
Analysis of PAHs and their transformation products in contaminated soil...
64
Analysis of PAHs and their transformation products in contaminated soil and remedial processes Staffan Lundstedt Akademisk avhandling Som med tillstånd av rektorsämbetet vid Umeå universitet för erhållande av Filosofie Doktorsexamen vid Teknisk-naturvetenskapliga fakulteten i Umeå, framlägges till offentlig granskning vid Kemiska institutionen, hörsal KB3B1 i KBC-huset, lördagen den 24/5, 2003, klockan 10.00. Fakultetsopponent: Dr. John Parsons, Department of Environmental and Toxicological Chemistry, University of Amsterdam, The Netherlands.
Transcript of Analysis of PAHs and their transformation products in contaminated soil...

Analysis of PAHs and their transformation products in contaminated soil and
remedial processes
Staffan Lundstedt
Akademisk avhandling
Som med tillstånd av rektorsämbetet vid Umeå universitet för erhållande av Filosofie Doktorsexamen vid Teknisk-naturvetenskapliga fakulteten i Umeå,
framlägges till offentlig granskning vid Kemiska institutionen, hörsal KB3B1 i KBC-huset, lördagen den 24/5, 2003, klockan 10.00. Fakultetsopponent:
Dr. John Parsons, Department of Environmental and Toxicological Chemistry, University of Amsterdam, The Netherlands.

Copyright © 2003 Staffan Lundstedt
Department of Chemistry Environmental Chemistry
Umeå University SE-901 87 Umeå
SWEDEN
ISBN 91-7305-452-6
Printed in Sweden by Solfjädern Offset AB
Umeå 2003

Abstract Soil that is heavily contaminated with polycyclic aromatic hydrocarbons (PAHs) is often found at the sites of former gasworks and wood-impregnation plants. Since PAHs are toxic these sites represent a hazard to human health and the environment, and therefore they need to be treated, preferably by a method that destroys the contaminants, and thus eliminates the problem permanently. However, during biological and chemical degradation of PAHs other toxic compounds may be formed. If these transformation products are sufficiently persistent they could potentially accumulate during remedial processes.
In the work underlying this thesis the degradation and transformation of PAHs were studied in three remedial processes: viz. a pilot-scale bioslurry reactor, microcosms with wood-rotting fungi and lab-scale treatments with Fenton’s reagent. A group of transformation products referred to as oxygenated-PAHs (oxy-PAHs) was found to be particularly important, as these compounds are toxic and were shown to be relatively persistent in the environment. The oxy-PAHs were, for instance, found at significant concentrations in the gasworks soil used in most of the studies. This soil was highly weathered and had therefore been depleted of the more readily degradable compounds. In addition, experiments in which earthworms were exposed to the gasworks soil showed that the oxy-PAHs were more easily taken up in living organisms than PAHs.
To facilitate the studies, new extraction and fractionation methods were developed. For instance, pressurized liquid extraction (PLE) was investigated for its reliability and efficiency to extract PAHs and oxy-PAHs from soil. Furthermore, a selective PLE-method was developed that can simultaneously extract and separate the PAHs and oxy-PAHs into two different fractions. This was accomplished by adding a chromatographic material (silica or Florisil) to the extraction cell.
Under certain conditions all three remedial processes resulted in increasing amounts of oxy-PAHs in the soil. For example, 1-acenaphthenone and 4-oxapyrene-5-one accumulated in the bioslurry reactor. Similarly, in the soil inoculated with a white-rot fungus 9-fluorenone, benzo[a]anthracene-7,12-dione, 4-hydroxy-9-fluorenone and 4-oxapyrene-5-one accumulated. Finally, in an ethanol-Fenton treatment the concentration of some PAH-quinones increased in the soil.
The results show that it might be necessary to monitor oxy-PAHs as well as PAHs during the remediation of PAH-contaminated sites. Otherwise, the soil may be considered detoxified too early in the process. In the long term it would be desirable to include analyses with sufficient marker compounds to follow the possible production and elimination of the oxy-PAHs. However, until such compounds can be identified it is suggested that contaminated soil should be screened for oxy-PAHs in general. The selective PLE-method presented in this thesis could be a useful tool for this.
Keywords: Polycyclic aromatic hydrocarbons, PAHs, degradation, transformation, formation, accumulation, oxidation products, metabolites, oxygenated polycyclic aromatic hydrocarbons, oxy-PAHs, soil, gasworks, remediation, bioremediation, bioslurry, wood-rotting fungi, Fenton’s reagent, availability, bioavailability, chemical analysis, extraction, pressurized liquid extraction, PLE, accelerated solvent extraction, ASE


i
Table of contents
List of abbreviations .................................................................................................... ii
List of papers ............................................................................................................... iii
1. Introduction..............................................................................................................1
2. Polycyclic Aromatic Hydrocarbons.......................................................................3 2.1 Properties and environmental fate ............................................................................................4 2.2 Contaminated sites ......................................................................................................................5 2.3 PAH toxicity.................................................................................................................................6
3. Degradation of PAHs..............................................................................................9 3.1 Biological degradation.................................................................................................................9 3.2 Chemical degradation of PAHs .............................................................................................. 12 3.3 Transformation products of PAHs........................................................................................ 14
4. Analysis of PAHs and oxy-PAHs in soil ............................................................16 4.1 Sample pretreatment ................................................................................................................ 16 4.2 Pressurized liquid extraction ................................................................................................... 17 4.3 Clean up ..................................................................................................................................... 21 4.4 Selective extraction of PAHs and oxy-PAHs........................................................................ 23 4.5 Instrumental analysis ................................................................................................................ 24
5. The gasworks site at Husarviken .........................................................................27 5.1 Characterization ........................................................................................................................ 27 5.2 Availability of the contaminants ............................................................................................. 29
6. Degradation and formation in remedial processes ...........................................32 6.1 Bioremediation.......................................................................................................................... 32 6.2 Chemical remediation methods .............................................................................................. 36 6.3 Oxy-PAHs in remedial processes ........................................................................................... 39
7. Concluding remarks and future perspectives.....................................................42
8. Acknowledgement .................................................................................................44
9. References ...............................................................................................................45
ii
List of abbreviations
BSTFA N,O-bis(trimethylsilyl)trifluoroacetamide DCM dichloromethane FID flame ionization detector GC gas chromatography HMW high molecular weight HPLC high performance liquid chromatography Hx Hexane IARC International Agency for Research on Cancer IS internal standard Kow octanol-water partitioning coefficient LMW low molecular weight MGPs manufactured gas plants MS mass spectrometry NIST National Institute of Standards and Technology Oxy-PAHs oxygenated polycyclic aromatic hydrocarbons PACs polycyclic aromatic compounds PAHs polycyclic aromatic hydrocarbons PCBs polychlorinated bipenyls PLE pressurized liquid extraction SIM selected ion monitoring SPE solid phase extraction SRM standard reference material TMCS trimethylchlorosilane Tol toluene US-EPA United States Environmental Protection Agency

