
ANATOMY OF MECHANICAL VENTILATION - … · P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet...
16
e281 CHAPTER 127 Liver Failure: Acute and Acute-on-Chronic CONSTANTINE KARVELLAS and R. TODD STRAVITZ ACUTE LIVER FAILURE Definitions Acute liver failure (ALF) is defined as the development of hepatic encephalopathy (HE) and coagulopathy in a patient with no history of previous liver disease, with the onset HE within 26 weeks of jaundice (1). It should be stressed that ALF is not a disease, but rather a clinical syndrome triggered by numerous etiologic agents. There are three possible out- comes after ALF: spontaneous survival without orthotopic liver transplantation (OLT), OLT, or death. In the U.S. ALF Study Group Registry consisting of more than 2,000 enrollees between 1998 and 2013, 30% of patients died, one-quarter underwent OLT, and 50% recovered spontaneously (W. M. Lee, personal communication). Etiology of ALF is the single, most important factor in determining outcome. In patients with acetaminophen (APAP) overdose, spontaneous recovery is the rule (63% in the ALF Study Group Registry). Patients with acute hepatitis A, ischemic hepatitis, and pregnancy-related ALF—assuming prompt delivery of the fetus—also have high rates of spontaneous survival (>50%). Patients with ALF from all other causes have very poor rates of recovery without OLT, approximately 25% for those with indeterminate cause, acute hepatitis B, and idiosyncratic drug reactions (2). The shorter the interval between jaundice and the development of HE, the higher the likelihood of spontaneous survival; conversely, the longer this interval, the more likely is death without OLT (3). Epidemiology In the ALF Study Group Registry, APAP accounts for approxi- mately 45% of cases, half of those due to ingestion of a single large dose with suicidal intent, and the other half as “thera- peutic misadventures” (3). The second most common cause of ALF remains indeterminate even after extensive serologic and historical evaluation (12% of cases), followed by idiosyncratic drug reactions (11%) and acute hepatitis B (7%), with auto- immune hepatitis, acute hepatitis A, hepatic vein thrombosis (Budd–Chiari syndrome), ischemic hepatitis, fulminant Wilson disease, malignant infiltration of the liver, and pregnancy-asso- ciated ALF (acute fatty liver and HELLP [hemolysis, elevated liver enzymes, and low platelets] syndrome) constituting fewer than 5% of cases each. The initial laboratory and procedural evaluation of patients with ALF to determine cause is indi- cated in Table 127.1 (4). Pathophysiology Multiorgan System Failure Multiorgan system failure (MOSF) is the most common cause of death in patients with ALF. The pathogenesis of MOSF in ALF is not well understood, but early activation of proinflam- matory cytokine pathways followed by compensatory anti- inflammatory responses are both involved (5). Although the systemic inflammatory response syndrome (SIRS) is the clini- cal expression of the proinflammatory response and accom- panies MOSF, the compensatory anti-inflammatory response may be responsible for the subsequent development of sepsis and death (5). Highly prothrombotic microparticles derived from multiple parent cells (platelets, hepatocytes, endothelial cells, and monocytes) may also play a role in the MOSF of ALF, and may explain why ALF is a relatively prothrombotic state despite the general assumption by clinicians that these patients had a bleeding tendency (6). Hepatic Encephalopathy and Intracranial Hypertension A manifestation of MOSF peculiar to ALF is the develop- ment of HE, which may signify progression to cerebral edema. Cerebral edema becomes increasingly likely as the level of con- sciousness declines, and occurs in 38% to 81% of cases of grade 3 or 4 HE, respectively (7). The pathophysiology of cere- bral edema in ALF has been studied extensively and is unique compared to other causes of raised intracranial pressure (ICP) (Fig. 127.1). Although both cytotoxic (intracellular) and vaso- genic (extracellular) edema coexist, the former predominates, coinciding with the observation that most swelling localizes to gray matter astrocytes. Although there may be increased permeability to water and various other molecules, there is no widespread breakdown of the blood–brain barrier (8). Elevated serum ammonia, produced primarily by gut micro- organisms and inadequately cleared by the liver, has long been recognized to contribute to the development of cerebral edema in ALF. Patients who develop cerebral herniation have sub- stantially higher serum ammonia levels—usually more than 200 μmol/L—and greater cerebral ammonia uptake. Con- versely, herniation rarely occurs when serum ammonia levels remain below 150 μmol/L (9). Ammonia readily crosses the blood–brain barrier and is taken up by astrocytes, where it combines with glutamate to form glutamine, which in turn contributes to an osmotic gradient that draws water into the intracellular space. Although the brain usually compensates for such an osmotic challenge by extruding organic osmo- lytes—for example, myoinositol—the rapidity of its develop- ment in ALF often does not allow time for compensation to occur, as it would with chronic liver failure (10,11). Increased cerebral blood flow (CBF) is an important fac- tor in the formation of cerebral edema. Although the cerebral metabolic rate is low in patients with ALF, CBF rises with the accumulation of ammonia and is frequently excessive rela- tive to energy expenditure (12). Early in the course of disease, CBF may be appropriately low to match the reduced cere- bral metabolic rate, but with progression of hepatic failure, hyperemia ensues (13). Systemic inflammation often coincides LWBK1580_C127e_p281-296.indd 281 29/07/17 12:02 PM
Transcript of ANATOMY OF MECHANICAL VENTILATION - … · P1: OSO/OVY P2: OSO/OVY QC: OSO/OVY T1: OSO Printer: Yet...

e281
CHAPTER
127Liver Failure: Acute and Acute-on-ChronicConstantine Karvellas and r. todd stravitz
ACUTE LIVER FAILURE
Definitions
Acute liver failure (ALF) is defined as the development of hepatic encephalopathy (HE) and coagulopathy in a patient with no history of previous liver disease, with the onset HE within 26 weeks of jaundice (1). It should be stressed that ALF is not a disease, but rather a clinical syndrome triggered by numerous etiologic agents. There are three possible out-comes after ALF: spontaneous survival without orthotopic liver transplantation (OLT), OLT, or death. In the U.S. ALF Study Group Registry consisting of more than 2,000 enrollees between 1998 and 2013, 30% of patients died, one-quarter underwent OLT, and 50% recovered spontaneously (W. M. Lee, personal communication). Etiology of ALF is the single, most important factor in determining outcome. In patients with acetaminophen (APAP) overdose, spontaneous recovery is the rule (63% in the ALF Study Group Registry). Patients with acute hepatitis A, ischemic hepatitis, and pregnancy-related ALF—assuming prompt delivery of the fetus—also have high rates of spontaneous survival (>50%). Patients with ALF from all other causes have very poor rates of recovery without OLT, approximately 25% for those with indeterminate cause, acute hepatitis B, and idiosyncratic drug reactions (2). The shorter the interval between jaundice and the development of HE, the higher the likelihood of spontaneous survival; conversely, the longer this interval, the more likely is death without OLT (3).
Epidemiology
In the ALF Study Group Registry, APAP accounts for approxi-mately 45% of cases, half of those due to ingestion of a single large dose with suicidal intent, and the other half as “thera-peutic misadventures” (3). The second most common cause of ALF remains indeterminate even after extensive serologic and historical evaluation (12% of cases), followed by idiosyncratic drug reactions (11%) and acute hepatitis B (7%), with auto-immune hepatitis, acute hepatitis A, hepatic vein thrombosis (Budd–Chiari syndrome), ischemic hepatitis, fulminant Wilson disease, malignant infiltration of the liver, and pregnancy-asso-ciated ALF (acute fatty liver and HELLP [hemolysis, elevated liver enzymes, and low platelets] syndrome) constituting fewer than 5% of cases each. The initial laboratory and procedural evaluation of patients with ALF to determine cause is indi-cated in Table 127.1 (4).
Pathophysiology
Multiorgan System Failure
Multiorgan system failure (MOSF) is the most common cause of death in patients with ALF. The pathogenesis of MOSF in
ALF is not well understood, but early activation of proinflam-matory cytokine pathways followed by compensatory anti-inflammatory responses are both involved (5). Although the systemic inflammatory response syndrome (SIRS) is the clini-cal expression of the proinflammatory response and accom-panies MOSF, the compensatory anti-inflammatory response may be responsible for the subsequent development of sepsis and death (5). Highly prothrombotic microparticles derived from multiple parent cells (platelets, hepatocytes, endothelial cells, and monocytes) may also play a role in the MOSF of ALF, and may explain why ALF is a relatively prothrombotic state despite the general assumption by clinicians that these patients had a bleeding tendency (6).
Hepatic Encephalopathy and Intracranial Hypertension
A manifestation of MOSF peculiar to ALF is the develop-ment of HE, which may signify progression to cerebral edema. Cerebral edema becomes increasingly likely as the level of con-sciousness declines, and occurs in 38% to 81% of cases of grade 3 or 4 HE, respectively (7). The pathophysiology of cere-bral edema in ALF has been studied extensively and is unique compared to other causes of raised intracranial pressure (ICP) (Fig. 127.1). Although both cytotoxic (intracellular) and vaso-genic (extracellular) edema coexist, the former predominates, coinciding with the observation that most swelling localizes to gray matter astrocytes. Although there may be increased permeability to water and various other molecules, there is no widespread breakdown of the blood–brain barrier (8).
Elevated serum ammonia, produced primarily by gut micro-organisms and inadequately cleared by the liver, has long been recognized to contribute to the development of cerebral edema in ALF. Patients who develop cerebral herniation have sub-stantially higher serum ammonia levels—usually more than 200 μmol/L—and greater cerebral ammonia uptake. Con-versely, herniation rarely occurs when serum ammonia levels remain below 150 μmol/L (9). Ammonia readily crosses the blood–brain barrier and is taken up by astrocytes, where it combines with glutamate to form glutamine, which in turn contributes to an osmotic gradient that draws water into the intracellular space. Although the brain usually compensates for such an osmotic challenge by extruding organic osmo-lytes—for example, myoinositol—the rapidity of its develop-ment in ALF often does not allow time for compensation to occur, as it would with chronic liver failure (10,11).
Increased cerebral blood flow (CBF) is an important fac-tor in the formation of cerebral edema. Although the cerebral metabolic rate is low in patients with ALF, CBF rises with the accumulation of ammonia and is frequently excessive rela-tive to energy expenditure (12). Early in the course of disease, CBF may be appropriately low to match the reduced cere-bral metabolic rate, but with progression of hepatic failure, hyperemia ensues (13). Systemic inflammation often coincides
LWBK1580_C127e_p281-296.indd 281 29/07/17 12:02 PM

e282 SECTion 13 Gastrointestinal disease and dysfunCtion
CBF tends to vary directly with blood pressure (15). Thus, although excessive CBF contributes to worsening cerebral edema, patients are also especially vulnerable to ischemia if hypotension occurs. Furthermore, despite global increases in CBF, there may be important regional variations, such that blood flow is excessive in some areas and insufficient in others (16). Thus, it is important that CPP be carefully controlled, and extremes avoided.
Diagnosis
Prognostic schemes have been developed to predict death with-out OLT in patients with ALF. The King’s College Criteria (17), retrospectively analyzed outcomes in patients with ALF accord-ing to cause and subsequently validated the model in a test population. For patients with APAP-induced ALF, predictors of death included acidosis on admission (arterial pH < 7.30), or azotemia, severe coagulopathy, and high-grade HE (grade 3 or 4). In patients with ALF due to other causes, severe coagulopa-thy or any three of the criteria listed in Table 127.2 also pre-dicted death. Although the original series using these criteria reported a predictive accuracy for death without OLT of more than 85%, subsequent analyses have suggested that they are less accurate (18). Consequently, other prognostic parameters continue to be applied to individual patients with ALF during the weighty decision of whether to proceed with OLT.
