Antibacterial-Resistant Pseudomonas aeruginosa: Clinical ... · Antibacterial-Resistant Pseudomonas...
29
CLINICAL MICROBIOLOGY REVIEWS, Oct. 2009, p. 582–610 Vol. 22, No. 4 0893-8512/09/$08.000 doi:10.1128/CMR.00040-09 Copyright © 2009, American Society for Microbiology. All Rights Reserved. Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms Philip D. Lister, 1 * Daniel J. Wolter, 2 and Nancy D. Hanson 1 Department of Medical Microbiology and Immunology, Creighton University School of Medicine, Omaha, Nebraska, 1 and Department of Pediatrics, University of Washington, Seattle, Washington 2 INTRODUCTION .......................................................................................................................................................582 HISTORICAL AND CLINICAL SIGNIFICANCE OF P. AERUGINOSA...........................................................583 RESISTANCE CHALLENGES FOR TREATMENT OF P. AERUGINOSA .......................................................583 Antibacterial Resistance Trends ...........................................................................................................................583 Imported Resistance Mechanisms ........................................................................................................................585 Chromosomally Encoded Resistance Mechanisms ............................................................................................585 AmpC-MEDIATED RESISTANCE...........................................................................................................................587 AmpC and Resistance to -Lactams....................................................................................................................587 Clinical Significance of AmpC Overproduction..................................................................................................589 Pathways for AmpC Overproduction ...................................................................................................................590 Factors Involved in Regulation of ampC Expression .........................................................................................590 Mechanism of ampC Induction .............................................................................................................................590 Mechanisms of ampC Derepression .....................................................................................................................591 AmpD Homologues and Regulation of ampC Expression .................................................................................591 PBP4 and Regulation of ampC Expression .........................................................................................................592 OprD-MEDIATED RESISTANCE ...........................................................................................................................592 OprD and P. aeruginosa Susceptibility to Carbapenems...................................................................................593 Characterization of oprD Promoter Elements ....................................................................................................593 Molecular Mechanisms of OprD-Mediated Resistance.....................................................................................593 Discordance between oprD Expression and Susceptibility to Imipenem ........................................................594 EFFLUX-MEDIATED RESISTANCE......................................................................................................................595 MexAB-OprM Efflux Pump ...................................................................................................................................596 MexCD-OprJ Efflux Pump ....................................................................................................................................598 MexEF-OprN Efflux Pump ....................................................................................................................................598 MexXY Efflux Pump ...............................................................................................................................................599 MexJK Efflux Pump ...............................................................................................................................................601 Additional RND Efflux Pumps ..............................................................................................................................601 COREGULATION OF RESISTANCE MECHANISMS ........................................................................................601 MexCD-OprJ Overproduction and Hypersusceptibility to Antibacterials......................................................601 Coregulation of MexEF-OprN and OprD ...........................................................................................................602 PREVENTING EMERGENCE OF CHROMOSOMALLY ENCODED RESISTANCE ...................................603 CONCLUDING COMMENTS ..................................................................................................................................603 ACKNOWLEDGMENTS ...........................................................................................................................................603 REFERENCES ............................................................................................................................................................603 INTRODUCTION Infectious diseases have been an important cause of mor- bidity and mortality throughout our history. With the expan- sion of the antibiotic era during the 20th century, there was a growing confidence that the need for infectious disease spe- cialists would all but disappear. However, no one could have predicted the impact that an increasing immunocompromised population would have on the resurgence of infectious diseases during the last 3 decades. Furthermore, the ability of bacterial pathogens to adapt and to overcome the challenges of antibi- otics in their environment has been nothing short of impres- sive. We are now faced with a growing population of pan- resistant bacteria that threaten to move us into what some consider the “postantibiotic era” of infectious diseases. Some of the more problematic drug-resistant pathogens en- countered today include methicillin-resistant Staphylococcus au- reus, multidrug-resistant Streptococcus pneumoniae, and vancomy- cin-resistant Enterococcus spp. among the gram-positive bacteria and multidrug-resistant Acinetobacter baumannii, Klebsiella pneu- moniae, Escherichia coli, and Pseudomonas aeruginosa among the gram-negative bacteria. This review focuses specifically on the resistance problems associated with P. aeruginosa, with a special emphasis on the complexity by which key chromosomally en- coded resistance mechanisms are regulated and coregulated to make P. aeruginosa one of our greatest therapeutic challenges. * Corresponding author. Mailing address: Department of Medical Microbiology and Immunology, Creighton University School of Med- icine, 2500 California Plaza, Omaha, NE 68178. Phone: (402) 280- 1224. Fax: (402) 280-1875. E-mail: [email protected]. 582 on October 12, 2020 by guest http://cmr.asm.org/ Downloaded from
Transcript of Antibacterial-Resistant Pseudomonas aeruginosa: Clinical ... · Antibacterial-Resistant Pseudomonas...

CLINICAL MICROBIOLOGY REVIEWS, Oct. 2009, p. 582–610 Vol. 22, No. 40893-8512/09/$08.00�0 doi:10.1128/CMR.00040-09Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact andComplex Regulation of Chromosomally Encoded
Resistance MechanismsPhilip D. Lister,1* Daniel J. Wolter,2 and Nancy D. Hanson1
Department of Medical Microbiology and Immunology, Creighton University School of Medicine, Omaha, Nebraska,1 andDepartment of Pediatrics, University of Washington, Seattle, Washington2
INTRODUCTION .......................................................................................................................................................582HISTORICAL AND CLINICAL SIGNIFICANCE OF P. AERUGINOSA...........................................................583RESISTANCE CHALLENGES FOR TREATMENT OF P. AERUGINOSA .......................................................583
Antibacterial Resistance Trends...........................................................................................................................583Imported Resistance Mechanisms........................................................................................................................585Chromosomally Encoded Resistance Mechanisms ............................................................................................585
AmpC-MEDIATED RESISTANCE...........................................................................................................................587AmpC and Resistance to �-Lactams....................................................................................................................587Clinical Significance of AmpC Overproduction..................................................................................................589Pathways for AmpC Overproduction ...................................................................................................................590Factors Involved in Regulation of ampC Expression.........................................................................................590Mechanism of ampC Induction .............................................................................................................................590Mechanisms of ampC Derepression .....................................................................................................................591AmpD Homologues and Regulation of ampC Expression .................................................................................591PBP4 and Regulation of ampC Expression .........................................................................................................592
OprD-MEDIATED RESISTANCE ...........................................................................................................................592OprD and P. aeruginosa Susceptibility to Carbapenems...................................................................................593Characterization of oprD Promoter Elements ....................................................................................................593Molecular Mechanisms of OprD-Mediated Resistance.....................................................................................593Discordance between oprD Expression and Susceptibility to Imipenem ........................................................594
EFFLUX-MEDIATED RESISTANCE......................................................................................................................595MexAB-OprM Efflux Pump ...................................................................................................................................596MexCD-OprJ Efflux Pump ....................................................................................................................................598MexEF-OprN Efflux Pump ....................................................................................................................................598MexXY Efflux Pump ...............................................................................................................................................599MexJK Efflux Pump ...............................................................................................................................................601Additional RND Efflux Pumps..............................................................................................................................601
COREGULATION OF RESISTANCE MECHANISMS........................................................................................601MexCD-OprJ Overproduction and Hypersusceptibility to Antibacterials......................................................601Coregulation of MexEF-OprN and OprD ...........................................................................................................602
PREVENTING EMERGENCE OF CHROMOSOMALLY ENCODED RESISTANCE ...................................603CONCLUDING COMMENTS ..................................................................................................................................603ACKNOWLEDGMENTS ...........................................................................................................................................603REFERENCES ............................................................................................................................................................603
INTRODUCTION
Infectious diseases have been an important cause of mor-bidity and mortality throughout our history. With the expan-sion of the antibiotic era during the 20th century, there was agrowing confidence that the need for infectious disease spe-cialists would all but disappear. However, no one could havepredicted the impact that an increasing immunocompromisedpopulation would have on the resurgence of infectious diseasesduring the last 3 decades. Furthermore, the ability of bacterialpathogens to adapt and to overcome the challenges of antibi-
otics in their environment has been nothing short of impres-sive. We are now faced with a growing population of pan-resistant bacteria that threaten to move us into what someconsider the “postantibiotic era” of infectious diseases.
Some of the more problematic drug-resistant pathogens en-countered today include methicillin-resistant Staphylococcus au-reus, multidrug-resistant Streptococcus pneumoniae, and vancomy-cin-resistant Enterococcus spp. among the gram-positive bacteriaand multidrug-resistant Acinetobacter baumannii, Klebsiella pneu-moniae, Escherichia coli, and Pseudomonas aeruginosa among thegram-negative bacteria. This review focuses specifically on theresistance problems associated with P. aeruginosa, with a specialemphasis on the complexity by which key chromosomally en-coded resistance mechanisms are regulated and coregulated tomake P. aeruginosa one of our greatest therapeutic challenges.
* Corresponding author. Mailing address: Department of MedicalMicrobiology and Immunology, Creighton University School of Med-icine, 2500 California Plaza, Omaha, NE 68178. Phone: (402) 280-1224. Fax: (402) 280-1875. E-mail: [email protected].
582
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

HISTORICAL AND CLINICAL SIGNIFICANCEOF P. AERUGINOSA
The opportunistic bacterial pathogen currently known as P.aeruginosa has received several names throughout its historybased on the characteristic blue-green coloration producedduring culture. Sedillot in 1850 was first to observe that thediscoloration of surgical wound dressings was associated with atransferable agent (196). The pigment responsible for the bluecoloration was extracted by Fordos in 1860, and in 1862 Luckewas the first to associate this pigment with rod-shaped organ-isms (196). P. aeruginosa was not successfully isolated in pureculture until 1882, when Carle Gessard reported in a publica-tion entitled “On the Blue and Green Coloration of Bandages”the growth of the organism from cutaneous wounds of twopatients with bluish-green pus (65). In several additional re-ports between 1889 and 1894, P. aeruginosa (Bacillus pyocya-neus) was described as the causative agent of blue-green pu-rulence in the wounds of patients (261). A more thoroughpresentation on the routes of invasion and dissemination of P.aeruginosa leading to acute or chronic infection was providedby Freeman in a 1916 article (56).
P. aeruginosa is a ubiquitous organism present in many di-verse environmental settings, and it can be isolated from var-ious living sources, including plants, animals, and humans. Theability of P. aeruginosa to survive on minimal nutritional re-quirements and to tolerate a variety of physical conditions hasallowed this organism to persist in both community and hos-pital settings. In the hospital, P. aeruginosa can be isolatedfrom a variety of sources, including respiratory therapy equip-ment, antiseptics, soap, sinks, mops, medicines, and physio-therapy and hydrotherapy pools (199). Community reservoirsof this organism include swimming pools, whirlpools, hot tubs,contact lens solution, home humidifiers, soil and rhizosphere,and vegetables (77, 196, 199).
P. aeruginosa is seldom a member of the normal microbialflora in humans. Representative colonization rates for specificsites in humans are 0 to 2% for skin, 0 to 3.3% for the nasalmucosa, 0 to 6.6% for the throat, and 2.6 to 24% for fecalsamples (164). However, colonization rates may exceed 50%during hospitalization (199), especially among patients whohave experienced trauma to or a breach in cutaneous or mu-cosal barriers by mechanical ventilation, tracheostomy, cathe-ters, surgery, or severe burns (17, 49, 182, 252, 257). Patientswith impaired immunity have higher risks for colonization bythis organism (164, 199), and disruption in the normal micro-bial flora as a result of antimicrobial therapy has also beenshown to increase colonization by P. aeruginosa (17, 18, 250).
Despite the wide distribution of P. aeruginosa in nature andthe potential for community-acquired infections, serious infec-tions with P. aeruginosa are predominantly hospital acquired.A review of surveillance data collected by the CDC NationalNosocomial Infections Surveillance System from 1986 to 1998shows that P. aeruginosa was identified as the fifth most fre-quently isolated nosocomial pathogen, accounting for 9% of allhospital-acquired infections in the United States (48, 171). P.aeruginosa was also the second leading cause of nosocomialpneumonia (14 to 16%), third most common cause of urinarytract infections (7 to 11%), fourth most frequently isolatedpathogen in surgical site infections (8%), and seventh leading
contributor to bloodstream infections (2 to 6%). Data frommore recent studies continue to show P. aeruginosa as thesecond most common cause of nosocomial pneumonia, healthcare-associated pneumonia, and ventilator-associated pneu-monia (64, 106) and the leading cause of pneumonia amongpediatric patients in the intensive care unit (ICU) (214).
P. aeruginosa is especially problematic for seriously ill pa-tients in ICUs. From 1992 to 1997, data from the NationalNosocomial Infections Surveillance System showed that P.aeruginosa was responsible for 21% of pneumonias, 10% ofurinary tract infections, 3% of bloodstream infections, and13% of eye, ear, nose, and throat infections within ICUs in theUnited States (213). A similar study conducted in Europeidentified P. aeruginosa as the second most frequently isolatedorganism in reported cases of ICU-acquired infections (242).In this surveillance study, P. aeruginosa was accountable for30% of pneumonias, 19% of urinary tract infections, and 10%of bloodstream infections.
RESISTANCE CHALLENGES FOR TREATMENTOF P. AERUGINOSA
P. aeruginosa presents a serious therapeutic challenge fortreatment of both community-acquired and nosocomial infec-tions, and selection of the appropriate antibiotic to initiatetherapy is essential to optimizing the clinical outcome (15,156). Unfortunately, selection of the most appropriate antibi-otic is complicated by the ability of P. aeruginosa to developresistance to multiple classes of antibacterial agents, even dur-ing the course of treating an infection. Epidemiological out-come studies have shown that infections caused by drug-resis-tant P. aeruginosa are associated with significant increases inmorbidity, mortality, need for surgical intervention, length ofhospital stay and chronic care, and overall cost of treating theinfection (7, 25, 62). Even more problematic is the develop-ment of resistance during the course of therapy, a complicationwhich has been shown to double the length of hospitalizationand overall cost of patient care (41). P. aeruginosa can developresistance to antibacterials either through the acquisition ofresistance genes on mobile genetic elements (i.e., plasmids) orthrough mutational processes that alter the expression and/orfunction of chromosomally encoded mechanisms. Both strate-gies for developing drug resistance can severely limit the ther-apeutic options for treatment of serious infections.
Antibacterial Resistance Trends
Presented in Table 1 are rates of P. aeruginosa resistance toseveral antipseudomonal drugs (54, 95, 99, 100, 178, 211, 212).This summary is not meant to be inclusive of all of the pub-lished literature, but rather highlights data reported for iso-lates from several U.S. surveillance studies since January 2000.If multiple years were included in a study, the resistance ratesfor the most recent year are presented in Table 1.
P. aeruginosa exhibits the highest rates of resistance for thefluoroquinolones, with resistance to ciprofloxacin and levo-floxacin ranging from 20 to 35%. Although not surprising, thehighest rates were reported for isolates obtained from patientsin ICUs (Table 1). P. aeruginosa isolates from ICU patientsalso trend toward higher rates of �-lactam resistance than
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 583
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

general trends for hospitalized patients. Based on the data inTable 1, it is difficult to draw any strong conclusions abouttrends of resistance to various �-lactams. Among the amino-glycosides, most studies have focused on gentamicin, with re-sistance rates ranging from 12 to 22%. Gentamicin was theleast active of the aminoglycosides, with lower rates of resis-tance being reported for tobramycin and amikacin in moststudies (Table 1).
Although the resistance trends from large national surveil-lance studies provide important data for consideration, thesestudies do not address the potential for much higher rates ofresistance within individual communities and hospitals. Forexample, during the years 2001 and 2006, rates of nonsuscep-tibility among P. aeruginosa isolates in Brooklyn, NY, rangedfrom 27 to 29% for cefepime, 30 to 31% for imipenem, 23%for meropenem, and 41 to 44% for ciprofloxacin (113). Theserates are substantially higher than national trends focusing onall hospital isolates of P. aeruginosa (Table 1).
Not only are rates of resistance to individual drugs or drug
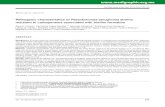
classes a concern, but the prevalence of multidrug-resistantstrains (resistant to three or more drug classes) is an even moreserious therapeutic challenge. A national surveillance of 13,999nonduplicate P. aeruginosa isolates from ICU patients showedthat multidrug resistance increased significantly, from 4% in1993 to 14% in 2002 (Fig. 1A) (178). For comparison, anotherICU surveillance study evaluated over 37,000 P. aeruginosaisolates from 1997 to 2002 and reported an increase in preva-lence of multidrug-resistant strains from 13% to 21% (Fig. 1B)(132). Finally, Flamm et al. reported rates of multidrug-resis-tant P. aeruginosa ranging from 23 to 26% among 52,000 P.aeruginosa isolates collected in the United States from 1999 to2002 (54). The highest prevalence of multidrug-resistantstrains was observed among isolates from lower respiratorytract infections, whereas the lowest prevalence was observedamong isolates from upper respiratory tract infections. Notsurprisingly, multidrug-resistant strains were isolated more fre-quently from ICU and nursing home patients.
A multidrug-resistant phenotype can arise in P. aeruginosa
FIG. 1. Increasing prevalence of multidrug resistance among P. aeruginosa isolates from ICU patients in the United States. (A) Data for 13,999nonduplicate isolates collected from 1993 to 2002 (178); (B) data for 37,390 isolates collected from 1997 to 2000 (132). Data represent thepercentage of P. aeruginosa isolates that expressed a phenotype of multidrug resistance (resistance to three or more drug classes) during each yearof the studies. (Panel A is adapted from reference 178 with permission; panel B is based on data from reference 132.)
TABLE 1. Rates of antibacterial resistance among P. aeruginosa isolates from hospitals and ICUs
Antibiotic
% of strains exhibiting resistancea
Hospital study,2006
(n � 606)(211)
Hospital study,2005
(n � 589)(212)
Hospital study,2002
(n � 9,896)(54)
ICU study, 2002(n � 951)
(178)
ICU study,2000–2002
(n � 7,500)(95)
Hospital study,2001
(n � 2,157)(99)
ICU study,2001
(n � 543)(99)
Hospital study,2000
(n � 882)(100)
�-LactamsCefepime 6 5 9 25 12 8 10 9Ceftazidime 13 10 13 19 17 9 9 13Piperacillin-tazobactam 11 9 11 10 14 8 8 13Aztreonam 12 32Imipenem 11 7 16 23 22 12 16 16Meropenem 6 7 18 11 16 10
FluoroquinolonesCiprofloxacin 21 22 35 32 33 26 25 25Levofloxacin 22 22 34 32 27 25 27
AminoglycosidesAmikacin 5 10 4 3Tobramycin 8 10 12 16Gentamicin 12 12 16 22 15 15 14
a Based upon CLSI interpretive breakpoints.
584 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

through the acquisition of multiple imported resistance mech-anisms on mobile genetic elements, a combination of importedand chromosomally encoded resistance mechanisms, accumu-lation of multiple chromosomal changes over time, and/or asingle mutational event leading to the overexpression of amultidrug resistance mechanism, i.e., an efflux pump. This re-view focuses primarily on the key chromosomally encodedresistance mechanisms, their clinical significance, and theircomplex mechanisms of regulation. However, a brief overviewof important imported resistance mechanisms is presentedfirst.
Imported Resistance Mechanisms
In relation to the antipseudomonal drug classes presented inTable 1, imported resistance among P. aeruginosa isolates im-pacts the �-lactams and aminoglycosides but not the fluoro-quinolones. As discussed below, fluoroquinolone resistanceamong P. aeruginosa isolates has been linked only to chromo-somal genes, with mutational changes in the fluoroquinolonetargets DNA gyrase (gyrA and gyrB) and/or topoisomerase IV(parC and parE) and/or overexpression of multidrug effluxpumps (Fig. 2). Although the plasmid-encoded DNA gyraseprotection protein Qnr and the fluoroquinolone-modifying en-zyme AAC(6�)Ib-cr can contribute to fluoroquinolone resis-tance among strains of Enterobacteriaceae (89, 186, 215), thesetwo plasmid-encoded mechanisms have not been found in clin-ical isolates of P. aeruginosa (89, 198).
Imported resistance to the �-lactams involves the produc-tion of inactivating �-lactamases, for which several familieshave been identified among clinical isolates of P. aeruginosa.The variety, prevalence, and clinical significance of imported�-lactamases in P. aeruginosa have been addressed in severalreviews over the last decade (74, 132–134). The most commonimported �-lactamases found among P. aeruginosa isolates arepenicillinases belonging to the molecular class A serine �-lac-tamases (PSE, CARB, and TEM families). Within this group,enzymes belonging to the PSE family appear to be the mostprevalent (14). The therapeutic impact of these penicillinasesis relatively limited since they do not impact the clinical efficacyof extended-spectrum cephalosporins, monobactams, or car-bapenems. Although less frequent, class A extended-spectrum�-lactamases have also been detected in strains of P. aerugi-nosa and have included enzymes from the TEM, SHV,CTX-M, PER, VEB, GES, and IBC families (27, 36, 155, 175,192, 197, 268, 284). Extended-spectrum �-lactamases from theclass D, OXA-type enzymes have also been encountered withinP. aeruginosa (170, 191).
Similar to the case for the Enterobacteriaceae, extended-spectrum �-lactamases alone do not provide P. aeruginosa re-sistance to the carbapenems. However, the prevalence of dif-ferent classes of carbapenem-hydrolyzing enzymes has beenincreasing globally. The first class B metallo-�-lactamases in P.aeruginosa were identified in 1991 in Japan (266). Since thatinitial report, metallo-�-lactamases have been reported for P.aeruginosa isolates from nearly all regions of the globe (57, 96,117, 176, 187, 281), and four major families have been identi-fied (IMP, VIM, SPM, and GIM families) (28, 59, 135, 176,253). Recently, class A carbapenemases of the KPC familyhave been identified. The first characterized KPC-producing P.
aeruginosa isolate was collected in Colombia and reported in2007 (262). The most recent report identifies the spread ofKPC genes into clonally related and unrelated strains of P.aeruginosa from Puerto Rico (281). The first identification ofan imported OXA-type carbapenemase in P. aeruginosa wasreported in 2008, and it was shown to be the same OXA-40carbapenemase previously described for A. baumannii (237).
Imported resistance to aminoglycosides most commonly in-volves enzymatic inactivation of the drug molecule throughchemical modification. The history, molecular characteriza-tion, prevalence, and clinical significance of aminoglycoside-inactivating enzymes in P. aeruginosa were recently coveredin an excellent review (200). These enzymes are categorizedinto the following three families, based upon the chemicalmodification they mediate: (i) aminoglycoside phosphoryl-transferase enzymes phosphorylate the drug molecule, (ii) ami-noglycoside acetyltransferase enzymes acetylate the drug mol-ecule, and (iii) aminoglycoside nucleotidyltransferase enzymesadenylate the drug molecule. Although the range of aminogly-cosides inactivated by specific enzymes within this family candiffer, the ability of P. aeruginosa to carry the genes for multi-ple aminoglycoside-inactivating enzymes provides individualstrains with the potential to develop resistance to all aminogly-cosides.
In addition to the variety of aminoglycoside-modifying en-zymes, high-level resistance to multiple aminoglycosides can beassociated with methylation of the 16S rRNA. This mechanismwas first reported for P. aeruginosa in 1993, and the methylase-encoding gene was designated rmtA (287). There are currentlyfive characterized ribosomal methyltransferase enzymes (RmtA,RmtB, RmtC, RmtD, and ArmA) that have been found world-wide among clinical isolates of P. aeruginosa and Enterobacte-riaceae (42, 60, 61, 265, 286, 287). Although methyltransferasesdo not appear to be as common as the aminoglycoside-modi-fying enzymes, all of the genes encoding these enzymes havebeen associated with mobile genetic elements (43, 61, 68, 264,265, 285), raising concern about their widespread dissemina-tion among P. aeruginosa isolates and other gram-negativebacilli.
Chromosomally Encoded Resistance Mechanisms
Similar to imported resistance mechanisms, there are a va-riety of resistance mechanisms encoded on the P. aeruginosachromosome. These mechanisms include several aminoglyco-side-inactivating enzymes (200) and a class D oxacillinase,OXA-50 (67). As mentioned above, characterized mechanismsof fluoroquinolone resistance among P. aeruginosa isolateshave been restricted to chromosomal genes, including targetmutations and active efflux (Fig. 2). Similar to the case forother gram-negative bacteria, DNA gyrase is the primary tar-get for the fluoroquinolones in P. aeruginosa (84). Therefore,the first target-specific mutations are typically observed withinthe quinolone resistance determining region (QRDR) of gyrA(6, 116, 165, 249, 283). The highest levels of resistance areobserved in strains that have mutations in the QRDR of bothgyrA and the topoisomerase IV gene parC (6, 90, 91, 116, 165).Although mutational changes within the other two genes, gyrBand parE, have been described, the prevalence of these muta-tions appears to be much lower (6). Recent studies involving
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 585
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