iii
List of papers
This thesis is based on the following papers, which will be referred to by their respective roman numerals.
I. Lundstedt S, van Bavel B, Haglund P, Tysklind M, Öberg LG,
Pressurised liquid extraction of polycyclic aromatic hydrocarbons from contaminated soils. Journal of Chromatography A (2000) 883, 151-162
II. Matscheko N, Lundstedt S, Svensson L, Harju M, Tysklind M
Accumulation and elimination of 16 polycyclic aromatic compounds in the earthworm (Eisenia fetida). Environmental Toxicology and Chemistry (2002) 21(8), 1724-1729
III. Andersson BE, Lundstedt S, Tornberg K, Schnürer Y, Öberg LG,
Mattiasson B, Incomplete degradation of polycyclic aromatic hydrocarbons in soil inoculated with wood-rotting fungi and their effect on the indigenous soil bacteria. Environmental Toxicology and Chemistry (2003) 22(6), 1238-1243
IV. Lundstedt S, Haglund P, Öberg LG, Degradation and formation of
polycyclic aromatic compounds during bioslurry treatment of an aged gasworks soil. Environmental Toxicology and Chemistry (2003) 22(7), 1413-1420
V. Lundstedt S, Ong R, Haglund P, Öberg LG, Simultaneous
extraction and fractionation of polycyclic aromatic hydrocarbons and their oxygenated derivatives in soil using selective pressurized liquid extraction. Submitted Manuscript
VI. Lundstedt S, Jonsson S, Persson Y, Öberg LG
Transformation of PAHs during ethanol-Fenton treatment of an aged gasworks soil. Manuscript
The published papers are reproduced with the kind permission of the respective journals.

iv

1
1. Introduction
According to the Swedish Environmental Protection Agency there are at least 38 000 contaminated sites in Sweden, most of which are associated with past or present industrial activities. The contamination mainly originates from the time before 1970 – before the days of widespread environmental concern and before strict environmental regulations were established. However, at many sites the contaminant levels remain high. People living in the vicinity may be exposed and the contaminants may spread to the surroundings. Consequently, contaminated soil and groundwater constitute a major environmental problem that needs to be resolved.
The term ‘soil remediation’ refers to actions designed to eliminate or minimize the risk associated with contaminated soil. This goal may be achieved in several different ways and the selected method depends on factors such as the contaminants present, the site conditions and the cost. The most common method in Sweden is, still, to excavate the contaminated soil and transport it to a landfill that is considered to be safe from an environmental point of view. However, for organic contaminants this is not the preferred solution. Instead, an environmentally sustainable policy should ideally be based on methods that permanently destroy the contaminants, i.e. destruction methods.
The most effective and reliable method to destroy organic contaminants in soil is incineration. However, this method is expensive, it makes the soil sterile and depletes it of all organic matter. Hence, other methods have been developed. Biological and chemical remediation methods utilize microorganisms and reactive chemicals, respectively, to accomplish the degradation. These methods also have the potential to degrade a wide variety of soil contaminants, but they usually need careful optimization.
The ultimate goal of any degradation process is complete mineralization of the organic contaminants, resulting in carbon dioxide, water and other inorganic compounds. However, during biological and chemical degradation processes partial transformation may lead to the formation of other organic compounds. This may cause problems if the transformation products are also hazardous and persistent. In the worst case it could lead to increased toxicity, even if the original contaminants have been degraded. This potential problem is seldom considered during remedial monitoring programs, in which only the original contaminants are usually analyzed.

Introduction
2
In the work described in this thesis the transformations of polycyclic aromatic hydrocarbons (PAHs) in contaminated soil have been studied. New analytical methods have been developed for improved chemical analysis of PAHs and related compounds, including transformation products (of which oxygenated PAHs were found to be particularly interesting). For the purpose of our studies, a soil from an old gasworks site was chosen. It has been thoroughly characterized with respect to PAHs and related compounds, and the bioavailability of the contaminants has been studied by exposing earthworms to the soil. Moreover, the analytical methods have been used to monitor PAHs and their transformation products in three different remedial processes: a pilot-scale bioslurry with adapted bacteria, a microcosm with wood-rotting fungi, and finally a lab-scale Fenton degradation.

3
2. Polycyclic Aromatic Hydrocarbons
Polycyclic aromatic hydrocarbons (PAHs) comprise a large and heterogeneous group of organic contaminants that are formed and emitted as a result of the incomplete combustion of organic material. Anthropogenic sources, e.g. road traffic and combustion of fossil fuels, predominate but there are also natural sources, e.g. volcanic eruptions and forest fires. PAHs consist of fused benzene rings in linear, angular or clustered arrangements (Figure 1), and contain by definition only carbon and hydrogen atoms. However, nitrogen, sulfur and oxygen atoms may readily substitute in the benzene rings to form heterocyclic aromatic compounds, which are commonly grouped with the PAHs. Furthermore, PAHs substituted with alkyl groups are normally found together with the PAHs in the environment. The whole group of PAHs and related compounds are sometimes referred to as polycyclic aromatic compounds (PACs). PAHs have been thoroughly studied due to their toxicity, persistency and environmental prevalence [1,2]. Such studies are often limited to 16 PAHs, designated as priority pollutants by the United States Environmental Protection Agency (US-EPA).
Figure 1. Structures of the 16 US-EPA PAHs and selected alkyl-PAHs and heterocyclic compounds.
CH3 CH3
CH3
O
N
S
Naphthalene Acenaphthene Acenaphthylene Fluorene Phenanthrene
1-Methylanthracene
Pyrene Fluoranthene Benzo[a]anthracene Chrysene Benzo[k]fluoranthene
Benzo[b]fluoranthene Benzo[a]pyrene Dibenz[a,h]anthracene Indeno[1,2,3-cd]pyrene Benzo[ghi]perylene
7,12-Dimethylbenzo[a]anthracene
Phenanthridine
Anthracene
Benzo[b]naphtho[2,3-d]thiopheneDibenzofuran

