Reports should be specified in a controlled manner as an integral part of the Quality Management System.
This section should describe these or refer to the relevant quality system documents. It is common practice to produce validation records as scripts (See Appendix 10) using a controlled pro-forma.
Validation, change control & project management.
The purpose of Change Control (See Section 7) is to maintain the valid state of critical laboratory process, equipment, facilities or systems as changes are proposed and implemented.
This section of the VMP can be used to either describe the change control process or to refer to separate change control procedures within the quality system.
It should show how validation process fits into the overall change control process.
The implementation of entirely new laboratory process, equipment, facilities or systems may be managed through change control, but these, and possibly more extensive changes, may be large in scope, involving significant business risks. It may therefore be necessary to use formal project management arrangements available to, or imposed upon the laboratory.
This section should make it clear how any formal project management arrangements ensure that the validation requirements are identified and met. The project management methodology should require individual VMPs to be produced (See Above).
Links to other Quality System Processes. Procurement:
These links may be shown diagrammatically. Validation is usually focussed on validating operational processes and systems and qualifying the facilities, equipment and materials used in the process or by the system. Clearly the facilities, equipment and materials are usually supplied by third parties. Therefore:
The laboratory quality system should control the procurement and supply of quality critical goods and services.
This should include the qualification of suppliers and possibly trials or evaluation of equipment or materials prior to purchase.

Appendix 1: Information to be included in the validation master plan
6
Training & Document Control: Facilities & Equipment management:
It is also possible that an important part of the qualification of new facilities, equipment etc. known as Design Qualification and will relate to the Functional Design Specification (See Section 10), is performed as part of the procurement process.
Clearly, the outcomes from this activity will influence subsequent validation before these goods and services are put into use and therefore, this section of the VMP should describe, or refer to, the supplier control procedures. Development of SOPs and training in the use of these SOPs for operating any new system will be crucial before it is finally approved for use. This requirement will normally be included in the PQ protocol. Once the laboratory process, equipment, facilities or systems has been approved for use, it is essential that documentation is maintained in a current state. Therefore, part of maintaining processes and systems in a valid state is the qualification of operational staff and of SOPs used. As these are usually described in separate training and document control procedures they should simply be referenced in this section. It would be appropriate to describe or reference any staff proficiency schemes operated by the laboratory in this section. The management and control of facilities and equipment is critical to maintaining the valid state. In particular servicing, calibration of instrumentation and re-qualification should be planned and managed within the laboratory quality system. These arrangements may be described in this section or reference made to the appropriate procedures. Automated test systems may be subject to proficiency or EQA Schemes and these should be mentioned here.

Appendix 2: Example of a VMP currently at use in an NHS hospital
7
Example of a VMP currently in use at an NHS Hospital
Department of Blood Transfusion
Code: enter details Page Insert numbers
Title: Validation Master Plan (VMP)
Area of application: Blood Transfusion Hospital A
Blood Transfusion Hospital B Blood Transfusion Hospital C
Index code:
Implementation date:
This copy issued to:
Related CPA standard/key words:
This document is under the department document control system.
To comply with CPA standard A8 the document control system records the reason for change, current revision status of documents, dates of review, document owner and approver and locations of printed copies.
It is forbidden to photocopy from authorised printed copies which have been issued to locations as recorded in the
software. Authorised printed copies can be identified by the authorisation signature and stamp present in the space
below.
SIGNATURE
LIST OF CONTENTS
Section Page No
1. Validation Policy 2 2. Organisational structure of validation activities 2 3. Summary of the facilities, systems, equipment and processes to be validated 3 4. Documentation format 4 5. Planning and scheduling 4 6. Change control 5 7. References 5
Appendix 2

Appendix 2: Example of a VMP currently at use in an NHS hospital
8
1. Validation Policy
1.1 The validation policy of the Insert name Hospitals blood Transfusion Department is set out in
the policy document insert number/link. This policy applies when a change is proposed to a
starting material, product component, process equipment, process environment (or site),
method of production or testing or any other change that may affect product quality (or result quality) or reproducibility of the process
2 Organisational structure of validation activities
2.1 Planning validation
2.1.1 All validation planning is the responsibility of the Blood Transfusion Quality Manager who
liaises with the appropriate Site Coordinator or his designated deputy to ensure that
appropriate validation takes place.
2.1.2 The validation planning should also involve experts in the area being validated.
Major IT validation should involve the IT coordinator
Validation in Ante-Natal Testing should involve the Ante-Natal coordinator 2.1.3 For each validation there will be a validation team comprising;
The Quality Manager
A Site Coordinator or designated deputy
Other experts as appropriate
2.1.4 The quality Manager will be responsible for assembling the validation team
2.1.5 For large scale changes there may need to be separate validation plans covering each area
2.2 Performing validation
2.2.1 All validations should be performed by staff familiar with the processes being validated. The
laboratory validations must be performed by a Health Professions Council Registered Biomedical Scientist or above and overseen by an Advanced Biomedical Scientist or above
2.3 Approving validation
2.3.1 Normally all validations will be approved by the Quality Manager
2.3.2 Occasionally validations may be approved Department Manager or appropriate Site-
Coordinator in the absence of the Quality Manager
2.4 Final validation summary report
2.4.1 There should be a document to indicate whether approval for release has been given, this should include any conditions on release.
2.4.2 Final sign off of the validation must be by the Department Manager, Quality Manager or
appropriate Site-Coordinator
3 Summary of the laboratory process, equipment, facilities or systems to be validated
3.1.1 Any changes to the systems or processes in the following areas need to be validated
Sample labelling reception and booking-in
Sample storage and transport.
Automated Sample handling systems o Insert machines
Controlled temperature storage of critical reagents and controls.
Critical Test methods including result reporting.
o Electronic issue
o Crossmatching
o Grouping and antibody screening
o Phenotyping
o DAT testing

Appendix 2: Example of a VMP currently at use in an NHS hospital
9
o Antibody investigation
o Transfusion Reaction investigation
o MAJAX procedures
o Ante-natal Testing
Blood component processing labelling & tracking.
o Secondary processing systems Plasma thawers
Platelet agitators
o Labelling procedures
o Release procedures
Blood Component cold storage.
o Receipt procedures
o Storage procedures
o Monitoring
o Alarms
Blood Component Distribution.
o Blood Track procedures o Traceability procedures
4 Documentation format
4.1 All validation documentation should take the same format this will be:
4.2 Validation plan
4.2.1 Approval sheet
4.2.2 Document change control sheet
4.2.3 Purpose and scope
4.2.4 Background References 4.2.5 Definitions and acronyms
4.2.6 System definition and description
4.2.7 System maintenance and support strategy
4.2.8 Validation approach
4.2.9 Implementation strategy
4.2.10 Training requirements related to responsibilities
4.2.11 Appendices
4.3 Validation summary report (see template in Appendix 11)
4.3.1 Introduction; to include
Validation plan details
4.3.2 Validation results
4.3.3 Unexpected results / problems
4.3.4 Recommendations / Further action
4.3.5 Continuing Validation
4.3.6 References; to include
Validation master plan
Validation Plan
Change control
5 Planning and scheduling
5.1 All validations should be planned
5.2 The validation plan should incorporate any planning and a timescale for implementation. The
decisions on timescale will be down to the validation team.
6 Change control

Appendix 2: Example of a VMP currently at use in an NHS hospital
10
6.1 For any proposed change to anything which could affect the quality or reproducibility of test
results or components is completed.
6.2 Change control is needed for changes to:
6.2.1 Starting material (e.g. reagents, consumables)
6.2.2 Procedure / method
6.2.3 Environment 6.2.4 Equipment
6.3 Changes are assessed by the Quality Manager and any other appropriate officers. The result of
this assessment can be either:
6.3.1 Change approved no validation needed
6.3.2 Change approved validation required
6.3.3 Change not approved
7 References
7.1 Policies
7.2 Procedures
7.3 Forms
7.4 Add templates

Appendix 3: Change control request form
11
CHANGE CONTROL REQUEST FORM
1 Title of Change
1.1 Change to new panel cell supplier
2 Reason for Change
2.1 Investigation of a failure to identify a combination of antibodies in a NEQAS exercise suggested that one of the
contributory factors in this identification was the poor antigen profile of the panel cells currently used
2.2 Examples of supplier B panel cell antigen profiles were sought and these would have produced unequivocal results and
would have aided identification
2.3 The department would like to source its antibody identification panels from supplier B
3 Description of Change
3.1 Change of panel cell supplier from Supplier A to supplier B
3.2 Cells from supplier B will be provided in modified Alsevers solution at 3%. In order to use these cells by the current
technique the cells will need to be washed and prepared to 0.8% in supplier A’s diluent.
3.3 The diluent product insert indicates a method for preparing cells to 0.8%. The insert indicates that fresh cells (from patient’s
or donor blood) prepared in this manner will remain stable for 7 days. The insert indicated that commercial panel cells
prepared in this manner are only guaranteed to be stable for 24 hours. Cells used in this manner do not need any validation
3.4 Daily preparation and use is not practical within the department. The department would like to demonstrate that supplier B’s
cells can be prepared using the method on the diluent product insert and remain suitable for use for 7 days or longer.
Appendix 3

Appendix 3: Change control request form
12
4 Impact//Quality Risk Assessment
4.1 Risk matrix used in assessment: Risk score = Impact x Likelihood Impact Description Likelihood Description Risk
Score
Risk Level - Treatment Timeframe
1 Insignificant 5 Almost Certain
Score Risk Rating
2 Low 4 Likely 1-3 Low These risks are considered acceptable, no action over and above
existing procedures
3 Moderate 3 Possible 4-6 Moderate Monitoring of risks with view to
effort being made to reduce these
within a 12 month period
4 Severe 2 Unlikely 8-12 Significant Management consideration of risks
and reduction of these within 6 month
period
5 Catastrophic 1 Rare 15-25 Critical Senior management attention
immediately with view to action
being taken to reduce risk
4.2 Antibody Identification has a number of potential risks associated with it
4.2.1 Failure to detect clinically significant antibody can result from
An inadequately prepared panel
o Incorrect cell suspensions
o Incorrect tubing out of panel cells
o Failure to add patient’s plasma
Deterioration of red cell antigens through storage
Failure to provide all relevant clinically significant antigens on the profile particularly those with homozygous
expression
4.2.2 Failure to identify antibody can result from
Deterioration of red cell antigens through storage