586 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

screening of the Harvard PA14 library of P. aeruginosa mutantshave identified a number of other chromosomal genes that maybe involved in antibacterial resistance or in increasing thefrequency of mutation to resistance (20, 44, 233, 271).
The three most intensely studied chromosomally encodedresistance mechanisms in P. aeruginosa are the AmpCcephalosporinase, the OprD outer membrane porin, and themultidrug efflux pumps (Fig. 2). The remainder of this re-view focuses specifically on the clinical relevance of thesethree resistance mechanisms and the complex pathways P.aeruginosa utilizes to regulate their expression. The abilityof P. aeruginosa to coregulate different resistance mecha-nisms makes this pathogen a constantly moving target thatcontinues to challenge therapeutic strategies.
AmpC-MEDIATED RESISTANCE
AmpC and Resistance to �-Lactams
P. aeruginosa carries an inducible AmpC cephalosporinasewhich is similar to the chromosomally encoded AmpC found inseveral members of the Enterobacteriaceae (23, 34, 76, 222,227). Wild-type strains of P. aeruginosa produce only low basallevels of AmpC and are susceptible to the antipseudomonalpenicillins, penicillin-inhibitor combinations, cephalosporins,and carbapenems (227). However, when AmpC production issignificantly increased, P. aeruginosa develops resistance to all�-lactams, with the exception of the carbapenems, to be discussedlater (34, 227). This is in contrast to some AmpC-overproducingmembers of the Enterobacteriaceae, which remain susceptible tocefepime and require additional mechanisms to developcefepime resistance (i.e., downregulation of porin production)(13, 45, 78, 194, 223). Although it is possible that variability inthe hydrolytic activity of AmpCs from P. aeruginosa and theEnterobacteriaceae could play a role in these differences,cefepime hydrolysis data obtained with purified AmpC en-zymes do not support this hypothesis (208). Rather, the greaterimpermeability of the P. aeruginosa outer membrane may playan important role in allowing AmpC overproduction to pushcefepime MICs above the resistance breakpoint (75).
Whereas resistance to most of the �-lactams emerges as aresult of AmpC overproduction, a definitive relationship be-tween P. aeruginosa AmpC and carbapenem susceptibility re-mains convoluted. AmpC-deficient strains of P. aeruginosa cre-ated through allelic exchange exhibit significant, �4-foldincreases in susceptibility to imipenem and panipenem but notto meropenem (148). Additional P. aeruginosa isolates, definedas AmpC deficient yet still producing AmpC, also exhibit in-
creased susceptibility to imipenem and doripenem but not tomeropenem (131, 136, 169). These data suggest that AmpCmay play a role in the level of intrinsic susceptibility of P.aeruginosa to carbapenems. In contrast, published data havesuggested that overproduction of AmpC does not play a dis-cernible role in the development of carbapenem resistanceamong P. aeruginosa isolates. AmpC overproduction amongisogenic mutants selected with �-lactams does not significantlydecrease P. aeruginosa susceptibility to carbapenems (63, 131,169, 274). In addition, AmpC overproduction in carbapenem-susceptible clinical isolates of P. aeruginosa has been reported.Data from one study of 47 characterized AmpC-overproducingclinical isolates showed that only 7 (15%) were resistant toimipenem (calculated from the data in Table 2 of reference225). Gutierrez et al. recently reported that 51% of carbap-enem-resistant clinical isolates of P. aeruginosa in their studyoverproduced AmpC (73). Although statistical analysis sug-gested that meropenem-resistant strains were more likely tooverproduce AmpC than meropenem-susceptible strains, Gu-tierrez et al. concluded that “AmpC hyperproduction is neithersufficient nor necessary for meropenem clinical resistance”.
The challenge of specifically linking AmpC overproductionto carbapenem resistance is our own limited knowledge of thecomplex interplay between resistance mechanisms in P. aerugi-nosa and the multitude of pathways by which P. aeruginosacoregulates resistance mechanisms. How do we specifically linkAmpC overproduction to carbapenem resistance among un-controlled clinical isolates with diverse genetic and environ-mental backgrounds? Even with isogenic laboratory strains,how do we specifically link AmpC production to a particularphenotype, knowing that altered AmpC production could beaccompanied by changes in the expression of other resistancemechanisms through coregulatory pathways? Despite howmuch we have yet to learn about the resistance potential of P.aeruginosa, the data generated thus far suggest that AmpCoverproduction alone does not significantly alter P. aerugino-sa’s susceptibility to the carbapenems but could certainly con-tribute to resistance if accompanied by additional resistancemechanisms (e.g., efflux pump overproduction, decreasedOprD, and/or production of a class A/class B carbapenemase).Adding even more complexity is the potential for mutationalvariants of the chromosomally encoded AmpC enzyme (ex-tended-spectrum AmpC) to provide P. aeruginosa with carbap-enem resistance (216). Extended-spectrum AmpCs were firstidentified in Serratia marcescens and Enterobacter spp. (10, 143,153) and, most recently, in E. coli (141, 142). Amino acidmodifications near the active sites of these enzymes can lead to
FIG. 2. Mutational resistance to fluoroquinolones and carbapenems involving chromosomally encoded mechanisms expressed by P. aeruginosa.(A) Interactions of fluoroquinolones and carbapenems with “wild-type” susceptible P. aeruginosa expressing basal levels of AmpC, OprD, andnonmutated fluoroquinolone target genes (gyrA, gyrB, parC, and parE). Fluoroquinolone molecules pass through the outer membrane, pepti-doglycan, periplasmic space, and cytoplasmic membrane and interact with DNA gyrase and topoisomerase IV (Topo IV) targets in the cytoplasmwhen these enzymes are complexed with DNA. Carbapenem molecules pass through the outer membrane-specific porin OprD and interact withtheir target PBPs, located on the outside of the cytoplasmic membrane. (B) Chromosomally encoded mechanisms of resistance to fluoroquinolonesand carbapenems. Fluoroquinolone resistance is mediated by (i) overexpression of RND efflux pumps extruding the drug molecules from theperiplasmic and cytoplasmic spaces and/or (ii) mutational changes within the target genes. Locations of the QRDRs within target genes arehighlighted in yellow. Carbapenem resistance is mediated primarily by (i) decreased production or loss of functional OprD in the outer membraneand/or (ii) overproduction of RND efflux pumps (with the exception of imipenem). Minor changes in susceptibility can be observed due tooverexpression of AmpC, adding to the resistance potential.
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 587
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

588 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

increased hydrolytic activity against cefepime, ceftazidime, andimipenem. However, increased catalytic activity of these en-zymes only reduces susceptibility to cefepime and imipenem.Overproduction of these extended-spectrum AmpCs seems tobe a requirement for cefepime (10) and/or carbapenem (216)resistance.
Clinical Significance of AmpC Overproduction
Similar to the discussion above, the clinical impact ofAmpC overproduction by P. aeruginosa is difficult to assessdue to the complex interplay of multiple resistance mecha-nisms. Nevertheless, Tam et al. reported that patients were67.5 times more likely to be given inappropriate antibioticswhen the infections were caused by AmpC-overproducing P.aeruginosa than when they were caused by P. aeruginosa thatdid not overproduce AmpC (251). In addition, these pa-
tients were more likely to have persistent bacteremia (45%versus 6%), underscoring the need to rapidly identifyAmpC-overproducing P. aeruginosa isolates in the clinicallaboratory and to understand the mechanisms of AmpCoverproduction in this pathogen.
The ability of resistant P. aeruginosa to emerge during thecourse of therapy presents an even greater challenge. In thisscenario, patients are treated with an appropriate �-lactambased on initial susceptibility data, only to fail therapy due tothe emergence of AmpC-mediated resistance. This phenome-non has been observed in 14 to 56% of patients treated withantipseudomonal penicillins, penicillin-inhibitor combinations,aztreonam, and extended-spectrum cephalosporins (50, 71, 94,97, 118, 147, 226, 228, 235), and emergence of resistance/clinical failure is observed most frequently with infections out-side the urinary tract and in patients with underlying cysticfibrosis and neutropenia. Although combining an aminoglyco-
FIG. 3. Mechanisms involved in regulation of ampC expression. These figures represent the current knowledge obtained from studies withmembers of the Enterobacteriaceae and appear to parallel events in P. aeruginosa. (A) Wild-type basal expression of ampC. During normal cell wallrecycling, 1,6-anhydromuropeptides are removed from the cell wall and transported into the cytoplasm via the AmpG permease. The 1,6-anhydromuropeptides are cleaved by AmpD to generate free tripeptides, which are later converted into UDP-MurNAc-pentapeptides. UDP-MurNAc-pentapeptide interacts with AmpR bound to the ampR-ampC intergenic region, creating a conformation that represses transcription ofampC. Low basal levels of AmpC are produced, and the enzyme is localized to the periplasmic space. (B) �-Lactam induction of ampC expression.Inducing �-lactams, such as cefoxitin and imipenem, cross the outer membrane through porins, enter the periplasmic space, and interact with targetPBPs. An increase in pools of 1,6-anhydromuropeptides is observed, and AmpD is unable to efficiently process the higher levels of cell wallfragments. The anhydro-MurNAc-peptides (inducing peptides) replace UDP-MurNAc-pentapeptides (suppressing peptides) bound to AmpR,causing a conformational change in the protein. AmpR is converted into a transcriptional activator, ampC is expressed at higher levels, and levelsof AmpC increase in the periplasmic space. When the amount of �-lactam decreases below “inducing levels,” the cytoplasmic pool of anhydro-MurNAc-peptides also decreases, and AmpD is able to efficiently cleave these peptides, restoring wild-type ampC expression, as shown in panelA. (C) AmpD-associated derepression of ampC expression. Mutations leading to the inactivation of AmpD or decreased expression of ampDimpair the processing of cell wall recycled products and lead to increased levels of anhydro-MurNAc-peptides (inducing peptides) in the cytoplasm.As a result, the binding of inducing peptides to AmpR is favored, AmpR is “locked” in a conformation for transcriptional activation of ampCexpression, and high-level constitutive expression of ampC is observed.
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 589
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

side with an antipseudomonal �-lactam is one strategy forpreventing the emergence of AmpC-mediated resistance, thiscombination is not always effective in achieving that goal (97,118, 147, 226, 228, 235).
As an initial step toward the future identification of novelstrategies for combating AmpC-mediated resistance, it is es-sential that we elucidate the mechanisms by which the expres-sion of this resistance mechanism is regulated. The followingsections review the current understanding of mechanisms in-volved in regulation of AmpC-mediated resistance among P.aeruginosa isolates and, where appropriate, compare and con-trast them with mechanisms of AmpC regulation among En-terobacteriaceae.
Pathways for AmpC Overproduction
Overproduction of the chromosomally encoded AmpC en-zyme in P. aeruginosa and some members of the Enterobacte-riaceae can occur either by induction of the ampC gene orthrough a process of derepression leading to constitutivehigh-level expression. Overexpression through the inductionpathway occurs during exposure to specific �-lactams and�-lactamase inhibitors (e.g., cefoxitin, imipenem, and clavu-lanate) (112, 123, 128, 247, 267), but the process is reversibleafter removal of the inducing agent. In contrast, AmpCderepression occurs when proteins involved in the inductionpathway are compromised through chromosomal mutations(83, 97, 108, 111, 114, 124) and the cephalosporinase isconstitutively produced at an elevated level, even in theabsence of an inducing �-lactam (9, 83, 114, 115). As dis-cussed later in this section, phenotypes of AmpC derepres-sion are more diverse among P. aeruginosa isolates thanamong the Enterobacteriaceae. Strains of P. aeruginosa cantransition through a phenotype of partial derepession beforeachieving full derepression of AmpC.
Factors Involved in Regulation of ampC Expression
Figure 3 illustrates the key components involved in the reg-ulation of ampC expression. The ampC induction pathwayinvolves the following three major gene products: (i) an innermembrane permease known as AmpG; (ii) a cytosolic amidase,AmpD; and (iii) a transcription factor, AmpR, belonging tothe LysR family of regulatory proteins (11, 138, 139). Thesethree proteins are required for induction of the ampC gene inboth Enterobacteriaceae and P. aeruginosa, although there is nodirect evidence linking AmpG to the ampC induction pathwayof P. aeruginosa (11, 81, 88, 109, 110, 127, 138, 139).
The P. aeruginosa ampC and ampR genes and their corre-sponding intergenic region were first described in 1990 (139),and the gene organization is identical to that for members ofthe Enterobacteriaceae with inducible ampC genes. In both P.aeruginosa and Enterobacteriaceae, the ampR and ampC genesare divergently transcribed, and the binding of AmpR to theintergenic region between ampC and ampR is required forampC induction (Fig. 3) (12, 16, 86, 126, 138, 139, 217). In theEnterobacteriaceae, AmpR negatively regulates ampC expres-sion as well as the expression of its own gene, ampR (125, 126).In studies with P. aeruginosa, the data have been more con-flicting and difficult to compare. Using an ampR knockout of P.
aeruginosa PAO1, Kong et al. reported a 12-fold increase innitrocefin hydrolysis in the absence of AmpR (107). However,interpretation of these data was complicated by the presence ofanother chromosomal �-lactamase, PoxB, which also hydro-lyzes nitrocefin and appears to be regulated by AmpR (107).Further experiments with a PampC-lacZ promoter gene con-struct inserted into the chromosome of P. aeruginosa suggestedthat AmpR does not negatively regulate ampC (107). Morerecently, however, Moya et al. concluded that AmpR is anegative regulator of ampC, based upon their more directanalysis of ampC expression in an ampR knockout of P. aerugi-nosa PAO1 (166).
Recent reporter gene studies have also suggested that P.aeruginosa AmpR is a global regulator affecting the expressionof multiple genes (lasB, rhlR, poxB, lasA, lasI, and lasR) inaddition to ampC (107). Although direct RNA expression ex-periments are needed to confirm a global regulatory function,these data raise the possibility that regulation by AmpR maybe more complex in P. aeruginosa than what has been shownfor members of the Enterobacteriaceae (107).
Mechanism of ampC Induction
Whether ampC is expressed at a low constitutive level orelevated through induction is dependent upon the type ofcofactor (i.e., cell wall precursor peptide) that binds to AmpR(Fig. 3). In this respect, the AmpC regulatory pathway is inti-mately linked to the cell wall recycling pathway via AmpD andAmpR. The general mechanism for regulation of low basalampC expression in a wild-type P. aeruginosa strain is extrap-olated from data obtained from the Enterobacteriaceae andshown in Fig. 3A. During normal growth, cell wall synthesisrequires the addition and subtraction of cell wall components,resulting in the release of muropeptides (39, 40, 69, 80, 87).These muropeptides are transported into the cell via theAmpG permease (30, 87). Once inside the cell, these muropep-tides are modified by AmpD into free peptide and anhydro-muramic acid, with the resulting peptide recycled back into thecell wall synthesis pathway (80, 81, 87, 88). During peptideprocessing for use in cell wall synthesis, UDP is added tothe pentapeptide (80). Excess “repressing” UDP-pentapeptidebinds to AmpR and keeps AmpR in a conformational statethat does not allow efficient transcription from the ampC pro-moter (86). It has been hypothesized that this conformation ofAmpR prevents RNA polymerase from interacting efficientlywith the ampC promoter, resulting in a low basal level of ampCexpression.
The process of AmpC induction requires binding of an in-ducing �-lactam or �-lactamase inhibitor (e.g., cefoxitin, imi-penem, or clavulanate) to penicillin binding proteins (PBPs)(Fig. 3B) (183, 190, 224). Since induction of AmpC does notresult from the interactions of all �-lactams with PBPs, thereclearly is something specific about the interactions of inducingcompounds such as cefoxitin, imipenem, and clavulanate. Stud-ies with the Enterobacteriaceae have shown that inducing �-lac-tams have a higher affinity for the low-molecular-weight PBPs(72, 82, 177), and genetic knockout of the genes for these PBPsprovides additional support for their role (183, 190, 224). How-ever, these studies were performed in E. coli, which does notpossess an intrinsic inducible ampC system, making the data
590 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

difficult to interpret (1). Data from a recently published studywith P. aeruginosa have demonstrated that loss of the nones-sential low-molecular-weight protein PBP4 is associated withincreased expression of ampC and partial derepression ofampC (166). Although these experiments clearly demonstratedan association between PBP4 and derepression of ampC (de-scribed in more detail later in the review), the authors con-cluded from their study that PBP4 plays a role in ampC induc-tion as well. However, strains lacking functional PBP4 stilldemonstrated induction of ampC, suggesting that PBP4 is notessential for the induction pathway (166). The role of otherlow-molecular-weight PBPs was not addressed in this studyand should still be evaluated to determine their selective rolein induction of ampC by inducing �-lactams.
Regardless of the precise mechanism responsible for selec-tive AmpC induction with specific �-lactams, the result is anincrease in the concentration of “inducing” muropeptides com-pared to the amount of “repressing” UDP-pentapeptide foundin the cytoplasm (39, 40, 87). The “repressing” UDP-pentapep-tide bound to AmpR is believed to be replaced by the “induc-ing” muropeptide form, changing the conformation of AmpRas it binds to the ampC promoter (76, 86). This conformationalchange has been suggested to provide a more efficient inter-action with RNA polymerase, resulting in a significant increasein ampC expression (86). When the inducing �-lactam or �-lac-tamase inhibitor is removed, normal cell wall synthesis and cellwall recycling are restored. The result is a restoration of basalampC expression through the replacement of the AmpR-bound “inducing” muropeptide with the “repressing” UDP-pentapeptide that is now present at a higher concentration.
Mechanisms of ampC Derepression
Modification of any protein involved in the induction path-way or modification of the ampC promoter could conceivablylead to derepression of ampC expression. Although studieswith the Enterobacteriaceae have identified mutations withinampR (111), the majority of changes observed in clinical iso-lates have been associated with ampD (46, 83, 108, 231, 246).AmpD-associated derepression has been linked to mutationswithin the ampD structural gene and mutations leading todecreased ampD expression (231). In these strains, the AmpDamidase that cleaves the muropeptides entering the cell duringcell wall synthesis has been modified or decreased, and normalprocessing of the muropeptides would be compromised (Fig.3C). As a result, the concentration of “inducing” muropeptidesin the cytoplasm would permanently increase, which favorstheir binding to AmpR due to the stoichiometric effect de-scribed above for the pathway of induction (39, 40, 86, 87).This culminates in a constitutive elevation of ampC expression.
Paralleling what is observed for the Enterobacteriaceae, P.aeruginosa AmpD has been characterized as a negative regu-lator of AmpC (114), and ampD mutations are an importantmechanism of ampC derepression (9, 97, 98, 115, 232). Al-though not as common as ampD mutations, the potential roleof mutated AmpR in P. aeruginosa derepression of AmpC hasbeen described and correlated with a similar mutation in En-terobacter cloacae (9, 111, 232). In contrast to the case for theEnterobacteriaceae, full derepression of AmpC in P. aeruginosais not always a single-step process. Campbell et al. have de-
scribed the following three phenotypes of ampC expression: (i)low-basal-level expression that is inducible (wild type), (ii)moderate-basal-level expression that is inducible (partial dere-pression), and (iii) high-basal-level expression that is constitu-tive (full derepression) (23). A further complexity is that someAmpC-overproducing P. aeruginosa strains do not exhibit mu-tations in either ampR, ampD, or the ampR-ampC intergenicregion and do not exhibit changes in the level of ampD expres-sion (9, 23, 97, 232, 282). In one of these studies, mutants thatexpressed significant increases in the basal level of ampC wereselected from a partially derepressed P. aeruginosa strain thatlacked a functional AmpD protein (282). The mechanism ofincreased ampC expression did not involve additional muta-tions in ampR, ampD, or the ampR-ampC intergenic region orchanges in ampD expression. However, a functional ampDgene was able to completely transcomplement and restore thewild-type phenotype for ampC expression and ceftazidime sus-ceptibility (282). These observations suggest that undiscoveredfactors or pathways likely contribute to the regulation of ampCand the derepressed phenotype.
Additional candidates for regulation of ampC expressioninclude AmpE, homologues of AmpD, and PBP4. In P. aerugi-nosa and the Enterobacteriaceae, expression of ampD is linkedto the ampE gene (83, 114). Although some publications haveindicated a potential role for AmpE in the AmpC regulatorypathway (83, 98), a specific role has yet to be established. Otherstudies suggest that AmpE plays no role in the regulation ofAmpC (114, 166).
AmpD Homologues and Regulation of ampC Expression
The entire genome of P. aeruginosa PAO1 has been se-quenced, opening the door for further investigations into themechanisms involved in the regulation of ampC expression. In2006, Juan et al. reported the identification of two additional P.aeruginosa AmpD homologues (AmpDh2 and AmpDh3) (98).E. coli possesses an outer membrane lipoprotein designatedAmiD that was suggested to be a homologue of AmpDh2 bythe same investigators (167), and this was later confirmed bySchmidtke and Hanson (232). There are only two N-acetyl-anhydro-muramyl-L-alanine amidases in the E. coli genome, sono homologue for AmpDh3 has been determined. Althoughthe level of identity between AmpDh2 or AmpDh3 and theoriginal AmpD protein is only 25 to 27% at the amino acidlevel, this may be attributed to the fact that the two newhomologues possess membrane-spanning tails not observedwith AmpD (232). However, AmpDh2 and AmpDh3 retainnecessary conserved sequences within the active site comparedwith AmpD and are able to transcomplement an AmpD-defi-cient strain of P. aeruginosa PAO1 (98).
Using a series of ampD homologue knockout mutants, Juanet al. investigated the relative impact of each homologue onexpression of ampC (98). Their observations are summarizedin Table 2. The wild-type laboratory strain P. aeruginosa PAO1served as the parent strain in this study and exhibited theexpected low-basal-level inducible phenotype. The loss ofampD from P. aeruginosa PAO1 changed the phenotype tomoderate-basal-level inducible expression of ampC (partial de-repression). Surprisingly, deletion of ampDh2 or ampDh3alone or in combination with each other did not alter the
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 591
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