Polycyclic Aromatic Hydrocarbons
4
2.1 Properties and environmental fate Generally, PAHs are lipophilic compounds that show high affinity for
organic matter. However, individual PAHs differ substantially in their physico-chemical properties. As shown in Table 1, properties such as aqueous solubility and vapor pressure range in five and twelve orders of magnitude, respectively, moving from two to six benzene rings in the PAH-molecule. Thus, low molecular weight (LMW) PAHs are much more water soluble and volatile than their high molecular weight (HMW) relatives, while the HMW PAHs show higher hydrophobicity than the LMW compounds [3]. The difference in hydrophobicity is also reflected by the octanol-water partitioning coefficient (KOW) shown in Table 1. These physico-chemical properties largely determine the environmental behavior of PAHs, and indicate that transfer and turnover will be more rapid for LMW PAHs than for the heavier PAHs [4]. The semi-volatile nature of the LMW PAHs means that they exist in the atmosphere partly as vapors and are therefore highly susceptible to atmospheric degradation processes. Similarly, in aqueous environments, the LMW PAHs are partly dissolved, making them highly available for various degradation processes. The HMW PAHs, on the other hand, are primarily associated with particles in the atmosphere and water, and are therefore less available for degradation. Furthermore, PAHs adsorbed to particles may be transported over long distances in the atmosphere and are therefore ubiquitous in the environment [1,5].
Table 1. Properties of the 16 US-EPA PAHs, from Mackay et al. 1992 [3]. Number
of rings Molecular weight
Aqueous solubility (mg/l)
Vapor press. (Pa)
Log Kow
Naphthalene 2 128 31 1.0x102 3.37 Acenaphthylene 3 152 16 9.0x10-1 4.00 Acenaphthene 3 154 3.8 3.0x10-1 3.92 Fluorene 3 166 1.9 9.0x10-2 4.18 Phenanthrene 3 178 1.1 2.0x10-2 4.57 Anthracene 3 178 0.045 1.0x10-3 4.54 Pyrene 4 202 0.13 6.0x10-4 5.18 Fluoranthene 4 202 0.26 1.2x10-3 5.22 Benzo[a]anthracene 4 228 0.011 2.8x10-5 5.91 Chrysene 4 228 0.006 5.7x10-7 5.91 Benzo[b]fluoranthene 5 252 0.0015 - 5.80 Benzo[k]fluoranthene 5 252 0.0008 5.2x10-8 6.00 Benzo[a]pyrene 5 252 0.0038 7.0x10-7 5.91 Dibenzo[a,h]anthracene 6 278 0.0006 3.7x10-10 6.75 Indeno[1,2,3-cd]pyrene 6 276 0.00019 - 6.50 Benzo[ghi]perylene 6 276 0.00026 1.4x10-8 6.50

Polycyclic Aromatic Hydrocarbons
5
PAHs in soil In soil most PAHs are strongly sorbed to the organic matter, making them
relatively unavailable for degradation processes [4]. PAHs can therefore remain in the soil for many centuries, posing a long-term threat to the environment [1], although LMW PAHs are partly lost through degradation processes, volatilization and leaching [4-6]. The effect of sorption generally increases as the number of benzene rings in the PAH-molecule increases [7,8], since this implies higher lipophilicity. Furthermore, it has been shown that the degradability and extractability of organic compounds in soil decreases with the time they have been in contact with the soil: a phenomenon referred to as ‘aging’ or ‘weathering’ [9-12]. Aging is mainly a result of slow diffusion into the soil organic matter, but other mechanisms involved include the formation of bound residues and physical entrapment within soil micro pores [13-15]. The processes of sorption and aging limit, on one hand, the degradability of the contaminants. On the other hand, these processes reduce the toxicity of the soil contaminants, by lowering the fraction available for uptake by living organisms [11,13].
2.2 Contaminated sites Although PAHs are ubiquitous environmental pollutants, concentrations
are generally higher near the emission sources. Thus, elevated concentrations have been found in urban soils [16] and roadside soils [17], while very high concentrations (>10 000 mg/kg soil) have been reported for contaminated sites [Paper IV; 5,18,19]. The activities at these sites have usually involved production or use of fossil fuels or products derived from fossil fuels, such as coal tar and creosote. They include the following industrial activities [4,5]:
• Gasification/liquification of fossil fuels (gasworks) • Coke production • Coal-tar production • Wood-treatment processes • Wood-preservative production • Asphalt production • Fuel processing
Gasworks sites The PAH-contamination at old gasworks sites (Chapter 5), or
manufactured gas plants (MGPs) as they are also called, mainly originates from coal tar, which was a byproduct during the production of town gas from coal. In this process, combustible gases were drawn off from coal under pyrolytic

Polycyclic Aromatic Hydrocarbons
6
conditions, at temperatures higher than 1000°C [20]. Coal tar and other impurities, e.g. ammonia, cyanides and hydrogen sulfide, were separated from the gas before it was delivered to the cities. There was initially no use for the coal tar, which was therefore dumped at the sites, often directly on the ground or in neighboring watercourses. However, gradually a market was developed for several by-products derived from coal tar, which were obtained through distillation of the tar. One of the fractions was creosote, constituting about 10% of the original coal tar. Creosote has been used as a wood preservative domestically and industrially for many years [1]. Consequently, numerous wood-impregnation sites are also highly contaminated with PAHs [21].
Coal-tar and creosote Coal tar is a complex mixture of hydrocarbons, phenols and heterocyclic
compounds. Its composition is mainly a function of the temperature of the gasification process used and, to a lesser extent, of the nature of the coal used as feedstock [20]. More than 400 compounds have been identified in coal tars, although as many as several thousands may actually be present [22]. Most of the extractable organic compounds are PAHs [18], and the proportion of HMW compounds is higher than in creosote [1]. Creosote is, thus, primarily a distillate of the LMW PAHs in coal tar. It contains more than 200 compounds, of which approximately 85% are PAHs. The other components in creosote are N-, S-, and O-heteocyclics (5%) and phenolic compounds (10%) [21].
2.3 PAH toxicity A wide range of PAH-induced ecotoxicological effects in a diverse suite of
biota, including microorganisms, terrestrial plants, aquatic biota, amphibians, reptiles, birds and terrestrial mammals have been reported [23]. Effects have been documented on survival, growth, metabolism and tumor formation, i.e. acute toxicity, developmental and reproductive toxicity, cytotoxicity, genotoxicity and carcinogenicity. However, the primary focus of the toxicological research on PAHs has been on genotoxicity and carcinogenicity.
In these studies, several PAHs have been shown to damage DNA and cause mutations, which in some cases may result in cancer. However, for the unsubstituted PAHs it is not the original compound that reacts with DNA. The PAHs require metabolic activation and conversion to display their genotoxic and carcinogenic properties [24]. This happens as the PAHs are metabolized in higher organisms. PAHs do not accumulate in the same manner as some other lipophilic organic compounds, e.g. PCBs. Instead, they are converted to more water-soluble forms, which facilitates their subsequent excretion from the organism. Unfortunately, this may also lead to the

Polycyclic Aromatic Hydrocarbons
7
formation of reactive intermediates that may react with DNA to form adducts, preventing the gene involved from functioning normally. The DNA-damage may be repaired, but if the repair fails a mutation, i.e. an irreparable genetic damage, will have occurred. Mutations may affect many different functions of a cell, but above all they may induce cancer [25].
In Figure 2 the metabolic activation of benzo[a]pyrene is shown. This compound is probably the most thoroughly studied PAH, and is also one of the most carcinogenic compounds known. The initial step in the metabolism of PAHs involves the multifunctional P-450 enzyme system forming different epoxides through the addition of one atom of oxygen across a double bond. The epoxides are short-lived compounds and may rearrange spontaneously to phenols or undergo hydrolysis to dihydrodiols. These products may then be conjugated with glutathione, glucuronic acid or sulfuric acid, to form products that can be excreted by the organism. This conjugation process is, thus, regarded as the true detoxification and excretion process. However, the dihydrodiols may also act as a substrate for cytochrome P-450 once again to form new dihydrodiol epoxides, e.g. trans-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene-9,10-oxide, which unfortunately are poor substrates for further hydrolysis. These dihydrodiol epoxides may instead react with proteins, RNA and, most seriously, DNA, thus causing mutations and possibly cancer [24,25].
Figure 2. Metabolic activation of benzo[a]pyrene, from IARC 1983 [25].
O OH
OH
O
OHOH
OHSulphate ester
Glucuronide
Gluthathione conjugate
Binds to DNA
Benzo[a]pyrene Benzo[a]pyrene-7,8-oxide7-Hydroxybenzo[a]pyrene
trans-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene-9,10-oxide
trans-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene
cyt P-450
cyt P-450