Appendix 3: Change control request form
13
Failure to provide all relevant clinically significant antigens on the profile particularly those with homozygous
expression
Antigen profiles on panels providing insufficient antigen negative cells making distinguishing of antibody mixtures
particularly difficult
4.2.3 All of the above risks can cause potential serious problems to patients including:
Failure to provide compatible blood Risk score = 4x3 = 12
Failure to provide compatible blood in a timely fashion – caused by additional testing or need to refer samples where
the panels cannot provide antibody identification Risk score = 4x3 = 12
5 Mitigation of risks
5.1 Incorrect Cell Suspensions: can be mitigated by the production of a robust standard operating procedure which ensures
that the cell suspensions are prepared by the same method as indicated on the diluent product insert.
5.2 Incorrect tubing out of cells: can be mitigated by the production of a robust standard operating procedure which ensures
that cells are prepared and labelled in the same way each time
5.3 Failure to provide an inadequate antigen profile: this is mitigated by ensuring that the cells meet the Red Book
Guidelines
5.4 Deterioration of red cell antigens on storage: This can be mitigated by validation of the activity of a prepared panel from
date of preparation (as soon after receipt as possible) until its expiry.
5.5 Activity of Panel cells: The panel cells should be quality controlled after preparation to demonstrated that they are working
correctly
6 Validation requirements
6.1 A validation plan must be prepared and validation performed to demonstrate that supplier B’s panel cells do not deteriorate
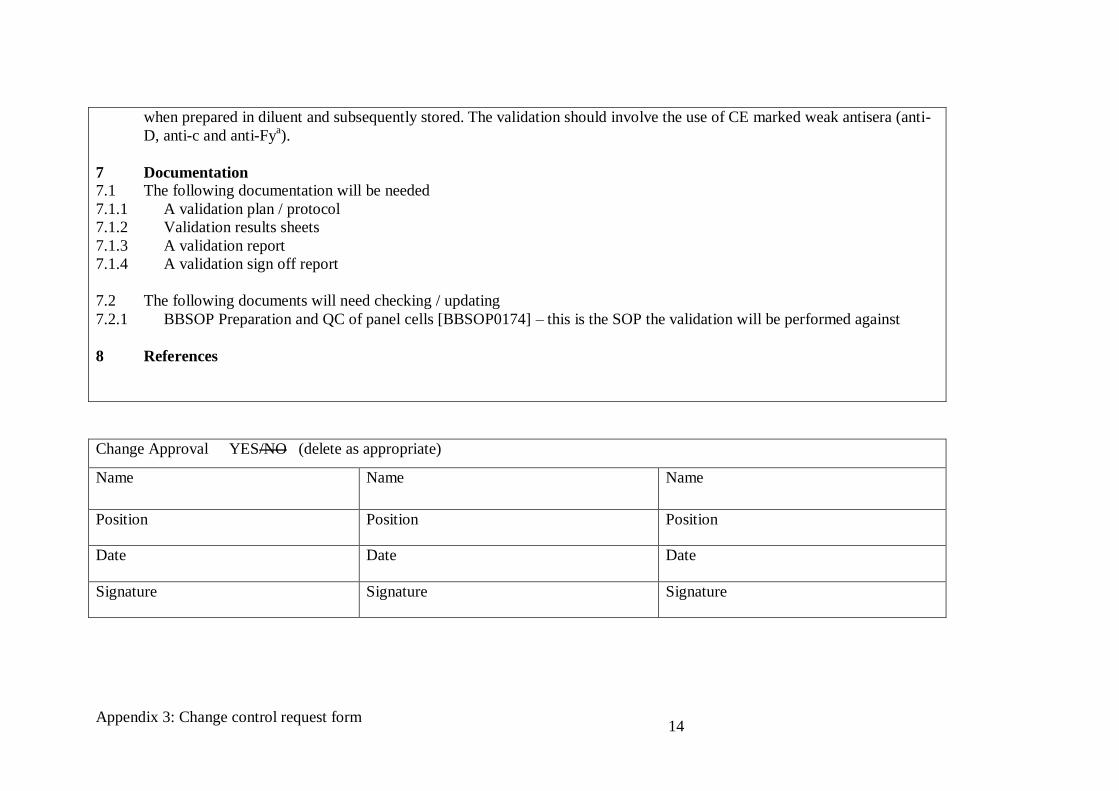
Appendix 3: Change control request form
14
when prepared in diluent and subsequently stored. The validation should involve the use of CE marked weak antisera (anti-
D, anti-c and anti-Fya).
7 Documentation
7.1 The following documentation will be needed
7.1.1 A validation plan / protocol
7.1.2 Validation results sheets
7.1.3 A validation report
7.1.4 A validation sign off report
7.2 The following documents will need checking / updating
7.2.1 BBSOP Preparation and QC of panel cells [BBSOP0174] – this is the SOP the validation will be performed against
8 References
Change Approval YES/NO (delete as appropriate)
Name Name Name
Position Position Position
Date Date Date
Signature Signature Signature
Appendix 4: specifications of what could be included in a User Requirement
Specification
15
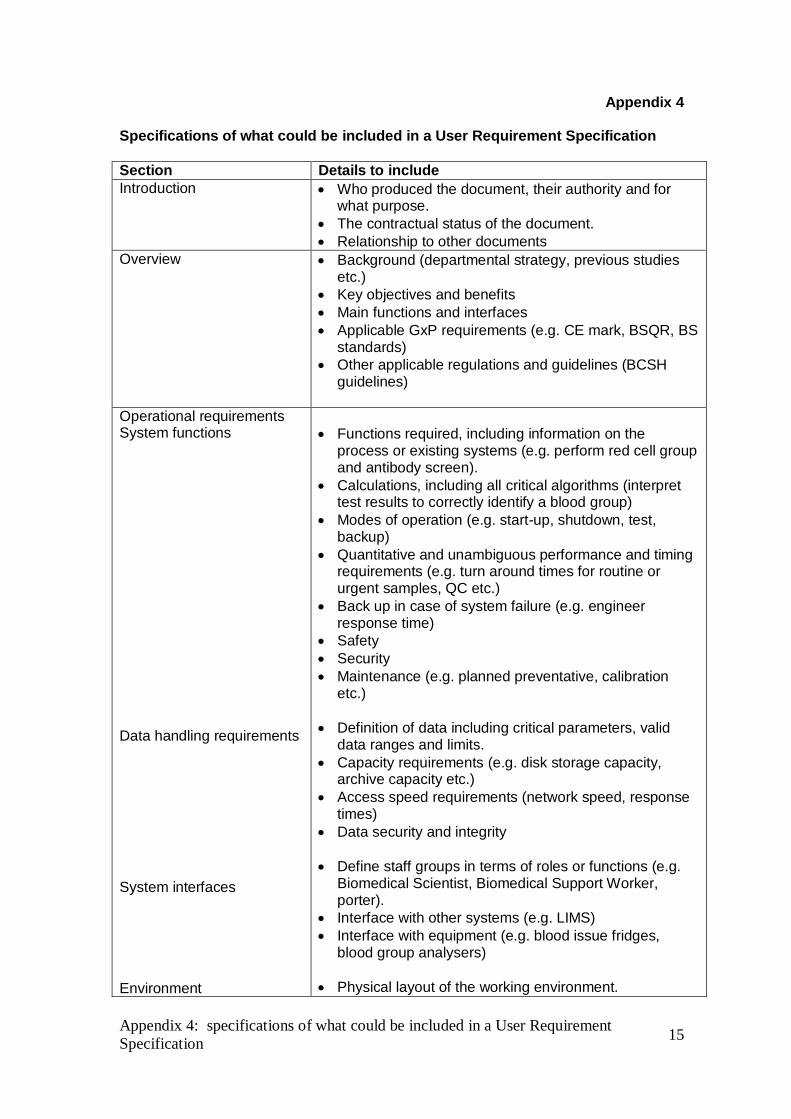
Appendix 4
Specifications of what could be included in a User Requirement Specification
Section Details to include
Introduction
Who produced the document, their authority and for what purpose.
The contractual status of the document.
Relationship to other documents
Overview
Background (departmental strategy, previous studies etc.)
Key objectives and benefits
Main functions and interfaces
Applicable GxP requirements (e.g. CE mark, BSQR, BS standards)
Other applicable regulations and guidelines (BCSH guidelines)
Operational requirements System functions Data handling requirements System interfaces Environment
Functions required, including information on the process or existing systems (e.g. perform red cell group and antibody screen).
Calculations, including all critical algorithms (interpret test results to correctly identify a blood group)
Modes of operation (e.g. start-up, shutdown, test, backup)
Quantitative and unambiguous performance and timing requirements (e.g. turn around times for routine or urgent samples, QC etc.)
Back up in case of system failure (e.g. engineer response time)
Safety
Security
Maintenance (e.g. planned preventative, calibration etc.)
Definition of data including critical parameters, valid data ranges and limits.
Capacity requirements (e.g. disk storage capacity, archive capacity etc.)
Access speed requirements (network speed, response times)
Data security and integrity
Define staff groups in terms of roles or functions (e.g. Biomedical Scientist, Biomedical Support Worker, porter).
Interface with other systems (e.g. LIMS)
Interface with equipment (e.g. blood issue fridges, blood group analysers)
Physical layout of the working environment.

Appendix 4: specifications of what could be included in a User Requirement
Specification
16
Physical conditions (e.g. dusty, sterile, air conditioned)
Constraints
Timescales and milestones (e.g. speed of delivery, commissioning time etc.)
Compatibility (e.g. will the software work on your current server / IT system)
Availability (e.g. required 24/7 or 23 hrs per day)
Procedural constraints, these include external but inter related factors (e.g. specimen tube type, workforce skill mix)
Cost
Glossary
Definitions of any terms that may be unfamiliar to the readers of the document.