Treatment
Etiology-Specific Management
N-acetylcysteine (NAC) has become widely administered to patients with ALF due to APAP on the basis of both labo-ratory and clinical data (29,30); however, randomized, pla-cebo-controlled studies documenting the efficacy of NAC in APAP overdose have never been performed. Several rules of administration require emphasis. First, although nomograms describing the probability of hepatotoxicity after a single APAP ingestion have been used widely to determine whether NAC should be administered, the time of ingestion frequently cannot be determined accurately, and ingestions are often multiple. Therefore, NAC should be administered whenever there is uncertainty about timing or dose. Second, intravenous NAC should be administered when a patient has higher than grade 1 HE, or in patients who do not tolerate oral dosing
Acute Liver Failure
↑Ammonia
Astrocytes ↑ GlutamineMitochondrial dysfunction Oxidative stress; ↑HO-1 ? Ischemia,↑lactate
Extracellular space, neurons ↑ Glutamate →
↑ neuronal Ca2+, abnormal depolarization, seizures,↑NO
↑ Aquaporin expression, BBB permeability
Inflammation /SIRS
↑CBF
Cytotoxic Edema ↑ ICP↓ CPP
Vasogenic Edema
TablE 127.1 Cause, Prevalence, and Evaluation of acute liver Failure
Cause Prevalencea (%) Evaluation
acetamino-phen
45 History; acetominophen level
HBv 7 anti-HBc (igM and total), HBsag, anti-HBs, HBv dna, anti-Hdv
autoimmune hepatitis
5 ana, asMa, anti-lKM, immunoglobulins
Hepatitis a virus
4 anti–hepatitis a virus (igM and total)
shock liver 4 echocardiogram, brain natriuretic peptide
Wilson disease 3 serum ceruloplasmin and copper; urine copper; slit lamp eye exam
Budd–Chiari syndrome
2 doppler ultrasound of liver
Malignancy 1 Contrast-enhanced Ct; Mri (preferred)
aflP/HellP 1 Pregnancy test
Hsv 0.5 anti-Hsv 1/2, Hsv dna
other 4 anti-HCv/HCv rna, anti-CMv/CMv dna, anti-eBv/eBv dna, toxicology screen
indeterminate 14 all of above negative
idiosyncratic drug
13 History; all of above negative
HBv, hepatitis B virus; Hdv, hepatitis d virus; ana, anti-nuclear antibody; asMa, anti-smooth muscle antibody; anti-lKM, anti-liver kidney microsomal antibody; Ct, computed tomography; Mri, magnetic resonance imaging; aflP/HellP, acute fatty liver of pregnancy/hemolysis-elevated liver enzymes–low platelet syn-drome; Hsv, herpes simplex virus; HCv, hepatitis C virus; CMv, cytomegalovirus; eBv, epstein–Barr virus.
suggested evaluation of patients with idiosyncratic drug hepatotoxicity and inde-terminate alf should include all of the tests listed, because these are diagnoses of exclusion.
aPrevalence of etiologies are estimates based on the u.s. acute liver failure study Group Cohort (personal communication).
FIgURE 127.1 Pathogenesis of cerebral edema and intracranial hypertension in acute liver failure. BBB, blood–brain barrier; Ca2+, calcium; CBf, cerebral blood flow; CPP, cerebral perfusion pressure; Ho-1, heme-oxidase-1; iCP, intracranial pressure; no, nitric oxide; sirs, systemic inflamma-tory response syndrome.
with increases in CBF and ICP, suggesting that it may have a pathogenic role (14). Abnormal CBF in patients with ALF also results from impairment in cerebrovascular autoregulation. Rather than remaining constant in the face of varying levels of cerebral perfusion pressure:
CPP = mean arterial pressure (MAP) - ICP
LWBK1580_C127e_p281-296.indd 282 29/07/17 12:02 PM
CHAPTER 127 liver failure: acute and acute-on-Chronic e283
(31). Finally, since the administration of NAC, even late after ingestion, appears to confer survival benefit, dosing should continue until evidence of severe liver injury resolves (inter-national normalized ratio [INR] less than 1.5 and resolution of HE) (32).
Other Etiology-Specific Treatments
Specific medications may be considered in patients with ALF due to non-APAP etiologies, and are outlined in Table 127.3. It should be emphasized that studies to support the use of these therapies do not exist; one using lamivudine for acute hepatitis B found no benefit versus placebo (33). Another randomized, placebo-controlled trial suggested improved transplant-free survival of patients with non-APAP ALF and low-grade (stage I-II) HE who received NAC (34).
Management of HE, Cerebral Edema, and Seizures
Considering the central importance of ammonia in the patho-genesis of HE and cerebral edema, clinicians often use thera-pies aimed at lowering serum ammonia levels in patients with ALF. The most commonly used agents in chronic liver disease, nonabsorbable disaccharides (e.g., lactulose) and oral antibi-otics (neomycin, rifaximin, or metronidazole), have not been studied in patients with ALF but probably have little effect on outcome. A computed tomographic (CT) scan of the head should be considered early in the course of ALF to exclude other causes of altered mental status, especially intracerebral hemorrhage. Although a head CT may also detect cerebral edema, a normal scan does not rule out clinically important
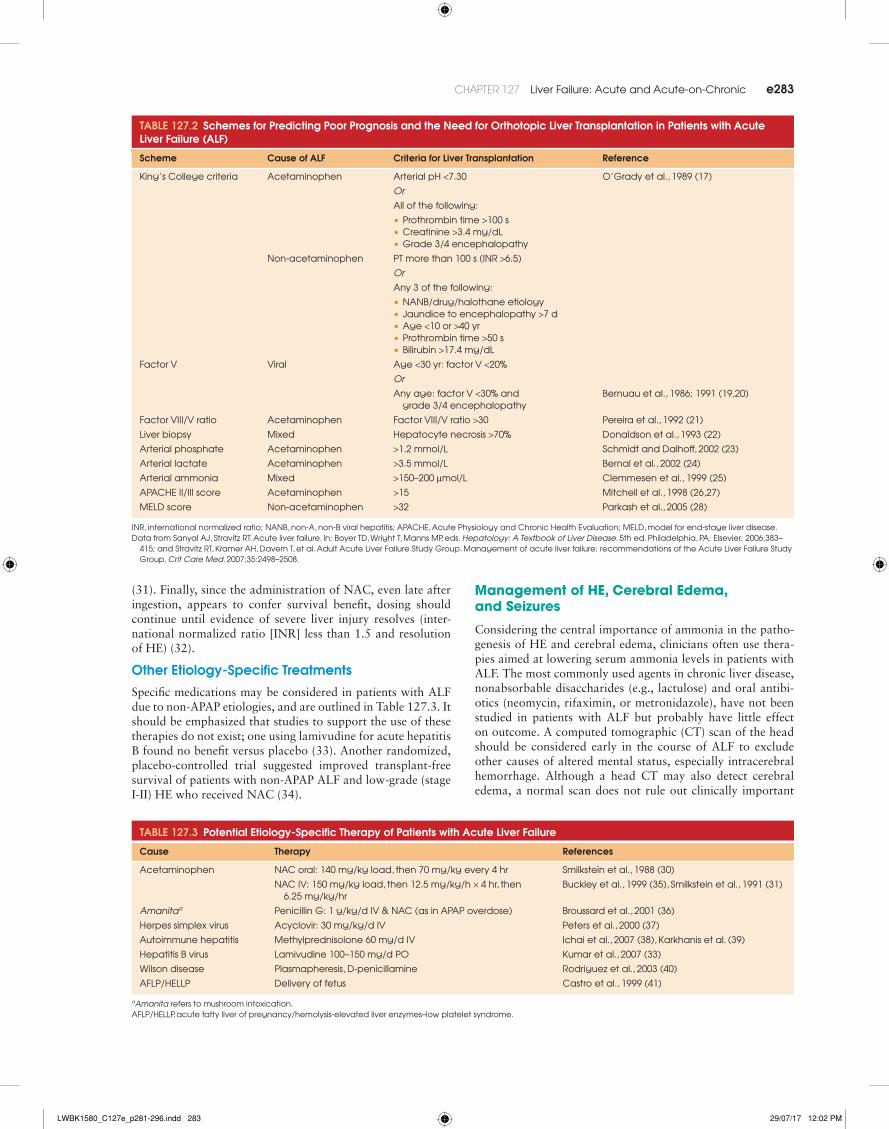
TablE 127.2 Schemes for Predicting Poor Prognosis and the Need for Orthotopic liver Transplantation in Patients with acute liver Failure (alF)
Scheme Cause of alF Criteria for liver Transplantation Reference
King’s College criteria acetaminophen arterial pH <7.30 o’Grady et al., 1989 (17)
Or
all of the following:
•Prothrombin time >100 s•Creatinine >3.4 mg/dl•Grade 3/4 encephalopathy
non-acetaminophen Pt more than 100 s (inr >6.5)
Or
any 3 of the following:
•nanB/drug/halothane etiology•Jaundice to encephalopathy >7 d•age <10 or >40 yr•Prothrombin time >50 s•Bilirubin >17.4 mg/dl
factor v viral age <30 yr: factor v <20%
Or
any age: factor v <30% and grade 3/4 encephalopathy
Bernuau et al., 1986; 1991 (19,20)
factor viii/v ratio acetaminophen factor viii/v ratio >30 Pereira et al., 1992 (21)
liver biopsy Mixed Hepatocyte necrosis >70% donaldson et al., 1993 (22)
arterial phosphate acetaminophen >1.2 mmol/l schmidt and dalhoff, 2002 (23)
arterial lactate acetaminophen >3.5 mmol/l Bernal et al., 2002 (24)
arterial ammonia Mixed >150–200 μmol/l Clemmesen et al., 1999 (25)
aPaCHe ii/iii score acetaminophen >15 Mitchell et al., 1998 (26,27)
Meld score non-acetaminophen >32 Parkash et al., 2005 (28)
inr, international normalized ratio; nanB, non-a, non-B viral hepatitis; aPaCHe, acute Physiology and Chronic Health evaluation; Meld, model for end-stage liver disease.data from sanyal aJ, stravitz rt. acute liver failure. in: Boyer td, Wright t, Manns MP, eds. Hepatology: A Textbook of Liver Disease. 5th ed. Philadelphia, Pa: elsevier; 2006:383–
415; and stravitz rt, Kramer aH, davern t, et al. adult acute liver failure study Group. Management of acute liver failure: recommendations of the acute liver failure study Group. Crit Care Med. 2007;35:2498–2508.
TablE 127.3 Potential Etiology-Specific Therapy of Patients with acute liver Failure
Cause Therapy References
acetaminophen naC oral: 140 mg/kg load, then 70 mg/kg every 4 hr smilkstein et al., 1988 (30)
naC iv: 150 mg/kg load, then 12.5 mg/kg/h × 4 hr, then 6.25 mg/kg/hr
Buckley et al., 1999 (35), smilkstein et al., 1991 (31)
Amanitaa Penicillin G: 1 g/kg/d iv & naC (as in aPaP overdose) Broussard et al., 2001 (36)
Herpes simplex virus acyclovir: 30 mg/kg/d iv Peters et al., 2000 (37)
autoimmune hepatitis Methylprednisolone 60 mg/d iv ichai et al., 2007 (38), Karkhanis et al. (39)
Hepatitis B virus lamivudine 100–150 mg/d Po Kumar et al., 2007 (33)
Wilson disease Plasmapheresis, d-penicillamine rodriguez et al., 2003 (40)
aflP/HellP delivery of fetus Castro et al., 1999 (41)
aAmanita refers to mushroom intoxication.aflP/HellP, acute fatty liver of pregnancy/hemolysis-elevated liver enzymes–low platelet syndrome.
LWBK1580_C127e_p281-296.indd 283 29/07/17 12:02 PM
e284 SECTion 13 Gastrointestinal disease and dysfunCtion
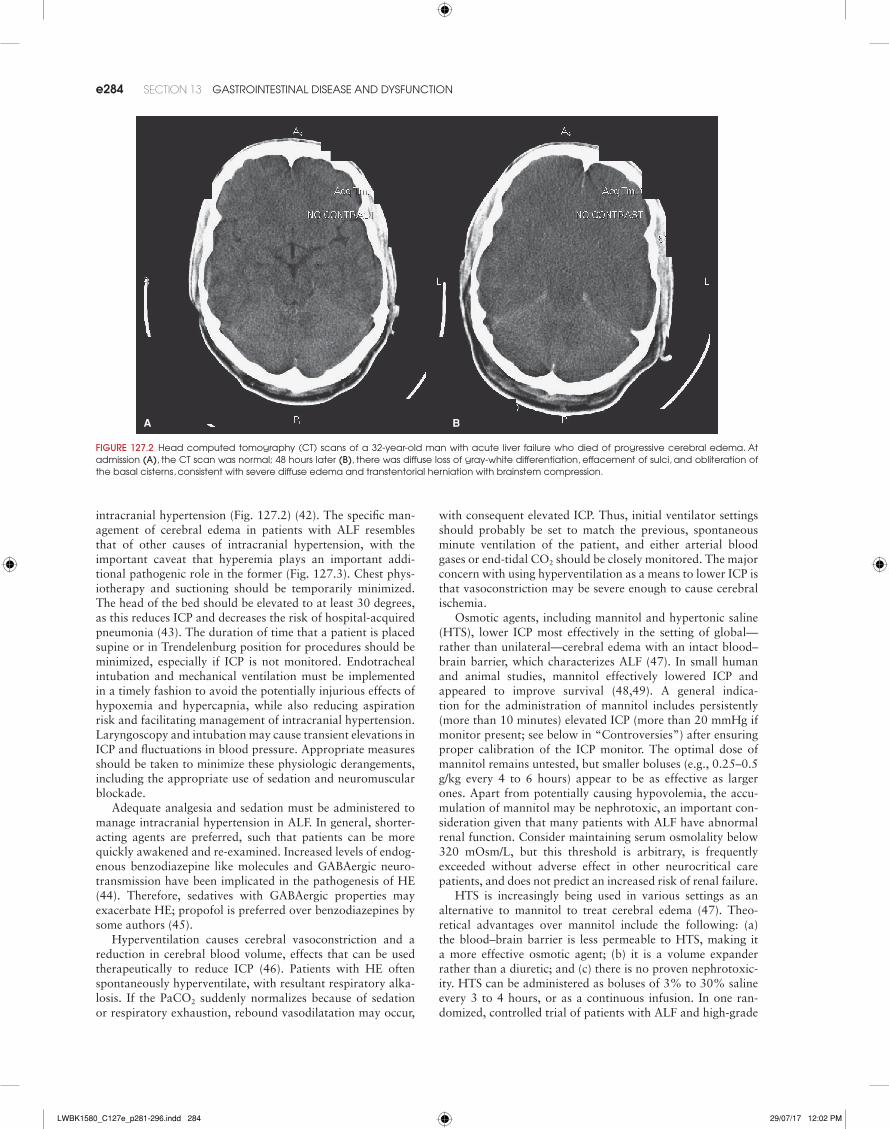
intracranial hypertension (Fig. 127.2) (42). The specific man-agement of cerebral edema in patients with ALF resembles that of other causes of intracranial hypertension, with the important caveat that hyperemia plays an important addi-tional pathogenic role in the former (Fig. 127.3). Chest phys-iotherapy and suctioning should be temporarily minimized. The head of the bed should be elevated to at least 30 degrees, as this reduces ICP and decreases the risk of hospital-acquired pneumonia (43). The duration of time that a patient is placed supine or in Trendelenburg position for procedures should be minimized, especially if ICP is not monitored. Endotracheal intubation and mechanical ventilation must be implemented in a timely fashion to avoid the potentially injurious effects of hypoxemia and hypercapnia, while also reducing aspiration risk and facilitating management of intracranial hypertension. Laryngoscopy and intubation may cause transient elevations in ICP and fluctuations in blood pressure. Appropriate measures should be taken to minimize these physiologic derangements, including the appropriate use of sedation and neuromuscular blockade.