low-basal-level inducible phenotype. Although deletion ofampD in combination with deletion of ampDh2 or ampDh3 didnot alter the partially derepressed phenotype observed withampD deletion alone, the double knockout of ampD andampDh3 did exhibit significant increases in both basal andcefoxitin-induced ampC expression. Finally, full derepressionto high-basal-level, constitutive expression of ampC requiredthe combined deletion of all three homologues. From thesedata, Juan et al. concluded that AmpD exhibits the greatestinfluence on ampC expression, followed by AmpDh3 and thenAmpDh2.
The discovery and characterization of the ampD homo-logues, as well as their coordinated regulation of ampC expres-sion, were important contributions to understanding the com-plex regulation of ampC expression in this pathogen. Notsurprisingly, data from the analysis of clinical isolates suggestthe use of additional pathways for regulation of ampC. Forexample, the same researchers analyzed 10 clinical isolates ofP. aeruginosa that overproduced AmpC and found that 4 of theisolates did not exhibit any mutations in ampD (97), suggestingthat loss of ampD is not an absolute requirement for ampCderepression in P. aeruginosa. Although potential decreases inampD homologue expression were not assessed, ampC dere-pression in these four strains was later shown to be associatedwith mutations in the nonessential low-molecular-weight PBP4gene, dacB (see below) (166). Schmidtke and Hanson reportedthat three clinical isolates of P. aeruginosa exhibiting full de-repression of ampC (constitutive high-basal-level expression)exhibited inactivating mutations within ampD only, with nomutations or changes in the expression of ampDh2 or ampDh3(232). Although the potential role of PBP4 was not evaluatedby Schmidtke and Hanson, full derepression of ampC has beenobserved in a spontaneous dacB (PBP4) mutant of P. aerugi-nosa PAO1 with subsequent knockout of ampD (166).
PBP4 and Regulation of ampC Expression
PBP4 was recently identified as an important component ofampC regulation (166). Genetic knockout of dacB, which en-codes PBP4, results in a partially derepressed phenotype in P.aeruginosa PAO1, similar to that of a knockout mutant of
ampD (166). Furthermore, PBP4 mutations have also beenassociated with derepression of ampC in clinical isolates, andthe association appears to be at least as common as mutationsin ampD (166).
PBP4-associated derepression of ampC is also associatedwith the two-component global regulator CreBC, which is ahomologue of the Aeromonas sp. regulator BrlAB (8, 174).Laboratory mutants and clinical isolates that exhibit PBP4-associated derepression of ampC also exhibit a significant in-crease in the expression of creD (166), a gene regulated byCreBC. However, the protein encoded by creD has not beencharacterized, and its role in ampC regulation is questionable.Elimination of creD in PBP4-associated derepressed mutantsdid not significantly alter �-lactam susceptibility (166). In con-trast, the knockout of creBC significantly increased susceptibil-ity to penicillins, cephalosporins, and aztreonam in ampC-derepressed dacB mutants. This link between CreBC andampC regulation appears to be specific for PBP4, as no corre-lation was noted for ampD-associated derepressed mutants.Perhaps the most interesting observation is that increased�-lactam susceptibility was not associated with any changes inthe level of ampC expression, suggesting that CreBC enhancesresistance through a pathway other than AmpC.
Each of the studies discussed above provides important in-formation on how P. aeruginosa regulates expression of ampC.However, it is clear that we have much to learn about thecomplex mechanisms by which AmpR, AmpD homologues,and PBP4 interact to control this important resistance mech-anism. Furthermore, the pathway that P. aeruginosa uses toselectively induce ampC in the presence of �-lactams such ascefoxitin and imipenem remains uncharacterized, despite ouradvances in understanding the steps to derepression. Of par-ticular interest would be further investigation into other low-molecular-weight PBPs and their role as initial triggers forampC induction. A more complete understanding of the fac-tors and complex pathways regulating ampC expression couldlead to the identification of potential targets for controllingAmpC-mediated resistance and preserving the antibacterialactivity of the �-lactam class.
OprD-MEDIATED RESISTANCE
The outer membrane of gram-negative bacteria constitutes asemipermeable barrier that slows the penetration of antibiot-ics, and specific to this review, the outer membrane of P.aeruginosa is only 8% as permeable as the outer membrane ofEscherichia coli (75). However, in order to survive, P. aerugi-nosa must allow the passage of nutrients into the cell, and thisis accomplished through a collection of water-filled proteinchannels called porins. Sequence analysis of the P. aeruginosagenome has identified 163 known or predicted outer mem-brane proteins, with 64 of these outer membrane proteinsgrouped into three families of porins (75). These porins play animportant physiological role in the transport of sugars, aminoacids, phosphates, divalent cations, and siderophores (75).Certain hydrophilic antibiotics, such as �-lactams, aminoglyco-sides, tetracyclines, and some fluoroquinolones, have also beenshown to transverse the outer membrane through porin chan-nels (173, 289). Not surprisingly, the loss of specific porinchannels can decrease the susceptibility of P. aeruginosa to
TABLE 2. Genetic knockout of ampD homologue genes and ampCexpression in P. aeruginosa PAO1a
StrainAvg level of ampC expression � SDb
Basal Induced
PAO1 1c 43 � 9PA�D 60 � 19 152 � 38PA�Dh2 1c 48 � 15PA�Dh3 1c 55 � 5PA�Dh2Dh3 2 � 0.14 81 � 26PA�DDh2 62 � 9 234 � 58PA�DDh3 191 � 52 1,014 � 297PA�DDh2Dh3 1,020 � 87 1,105 � 88
a Modified from reference 91 with permission.b Real-time reverse transcription-PCR was used to measure ampC expression
in P. aeruginosa grown in the presence of 50 �g/ml of cefoxitin (induced) or theabsence of cefoxitin (basal).
c Actual ampC expression data were not presented in the original report (91).Based on the methodology described and interpretation of the data presented,basal ampC expression was set to a value of 1 in this table.
592 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

certain antibacterial agents. This section of the review focusesspecifically on the association of the outer membrane porinOprD and the susceptibility of P. aeruginosa to carbapenemantibiotics.
OprD and P. aeruginosa Susceptibility to Carbapenems
The P. aeruginosa porin OprD is a substrate-specific porinthat has been shown to facilitate the diffusion of basic aminoacids, small peptides that contain these amino acids, and car-bapenems into the cell (254, 255). This aqueous porin sharesclose homology to the nonspecific porin OmpF in E. coli.Pirnay et al. evaluated oprD genes from 55 strains of P. aerugi-nosa (environmental and clinical) and found evidence that thegenetic sequence of oprD and the amino acid sequence ofOprD are diverse across individual strains (195). DNA se-quence identities ranged from 91 to 93%, whereas amino acidsequence identities varied from 88 to 93%. There was evidenceof intraspecies recombinational events.
OprD appears to serve as the preferred portal of entry forthe carbapenems, and loss of OprD from the outer membranesignificantly decreases the susceptibility of P. aeruginosa toavailable carbapenems (103, 132, 140, 144, 169, 209, 210, 221,229, 254). However, data presented later in this section high-light how much we have yet to learn about the dynamic inter-actions of carbapenems with P. aeruginosa.
The impact of OprD-mediated resistance on the carbapen-ems can be analyzed in two ways. The first consideration is therelative impact on the antibacterial potency of the carbapen-ems, as measured by increases in MICs. Data from a recentstudy of isogenic “wild-type” and OprD-deficient mutant pairsdemonstrated that the loss of OprD decreases the susceptibil-ity of P. aeruginosa to meropenem 4- to 32-fold, compared to 4-to 16-fold for imipenem and 8- to 32-fold for doripenem (221).For several OprD-deficient mutants in this study, the impacton meropenem potency was greater than that on the potencyof other carbapenems. These data appear to conflict with theconclusions of Perez et al., who suggested that meropenemutilizes alternative pathways for entry across the outer mem-brane of P. aeruginosa (188). However, Perez et al. did notevaluate isogenic mutant pairs in their analysis of OprD defi-ciency and meropenem susceptibility. Rather, these investiga-tors focused their study on unrelated clinical isolates that ex-hibited a phenotype of imipenem resistance and meropenemsusceptibility. Although the P. aeruginosa isolates were suscep-tible to meropenem, MICs ranged from 2 to 4 �g/ml, wellabove expected meropenem MICs against “wild-type” strains(21, 256). Therefore, it is likely that decreased OprD in theclinical isolates was in fact responsible for elevating mero-penem MICs to the susceptible breakpoint.
The second aspect of OprD-mediated resistance to consideris the clinical impact on the carbapenems. Although loss ofOprD may impact susceptibility to imipenem less than that tomeropenem (based on changes in MICs), this resistance mech-anism frequently pushes MICs of imipenem above the resis-tance breakpoint. For example, in a study by Sakyo et al., all 10OprD-deficient mutants lost susceptibility to imipenem, withMICs of �4 �g/ml (221). This is not surprising, since theMIC50 for imipenem against P. aeruginosa is already 1 �g/ml(21, 256), requiring only an eightfold decrease in potency to
push the MIC into the intermediate range. In contrast to thecase for imipenem, MICs for meropenem remained below 4�g/ml for 4 of 10 mutants, and MICs for doripenem remainedbelow 4 �g/ml for 8 of 10 mutants (221). Meropenem anddoripenem exhibit an intrinsic potency that is fourfold greaterthan that of imipenem (21, 256). Therefore, the impact ofOprD deficiency on the potency of these carbapenems doesnot always push the MICs above the susceptible breakpoint,and additional resistance mechanisms (efflux pump and/or car-bapenemase) may be required to provide resistance to thesetwo carbapenems.
The following sections review the current understanding ofhow P. aeruginosa regulates the expression of oprD. Publisheddata to this point have identified mechanisms influencing thetranscriptional expression of oprD and the translation of afunctional porin protein.
Characterization of oprD Promoter Elements
Although the relationship between OprD and resistance tothe carbapenems has been recognized for 2 decades, charac-terization of the oprD promoter and understanding of theregulation of OprD remain limited compared to our under-standing of AmpC- and efflux-mediated resistance mecha-nisms. Figure 4 depicts the key promoter elements for oprD,based upon 5� rapid amplification of cDNA ends of the oprDpromoter of P. aeruginosa PAO1 grown in Mueller-Hintonbroth (278). In these studies, two start sites for oprD transcrip-tion were identified, with transcription initiating at similar fre-quencies from an adenine 71 bases and a thymine 23 basesupstream of the translational start codon for oprD. Putative10 and 35 elements are also presented in Fig. 4.
The transcriptional start sites and putative promoter ele-ments observed for P. aeruginosa grown in Mueller-Hintonbroth are different from those described by Ochs et al. (179).When P. aeruginosa was cultured with arginine, histidine, glu-tamate, or alanine as the sole source of carbon and with am-monium sulfate as the nitrogen source, the primary start site oftranscription was reported to be a thymine located 89 basesupstream of the oprD translational start codon. Furthermore,induction of oprD expression by arginine was shown to bedependent on the arginine-responsive regulatory proteinArgR, which binds to an operator in the promoter region ofoprD (179). In contrast, induction with the amino acid gluta-mate was independent of ArgR activation. Therefore, it ap-pears that P. aeruginosa utilizes multiple transcriptional startsites and mechanisms to regulate expression of oprD, depend-ing upon the growth conditions encountered.
Molecular Mechanisms of OprD-Mediated Resistance
The pathway to OprD-mediated resistance can involvemechanisms that decrease the transcriptional expression ofoprD and/or mutations that disrupt the translational produc-tion of a functional porin for the outer membrane. At the levelof oprD transcription, characterized mechanisms include (i)disruptions of the oprD promoter, (ii) premature terminationof oprD transcription, (iii) coregulation with mechanisms oftrace metal resistance, (iv) salicylate-mediated reduction, and(v) decreased transcriptional expression through mechanisms
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 593
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

of coregulation with the multidrug efflux pump encoded bymexEF-oprN. oprD promoter disruptions have occurred as aresult of deletions or insertions within the upstream region ofoprD. Yoneyama and Nakae reported the association of a largedeletion encompassing the putative promoter and initiationcodon that prevented transcription of oprD (288). IS1394 andan ISPa16-like insertion (IS) element have been describedupstream of the oprD coding region for imipenem-resistantisolates of P. aeruginosa exhibiting decreased oprD expression(273, 281).
El Amin et al. evaluated the transcription of oprD amongclinical isolates of P. aeruginosa by using two sets of primersthat amplified either the upstream or downstream regions ofthe structural gene (47). Two strains of P. aeruginosa exhibitednormal levels of oprD transcription in experiments with theupstream primers, but oprD transcripts were undetectable withthe downstream primers. Four additional strains demonstratedsignificant differences in the amounts of oprD transcript mea-sured with the two primer sets. El Amin et al. concluded thatpremature termination of transcription was occurring in thesestrains, potentially due to mutations within the structural genesequence.
The trace metals zinc and copper have been shown to de-crease the expression of oprD in P. aeruginosa, leading toimipenem resistance (22, 189). This negative regulation is me-diated through the regulatory proteins CzcR and CopR, whichrespond to the presence of zinc and copper, respectively, toactivate mechanisms of metal resistance. CzcR and CopR bothdownregulate oprD transcription directly or indirectly, throughunidentified factors. The weak aromatic acid salicylate was alsoshown to repress the transcription of oprD through an unchar-acterized mechanism, leading to imipenem resistance amongsalicylate-exposed P. aeruginosa isolates (180).
Perhaps the most complex and intriguing mechanisms im-pacting the transcription of oprD are those that are linked tothe regulation of expression of the mexEF-oprN efflux pump(101, 180). These mechanisms of coregulation are discussed indetail later in the review, but they highlight the complexity bywhich P. aeruginosa is able to regulate expression of resistancemechanisms and why it is sometimes so difficult to definitivelylink phenotypes to changes in one specific mechanism. Finally,
mechanisms of OprD deficiency related to translation of anactive porin include (i) mutations, insertions, and/or deletionscreating frameshifts and premature stop codons (195) and (ii)disruption of the oprD structural gene by insertion of large ISelements (51, 277).
Discordance between oprD Expression andSusceptibility to Imipenem
Although the relationship between OprD deficiency and imi-penem resistance has been well established in the literature, itshould not come as a surprise that P. aeruginosa does notalways follow expected rules. The genetic versatility of thispathogen and its ability to coregulate multiple resistancemechanisms make P. aeruginosa a constantly moving target andone of our greatest therapeutic challenges. Studies from ourlaboratory have identified intriguing strains that exhibit discor-dance between oprD expression and susceptibility to carbap-enems.
The first example is an isogenic mutant, P. aeruginosa 410L,that was selected with levofloxacin from P. aeruginosa Tokai#1(275). As background, strain Tokai#1 is an isogenic mutant ofP. aeruginosa PAO1 that lacks susceptibility to both levofloxa-cin and imipenem (145). The MIC for imipenem againstTokai#1 is 8 �g/ml, compared to 1 �g/ml against the parentalPAO1 strain, and this decreased susceptibility correlates with afivefold decrease in the level of oprD expression (275). Expres-sion of oprD in mutant strain 410L decreased further, to a level3-fold below that of Tokai#1 and 17-fold below that of thewild-type parent, PAO1. However, despite the further de-crease in oprD expression and the inability to detect OprD inouter membrane preparations, mutant 410L lost its resistanceto imipenem and reverted back to a level of susceptibilitysimilar to that of the original PAO1 parent.
The second example is an isogenic imipenem-hypersuscep-tible mutant, P. aeruginosa 244-921C, that was selected from animipenem-resistant clinical isolate, P. aeruginosa strain 244,using ciprofloxacin (274). The mechanism of imipenem resis-tance for P. aeruginosa 244 (MIC � 16 �g/ml) was shown to bea base transition creating a premature stop codon and prevent-ing translation of full-length OprD. This mutation was retained
FIG. 4. Characterization of oprD promoter elements. Transcription of oprD in P. aeruginosa PAO1 initiates with equal frequencies from twostart sites, located 23 bases (SS1) and 71 bases (SS2) upstream of the structural gene (ATG translation start codon is highlighted in orange). Theputative 10 and 35 promoter elements for SS1 are highlighted in red, and the putative 10 and 35 promoter elements for SS2 are highlightedin blue.
594 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

in the isogenic 244-921C mutant. Therefore, the eightfold in-crease in susceptibility of P. aeruginosa 244-921C to imipenem(MIC � 2 �g/ml) occurred despite the absence of an activeOprD porin in the outer membrane.
The discordance between OprD and susceptibility to imi-penem described above highlights how much we have yet tolearn about the dynamic interactions of carbapenems with P.aeruginosa. The mechanism(s) responsible for increased sus-ceptibility to imipenem in these strains has yet to be elucidated,but characterization of these mechanisms could provide path-ways for development of therapeutic strategies to enhance theefficacy of carbapenems.
EFFLUX-MEDIATED RESISTANCE
While the loss of porins such as OprD represents an effectivebarrier for drug entry into the cell, a reduction in drug accu-mulation can also be achieved through active export by mem-
brane-associated pumps. Efflux pumps have been categorizedinto five superfamilies, based primarily on amino acid se-quence identity, the energy source required to drive export,and substrate specificities of the different pumps (218, 259).The superfamilies include (i) the ATP-binding cassette (ABC)family, (ii) the small multidrug resistance family, (iii) the majorfacilitator superfamily, (iv) the resistance-nodulation-division(RND) family, and (v) the multidrug and toxic compoundextrusion family. Although sequence analysis of the P. aerugi-nosa genome has revealed the presence of efflux systems fromall five superfamilies, the largest number of predicted pumpsbelong to the RND family, with a total of 12 RND systems(including two divalent metal cation transporters) (248). Un-like the primary active transporters of the ABC superfamily,which utilize ATP hydrolysis for energy, the RND family (aswell as the remaining superfamilies) are secondary activetransporters (symporters, antiporters, and uniporters) that de-rive the energy required for compound extrusion by protonmotive force. Disruption of the proton gradient through theaddition of a proton uncoupler, carbonyl cyanide m-chlorophe-nylhydrazone, increases the accumulation in these bacteria ofsubstrates that are normally exported (172, 201).
RND pumps typically exist as a tripartite system consistingof a periplasmic membrane fusion protein (MFP), an outermembrane factor (OMF), and a cytoplasmic membrane(RND) transporter (Fig. 5). This complex forms a channelspanning the entire membrane, allowing for the transportationof lipophilic and amphiphilic drugs from the periplasmic spaceand cytoplasm to the extracellular environment. The genesencoding these pumps are organized into operons on the P.aeruginosa chromosome (Fig. 6). Each operon is composed ofat least two genes, coding for the MFP and the RND trans-porter. Six of the 12 operons possess an OMF gene, completingthe tripartite system, while the remaining operons are devoidof an OMF gene. Several operons have an adjacent regulatorygene transcribed in the same orientation or divergently fromthe operon, whose product functions as a repressor or activatorof pump expression. Operons may contain additional genesbesides those coding for the efflux pump. For example, mexGin the mexGHI-opmD operon encodes an integral membraneprotein, and PA2528-PA2527-PA2526-opmB possesses a sec-ond RND transporter gene, PA2526.
The 10 RND pumps in P. aeruginosa (excluding the metalcation transporters) are named MexAB-OprM, MexCD-OprJ,MexEF-OprN, MexXY, MexJK, MexGHI-OpmD, MexVW,MexPQ-OpmE, MexMN, and TriABC (Table 3). Mex is anacronym for multiple efflux, and “Tri” refers to triclosan efflux.While several of these pumps share common substrates, theyare also responsible for unique phenotypes inherent to theirexpression. Substrates of these pumps include antibiotics, bio-cides, dyes, detergents, organic solvents, aromatic hydrocar-bons, and homoserine lactones (Table 3) (205, 234). Althoughnot discussed in this review, these pumps may also have aphysiological role in P. aeruginosa (e.g., cell-to-cell communi-cation and pathogenicity) (193), besides their protective effectsagainst antimicrobials. This section of the review focuses onthe complex regulation of these pumps and the antibiotic phe-notypes associated with their expression in planktonic cells.While P. aeruginosa can also live as a community encompassedin an exopolysaccharide matrix (i.e., biofilm), recent evidence
FIG. 5. Structure and function of RND efflux pumps in P. aerugi-nosa. RND pumps typically exist in a tripartite system consisting ofan RND cytoplasmic membrane transporter (RND), an MFP, andan OMF. This complex forms a channel spanning the entire mem-brane, allowing for the proton-driven transport of lipophilic andamphiphilic drugs from the cytoplasm of the cell across the cyto-plasmic membrane, through the periplasmic space, across the pep-tidoglycan, and across the outer membrane. The RND efflux pumpscan also extrude drugs from the periplasmic space before they crossthe cytoplasmic membrane.
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 595
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