Appendix 5: Example of a user requirement specification
17
Example of a User Requirement Specification
Provision of Blood Grouping analysers and reagents
The proposed equipment must be able to meet the current workloads with capacity to increase
these by xx%. The current annual blood transfusion workload is approximately
Blood Groups and screens: enter number
DAGT: enter number
Neonatal grouping with DAGT: enter number
Antibody panel‟s approx: enter number
Full crossmatch on cards: enter number Proposals are required for processing of the current workload as stated. The requirement
must address but not be limited to:
Delivery,
Installation
Commissioning
Consumables
Reagents
Quality Control
Maintenance of the equipment Bi-directional interface to the laboratory computer system (Enter system)
Training
Disposal of equipment at the end of the life. The Tenderer must detail how they will comply with the requirement.
It is a requirement that the current laboratory output must be maintained during installation,
acceptance testing and qualification of the automation.
Proposals must be able to show from current users a high level of satisfaction regarding the
automation, product and technical support and customer care.
The supplier and their automated users must have a proven track record with regards to
NEQAS returns.
The supplier must have adequate support facilities.
2. General Analyser Specification
Proposals must specify the proposed equipment, hardware, uninterruptible power supply
etc.
All equipment proposed must be automated, and capable of meeting the volumes provide in
this document with the ability to increase by at least xx%.
Due to the nature of the work the User requires equipment to be operable 24 hours a day
seven days a week. Currently the User has four analysers installed to manage any
inoperable time, and must state how they will ensure equipment will be operable 24/7.
Details of guaranteed uptime (and its definition) must be provided, details on; how uptime is
measured, how this will be attained, and remedies to the Authority if the uptime is not
maintained are required to be submitted.
Guaranteed uptime must be 24 hours a day seven days a week provided over a calendar month, tenderer’s must provide details of how this will be achieved.
(Mixed field reaction for post BMT relapses and mixed ABO transfusion should be
recognised).
Appendix 5

Appendix 5: Example of a user requirement specification
18
The range of tests that must be available on the machine are (but not limited to) the following;
ABO and Rh D grouping both full ABO group 3 cell antibody screens by IAT DAT‟s including monospecific typing Antibody identification panel with enzyme treated and IAT cells Secondary antibody identification panel with enzyme treated and IAT cells Miscellaneous red cell phenotyping
The analyser must be capable of running without continual operator presence. The proposed system must allow customer definable password protection levels and users.
User friendly and safe operation is expected. Start up, shut down, calibration, QC, local maintenance and general cleaning procedures must be stated and the length of time involved and required frequency of these procedures. Requirements and consumption rates for power, water, saline, drainage and air conditioning must be stated and installation costs included.
Details of any additional consumables, special waste containers must be provided and full
costs provided. Proposed system must conform to current blood transfusion guidelines as defined by the
British Committee for Standards in Haematology (BCSH) – Blood Transfusion Task Force or equivalent.
Proposed system must conform to current EC directives for in vitro diagnostics (IVD) electrical safety (CE) and CPA guidelines or equivalent.
The tendered should state whether they have a software package to assist in the identification of atypical antibodies and whether this attracts an additional cost.
Fully detailed operator manual must be provided. Such manuals must be renewed as and
when the instrument software or hardware is updated and must be supplied in English. The User will expect all safety upgrades or enhancements to the equipment to be
undertaken free of charge. 3. Interfacing Proposed equipment must be compatible with the laboratory‟s LIMS (currently insert
system). Tenderers should state how many installations of the proposed system are interfaced with this LIMS, giving location and contact information for each.
Interfaces must be operable before “go live” and noted in a project plan or key stage
document with the submission, Tenderer‟s must also advise of any remedies if the proposed project plan is delayed.
Tenderer‟s must state details of any Laboratory information systems the proposed system is
interfaced with, providing relevant contact information. The Tenderer must state how it will achieve the interface to the LIMS and timescales to
complete the interface. Data transfer must be automatic and on-line but must also be able to cope with LIMS
downtime. Provide details that this is possible within the proposed equipment. The cost of interface development, installation, licence and maintenance must be included
in the system cost and set out in the pricing schedule. The pricing must include both sides of the interface.

Appendix 5: Example of a user requirement specification
19
4. Sampling requirements Cap piercing facility is desirable, proposals must state if cap piercing is available by the
proposed equipment. The system must be capable of reading and sampling from bar coded primary tubes. The
system must be compatible with Codabar and ISBT128 bar codes. Please state all other bar code configurations that are readable by the proposed equipment.
Small volume paediatric samples must be accommodated. The minimum volume
requirements for all sample tube sizes must be stated. The system must have the capability to accept a wide variety of sample tubes. Sample tube sizes and types that are not compatible with the proposed equipment must be
clearly stated STAT/Urgent facility should be available. A rapid ABO and Rh D group should be available
in less than 10 minutes. A full group and antibody screen must be completed in less than 40 minutes.
Samples should be able to be removed from the proposed equipment either; prior or post
sampling in case urgent testing is required. Varying length of time in which samples can be removed must be stated.
The equipment should be able to display time until the results of test/s will be reportable. The system must validate that appropriate volumes of red cells, plasma or reagent have
been added to the test. Any deficiencies must be highlighted to the operator. Please state how the system reports such occurrences.
The sampling system should have level sensing, clot detection, bubble sensing and short
sample alerts both audible and visual. Warnings should be given when there is an error. Known interferences including icterus, lipaemia, and haemolysis must be stated and how
any compensation if any is made. Details of reagent and sample carry-over must be provided. The tests should be accurate on fresh samples for up to 72 hours and normally observed
storage temperatures must not affect them. It should not be necessary to equilibrate refrigerated samples to room temperature.
The equipment should be able to process plasma/serum that has already been separated
from the red cells for antibody screening. The equipment should be able to process different tests within the 1 batch i.e. adult group,
DAT, Rh phenotype. 5. Reagent/Cell Requirements
The red cells provided for antibody screening must always conform to the BCSH published guidelines regarding required antigen phenotypes and homozygozity.
Please state whether the proposed solution can provide a Cw and a Kpa positive cell on
your standard screening cells. If so please state the number of screening cells used to provide this guaranteed expression.

Appendix 5: Example of a user requirement specification
20
Please state the number of cells in the primary and secondary antibody identification panel and the medium the cells are suspended in.
Please state whether an antibody identification software package is provided with each
panel. Please provide specifics of the package. The equipment must have level detection and be able to calculate if there are any shortfalls
in either regents or consumables to complete a batch of work and alert the Biomedical Scientist (BMS) immediately. The alert must be both audible and visual.
No reagent or cell preparation must be required. All reagents or cells must have “load and
run” facility. All reagents containing red cells must be agitated by such methods as required to prevent
settling out. All reagents must be bar coded. The equipment must be capable of reading bar coded
information from reagent packs. As a minimum, batch number, expiry date and date of placing on the equipment must be
recorded and it should also warn the BMS of expiring reagents. State storage requirements for one month and six weekly supply of red cells, reagents and
consumables including space required at room temperature, refrigerated or deep frozen. State guaranteed minimum shelf-life of products provided. Provide details of standard and emergency orders for red cells or reagents and the lead-
time and cost. Details of any third party consumables that are compatible with the proposed systems must
be provided.
6. Quality Control (QC) The system must have monitoring of all aspects of instrument performance (incubation
temperature, centrifuge speed, pipette volumes etc). Submissions must include details of the quality control material (QC) proposed and any
associated cost. Proposals must specify the recommended frequency of QC. All QC material must be bar coded and must not require any preparation. QC results should be clearly indicated with appropriate status tags against defined results. The system should not normally allow testing to proceed where the calibration and QC data
are outside the prescribed limits or where the calibration and / or QC has not been performed in accordance with the system configuration. There should be a security protected override for this. Any results generated with the override activated should be flagged to show this.
Details of QC handling programmes on the equipment must be given. The onboard storage
capacity of QC data must be given. The QC batch numbers, targets and results should be available for storage suitable for
accreditation purposes.

Appendix 5: Example of a user requirement specification
21
Details of onboard validation, approval and checking of patient results must be given. Automatic validation of results within user-defined limits should be available.
7. Data Processing and Storage The equipment must be capable of interfacing with the laboratory computer system i.e.
insert system. The interface must be bi-directional. Provide details of when the proposed interface will be operational and what functionality will
be available for go live. Where necessary it must be possible to use the equipment in a stand-alone mode.
Automatic reconnection to the host computer should be available and transmission of results from stand-alone running.
State the capability of the equipment to continue to process samples and generate reports
during periods of unavailability of the computer host system and the mechanism for doing this.
The requirement for a data manager, either supplied as original equipment or as an adjunct
to the equipment must be stated. The precise specification and functionality of such a data manager must be clearly stated.
Provide details of the data handling and management capabilities of the system including
inputting of any additional tests and storage facilities/capacity for patient records. If the equipment proposed has several linked analysers it must be possible for the other
analysers to continue operating if one or more of the analysers are in-operable for whatever reason.
Stored data must be easily retrievable. A full audit trail must be available of all tests performed including QC. Please state what
information is stored and is retrievable. A pictorial representation of all tests performed must be stored. Please state the format that the audit trail information and pictures will be stored and what
capacity of data/pictures can be stored. There should be a facility to operate and monitor the analysers remotely using a handheld
Wi-Fi device.
8. Maintenance
Routine maintenance must be able to be performed by the BMS staff. The daily, weekly and yearly maintenance procedures must be described. The quantity, frequency and duration of preventative maintenance visits per annum must be
stated. Maintenance contracts available must be described along with the guaranteed response
times for callouts. State the support available at night, at weekends and public holidays. The times during which technical support is available must be stated.
Fully detailed operator manual must be provided in English. Please state the level of “self-help” available from the manuals.