Adequate analgesia and sedation must be administered to manage intracranial hypertension in ALF. In general, shorter-acting agents are preferred, such that patients can be more quickly awakened and re-examined. Increased levels of endog-enous benzodiazepine like molecules and GABAergic neuro-transmission have been implicated in the pathogenesis of HE (44). Therefore, sedatives with GABAergic properties may exacerbate HE; propofol is preferred over benzodiazepines by some authors (45).
Hyperventilation causes cerebral vasoconstriction and a reduction in cerebral blood volume, effects that can be used therapeutically to reduce ICP (46). Patients with HE often spontaneously hyperventilate, with resultant respiratory alka-losis. If the PaCO2 suddenly normalizes because of sedation or respiratory exhaustion, rebound vasodilatation may occur,
A B
FIgURE 127.2 Head computed tomography (Ct) scans of a 32-year-old man with acute liver failure who died of progressive cerebral edema. at admission (A), the Ct scan was normal; 48 hours later (B), there was diffuse loss of gray-white differentiation, effacement of sulci, and obliteration of the basal cisterns, consistent with severe diffuse edema and transtentorial herniation with brainstem compression.
with consequent elevated ICP. Thus, initial ventilator settings should probably be set to match the previous, spontaneous minute ventilation of the patient, and either arterial blood gases or end-tidal CO2 should be closely monitored. The major concern with using hyperventilation as a means to lower ICP is that vasoconstriction may be severe enough to cause cerebral ischemia.
Osmotic agents, including mannitol and hypertonic saline (HTS), lower ICP most effectively in the setting of global—rather than unilateral—cerebral edema with an intact blood–brain barrier, which characterizes ALF (47). In small human and animal studies, mannitol effectively lowered ICP and appeared to improve survival (48,49). A general indica-tion for the administration of mannitol includes persistently (more than 10 minutes) elevated ICP (more than 20 mmHg if monitor present; see below in “Controversies”) after ensuring proper calibration of the ICP monitor. The optimal dose of mannitol remains untested, but smaller boluses (e.g., 0.25–0.5 g/kg every 4 to 6 hours) appear to be as effective as larger ones. Apart from potentially causing hypovolemia, the accu-mulation of mannitol may be nephrotoxic, an important con-sideration given that many patients with ALF have abnormal renal function. Consider maintaining serum osmolality below 320 mOsm/L, but this threshold is arbitrary, is frequently exceeded without adverse effect in other neurocritical care patients, and does not predict an increased risk of renal failure.
HTS is increasingly being used in various settings as an alternative to mannitol to treat cerebral edema (47). Theo-retical advantages over mannitol include the following: (a) the blood–brain barrier is less permeable to HTS, making it a more effective osmotic agent; (b) it is a volume expander rather than a diuretic; and (c) there is no proven nephrotoxic-ity. HTS can be administered as boluses of 3% to 30% saline every 3 to 4 hours, or as a continuous infusion. In one ran-domized, controlled trial of patients with ALF and high-grade
LWBK1580_C127e_p281-296.indd 284 29/07/17 12:02 PM

CHAPTER 127 liver failure: acute and acute-on-Chronic e285
encephalopathy, an HTS infusion (30% saline, 5 to 20 mL/hr) to maintain serum sodium levels of 145 to 155 mmol/L effec-tively acted as prophylaxis against intracranial hypertension. The presence of hyponatremia may contribute to the develop-ment of cerebral edema early in the course of ALF (50) and is an independent predictor of worse outcome (51). Hyponatre-mia should therefore be corrected, but serum sodium levels should not be raised more quickly than 0.3 to 0.5 mmol/L/hr to minimize the risk of osmotic demyelination (Central Pontine Myelinolysis).
Nonconvulsive seizures are a potential cause of worsening cerebral edema and secondary brain injury. A small study using two-channel EEG—rather than the usual 16 to 20 channels—suggested that nonconvulsive seizures are relatively common with ALF, although the criteria used for electrographic seizures were not described (52). Given the frequency of nonconvul-sive seizures and status epilepticus among comatose patients in general, routinely obtaining an EEG in patients with hepatic encephalopathy, particularly when there is a clinical change in neurologic status, may be useful. However, there is currently insufficient evidence to justify the routine use of prophylactic phenytoin (52,53). Furthermore, sedation with propofol or
benzodiazepines is likely to provide more effective antiseizure prophylaxis than phenytoin.
Management of Cardiopulmonary Complications
Patients with ALF frequently develop SIRS, regardless of whether or not their course is complicated by infection (54). Vasoplegia is common in ALF and can be complicated by seda-tives, mechanical ventilation, and relative adrenal insufficiency (55). Norepinephrine is currently the preferred vasopressor for vasodilatory shock (56). The goal MAP should be individu-alized to optimize organ perfusion, rather than choosing an arbitrary number, but is generally kept above 65 mmHg. CPP should be maintained above 50 to 60 mmHg, since CBF auto-regulation fails below these levels in most individuals; if an ICP monitor is not placed, clinicians should err on the side of higher blood pressure. Although there are theoretical con-cerns that vasopressin may exacerbate ICH, it may be used as a second-line vasopressor (57). In patients with relative adrenal insufficiency, treatment with low-dose corticosteroids (e.g., hydrocortisone 50 to 100 mg IV every 6 to 8 hours, or 8 to 10 mg/hr as a continuous IV infusion) reduces vasopressor requirements, although the impact on outcome is uncertain.
As many as 37% of patients with ALF develop acute lung injury (ALI) or acute respiratory distress syndrome (ARDS) (58). With established ALI or ARDS, tidal volumes should be limited to 6 mL/kg of predicted body weight, although increases in PaCO2 cannot be tolerated in the setting of cere-bral edema and intracranial hypertension. Given the high risk of developing ARDS, it may be advisable to limit tidal vol-umes even in the absence of established ALI/ARDS. Positive end-expiratory pressure (PEEP/CPAP) settings should be suf-ficient to achieve adequate oxygenation, while concomitantly ensuring that ICP, blood pressure, and cardiac output are not compromised.
Management of Acute Kidney Injury
Acute kidney injury (AKI) complicates up to 50% of cases of ALF, and is more prevalent in acetaminophen-induced ALF (59,60). Hypovolemia, hypotension, and the use of nephrotoxins—including aminoglycosides, nonsteroidal anti-inflammatory drugs, and intravenous contrast—should be minimized. Patients with ALF can develop hepatorenal syn-drome (HRS) as a consequence of intense renal vasoconstric-tion (see Acute-on-Chronic Liver Failure, below). The decision to initiate renal replacement therapy (RRT) must consider the magnitude of renal dysfunction, metabolic derangements, and volume overload. Continuous renal replacement therapy (CRRT) is preferred over intermittent hemodialysis (IHD) by many clinicians, largely because of more stable volume man-agement and greater time-averaged dialysis dose. Even tran-sient hypotension is poorly tolerated in patients with cerebral edema; not only does the CPP decrease, but cerebral vasodi-latation may increase, with further increases in CBF and ICP (61). Excessively rapid correction of metabolic acidosis with bicarbonate-based dialysate may transiently increase cerebro-spinal fluid CO2, reduce central nervous system (CNS) pH, and promote more cerebral vasodilatation (62). Good results have been achieved with CRRT in patients with ALF (63). Regardless of the mode of RRT, an adequate hemofiltration or
Minimize stimulation, quiet environment Head of bed elevation to (at least) 30° if not hypotensive Early consideration of endotracheal intubation Avoid increases in PCO2 compared with baseline, especially post-ETT Correct hyponatremia, if necessary with HTS (raise Na+ <0.5 mmol/L/hr) Maintain normal serum glucose Keep temperature <37.5°C
CT to rule out hemorrhage/infarct Consider EEG to rule out nonconvulsive seizures Place ICP monitor If INR >1.5, consider pretreatment with rFVIIa
Goals: ICP <20 to 25 mmHg, CPP >60 mmHg (but avoid >80)
Rescue interventions: (1) Mild hypothermia (<35°C) (2) Hyperventilation: - Perform with surrogate for CBF/perfusion (TCD, SjO2, PbtO2) - If SjO2 used, keep >60%; if PbtO2, keep >15–20 mmHg (3) Deeper sedation with goal of burst suppression
- EEG to guide dosing (4) Indomethacin?
(1) Mannitol 0.25–0.5 g/kg every 3–4 hr—ensure osmolar gapnormalizes (<15–20) between doses (2) If (1) ineffective or contraindicated, use hypertonic saline to
- May use boluses (every 3 hr) or infusion - Caution when therapy is weaned to avoid rebound ↑ICP
maintainserum Na+ 145–155 mmol/L:
FIgURE 127.3 algorithm for management of cerebral edema and raised intracranial pressure in acute liver failure (alf) (see text for details). CBf, cerebral blood flow; CPP, cerebral perfusion pressure; Ct, computed tomography; eeG, electroencephalogram; ett, endotracheal tube; rfviia, recombinant factor 7 activated; Hts, hypertonic saline; iCP, intra-cranial pressure; inr, international normalized ratio; Pbto2, brain tissue oxygen tension; sjo2, jugular venous saturation; tCd, transcranial doppler.
LWBK1580_C127e_p281-296.indd 285 29/07/17 12:02 PM

e286 SECTion 13 Gastrointestinal disease and dysfunCtion
dialysis dose should be used, while blood pressure—and pref-erably ICP—are carefully monitored and maintained CRRT. Hemofiltration may also impact serum ammonia levels (64).
Management of Infections
ALF is associated with reticuloendothelial dysfunction and impaired immunity, with reduced complement levels, abnor-mal opsonization, and ineffective phagocytosis. ALF patients are, therefore, at high risk of nosocomial infections with both bacterial and fungal pathogens, which occur in almost 40% of these patients (65). Early diagnosis can be difficult since patients often have subtle manifestations of infection, but is vital because of the high associated morbidity and mortality. Daily surveillance cultures (urine, blood, sputum) and chest radiography should be considered, as they may improve early diagnosis of infection and guide selection of antimicrobial agents (66). Prophylactic antibiotics (enteral and parenteral) have not shown to consistent impact rates of bacteremia or sur-vival in ALF (67). Nevertheless, many clinicians prefer to use them, especially in patients listed for transplantation. Empiric broad-spectrum antibiotics (including vancomycin and an anti-fungal agent, as indicated) should be considered in any patient with ALF who develops significant isolates on surveillance cul-tures, unexplained progression of HE, or signs of SIRS, as these frequently predict sepsis in patients with ALF (14,54).
Management of Coagulopathy
Despite a deficiency of clotting factors, low fibrinogen, throm-bocytopenia, and platelet dysfunction, clinically important spontaneous bleeding is relatively infrequent in patients with ALF, being seen in less than 10% of patients (68). Therefore, the routine use of blood products to correct these abnormalities is not justified since they are unnecessary, ineffective, and per-haps most importantly, interfere with the prognostic utility of the INR. Although treatment of coagulopathy may be consid-ered in anticipation of invasive procedures, global hemostasis in patients with ALF is usually normal or even hypercoagulable (69), and transfusion of plasma may exacerbate a prothrom-botic state characterized by profoundly high von Willebrand factor and factor VIII, and deficient fibrinolysis (6,70,71). Vitamin K deficiency has been reported to contribute to the coagulopathy of ALF (72) and should be repleted parenterally (10 mg subcutaneously [SC] or slow [over 30 minutes] IV). Coagulopathy and mechanical ventilation are well-established indications for gastrointestinal (GI) stress ulcer prophylaxis, which has been shown to decrease the risk of GI bleeding in ALF patients (73). Deep venous thrombosis prophylaxis is rec-ommended; although there are no published data, low-dose unfractionated heparin or low–molecular-weight heparin can be safely used (Richard T. Stravitz, personal observations).