Polycyclic Aromatic Hydrocarbons
8
PAHs may also interact in other steps of the carcinogenic process. For example, they may promote cellular proliferation [23]. As a group, PAHs show varying ability to induce cancer and it is difficult to identify the structural features associated with their carcinogenic activity. However, for unsubstituted PAHs, it seems that a minimum of four benzene rings is required for carcinogenic activity [24], but that does not mean that all PAHs with four benzene rings are carcinogenic. Some are very weak in this respect while others are among the most carcinogenic compounds known, e.g. benzo[a]pyrene. Structure-activity relationships become even more complex when substitution of the molecular structure occurs. For example, although benz[a]anthracene is a fairly weak carcinogen, 7,12-dimethylbenz[a]anthracene is a very potent carcinogen [24]. Furthermore, some environmental transformation products of PAHs may react directly with DNA, causing mutations and possibly cancer, without the need for metabolic activation [26,27]. This is further discussed in chapter 3.

9
3. Degradation of PAHs
Degradation of PAHs in the environment occurs through biological, chemical and photochemical processes. These processes may also be utilized for remedial purposes. However, the degradation may result in a variety of transformation products some of which could potentially accumulate. In this chapter the degradation of PAHs will be discussed. Furthermore, the reasons why the oxygenated PAHs were considered to be the most important group of transformation products to study will be explained.
3.1 Biological degradation Biological degradation appears to be the main process responsible for the
removal of PAHs in soil [5,8]. Microorganisms, such as bacteria and fungi, may transform the PAHs to other organic compounds or to inorganic end products such as carbon dioxide and water [28-30]. The latter process has been referred to as mineralization. Some PAH-degrading microorganisms, primarily bacteria, are capable to use the PAHs as a carbon and energy source, and may thus transform the contaminants into molecules that can enter the organisms’ central metabolic pathways [28,29]. Other microorganisms have the capacity to degrade PAHs, while living on a widely available substrate. Such cometabolism does not always result in growth of the microorganism, and sometime the cosubstrate, i.e. the PAH, is only transformed into another compound without any apparent benefit for the organism. This may lead to partial degradation, if no enzyme capable of transforming the metabolite is available [30]. For PAHs, the contribution of the cometabolic degradation processes increases as the number of rings in the PAH-molecule increases, since far fewer microorganisms are capable of using the HMW PAHs as carbon and energy sources [29,31,32].
Microbial degradation pathways PAH-degrading bacteria generally use the PAHs as a carbon and energy
source while fungi metabolize the PAHs to more water-soluble compounds, thereby facilitating their subsequent excretion. Bacteria and fungi therefore have different metabolic pathways (Figure 4) [28,29]. The general fungal pathway is quite similar to the transformation pathways found in humans and other mammals. Thus, as can be seen in Figure 4, fungi oxidize PAHs via the cytochrome P-450 enzyme system to form phenols and trans-dihydrodiols, which can be conjugated and excreted from the organism. The bacterial degradation of PAHs generally begins with a dioxygenase attack on one of the aromatic rings to form a cis-dihydrodiol, which is subsequently dehydrated to catechol (Figure 4). Catechol is a key intermediate from which ring cleavage

Degradation of PAHs
10
can occur. The aromatic ring is cleaved between the hydroxyl groups (ortho fission) or adjacent to one of the hydroxyl groups (meta fission). Successive ring degradation may then occur, so that the structure is ultimately degraded to molecules that can enter the central metabolic pathways of the bacteria [28,29].
Figure 4. General pathways for the microbial degradation of PAHs, based on Cerniglia 1992 [29].
These general metabolic pathways have been verified for a large number of bacteria and fungi, in pure cultures, during the degradation of a variety of individual PAHs, as reviewed by Cerniglia, Sutherland et al., and Kanaly & Harayama [29,32-34]. They are illustrated in Figures 5 and 6, which show fungal and bacterial metabolic pathways proposed for pyrene, respectively. However, these and other proposed pathways [35-37] also show that many other reactions may occur in parallel, especially during cometabolic degradation of PAHs. For example, as shown in Figures 5 and 6, the pyrene quinones and the 4-hydroxyperinaphthenone may be formed as dead-end products in parallel with the general degradation pathways used by the microorganisms in question.
H
O
H
OH
OH
H
H
OH
OH
OH
H
H
OH
OH
COOHCOOH
CHOCOOH
OH
O
O
O-GlucosideO-GlucuronideO-SulfateO-Xyloside
PAH
Ring fisson
O2, Cyt P-450Monooxygenase
Fungi
Bacteria
White-rot fungiH2O2, Lignin peroxidases, laccases
O2, Dioxygenase
cis-Dihydrodiol
Dihydrogenase
Catechol
cis,cis-Muconic acid
2-Hydroxymuconic semialdehyd
Orthofission
Meta fission
Epoxide hydrolase
Non-enzymaticrearrangement
Arene oxide trans-Dihydrodiol
Phenol
Quinone

Degradation of PAHs
11
Figure 5. Proposed pathways for the fungal metabolism of pyrene, by Penicillium glabrum strain TW 9424, based on Wunder et al. 1997 [35].
Figure 6. Proposed pathways for the bacterial metabolism of pyrene, by Mycobacterium sp. PYR-1, based on Cerniglia 1992 [29].
OH
OSO3H
OH
OH
OH
OH
O
O
O
O
OMe
Pyrene
1-Pyrenyl sulfate1-Methoxy pyrene
1,6-Dihydroxypyrene
1,8-Dihydroxypyrene
1,8-Pyrenequinone
1,6-Pyrenequinone
OHOH
H
HOH
OHOH
OH
O OH
COOH
OH
O
COOHCOOH
COOH
OHOH
HOH
OH
COOHO OHCOOH
OH
COOH
OH
OH
COOH
COOH
COOH
Pyrene
4-Hydroxyperinaphthenone
cis-4,5-Pyrene dihydrodiol
4-Phenanthroate
Phthalate
Cinnamate
O2
O2
CO2
CO2