has suggested that the RND efflux pumps do not participate inbiofilm resistance to typical antipseudomonal agents (38) butmay be involved with azithromycin resistance in biofilms (66).
MexAB-OprM Efflux Pump
During a study examining siderophore-mediated iron trans-port, the first multidrug efflux pump in P. aeruginosa, MexAB-OprM, was discovered by Poole et al. (204). MexAB-OprM isable to export drugs from several different classes, includingfluoroquinolones, tetracyclines, chloramphenicol, �-lactamsand �-lactamase inhibitors, macrolides, novobiocin, tri-methoprim, and sulfonamides (102, 120, 121, 243, 244). Of theRND efflux pumps, MexAB-OprM has the broadest substrateprofile for the �-lactam class, with an ability to export �-lac-tams such as the carboxypenicillins, aztreonam, extended-spec-trum cephalosporins (e.g., ceftazidime and cefotaxime), pen-ems (e.g., faropenem), and the carbapenems meropenem andpanipenem (but not imipenem and biapenem). MexAB-OprMparticipates in the intrinsic resistance of P. aeruginosa to theagents listed above through its constitutive production in wild-type cells (205). Knockout studies of mexAB-oprM alone or incombination with other resistance mechanisms have confirmedthe pump’s role in intrinsic resistance, as these mutants be-come hypersensitive (120, 148, 161).
Two additional characteristics are associated with mexAB-oprM expression. First, growth-phase-dependent expression ofmexAB-oprM has been demonstrated (53). As the growth cycleprogressed and cell density increased, mexAB-oprM transcrip-tion also increased until maximum expression occurred in late
log phase/early stationary phase. The growth-phase-dependentupregulation was suggested to involve a quorum sensing signal.Quorum sensing is a mechanism by which bacteria monitor celldensity through cell-to-cell communication, allowing for thecoordinated expression of certain genes (e.g., virulence fac-tors) in a cell density-dependent manner (260). Cell-to-cellsignaling is mediated by diffusible autoinducers, known as ho-moserine lactone molecules, which interact with their cognatereceptors to activate gene expression. N-Butyryl-L-homoserinelactone (C4-HSL) is synthesized as part of the rhl quorumsensing system and was shown to enhance expression ofmexAB-oprM (146, 230). Second, the OMF gene, oprM, wasshown to be expressed independently of mexAB (290). A weakpromoter was discovered upstream of oprM in the coding re-gion of mexB. This promoter was suggested to be less activethan the promoter upstream of mexA, contributing to only afraction of the total amount of transcript. OprM may serve asan OMF for the MexXY (5, 152, 159), MexJK (33), MexVW(122), and MexMN (158) systems, and possibly other RNDpumps. Independent expression of oprM may allow for suffi-cient levels of OMF to accommodate multiple pumps or toensure its presence in case expression from the mexAB-oprMpromoter is compromised.
Several regulatory loci influence the expression of themexAB-oprM operon. The mexR gene is located directly up-stream of but transcribed divergently from mexAB-oprM andencodes a repressor belonging to the MarR family of regula-tory proteins (Fig. 6) (206). MexR binds as a stable homodimerto two sites, each consisting of inverted repeat sequences,
FIG. 6. RND efflux operons in P. aeruginosa. Genes which encode protein components or characterized pumps are denoted by their genenames, and genes encoding protein components of uncharacterized pumps are designated with the P. aeruginosa (PA) numbers assigned in theannotated P. aeruginosa genome sequence (GenBank). Genes are depicted with the following color scheme: dark red arrow, transcriptionalregulator-encoding gene; dark blue arrow, membrane fusion protein-encoding gene; light blue arrow, RND transporter-encoding gene; red arrow,outer membrane protein-encoding gene; and gold arrow, gene encoding a protein with unknown function. (Adapted from reference 234 withpermission of the publisher.)
596 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

within the mexR-mexA intergenic region (52). The bindingregion encompasses overlapping mexR and mexA promoters,and association of MexR dimers with these sites repressestranscription of the mexAB-oprM operon and negatively auto-regulates its own expression (52, 220). A second regulatoryfactor of the mexAB-oprM operon, NalD, was discovered fol-lowing insertional mutagenesis of a gene, nalD, located adja-cent to a putative pump gene of the major facilitator super-family (239). NalD is a repressor of the TetR family that bindsto a sequence upstream of mexAB-oprM but downstream of themexR binding sites (160). The NalD operator overlaps a secondpromoter for mexAB-oprM, and binding of NalD restricts ex-
pression from this proximal mexA promoter. The C4-HSL-mediated growth phase regulation of mexAB-oprM describedabove occurs independently of MexR (230). However, the pos-sibility that C4-HSL directly or indirectly influences NalD-mediated regulation of mexAB-oprM to promote growth phaseexpression has yet to be determined experimentally.
Overexpression of mexAB-oprM has been detected in nalB-,nalC-, and nalD-type multidrug-resistant mutants and selectedboth in vivo (2, 137, 181, 184, 239, 273) and in vitro (2, 24, 239,245). In nalB-type strains, mutations within mexR disrupt thetranslation of full-length protein or compromise the repressoractivity of MexR by causing a loss of dimerization, defects in
TABLE 3. Characteristics of RND efflux pumps in P. aeruginosa
Operon Component FunctionaRegulatore Substrate(s)
Primaryb Secondaryc Antibiotics Additional compounds
mexAB-oprM MexA MFP MexR NalC Fluoroquinolones, �-lactams, Biocides (e.g., triclosan),MexB RND NalD �-lactamase inhibitors, detergents, dyes, HSL,f
OprM OMF tetracyclines, chloramphenicol,macrolides, novobiocin,trimethoprim, sulfonamides
aromatic hydrocarbons
mexCD-oprJ MexC MFP NfxB Fluoroquinolones, �-lactams, Biocides (e.g., triclosan),MexD RND tetracycline, chloramphenicol, detergents, dyes,OprJ OMF macrolides, trimethoprim,
novobiocinaromatic hydrocarbons
mexEF-oprN MexE MFP MexT MexS Fluoroquinolones, Biocides (e.g., triclosan),MexF RND MvaT chloramphenicol, trimethoprim aromaticOprN OMF hydrocarbons
mexXY MexX MFP MexZ Fluoroquinolones, �-lactams,MexY RND tetracycline, aminoglycosides,OprM/Opm-d OMF macrolides, chloramphenicol
mexJK MexJ MFP MexL Tetracycline, erythromycin Biocides (e.g., triclosan)MexK RNDOprM/OpmH OMF
mexGHI-opmD MexG ? SoxR Fluoroquinolones VanadiumMexH MFPMexI RNDOpmD OMF
mexVW MexV MFP — Fluoroquinolones, tetracycline,MexW RND chloramphenicol, erythromycinOprM OMF
mexPQ-opmE MexP MFP — Fluoroquinolones, tetracycline,MexQ RND chloramphenicol, macrolidesOpmE OMF
mexMN MexM MFP — Chloramphenicol, thiamphenicolMexN RNDOprM OMF
triABC TriA MFP — TriclosanTriB MFPTriC RNDOpmH OMF
a MFP, membrane fusion protein; RND, resistance-nodulation-division transporter; OMF, outer membrane factor; ?, encodes a protein of unknown function.b Regulatory proteins that directly control expression of the efflux operons.c Proteins that indirectly activate operon expression when mutated.d MexXY may utilize OpmB, OpmG, OpmH, and/or OmpI as OMFs.e —, not identified.f HSL, homoserine lactones.
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 597
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

DNA binding, or, possibly, instability of the protein (2, 219).Several mutations in mexR have been described, including nu-cleotide changes (e.g., base substitutions, deletions, and inser-tions) and insertion of an IS element (2, 19, 219, 245). Theabsence of MexR from its operators leads to pump overexpres-sion from the distal mexA promoter (52). nalC-type strainshyperexpress mexAB-oprM, but at lower levels than those innalB-type mutants (245). Transposon insertional mutagenesisidentified the site for nalC-type mutations in the PA3721 gene(renamed nalC) (24). nalC encodes a putative repressor of theTetR/AcrR family, whose genes are located upstream of anoperon, PA3720-PA3719, that is negatively regulated by NalC(24). Loss of NalC resulted in overexpression of PA3720-PA3719, and subsequent experiments demonstrated thatPA3719 (renamed ArmR) upregulates mexAB-oprM by inter-acting with MexR (24, 35). ArmR, a 53-amino-acid antirepres-sor, allosterically inhibits MexR dimer-DNA binding by occu-pying a hydrophobic binding cavity within the center of theMexR dimer (272). As implied by the name, overexpression ofmexAB-oprM in nalD-type mutants occurs in response to dis-ruption of NalD (239). Complementation of nalD-type mu-tants with a functional nalD gene reduced mexAB-oprM hyper-expression and drug resistance (239). Mutations within NalDwere believed to alleviate repression of the proximal mexApromoter (160), presumably by an inability to bind to its op-erator. Interestingly, maximum expression from the proximalmexA promoter in NalD-negative strains requires the presenceof MexR (160).
A mutational event within one of the known regulatorygenes may not be the sole mechanism to increase mexAB-oprMtranscription. Chen et al. recently described the effect of oxi-dative stress on operon expression (29). Oxidation of two cys-teine residues within MexR causes the formation of an inter-monomer disulfide bond which alters the conformation ofMexR. Oxidized MexR dissociates from the mexR-mexA inter-genic region, allowing access of RNA polymerase to the distalmexA promoter. In this situation, the MexR protein serves as asensor of oxidative stress, and the response culminates in ac-tivation of the efflux defense mechanism, possibly to removethe agent responsible for inducing the oxidative stress.
MexCD-OprJ Efflux Pump
The operon coding for MexCD-OprJ was cloned and se-quenced by Poole et al. in 1996 and showed a high degree ofhomology to MexAB-OprM (203). MexCD-OprJ can ex-trude a variety of antimicrobial agents, including fluoro-quinolones, �-lactams, chloramphenicol, tetracycline, novo-biocin, trimethoprim, and macrolides (70, 102, 149, 243).Unlike MexAB-OprM, MexCD-OprJ does not have an exten-sive substrate profile for the �-lactams, but rather, it preferen-tially exports the fourth-generation cephalosporins (e.g.,cefepime, cefpirome, and cefozopran) (149, 203). Transcrip-tion of mexCD-oprJ can be observed in wild-type cells (276),but the levels are most likely not sufficient to produce detect-able levels of protein (70, 203). In addition, deletion of mexCD-oprJ has no impact on wild-type susceptibility, indicating thatthis pump does not contribute to intrinsic resistance (162, 244).Expression of mexCD-oprJ was shown to be inducible in re-sponse to benzalkonium chloride, chlorhexidine gluconate, tet-
raphenylphosphonium chloride, ethidium bromide, rhodamine6G, and acriflavine but not in response to clinically relevantantibiotics (162). Induction of mexCD-oprJ by membrane dam-aging agents (i.e., chlorhexidine) was dependent upon thestress response sigma factor AlgU (55).
Expression of mexCD-oprJ is governed by the product of agene, nfxB, located upstream of mexCD-oprJ but transcribeddivergently from the operon (Fig. 6) (203). NfxB displays sim-ilarity to proteins of the LacI-GalR family who possess a helix-loop-helix motif characteristic of DNA binding (203). NfxBnegatively regulates expression of mexCD-oprJ, as well as itsown expression (203, 238), by binding to a site composed oftwo 39-bp repeats within the nfxB-mexC intergenic region(238). Mutations within nfxB are suggested to alter the repres-sor activity of NfxB, leading to hyperexpression of mexCD-oprJin so-called nfxB-type mutants (203, 238). Several mutationshave been described for nfxB from laboratory strains and clin-ical isolates, such as base substitutions, deletions, and inter-ruption with an IS element (37, 90, 203). Two types of hyper-expressing mutants, types A and B, have been identified in P.aeruginosa, with differing levels of MexCD-OprJ productionand susceptibility (149). Type B mutants produce largeramounts of MexCD-OprJ and have more substantial changesin susceptibility than do type A mutants. Complementation ofboth types of mutants with wild-type nfxB restored susceptibil-ities to wild-type levels, suggesting that mutations in NfxB wereresponsible (149). Regulation of mexCD-oprJ expression maynot be limited to NfxB. In mexAB-oprM and oprM deletionderivatives of wild-type strain PAO1, expression of mexCD-oprJ markedly increased, indicating that MexAB-OprM mayinfluence the expression of mexCD-oprJ (119). The mechanismresponsible for mexCD-oprJ upregulation in MexAB-OprM-deficient strains has yet to be determined.
Whereas the overproduction of MexCD-OprJ in P. aerugi-nosa provides resistance to several antibacterial drugs, an in-teresting phenotype of hypersusceptibility (�4-fold increase insusceptibility) to some �-lactams and aminoglycosides is ob-served in the same strains. The correlation between MexCD-OprJ overproduction and hypersusceptibility and the mecha-nisms responsible for hypersusceptibility are discussed later inthis section.
MexEF-OprN Efflux Pump
Three open reading frames in an operon named mexEF-oprN were characterized by Kohler et al., and the products ofmexEF-oprN exhibit amino acid identity to the components ofMexAB-OprM and MexCD-OprJ (104). Substrates recognizedby MexEF-OprN include fluoroquinolones, chloramphenicol,and trimethoprim (104), but the pump has no apparent affinityfor currently available �-lactams. Expression of mexEF-oprNwas originally reported as quiescent (104), but later studiesdetected low-level expression in strains with wild-type suscep-tible phenotypes (119, 276). Disruption of mexEF-oprN in wild-type strains did not affect susceptibility, excluding its partici-pation in intrinsic resistance (104).
Regulation of mexEF-oprN differs from that of mexAB-oprMand mexCD-oprJ in that operon expression is not suppressed bya negative regulator. Multiple factors are capable of control-ling mexEF-oprN transcription. mexT, located upstream of and
598 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

transcribed in the same orientation as mexEF-oprN (Fig. 6 and7A), encodes a protein belonging to the LysR family of tran-scriptional activators that is capable of positively regulatingmexEF-oprN expression (Fig. 7A) (101, 180). Because of se-quence variations within mexT genes of different wild-type P.aeruginosa strains, activation of MexT can occur through dif-ferent pathways (145). In some P. aeruginosa strains, MexTexists in a dormant form because of suppressing mutationswithin the coding region, but additional cis-acting mutations ordeletions within mexT convert inactive MexT into an activeform (145).
In other strains, suppressing mutations are not presentwithin mexT (145), but stimulation of MexT requires potentialchanges in the levels of cognate effector molecules (101, 145).Mutations in these strains occur within a gene, mexS, locatedupstream of mexT that encodes a putative oxidoreductase/dehydrogenase homologue (Fig. 7C) (241). Inactivation ofMexS is believed to cause a buildup of metabolites that serveas effector molecules for MexT, which, in turn, upregulatesmexEF-oprN expression to remove the toxic metabolites (241).Therefore, an active MexT protein is required for upregulationof mexEF-oprN by this pathway. While mexEF-oprN-overex-pressing strains have been encountered among nosocomial P.aeruginosa strains (47, 181, 207, 273), a correlation betweenpump overexpression and genetic changes within mexT and/ormexS in clinical isolates has, to the best of our knowledge, beenlacking.
Expression of mexEF-oprN is also controlled by a member ofthe histone-like nucleoid structuring protein family, MvaT,which serves as a global regulator of genes involved in viru-lence, housekeeping, and biofilm formation (Fig. 7D) (26, 35,258, 270). MvaT binds to and oligomerizes across AT-richregions of DNA with a high affinity and, as a result, silences theexpression of certain genes (26). Deletion of mvaT caused anincrease in mexEF-oprN expression that was not dependent onmexT or mexS (270). MvaT does not bind upstream of mexEF-oprN (270). Thus, the loss of this regulatory protein most likelyhas an indirect effect on regulating mexEF-oprN expression.
Data from a more recent study suggest that regulation ofmexEF-oprN is even more complex and that more uncharac-terized pathways are likely operative in P. aeruginosa (279). Inthis study, an isogenic mutant overexpressing mexEF-oprN wasselected from a clinical isolate of P. aeruginosa by use of levo-floxacin. Analysis of gene sequences and transcriptional ex-pression demonstrated that the overexpression of mexEF-oprNdid not involve changes in mexT, mexS, or mvaT. The mecha-nism of mexEF-oprN regulation in this isogenic mutant remainsto be characterized.
Similar to nfxB-type mutants, nfxC-type mutants becomehypersusceptible to certain �-lactams and aminoglycosides (58,202). �-Lactam hypersusceptibility was associated with repres-sion of the quorum sensing-mediated enhancement of mexAB-oprM expression by MexT (146). Transcription of the C4-HSLautoinducer synthase gene, rhlI, was previously shown to bereduced in nfxC-type mutants (105). As mentioned above,growth phase expression of mexAB-oprM is dependent on thisquorum sensing molecule. Therefore, MexT appears to act asa global regulator controlling expression of mexEF-oprN, oprD,and genes regulated by the autoinducer C4-HSL, such asmexAB-oprM.
MexXY Efflux Pump
mexXY was cloned from the P. aeruginosa chromosome byMine et al. in 1999, and the pump displays properties similar tothose of the efflux systems described above (159). Unlike theoperons described above, mexXY lacks a gene coding for anouter membrane protein (Fig. 6). Instead, MexXY is able toassociate with OprM (5, 32, 159), and possibly other outermembrane proteins, such as OpmB, OpmG, OpmH, andOpmI, to form a functional tripartite system (32, 168). Fluo-roquinolones, specific �-lactams (i.e., cefepime), aminoglyco-sides, tetracycline, chloramphenicol, and erythromycin are allsubstrates for MexXY (202, 234). mexXY expression is inducedwhen cells are grown in the presence of tetracycline, erythro-mycin, and gentamicin (152). Deletion of mexXY in wild-typestrains increases their susceptibility to these antibiotics, sug-gesting that this pump contributes to intrinsic resistance (152,161). MexXY does not contribute to the intrinsic resistance tofluoroquinolones, a pump substrate, because these agents failto induce mexXY expression (152).
The product of a gene, mexZ, located upstream of but tran-scribed divergently from mexXY, negatively regulates the ex-pression of the operon (Fig. 6) (5, 154). MexZ belongs to theTetR family of transcriptional regulators and contains a char-acteristic N-terminal helix-turn-helix DNA binding domain.MexZ binds as a homodimer to an inverted repeat region,located in the mexZ-mexX intergenic region, which encom-passes the putative mexXY promoter (154). Mutations in mexZor the mexZ-mexX intergenic region have been associated withhyperexpression of mexXY (85, 263). However, overexpressionof mexXY has also been detected in mutants not harboringmutations within mexZ or the mexZ-mexX intergenic region(137, 240, 269). Llanes et al. proposed the names agrZ-typeand agrW-type mutants to describe mutants hyperexpressingmexXY with alterations within mexZ and outside mexZ, respec-tively (137). Thus, mexXY may be regulated by multiple factors,similar to mexAB-oprM, and mutations within as yet unidenti-fied genes are necessary for mexXY overexpression. MexXYhyperexpression has been well documented for agrZ-type andagrW-type clinical isolates resistant to fluoroquinolones,cefepime, and aminoglycosides (79, 137, 240, 263, 283).
Multiple pathways also participate in the regulation ofmexXY induction. Interaction of bacterial ribosomes with pro-tein synthesis inhibitors (e.g., tetracycline, chloramphenicol,and aminoglycosides) is necessary to upregulate mexXY expres-sion, as plasmid-encoded ribosomal protection mechanismsreduce mexXY drug inducibility (93). Induction may not alwaysrequire the presence of MexZ (93, 163). Inducing antibioticshad no effect on the binding of MexZ to its operator (154) andcould further enhance mexXY expression in a mexZ-deficientmutant (93), suggesting that mexXY induction was independentof MexZ. A more recent study by Morita et al. implicated thePA5471 gene in mediating mexXY induction (163). Disruptionof PA5471 compromised mexXY drug inducibility, while in-creased expression of PA5471 alone stimulated mexXY expres-sion. PA5471 transcription is also induced by ribosome inhib-itors (the same drugs which induce mexXY), and PA5471induction indirectly or directly alters MexZ activity, but notmexZ expression, resulting in mexXY upregulation (163).
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 599
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

FIG. 7. Coregulation of mexEF-oprN and oprD in P. aeruginosa. The models represent the proposed mechanisms of coregulation of mexEF-oprN andoprD in P. aeruginosa. Each panel represents the chromosome of P. aeruginosa, highlighting the mexE, mexF, oprN, and oprD structural genes and theproposed genes involved in coregulation, mexT, mexS, and mvaT. (A) Basal expression of mexEF-oprN and oprD in wild-type P. aeruginosa. In wild-typeP. aeruginosa, MexT is functionally inactive due to either the presence of suppressing mutations or the lack of a secondary effector molecule. As a result,expression of mexEF-oprN occurs at a low basal level, and expression of oprD occurs at a level sufficient to provide quantities of OprD in the outermembrane sufficient for normal cellular function. (B) MexT-associated coregulation of mexEF-oprN and oprD. In nfxC-type mutants, MexT becomesactive through a mutation within the structural gene. The activated MexT protein positively regulates (green arrow) transcription of mexEF-oprN, leadingto overexpression of the efflux operon and overproduction of the MexEF-OprN efflux pump. Simultaneously, MexT negatively regulates (red arrow) oprDat the transcriptional and posttranscriptional levels, leading to decreased production of OprD. (C) MexS-associated coregulation of mexEF-oprN andoprD. Loss of MexS, a putative oxidoreductase/dehydrogenase, has been suggested to cause a buildup of secondary metabolites which may serve aseffector molecules for MexT. These effector molecules could bind to MexT, alter the conformational state of the regulatory protein, and transform MexTinto an activating transcriptional regulator. As a result, MexT can positively regulate (green arrow) the expression of mexEF-oprN and negatively regulate(red arrow) the expression of oprD, similar to what is described for panel B. (D) MvaT-associated coregulation of mexEF-oprN and oprD. Loss of theglobal regulatory protein MvaT is also associated with the upregulation of the mexEF-oprN operon. The mechanism of MvaT-associated regulation hasnot been elucidated, but it functions independent of MexT and MexS. In contrast to the case for the MexT- and MexS-associated regulatory pathways,loss of MvaT causes an upregulation of both mexEF-oprN and oprD expression.
600
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