Management of Metabolic Derangements
ALF is a catabolic state, with increased energy requirements and negative nitrogen balance, which may in turn contribute to immunosuppression (74). Higher-than-usual caloric intake is therefore recommended, with 35 to 40 kcal/kg/day and 0.8 to 1 g/kg/day of protein, preferably provided via the enteral route. Reduced hepatic glycogen stores and impaired gluco-neogenesis are responsible for the frequent development of hypoglycemia, which often requires treatment with intrave-nous dextrose. Conversely, hyperglycemia may contribute to increases in ICP and other complications. Thus, blood glucose
values must be closely monitored and maintained within the normal range with intravenous short-acting insulin.
Liver Transplantation for ALF
OLT remains the treatment of last resort for ALF. The decision to list a patient with ALF for OLT requires careful clinical and psychosocial assessment and should be started immediately on recognition of poor prognosis as discussed above. In addition to usual clinical evaluation, patients with ALF due to APAP overdose often present with histories of suicidal ideation or substance abuse, which may preclude their consideration as viable OLT candidates. Because OLT candidates with ALF are generally younger and healthier than their counterparts with chronic liver disease, the pretransplant evaluation can usually be abbreviated to include echocardiography, duplex ultraso-nography of the liver, and routine pretransplant laboratories (e.g., total anti-CMV, HIV antibody).
Criteria for listing a patient with ALF for OLT change and current criteria may be found at UNOS.ORG in Policy 3.6. Presently, patients with ALF are given priority to receive a cadaveric organ over all patients with chronic liver disease (Status 1). Candidates must have a life expectancy without OLT of less than 7 days, have onset of HE within 8 weeks of the first symptoms of liver disease, and no history of pre-exist-ing liver disease. In addition, patients must be in the ICU and must fulfill one of the following three criteria: (i) be ventilator dependent; (ii) require dialysis or CRRT; or (iii) have an INR more than 2.0. Patients with acute decompensated Wilson dis-ease may also be listed as Status 1 in consideration with their extremely poor prognosis for spontaneous survival. Before transporting a patient with ALF to the operating room for OLT, a detailed review of the patient’s neurologic status must be made so that OLT is not performed when likelihood of neu-rologic recovery is poor. Specifically, it has been observed that severe, sustained intracranial hypertension predicts brainstem herniation during OLT or poor neurologic recovery after OLT, and patients with ICP greater than 40 mmHg or CPP less than 40 mmHg for more than 2 hours appear to be particularly vulnerable to these disastrous outcomes (75).
Early (3-month) mortality after OLT for ALF is higher than for patients transplanted for all causes of chronic liver disease—about 20% versus 10%, respectively, reflecting the acuity and severity of disease at the time of transplant. There-after, however, 5- and 10-year survival after OLT for ALF approximates 70% and 65%, respectively (76). Well-selected APAP-ALF patients with careful review of psychosocial fac-tors potentially have outcomes similar to patients with chronic liver disease patients (77).
Controversies
Intracranial Pressure Monitoring
ICP cannot accurately be determined noninvasively and carries important prognostic implications for spontaneous survival and neurologic recovery after OLT, many experts advocate ICP monitor placement in OLT candidates with stage III or IV HE (78). Although there is a perception that ICP monitoring significantly improves the outcome of patients with ALF, no prospective, randomized studies exist to support the practice, and retrospective studies in fact refute this perception (79).
LWBK1580_C127e_p281-296.indd 286 29/07/17 12:02 PM

CHAPTER 127 liver failure: acute and acute-on-Chronic e287
The invariable presence of coagulopathy in patients with ALF increases the bleeding risk of ICP monitor placement, and earlier studies reported bleeding complications in up to 20%. However, the clinical significance of many of these bleeding complications was probably negligible (78), and more recent series have found lower bleeding rates (5%) (79). Although placement of ICP monitors into the epidural space may min-imize the risk of hemorrhage, the accuracy of this practice, compared with other methods of ICP monitoring, has not been well studied.
Therapeutic Hypothermia in ALF
Fever increases ICP and is an independent predictor of worse outcome in brain-injured patients, such that hyperthermia should be avoided (80). The induction of mild hypothermia may interfere with several steps in the pathogenesis of cere-bral edema. Specifically, hypothermia attenuates the osmotic gradient created by increased astrocytic glutamine, normalizes extracellular glutamate and lactate, decreases CBF, restores autoregulation, and reduces ICP (81). Temperatures of 32° to 33°C have been used to control intracranial hypertension in patients with ALF refractory to standard care. However, a recent controlled study failed to show significant benefit with the potential exception of younger acetaminophen patients (82). Important potential adverse effects of hypothermia in the setting of ALF include interference with coagulation, an increased risk of infection, and cardiac dysrhythmias.
ACUTE-ON-CHRONIC LIVER FAILURE
Definitions
Patients with cirrhosis, the fibro-inflammatory alteration of hepatic architecture resulting in portal hypertension, usually die from acute hepatic decompensation (AD), which triggers MOSF. The term AD is often invoked in a patient with cirrhosis who develops ascites, hepatic encephalopathy, GI hemorrhage, and/or bacterial infection, without progression to MOSF (83). By contrast, acute-on-chronic liver failure (ACLF) has recently been defined as AD with MOSF (83,84). The natural his-tory and clinical manifestations of ACLF have been recently described in order to study the entity, and a prognostic score for ACLF was later developed and validated, which included entries for the number of organs failed, age, and white blood cell (WBC) count (85,86) (available online at http://www.clif-consortium.com). A North American consortium subsequently proposed a second definition of ACLF, including grade III/IV hepatic encephalopathy, hypotension despite adequate volume repletion, and/or a requirement for mechanical ventilation or RRT (87). Transplant-free mortality at 28 and 90 days is 33% and 51%, respectively, in patients with ACLF, but only 2% and 10%, respectively, in those with AD (86). The most common extrahepatic manifestation of ACLF is renal failure, which also portends the worst prognosis.
Pathophysiology
According to the Scientific Registry of Transplant Recipients, the most common cause of end-stage liver disease in the United
States is chronic hepatitis C, with or without a contribution from alcohol abuse (seen in about 40% of cases). Patients with alcoholic cirrhosis and indeterminate (cryptogenic) causes, many of whom have nonalcoholic steatohepatitis (NASH), occupy second and third most frequent causes, respectively. Other less common etiologies include chronic hepatitis B, immune-mediated liver diseases—autoimmune hepatitis, primary biliary cirrhosis, primary sclerosing cholangitis—and hereditary liver diseases—hemochromatosis, alpha-1-antitrypsin deficiency.
Patients with cirrhosis decompensate as a result of contribu-tions from two basic pathogenic mechanisms: portal hyperten-sion and hepatocellular insufficiency. Complications of portal hypertension include hemodynamic alterations, functional renal failure, ascites, and GI bleeding, most commonly from variceal hemorrhage. Complications of hepatocellular insuf-ficiency include coagulopathy and hepatic encephalopathy, although it should be appreciated that the latter also occurs as a result of portosystemic shunting.
The pathophysiology of ACLF is the subject of intense ongoing research; however, the topic remains poorly under-stood largely because the syndrome required formal definition and recognition as a clinical entity. Although the definitions of ACLF noted above were not used in most of the studies explor-ing pathogenesis, a consensus of experts has summarized the available data (86). ACLF usually occurs after a triggering event in a patient with stable cirrhosis, such as alcoholic hepa-titis, superimposed viral hepatitis, drug-induced liver injury, or a complication of cirrhosis itself, most often infection, in turn, often due to bacterial translocation from the gut.
A fundamental feature of cirrhosis critical to the pathogen-esis of AD and ACLF is an abnormal hemodynamic state char-acterized by low systemic vascular resistance (SVR), systemic hypotension, and splanchnic vasodilation. Consequently, arte-rial underfilling of critical regulatory vascular beds in the renal arterial and hypothalamic circulations result in the elabora-tion of compensatory neurohumoral effectors, such as renin, angiotensin, vasopressin, and norepinephrine. The primary mechanism underlying low SVR includes release of vasodi-latory mediators such as endothelins and nitric oxide by the portal endothelium, which is exacerbated by triggers of ACLF (86). The normal compensatory mechanisms which occur in response to low SVR underlie the formation of ascites, the functional renal failure of cirrhosis—HRS—and hyponatre-mia. HRS and cirrhotic ascites have the same basic pathogen-esis (Fig. 127.4). As outlined above, renal arterial constriction occurs in normal compensation for systemic hypotension. Poor renal perfusion results in sodium retention, plasma vol-ume expansion, and, in the presence of hepatic sinusoidal hypertension, the transudation of lymph across the Glisson capsule as low-protein ascites (88). HRS can be considered an exaggeration of this renal vasoconstriction, often in the setting of cardiac hypocontractility (89).
Diagnosis
Cardiovascular Manifestations
In addition to the circulatory and neurohumoral features of AD and ACLF discussed above, patients with decompensated cirrhosis have impaired cardiac contractility, particularly after infection or GI bleeding (90). Myocardial failure was
LWBK1580_C127e_p281-296.indd 287 29/07/17 12:02 PM

e288 SECTion 13 Gastrointestinal disease and dysfunCtion
formerly ascribed to the ethanol toxicity or iron deposition in myocardium; however, cirrhotic cardiomyopathy, depressed myocardial contractility as a complication of cirrhosis per se, has been recognized as a distinct clinical entity. Diag-nostic criteria of cirrhotic cardiomyopathy include blunted inotropic and chronotropic responses to stress, diastolic dysfunction, and prolonged QT interval on electrocardio-gram (ECG). A pathogenic role of cirrhotic cardiomyopathy has been documented in patients with HRS, particularly in the setting of infection, and in the circulatory dysfunction after large volume paracentesis without adequate plasma expansion (89,91,92).
Renal Manifestations
AKI in patients with decompensated cirrhosis is an indepen-dent predictor of death in the ICU (93), frequently signals the onset of infection (94), and is an integral component of ACLF (86,95). The definition of AKI in cirrhosis is based upon the modified Risk, Injury, Failure, Loss, End-stage renal disease (RIFLE) criteria rather than a fixed serum creatinine (96). The differential diagnosis of AKI in patients with cir-rhosis includes prerenal azotemia, HRS, and acute tubular necrosis (ATN). Analysis of urine sediment and sodium differ-entiate the above possibilities: the former two diagnoses pres-ent with normal urine sediment and low (<10 mEq/L) urine sodium, and the latter with renal tubular cell debris and high urine sodium. The distinction of these causes of renal failure remains paramount, since in its late stages, HRS portends a very poor prognosis and is generally irreversible without OLT, in contrast to prerenal azotemia and ATN. In practical terms, the diagnosis of HRS is often made after the exclusion of sep-tic shock, intrinsic renal disease, obstructive uropathy, and most important, prerenal azotemia, the latter after a 1.5-L IV fluid challenge—normal saline with or without colloid (Table 127.4) (97).
Infectious Manifestations
Bacterial infections represent the most common trigger for ACLF and remain one of the two primary causes of death (84,95). Risk factors for bacterial infections in hospitalized
patients with cirrhosis include ICU admission and GI bleeding (98). Patients with cirrhosis are relatively immunocompromised as a result of portal hypertension and immune dysfunction. Portal hypertension results in the formation of a low-protein ascites, which is susceptible to infection because of its low complement concentration and, thus, low opsonic activity (99). In addition, gut congestion from portal hypertension increases the likelihood of bacterial translocation into blood, which seeds the ascites secondarily, the so-called spontaneous bacterial peritonitis (SBP) (100).
Most studies of bacterial infections in patients with cirrho-sis were performed in the 1980s, during which community-acquired, gram-negative infections (urinary tract infections and SBP) predominated. More recent studies, however, have documented an evolution of the epidemiology of infection in patients with cirrhosis. SBP remains the most common bacte-rial infection in patients admitted to the ICU, but a shift toward gram-positive infections has occurred. In one major hepatic disease ICU, 77% of isolates were gram-positive, which was ascribed to the widespread use of prophylactic fluoroquino-lones in cirrhotic patients with low-protein ascites (101), and to the frequent use of invasive procedures, including IV cath-eter insertion and variceal band ligation (102).
Na/waterretention
Systemicvasodilation
Arterialunderfilling
ADHAldosterone
Free waterretention
Renalvasoconstriction
Expandedplasma volume
Hyponatremia HRS
Portal/sinusoidalhypertension
Ascites
Cirrhosis
NorepinephrineAngiotensin
NO
Lymph formation> absorption
Release ofhormones
FIgURE 127.4 Pathogenesis of ascites, hepatorenal syn-drome, and hyponatremia in patients with decomposed cirrhosis. vasoactive substances are shown in italics. adH, antidiuretic hormone/vasopressin; Hrs, hepatorenal syndrome; no, nitric oxide. (adapted from sandhu Bs, sanyal aJ. Management of ascites in cirrhosis. Clin Liver Dis. 2005;9[4]:715–732.)