Degradation of PAHs
12
Wood-rotting fungi One group of fungi, referred to as wood-rotting fungi, has the ability to
degrade PAHs by alternative pathways [29]. These fungi excrete extracellular enzymes capable of degrading lignin in wood. However, the enzymes are not very specific and can also transform organic pollutants, such as PAHs [38]. In this process, the PAHs are transformed to quinones via aryl cation radicals [33]. Some wood-rotting fungi may then cleave the rings and continue the degradation to carbon dioxide and water [39,40]. However, for other fungi the quinones appear to be dead-end products [Paper III; 41]. In Figure 7 the oxidation pathways of selected PAHs by wood-rotting fungi is presented.
Figure 7. Oxidation of PAHs by wood-rotting fungi, from Cerniglia 1997 [33]
3.2 Chemical degradation of PAHs PAHs in soil are also degraded through abiotic processes. Oxidation
reactions are the most important in this context, although photochemical reactions may contribute significantly to the degradation on the surface of soils [42,43]. In addition, most of the oxidants that commonly initiate the oxidation reactions in the environment, i.e. singlet oxygen (1O2), organic peroxides, hydrogen peroxide, ozone and radicals such as alkoxy radicals (RO•), peroxy radicals (RO2•) and hydroxyl radicals (HO•), are directly or indirectly generated through photochemical processes [44]. However, some can also be produced from inorganic salts and oxides, especially those of iron and manganese [42].
Chemical oxidation reactions involving hydroxyl radicals, generated from hydrogen peroxide, and ozone, have been most widely studied. Hydroxyl
O
O
O
O
O
O
OH O
O
O
COOH
COOH
CO2
+ +
Fluorene 9-Hydroxy fluorene 9-Fluorenone
Anthracene 9,10-Anthraquinone
Benzo[a]pyrene 1,6-Benzo[a]pyrene quinone 3,6-Benzo[a]pyrene quinone 6,12-Benzo[a]pyrene quinone
Phthalic acid

Degradation of PAHs
13
radicals are strong, relatively unspecific oxidants that react with aromatic compounds, such as PAHs, at near diffusion-controlled rates (i.e., kOH*> 109 M-1s-1) [45] by abstracting hydrogen atoms or by addition to double bonds [46]. The ozone molecule may attack double bonds directly, but it can also form reactive hydroxyl radicals by decomposing water [47-49]. The reaction pathways that follow are very complex, and numerous intermediates are formed. The final reaction products include, for both oxidants, a mixture of ketones, quinones, aldehydes, phenols and carboxylic acids [42,48-53].
Photochemical degradation of PAHs often involves the same oxidative species that are produced during the pure chemical oxidation of PAHs, i.e. oxygen, hydroxyl radicals and other radicals. Consequently, the reaction products include similar complex mixtures of ketones, quinones, aldehydes, phenols and carboxylic acids [42,51,54-56].
Reactivity of PAHs During chemical reactions, PAHs are largely transformed into other PACs,
i.e. they do not lose their aromatic character. The aromaticity is conserved since the non-aromatic hydrocarbons are much richer in energy. Thus, considerable amounts of energy are required to change an aromatic compound into a non-aromatic.
The electron distribution over the PAH molecule determines the positions of the molecule that are most reactive. For addition reactions, Whelands’s concept [57] of localization energy has proven useful. The localization energy (Lu) is the energy required to isolate a π-electron at the center of u of a PAH from the remaining π-system. In this process, the attacking species may be a nucleophile, an electrophile or a radical. A number of reactivity indices have been developed that reflect the localization energy, one of which is Dewar’s reactivity number, Nu [58]. The magnitude of Nu reflects the localization energy and the reactivity at a certain position of an aromatic compound. The smaller the value of Nu the lower the activation energy and the greater the reaction rate of an addition at the center of u. Nu values for a number of aromatic compounds are given in Figure 8. The values shown in this figure indicate that benzene should be the least reactive species. For naphthalene, higher reactivity is expected for the 1-position than the 2-position. Similarly, anthracene and phenanthrene should preferably react at the 9,10-positions, although the reactivity of phenanthrene should be markedly lower than that of anthracene. Thus, the localization energy concept may be helpful in clarifying the mechanisms of PAH-degradation and PAH-derivative formation.

Degradation of PAHs
14
Figure 8. Electron localization energies for benzene and selected PAHs, based on the work of Zander 1979 [59].
3.3 Transformation products of PAHs The ultimate result of a degradation process is total mineralization of the
organic contaminants, resulting in carbon dioxide, water and other inorganic compounds. However, as shown above for PAHs, both biological and chemical degradation processes may produce a variety of other compounds that may include temporary intermediates and compounds that are resistant to further degradation. Such dead-end products could potentially accumulate during a remedial process if no mechanism is available for their further degradation. This may happen if, for instance, the original contaminant has been transformed co-metabolically, and no microorganisms is present that could metabolize the transformation product, or if a chemical oxidizing agent has been added in insufficient quantity or strength [60].
From the literature and pathways described above, the dead-end products appear to be primarily carbonyl compounds, such as ketones, quinones, dicarboxylic acid anhydrides and coumarins, while carboxylic acids and phenols seems to be more easily degraded or transformed. The higher stability of the carbonyl compounds is also indicated by the fact that these compounds are found more frequently than the hydroxylated and carboxylated compounds in environmental samples, including contaminated soil [Papers II, IV-VI; 61-68]. This could be due in some cases to restriction in the analytical methods used, since the hydroxylated and carboxylated compounds are more polar and more difficult to analyze. However, similar results have been obtained with methods carefully developed for all these classes of compounds [69,70]. Consequently, the studies this thesis is based upon focused on the carbonylic PAH-
2.31 1.812.12
1.57 1.261.89
1.35
1.43
1.51
1.962.04
2.18
1.86
1.80
1.67
2.002.12
1.68
1.51
2.31
1.622.21
1.55
1.64
1.79 1.82
benzene naphthalene anthracene benzo[a]anthraceneDibenz[a,h]anthracene
Phenanthrene Chrysene Triphenylene Pyrene Benzo[ghi]perylene

Degradation of PAHs
15
derivatives, which will be referred to as oxygenated PAHs (oxy-PAHs) in the following sections.
Another motive for studying the oxy-PAHs is that many of these compounds are toxic and mutagenic [26,27,71-74]. Some are even more toxic than their parent PAHs [71-73]. For example, anthracene-9,10-dione and phenanthrene-9,10-dione have been shown to inhibit the growth of duckweed, Lemna gibba, and the marine bacterium Photobacterium phosphoreum [72,73]. Furthermore, 7H-benz[de]anthracen-7-one, 4-oxapyren-5-one and several benzofluorenones, benzo[a]pyrene quinones and pyrene quinones have all shown mutagenic activities in biological assays, such as the Ames test [26,27,75]. In contrast to the unsubstituted PAHs some of these oxy-PAHs do not require metabolic activation to exhibit their mutagenic properties, but may react with DNA directly (cf. section 2.3). Further, oxy-PAHs have also been identified as the predominant class of compounds in the most mutagenic fractions obtained during bioassay-based chemical fractionation of various environmental samples [62,65,68].