MexJK Efflux Pump
The MexJK efflux pump was discovered in a �mexAB-oprM�mexCD-oprJ background strain following selection with thebroad-spectrum biocide triclosan (33). An OMF-encodinggene is also absent from the mexJK operon (Fig. 6), but athree-component system is formed with the addition of eitherOprM (32, 33) or OpmH (32). MexJK-OprM exports erythro-mycin and tetracycline from the cell, while MexJK-OpmH re-moves triclosan (32). mexJK was reported not to be expressedin wild-type cells (33).
A member of the TetR regulatory family, mexL, is presentupstream of mexJK but transcribed in the opposite orientation(Fig. 6) (33). MexL binds as a multimer to inverted repeatswithin the mexL-mexJ intergenic region encompassing overlap-ping mexJK and mexL promoters (31). MexL binding to thissite represses the transcription of both mexJK and mexL,thereby negatively autoregulating its own expression. A muta-tion within the helix-turn-helix motif of MexL prevented bind-ing to its operator site and led to mexJK overexpression in alaboratory mutant (31). Overexpression of mexJK has also beendetected in a few clinical isolates, but the pump’s contributionto resistance and potential mutations in mexL were not eval-uated (79).
Additional RND Efflux Pumps
The contributions of the remaining RND efflux pumps toresistance and the factor(s) governing their expression havejust begun to be elucidated. Their up- or downregulation inclinical isolates with various phenotypes has yet to be reported.mexGHI-opmD expression is detectable in wild-type cells andis under the control of a redox-active protein, SoxR, belongingto the MerR family of transcriptional regulators (4, 185). Nor-floxacin was the only antibiotic reported to be a substrate ofMexGHI-OpmD (236), but this pump was also implicated inthe export of other nonantibiotic compounds, including vana-dium (4). Interestingly, a mutant incapable of expressingmexGHI-opmD showed decreased susceptibility to tetracyclineand ticarcillin-clavulanate (4). mexI and opmD knockout mu-tants in a subsequent study also exhibited decreased suscepti-bility to kanamycin, spectinomycin, carbenicillin, nalidixic acid,tetracycline, and chloramphenicol (3). Perhaps the loss ofmexGHI-opmD triggered the overexpression of another RNDefflux pump, similar to the overexpression of mexCD-oprJ andmexEF-oprN observed in P. aeruginosa knockout mutants ofmexAB-oprM (119).
The substrate profiles of MexVW, MexPQ-OpmE, andMexMN were all examined in an efflux-deficient strain(�mexAB-oprM �mexCD-oprJ �mexEF-oprN �mexXY) harbor-ing a plasmid coding for each system individually (122, 158).MexVW used OprM and other, as yet unidentified OMF pro-teins for the export of fluoroquinolones, tetracycline, chloram-phenicol, and erythromycin (122). MexPQ-OpmE elevated theMICs of fluoroquinolones, tetracycline, chloramphenicol, andseveral macrolides in the recipient strain (158). Susceptibilityto chloramphenicol and thiamphenicol was decreased in therecipient containing MexMN in combination with OprM (158).
TriABC was the last RND efflux pump to be characterizedfor P. aeruginosa. triABC differs from the other RND operons
in that triA and triB each code for MFPs, both of which arerequired for substrate export (157). The OMF OpmH associ-ates with TriABC to assemble a functional pump capable ofexporting triclosan (157). No antibiotics were reported as sub-strates for this pump.
COREGULATION OF RESISTANCE MECHANISMS
MexCD-OprJ Overproduction and Hypersusceptibilityto Antibacterials
While the overexpression of mexCD-oprJ provides resistanceto several classes of antibacterial drugs, hypersusceptibility toaminoglycosides and other �-lactams is also observed (149,203). Hypersusceptibility is defined as a fourfold or greaterincrease in susceptibility, and the level of hypersusceptibility isrelated to the level of MexCD-OprJ production. Masuda et al.demonstrated that type B mutants (most production ofMexCD-OprJ) exhibit larger increases in susceptibility than dotype A mutants (moderate production of MexCD-OprJ) (149).However, the mechanism(s) responsible for hypersusceptibilityis much more complex than simply being associated with pro-duction of MexCD-OprJ. Phenotypic reversion studies withmexCD-oprJ-overexpressing mutants have demonstrated thathypersusceptibility to some antibiotics is not always associatedwith the level of mexCD-oprJ overexpression (280). Further-more, as discussed below, a single mechanism does not accountfor hypersusceptibility to all affected antibiotics, and the mech-anism(s) responsible for hypersusceptibility to imipenem re-mains uncharacterized.
Hypersusceptibility of laboratory-generated nfxB-type mu-tants to carbenicillin and aztreonam (MexAB-OprM sub-strates) has previously been linked to a concurrent downregu-lation of MexAB-OprM production (70). However, during theanalysis of nfxB-type clinical isolates, mexB transcript andMexB protein levels were not downregulated, despite hyper-susceptibility to these drugs (92). Instead, a decrease inMexAB-OprM pump activity in these strains was proposed asthe mechanism (92). These results do not necessarily exclude apossible coregulation of mexCD-oprJ and oprM expression,since transcription of oprM can occur independently of that ofmexAB (290).
MexCD-OprJ-overproducing mutants become hypersuscep-tible to additional �-lactams, including sulbenicillin, cefpo-doxime, ceftriaxone, imipenem, and biapenem (151), but noneof the aforementioned drugs are exported by MexAB-OprM,implying an additional mechanism(s). A disruption in the in-duction pathway of AmpC was suggested to cause hypersus-ceptibility to these �-lactams (151). Although data from ourown studies also showed decreased induction of ampC expres-sion associated with overexpression of mexCD-oprJ (274), thisassociation was observed only in isogenic mutants from a clin-ical isolate with wild-type ampC expression. When the parentstrains were derepressed for ampC expression, overexpressionof mexC was not associated with any change in ampC expres-sion among the isogenic mutants selected (274).
The mechanism(s) involved in hypersusceptibility to imi-penem remains uncharacterized. Although other investiga-tors have suggested that imipenem hypersusceptibility is aresult of disrupted ampC induction (151), the observation
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 601
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

that AmpC overproduction does not significantly increaseimipenem MICs (63, 131, 169, 274) argues against this hy-pothesis. Furthermore, our own studies have shown thatimipenem-hypersusceptible mutants can be selected from P.aeruginosa parent strains that are partially and fully dere-pressed for ampC expression, without any changes in eitherthe expression of ampC or AmpC hydrolysis activity (274).These studies also demonstrated that imipenem hypersus-ceptibility is unrelated to the level of OprD, as hypersus-ceptible mutants failed to exhibit any changes in oprD ex-pression or OprD protein in the outer membrane (274).Furthermore, imipenem-hypersusceptible mutants were se-lected from a P. aeruginosa strain that lacked a functionalOprD protein (274). Characterization of the mechanism(s)responsible for imipenem hypersusceptibility amongmexCD-oprJ-overexpressing P. aeruginosa strains may un-cover potential drug targets to enhance the activity of car-bapenems.
The mechanism responsible for hypersusceptibility to amino-glycosides remains unknown. As previously discussed, amino-glycosides are substrates for the MexXY efflux pump, andsuppression of MexXY in strains overproducing MexCD-OprJhas been suggested (205), but this inverse relationship has notbeen observed in clinical isolates (92).
Coregulation of MexEF-OprN and OprD
One of the most intriguing and complex coregulatory path-ways involves the concurrent overproduction of the MexEF-OprN efflux pump and downregulation/upregulation of theOprD porin. Clinically, this coregulatory process allows a sin-gle mutational process to impact the potencies of both thefluoroquinolones and carbapenems. To date, three proteins,MexT, MexS, and MvaT, have been linked directly or indirectlyto the coregulation of MexEF-OprN and OprD in P. aerugi-nosa (Fig. 7).
P. aeruginosa strains that overexpress mexEF-oprNthrough MexT become resistant to antibiotics which aresubstrates for the pump (listed above) but also lose suscep-tibility to imipenem, which is not extruded by MexEF-OprN.Rather, the loss of susceptibility to imipenem is associatedwith a concurrent decrease of oprD expression and OprD(104, 150, 180). The link between pump hyperexpressionand porin loss involves coregulation by MexT (Fig. 7B).Studies have shown that MexT alone is capable of down-regulating oprD at the transcriptional and posttranscrip-tional levels, causing a significant reduction in the amount ofOprD (101, 180). Further study into this mechanism of co-regulation has shown that neither premature termination oftranscription nor changes in transcript stability are respon-sible for the reduced levels of oprD transcript (278). Instead,the MexT-associated decrease in oprD expression involvesdecreased transcription initiation from the start-site-proxi-mal oprD promoter (Fig. 8) (278). It is possible that MexT oranother regulatory factor(s) and/or cofactors bind to theregion between SS1 and SS2 and inhibit transcription initi-ation from SS1.
Mutations within mexS, encoding a putative oxidoreduc-tase/dehydrogenase, are also associated with concurrent
overexpression of mexEF-oprN and downregulation of oprDexpression (Fig. 7C) (241). As discussed above for regula-tion of MexEF-OprN, inactivation of MexS is believed tocause a buildup of metabolites that serve as effector mole-cules for MexT (241), which mediates the downregulation ofoprD expression following activation. In contrast to MexT-and MexS-associated downregulation of oprD expression,inactivation of the global regulator MvaT causes an upregu-lation in both mexEF-oprN and oprD transcription (Fig. 7D)(270). However, the mechanistic pathway responsible foroprD upregulation has not been characterized.
Regulation of mexEF-oprN expression by MexT (with orwithout mexS inactivation) and MvaT can impact suscepti-bility to imipenem through oprD coregulation. However,coregulation of mexEF-oprN and oprD is not always ob-served, further highlighting the complexity of resistancemechanism regulation in P. aeruginosa. For example, in arecent study, an isogenic mutant overexpressing mexEF-oprN showed no phenotypic change in imipenem suscepti-bility, oprD transcript levels, or levels of OprD in the outermembrane (279). Sequence and expression analysis of theregulatory genes mexT, mexS, and mvaT failed to show anygenetic changes in the mutant, suggesting an alternativepathway of mexEF-oprN regulation without concurrent oprDregulation (279). Reminiscent of increased mexCD-oprJ ex-pression in mexAB-oprM and oprM knockout mutants,mexEF-oprN was also hyperexpressed in mexAB-oprM-inac-tivated mutants, but a change in imipenem susceptibility wasnot observed in the deletion mutants (119).
FIG. 8. MexT-associated downregulation of oprD expression. Tran-scription of oprD in wild-type P. aeruginosa PAO1 initiates from twostart sites, SS1 and SS2. MexT-associated downregulation of oprDexpression is associated with a selective inhibition of transcription fromSS1 (278). The model proposed in this figure involves the binding of aregulatory protein, potentially MexT, with or without cofactors, withinthe promoter region between SS1 and SS2, blocking efficient initiationof transcription from SS1.
602 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

PREVENTING EMERGENCE OF CHROMOSOMALLYENCODED RESISTANCE
This review has highlighted the impressive ability of P.aeruginosa to develop antibacterial resistance through mu-tational changes in the function and/or production of chro-mosomally encoded resistance mechanisms. Furthermore,the most difficult challenge with this pathogen is the abilityof P. aeruginosa to become resistant during treatment of aninfection. Unfortunately, the prospect for bringing new, andspecifically novel, antipseudomonal drugs to clinical use inthe near future is not promising. Therefore, the challengefacing us today is to slow the emergence of resistancethrough optimizing therapy with currently available drugs.
The two most common strategies considered to address thisneed are (i) optimizing therapy through our understanding ofbasic antibacterial pharmacodynamic principles and (ii) treat-ing P. aeruginosa with a combination of antibacterial drugs.Although our understanding of antibacterial pharmacodynam-ics can help in the selection of the best antibiotic and/or dosingstrategy to optimize therapy, preventing the emergence of re-sistance requires the inclusion of potential resistant subpopu-lations into the equation. Unfortunately, this is not alwaysstraightforward, since the shifts in susceptibility associated withmutations can vary widely depending upon the resistancemechanism involved, the differential impact of resistancemechanisms on specific antibiotics, and the potential cooper-ation of multiple resistance mechanisms. Therefore, pharma-codynamic optimization based upon the susceptibility of theoriginal clinical isolate does not always address the risk ofresistance emerging during therapy. Some investigators havepromoted replacing the MIC with the mutation preventionconcentration, but this approach has not been adopted byclinical laboratories.
A more accepted approach is to treat serious P. aeruginosainfections with a combination of antibacterial agents. Althoughsynergistic interactions are an important aspect for some drugcombinations (e.g., trimethoprim-sulfamethoxazole), the pri-mary focus of combination therapy against P. aeruginosa ispreventing the emergence of resistance. The combination of anantipseudomonal �-lactam with an aminoglycoside has oftenbeen the treatment of choice for this pathogen. However, thiscombination does not always prevent the emergence of AmpC-mediated resistance to the �-lactams, and clinical failures arestill a risk (97, 118, 147, 226, 228, 235). Therefore, the searchfor more effective combinations must be a priority. The com-bination of levofloxacin-imipenem has been shown to preventthe emergence of resistance during “therapy” of P. aeruginosain a two-compartment pharmacodynamic model (129, 130). Inthese in vitro studies, the levofloxacin-imipenem combinationprevented emergence of resistance in susceptible clinical iso-lates, as well as in strains with characterized mechanisms thatprovided resistance to one or both drugs in the combination.Although these two studies focused specifically on levofloxa-cin-imipenem, it is expected that ciprofloxacin, meropenem,and doripenem could be used as alternatives in this combina-tion without compromising efficacy. At this time, clinical eval-uation of the combination is needed.
CONCLUDING COMMENTS
Treatment of infectious diseases becomes more challengingwith each passing year. Continued increases in immunosup-pressed/compromised patient populations and the evolu-tionary advantage of bacteria to rapidly mutate and adapt toantibacterial/biocide threats in their environment make thetreatment of infectious diseases a serious challenge. This isespecially true for the opportunistic pathogen P. aeruginosaand its ability to develop a multidrug-resistant phenotype. Al-though the potential import of resistance mechanisms on mo-bile genetic elements is a continuing threat, perhaps the mostdifficult challenge we face with P. aeruginosa is its ability torapidly develop resistance to multiple classes of antibioticsduring the course of treating a patient. The chromosomalAmpC cephalosporinase, the outer membrane porin OprD,and the multitude of efflux pumps are particularly relevant tothis therapeutic challenge, and the discussion presented in thisreview highlights the complex mechanisms and pathways bywhich P. aeruginosa regulates and/or coregulates their expres-sion. As the pipeline of new drugs achieving FDA approvalcontinues to diminish, it is critical that we look for novelstrategies to combat the threat of antibacterial resistance.
One potential strategy is to target the regulation of bacterialresistance mechanisms as a pathway to enhance the potency ofavailable drugs and, perhaps, restore the efficacy of availabledrugs. In addition to the development of direct inhibitors ofresistance mechanisms, i.e., �-lactamase inhibitors, anotherstrategy is to target the regulation of gene expression. Al-though a great deal of knowledge has been gained towardunderstanding the mechanisms by which P. aeruginosa regu-lates AmpC, OprD, and efflux pumps, it is clear that we havea long road ahead and have much to learn about how thisresourceful pathogen coregulates different resistance mecha-nisms to meet the antibacterial challenges it faces.
ACKNOWLEDGMENTS
Daniel Wolter created Fig. 2 through 8. We acknowledge PatrickLane for his enhancement of the figures.
Philip Lister received research grant support from the NationalInstitutes of Health (5R21AI070188-02), Ortho-McNeil, Merck,Wyeth-Ayerst, and Astra-Zeneca. Nancy Hanson received researchgrant support from the National Institutes of Health (5R21AI070188-02), Merck, Astra-Zeneca, Johnson & Johnson, and Becton-Dickin-son.
REFERENCES
1. Abdalhamid, B., P. A. Wickman, and N. D. Hanson. 2005. Correlation ofampC induction with PBP binding in Enterobacter cloacae, abstr. C1-2211.Abstr. 45th Intersci. Conf. Antimicrob. Agents Chemother., Washington,DC.
2. Adewoye, L., A. Sutherland, R. Srikumar, and K. Poole. 2002. The mexRrepressor of the mexAB-oprM multidrug efflux operon in Pseudomonasaeruginosa: characterization of mutations compromising activity. J. Bacte-riol. 184:4308–4312.
3. Aendekerk, S., S. P. Diggle, Z. Song, N. Hoiby, P. Cornelis, P. Williams, andM. Camara. 2005. The MexGHI-OpmD multidrug efflux pump controlsgrowth, antibiotic susceptibility and virulence in Pseudomonas aeruginosavia 4-quinolone-dependent cell-to-cell communication. Microbiology 151:1113–1125.
4. Aendekerk, S., B. Ghysels, P. Cornelis, and C. Baysse. 2002. Characteriza-tion of a new efflux pump, MexGHI-OpmD from Pseudomonas aeruginosathat confers resistance to vanadium. Microbiology 148:2371–2381.
5. Aires, J. R., T. Kohler, H. Nikaido, and P. Plesiat. 1999. Involvement of anactive efflux system in the natural resistance of Pseudomonas aeruginosa toaminoglycosides. Antimicrob. Agents Chemother. 43:2624–2628.
6. Akasaka, J. R., M. Tanaka, A. Yamaguchi, and K. Sato. 2001. Type II
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 603
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

topoisomerase mutations in fluoroquinolone-resistant clinical strains ofPseudomonas aeruginosa isolated in 1998 and 1999: role of target enzyme inmechanism of fluoroquinolone resistance. Antimicrob. Agents Chemother.45:2263–2268.
7. Aloush, V., S. Navon-Venezia, Y. Seigman-Igra, S. Cabili, and Y. Carmeli.2006. Multidrug-resistant Pseudomonas aeruginosa: risk factors and clinicalimpact. Antimicrob. Agents Chemother. 50:43–48.
8. Avison, M. B., P. Niumpsup, K. Nurmahomed, T. R. Walsh, and P. M.Bennett. 2004. Role of the ‘cre/blr-tag’ DNA sequence in regulation of geneexpression by the Aeromonas hydrophila beta-lactamase regulator, BlrA. J.Antimicrob. Chemother. 53:197–202.
9. Bagge, N., O. Ciofu, M. Hentzer, J. I. Cambell, M. Givskov, and N. Hoiby.2002. Constitutive high expression of chromosomal beta-lactamase inPseudomonas aeruginosa caused by a new insertion sequence (IS1669) lo-cated in ampD. Antimicrob. Agents Chemother. 46:3406–3411.
10. Barnaud, G., R. Labia, L. Raskine, M. J. Sanon-Le Pors, A. Philippon, and G.Arlet. 2001. Extension of resistance to cefepime and cefpirome associated to asix amino acid deletion in the H-10 helix of the cephalosporinase of an Enter-obacter cloacae clinical isolate. FEMS Microbiol. Lett. 195:185–190.
11. Bartowsky, E., and S. Normark. 1993. Interactions of wild-type and mutantAmpR of Citrobacter freundii with target DNA. Mol. Microbiol. 10:555–565.
12. Bartowsky, E., and S. Normark. 1991. Purification and mutant analysis ofCitrobacter freundii AmpR, the regulator for chromosomal AmpC beta-lactamase. Mol. Microbiol. 5:1715–1725.
13. Bellido, F., J. C. Pechere, and R. E. W. Hancock. 1991. Reevaluation of thefactors involved in the efficacy of new beta-lactams against Enterobactercloacae. Antimicrob. Agents Chemother. 35:73–78.
14. Bert, F., C. Branger, and N. Lambert-Zechovsky. 2002. Identification of PSEand OXA beta-lactamase genes in Pseudomonas aeruginosa using PCR-restric-tion fragment length polymorphism. J. Antimicrob. Chemother. 50:11–18.
15. Bisbe, J., J. M. Gatell, and J. Puig. 1988. Pseudomonas aeruginosa bacte-remia: univariate and multivariate analyses of factors influencing the prog-nosis in 133 episodes. Rev. Infect. Dis. 10:629–635.
16. Bishop, R. E., and J. H. Weiner. 1993. Overproduction, solubilization,purification, and DNA-binding properties of AmpR from Citrobacter freun-dii. Eur. J. Biochem. 213:405–412.
17. Blanc, D. S., C. Petignat, B. Janin, J. Bille, and P. Fancioli. 1998. Fre-quency and molecular diversity of Pseudomonas aeruginosa upon admissionand during hospitalization: a prospective epidemiologic study. Clin. Micro-biol. Infect. 4:242–247.
18. Bonten, M. J., D. C. Bergmans, H. Speijer, and E. E. Stobberingh. 1999.Characteristics of polyclonal endemicity of Pseudomonas aeruginosa colo-nization in intensive care units. Implications for infection control. Am. J.Crit. Care Med. 160:1212–1219.
19. Boutoille, D., S. Corvec, N. Caroff, C. Giraudeau, E. Espaze, J. Caillon, P.Plesiat, and A. Reynaud. 2004. Detection of an IS21 insertion sequence inthe mexR gene of Pseudomonas aeruginosa increasing beta-lactam resis-tance. FEMS Microbiol. Lett. 230:143–146.
20. Breidenstein, E. B. M., B. K. Khaira, I. Wiegand, J. Overhage, and R. E.Hancock. 2008. Complex ciprofloxacin resistome revealed by screening aPseudomonas aeruginosa mutant library for altered susceptibility. Antimi-crob. Agents Chemother. 52:4486–4491.
21. Brown, S. D., and M. M. Traczewski. 2005. Comparative in vitro antimicro-bial activity of a new carbapenem, doripenem: tentative disc diffusion cri-teria and quality control. J. Antimicrob. Chemother. 55:944–949.
22. Caille, O., C. Rossier, and K. Perron. 2007. A copper-activated two-com-ponent system interacts with zinc and imipenem resistance in Pseudomonasaeruginosa. J. Bacteriol. 189:4561–4568.
23. Campbell, J. I. A., O. Cioufu, and N. Hoiby. 1997. Pseudomonas aeruginosaisolates from patients with cystic fibrosis have different beta-lactamaseexpression phenotypes but are homogenous in the ampC-ampR geneticregion. Antimicrob. Agents Chemother. 41:1380–1384.
24. Cao, L., R. Srikumar, and K. Poole. 2004. MexAB-OprM hyperexpressionin NalC-type multidrug-resistant Pseudomonas aeruginosa: identificationand characterization of nalC gene encoding a repressor of PA3720-PA3719.Mol. Microbiol. 53:1423–1436.
25. Carmeli, Y., N. Troillet, A. W. Karchmer, and M. H. Samore. 1999. Healthand economic outcomes of antibiotic resistant Pseudomonas aeruginosa.Arch. Intern. Med. 159:1127–1132.
26. Castang, S., H. R. McManus, K. H. Turner, and S. L. Dove. 2008. H-NSfamily members function coordinately in an opportunistic pathogen. Proc.Natl. Acad. Sci. USA 105:18947–18952.
27. Castanheira, M., R. E. Mendes, T. R. Walsh, A. C. Gales, and R. N. Jones.2004. Emergence of the extended-spectrum beta-lactamase GES-1 in a Pseudo-monas aeruginosa strain from Brazil: report from the Sentry antimicrobialsurveillance program. Antimicrob. Agents Chemother. 48:2344–2345.
28. Castanheira, M., M. A. Toleman, R. N. Jones, F. J. Schmidt, and T. R.Walsh. 2004. Molecular characterization of a beta-lactamase gene,blaGIM-1, encoding a new subclass of metallo-beta-lactamase. Antimicrob.Agents Chemother. 48:4654–4661.
29. Chen, H., J. Hu, P. R. Chen, L. Lan, Z. Li, L. M. Hicks, A. R. Dinner, andC. He. 2008. The Pseudomonas aeruginosa multidrug efflux regulator MexR
uses an oxidation-sensing mechanism. Proc. Natl. Acad. Sci. USA 105:13586–13591.
30. Cheng, Q., and J. T. Park. 2002. Substrate specificity of the AmpG per-mease required for recycling of cell wall anhydro-muropeptides. J. Bacte-riol. 184:6434–6436.
31. Chuanchuen, R., J. B. Gaynor, R. Karkhoff-Schweizer, and H. P. Schweizer.2005. Molecular characterization of MexL, the transcriptional repressor ofthe mexJK multidrug efflux operon in Pseudomonas aeruginosa. Antimicrob.Agents Chemother. 49:1844–1851.
32. Chuanchuen, R., T. Murata, N. Gotoh, and H. P. Schweizer. 2005. Substrate-dependent utilization of OprM and OpmH by the Pseudomonas aeruginosaMexJK efflux pump. Antimicrob. Agents Chemother. 49:2133–2136.
33. Chuanchuen, R., C. T. Narasaki, and H. P. Schweizer. 2002. The MexJKefflux pump of Pseudomonas aeruginosa requires OprM for antibiotic effluxbut not for efflux of triclosan. J. Bacteriol. 184:5036–5044.
34. Colom, K., A. Fdz-Aranguiz, E. Suinaga, and R. Cisterna. 1995. Emergenceof resistance to beta-lactam agents in Pseudomonas aeruginosa with group1 beta-lactamases in Spain. Eur. J. Clin. Microbiol. Infect. Dis. 14:964–971.
35. Daigle, D. M., L. Cao, S. Fraud, M. S. Wilke, A. Pacey, R. Klinoski, N. C.Strynadka, C. R. Dean, and K. Poole. 2007. Protein modulator of multidrugefflux gene expression in Pseudomonas aeruginosa. J. Bacteriol. 189:5441–5451.
36. Danel, F., L. M. Hall, D. Gur, H. E. Akalin, and D. M. Livermore. 1995.Transferable production of PER-1 beta-lactamase in Pseudomonas aerugi-nosa. J. Antimicrob. Chemother. 35:281–294.
37. Dean, C. R., M. A. Visalli, S. J. Projan, P. E. Sum, and P. A. Bradford. 2003.Efflux-mediated resistance to tigecycline (GAR-936) in Pseudomonasaeruginosa PAO1. Antimicrob. Agents Chemother. 47:972–978.
38. De Kievit, T. R., M. D. Parkins, R. J. Gillis, R. Srikumar, H. Ceri, K. Poole,B. H. Iglewski, and D. G. Storey. 2001. Multidrug efflux pumps: expressionpatterns and contribution to antibiotic resistance in Pseudomonas aerugi-nosa biofilms. Antimicrob. Agents Chemother. 45:1761–1770.
39. Dietz, H., D. Pfeifle, and B. Wiedemann. 1996. Location of N-acetyl-muramyl-L-alanyl-D-glutamylmesodiaminopimelic acid, presumed signalmolecule for beta-lactamase induction, in the bacterial cell. Antimicrob.Agents Chemother. 40:2173–2177.
40. Dietz, H., D. Pfeifle, and B. Wiedemann. 1997. The signal molecule forbeta-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob. Agents Chemother. 41:2113–2120.
41. Dimatatac, E. L., M. M. Alejandria, C. Montalban, C. Pineda, C. Ang, and R.Delino. 2003. Clinical outcomes and costs of care of antibiotic resistant Pseudo-monas aeruginosa infections. Philipp. J. Microbiol. Infect. Dis. 32:159–167.
42. Doi, Y., D. de Oliveira Garcia, J. Adams, and D. Paterson. 2007. Copro-duction of novel 16S rRNA methylase RmtD and metallo-beta-lactamaseSPM-1 in a panresistant Pseudomonas aeruginosa isolate from Brazil. An-timicrob. Agents Chemother. 51:852–856.
43. Doi, Y., K. Yokoyama, K. Yamane, J. Wachino, N. Shibata, T. Yagi, K.Shibayama, H. Kato, and Y. Arakawa. 2004. Plasmid-mediated 16S rRNAmethylase in Serratia marcescens conferring high-level resistance to amin-oglycosides. Antimicrob. Agents Chemother. 48:491–496.
44. Dotsch, A., T. Becker, C. Pommerenke, Z. Magnowska, L. Jansch, and S.Haussler. 2009. Genomewide identification of genetic determinants of an-timicrobial drug resistance in Pseudomonas aeruginosa. Antimicrob. AgentsChemother. 53:2522–2531.
45. Ehrhardt, A. F., and C. C. Sanders. 1993. Beta-lactam resistance amongstEnterobacter species. J. Antimicrob. Chemother. 32(Suppl. B):1–11.
46. Ehrhardt, A. F., C. C. Sanders, J. R. Romero, and J. S. Leser. 1996.Sequencing and analysis of four new Enterobacter ampD alleles. Antimi-crob. Agents Chemother. 40:1953–1956.
47. El Amin, N., C. G. Giske, S. Jalal, B. Keijser, G. Kronvall, and B. Wretlind.2005. Carbapenem resistance mechanisms in Pseudomonas aeruginosa: al-terations of porin OprD and efflux proteins do not fully explain resistancepatterns observe in clinical isolates. APMIS 113:187–196.
48. Emori, T. G., and R. P. Gaynes. 1993. An overview of nosocomial infec-tions, including the role of the microbiology laboratory. Clin. Microbiol.Rev. 6:428–442.
49. Erol, S., U. Altoparlak, M. N. Akcay, F. Celebi, and M. Parlak. 2004.Changes of microbial flora and wound colonization in burned patients.Burns 30:357–361.
50. Eron, L. J., R. I. Goldenberg, D. M. Poretz, and C. H. Park. 1983. Pip-eracillin therapy for Pseudomonas infections. South. Med. J. 76:859–862.
51. Evans, J. C., and H. Segal. 2007. A novel insertion sequence, ISPA26, inoprD of Pseudomonas aeruginosa is associated with carbapenem resistance.Antimicrob. Agents Chemother. 51:3776–3777.
52. Evans, K., L. Adewoye, and K. Poole. 2001. MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa: identification ofMexR binding sites in the mexA-mexR intergenic region. J. Bacteriol. 183:807–812.
53. Evans, K., and K. Poole. 1999. The MexA-MexB-OprM multidrug effluxsystem of Pseudomonas aeruginosa is growth-phase regulated. FEMS Mi-crobiol. Lett. 173:35–39.
54. Flamm, R. K., M. K. Weaver, C. Thornsberry, M. E. Jones, J. A. Karlowsky,and D. F. Sahm. 2004. Factors associated with relative rates of antibiotic
604 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