TablE 127.4 Diagnostic Criteria of Hepatorenal Syndrome
Major criteria•low Gfr (creatinine >2.5 mg/dl; CrCl <20 ml/min)•absence of shock, infection, nephrotoxins•absence of improvement after 1.5-l fluid challenge•absence of intrinsic renal disease•Proteinuria less than 500 mg/d•normal renal ultrasound
Minor criteria•oliguria (less than 500 ml/d)•urine sodium less than 10 meq/l•serum sodium less than 130 meq/l
Gfr, glomerular filtration rate; CrCl, creatinine clearance.data from arroyo v, Gines P, Gerbes al, et al. definition and diagnostic criteria of
refractory ascites and hepatorenal syndrome in cirrhosis. international ascites Club. Hepatology. 1996;23(1):164–176.
LWBK1580_C127e_p281-296.indd 288 29/07/17 12:02 PM

CHAPTER 127 liver failure: acute and acute-on-Chronic e289
Gastrointestinal Manifestations
Acute upper gastrointestinal (UGI) bleeding, presenting as hematemesis and/or melena, remains one of the three most common indications for admission of patients with cirrhosis to the ICU and a common precipitant of ACLF. Esophageal varices account for most UGI bleeds in patients with cirrhosis, with gastric varices accounting for approximately 5% to 10% (103), and nonvariceal UGI pathology (gastric or duodenal mucosal lesions) noted in up to 30%. Other uncommon causes of UGI bleeding associated with cirrhosis include portal hyper-tensive gastropathy and gastric antral vascular ectasia, which more often present with occult GI bleeding and anemia (104). Therefore, upper endoscopy must be performed in all patients admitted to the ICU with acute UGI bleeding to identify its source as well as administer therapy.
Pulmonary Manifestations
Respiratory failure accounts for up to 40% of admissions of cirrhotic patients to the ICU. The differential diagnosis of respi-ratory distress in patients with cirrhosis may be categorized into complications of cirrhosis, pulmonary vascular diseases resulting from portal hypertension, and primary liver diseases with cardiopulmonary manifestations (Table 127.5) (105). Pulmonary complications of cirrhosis include massive ascites with compression of the lungs and diaphragm, and hepatic hydrothorax (HH), the accumulation of extracellular fluid with similar protein characteristics as ascites (low-protein) within the pleural space (106). HH usually occurs in the right pleural space (85%) and may occur in the absence of obvious ascites as a result of negative intrathoracic pressure during inspira-tion. Pulmonary vascular complications may also cause respi-ratory failure in patients with cirrhosis. The hepatopulmonary syndrome (HPS) is defined as a widened alveolar–arterial oxy-gen gradient due to intrapulmonary vasodilation in a patient with liver disease. The pathogenesis of HPS remains obscure but likely results from the release of vasoactive mediators from the liver, which increase intrapulmonary nitric oxide produc-tion (Fig. 127.5). In a patient with cirrhosis and resting hypox-emia (PaO2 <70 mmHg while breathing an FiO2 of 0.21), the
diagnosis is confirmed by a contrast echocardiogram, in which agitated saline administered intravenously delivers microbub-bles into the left ventricle at least three heartbeats after their appearance in the right ventricle (107).
In contrast to HPS, portopulmonary hypertension (PPH) is the development of increased pulmonary vascular resistance due to vasoconstriction and subsequent vascular remodeling in a patient with portal hypertension (Fig. 127.6) (108). Screen-ing with transthoracic echocardiography reveals evidence of pulmonary hypertension—right ventricular systolic pressure greater than 50 mmHg—but the diagnosis must be confirmed with right heart catheterization showing elevated mean pulmo-nary artery (PA) pressure (PAP >25 mmHg) as well as high pul-monary vascular resistance—more than 240 dynes/second per cm–5—and normal pulmonary capillary wedge pressure (109).
Neurologic Manifestations
Changes in mental status frequently accompany admission of patients with cirrhosis to the ICU and should not automatically suggest the presence of HE (Table 127.6). Usually, HE presents as a global decline in cognition and intellect, but focal neurologic
TablE 127.5 Differential Diagnosis of Respiratory Distress and Hypoxemia in Patients with Cirrhosis
Complications of cirrhosis•Massive ascites•Hepatic hydrothorax•Muscle wasting•aspiration pneumonia
Pulmonary vascular disorders due to portal hypertension•Hepatopulmonary syndrome•Portopulmonary hypertension
liver diseases with cardiopulmonary manifestations•alpha-1-antitrypsin deficiency (basilar emphysema)•sarcoidosis (restrictive lung disease, cardiomyopathy)•Hemochromatosis (cardiomyopathy)•ethanol (cardiomyopathy)
data from arguedas Mr, fallon MB. Hepatopulmonary syndrome. Clin Liver Dis. 2005;9(4):733–746.
Vasoactive mediators(e.g., endothelins)
Precapillary vasodilation
A-V shunting
Hypoxemia
DiagnosisPathophysiology
DyspneaClubbingCyanosis
ABG: PaO2 <70 mmHg CXR: clearPFT: normal spirometry
Contrast echo: shuntingChest CT: peripheral vasodilation
Pulmonary NO
Portal hypertension
FIgURE 127.5 Pathophysiology and diagnostic algorithm of hepatopulmonary syndrome. aBG, arterial blood gas; a-v, arteriovenous; Ct, computed tomography; CXr, chest x-ray film; echo, echocardiogram; no, nitric oxide; Pft, pulmo-nary function testing.
LWBK1580_C127e_p281-296.indd 289 29/07/17 12:02 PM

e290 SECTion 13 Gastrointestinal disease and dysfunCtion
deficits and signs of cerebral edema—decerebrate posturing and seizures—have also been described (110). After screening for toxic and metabolic derangements, a severely obtunded patient should undergo non–contrast-enhanced head CT to rule out intracranial bleeding. HE may then be diagnosed on clinical grounds after ruling out the above; high serum ammonia levels may help confirm, but are not necessary to make, the diagnosis. The presentation of a patient with advanced-grade HE to the ICU should prompt a search for precipitating factors, particu-larly infection and UGI bleeding (Table 127.7). The administra-tion of broad-spectrum antibiotics should be considered until negative cultures have returned, and fluid and electrolyte abnor-malities should be corrected. If sedation is required for proce-dures, benzodiazepines should be used with caution, since they can exacerbate even subclinical HE.
Treatment
Cardiovascular Complications
The treatment of heart failure in the setting of decompen-sated cirrhosis remains poorly defined; due to the underlying
hemodynamic abnormalities, afterload reduction with angiotensin-converting enzyme inhibitors may precipitate pro-found hypotension and renal failure, and cardiac glycosides and β-adrenergic agonists have been shown to be relatively ineffective.
Renal Complications and ascites
The treatment of ascites and azotemia will be considered together, since they share common pathogenic mechanisms. Ascites in the ICU is usually approached first with sodium restriction, and the judicious administration of diuretics, as long as the patient does not have marked azotemia (creatinine >2 g/dL), electrolyte abnormalities, or hypotension. A combina-tion of furosemide and spironolactone has been shown to effect a diuresis better than either agent alone; a ratio of 40 mg of the former to 100 mg of the latter administered by mouth has been shown empirically to preserve potassium balance in most patients with cirrhosis (111); the administration of IV albumin (12.5 g/day) may improve the efficacy of diuretics. Large-vol-ume paracentesis should be performed if ascites interferes with ventilation of the patient or if diuretics result in azotemia or
ECG: RVHCXR: heart size PFT: normal spirometry
Right heart cath:- PAP- PVR- normal PCWP
Vasoactive mediators
DiagnosisPathophysiology
DyspneaChest painFatigueJVD
Portal hypertension
Pulmonary arteriolarconstriction
Echo: TR, RVH
Medial hypertrophy ofpulmonary arterioles
Obliteration of
pulmonary arteriolesFIgURE 127.6 Pathophysiology and diagnostic algorithm of portopulmonary hypertension. eCG, electrocardiogram; Jvd, jugular venous distention; PaP, pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; Pft, pulmonary function testing; Pvr, pulmonary vascular resistance; rvH, right ventricu-lar hypertrophy; tr, tricuspid valve regurgitation.
TablE 127.6 Differential Diagnosis of altered Mental Status in Patients with Cirrhosis
•Hepatic encephalopathy•electrolyte abnormalities
• Hyponatremia• Hypokalemia• Hypomagnesemia
•Hypoglycemia• uremia• intracranial bleeding
• subdural hematoma• subarachnoid hemorrhage
•alcohol and/or drugs• intoxication• Withdrawal
TablE 127.7 Precipitating Events and Mechanisms of Hepatic Encephalopathy in Patients with Cirrhosis
Event Mechanism
•excessive protein ingestion ↑ Gut ammonia production
•Constipation
•Gastrointestinal bleeding
•Portosystemic shunting ↓ neurotoxin clearance
•fever, infection (ammonia, endogenous benzodiazepines)
•dehydration, azotemia ↓ renal excretion of ammonium
•Hypokalemia
•sedatives (benzodiazepines)
↑ inhibitory neurotransmission (gamma-aminobutyric acid)
LWBK1580_C127e_p281-296.indd 290 29/07/17 12:02 PM

CHAPTER 127 liver failure: acute and acute-on-Chronic e291
electrolyte abnormalities. IV colloid administration—albumin 6 to 8 g for each liter of ascites removed—should accompany paracentesis of 5 L or more to prevent postparacentesis circula-tory dysfunction (112). The insertion of a transjugular intrahe-patic portosystemic shunt (TIPS) may be considered in patients who have failed medical therapy (113). In an ICU setting, how-ever, patients are often too ill to consider TIPS for this indica-tion, although not necessarily for the indication of refractory variceal bleeding (see below).
The optimal treatment of HRS remains undefined. Figure 127.7 outlines potential treatments, not mutually exclusive, related to the pathogenic mechanisms of HRS. The ultimate treatment of the hemodynamic abnormalities of cirrhosis is OLT, which can completely reverse HRS if performed rela-tively soon after its onset (114). Albumin infusions (25 to 50 g/day) are commonly administered. Vasoconstrictor ther-apy holds promise for reversing HRS by reversing the state of systemic vasodilation. The oral α-adrenergic agonist mido-drine (5 to 10 mg orally thrice daily) was found to be effective in one widely cited but preliminary study, when used in com-bination with octreotide (100 μg subcutaneously twice daily) which increases systemic blood volume and thus renal blood flow by counteracting splanchnic vasodilation (115). Terlip-ressin, an intravenously administered vasopressin analog with fewer ischemic complications, also reverses systemic vasodi-lation and has been found to effectively reverse HRS, but is not yet available in the United States; it may be more effec-tive than midodrine/octreotide (116); norepinephrine, which is readily available, may be an effective alternative (117). Elec-trolyte abnormalities should be corrected; hyponatremia and hypomagnesemia result from furosemide administration, and hyperkalemia from spironolactone administration. Hypona-tremia also results from hemodilution in the setting of high vasopressin release from the neurohypophysis, exacerbates hepatic encephalopathy, and portends a poor prognosis (118).
Infectious Complications
Any patient with cirrhosis admitted to an ICU with clinical suspicion of sepsis should immediately receive empiric IV
antibacterial agents to cover gram-positive as well as gram-negative organisms until cultures and sensitivities allow narrowing of the regimen; empiric vancomycin should be con-sidered in patients who have been instrumented. The choice of coverage for gram-negative bacilli should be a third-genera-tion cephalosporin—cefotaxime (2 g IV every 8 hours) or cef-triaxone (1 g IV every 24 hours) (111,119)—aminoglycosides should be avoided except in serious infections with a multiply resistant organism because of the susceptibility of cirrhotic patients to aminoglycoside nephrotoxicity. Determinants of in-hospital mortality of cirrhotic patients with sepsis include inappropriate or delayed empiric antimicrobial therapy, and the use of a single rather than combined agents (120).
Diagnostic paracentesis should be performed on all cir-rhotic patients admitted to the ICU with ascites and renal failure, HE, or any evidence of infection (111). Localizing symptoms and signs of peritonitis—abdominal pain, fever, rebound tenderness—may be absent in up to 30% of patients with SBP. Ascites should be immediately inoculated into cul-ture bottles at the bedside, which has been shown to increase culture yields (121); however, even bedside inoculation with a large volume of ascites (20 mL) yields positive cultures in only 40% to 50% (102). Therefore, the diagnosis of SBP should rely on a PMN count 250 cells/μL or more; culture-negative neutrocytic ascites—with 250 or more PMNs/μL—should be considered the equivalent of SBP (122). Patients with ascites and this PMN count should receive a third-generation cepha-losporin and IV albumin—1.5 g/kg at diagnosis and 1.0 g/kg 48 hours after diagnosis—which has been shown both to decrease the incidence of HRS and to improve mortality (123). A similar diagnostic and therapeutic algorithm should be followed for spontaneous bacterial empyema, the infectious equivalent of SBP in patients with HH. The incidence of fun-gal infections also increases in cirrhotic patients admitted to the ICU, including with invasive Aspergillus; Candida species also rarely infect cirrhotic ascites, but with disastrous outcome when it occurs.