16
4. Analysis of PAHs and oxy-PAHs in soil
As shown in the previous chapter it may be relevant to monitor oxy-PAHs as well as the unsubstituted PAHs during the assessment and remediation of contaminated soil. This will give more information about the soil contamination and the levels of toxicity than an ordinary PAH analysis. However, it also makes further demands on the analytical methods used, since the transformation products are often present in lower concentrations than the parent PAHs and they may be difficult to identify in the complex mixtures found in these samples. It is therefore essential to use powerful analytical tools to fractionate, separate and identify the analytes in the samples. Furthermore, analysis of organic contaminants in soil always requires powerful extraction techniques to release the strongly sorbed contaminants from the soil material.
In the work underlying this thesis the analytical development has focused on the extraction and fractionation procedures, since these are the most time-consuming and labor-intensive steps during environmental analysis. However, in the following sections all the procedures used are discussed. The steps in the analytical chain may be summarized as follows:
*Pretreatment, which is performed to increase the homogeneity of the soil and to increase the extractability of the analytes in the soil.
*Extraction, which is performed to release the contaminants from the solid matrix and quantitatively transfer them to another medium, usually an organic solvent.
*Clean up, which is performed to remove co-extracted compounds that could interfere during the subsequent analysis, and to separate different classes of analytes prior to analysis.
*Instrumental analysis, which is performed to separate, identify and quantify the individual analytes in the sample.
4.1 Sample pretreatment The sample pretreatment included sieving, air-drying and grinding. In some
studies [Paper III] the soil was also acidified prior to extraction to improve the extractability of acidic transformation products [76]. Samples were sieved to remove the larger particles, partly because they reduce the homogeneity of the soil and partly because the contaminants are primarily associated with the smaller particles in the soil [15]. Furthermore, the use of a specific particle fraction facilitates comparison with other soil samples. The drying was performed to facilitate the subsequent grinding and to increase the contact

Analysis of PAHs and oxy-PAHs in soil
17
between the soil and the organic solvent used for extraction. Drying at elevated temperatures was avoided since this may result in losses of volatile analytes, e.g. naphthalene. The samples were ground to further increase the homogeneity of the samples and to increase the extractability of the analytes, by increasing the exposed surface area in the soil.
In the studies described in Papers I, II and V, a rather large particle fraction (< 5mm) of the original soils were used. This fraction was homogenized in a ball mill so that representative sub-samples could be taken for the subsequent extractions. In other cases [Papers III, IV and VI] a finer particle fraction of the original soil was used (2.5, 2.3 and 2.0 mm respectively), from which it was easier to take representative samples directly. The selected samples were still ground in a mortar prior to extraction, to improve the extraction efficiency. However, a recent study by Bergknut et al. 2003 [77] shows that this may not have been necessary with this particular soil. In the cited study, grinding had no effect on the extractability of the PAHs and oxy-PAHs, showing that the contaminants were not physically trapped in the solid material.
4.2 Pressurized liquid extraction The soil samples were extracted by pressurized liquid extraction (PLE).
This relatively new technique utilizes organic solvent at elevated temperature and pressure, which dramatically accelerates the extraction process compared to traditional techniques, such as Soxhlet and ultrasonic extraction. The theoretical basis of this method is described in more detail in Paper I and in a paper by Richter et al. 1996 [78], who first introduced the PLE-technique. Since then, a number of studies have shown the PLE-technique to be equally or even more effective than Soxhlet and ultrasonic extraction for extracting many compounds from soil samples [Paper I; 79-81]. The PLE-technique also has the advantages of being less time consuming and labor intensive than Soxhlet and ultrasonic extraction, and it uses lower amounts of organic solvent.
A schematic diagram of the PLE-system is presented in Figure 9. The sample is loaded into the extraction cell, which then is placed into the oven of the extraction system. Solvent is pumped from the solvent bottle into the cell, which is pressurized and heated. The extraction sequence starts with a dynamic phase during which temperature and pressure are equilibrated. A certain amount of solvent is then passing through the cell while the pump, which fills the cell, and the valve, which releases solvent from the cell, control the pressure. The dynamic phase is followed by one or more static phases, during which the same portion of solvent is held pressurized inside the cell. When

Analysis of PAHs and oxy-PAHs in soil
18
these phases are concluded the solvent is allowed to flow to the collection vial and the sample is flushed with fresh solvent. Finally, the sample is purged with a suitable gas (e.g. N2) to recover the last solvent residues. All the solvent that passes through the cell is collected in the collection vial.
Figure 9. Schematic diagram of the pressurized liquid extraction (PLE) system.
Method optimization In Paper I the reliability and efficiency of the PLE technique for extracting
PAHs from contaminated soil were investigated. The influence of several variables, including sample load, pressure, temperature, extraction time, choice of solvents, ratio of binary solvents, and solvent volume, were studied using experimental design. The experimental design approach maximizes the information obtained from a minimum number of experiments by systematically varying more than one variable between different experiments [82]. The study showed that PLE is a robust, reliable and exhaustive technique for extracting PAHs from contaminated soil. The extraction pressure and extraction time had no influence on the extraction efficiency in the studied intervals (7-19 MPa and 2-20 min respectively), and there was only a slight indication that temperatures below 100°C might affect the extraction result for some PAHs. Furthermore, the choice of extraction solvent was not critical for the extraction result, provided that the solvent volume/sample load ratio was sufficiently high, i.e. by using two static extraction cycles followed by a rinse with a whole cell volume. This may explain the previously observed benefit gained by using two short extraction cycles compared to one longer cycle [83], since the former results in a larger volume of solvent. It was thus concluded
Gas cylinder
Collection vial
Solvent
Pump
Oven Extraction
cell
Valve