resistance in Pseudomonas aeruginosa isolates tested in clinical laboratoriesin the United States from 1999 to 2002. Antimicrob. Agents Chemother.48:2431–2436.
55. Fraud, S., A. J. Campigotto, Z. Chen, and K. Poole. 2008. MexCD-OprJmultidrug efflux system of Pseudomonas aeruginosa: involvement in chlo-rhexidine resistance and induction by membrane-damaging agents depen-dent upon the AlgU stress response sigma factor. Antimicrob. AgentsChemother. 52:4478–4482.
56. Freeman, L. 1916. Chronic general infection with the Bacillus pyocyaneus.Ann. Surg. 64:195–202.
57. Fritsche, T. R., H. S. Sader, M. A. Toleman, T. R. Walsh, and R. N. Jones.2005. Emerging metallo-beta-lactamase-mediated resistances: a summaryreport from the worldwide SENTRY antimicrobial surveillance program.Clin. Infect. Dis. 41(Suppl. 4):S276–S278.
58. Fukuda, H., M. Hosaka, S. Iyobe, N. Gotoh, T. Nishino, and K. Hirai. 1995.nfxC-type quinolone resistance in a clinical isolate of Pseudomonas aerugi-nosa. Antimicrob. Agents Chemother. 39:790–792.
59. Gales, A. C., L. C. Menezes, S. Silbert, and H. S. Sader. 2003. Dissemina-tion in distinct Brazilian regions of an epidemic carbapenem-resistantPseudomonas aeruginosa producing SPM metallo-beta-lactamase. J. Anti-microb. Chemother. 52:699–702.
60. Galimand, M., P. Courvalin, and T. Lambert. 2003. Plasmid-mediatedhigh-level resistance to aminoglycosides in Enterobacteriaceae due to 16SrRNA methylation. Antimicrob. Agents Chemother. 47:2565–2571.
61. Galimand, M., P. Sabtcheva, P. Courvalin, and T. Lambert. 2005. Worldwidedisseminated armA aminoglycoside resistance methylase gene borne by com-posite transposon Tn1548. Antimicrob. Agents Chemother. 49:2949–2953.
62. Gasink, L. B., N. O. Fishman, M. G. Weiner, I. Nachamkin, W. B. Bilker,and E. Lautenbach. 2006. Fluoroquinolone-resistant Pseudomonas aerugi-nosa: assessment of risk factors and clinical impact. Am. J. Med. 119:526e19–526e25.
63. Gates, M. L., C. C. Sanders, R. V. Goering, and W. E. Sanders. 1986.Evidence for multiple forms of type I chromosomal beta-lactamase inPseudomonas aeruginosa. Antimicrob. Agents Chemother. 30:453–457.
64. Gaynes, R., and J. R. Edwards. 2005. Overview of nosocomial infectionscaused by gram-negative bacilli. Clin. Infect. Dis. 41:848–854.
65. Gessard, C. 1984. Classics in infectious diseases. On the blue and green col-oration that appears on bandages. Rev. Infect. Dis. 6(Suppl. 3):S775–S776.
66. Gillis, R. J., K. G. White, K. H. Choi, V. E. Wagner, H. P. Schweizer, andB. H. Iglewski. 2005. Molecular basis of azithromycin-resistant Pseudomo-nas aeruginosa biofilms. Antimicrob. Agents Chemother. 49:3858–3867.
67. Girlich, D., T. Naas, and P. Nordmann. 2004. Biochemical characterizationof the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa.Antimicrob. Agents Chemother. 48:2043–2048.
68. Gonzalez-Zorn, B., A. Catalan, J. A. Escudero, L. Dominguez, T. Teshager,C. Porrero, and M. A. Moreno. 2005. Genetic basis for dissemination ofarmA. J. Antimicrob. Chemother. 56:583–585.
69. Goodell, E. W. 1985. Recycling of murein by Escherichia coli. J. Bacteriol.163:305–310.
70. Gotoh, N., H. Tsujimoto, M. Tsuda, K. Okamoto, A. Nomura, T. Wada, M.Nakahashi, and T. Nishino. 1998. Characterization of the MexC-MexD-OprJ multidrug efflux system in delta-mexA-mexB-oprM mutants of Pseudo-monas aeruginosa. Antimicrob. Agents Chemother. 42:1938–1943.
71. Gribble, M. J., A. W. Chow, S. C. Naiman, J. A. Smith, W. R. Bowie, S. L.Sacks, L. Grossman, N. Buskard, G. H. Growe, and L. H. Plenderleith. 1983.Prospective randomized trial of piperacillin monotherapy versus carboxypeni-cillin-aminoglycoside combination regimens in the empirical treatment of se-rious bacterial infections. Antimicrob. Agents Chemother. 24:388–393.
72. Grundstrom, T., and S. Normark. 1985. Initiation of translation makesattenuation of ampC in E. coli dependent on growth rate. Mol. Gen. Genet.198:411–415.
73. Gutierrez, O., C. Juan, E. Cercenado, F. Navarro, E. Bouza, P. Coll, J. L.Perez, and A. Oliver. 2007. Molecular epidemiology and mechanisms ofcarbapenem resistance in Pseudomonas aeruginosa isolates from Spanishhospitals. Antimicrob. Agents Chemother. 51:4329–4335.
74. Hancock, R. E. W. 1998. Resistance mechanisms in Pseudomonas aerugi-nosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis.27(Suppl. 1):S93–S99.
75. Hancock, R. E. W., and F. S. Brinkman. 2002. Function of pseudomonasporins in uptake and efflux. Annu. Rev. Microbiol. 56:17–38.
76. Hanson, N. D., and C. C. Sanders. 1999. Regulation of inducible AmpCbeta-lactamase expression among Enterobacteriaceae. Curr. Pharm. Des.5:881–894.
77. Harris, A. A., L. Goodman, and S. Levin. 1984. Community-acquiredPseudomonas aeruginosa pneumonia associated with the use of a homehumidifier. West. J. Med. 141:521–523.
78. Hiraoka, M., S. Masuyoshi, S. Mitsuhashi, K. Tomatsu, and M. Inoue.1988. Cephalosporinase interactions and antimicrobial activity of BMY-28142, ceftazidime, and cefotaxime. J. Antibiot. 41:86–93.
79. Hocquet, D., P. Nordmann, F. El Garch, L. Cabanne, and P. Plesiat. 2006.Involvement of the MexXY-OprM efflux system in emergence of cefepime
resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob.Agents Chemother. 50:1347–1351.
80. Holtje, J. V. 1998. Growth of the stress-bearing and shape-maintainingmurein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 62:181–203.
81. Holtje, J. V., U. Kopp, A. Ursinus, and B. Wiedemann. 1994. The negativeregulator of beta-lactamase induction AmpD is a N-acetyl-anhydro-muramyl-L-alanine amidase. FEMS Microbiol. Lett. 122:159–164.
82. Honore, N., M. H. Nicolas, and S. T. Cole. 1986. Inducible cephalosporinaseproduction in clinical isolates of Enterobacter cloacae is controlled by aregulatory gene that has been deleted from Escherichia coli. EMBO J.20:3709–3714.
83. Honore, N., M. H. Nicolas, and S. T. Cole. 1989. Regulation of enterobac-terial cephalosporinase production: the role of a membrane-bound sensorytransducer. Mol. Microbiol. 3:1121–1130.
84. Hooper, D. C. 2000. Mechanisms of action and resistance of older andnewer fluoroquinolones. Clin. Infect. Dis. 31:S24–S28.
85. Islam, S., S. Jalal, and B. Wretlind. 2004. Expression of MexXY effluxpump in amikacin-resistant isolates of Pseudomonas aeruginosa. Clin. Mi-crobiol. Infect. 10:877–883.
86. Jacobs, C., J. M. Frere, and S. Normark. 1997. Cytosolic intermediates forcell wall biosynthesis and degradation control inducible beta-lactam resis-tance in gram-negative bacteria. Cell 88:823–832.
87. Jacobs, C., L. J. Huang, E. Bartowsky, S. Normark, and J. T. Park. 1994.Bacterial cell wall recycling provides cytosolic muropeptides as effectors forbeta-lactamase induction. EMBO J. 13:4684–4694.
88. Jacobs, C., B. Joris, M. Jamin, K. Klarsov, J. Van Beeumen, D. Mengin-Lecreuix, J. van Heijenoort, J. T. Park, S. Normark, and J.-M. Frere. 1995.AmpD, essential for both beta-lactamase regulation and cell wall recycling,is a novel cytosolic N-acetylmuramyl-L-alanine amidase. Mol. Microbiol.15:533–539.
89. Jacoby, G. A., N. Chow, and K. B. Waites. 2003. Prevalence of plasmid-mediated quinolone resistance. Antimicrob. Agents Chemother. 47:559–562.
90. Jalal, S., O. Ciofu, N. Hoiby, N. Gotoh, and B. Wretlind. 2000. Molecularmechanisms of fluoroquinolone resistance in Pseudomonas aeruginosa isolatesfrom cystic fibrosis patients. Antimicrob. Agents Chemother. 44:710–712.
91. Jalal, S., and B. Wretlind. 1998. Mechanisms of quinolone resistance inclinical strains of Pseudomonas aeruginosa. Microb. Drug Resist. 4:257–261.
92. Jeannot, K., S. Elsen, T. Kohler, I. Attree, C. van Delden, and P. Plesiat.2008. Resistance and virulence of Pseudomonas aeruginosa clinical strainsoverproducing the MexCD-OprJ efflux pump. Antimicrob. Agents Che-mother. 52:2455–2462.
93. Jeannot, K., M. L. Sobel, F. El Garch, K. Poole, and P. Plesiat. 2005.Induction of the MexXY efflux pump in Pseudomonas aeruginosa is depen-dent on drug-ribosome interaction. J. Bacteriol. 187:5341–5346.
94. Jimenez-Lucho, V. E., L. D. Saravolatz, A. A. Meideros, and D. Pohlod.1986. Failure of therapy in Pseudomonas endocarditis: selection of resistantmutant. J. Infect. Dis. 154:64–68.
95. Jones, M. E., D. C. Draghi, C. Thornsberry, J. A. Karlowsky, D. F. Sahm,and R. P. Wenzel. 2004. Emerging resistance among bacterial pathogens inthe intensive care unit—a European and North American surveillancestudy (2000–2002). Ann. Clin. Microbiol. Antimicrob. 3:14–26.
96. Jones, R. N., D. J. Biedenbach, H. S. Sader, T. R. Fritsche, M. A. Toleman,and T. R. Walsh. 2005. Emerging epidemic of metallo-beta-lactamase-mediated resistances. Diagn. Microbiol. Infect. Dis. 51:77–84.
97. Juan, C., M. D. Macia, O. Gutierrez, C. Vidal, J. L. Perez, and A. Oliver.2005. Molecular mechanisms of beta-lactam resistance mediated by AmpChyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob.Agents Chemother. 49:4733–4738.
98. Juan, C., B. Moya, J. L. Perez, and A. Oliver. 2006. Stepwise upregulationof the Pseudomonas aeruginosa chromosomal cephalosporinase conferringhigh-level beta-lactam resistance involves three AmpD homologues. Anti-microb. Agents Chemother. 50:1780–1787.
99. Karlowsky, J. A., D. C. Draghi, M. E. Jones, C. Thornsberry, I. R. Fried-land, and D. F. Sahm. 2003. Surveillance for antimicrobial susceptibilityamong clinical isolates of Pseudomonas aeruginosa and Acinetobacter bau-mannii from hospitalized patients in the United States, 1998–2001. Anti-microb. Agents Chemother. 47:1681–1688.
100. Karlowsky, J. A., L. J. Kelly, C. Thornsberry, M. E. Jones, A. T. Evange-lista, I. A. Critchley, and D. F. Sahm. 2002. Susceptibility to fluoroquino-lones among commonly isolated gram-negative bacilli in 2000: TRUST andTSN data for the United States. Int. J. Antimicrob. Agents 19:21–31.
101. Kohler, T., S. F. Epp, L. K. Curty, and J. C. Pechere. 1999. Characterizationof MexT, the regulator of the MexE-MexF-OprN multidrug efflux system ofPseudomonas aeruginosa. J. Bacteriol. 181:6300–6305.
102. Kohler, T., M. Kok, M. Michea-Hamzehpour, P. Plesiat, T. Gotoh, T.Nishino, L. K. Curty, and J. C. Pechere. 1996. Multidrug efflux in intrinsicresistance to trimethoprim and sulfamethoxazole in Pseudomonas aerugi-nosa. Antimicrob. Agents Chemother. 40:2288–2290.
103. Kohler, T., M. Michea-Hamzehpour, S. F. Epp, and J. C. Pechere. 1999.Carbapenem activities against Pseudomonas aeruginosa: respective contribu-tions of OprD and efflux systems. Antimicrob. Agents Chemother. 43:424–427.
104. Kohler, T., M. Michea-Hamzehpour, U. Henz, N. Gotoh, L. K. Curty, and
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 605
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