The mortality of cirrhotic patients admitted to the ICU with infection remains high, with death usually from hepatic failure, HRS, or refractory septic shock. As with patients with ALF, rel-ative adrenal insufficiency commonly accompanies decompen-sated cirrhosis and sepsis, and refractory shock in such patients may respond to stress doses of corticosteroids (124).
Gastrointestinal Complications
Despite the trend toward improved survival after variceal hem-orrhage in the last two decades (125), each episode still carries a mortality risk of 10% to 20%, and rebleeding remains an important concern especially during the first few days after the index bleed (Fig. 127.8). General resuscitative measures on admission to the ICU should include correction of hypo-tension, repletion of blood—not to exceed a hemoglobin of approximately 8 to 9 g/dL, as further repletion increases por-tal pressure—and consideration of endotracheal intubation before endoscopy (Table 127.8). A recent prospective, random-ized study of transfusion strategies in patients with UGI bleed-ing has suggested that a restrictive strategy, in which patients received red blood cell transfusion only when hemoglobin dropped to less than 7 g/dL, was associated with significantly higher survival, fewer complications, and lower portal pres-sures in patients with Child-Pugh A/B class cirrhosis compared to a more liberal transfusion strategy, administered when the
Systemicvasodilation
Renal arterialunderfilling
Angiotensin
Renal vasoconstriction HRS
avoidNSAIDs
[albumin(in SBP)]
[octreotide]
Vasodilatory renalprostaglandins
Sympathetictone
Norepinephrine
NO
[liver transplantation]
[midodrine][Terlipressin]
RAAsystem
Cirrhosis
FIgURE 127.7 Pathogenesis and treatment of hepatorenal syndrome (Hrs). interventions are indicated in bracketed italics. Hrs, hepatorenal syndrome; nsaids, nonsteroidal anti-inflammatory drugs; raa, renin–angiotensin–aldosterone axis; sBP, spontaneous bacterial peritonitis.
LWBK1580_C127e_p281-296.indd 291 29/07/17 12:02 PM

e292 SECTion 13 Gastrointestinal disease and dysfunCtion
hemoglobin dropped to less than 9 g/dL (126). Endotracheal intubation prior to endoscopy may decrease the risk of fatal massive aspiration of blood, but does not decrease the risk of aspiration pneumonia (127). Suppression of gastric acid secre-tion with proton pump inhibitors has been shown to decrease the number, size, and complications from post–band ligation esophageal ulcers (128).
In the last 20 years, three major improvements in the man-agement of acute variceal hemorrhage have increased survival of patients with cirrhosis: the early administration of vasoac-tive agents to decrease portal pressure; the widespread use of variceal band ligation; and antibiotic prophylaxis at the time of acute bleed (Table 127.8). Vasopressin, formerly used for lowering portal pressure for this indication, has fallen from favor due to vasospastic adverse effects; the vasopressin analog terlipressin has a better safety profile and appears to improve control of acute variceal bleeding (129). The soma-tostatin analog octreotide—100 μg IV bolus followed by 50 μg/hr as an IV infusion—also has a favorable safety pro-file, and a meta-analysis has demonstrated improved rates of sustained bleeding control (130); it should be administered as early as possible.
Endoscopic therapy after stabilization of the patient remains the definitive treatment for bleeding esophageal
varices and controls active bleeding in more than 75% of cases when combined with vasoactive therapy (131); recur-rent bleeding should prompt a second attempt at endoscopic treatment in most cases. In patients with recurrence after a second endoscopic treatment, or in any recurrence with hemo-dynamic instability, emergent insertion of a TIPS should be considered. Recent series have also shown that early TIPS in cirrhotics with a high risk of failure of medical management—Child C cirrhosis, or Child B cirrhosis with active bleeding on EGD—is associated with much lower failure to control bleed-ing, rebleeding, and borderline higher survival than continued medical management (132). Patients with acute hemorrhage from fundic gastric varices present a particular therapeutic challenge because the bleeding is more profuse and interven-tions have been less successful in controlling the acute bleed (103). Vasoactive therapy should be administered as for bleed-ing from esophageal varices, and tissue adhesive therapy may be attempted with cyanoacrylate glue (133). In the absence of endoscopic and vasoactive control of gastric variceal bleeding, insertion of a large tamponade balloon (Linton tube) can tem-porize control before a TIPS procedure.
In previous periods, bacterial infection complicated acute variceal bleeding in 40% of patients and contributed to renal failure and recurrent early bleeding (134). Antibiotic prophy-laxis after variceal hemorrhage has been shown to decrease the incidence of infection, as well as variceal rebleeding, result-ing in improved survival (134,135). Although oral or IV fluo-roquinolones—norfloxacin 400 mg orally/day or ofloxacin 200 mg IV twice daily—have been more thoroughly studied for this purpose, a randomized trial has suggested superior efficacy of cephalosporins—for example, ceftriaxone, 1 g IV daily—because of the widespread development of resistance to the former (136); however, local resistance patterns should be considered.
Pulmonary Complications
The prognosis of patients who develop refractory HH, which is diagnosed by the serum-to-pleural fluid albumin gradi-ent similar to the serum-to-ascites albumin gradient (SAAG) (137), is very poor unless the patient undergoes OLT (137). The basic treatment of HH includes diuretic administration as for ascites, and therapeutic thoracentesis. Placement of a TIPS in patients with refractory HH may be considered but is not universally effective, and relapse-free 1-year survival is only 35% (138). Chest tube placement and pleurodesis are relatively contraindicated in refractory HH as they often contaminate the pleural space, precluding OLT (139). An infectious complication of HH, spontaneous bacterial empy-ema, should be diagnosed and managed similarly to SBP, and has a very high mortality (140).
Stops spontaneously(50–60%)
Doesn’t stop(40–50%)
Stops withtherapy
Exsanguination(5–8%)
Rebleeding(30–40%)
<1 wk fromindex bleed
50%
1–6 wk fromindex bleed
50%
Overall mortality:15–20%/bleeding episode
Bleeding
FIgURE 127.8 natural history of acute esophageal variceal hemorrhage in patients with cirrhosis. (adapted from de franchis r, Primignani M. nat-ural history of portal hypertension in patients with cirrhosis. Clin Liver Dis. 2001;5[3]:645–663.)
TablE 127.8 Specific Management of acute Variceal Hemorrhage and Its Complications
Therapeutic Maneuver Dose/Route/Indication References
transfusion of rBC to hemoglobin of 7–8 g/dl villanueva et al., 2013 (152)
octreotide 100 μg iv bolus, then 50 μg/h for 5 d Corley et al., 2001 (130)
endoscopy evl preferred over evs de franchis and Primignani, 1999 (153)
Proton pump inhibitors Pantoprazole 40 mg iv or Po/d for 9 d shaheen et al., 2005 (128)
antibiotic prophylaxis norfloxacin 400 mg/d for 7 d, or ceftriaxone 1 g/d for 7 d
Bernard et al., 1999 (135); fernandez et al., 2006 (136)
tiPs after two failed therapeutic endoscopies Mihas and sanyal, 2004 (154)
rBC, red blood cells; evl, esophageal variceal ligation; evs, endoscopic variceal sclerotherapy; tiPs, transjugular intrahepatic portosystemic shunt.
LWBK1580_C127e_p281-296.indd 292 29/07/17 12:02 PM

CHAPTER 127 liver failure: acute and acute-on-Chronic e293
Supplemental oxygen usually bridges patients with HPS to OLT, which improves or reverses the process in 85% of patients (141). Patients with HPS have increased transplant waiting-list mortality when compared to patients with normal gas exchange; consequently, patients with HPS and PaO2 less than 60 mmHg on room air are allowed increased priority for OLT under the current organ allocation system in the United States. Perioperative mortality after OLT in patients with HPS varies according to the degree of shunting and hypoxemia (141,142). The optimal treatment of PPH—indicated when mean PAP is more than 35 mmHg—has not been well defined (109); prostacyclin analogs (epoprostenol titrated via PA cath-eter; inhaled iloprost [5 μg six times daily]) phosphodiesterase inhibitors, endothelin receptor antagonists, or combination therapy appear to be effective (143). PPH does not always reverse after OLT, and therefore, patients with mean PAP more than 35 mmHg after maximal medical treatment are often not offered transplant.
Management of Neurologic Complications
The specific treatment of HE poses special challenges in the ICU. The standard therapy, oral lactulose, must be adminis-tered via nasogastric tube in an intubated patient, cannot be given if there is an ileus, and its overzealous administration risks aspiration pneumonia, gaseous distention of the bowel, toxic megacolon, and electrolyte imbalance. Rectal lactulose offers an alternative route of administration, but its efficacy over tap water or saline enemas is unknown. The “nonabsorb-able” antibiotic neomycin should be avoided, as the absorp-tion of even small quantities from the gut risks renal injury. Rifaximin, a rifampin derivative that also decreases gut flora production of neurotoxins, appears to be as effective as lactu-lose, and has a good safety profile (144). The benzodiazepine receptor antagonist flumazenil (1 mg IV) improves HE, but the benefit wanes within 2 hours (145). Extracorporeal albumin dialysis also improves HE in refractory cases and may be con-sidered as a bridge to OLT (146).
Controversies
A major source of controversy in managing patients with decompensated cirrhosis revolves around the perception of bleeding risk. Patients with stable cirrhosis develop AD and ACLF as a result of bleeding, usually from the UGI tract. However, management of this type of bleeding, which results from portal hypertension, is not very controversial. The con-troversy lies in whether patients who present with a variceal bleed should have their INR and platelet count treated, and if so, the goals of treatment, especially since the transfusion of high volumes of plasma raises portal pressures, and may increase the risk of rebleeding (147). Major controversy con-tinues to involve the prophylactic use of platelets and plasma in patients with cirrhosis before invasive procedures. Research in the last 10 years has started to help make clinicians more comfortable with conservative repletion of factors and blood products, beginning with the seminal observations by Tripodi et al. (148), who described in vitro assay conditions that demon-strated that patients with cirrhosis generate as much thrombin as normal healthy controls. A second landmark study by the same group showed that a platelet count of approximately 60 × 109 cells/L was sufficient to preserve thrombin generation at or near the 90th percentile of normal healthy controls (149).
Subsequent studies in patients with cirrhosis have suggested that, despite the consequences of portal hypertension that lead to bleeding, patients may in fact be hypercoagulable (150). Although many questions remain unanswered, and guidelines to help the clinician remain undefined, the area is being vigor-ously studied.
• ALF is a clinical syndrome with more than a dozen causes, few of which have etiology-specific treatments. N-acetylcysteine may be an appropriate treatment for all etiologies.
• The roles of direct ICP monitoring and therapeutic hypothermia in managing patients with ALF and cere-bral edema remain very controversial.
• The three most common causes of death in patients with ALF are cerebral edema/intracranial hypertension/brainstem herniation, infection, and multiorgan system failure.
• OLT is a highly effective treatment for ALF but must be judiciously applied, as many patients recover spontane-ously, organs are scarce, and long-term complications of OLT remain considerable. Therefore, prediction of death without OLT is of paramount importance.
• Patients with cirrhosis are admitted to the ICU most commonly as the result of acute hepatic decompensa-tion, presenting as infection and/or portal hypertensive UGI bleeding.
• ACLF has recently been recognized as a distinct clini-cal entity consisting of acute hepatic decompensation in the presence of extrahepatic organ failure.
• ACLF has a high short-term mortality, which parallels the degree of multiorgan system failure. The most com-mon extrahepatic manifestation of ACLF, renal failure, portends the worst prognosis.
• Patients with ACLF should receive empiric, multiple antimicrobial agents as soon as possible, until screening tests for infection (including diagnostic paracentesis) have either returned negative or identified a responsible source and organism.
Key Points
References 1. Trey C, Davidson C. The management of fulminant hepatic failure. In: eds.
Progress in Liver Disease. 1970:282–298. 2. Lee WM. Acute liver failure in the United States. Semin Liver Dis.
2003;23(3):217–226. 3. Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute
liver failure: results of a United States multicenter, prospective study. Hep-atology. 2005;42(6):1364–1372.
4. Lee WM, Stravitz RT, Larson AM. Introduction to the revised American Association for the Study of Liver Diseases Position Paper on acute liver failure 2011. Hepatology. 2012;55(3):965–967.
5. Antoniades CG, Berry PA, Wendon JA, Vergani D. The importance of immune dysfunction in determining outcome in acute liver failure. J Hepa-tol. 2008;49(5):845–861.
6. Stravitz RT, Bowling R, Bradford RL, et al. Role of procoagulant mic-roparticles in mediating complications and outcome of acute liver injury/acute liver failure. Hepatology. 2013;58(1):304–313.
7. Jalan R. Intracranial hypertension in acute liver failure: pathophysiologi-cal basis of rational management. Semin Liver Dis. 2003;23(3):271–282.
8. Blei AT. The pathophysiology of brain edema in acute liver failure. Neuro-chem Int. 2005;47(1-2):71–77.
LWBK1580_C127e_p281-296.indd 293 29/07/17 12:02 PM

e294 SECTion 13 Gastrointestinal disease and dysfunCtion
9. Bernal W, Hall C, Karvellas CJ, et al Arterial ammonia and clinical risk factors for encephalopathy and intracranial hypertension in acute liver failure. Hepatology. 2007;46(6):1844–1852.