Appendix 5: Example of a user requirement specification
22
Please state whether on-line manuals are available. Modem links for remote access for problem solving should be available. Please provide details of the locality of engineers and spare parts relative to the Authority‟s
normal place of business. A guarantee must be provided that the proposed equipment will be supported and spares
available for the period.
9. Training
Provide details of the initial on-site training for staff during the set-up period. Proposals must include details of the training courses included with the supply of
automation, including the number of places available and the duration and location of the courses. Please provide an example of a training prospectus for the system.
State whether additional courses are available at a later date and whether any on-site
training is included. Details of any user groups in the UK and the frequency of meetings should be provided;
proposal of support should be included. 10. Health and Safety The proposed equipment must comply with relevant regulations for electrical, mechanical
and biological safety. All reagents and cells proposed must confirm comply with relevant regulations regarding
shipping, labelling and information on hazardous substances. COSHH data must be confirmed as available and must be supplied in advance of installation.
Provide details of waste disposal requirements including any special precautions for
handling “High Risk” samples or waste. A decontamination procedure for the equipment must be provided with recommendations
(including recommended cleaning products) of when it should be used.
Appendix 6: Specifications for inclusion of FDS
23
Appendix 6
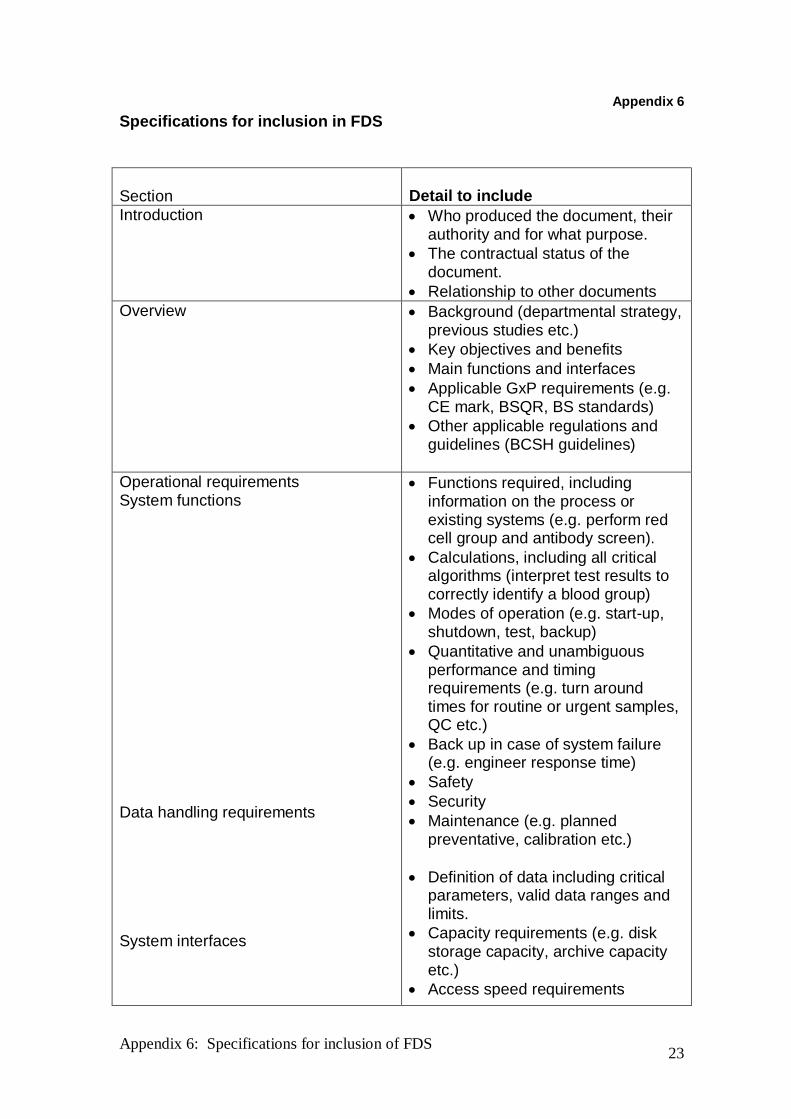
Specifications for inclusion in FDS
Section
Detail to include
Introduction
Who produced the document, their authority and for what purpose.
The contractual status of the document.
Relationship to other documents
Overview
Background (departmental strategy, previous studies etc.)
Key objectives and benefits
Main functions and interfaces
Applicable GxP requirements (e.g. CE mark, BSQR, BS standards)
Other applicable regulations and guidelines (BCSH guidelines)
Operational requirements System functions Data handling requirements System interfaces
Functions required, including information on the process or existing systems (e.g. perform red cell group and antibody screen).
Calculations, including all critical algorithms (interpret test results to correctly identify a blood group)
Modes of operation (e.g. start-up, shutdown, test, backup)
Quantitative and unambiguous performance and timing requirements (e.g. turn around times for routine or urgent samples, QC etc.)
Back up in case of system failure (e.g. engineer response time)
Safety
Security
Maintenance (e.g. planned preventative, calibration etc.)
Definition of data including critical parameters, valid data ranges and limits.
Capacity requirements (e.g. disk storage capacity, archive capacity etc.)
Access speed requirements

Appendix 6: Specifications for inclusion of FDS
24
Environment
(network speed, response times)
Data security and integrity
Define staff groups in terms of roles or functions (e.g. Biomedical Scientist, Biomedical Support Worker, porter).
Interface with other systems (e.g. LIMS)
Interface with equipment (e.g. blood issue fridges, blood group analysers)
Physical layout of the working environment.
Physical conditions (e.g. dusty, sterile, air conditioned)

Appendix 7: Example of a validation plan
25
Appendix 7
Example of a Validation Plan
Department of Blood Transfusion
VALIDATION PLAN
{Insert Title of Validation}
Validation Plan Reference number
{Insert title of Validation}
Validation Plan Prepared by: {Insert details}
Date:
{Insert date of plan preparation}

Appendix 7: Example of a validation plan
26
Department of Blood Transfusion
VALIDATION PLAN
{Insert Title of Validation}
Validation Plan Reference number Page x of y
I recommend approval of this validation plan; {insert title) Signature____________________ Date_________ {Insert name} {Insert position}

Appendix 7: Example of a validation plan
27
Department of Blood Transfusion
VALIDATION PLAN
{Insert Title of Validation}
Validation Plan Reference number Page x of y
1 Purpose and scope 1.1 Introduction
1.1.1 1.2 Goals
1.2.1 1.3 Scope
1.3.1 1.4 Specific procedures and processes covered
1.4.1 1.5 Assumptions
1.5.1
2 Background References
2.1 References to legal documents
2.1.1 2.2 References to Guidelines
2.2.1 2.3 References to other documents 2.3.1
3 Definitions and acronyms
3.1

Appendix 7: Example of a validation plan
28
Department of Blood Transfusion
VALIDATION PLAN
{Insert Title of Validation}
Validation Plan Reference number Page x of y
4 System description
4.1 5 System maintenance and support strategy
5.1 6 Validation approach 6.1 Schedule
6.1.1 6.2 Resource summary
6.2.1 Staffing 6.2.2 Facilities 6.2.3 Equipment 6.2.4 Finance 6.3 Responsibilities
6.3.1 6.4 Method of validation 6.4.1 Tools 6.4.2 Techniques 6.4.3 Method 6.4.3.1 Design Qualification 6.4.3.2 Installation Qualification 6.4.3.3 Operational Qualification 6.4.3.4 Performance Qualification

Appendix 7: Example of a validation plan
29
Department of Blood Transfusion
VALIDATION PLAN
{Insert Title of Validation}
Validation Plan Reference number Page x of y
7 Implementation strategy
7.1 8 Training requirements related to responsibilities
8.1 9 Appendices 9.1 Appendix I: System hardware configurations if applicable 9.2 Appendix II: Software Components if applicable 9.3 Appendix III: Documents that Form the Validation Record and their
Approval Requirements (e.g. checklists)
Appendix 8: Installation, Operational and Process Qualification; IQ, OQ and PQ
30
Appendix 8
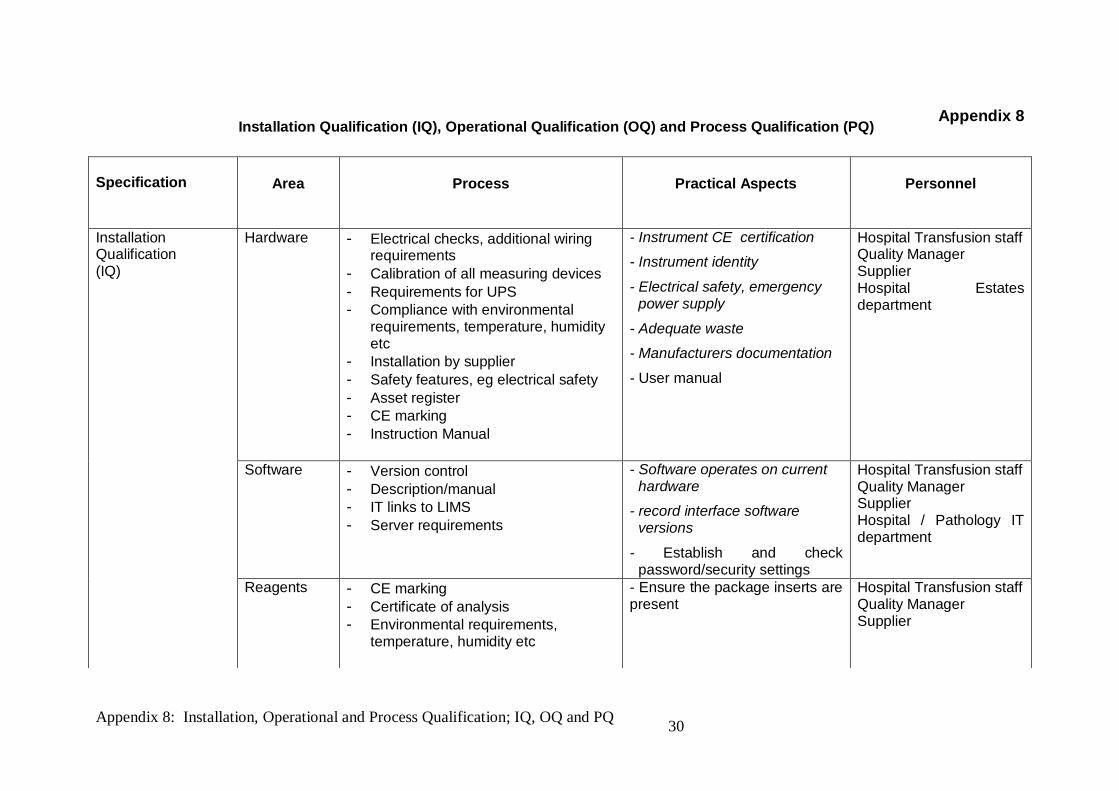
Specification
Area
Process
Practical Aspects
Personnel
Installation Qualification (IQ)
Hardware
- Electrical checks, additional wiring requirements
- Calibration of all measuring devices - Requirements for UPS - Compliance with environmental
requirements, temperature, humidity etc
- Installation by supplier - Safety features, eg electrical safety - Asset register - CE marking - Instruction Manual
- Instrument CE certification
- Instrument identity
- Electrical safety, emergency power supply
- Adequate waste
- Manufacturers documentation
- User manual
Hospital Transfusion staff Quality Manager Supplier Hospital Estates department
Software - Version control - Description/manual - IT links to LIMS - Server requirements
- Software operates on current hardware
- record interface software versions
- Establish and check password/security settings
Hospital Transfusion staff Quality Manager Supplier Hospital / Pathology IT department
Reagents - CE marking - Certificate of analysis - Environmental requirements,
temperature, humidity etc
- Ensure the package inserts are present
Hospital Transfusion staff Quality Manager Supplier
Installation Qualification (IQ), Operational Qualification (OQ) and Process Qualification (PQ)
Appendix 8: Installation, Operational and Process Qualification; IQ, OQ and PQ
31
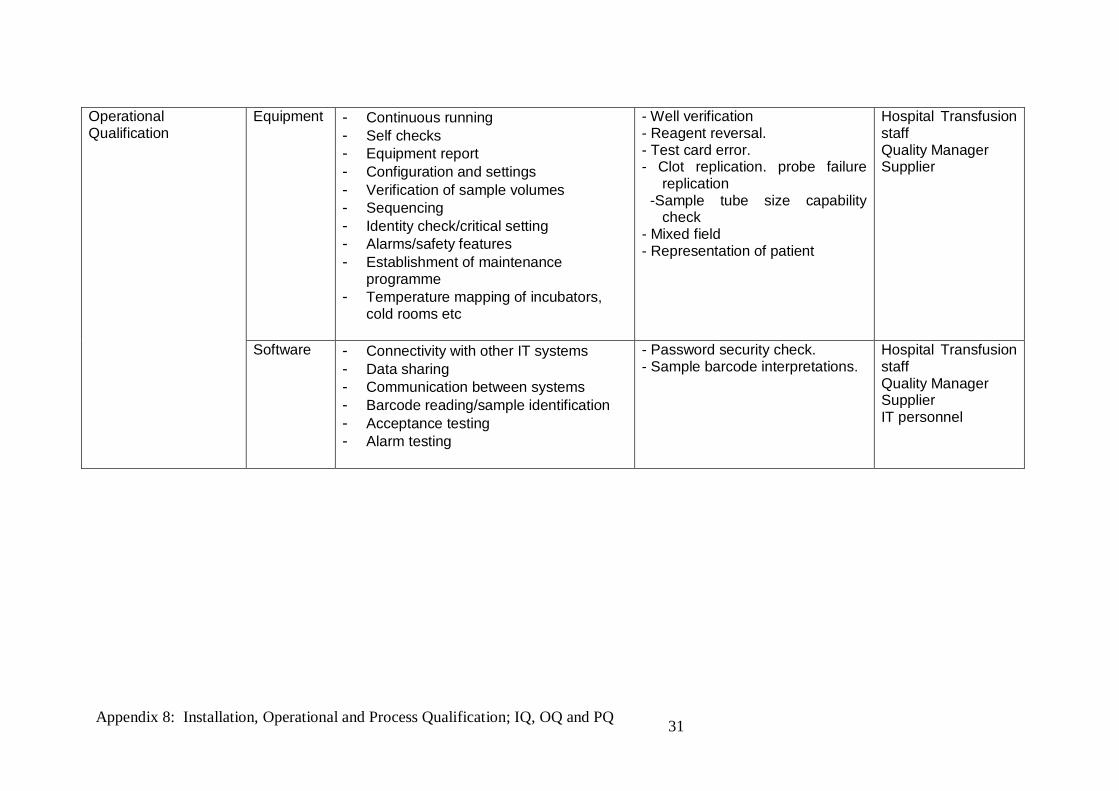
Operational Qualification
Equipment - Continuous running - Self checks - Equipment report - Configuration and settings - Verification of sample volumes - Sequencing - Identity check/critical setting - Alarms/safety features - Establishment of maintenance
programme - Temperature mapping of incubators,
cold rooms etc
- Well verification - Reagent reversal. - Test card error. - Clot replication. probe failure
replication -Sample tube size capability
check - Mixed field - Representation of patient
Hospital Transfusion staff Quality Manager Supplier
Software - Connectivity with other IT systems - Data sharing - Communication between systems - Barcode reading/sample identification - Acceptance testing - Alarm testing
- Password security check. - Sample barcode interpretations.
Hospital Transfusion staff Quality Manager Supplier IT personnel