Analysis of PAHs and oxy-PAHs in soil
19
that PLE could be successfully used for rapid and exhaustive extraction of PAHs in contaminated soil with relatively low solvent consumption.
The PLE-cell as a chromatographic column The influence of the sample load and solvent volume was discovered
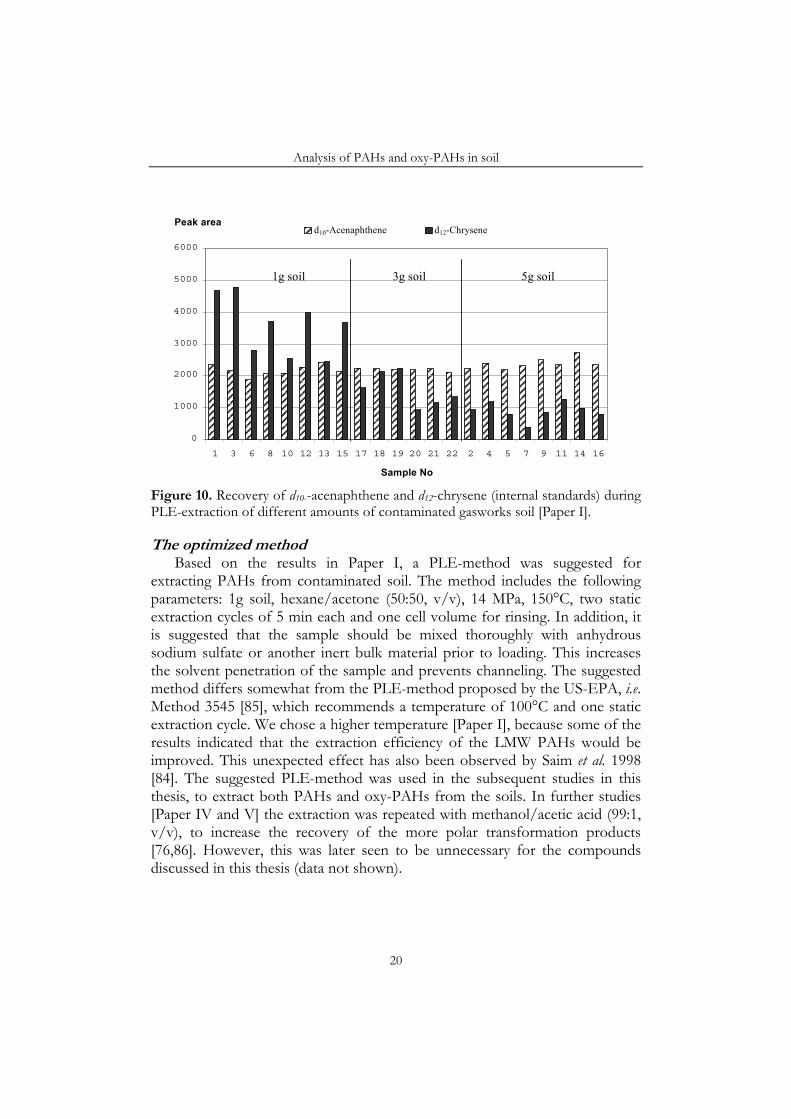
during the screening experiment in Paper I. In these experiments, the internal standard (IS) was added on top of the soil samples in the extraction cells, which considerably affected the final results. For instance, the recovery of the HMW compounds in the internal standard was much higher in experiments with a sample load of 1g than in experiments with a sample load of 5 g. Figure 10 illustrates this, by comparing the recoveries of two IS-compound, d12-chrysene, which was strongly affected by the sample load, and d10-acenaphthene, which was affected much less strongly. Furthermore, the HMW-compounds appeared to be recovered to a larger extent when the samples were extracted with a toluene-rich solvent. However, the LMW compounds in the IS and the native analytes were not affected to the same extent. This is because the PLE-cell resembles a chromatographic column, with the soil as the stationary phase, which retains the HMW PAHs more strongly than the LMW compounds. Furthermore, the IS added on top of the soil must travel through the whole soil layer before reaching the collection vial, while the native compounds, distributed evenly throughout the soil bed, have a shorter average distance to travel. Toluene seems to be better suited than hexane to elute the PAHs through the soil bed, which explains why toluene has shown higher extraction efficiency than other solvents in some previous studies [81]. The negative effect of n-hexane has also been observed previously [84], and is attributed to its very low polarity. However, the subsequent experiments presented in Paper I showed that the negative effect of a weaker solvent could be compensated by using a higher solvent volume/sample load ratio.
Analysis of PAHs and oxy-PAHs in soil
20
Figure 10. Recovery of d10--acenaphthene and d12-chrysene (internal standards) during PLE-extraction of different amounts of contaminated gasworks soil [Paper I].
The optimized method Based on the results in Paper I, a PLE-method was suggested for
extracting PAHs from contaminated soil. The method includes the following parameters: 1g soil, hexane/acetone (50:50, v/v), 14 MPa, 150°C, two static extraction cycles of 5 min each and one cell volume for rinsing. In addition, it is suggested that the sample should be mixed thoroughly with anhydrous sodium sulfate or another inert bulk material prior to loading. This increases the solvent penetration of the sample and prevents channeling. The suggested method differs somewhat from the PLE-method proposed by the US-EPA, i.e. Method 3545 [85], which recommends a temperature of 100°C and one static extraction cycle. We chose a higher temperature [Paper I], because some of the results indicated that the extraction efficiency of the LMW PAHs would be improved. This unexpected effect has also been observed by Saim et al. 1998 [84]. The suggested PLE-method was used in the subsequent studies in this thesis, to extract both PAHs and oxy-PAHs from the soils. In further studies [Paper IV and V] the extraction was repeated with methanol/acetic acid (99:1, v/v), to increase the recovery of the more polar transformation products [76,86]. However, this was later seen to be unnecessary for the compounds discussed in this thesis (data not shown).
0
1000
2000
3000
4000
5000
6000
1 3 6 8 10 12 13 15 17 18 19 20 21 22 2 4 5 7 9 11 14 16
Sample No
Peak aread10-Acenaphthene d12-Chrysene
1g soil 3g soil 5g soil

Analysis of PAHs and oxy-PAHs in soil
21
4.3 Clean up Clean up techniques may be divided into pre-separation techniques, which
are used to remove the bulk of the co-extracted (biogenic) material, and fractionation techniques, which are used to separate the target analytes in different fractions, and to remove similar anthropogenic compounds. However, for samples that have low organic contents, the pre-separation and fractionation steps are often combined into a single fractionation step. The fractionation of PAHs and PAH-transformation products may be performed by adsorption chromatography, using open column chromatography [87-89], solid phase extraction (SPE) [63,69,70,76] or high performance liquid chromatography (HPLC) [64,67,90,91]. The HPLC-techniques have the greatest resolution and reproducibility, and they may be coupled to sensitive instruments for analyte detection. However, the other techniques are simpler to use, less costly and have higher sample capacity than HPLC, and are therefore widely used in environmental analysis [92]. The stationary phases most commonly used for organic contaminants are silica gel, alumina and Florisil [92]. These are highly active adsorbents, which are often deactivated with water prior to use. This reduces their adsorption capacity and improves their reproducibility. The analytes are eluted from the columns with organic solvents of increasing polarity.
Silica gel fractionation The clean up procedures used in our studies mainly involved open column
chromatography with silica gel packed columns. Pre-studies with activated silica showed that many PAHs were insufficiently recovered from this adsorbent (data not shown), and some more polar oxidation products could not be eluted at all. Consequently, the silica was deactivated with 10% water prior to use. The columns were initially packed with 10 g silica [Paper I and II], but later 5 g was used [Paper III-VI]. The amount of silica was reduced to speed up the procedure and to save solvent. The elution scheme used for the open-column fractionations remained similar, but some modifications were made during the course of the investigations (Table 2). In pre-studies to Paper I, no pre-fraction was collected and discarded prior to the analyte fractions. Instead, all the eluted solvent was collected and pooled. However, it was discovered that sample components, probably aliphatic compounds, eluting from the silica column before the PAHs interfered during the subsequent gas chromatographic analysis of the PAHs, as shown in Figure 11. Therefore, a pre-PAH fraction was collected and discarded in all experiments described in the papers included in this thesis (Table 2). Initially [Papers I and II], the PAHs were eluted with hexane and a mixture of hexane/dichloro-methane. However, in Paper II, the volume of the second solvent was

Analysis of PAHs and oxy-PAHs in soil
22
increased to recover some oxy-PAH and heterocyclic compounds as well. The fractions were at this stage not separated but were analyzed together. The procedure was then refined [Paper III] so that the PAHs were eluted with a small volume of the hexane/dichloromethane mixture, followed by a separate oxy-PAH fraction eluted with pure dichloromethane. This elution scheme was then used in the subsequent studies [Papers IV-VI], except that the volume of the PAH-fraction was reduced still further to prevent the least strongly retained oxy-PAHs eluting in the PAH-fraction.
Figure 11. Chromatogram of PAHs in a gasworks soil extract, showing interfering compounds in the middle.
Table 2. Elution schemes for the silica gel columns used for fractionation of the soil extracts [Papers I-VI]. In all studies the silica gel was deactivated with 10% water prior to use. Paper I Paper II Paper III Paper IV-VI Silica gel 10 g 10 g 5 g 5 g Pre-fraction 10 ml hx 10 ml hx 5 ml hx 5 ml hx PAH-fraction 30 ml hx +
30 ml hx/DCM 20 ml hx/DCM 15 ml hx/DCM
Oxy-PAH fraction
30 ml hx + 130 ml hx/DCM 30 ml DCM 30 ml DCM
Hx=hexane, DCM=dichloromethane, Hx/DCM is a 3:1 mixture (v/v)