10. Cordoba J. Glutamine, myo-inositol, and brain edema in acute liver fail-ure. Hepatology. 1996;23(5):1291–1292.
11. Cordoba J, Gottstein J, Blei AT. Glutamine, myo-inositol, and organic brain osmolytes after portocaval anastomosis in the rat: implications for ammonia-induced brain edema. Hepatology. 1996;24(4):919–923.
12. Wendon JA, Harrison PM, Keays R, Williams R. Cerebral blood flow and metabolism in fulminant liver failure. Hepatology. 1994;19(6):1407–1413.
13. Aggarwal S, Obrist W, Yonas H, et al. Cerebral hemodynamic and meta-bolic profiles in fulminant hepatic failure: relationship to outcome. Liver Transpl. 2005;11(11):1353–1360.
14. Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology. 2003;125 (3):755–764.
15. Strauss G, Hansen BA, Kirkegaard P, et al. Liver function, cerebral blood flow autoregulation, and hepatic encephalopathy in fulminant hepatic fail-ure. Hepatology. 1997;25(4):837–839.
16. Strauss GI, Hogh P, Moller K, et al. Regional cerebral blood flow dur-ing mechanical hyperventilation in patients with fulminant hepatic failure. Hepatology. 1999;30(6):1368–1373.
17. O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prog-nosis in fulminant hepatic failure. Gastroenterology. 1989;97(2):439–445.
18. Halme L, Karkkainen P, Isoniemi H, et al. Carbohydrate 19-9 antigen as a marker of non-malignant hepatocytic ductular transformation in patients with acute liver failure: a comparison with alpha-fetoprotein and carcino-embryonic antigen. Scand J Gastroenterol. 1999;34(4):426–431.
19. Bernuau J. Criteria for emergency liver transplantation in patients with acute viral hepatitis and factor V below 50% of normal: a prospective study [Abstract]. Hepatology. 1991;14:49A.
20. Bernuau J, Rueff B, Benhamou JP. Fulminant and subfulminant liver fail-ure: definitions and causes. Semin Liver Dis. 1986;6(2):97–106.
21. Pereira LM, Langley PG, Hayllar KM, et al. Coagulation factor V and VIII/V ratio as predictors of outcome in paracetamol induced fulminant hepatic failure: relation to other prognostic indicators. Gut. 1992;33 (1):98–102.
22. Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indica-tors. Hepatology. 1993;18(6):1370–1376.
23. Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of out-come in severe acetaminophen-induced hepatotoxicity. Hepatology. 2002; 36(3):659–665.
24. Bernal W, Donaldson N, Wyncoll D, Wendon J. Blood lactate as an early predictor of outcome in paracetamol-induced acute liver failure: a cohort study. Lancet. 2002;359(9306):558–563.
25. Clemmesen JO, Larsen FS, Kondrup J, et al. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology. 1999;29(3):648–653.
26. Mitchell I, Bihari D, Chang R, et al. Earlier identification of patients at risk from acetaminophen-induced acute liver failure. Crit Care Med. 1998;26(2):279–284.
27. Fikatas P, Lee JE, Sauer IM, et al. APACHE III score is superior to King’s College Hospital criteria, MELD score and APACHE II score to predict outcomes after liver transplantation for acute liver failure. Transplant Proc. 2013;45(6):2295–2301.
28. Parkash O, Mumtaz K, Hamid S, et al. MELD score: utility and compari-son with King’s College criteria in non-acetaminophen acute liver failure. J Coll Physicians Surg Pak. 2012;22(8):492–496.
29. Makin A, Williams R. The current management of paracetamol overdos-age. Br J Clin Pract. 1994;48(3):144–148.
30. Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose: nalysis of the national multicenter study (1976 to 1985). N Engl J Med. 1988; 319(24):1557–1562.
31. Smilkstein MJ, Bronstein AC, Linden C, et al. Acetaminophen overdose: a 48-hour intravenous N-acetylcysteine treatment protocol. Ann Emerg Med. 1991;20(10):1058–1063.
32. Harrison PM, Keays R, Bray GP, et al. Improved outcome of paracetamol-induced fulminant hepatic failure by late administration of acetylcysteine. Lancet. 1990;335(8705):1572–1573.
33. Kumar M, Satapathy S, Monga R, et al. A randomized controlled trial of lamivudine to treat acute hepatitis B. Hepatology. 2007;45(1):97–101.
34. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856–864, 864.
35. Buckley NA, Whyte IM, O’Connell DL, Dawson AH. Oral or intrave-nous N-acetylcysteine: which is the treatment of choice for acetaminophen (paracetamol) poisoning? J Toxicol Clin Toxicol. 1999;37(6):759–767.
36. Broussard CN, Aggarwal A, Lacey SR, et al. Mushroom poisoning: from diar-rhea to liver transplantation. Am J Gastroenterol. 2001;96(11):3195–3198.
37. Peters DJ, Greene WH, Ruggiero F, McGarrity TJ. Herpes simplex-induced fulminant hepatitis in adults: a call for empiric therapy. Dig Dis Sci. 2000;45(12):2399–2404.
38. Ichai P, Duclos-Vallee JC, Guettier C, et al. Usefulness of corticosteroids for the treatment of severe and fulminant forms of autoimmune hepatitis. Liver Transpl. 2007;13(7):996–1003.
39. Karkhanis J, Verna EC, Chang MS, et al. Steroid use in acute liver failure. Hepatology. 2014;59(2):612–621.
40. Rodriguez FE, Tremosa LG, Xiol Q, et al. D-penicillamine and plasma-pheresis in acute liver failure secondary to Wilson’s disease. Rev Esp Enferm Dig. 2003;95(1):60–65.
41. Castro MA, Fassett MJ, Reynolds TB, et al. Reversible peripartum liver failure: a new perspective on the diagnosis, treatment, and cause of acute fatty liver of pregnancy, based on 28 consecutive cases. Am J Obstet Gyne-col. 1999;181(2):389–395.
42. Munoz SJ, Robinson M, Northrup B, et al. Elevated intracranial pressure and computed tomography of the brain in fulminant hepatocellular fail-ure. Hepatology. 1991;13(2):209–212.
43. Ng I, Lim J, Wong HB. Effects of head posture on cerebral hemodynam-ics: its influences on intracranial pressure, cerebral perfusion pressure, and cerebral oxygenation. Neurosurgery. 2004;54(3):593–597.
44. Basile AS, Hughes RD, Harrison PM, et al. Elevated brain concentra-tions of 1,4-benzodiazepines in fulminant hepatic failure. N Engl J Med. 1991;325(7):473–478.
45. Wijdicks EF, Nyberg SL. Propofol to control intracranial pressure in fulmi-nant hepatic failure. Transplant Proc. 2002;34(4):1220–1222.
46. Strauss G, Hansen BA, Knudsen GM, Larsen FS. Hyperventilation restores cerebral blood flow autoregulation in patients with acute liver failure. J Hepatol. 1998;28(2):199–203.
47. Murphy N, Auzinger G, Bernel W, Wendon J. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver fail-ure. Hepatology. 2004;39(2):464–470.
48. Canalese J, Gimson AE, Davis C, et al. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut. 1982;23(7):625–629.
49. Hanid MA, Davies M, Mellon PJ, et al. Clinical monitoring of intracranial pressure in fulminant hepatic failure. Gut. 1980;21(10):866–869.
50. Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol. 1998;29(4):589–594.
51. Tandon BN, Joshi YK, Tandon M. Acute liver failure: experience with 145 patients. J Clin Gastroenterol. 1986;8(6):664–668.
52. Ellis AJ, Wendon JA, Williams R. Subclinical seizure activity and prophy-lactic phenytoin infusion in acute liver failure: a controlled clinical trial. Hepatology. 2000;32(3):536–541.
53. Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure: a controlled clinical trial. J Hepatol. 2004;41(1):89–96.
54. Rolando N, Wade J, Davalos M, et al. The systemic inflammatory response syndrome in acute liver failure. Hepatology. 2000;32(4 Pt 1): 734–739.
55. Harry R, Auzinger G, Wendon J. The clinical importance of adrenal insuf-ficiency in acute hepatic dysfunction. Hepatology. 2002;36(2):395–402.
56. Beale RJ, Hollenberg SM, Vincent JL, Parrillo JE. Vasopressor and ino-tropic support in septic shock: an evidence-based review. Crit Care Med. 2004;32(11 Suppl):S455–S465.
57. Eefsen M, Dethloff T, Frederiksen HJ, et al. Comparison of terlipressin and noradrenalin on cerebral perfusion, intracranial pressure and cerebral extracellular concentrations of lactate and pyruvate in patients with acute liver failure in need of inotropic support. J Hepatol. 2007;47(3):381–386.
58. Trewby PN, Warren R, Contini S, et al. Incidence and pathophysiology of pulmonary edema in fulminant hepatic failure. Gastroenterology. 1978;74(5 Pt 1):859–865.
59. Ring-Larsen H, Palazzo U. Renal failure in fulminant hepatic failure and terminal cirrhosis: a comparison between incidence, types, and prognosis. Gut. 1981;22(7):585–591.
60. Moore K. Renal failure in acute liver failure. Eur J Gastroenterol Hepatol. 1999;11(9):967–975.
61. Davenport A, Will EJ, Davison AM. Effect of renal replacement therapy on patients with combined acute renal and fulminant hepatic failure. Kid-ney Int Suppl. 1993;41:S245–S251.
LWBK1580_C127e_p281-296.indd 294 29/07/17 12:02 PM

CHAPTER 127 liver failure: acute and acute-on-Chronic e295
62. Davenport A. Renal replacement therapy for patients with acute liver fail-ure awaiting orthotopic hepatic transplantation. Nephron. 1991;59(2): 315–316.
63. Naka T, Wan L, Bellomo R, et al. Kidney failure associated with liver transplantation or liver failure: the impact of continuous veno-venous hemofiltration. Int J Artif Organs. 2004;27(11):949–955.
64. Slack AJ, Auzinger G, Willars C, et al. Ammonia clearance with haemofil-tration in adults with liver disease. Liver Int. 2014;34(1):42–48.
65. Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis. 1996;16(4):389–402.
66. Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49–53.
67. Karvellas CJ, Cavazos J, Battenhouse H, et al. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol. 2014; 12(11):1942–1949.
68. Munoz SJ, Stravitz RT, Gabriel DA. Coagulopathy of acute liver failure. Clin Liver Dis. 2009;13(1):95–107.
69. Stravitz RT, Lisman T, Luketic VA, et al. Minimal effects of acute liver injury/acute liver failure on hemostasis as assessed by thromboelastogra-phy. J Hepatol. 2012;56(1):129–136.
70. Hugenholtz GC, Adelmeijer J, Meijers JC, et al. An unbalance between von Willebrand factor and ADAMTS13 in acute liver failure: implications for hemostasis and clinical outcome. Hepatology. 2013;58(2):752–761.
71. Lisman T, Bakhtiari K, Adelmeijer J, et al. Intact thrombin generation and decreased fibrinolytic capacity in patients with acute liver injury or acute liver failure. J Thromb Haemost. 2012;10(7):1312–1319.
72. Pereira SP, Rowbotham D, Fitt S, et al. Pharmacokinetics and efficacy of oral versus intravenous mixed-micellar phylloquinone (vitamin K1) in severe acute liver disease. J Hepatol. 2005;42(3):365–370.
73. MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute upper gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology. 1978;74(2 Pt 2):464–465.
74. Schneeweiss B, Pammer J, Ratheiser K, et al. Energy metabolism in acute hepatic failure. Gastroenterology. 1993;105(5):1515–1521.
75. Lidofsky SD, Bass NM, Prager MC, et al. Intracranial pressure monitor-ing and liver transplantation for fulminant hepatic failure. Hepatology. 1992;16(1):1–7.
76. Roberts MS, Angus DC, Bryce CL, et al. Survival after liver transplanta-tion in the United States: a disease-specific analysis of the UNOS database. Liver Transpl. 2004;10(7):886–897.
77. Karvellas CJ, Safinia N, Auzinger G, et al. Medical and psychiatric out-comes for patients transplanted for acetaminophen-induced acute liver failure: a case-control study. Liver Int. 2010;30(6):826–833.
78. Vaquero J, Fontana RJ, Larson AM, et al. Complications and use of intra-cranial pressure monitoring in patients with acute liver failure and severe encephalopathy. Liver Transpl. 2005;11(12):1581–1589.
79. Karvellas CJ, Fix OK, Battenhouse H, et al. Outcomes and complications of intracranial pressure monitoring in acute liver failure: a retrospective cohort study. Crit Care Med. 2014;42(5):1157–1167.
80. Munoz SJ, Moritz MJ, Bell R, et al. Factors associated with severe intra-cranial hypertension in candidates for emergency liver transplantation. Transplantation. 1993;55(5):1071–1074.