J. C. Pechere. 1997. Characterization of MexE-MexF-OprN, a positivelyregulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Micro-biol. 23:345–354.
105. Kohler, T., C. van Delden, L. K. Curty, M. M. Hamzehpour, and J. C. Pechere.2001. Overexpression of the MexEF-OprN multidrug efflux system affectscell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 183:5213–5222.
106. Kolleff, M. H., A. Shorr, Y. P. Tabak, V. Gupta, L. Z. Liu, and R. S.Johannes. 2005. Epidemiology and outcomes of health-care-associatedpneumonia: results from a large US database of culture-positive pneumo-nia. Chest 128:3854–3862.
107. Kong, K.-F., S. R. Jayawardena, S. D. Indulkar, A. del Puerto, C.-L. Koh,N. Hoiby, and K. Mathee. 2005. Pseudomonas aeruginosa AmpR is a globaltranscriptional factor that regulates expression of AmpC and PoxB beta-lactamases, proteases, quorum sensing, and other virulence factors. Anti-microb. Agents Chemother. 49:4567–4575.
108. Kopp, U., B. Wiedemann, S. Linquist, and S. Normark. 1993. Sequences ofwild-type and mutant ampD genes of Citrobacter freundii and Enterobactercloacae. Antimicrob. Agents Chemother. 37:224–228.
109. Korfmann, G., and C. C. Sanders. 1989. ampG is essential for high-levelexpression of AmpC beta-lactamase activity in Enterobacter cloacae. Anti-microb. Agents Chemother. 33:1946–1951.
110. Korfmann, G., C. C. Sanders, and E. S. Moland. 1991. Altered phenotypesassociated with ampD mutations in Enterobacter cloacae. Antimicrob.Agents Chemother. 35:358–364.
111. Kuga, A., R. Okamoto, and M. Inoue. 2000. ampR gene mutations thatgreatly increase class C beta-lactamase activity in Enterobacter cloacae.Antimicrob. Agents Chemother. 44:561–567.
112. Labia, R., A. Morand, and J. Peduzzi. 1986. Timentin and beta-lactamases.Antimicrob. Agents Chemother. 17(Suppl. C):17–26.
113. Landman, D., S. Bratu, S. Kochar, M. Panwar, M. Trehan, M. Doymaz, andJ. Quale. 2007. Evolution of antimicrobial resistance among Pseudomonasaeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae in Brook-lyn, N.Y. J. Antimicrob. Chemother. 60:78–82.
114. Langaee, T. Y., M. Dargis, and A. Huletsky. 1998. An ampD gene inPseudomonas aeruginosa encodes a negative regulator of AmpC beta-lac-tamase expression. Antimicrob. Agents Chemother. 42:3296–3300.
115. Langaee, T. Y., L. Gagnon, and A. Huletsky. 2000. Inactivation of the ampDgene in Pseudomonas aeruginosa leads to moderate-basal-level and hyper-inducible AmpC beta-lactamase expression. Antimicrob. Agents Che-mother. 44:583–589.
116. Lee, J. K., Y. S. Lee, Y. K. Park, and B. S. Kim. 2005. Alterations in theGyrA and GyrB subunits of topoisomerase II and the ParC and ParEsubunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates ofPseudomonas aeruginosa. Int. J. Antimicrob. Agents 25:290–295.
117. Lee, K., G. Y. Ha, B. M. Shin, J. J. Kim, J. O. Kang, S. J. Jang, D. Yong,and Y. Chong. 2004. Metallo-beta-lactamase-producing gram-negative ba-cilli in Korean Nationwide Surveillance of Antimicrobial Resistance grouphospitals 2003: continued prevalence of VIM-producing Pseudomonas spp.and increase of IMP-producing Acinetobacter spp. Diagn. Microbiol. Infect.Dis. 50:51–58.
118. Letendre, E. D., R. Mantha, and P. L. Turgeon. 1988. Selection of resis-tance by piperacillin during Pseudomonas aeruginosa endocarditis. J. Anti-microb. Chemother. 22:557–562.
119. Li, X. Z., N. Barre, and K. Poole. 2000. Influence of MexA-MexB-OprMmultidrug efflux system on expression of the MexC-MexD-OprJ and MexE-MexF-OprN multidrug efflux systems in Pseudomonas aeruginosa. J. Anti-microb. Chemother. 46:885–893.
120. Li, X. Z., H. Nikaido, and K. Poole. 1995. Role of mexA-mexB-oprM inantibiotic efflux in Pseudomonas aeruginosa. Antimicrob. Agents Che-mother. 39:1948–1953.
121. Li, X. Z., L. Zhang, R. Srikumar, and K. Poole. 1998. Beta-lactamaseinhibitors are substrates for the multidrug efflux pumps of Pseudomonasaeruginosa. Antimicrob. Agents Chemother. 42:399–403.
122. Li, Y., T. Mima, Y. Komori, Y. Morita, T. Kuroda, T. Mizushima, and T.Tsuchiya. 2003. A new member of the tripartite multidrug efflux pumps,MexVW-OprM, in Pseudomonas aeruginosa. J. Antimicrob. Chemother.52:572–575.
123. Lindberg, F., S. Lindquist, and S. Normark. 1988. Genetic basis of induc-tion and overproduction of chromosomal class I beta-lactamase in nonfas-tidious gram-negative bacilli. Rev. Infect. Dis. 10:782–785.
124. Lindberg, F., S. Lindquist, and S. Normark. 1987. Inactivation of the ampDgene causes semiconstitutive overproduction of the inducible Citrobacterfreundii beta-lactamase. J. Bacteriol. 169:1923–1928.
125. Lindberg, F., L. Westman, and S. Normark. 1985. Regulatory componentsin Citrobacter freundii ampC beta-lactamase induction. Proc. Natl. Acad.Sci. USA 82:4620–4624.
126. Lindquist, S., F. Lindberg, and S. Normark. 1989. Binding of theCitrobacter freundii AmpR regulator to a single DNA site provides bothautoregulation and activation of the inducible AmpC beta-lactamase. J.Bacteriol. 171:3746–3753.
127. Lindquist, S., K. Weston-Hafer, H. Schmidt, C. Pul, G. Korfmann, J.Erickson, C. C. Sanders, H. H. Martin, and S. Normark. 1993. AmpG, a
signal transducer in chromosomal beta-lactamase induction. Mol. Micro-biol. 9:703–715.
128. Lister, P. D., V. M. Gardner, and C. C. Sanders. 1999. Clavulanate inducesexpression of the Pseudomonas aeruginosa AmpC cephalosporinase at phys-iologically relevant concentrations and antagonizes the antibacterial activityof ticarcillin. Antimicrob. Agents Chemother. 43:882–889.
129. Lister, P. D., and D. J. Wolter. 2005. Levofloxacin-imipenem combinationprevents the emergence of resistance among clinical isolates of Pseudomo-nas aeruginosa. Clin. Infect. Dis. 40:S105–S114.
130. Lister, P. D., D. J. Wolter, P. A. Wickman, and M. D. Reisbig. 2006.Levofloxacin/imipenem prevents the emergence of high-level resistanceamong Pseudomonas aeruginosa strains already lacking susceptibility to oneor both drugs. J. Antimicrob. Chemother. 57:999–1003.
131. Livermore, D. M. 1992. Interplay of impermeability and chromosomal beta-lactamase activity in imipenem-resistant Pseudomonas aeruginosa. Antimi-crob. Agents Chemother. 36:2046–2048.
132. Livermore, D. M. 2002. Multiple mechanisms of antimicrobial resistance inPseudomonas aeruginosa: our worst nightmare. Clin. Infect. Dis. 34:634–640.
133. Livermore, D. M. 2003. The threat from the pink corner. Ann. Med. 35:226–234.
134. Livermore, D. M., and N. Woodford. 2006. The beta-lactamase threat inEnterobacteriaceae, Pseudomonas, and Acinetobacter. Trends Microbiol. 14:413–420.
135. Livermore, D. M., and N. Woodford. 2000. Carbapenemases: a problem inwaiting? Curr. Opin. Microbiol. 3:489–495.
136. Livermore, D. M., and Y.-J. Yang. 1987. Beta-lactamase lability and inducerpower of newer beta-lactam antibiotics in relation to their activity againstbeta-lactamase-inducibility mutants of Pseudomonas aeruginosa. J. Infect.Dis. 4:775–782.
137. Llanes, C., D. Hocquet, C. Vogne, D. Benali-Baitich, C. Neuwirth, and P.Plesiat. 2004. Clinical strains of Pseudomonas aeruginosa overproducingMexAB-OprM and MexXY efflux pumps simultaneously. Antimicrob.Agents Chemother. 48:1797–1802.
138. Lodge, J. M., S. J. W. Busby, and L. J. Piddock. 1993. Investigation of thePseudomonas aeruginosa ampR gene and its role at the chromosomal ampCbeta-lactamase promoter. FEMS Microbiol. Lett. 111:315–320.
139. Lodge, J. M., S. D. Minchin, L. J. Piddock, and S. J. W. Busby. 1990.Cloning, sequencing and analysis of the structural gene and regulatoryregion of the Pseudomonas aeruginosa chromosomal ampC beta-lactamase.Biochem. J. 272:627–631.
140. Lynch, M. J., G. L. Drusano, and H. L. Mobley. 1987. Emergence ofresistance to imipenem in Pseudomonas aeruginosa. Antimicrob. AgentsChemother. 31:1892–1896.
141. Mammeri, H., M. Galleni, and P. Nordmann. 2009. Role of the Ser-287-Asn replacement in the hydrolysis spectrum extension of AmpC beta-lactamases in Escherichia coli. Antimicrob. Agents Chemother. 53:323–326.
142. Mammeri, H., P. Nordmann, A. Berkani, and F. Eb. 2008. Contribution ofextended-spectrum AmpC (ESAC) beta-lactamases to carbapenem resis-tance in Escherichia coli. FEMS Microbiol. Lett. 282:238–240.
143. Mammeri, H., L. Poirel, P. Bemer, H. Drugeon, and P. Nordmann. 2004.Resistance to cefepime and cefpirome due to a 4-amino-acid deletion in thechromosome-encoded AmpC beta-lactamase of a Serratia marcescens clin-ical isolate. Antimicrob. Agents Chemother. 48:716–720.
144. Margaret, B. S., G. L. Drusano, and H. C. Standiford. 1989. Emergence ofresistance to carbapenem antibiotics in Pseudomonas aeruginosa. J. Anti-microb. Chemother. 24(Suppl. A):161–167.
145. Maseda, H., K. Saito, A. Nakajima, and T. Nakae. 2000. Variation of themexT gene, a regulator of the MexEF-OprN efflux pump expression inwild-type strains of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 192:107–112.
146. Maseda, H., I. Sawada, K. Saito, H. Uchiyama, T. Nakae, and N. Nomura.2004. Enhancement of the mexAB-oprM efflux pump expression by a quo-rum-sensing autoinducer and its cancellation by a regulator, MexT, of themexEF-oprN efflux pump operon in Pseudomonas aeruginosa. Antimicrob.Agents Chemother. 48:1320–1328.
147. Masterton, R. G., P. J. Garner, N. A. Harrison, and D. J. Rainford. 1987.Timentin resistance. Lancet ii:975–976.
148. Masuda, N., N. Gotoh, C. Ishii, E. Sakagawa, S. Ohya, and T. Nishino.1999. Interplay between chromosomal beta-lactamase and the MexAB-OprM efflux system in intrinsic resistance to beta-lactams in Pseudomonasaeruginosa. Antimicrob. Agents Chemother. 43:400–402.
149. Masuda, N., N. Gotoh, S. Ohya, and T. Nishino. 1996. Quantitative corre-lation between susceptibility and OprJ production in NfxB mutants ofPseudomonas aeruginosa. Antimicrob. Agents Chemother. 40:909–913.
150. Masuda, N., E. Sakagawa, and S. Ohya. 1995. Outer membrane proteinsresponsible for multiple drug resistance in Pseudomonas aeruginosa. Anti-microb. Agents Chemother. 39:645–649.
151. Masuda, N., E. Sakagawa, S. Ohya, N. Gotoh, and T. Nishino. 2001. Hy-persusceptibility of the Pseudomonas aeruginosa nfxB mutant to beta-lac-tams due to reduced expression of the AmpC beta-lactamase. Antimicrob.Agents Chemother. 45:1284–1286.
606 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

152. Masuda, N., E. Sakagawa, S. Ohya, N. Gotoh, H. Tsujimoto, and T.Nishino. 2000. Contribution of the MexX-MexY-OprM efflux system tointrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Che-mother. 44:2242–2246.
153. Matsumura, N., S. Minami, and S. Mitsuhashi. 1998. Sequences of homol-ogous beta-lactamases from clinical isolates of Serratia marcescens withdifferent substrate specificities. Antimicrob. Agents Chemother. 42:176–179.
154. Matsuo, Y., S. Eda, N. Gotoh, E. Yoshihara, and T. Nakae. 2004. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudo-monas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMSMicrobiol. Lett. 238:23–28.
155. Mavroidi, A., E. Tzelepi, A. Tsakris, V. Miriagou, D. Sofianou, and L. S.Tzouvelekis. 2001. An integron associated beta-lactamase (IBC-2) fromPseudomonas aeruginosa is a variant of the extended-spectrum beta-lacta-mase IBC-1. J. Antimicrob. Chemother. 48:627–630.
156. Micek, S. T., A. E. Lloyd, D. J. Ritchie, R. M. Reichley, V. J. Fraser, andM. H. Kollef. 2005. Pseudomonas aeruginosa bloodstream infection: impor-tance of appropriate initial antimicrobial treatment. Antimicrob. AgentsChemother. 49:1306–1311.
157. Mima, T., S. Joshi, M. Gomez-Escalada, and H. P. Schweizer. 2007. Iden-tification and characterization of TriABC-OpmH, a triclosan efflux pump ofPseudomonas aeruginosa requiring two membrane fusion proteins. J. Bac-teriol. 189:7600–7609.
158. Mima, T., H. Sekiya, T. Mizushima, T. Kuroda, and T. Tsuchiya. 2005.Gene cloning and properties of the RND-type multidrug efflux pump,MexPQ-OpmE and MexMN-OprM from Pseudomonas aeruginosa. Micro-biol. Immunol. 49:999–1002.
159. Mine, T., Y. Morita, T. Kataoka, T. Mizushima, and T. Tsuchiya. 1999.Expression in Escherichia coli of a new multidrug efflux pump, MexXY,from Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43:415–417.
160. Morita, Y., L. Cao, V. C. Gould, M. B. Avison, and K. Poole. 2006. nalDencodes a second repressor of the mexAB-oprM multidrug efflux operon ofPseudomonas aeruginosa. J. Bacteriol. 188:8649–8654.
161. Morita, Y., N. Kimura, T. Mima, T. Mizushima, and T. Tsuchiya. 2001.Roles of MexXY- and MexAB-multidrug efflux pumps in intrinsic multi-drug resistance of Pseudomonas aeruginosa. J. Gen. Appl. Microbiol. 47:27–32.
162. Morita, Y., Y. Komori, T. Mima, T. Kuroda, T. Mizushima, and T. Tsuch-iya. 2001. Construction of a series of mutants lacking all of the four majormex operons for multidrug efflux pumps or possessing each one of theoperons from Pseudomonas aeruginosa PAO1: MexCD-OprJ is an induciblepump. FEMS Microbiol. Lett. 202:139–143.
163. Morita, Y., M. L. Sobel, and K. Poole. 2006. Antibiotic inducibility of theMexXY multidrug efflux system of Pseudomonas aeruginosa: involvement ofthe antibiotic-inducible PA5471 gene product. J. Bacteriol. 188:1847–1855.
164. Morrison, A. J., and R. P. Wenzel. 1984. Epidemiology of infections due toPseudomonas aeruginosa. Rev. Infect. Dis. 6(Suppl. 3):S627–S642.
165. Mouneimne, H., J. Robert, V. Jarlier, and E. Cambau. 1999. Type IItopoisomerase mutations in ciprofloxacin-resistant strains of Pseudomonasaeruginosa. Antimicrob. Agents Chemother. 43:62–66.
166. Moya, B., A. Dotsch, C. Juan, J. Blazquez, L. Zamorano, S. Haussler, andA. Oliver. 2009. Beta-lactam resistance response triggered by inactivation ofa nonessential penicillin-binding protein. PLoS Pathog. 5:e1000353.
167. Moya, B., C. Juan, S. Alberti, J. L. Perez, and A. Oliver. 2008. Benefit ofhaving multiple ampD genes for acquiring beta-lactam resistance withoutlosing fitness and virulence in Pseudomonas aeruginosa. Antimicrob. AgentsChemother. 52:3694–3700.
168. Murata, T., N. Gotoh, and T. Nishino. 2002. Characterization of outermembrane efflux proteins OpmE, OpmD and OpmB of Pseudomonasaeruginosa: molecular cloning and development of specific antisera. FEMSMicrobiol. Lett. 217:57–63.
169. Mushtaq, S., Y. Ge, and D. M. Livermore. 2004. Doripenem versus Pseudo-monas aeruginosa in vitro: activity against characterized isolates, mutants,and transconjugants and resistance selection potential. Antimicrob. AgentsChemother. 48:3086–3092.
170. Naas, T., and P. Nordmann. 1999. OXA-type beta-lactamases. Curr.Pharm. Des. 5:865–879.
171. National Nosocomial Infections Surveillance System. 1998. National Nos-ocomial Infections Surveillance (NNIS) System report, data summary fromOctober 1986 to April 1998, issued June 1998. Am. J. Infect. Control26:522–533.
172. Nikaido, H. 1996. Multidrug efflux pumps of gram-negative bacteria. J.Bacteriol. 178:5853–5859.
173. Nikaido, H. 1989. Outer membrane barrier as a mechanism of antimicrobialresistance. Antimicrob. Agents Chemother. 33:1831–1836.
174. Niumsup, P., A. M. Simm, K. Nurmahomed, T. R. Walsh, P. M. Bennett,and M. B. Avison. 2003. Genetic linkage of the penicillinase gene, amp, andblrAB, encoding the regulator of beta-lactamase expression in Aeromonasspp. J. Antimicrob. Chemother. 51:1351–1358.
175. Nordmann, P., and T. Nass. 1994. Sequence analysis of PER-1 extended-
spectrum beta-lactamase from Pseudomonas aeruginosa and comparison withclass A beta-lactamases. Antimicrob. Agents Chemother. 38:104–114.
176. Nordmann, P., and L. Poirel. 2002. Emerging carbapenemases in gram-negative aerobes. Clin. Microbiol. Infect. 8:321–331.
177. Normark, S., T. Edlund, T. Grundstrom, S. Bergstrom, and H. Wolf-Watz.1977. Escherichia coli K-12 mutants hyperproducing chromosomal beta-lactamase by gene repetitions. J. Bacteriol. 132:912–922.
178. Obritsch, M. D., D. N. Fish, R. MacLaren, and R. Jung. 2004. Nationalsurveillance of antimicrobial resistance in Pseudomonas aeruginosa isolatesobtained from intensive care unit patients from 1993 to 2002. Antimicrob.Agents Chemother. 48:4606–4610.
179. Ochs, M. M., C.-D. Lu, R. E. W. Hancock, and A. T. Abdelal. 1999. Aminoacid-mediated induction of the basic amino acid-specific outer membraneporin OprD from Pseudomonas aeruginosa. J. Bacteriol. 181:5426–5432.
180. Ochs, M. M., M. P. McCusker, M. Bains, and R. E. Hancock. 1999. Neg-ative regulation of the Pseudomonas aeruginosa outer membrane porinOprD selective for imipenem and basic amino acids. Antimicrob. AgentsChemother. 43:1085–1090.
181. Oh, H., J. Stenhoff, S. Jalal, and B. Wretlind. 2003. Role of efflux pumps andmutations in genes for topoisomerases II and IV in fluoroquinolone-resistantPseudomonas aeruginosa strains. Microb. Drug Resist. 9:323–328.
182. Ohara, T., and K. Itoh. 2003. Significance of Pseudomonas aeruginosacolonization of the gastrointestinal tract. Intern. Med. 42:1072–1076.
183. Oliva, B., P. M. Bennett, and I. Chopra. 1989. Penicillin-binding protein 2is required for induction of the Citrobacter freundii class I chromosomalbeta-lactamase in Escherichia coli. Antimicrob. Agents Chemother.33:1116–1117.
184. Pai, H., J. Kim, J. H. Lee, K. W. Choe, and N. Gotoh. 2001. Carbapenemresistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimi-crob. Agents Chemother. 45:480–484.
185. Palma, M., J. Zurita, J. A. Ferraras, S. Worgall, D. H. Larone, L. Shi, F.Campagne, and L. E. Quadri. 2005. Pseudomonas aeruginosa SoxR does notconform to the archetypal paradigm for SoxR-dependent regulation of thebacterial oxidative stress adaptive response. Infect. Immun. 73:2958–2966.
186. Park, C. H., A. Robicsek, G. A. Jacoby, D. Sahm, and D. C. Hooper. 2006.Prevalence in the United States of aac(6�)-Ib-cr, encoding a ciprofloxacin-modifying enzyme. Antimicrob. Agents Chemother. 50:3953–3955.
187. Peleg, A. Y., C. Franklin, J. Bell, and D. W. Spelman. 2004. Emergence ofIMP-4 metallo-beta-lactamase in a clinical isolate from Australia. J. Anti-microb. Chemother. 54:699–700.
188. Perez, F. J., C. Gimeno, D. Navarro, and J. Garcia-de-Lomas. 1996. Mero-penem permeation through the outer membrane of Pseudomonas aerugi-nosa can involve pathways other than OprD porin channel. Chemotherapy42:210–214.
189. Perron, K., O. Caille, C. Rossier, C. Van Delden, J. L. Dumas, and T.Kohler. 2004. CzcR-CzcS, a two-component system involved in heavy metaland carbapenem resistance in Pseudomonas aeruginosa. J. Biol. Chem.279:8761–8768.
190. Pfeifle, D., E. Janas, and B. Wiedemann. 2000. Role of penicillin-bindingproteins in the initiation of AmpC beta-lactamase expression in Entero-bacter cloacae. Antimicrob. Agents Chemother. 44:169–172.
191. Philippon, L. N., T. Naas, A.-T. Bouthors, V. Barakett, and P. Nordmann.1997. OXA-18, a class D clavulanic acid-inhibited extended-spectrum beta-lactamase from Pseudomonas aeruginosa. Antimicrob. Agents Chemother.41:2188–2195.
192. Picao, R. C., L. Poirel, A. C. Gales, and P. Nordmann. 2009. Furtheridentification of CTX-M-2 extended-spectrum beta-lactamase in Pseudo-monas aeruginosa. Antimicrob. Agents Chemother. 53:2225–2226.
193. Piddock, L. J. 2006. Multidrug-resistance efflux pumps—not just for resis-tance. Nat. Rev. Microbiol. 4:629–636.
194. Piddock, L. J., and D. J. Griggs. 1991. Selection and characterization ofcefepime-resistant gram-negative bacteria. J. Antimicrob. Chemother. 28:669–676.
195. Pirnay, J.-P., D. De Vos, D. Mossialos, A. Vanderkelen, P. Cornelis, and M.Zizi. 2002. Analysis of the Pseudomonas aeruginosa oprD gene from clinicaland environmental isolates. Environ. Microbiol. 4:872–882.
196. Pitt, T. L. 1998. Pseudomonas, Burkholderia, and related genera, p. 1109–1138. In B. I. Duerden (ed.), Microbiology and microbial infections, vol. 2.Oxford University Press Inc., New York, NY.
197. Poirel, L., V. O. Rotimi, and E. M. Mokaddas. 2001. VEB-1-like extendedspectrum beta-lactamases in Pseudomonas aeruginosa, Kuwait. Emerg. In-fect. Dis. 17:468–470.
198. Poirel, L., M. Van De Loo, H. Mammeri, and P. Nordmann. 2005. Associationof plasmid-mediated quinolone resistance with extended-spectrum beta-lacta-mase VEB-1. Antimicrob. Agents Chemother. 49:3091–3094.
199. Pollack, M. 1995. Pseudomonas aeruginosa, p. 1820–2003. In G. L. Mandell,R. Dolan, and J. E. Bennett (ed.), Principles and practices of infectiousdiseases. Churchill Livingstone, New York, NY.
200. Poole, K. 2005. Aminoglycoside resistance in Pseudomonas aeruginosa. An-timicrob. Agents Chemother. 49:479–487.
201. Poole, K. 2000. Efflux-mediated resistance to fluoroquinolones in gram-negative bacteria. Antimicrob. Agents Chemother. 44:2233–2241.
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 607
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