Appendix 8: Installation, Operational and Process Qualification; IQ, OQ and PQ
32
Reagents - Controls – positive and negative - Red book requirements
Examples of all ABO and D groups
- Weak D and Dvi - Samples with negative antibody
screen - Samples with positive antibody
screens, to include weakly reacting antibodies -Specificity to be confirmed using samples containing anti-D,c,e,K,Fya, Jka and S Sensitivity check using antibody titration
Hospital Transfusion staff Quality Manager
Process Qualification (PQ)
Equipment - Parallel running with current system by all methods
- Maximum specification tested - Meaningful run time - Operation under worst case conditions - Tests under various load conditions
- Reliability The level and areas to be qualified
should be determined from a risk assessment
Hospital Transfusion staff Quality Manager
Software - Right interpretation - Back up - Interfaces - Consistency - Repeatability - Failures - Data archiving systems
- Record all false negatives and false positives
- Check download of all results
Hospital Transfusion staff Quality Manager IT staff
Appendix 8: Installation, Operational and Process Qualification; IQ, OQ and PQ
33
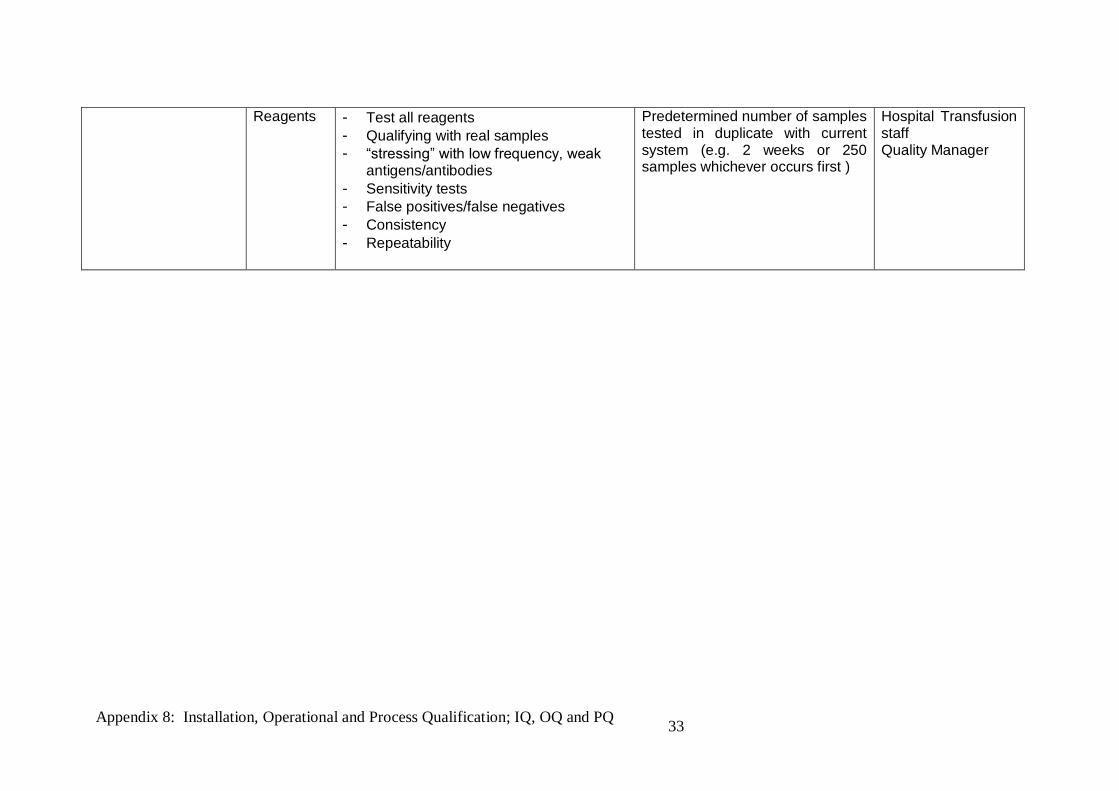
Reagents - Test all reagents - Qualifying with real samples - “stressing” with low frequency, weak
antigens/antibodies - Sensitivity tests - False positives/false negatives - Consistency - Repeatability
Predetermined number of samples tested in duplicate with current system (e.g. 2 weeks or 250 samples whichever occurs first )
Hospital Transfusion staff Quality Manager

Appendix 9: Example of validation protocol
34
Appendix 9
Example of Validation Protocol
NAME OF LABORATORY/INSTITUTION
DIRECTORATE
FUNCTION
VALIDATION PROTOCOL FOR Enter title of validation
Change Control Ref No:
Document approved by:
NAME: ......................................................... .................................................................... Quality Representative Date

Appendix 9: Example of validation protocol
35
1. Introduction
Introduction – This section must define such details as why the validation is required, who are the relevant stakeholders of the change, where this change will operate and in what timescale the changes will become effective.
2. Aims
Aims – This section will define the outcome of the validation for example, ensure that the particular piece of equipment is fit for purpose, or that a particular process gives the required output or functionality.
3. Applicable Documents
Applicable Documents – The scope of documentation will be defined and will comprise of at least a simple listing of the validation documents used (ie. Controlled document references), any supporting manufacturers documentation, instruction manuals, e-mails, SOP‟s used for the validation.
Change Control
Validation
4. Testing Protocol Description of tests required. May be detailed in IQ/OQ/PQ Validation report pro-formas
5. Documentation Documentation – This section will define the quality system requirements for logging the validation as to whether the validation is part of a wider change control process or if the validation plan is stand-alone. This section must define which documentation is required for final sign-off and where the validation documentation is stored and archived.
Appendix 10: Example of a Qualification Proforma
36
Appendix 10
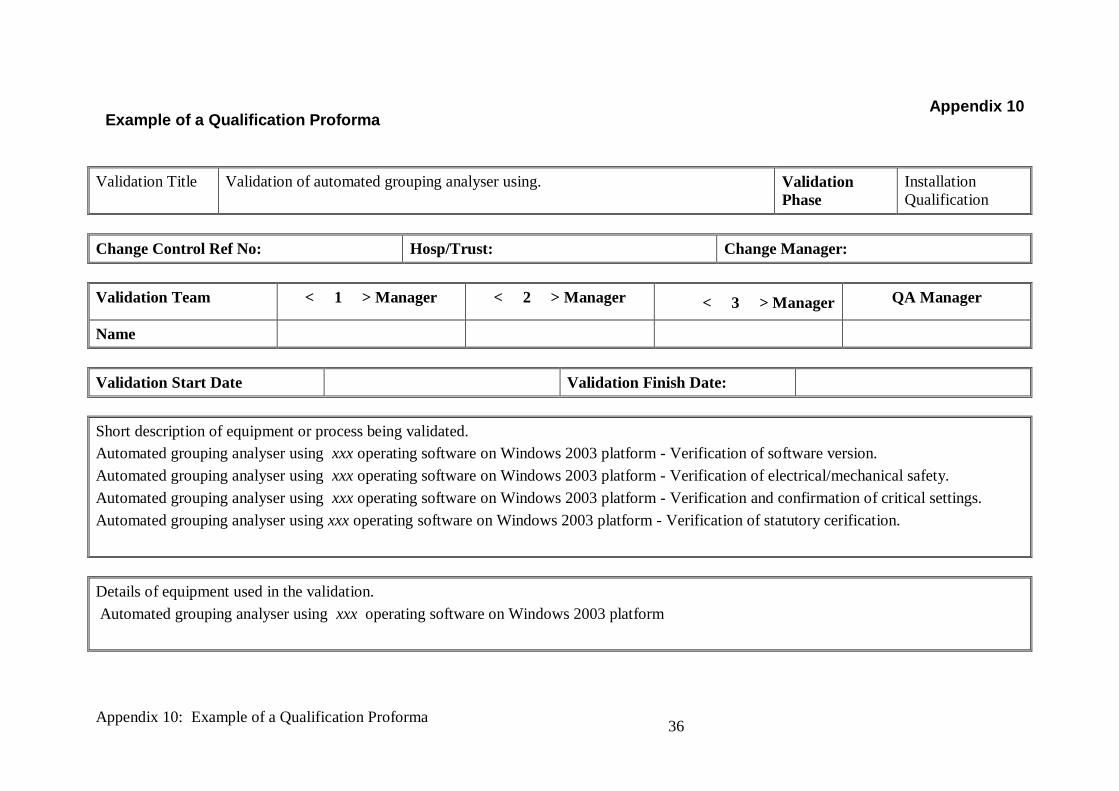
Example of a Qualification Proforma
Validation Title Validation of automated grouping analyser using. Validation
Phase
Installation
Qualification
Change Control Ref No: Hosp/Trust: Change Manager:
Validation Team < 1 > Manager < 2 > Manager < 3 > Manager QA Manager
Name
Validation Start Date Validation Finish Date:
Short description of equipment or process being validated.
Automated grouping analyser using xxx operating software on Windows 2003 platform - Verification of software version.
Automated grouping analyser using xxx operating software on Windows 2003 platform - Verification of electrical/mechanical safety.
Automated grouping analyser using xxx operating software on Windows 2003 platform - Verification and confirmation of critical settings.
Automated grouping analyser using xxx operating software on Windows 2003 platform - Verification of statutory cerification.
Details of equipment used in the validation.
Automated grouping analyser using xxx operating software on Windows 2003 platform
Example of a Qualification Proforma