81. Stravitz RT, Larsen FS. Therapeutic hypothermia for acute liver failure. Crit Care Med. 2009;37(7 Suppl):S258–S264.
82. Karvellas CJ, Todd SR, Battenhouse H, et al. Therapeutic hypothermia in acute liver failure: a multicenter retrospective cohort analysis. Liver Transpl. 2015;21(1):4–12.
83. Moreau R, Jalan R, Gines P, et al. Acute-on-chronic liver failure is a dis-tinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144(7):1426–37, 1437.
84. Moreau R, Arroyo V. Acute-on-chronic liver failure: a new clinical entity. Clin Gastroenterol Hepatol. 2015;13(5):836–841.
85. Jalan R, Pavesi M, Saliba F, et al. The CLIF Consortium Acute Decompen-sation score (CLIF-C ADs) for prognosis of hospitalised cirrhotic patients without acute-on-chronic liver failure. J Hepatol. 2015;62(4):831–840.
86. Jalan R, Saliba F, Pavesi M, et al. Development and validation of a prog-nostic score to predict mortality in patients with acute-on-chronic liver failure. J Hepatol. 2014;61(5):1038–1047.
87. Bajaj JS, O’Leary JG, Reddy KR, et al. Survival in infection-related acute-on-chronic liver failure is defined by extrahepatic organ failures. Hepatology. 2014;60(1):250–256.
88. Runyon BA, Montano AA, Akriviadis EA, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117(3):215–220.
89. Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Gines P, Moreira V, et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatol-ogy. 2005;42(2):439–447.
90. Gaskari SA, Honar H, Lee SS. Therapy insight: cirrhotic cardiomyopathy. Nat Clin Pract Gastroenterol Hepatol. 2006;3(6):329–337.
91. Ruiz-del-Arbol L, Monescillo A, Jimenez W, et al. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology. 1997;113(2):579–586.
92. Ruiz-del-Arbol L, Urman J, Fernandez J, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacte-rial peritonitis. Hepatology. 2003;38(5):1210–1218.
93. du Cheyron D, Bouchet B, Parienti JJ, et al. The attributable mortality of acute renal failure in critically ill patients with liver cirrhosis. Intensive Care Med. 2005;31(12):1693–1699.
94. Terra C, Guevara M, Torre A, et al. Renal failure in patients with cirrhosis and sepsis unrelated to spontaneous bacterial peritonitis: value of MELD score. Gastroenterology. 2005;129(6):1944–1953.
95. McPhail MJ. Improving MELD for use in acute liver failure. J Hepatol. 2011;54(6):1320–1321.
96. Wong F, Nadim MK, Kellum JA, et al. Working Party proposal for a revised classification system of renal dysfunction in patients with cirrhosis. Gut. 2011;60(5):702–709.
97. Arroyo V, Gines P, Gerbes AL, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology. 1996;23(1):164–176.
98. Deschenes M, Villeneuve JP. Risk factors for the development of bacte-rial infections in hospitalized patients with cirrhosis. Am J Gastroenterol. 1999;94(8):2193–2197.
99. Runyon BA, Morrissey RL, Hoefs JC, Wyle FA. Opsonic activity of human ascitic fluid: a potentially important protective mechanism against sponta-neous bacterial peritonitis. Hepatology. 1985;5(4):634–637.
100. Runyon BA, Hoefs JC. Ascitic fluid analysis in the differentiation of spon-taneous bacterial peritonitis from gastrointestinal tract perforation into ascitic fluid. Hepatology. 1984;4(3):447–450.
101. Gines P, Rimola A, Planas R, et al. Norfloxacin prevents spontaneous bac-terial peritonitis recurrence in cirrhosis: results of a double-blind, placebo-controlled trial. Hepatology. 1990;12(4 Pt 1):716–724.
102. Fernandez J, Navasa M, Gomez J, et al. Bacterial infections in cirrhosis: epidemiological changes with invasive procedures and norfloxacin pro-phylaxis. Hepatology. 2002 Jan;35(1):140–148.
103. Ryan BM, Stockbrugger RW, Ryan JM. A pathophysiologic, gastroentero-logic, and radiologic approach to the management of gastric varices. Gas-troenterology. 2004;126(4):1175–1189.
104. Payen JL, Cales P, Voigt JJ, et al. Severe portal hypertensive gastropathy and antral vascular ectasia are distinct entities in patients with cirrhosis. Gastroenterology. 1995 Jan;108(1):138–144.
105. Arguedas MR, Fallon MB. Hepatopulmonary syndrome. Clin Liver Dis. 2005;9(4):733–746.
106. Garcia N Jr, Mihas AA. Hepatic hydrothorax: pathophysiology, diagnosis, and management. J Clin Gastroenterol. 2004;38(1):52–58.
107. Velthuis S, Buscarini E, Gossage JR, et al. Clinical implications of pulmo-nary shunting on saline contrast echocardiography. J Am Soc Echocar-diogr. 2015;28(3):255–263.
108. Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology. 2006;44(6):1502–1510.
109. Krowka MJ. Evolving dilemmas and management of portopulmonary hypertension. Semin Liver Dis. 2006;26(3):265–272.
110. Cadranel JF, Lebiez E, DiMartino V, et al. Focal neurological signs in hepatic encephalopathy in cirrhotic patients: an underestimated entity? Am J Gastroenterol. 2001;96(2):515–518.
111. Runyon BA. Management of adult patients with ascites due to cirrhosis: an update. Hepatology. 2009;49(6):2087–2107.
112. Gines P, Tito L, Arroyo V, et al. Randomized comparative study of thera-peutic paracentesis with and without intravenous albumin in cirrhosis. Gastroenterology. 1988;94(6):1493–1502.
113. Salerno F, Camma C, Enea M, et al. Transjugular intrahepatic portosys-temic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology. 2007;133(3):825–834.
114. Cardenas A, Gines P. Therapy insight: management of hepatorenal syn-drome. Nat Clin Pract Gastroenterol Hepatol. 2006;3(6):338–348.
115. Angeli P, Volpin R, Gerunda G, et al. Reversal of type 1 hepatorenal syn-drome with the administration of midodrine and octreotide. Hepatology. 1999;29(6):1690–1697.
116. Cavallin M, Kamath PS, Merli M, et al. Terlipressin plus albumin versus midodrine and octreotide plus albumin in the treatment of hepatorenal syndrome: a randomized trial. Hepatology. 2015;62(2):567–574.
LWBK1580_C127e_p281-296.indd 295 29/07/17 12:02 PM

e296 SECTion 13 Gastrointestinal disease and dysfunCtion
117. Ghosh S, Choudhary NS, Sharma AK, et al. Noradrenaline vs terlipressin in the treatment of type 2 hepatorenal syndrome: a randomized pilot study. Liver Int. 2013;33(8):1187–1193.
118. Heuman DM, bou-Assi SG, Habib A, et al. Persistent ascites and low serum sodium identify patients with cirrhosis and low MELD scores who are at high risk for early death. Hepatology. 2004;40(4):802–810.
119. Runyon BA, McHutchison JG, Antillon MR, et al. Short-course versus long-course antibiotic treatment of spontaneous bacterial peritonitis: a randomized controlled study of 100 patients. Gastroenterology. 1991; 100(6):1737–1742.
120. Arabi YM, Dara SI, Memish Z, et al. Antimicrobial therapeutic determi-nants of outcomes from septic shock among patients with cirrhosis. Hepa-tology. 2012;56(6):2305–2315.
121. Runyon BA, Antillon MR, Akriviadis EA, McHutchison JG. Bedside inoc-ulation of blood culture bottles with ascitic fluid is superior to delayed inoculation in the detection of spontaneous bacterial peritonitis. J Clin Microbiol. 1990;28(12):2811–2812.
122. Runyon BA, Hoefs JC. Culture-negative neutrocytic ascites: a variant of spontaneous bacterial peritonitis. Hepatology. 1984;4(6):1209–1211.
123. Sort P, Navasa M, Arroyo V, Aldeguer X, et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrho-sis and spontaneous bacterial peritonitis. N Engl J Med. 1999;341(6): 403–409.
124. Fernandez J, Escorsell A, Zabalza M, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: Effect of treatment with hydrocortisone on survival. Hepatology. 2006;44(5):1288–1295.
125. Carbonell N, Pauwels A, Serfaty L, et al. Improved survival after variceal bleeding in patients with cirrhosis over the past two decades. Hepatology. 2004;40(3):652–659.
126. Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med. 2013;368(1):11–21.
127. Rudolph SJ, Landsverk BK, Freeman ML. Endotracheal intubation for air-way protection during endoscopy for severe upper GI hemorrhage. Gas-trointest Endosc. 2003;57(1):58–61.
128. Shaheen NJ, Stuart E, Schmitz SM, et al. Pantoprazole reduces the size of postbanding ulcers after variceal band ligation: a randomized, controlled trial. Hepatology. 2005;41(3):588–594.
129. Ioannou GN, Doust J, Rockey DC. Systematic review: terlipressin in acute oesophageal variceal haemorrhage. Aliment Pharmacol Ther. 2003;17(1):53–64.
130. Corley DA, Cello JP, Adkisson W, et al. Octreotide for acute esopha-geal variceal bleeding: a meta-analysis. Gastroenterology. 2001;120(4): 946–954.
131. Cardenas A, Gines P. Management of complications of cirrhosis in patients awaiting liver transplantation. J Hepatol. 2005;42 Suppl(1):S124–S133.
132. Garcia-Pagan JC, Di PM, Caca K, et al. Use of early-TIPS for high-risk variceal bleeding: results of a post-RCT surveillance study. J Hepatol. 2013;58(1):45–50.
133. Garcia-Pagan JC, Barrufet M, Cardenas A, Escorsell A. Management of gastric varices. Clin Gastroenterol Hepatol. 2014;12(6):919–928.
134. Hou MC, Lin HC, Liu TT, et al. Antibiotic prophylaxis after endoscopic therapy prevents rebleeding in acute variceal hemorrhage: a randomized trial. Hepatology. 2004;39(3):746–753.
135. Bernard B, Grange JD, Khac EN, et al. Antibiotic prophylaxis for the pre-vention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology. 1999;29(6):1655–1661.
136. Fernandez J, Ruiz del AL, Gomez C, et al. Norfloxacin vs ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hem-orrhage. Gastroenterology. 2006;131(4):1049–1056.
137. Badillo R, Rockey DC. Hepatic hydrothorax: clinical features, manage-ment, and outcomes in 77 patients and review of the literature. Medicine (Baltimore). 2014;93(3):135–142.
138. Dhanasekaran R, West JK, Gonzales PC, et al. Transjugular intrahepatic portosystemic shunt for symptomatic refractory hepatic hydrothorax in patients with cirrhosis. Am J Gastroenterol. 2010;105(3):635–641.
139. Orman ES, Lok AS. Outcomes of patients with chest tube insertion for hepatic hydrothorax. Hepatol Int. 2009;3(4):582–586.
140. Tu CY, Chen CH. Spontaneous bacterial empyema. Curr Opin Pulm Med. 2012;18(4):355–358.
141. Koch DG, Fallon MB. Hepatopulmonary syndrome. Curr Opin Gastroen-terol. 2014;30(3):260–264.
142. Collisson EA, Nourmand H, Fraiman MH, et al. Retrospective analysis of the results of liver transplantation for adults with severe hepatopulmonary syndrome. Liver Transpl. 2002;8(10):925–931.
143. DuBrock HM, Channick RN, Krowka MJ. What’s new in the treatment of portopulmonary hypertension? Expert Rev Gastroenterol Hepatol. 2015;1–10.
144. Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med. 2010;362(12):1071–1081.
145. Barbaro G, Di LG, Soldini M, et al. Flumazenil for hepatic encepha-lopathy grade III and IVa in patients with cirrhosis: an Italian multi-center double-blind, placebo-controlled, cross-over study. Hepatology. 1998;28(2):374–378.
146. Heemann U, Treichel U, Loock J, et al. Albumin dialysis in cirrhosis with superimposed acute liver injury: a prospective, controlled study. Hepatol-ogy. 2002;36(4 Pt 1):949–958.
147. Giannini EG, Stravitz RT, Caldwell SH. Correction of hemostatic abnor-malities and portal pressure variations in patients with cirrhosis. Hepatol-ogy. 2014;60(4):1442.
148. Tripodi A, Salerno F, Chantarangkul V, et al. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology. 2005;41(3):553–558.
149. Tripodi A, Primignani M, Chantarangkul V, et al. Thrombin gen-eration in patients with cirrhosis: the role of platelets. Hepatology. 2006;44(2):440–445.
150. Tripodi A, Primignani M, Chantarangkul V, et al. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroen-terology. 2009;137(6):2105–2111.
151. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273–275.
152. Villanueva C, Colomo A, Bosch A, et al. Transfusion for acute upper gastrointestinal bleeding. New Engl J Med. 2013;368:11–21.
153. De Franchis R, Primignani M. Endoscopic treatments for portal hyperten-sion. Semin Liver Dis. 1999;19:439–455.
154. Mihas A and Sanyal A. Recurrent variceal bleeding despite endoscopic and medical therapy. Gastroenterology. 2004;127:621–629.
LWBK1580_C127e_p281-296.indd 296 29/07/17 12:02 PM


