202. Poole, K. 2002. Outer membranes and efflux: the path to multidrug resis-tance in gram-negative bacteria. Curr. Pharm. Biotechnol. 3:77–98.
203. Poole, K., N. Gotoh, H. Tsujimoto, Q. Zhao, A. Wada, T. Yamasaki, S.Neshat, J. Yamagishi, X. Z. Li, and T. Nishino. 1996. Overexpression of themexC-mexD-oprJ efflux operon in nfxB-type multidrug resistant strains ofPseudomonas aeruginosa. Mol. Microbiol. 21:713–724.
204. Poole, K., D. E. Heinrichs, and S. Neshat. 1993. Cloning and sequenceanalysis of an EnvCD homologue in Pseudomonas aeruginosa: regulation byiron and possible involvement in the secretion of the siderophore pyover-dine. Mol. Microbiol. 10:529–544.
205. Poole, K., and R. Srikumar. 2001. Multidrug efflux in Pseudomonas aerugi-nosa: components, mechanisms, and clinical significance. Curr. Top. Med.Chem. 1:59–71.
206. Poole, K., K. Tetro, Q. Zhao, S. Neshat, D. E. Heinrichs, and N. Bianco.1996. Expression of the multidrug resistance operon mexA-mexB-oprM inPseudomonas aeruginosa: mexR encodes a regulator of operon expression.Antimicrob. Agents Chemother. 40:2021–2028.
207. Pumbwe, L., and L. J. V. Piddock. 2000. Two efflux systems expressedsimultaneously in multidrug-resistant Pseudomonas aeruginosa. Antimicrob.Agents Chemother. 44:2861–2864.
208. Queenan, A. M., W. Shang, M. Kania, M. G. P. Page, and K. Bush. 2007.Interactions of ceftobiprole with beta-lactamases from molecular classes Ato D. Antimicrob. Agents Chemother. 51:3089–3095.
209. Quinn, J. P., E. J. Dudek, C. A. DiVincenzo, D. A. Lucks, and S. A. Lerner.1986. Emergence of resistance to imipenem during therapy for Pseudomo-nas aeruginosa infections. J. Infect. Dis. 154:289–294.
210. Quinn, J. P., A. E. Studemeister, C. A. DiVincenzo, and S. A. Lerner. 1988.Resistance to imipenem in Pseudomonas aeruginosa: clinical experience andbiochemical mechanisms. Rev. Infect. Dis. 10:892–898.
211. Rhomberg, P. R., L. M. Deshpande, J. T. Kirby, and R. N. Jones. 2007.Activity of meropenem as serine carbapenemases evolve in US medicalcenters: monitoring report from the MYSTIC program (2006). Diagn. Mi-crobiol. Infect. Dis. 59:425–432.
212. Rhomberg, P. R., and R. N. Jones. 2007. Contemporary activity of mero-penem and comparator broad-spectrum agents: MYSTIC program reportfrom the United States component (2005). Diagn. Microbiol. Infect. Dis.57:207–215.
213. Richards, M. J., J. R. Edwards, D. H. Culver, R. P. Gaynes, et al. 1999.Nosocomial infections in medical intensive care units in the United States.Crit. Care Med. 5:887–892.
214. Richards, M. J., J. R. Edwards, D. H. Culver, R. P. Gaynes, et al. 1999.Nosocomial infections in pediatric intensive care units in the United States.Pediatrics 103:e39.
215. Robicsek, A., J. Strahilevitz, D. Sahm, G. A. Jacoby, and D. C. Hooper.2006. qnr prevalence in ceftazidime-resistant Enterobacteriaceae isolatesfrom the United States. Antimicrob. Agents Chemother. 50:2872–2874.
216. Rodriquez-Martinez, J.-M., L. Poirel, and P. Nordmann. 2009. Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob.Agents Chemother. 53:1766–1771.
217. Roh, I. K., I. J. Kim, J. H. Chung, and S. M. Byun. 1996. Affinity purifica-tion and binding characteristics of Citrobacter freundii AmpR, the transcrip-tional regulator of the ampC beta-lactamase gene. Biotechnol. Appl. Bio-chem. 23:149–154.
218. Saier, M. H., I. T. Paulsen, M. K. Sliwinski, S. S. Pao, R. A. Skurray, andH. Nikaido. 1998. Evolutionary origins of multidrug and drug-specific effluxpumps in bacteria. FASEB J. 12:265–274.
219. Saito, K., H. Akama, E. Yoshihara, and T. Nakae. 2003. Mutations affectingDNA-binding activity of the MexR repressor of mexR-mexA-mexB-oprMoperon expression. J. Bacteriol. 185:6195–6198.
220. Saito, K., S. Eda, H. Maseda, and T. Nakae. 2001. Molecular mechanism ofMexR-mediated regulation of MexAB-OprM efflux pump expression inPseudomonas aeruginosa. FEMS Microbiol. Lett. 195:23–28.
221. Sakyo, S., H. Tomita, K. Tanimoto, S. Fujimoto, and Y. Ike. 2006. Potencyof carbapenems for the prevention of carbapenem-resistant mutants ofPseudomonas aeruginosa. J. Antibiot. 59:220–228.
222. Sanders, C. C. 1992. Beta-lactamases of gram-negative bacteria: new chal-lenges for new drugs. Clin. Infect. Dis. 14:1089–1099.
223. Sanders, C. C. 1993. Cefepime: the next generation? Clin. Infect. Dis.17:369–379.
224. Sanders, C. C., P. A. Bradford, A. F. Ehrhardt, K. Bush, K. D. Young, T. A.Henderson, and W. E. Sanders. 1997. Penicillin-binding proteins and in-duction of AmpC beta-lactamase. Antimicrob. Agents Chemother.41:2013–2015.
225. Sanders, C. C., M. L. Gates, and W. E. Sanders. 1988. Heterogeneity ofclass I beta-lactamase expression in clinical isolates of Pseudomonas aerugi-nosa. Antimicrob. Agents Chemother. 32:1893–1895.
226. Sanders, C. C., and E. C. Sanders. 1983. Emergence of resistance duringtherapy with newer beta-lactam antibiotics: role of inducible beta-lactama-ses and implications for the future. Rev. Infect. Dis. 5:639–648.
227. Sanders, C. C., and W. E. Sanders. 1986. Type I beta-lactamases of gram-negative bacteria: interactions with beta-lactam antibiotics. J. Infect. Dis.154:792–800.
228. Sanders, W. E., and C. C. Sanders. 1988. Inducible beta-lactamases: clinicaland epidemiological implications for use of newer cephalosporins. Rev.Infect. Dis. 10:830–838.
229. Satake, S., H. Yoneyama, and T. Nakae. 1991. Role of OmpD2 and chro-mosomal beta-lactamase in carbapenem resistance in clinical isolates ofPseudomonas aeruginosa. J. Antimicrob. Chemother. 28:199–207.
230. Sawada, I., H. Maseda, T. Nakae, H. Uchiyama, and N. Nomura. 2004. Aquorum-sensing autoinducer enhances the mexAB-oprM efflux pump ex-pression without the MexR-mediated regulation of Pseudomonas aerugi-nosa. Microbiol. Immunol. 48:435–439.
231. Schmidtke, A. J., and N. D. Hanson. 2006. Model system to evaluate theeffect of ampD mutations on AmpC-mediated beta-lactam resistance. An-timicrob. Agents Chemother. 50:2030–2037.
232. Schmidtke, A. J., and N. D. Hanson. 2008. Role of ampD homologs inoverproduction of AmpC in clinical isolates of Pseudomonas aeruginosa.Antimicrob. Agents Chemother. 52:3922–3927.
233. Schurek, K. N., A. K. Marr, P. K. Taylor, I. Wiegand, L. Semenec, B. K.Khaira, and R. E. W. Hancock. 2008. Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob.Agents Chemother. 52:4213–4219.
234. Schweizer, H. P. 2003. Efflux as a mechanism of resistance to antimicrobialsin Pseudomonas aeruginosa and related bacteria: unanswered questions.Genet. Mol. Res. 2:48–62.
235. Scully, B. E., C. N. Ores, A. S. Prince, and H. C. Neu. 1985. Treatment oflower respiratory tract infections due to Pseudomonas aeruginosa in patientswith cystic fibrosis. Rev. Infect. Dis. 7(Suppl.):S669–S674.
236. Sekiya, H., T. Mima, Y. Morita, T. Kuroda, T. Mizushima, and T. Tsuchiya.2003. Functional cloning and characterization of a multidrug efflux pump,mexHI-opmD, from a Pseudomonas aeruginosa mutant. Antimicrob. AgentsChemother. 47:2990–2992.
237. Sevillano, E., L. Gallego, and J. M. Garcia-Lobo. 26 June 2008, postingdate. First detection of the OXA-40 carbapenemase in P. aeruginosa iso-lates, located on a plasmid also found in A. baumannii. Pathol. Biol. (Paris)[Epub ahead of print.] doi:10.1016/j.patbio.2008.05.002.
238. Shiba, T., K. Ishiguro, N. Takemoto, H. Koibuchi, and K. Sugimoto. 1995.Purification and characterization of the Pseudomonas aeruginosa NfxB protein,the negative regulator of the nfxB gene. J. Bacteriol. 177:5872–5877.
239. Sobel, M. L., D. Hocquet, L. Cao, P. Plesiat, and K. Poole. 2005. Mutationsin PA3574 (nalD) lead to increased MexAB-OprM expression and multi-drug resistance in laboratory and clinical isolates of Pseudomonas aerugi-nosa. Antimicrob. Agents Chemother. 49:1782–1786.
240. Sobel, M. L., G. A. McKay, and K. Poole. 2003. Contribution of the MexXYmultidrug transporter to aminoglycoside resistance in Pseudomonas aerugi-nosa clinical isolates. Antimicrob. Agents Chemother. 48:3202–3207.
241. Sobel, M. L., S. Neshat, and K. Poole. 2005. Mutations in PA2491 (mexS)promote MexT-dependent mexEF-oprN expression and multidrug resis-tance in a clinical strain of Pseudomonas aeruginosa. J. Bacteriol. 187:1246–1253.
242. Spencer, R. C. 1996. Predominant pathogens found in the European prev-alence of infection in intensive care study. Eur. J. Clin. Microbiol. Infect.Dis. 15:281–285.
243. Srikumar, R., T. Kon, N. Gotoh, and K. Poole. 1998. Expression of Pseudo-monas aeruginosa multidrug efflux pumps MexA-MexB-OprM and MexC-MexD-OprJ in a multidrug-sensitive E. coli strain. Antimicrob. AgentsChemother. 42:65–71.
244. Srikumar, R., X. Z. Li, and K. Poole. 1997. Inner membrane efflux com-ponents are responsible for beta-lactam specificity of multidrug effluxpumps in Pseudomonas aeruginosa. J. Bacteriol. 179:7875–7881.
245. Srikumar, R., C. J. Paul, and K. Poole. 2000. Influence of mutations in themexR repressor gene on expression of the MexA-MexB-OprM efflux systemof Pseudomonas aeruginosa. J. Bacteriol. 182:1410–1414.
246. Stapleton, P., K. Shannon, and I. Phillips. 1995. DNA sequence differencesof ampD mutants of Citrobacter freundii. Antimicrob. Agents Chemother.39:2494–2498.
247. Stobberingh, E. E. 1988. Induction of chromosomal beta-lactamase bydifferent concentrations of clavulanic acid in combination with ticarcillin.Antimicrob. Agents Chemother. 21:9–16.
248. Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J.Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L.Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, and Y. Yuan. 2000.Complete genome sequence of Pseudomonas aeruginosa PAO1, an oppor-tunistic pathogen. Nature 406:959–964.
249. Takenouchi, T., E. Sakagawa, and M. Sugawara. 1999. Detection of gyrAmutations among 335 Pseudomonas aeruginosa strains isolated in Japan andtheir susceptibilities to fluoroquinolones. Antimicrob. Agents Chemother.43:406–409.
250. Takesue, Y., T. Yokoyama, S. Akagi, H. Ohge, Y. Imamura, Y. Murakami,and T. Sueda. 2002. Changes in the intestinal flora after the administrationof prophylactic antibiotics to patients undergoing a gastrectomy. Surg. To-day 32:581–586.
251. Tam, V. M., K. T. Chang, A. N. Schilling, M. T. LaRocco, L. O. Gentry, andK. W. Garey. 2009. Impact of AmpC overexpression on outcomes of pa-
608 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

tients with Pseudomonas aeruginosa bacteremia. Diagn. Microbiol. Infect.Dis. 63:279–285.
252. Thuong, M., K. Arvaniti, R. Ruimy, P. de la Salmoniere, A. Scanvic-Hameg,J. C. Lucet, and B. Regnier. 2003. Epidemiology of Pseudomonas aeruginosaand risk factors for carriage acquisition in an intensive care unit. J. Hosp.Infect. 53:274–282.
253. Toleman, M. A., A. M. Simm, T. A. Murphy, A. C. Gales, D. J. Biedenbach,M. E. Jones, and T. R. Walsh. 2002. Molecular characterization of SPM-1,a novel metallo-beta-lactamase isolated in Latin America: report from theSENTRY antimicrobial surveillance program. J. Antimicrob. Chemother.50:673–679.
254. Trias, J., and H. Nikaido. 1990. Outer membrane protein D2 catalyzes facili-tated diffusion of carbapenems and penems through the outer membrane ofPseudomonas aeruginosa. Antimicrob. Agents Chemother. 34:52–57.
255. Trias, J., and H. Nikaido. 1990. Protein D2 channel of the Pseudomonasaeruginosa outer membrane has a binding site for basic amino acids andpeptides. J. Biol. Chem. 265:15680–15684.
256. Tsuji, M., Y. Ishii, A. Ohno, S. Miyazaki, and K. Yamaguchi. 1998. In vitroand in vivo antibacterial activities of S-4661, a new carbapenem. Antimi-crob. Agents Chemother. 42:94–99.
257. Valles, J., D. Mariscal, P. Cortes, P. Coll, A. Villagra, E. Diaz, A. Artigas,and J. Rello. 2004. Patterns of colonization by Pseudomonas aeruginosa inintubated patients: a 3-year prospective study of 1607 isolates using pulsed-field gel electrophoresis with implications for prevention of ventilator-associated pneumonia. Intensive Care Med. 30:1768–1775.
258. Vallet, I., S. P. Diggle, R. E. Stacey, M. Camara, I. Ventre, S. Lory, A.Lazdunski, P. Williams, and A. Filloux. 2004. Biofilm formation in Pseudo-monas aeruginosa: fimbrial cup gene clusters are controlled by the tran-scriptional regulator MvaT. J. Bacteriol. 186:2880–2890.
259. Van Bambeke, R., E. Balzi, and P. M. Tulkens. 2000. Antibiotic effluxpumps. Biochem. Pharmacol. 60:457–470.
260. Van Delden, C., and B. H. Iglewski. 1998. Cell-to-cell signaling and Pseudo-monas aeruginosa infections. Emerg. Infect. Dis. 4:551–560.
261. Villavicencio, R. T. 1998. The history of blue pus. J. Am. Coll. Surg. 187:212–216.
262. Villegas, M. V., K. Lolans, A. Correa, J. N. Kattan, J. A. Lopez, J. P. Quinn,and the Colombian Nosocomial Resistance Study Group. 2007. First iden-tification of Pseudomonas aeruginosa isolates producing a KPC-type car-bapenem-hydrolyzing beta-lactamase. Antimicrob. Agents Chemother. 51:1553–1555.
263. Vogne, C., J. R. Aires, C. Bailly, D. Hocquet, and P. Plesiat. 2004. Role of themultidrug efflux system MexXY in the emergence of moderate resistance toaminoglycosides among Pseudomonas aeruginosa isolates from patients withcystic fibrosis. Antimicrob. Agents Chemother. 48:1676–1680.
264. Wachino, J., K. Yamane, K. Kimura, N. Shibata, S. Suzuki, Y. Ike, and Y.Arakawa. 2006. Mode of transposition and expression of 16S rRNA meth-yltransferase gene rmtC accompanied by ISEcp1. Antimicrob. Agents Che-mother. 50:3212–3215.
265. Wachino, J., K. Yamane, K. Shibayama, H. Kurokawa, N. Shibata, S.Suzuki, Y. Doi, N. Kimura, Y. Ike, and Y. Arakawa. 2006. Novel plasmid-mediated 16S rRNA methylase, RmtC, found in a Proteus mirabilis isolatedemonstrating extraordinary high-level resistance against various aminogly-cosides. Antimicrob. Agents Chemother. 50:178–184.
266. Watanabe, M., S. Iyobe, M. Inoue, and S. Mitsuhashi. 1991. Transferableimipenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Che-mother. 35:147–151.
267. Weber, D. A., and C. C. Sanders. 1990. Diverse potential of beta-lactamaseinhibitors to induce class I enzymes. Antimicrob. Agents Chemother. 34:156–158.
268. Weldhagen, G. F., L. Poirel, and P. Nordmann. 2003. Ambler class A extended-spectrum beta-lactamases in Pseudomonas aeruginosa: novel developments andclinical impact. Antimicrob. Agents Chemother. 47:2385–2392.
269. Westbrock-Wadman, S., D. R. Sherman, M. J. Hickey, S. N. Coulter, Y. Q.Zhu, P. Warrener, L. Y. Nguyen, R. M. Shawar, K. R. Folger, and C. K.Stover. 1999. Characterization of a Pseudomonas aeruginosa efflux pumpcontributing to aminoglycoside impermeability. Antimicrob. Agents Che-mother. 43:2975–2983.
270. Westfall, L. W., N. L. Carty, N. Layland, P. Kuan, J. A. Colmer-Hamood,and A. N. Hamood. 2006. mvaT mutation modifies the expression of thePseudomonas aeruginosa multidrug efflux operon mexEF-oprN. FEMS Mi-crobiol. Lett. 255:247–254.
271. Wiegand, I., A. K. Marr, E. B. M. Breidenstein, K. N. Schurek, P. Taylor,and R. E. W. Hancock. 2008. Mutator genes giving rise to decreased anti-
biotic susceptibility in Pseudomonas aeruginosa. Antimicrob. Agents Che-mother. 52:3810–3813.
272. Wilke, M. S., M. Heller, A. L. Creagh, C. A. Haynes, L. P. McIntosh, K.Poole, and N. C. Strynadka. 2008. The crystal structure of MexR fromPseudomonas aeruginosa in complex with its antirepressor ArmR. Proc.Natl. Acad. Sci. USA 105:14832–14837.
273. Wolter, D. J., D. Acquazzino, R. V. Goering, P. Sammut, N. Khalaf, andN. D. Hanson. 2008. Emergence of carbapenem resistance in Pseudomonasaeruginosa isolates from a patient with cystic fibrosis in the absence ofcarbapenem therapy. Clin. Infect. Dis. 46:137–141.
274. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2005. AmpC and OprD arenot involved in the mechanism of imipenem hypersusceptibility amongPseudomonas aeruginosa isolates overexpressing the mexCD-oprJ effluxpump. Antimicrob. Agents Chemother. 49:4763–4766.
275. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2008. Discordance betweenimipenem resistance and oprD expression in a Pseudomonas aeruginosaoverexpressing mexEF-oprN, abstr. C1-1061. Abstr. 48th Intersci. Conf.Antimicrob. Agents Chemother., Washington, DC.
276. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2002. Efflux-mediated resis-tance among Pseudomonas aeruginosa: complex regulation of a complexresistance mechanism, abstr. A-132. Abstr. 102nd Gen. Meet. Am. Soc.Microbiol., Salt Lake City, UT.
277. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2004. Insertional inactivationof oprD in clinical isolates of Pseudomonas aeruginosa leading to carbap-enem resistance. FEMS Microbiol. Lett. 236:137–143.
278. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2008. MexT-associateddownregulation of oprD in Pseudomonas aeruginosa involves inhibition oftranscription initiation, abstr. C1-1056. Abstr. 48th Intersci. Conf. Antimi-crob. Agents Chemother., Washington, DC.
279. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2008. Novel mechanism ofmexEF-oprN efflux pump overexpression in Pseudomonas aeruginosa with-out coregulation of oprD expression, abstr. C1-1058. Abstr. 48th Intersci.Conf. Antimicrob. Agents Chemother., Washington, DC.
280. Wolter, D. J., N. D. Hanson, and P. D. Lister. 2005. Phenotypic reversion ofimipenem hypersusceptibility among mexCD-oprJ-overexpressing P. aerugi-nosa: a complex system becomes even more complex, abstr. C1-2214. Abstr.45th Intersci. Conf. Antimicrob. Agents Chemother., Washington, DC.
281. Wolter, D. J., N. Khalaf, I. E. Robledo, G. J. Vazquez, M. I. Sante, E. E.Aquino, R. V. Goering, and N. D. Hanson. 2009. Surveillance of carbap-enem-resistant Pseudomonas aeruginosa isolates from Puerto Rican medicalcenter hospitals: dissemination of KPC and IMP-18 beta-lactamases. An-timicrob. Agents Chemother. 53:1660–1664.
282. Wolter, D. J., A. J. Schmidtke, N. D. Hanson, and P. D. Lister. 2007.Increased expression of ampC in Pseudomonas aeruginosa mutants selectedwith ciprofloxacin. Antimicrob. Agents Chemother. 51:2997–3000.
283. Wolter, D. J., E. Smith-Moland, R. V. Goering, N. D. Hanson, and P. D.Lister. 2004. Multidrug resistance associated with mexXY expression inclinical isolates of Pseudomonas aeruginosa from a Texas hospital. Diagn.Microbiol. Infect. Dis. 50:43–50.
284. Woodford, N., J. Zhang, M. E. Kaufmann, S. Yarde, M. D. M. Tomas, C.Faris, M. S. Vardhan, S. Dawson, S. L. Cotterill, and D. M. Livermore.2008. Detection of Pseudomonas aeruginosa isolates producing VEB-typeextended-spectrum beta-lactamases in the United Kingdom. J. Antimicrob.Chemother. 62:1265–1268.
285. Yamane, K., Y. Doi, K. Yokoyama, T. Yagi, H. Kurokawa, N. Shibata, K.Shibayama, H. Kato, and Y. Arakawa. 2004. Genetic environments of thermtA gene in Pseudomonas aeruginosa clinical isolates. Antimicrob. AgentsChemother. 48:2069–2074.
286. Yamane, K., J. Wachino, Y. Doi, H. Kurokawa, and Y. Arakawa. 2005.Global spread of multiple aminoglycoside resistance genes. Emerg. Infect.Dis. 11:951–953.
287. Yokoyama, K., Y. Doi, K. Yamane, H. Kurokawa, N. Shibata, K. Shibayama,T. Yagi, H. Kato, and Y. Arakawa. 2003. Acquisition of 16S rRNA methylasegene in Pseudomonas aeruginosa. Lancet 362:1888–1893.
288. Yoneyama, H., and T. Nakae. 1993. Mechanism of efficient elimination ofprotein D2 in outer membrane of imipenem-resistant Pseudomonas aerugi-nosa. Antimicrob. Agents Chemother. 37:2385–2390.
289. Yoshimura, F., and H. Nikaido. 1985. Diffusion of beta-lactam antibioticsthrough the porin channels of Escherichia coli K-12. Antimicrob. AgentsChemother. 27:84–92.
290. Zhao, Q., X. Z. Li, R. Srikumar, and K. Poole. 1998. Contribution of outermembrane efflux protein OprM to antibiotic resistance in Pseudomonas aerugi-nosa independent of MexAB. Antimicrob. Agents Chemother. 42:1682–1686.
Continued next page
VOL. 22, 2009 ANTIBACTERIAL-RESISTANT PSEUDOMONAS AERUGINOSA 609
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from

Philip Lister is a Professor of Medical Mi-crobiology and Immunology and AssociateDirector of the Center for Research in Anti-Infectives and Biotechnology at CreightonUniversity. Dr. Lister received his B.S. inmicrobiology from Kansas State University(1986) and his Ph.D. in medical microbiol-ogy from Creighton University (1992). Aftera postdoctoral fellowship, Dr. Lister joinedthe faculty at Creighton University in 1994and was promoted to Professor of MedicalMicrobiology and Immunology in 2007, with a secondary appointmentfor the School of Pharmacy Science. Dr. Lister has been involved inantibacterial pharmacodynamics and resistance research for 15 yearsand currently serves as an Editor for the Journal of AntimicrobialChemotherapy and on the editorial boards of other journals. Dr. List-er’s research has focused on clinically important gram-positive andgram-negative pathogens, but it has been the therapeutic challenge ofP. aeruginosa that has been a primary focus.
Nancy Hanson is a Professor of Medical Mi-crobiology and Immunology and Director ofMolecular Biology for the Center for Re-search in Anti-Infectives and Biotechnology atCreighton University. Dr. Hanson receivedher Ph.D. in medical microbiology from theUniversity of Nebraska Medical Center in1991 and joined the faculty of Creighton Uni-versity in 1995. Her area of expertise involvesthe study of molecular mechanisms of antibi-otic resistance in gram-negative organisms,such as E. coli, K. pneumoniae, Salmonella spp., and Pseudomonas aerugi-nosa. Her research explores the following two aspects of antibiotic resis-tance mechanisms: (i) the regulation of the genes involved in resistanceand (ii) the development of PCR-based diagnostic tests that can be usedby clinical laboratories to detect resistance genes in clinical isolates. In2007, Dr. Hanson was awarded researcher of the year by the NebraskaChapter of the Cystic Fibrosis Foundation for her work on P. aeruginosa.
Daniel Wolter received his B.S. in biologyfrom Gonzaga University in 1997 and hisPh.D. in medical microbiology from Creigh-ton University in 2004. Dr. Wolter continuedhis training as a postdoctoral fellow in medicalmicrobiology at Creighton University until2008 and has since joined the Department ofPediatrics at the University of Washington asa Senior Fellow. A majority of his researchhas focused on the antimicrobial resistancemechanisms of Pseudomonas aeruginosa, withparticular emphasis on efflux-mediated drug resistance. His current re-search at the University of Washington explores the genotypic and phe-notypic adaptations of P. aeruginosa isolated from patients with cysticfibrosis (CF) and the polymicrobial interactions of this organism withother CF lung microbiota, such as Staphylococcus aureus.
610 LISTER ET AL. CLIN. MICROBIOL. REV.
on October 12, 2020 by guest
http://cmr.asm
.org/D
ownloaded from