Appendix 10: Example of a Qualification Proforma
37
Details of testing levels, methods, SOP’s used in validation
Check certification.
Check critical settings.
Check manufacturer supplied support documentation.
Validation Title Validation of Automated grouping analyser using xxx software.
No Description Acceptance Criteria Pass/Fai
l/ Re-
test
1.
Check instrument CE certification Record on receipt.
2.
Check instrument Identity. Record on receipt.
3.
Check manufacturer support documentation. Record on receipt..
4.
Check instrument for electrical safety. Sign-off by Facilities check.
5.
Check and record Windows operating
system version and Automated grouping
analyser operating system version.
Windows 2003, service pack 4.
Operating software xxx.
6.
Check and record interface software
versions
Advised by manufacturer.
Appendix 10: Example of a Qualification Proforma
38
7.
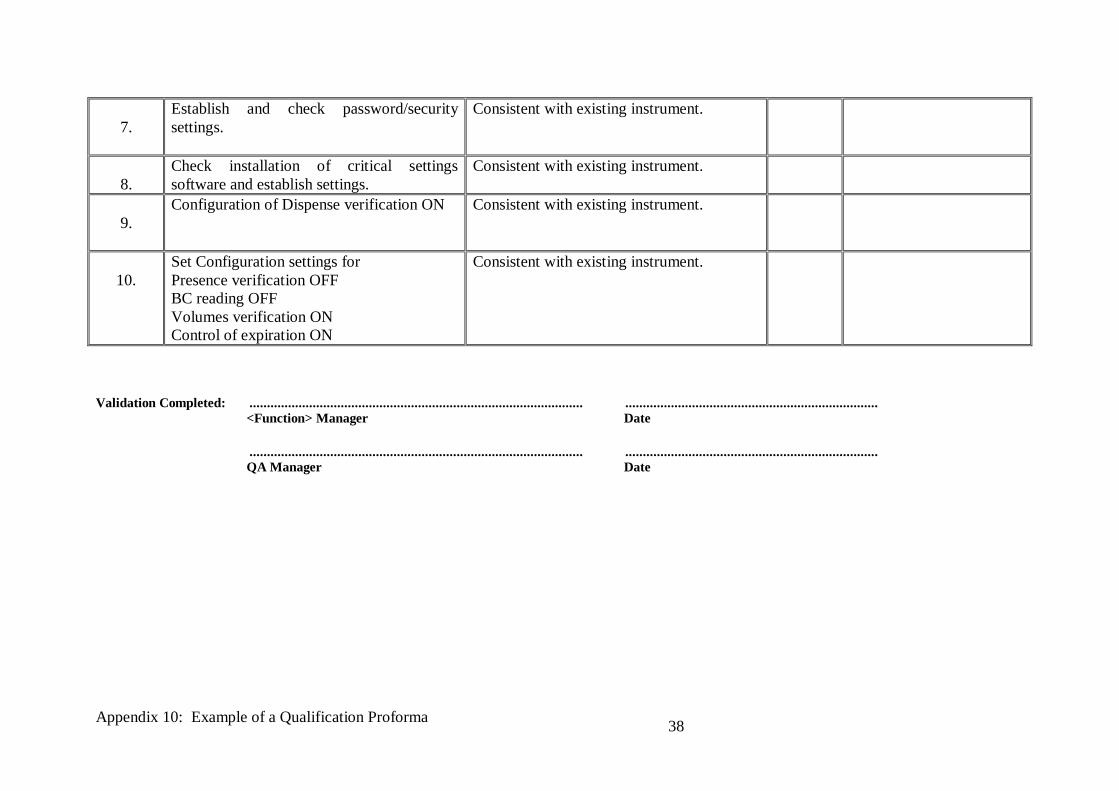
Establish and check password/security
settings.
Consistent with existing instrument.
8.
Check installation of critical settings
software and establish settings.
Consistent with existing instrument.
9.
Configuration of Dispense verification ON Consistent with existing instrument.
10.
Set Configuration settings for
Presence verification OFF
BC reading OFF
Volumes verification ON
Control of expiration ON
Consistent with existing instrument.
Validation Completed: ............................................................................................... ........................................................................
<Function> Manager Date
............................................................................................... ........................................................................
QA Manager Date

Appendix 10: Example of a Qualification Proforma
39
THIS PAGE TO BE REPLACED WITH XXXX (IMPLEMENTATION SIGN OFF FORM) FOR IMPLEMENTATION PHASE OF
VALIDATIONS
Recommendations/Comments: Validation Team Leader
Name ....................................... Signature: ............................................. Date: .........................................
Recommendations/Comments: Change Manager
Name ....................................... Signature: ............................................. Date: .........................................
Recommendations/Comments: on behalf of Review Board
<Title of Process Owner> ................................................ Name .............................................................. Signature:................................. Date:
QA Representative: Name ....................................... Signature: ............................................. Date: .........................................
<File Name and CCR Reference Number>
Appendix 10: Example of a Qualification Proforma
40
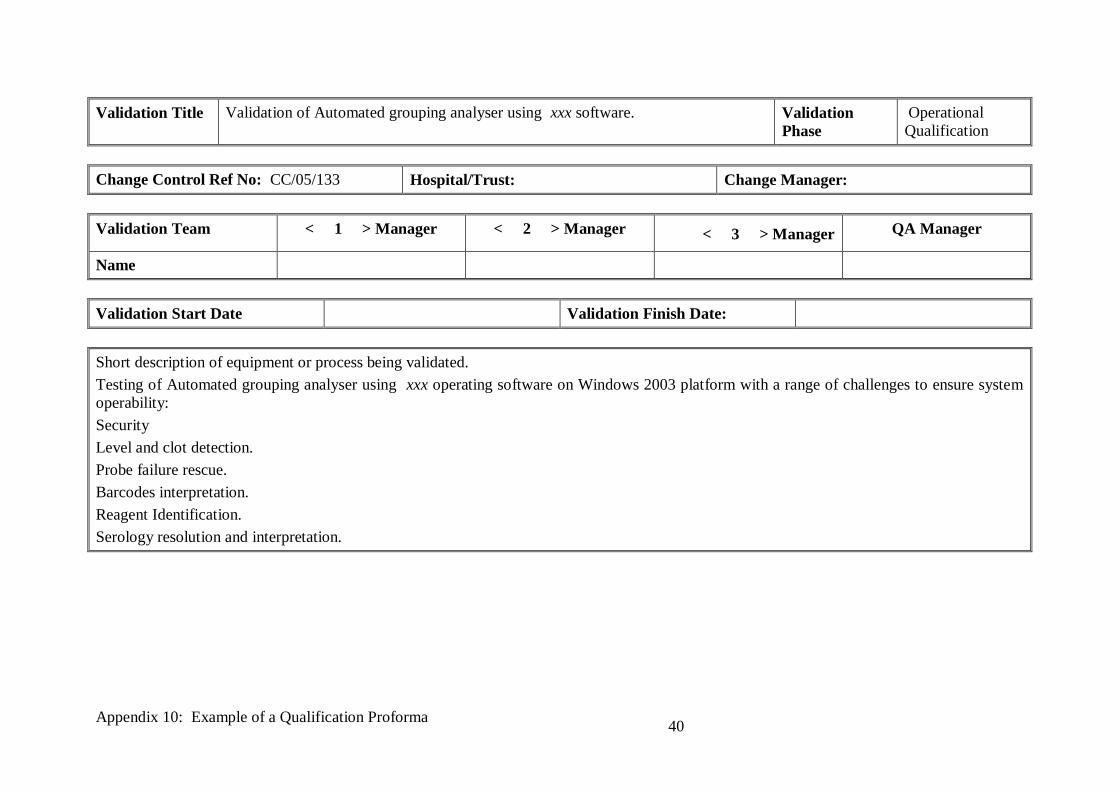
Validation Title Validation of Automated grouping analyser using xxx software. Validation
Phase
Operational
Qualification
Change Control Ref No: CC/05/133 Hospital/Trust: Change Manager:
Validation Team < 1 > Manager < 2 > Manager < 3 > Manager QA Manager
Name
Validation Start Date Validation Finish Date:
Short description of equipment or process being validated.
Testing of Automated grouping analyser using xxx operating software on Windows 2003 platform with a range of challenges to ensure system
operability:
Security
Level and clot detection.
Probe failure rescue.
Barcodes interpretation.
Reagent Identification.
Serology resolution and interpretation.

Appendix 10: Example of a Qualification Proforma
41
Details of equipment used in the validation.
Automated grouping analyser using xxx operating software on Windows 2003 platform
Automated grouping analyser using xxx operating software on Windows 2000 platform
Name of IT system data interface and host system
ABD/ABD ref:5005 grouping cards
ABDDAB ref:5009 grouping cards
Rh/K ref:5011 phenotyping cards
LISS IAT ref:**** IAT cards.
Details of testing levels, methods, SOP’s used in validation
x24 samples tested to ensure correct serological and sample barcode interpretations.
x2 ISBT donation barcode check.
x1 well verification check
x1 reagent reversal check.
x2 test card error check.
x1 clot replication. See 309cval.doc
x1 probe failure replication. See 309cval.doc
x1 sensitivity check using antibody titration.
x1 well verification check.
x2 test edit check
Sample tube size capability check
Password security check.
<File Name and CCR Reference Number>

Appendix 10: Example of a Qualification Proforma
42
Validation Title Validation of Automated grouping analyser using v3.11 software.
No Description Acceptance Criteria Pass/Fai
l/ Re-
test
1.
Test samples representing 8 commonly
encountered ABO/D combinations. Ie
A+,B+,O+,AB+,A-,B-,O-,AB-. with each
test card.
Consistent with ….. instrument. Compare printouts from
….. and Download
interface file host.pln
2.
Test samples representing a range of RhD
expression. X10. Including x1 CatVI. With
each test card
Consistent with ….. instrument. Compare printouts from
…… and Download
interface file host.pln
3.
Test x2 samples with 50% dual population
expression if forward typing tests. Ie. 50%
O- and 50% AB+ with each test card
Consistent with …… instrument.
No Download. Dp flag recorded for each
test well. Compare
printouts from ……. and
Download interface file
host.pln
4.
Test group A, B and O samples (x2 off)
where plasma is replaced with inert material
with each test card.
Consistent with …… instrument.
No Download Compare printouts from
……. and Download
interface file host.pln
5.
Test x2 samples simulating a DAT + case
where the control well result is POSITIVE,
with each test card.
Consistent with …… instrument.
No Download. Compare printouts from
…… and Download
interface file host.pln
6.
Check Sample test volume verification
functionality by replicating a sample
aspiration failure in a minimum of x1 well.
Check with manufacturer
7.
Check Sample clot detection functionality
by replicating a clotted sample failure in a
minimum of x1 sample.
Clot detection error message. Track and verify sequence.
See 309cval.doc
Appendix 10: Example of a Qualification Proforma
43
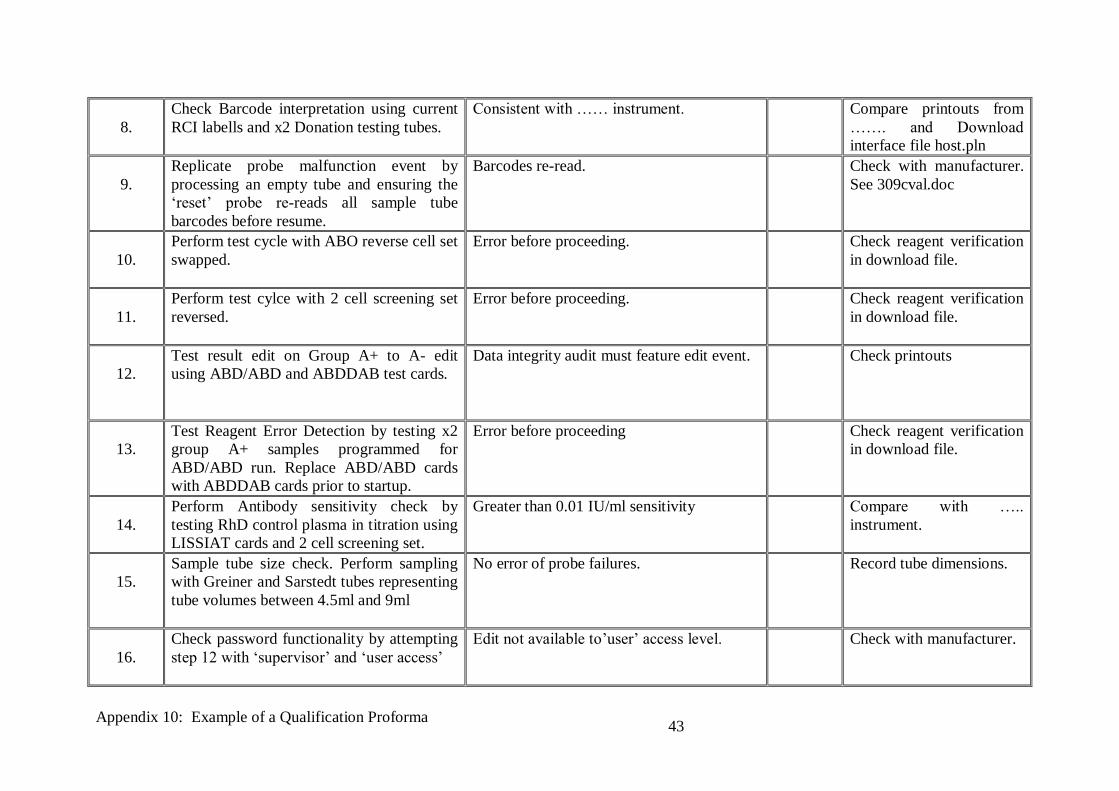
8.
Check Barcode interpretation using current
RCI labells and x2 Donation testing tubes.
Consistent with …… instrument. Compare printouts from
……. and Download
interface file host.pln
9.
Replicate probe malfunction event by
processing an empty tube and ensuring the
‘reset’ probe re-reads all sample tube
barcodes before resume.
Barcodes re-read. Check with manufacturer.
See 309cval.doc
10.
Perform test cycle with ABO reverse cell set
swapped.
Error before proceeding. Check reagent verification
in download file.
11.
Perform test cylce with 2 cell screening set
reversed.
Error before proceeding. Check reagent verification
in download file.
12.
Test result edit on Group A+ to A- edit
using ABD/ABD and ABDDAB test cards.
Data integrity audit must feature edit event. Check printouts
13.
Test Reagent Error Detection by testing x2
group A+ samples programmed for
ABD/ABD run. Replace ABD/ABD cards
with ABDDAB cards prior to startup.
Error before proceeding Check reagent verification
in download file.
14.
Perform Antibody sensitivity check by
testing RhD control plasma in titration using
LISSIAT cards and 2 cell screening set.
Greater than 0.01 IU/ml sensitivity Compare with …..
instrument.
15.
Sample tube size check. Perform sampling
with Greiner and Sarstedt tubes representing
tube volumes between 4.5ml and 9ml
No error of probe failures. Record tube dimensions.
16.
Check password functionality by attempting
step 12 with ‘supervisor’ and ‘user access’
Edit not available to’user’ access level. Check with manufacturer.

Appendix 10: Example of a Qualification Proforma
44
17.
Perform Sample switch check by loading x2
samples from step 12 and start run to ensure
barcode read. Open Machine and reverse
sample position.
Error or barcode re-check Compare with …….
instrument.
18.
19.
Validation Completed: ............................................................................................... ........................................................................
<Function> Manager Date
............................................................................................... ........................................................................
QA Manager Date
<File Name and CCR Reference Number>

Appendix 10: Example of a Qualification Proforma
45
THIS PAGE TO BE REPLACED WITH YYYY (IMPLEMENTATION SIGN OFF FORM) FOR IMPLEMENTATION PHASE OF
VALIDATIONS
Recommendations/Comments: Validation Team Leader
Name ....................................... Signature: ............................................. Date: .........................................
Recommendations/Comments: Change Manager
Name ....................................... Signature: ............................................. Date: .........................................
Recommendations/Comments: on behalf of Review Board
<Title of Process Owner> ................................................ Name .............................................................. Signature:................................. Date:
QA Representative: Name ....................................... Signature: ............................................. Date: .........................................
<File Name and CCR Reference Number>
Appendix 10: Example of a Qualification Proforma
46
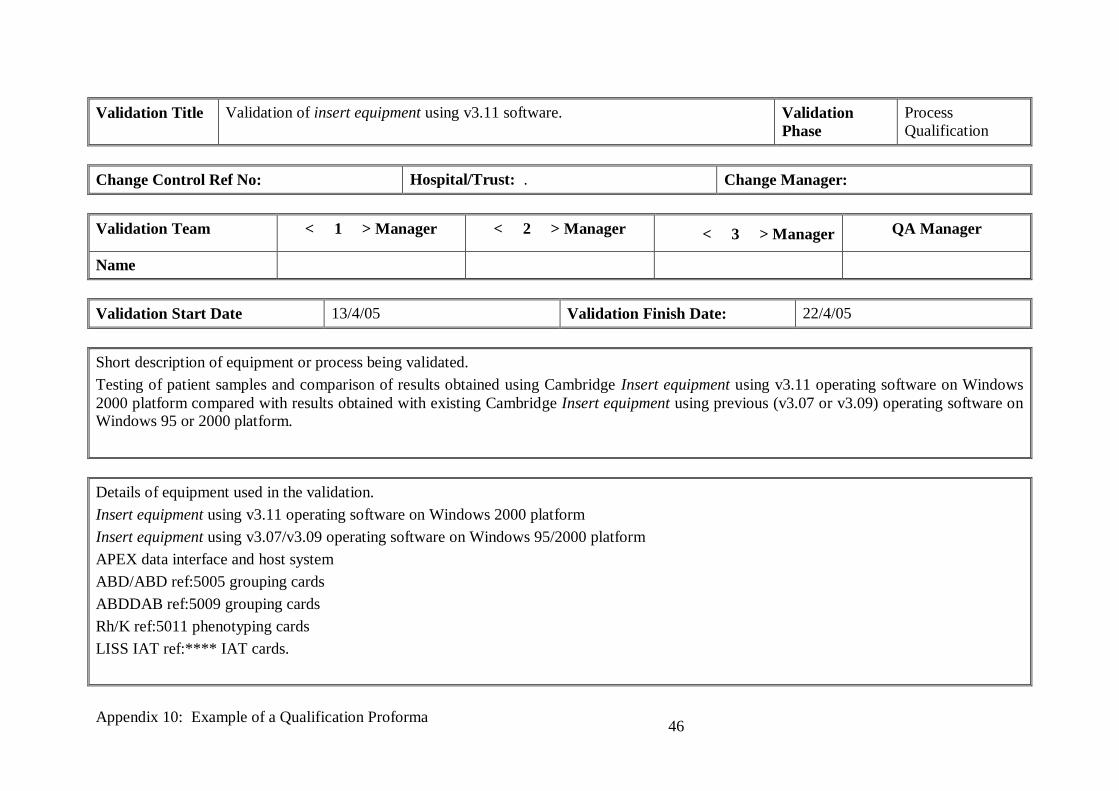
Validation Title Validation of insert equipment using v3.11 software. Validation
Phase
Process
Qualification
Change Control Ref No: Hospital/Trust: . Change Manager:
Validation Team < 1 > Manager < 2 > Manager < 3 > Manager QA Manager
Name
Validation Start Date 13/4/05 Validation Finish Date: 22/4/05
Short description of equipment or process being validated.
Testing of patient samples and comparison of results obtained using Cambridge Insert equipment using v3.11 operating software on Windows
2000 platform compared with results obtained with existing Cambridge Insert equipment using previous (v3.07 or v3.09) operating software on Windows 95 or 2000 platform.
Details of equipment used in the validation.
Insert equipment using v3.11 operating software on Windows 2000 platform
Insert equipment using v3.07/v3.09 operating software on Windows 95/2000 platform
APEX data interface and host system
ABD/ABD ref:5005 grouping cards
ABDDAB ref:5009 grouping cards
Rh/K ref:5011 phenotyping cards
LISS IAT ref:**** IAT cards.

Appendix 10: Example of a Qualification Proforma
47
Details of testing levels, methods, SOP’s used in validation
12 patient samples tested on v3.11 (new instrument) using ABDDAB cards and results compared against v3.07/v3.09 (existing instrument)
24 patient samples tested on v3.11 (new instrument) using ABD/ABD cards and results compared against v3.07/v3.09 (existing instrument)
48 patient samples tested on v3.11 (new instrument) using LISSIAT cards and results compared against v3.07 v3.09 / (existing instrument)
10 examples of significant antibodies tested on v3.11 (new instrument) using LISSIAT cards and results compared against v3.07/ v3.09
(existing instrument)
10 patient/tests samples tested on v3.11 (new instrument) using Rh/K cards and results compared against v3.07/ v3.09 (existing instrument) and manual results.
<File Name and CCR Reference Number>
Appendix 10: Example of a Qualification Proforma
48
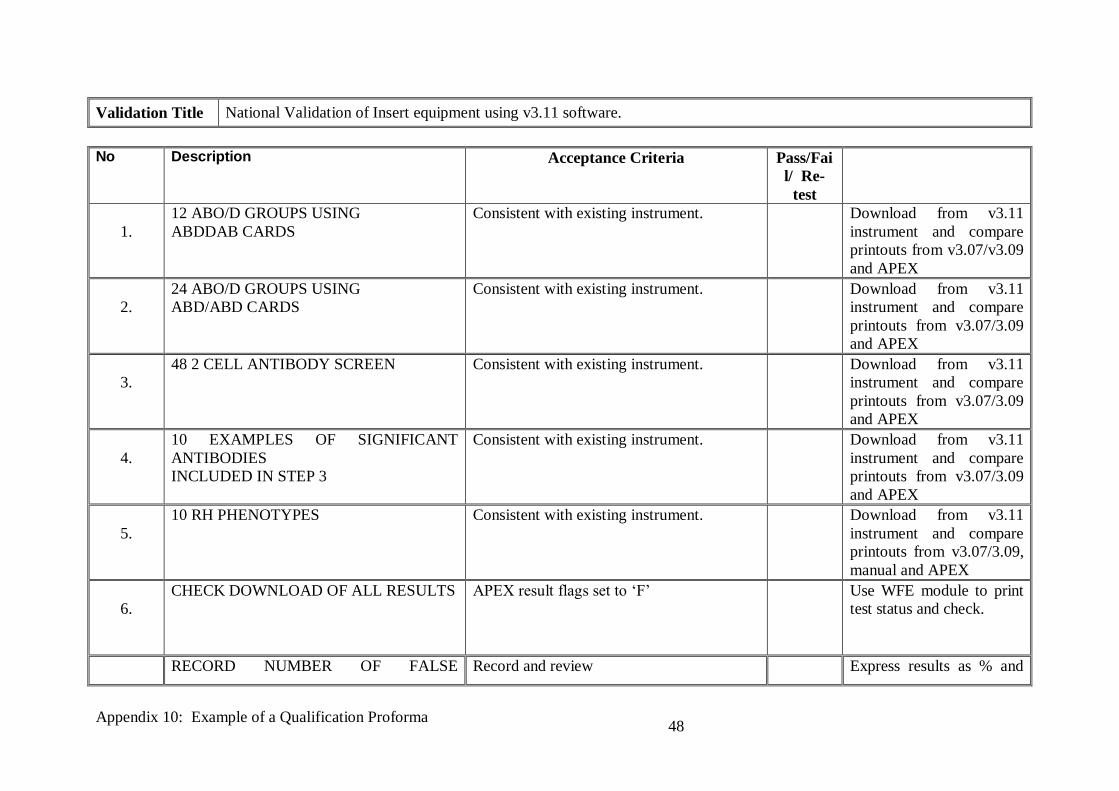
Validation Title National Validation of Insert equipment using v3.11 software.
No Description Acceptance Criteria Pass/Fai
l/ Re-
test
1.
12 ABO/D GROUPS USING
ABDDAB CARDS
Consistent with existing instrument. Download from v3.11
instrument and compare
printouts from v3.07/v3.09
and APEX
2.
24 ABO/D GROUPS USING
ABD/ABD CARDS
Consistent with existing instrument. Download from v3.11
instrument and compare
printouts from v3.07/3.09
and APEX
3.
48 2 CELL ANTIBODY SCREEN Consistent with existing instrument. Download from v3.11
instrument and compare
printouts from v3.07/3.09
and APEX
4.
10 EXAMPLES OF SIGNIFICANT
ANTIBODIES
INCLUDED IN STEP 3
Consistent with existing instrument. Download from v3.11
instrument and compare
printouts from v3.07/3.09
and APEX
5.
10 RH PHENOTYPES Consistent with existing instrument. Download from v3.11
instrument and compare
printouts from v3.07/3.09,
manual and APEX
6.
CHECK DOWNLOAD OF ALL RESULTS
APEX result flags set to ‘F’ Use WFE module to print
test status and check.
RECORD NUMBER OF FALSE Record and review Express results as % and

Appendix 10: Example of a Qualification Proforma
49
7.
POSITIVE/NEGATIVE AB SCREENS
FOR 20 WORKING DAYS.
RECORD NUMBER OF GROUP
FAILURES FOR 20 WORKING DAYS
provide summary.
Validation Completed: ............................................................................................... ........................................................................
<Function> Manager Date
............................................................................................... ........................................................................
QA Manager Date
<File Name and CCR Reference Number>

Appendix 10: Example of a Qualification Proforma
50
THIS PAGE TO BE REPLACED WITH ZZZZ (IMPLEMENTATION SIGN OFF FORM) FOR IMPLEMENTATION PHASE OF
VALIDATIONS
Recommendations/Comments: Validation Team Leader
Name ....................................... Signature: ............................................. Date: .........................................
Recommendations/Comments: Change Manager
Name ....................................... Signature: ............................................. Date: .........................................
Recommendations/Comments: on behalf of Review Board
<Title of Process Owner> ................................................ Name .............................................................. Signature:................................. Date:
QA Representative: Name ....................................... Signature: ............................................. Date: .........................................
Appendix 11: Validation sign-off report
51
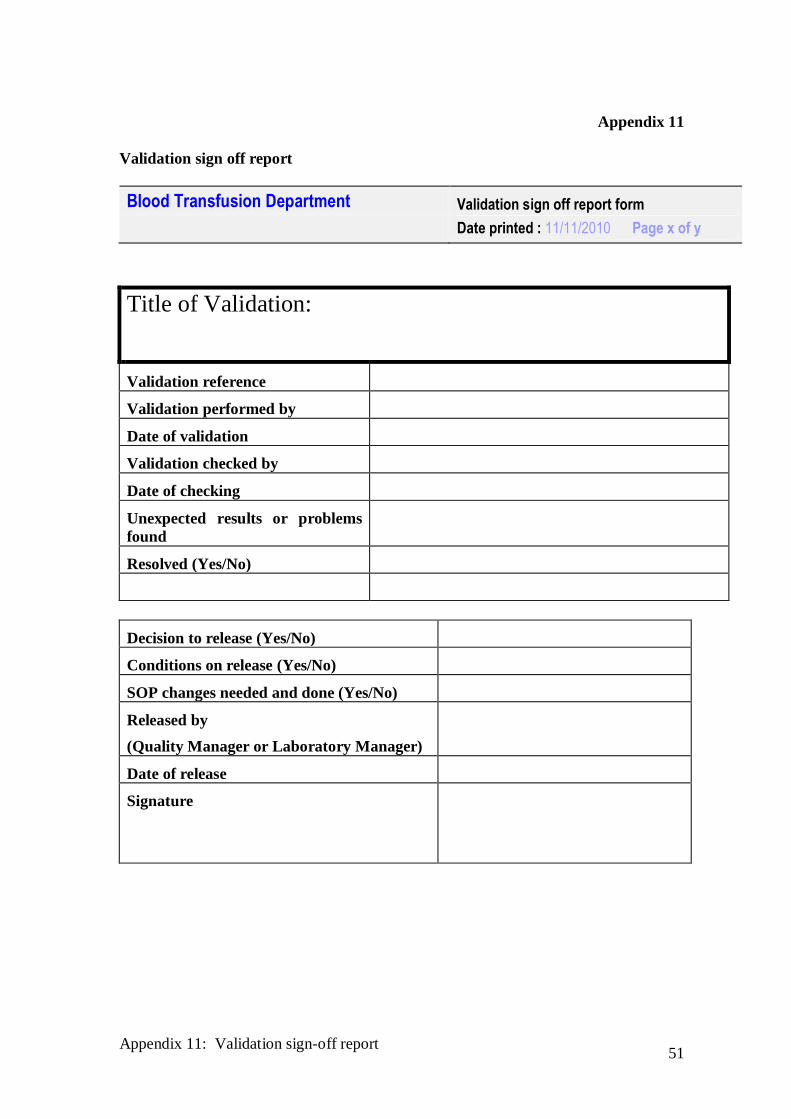
Appendix 11
Validation sign off report
Blood Transfusion Department Validation sign off report form
Date printed : 11/11/2010 Page x of y
Decision to release (Yes/No)
Conditions on release (Yes/No)
SOP changes needed and done (Yes/No)
Released by
(Quality Manager or Laboratory Manager)
Date of release
Signature
Title of Validation:
Validation reference
Validation performed by
Date of validation
Validation checked by
Date of checking
Unexpected results or problems
found
Resolved (Yes/No)

52
Acknowledgement and declaration of interest:
None of the authors have declared a conflict of interest. The Transfusion Task Force membership at the time of writing this guideline was: Derek Norfolk, Andrea Harris, Shubha Allard, Sarah Allford, Hafiz Qureshi, Joan Jones, Clare Taylor, Jenny White. These appendices are an accompaniment to the BCSH guidelines on validation and qualification, including change control, for hospital transfusion laboratories which can be downloaded from the BCSH website at www.bcshguidelines.com.


