Analysis of PAHs and oxy-PAHs in soil
23
4.4 Selective extraction of PAHs and oxy-PAHs To meet the continual demands for ever faster and less solvent-consuming
analytical procedures, a combined extraction and fractionation procedure was developed and described in Paper V. Here, pressurized liquid extraction with adsorbent-packed extraction cells was used to selectively extract PAHs and oxy-PAHs present in the gasworks soil in two separate fractions. A similar approach has been used previously to retain fat during the extraction of PCBs in adipose tissue [93-96]. Paper V reports the results of tests of the ability of three different adsorbents, i.e. silica gel, alumina and Florisil, to accomplish separate these compounds. The adsorbents and the soil were packed in the cells according to Figure 12, and the samples were then sequentially extracted with solvents of increasing polarity. Quantitative extraction and successful separation of the two compound classes was finally obtained by using extraction cells packed either with 2%-deactivated silica or with activated Florisil. These were extracted with hexane followed by hexane/dichloro-methane (1:1) and hexane/dichloromethane (3:1) followed by dichloro-methane/acetone (3:1), respectively. Only small amounts (mostly less than 1%) of PAHs and oxy-PAHs were extracted in the “wrong” fractions using these procedures. These amounts was considered acceptable for a screening method like this, which has several other advantages since it is fast while having less labor and solvent requirements than traditional methods. It may also be difficult to avoid a certain degree of co-elution between the PAHs and the oxy-PAHs with this experimental setup, since the extracted compounds are transferred to the chromatographic material without refocusing. The reliability of the fractionation could, however, be checked in each individual sample by analyzing selected compounds in both fractions.
The method involving silica-packed extraction cells was preferred to the alternative with Florisil-packed cells, because the former produced much purer extracts due to the lower polarity solvents used. These extracts could be analyzed directly without further clean up. This method was later successfully applied [Paper VI] as a rapid method for the concurrent analysis of PAHs and oxy-PAHs in contaminated soil.

Analysis of PAHs and oxy-PAHs in soil
24
Figure 12. Schematic diagram of the adsorbent-packed PLE-cell used for selective extraction of PAHs and oxy-PAHs from contaminated soil.
4.5 Instrumental analysis The samples have been exclusively analyzed by gas chromatography with
mass spectrometric detection (GC/MS). A standard set of PAHs could have been analyzed by GC with flame ionization detection (FID) or HPLC with UV- or fluorescence detection [92], but for a more thorough characterization of the contaminants present in soil, including individual oxy-PAHs, higher resolution and sensitivity is needed. GC/MS fulfils these requirements and facilitates the identification and quantification of a large number of compounds in the soil, as shown in Paper IV and Chapter 5. The GC can separate compounds with very similar properties, and many compounds may be identified solely by their GC-retention times in comparison with reference compounds and literature data. In Paper IV, retention indices were calculated for each compound in the chromatogram, according to Lee et al. [97], to facilitate this identification. However, for more reliable identification, the retention time data have been complemented with mass spectral data obtained from the MS. By using an electron-impact MS in the full scan mode, a relatively unique spectrum is provided for each compound in the chromato-gram [92,98]. These spectra could be compared with library spectra or with spectra obtained from reference compounds, which often results in very confident identifications. In Figure 13 the mass spectra for three oxy-PAHs identified in Paper III are shown.
Fraction 1: PAHssoil
adsorbent
O
O
O O
O
filter paper
Fraction 2: oxy-PAHs

Analysis of PAHs and oxy-PAHs in soil
25
Figure 13. Mass spectra of a) 4-hydroxy-9-fluorenone, b) 4-hydroxyperinaphthenone and c) 4-oxapyrene-5-one, identified in Paper III.
In the studies underlying this thesis, the identified compounds were quantified by comparing their peak areas in samples and reference standards using the internal standard technique [99]. In selected ion monitoring (SIM) mode the MS provides high sensitivity and selectivity, which facilitates accurate quantification of the analytes. MS-detection also allows the use of isotope labeled internal standards, which further improves the quantification procedure. The internal standards may be added prior to extraction or fractionation, and can then be used to compensate for the losses of analytes incurred during the analytical procedure [92,99]. In our studies, perdeuterated internal standards have been used. However, they have only been available for the PAHs. Thus, the determined oxy-PAH concentrations have not been compensated for losses caused by the analytical procedure. On the other hand, these determinations show relatively low variability [Papers IV-VI], and in most of the studies the differences between samples (remediated and unremediated soil, for instance) have been considered more important than the absolute concentrations in the individual samples. Furthermore, some oxy-PAHs were quantified by using the response factor for other oxy-PAHs [Papers V and VI], since authentic reference compounds were only available for a limited number of oxy-PAHs.
Derivatisation Although this thesis focuses on the carbonyl-containing transformation
products, hydroxylated PAHs have been analyzed in some cases. Paper III considered the formation of hydroxy-PAHs during the degradation of PAHs in an artificially contaminated soil. The possible presence of hydroxy-PAHs in the gasworks soil (chapter 5) has also been studied, but the results are not included in any of the papers, because no such compounds were identified in the soil. In
Rel. int.
OH
O
196168
139
139
196
O
OH
168
220
192
163
O
O
m/z 150 300 150 300
m/z150 300
m/z
100 % 100 %100 %
Rel. int. Rel. int.a b c

Analysis of PAHs and oxy-PAHs in soil
26
these studies, the hydroxy-PAHs were converted to trimethylsilyl derivatives through a reaction with N,O-bis(trimethylsilyl)trifluoroacetamide/trimethyl-chlorosilane (BSTFA/TMCS, 99:1) prior to GC-analysis. This is because hydroxylated compounds are often poorly suited to direct GC-analysis, due to their polarity, thermal instability and tendency to form hydrogen bonds [100]. However, by converting them into less reactive and more volatile derivatives their GC-properties may be improved. The silylation method used in the presented studies is described in Paper III. The efficiency of the method was tested in a pre-study, in which five hydroxy-PAHs (1-naphthol, 1-acenaphthenol, 9-fluorenol, 9-phenantrol and 9-pyrenol) were completely converted to their silyl-derivatives in less than an hour at room temperature.

27
5. The gasworks site at Husarviken
In five of the six papers included in this thesis the soil discussed came from a gasworks site at Husarviken in Stockholm, Sweden. At this site, gas was produced from coal throughout the period 1893-1972. Today, the subsurface of the site consists of a mixed fill (sandy soil, ash and demolition debris), contaminated with coal tar and its distillation products, heavy metals and cyanide. The contaminants are heterogeneously distributed, but are found throughout the area. The site conditions are typical of old gasworks sites [101,102]. In Paper IV, soil from this site was characterized with respect to PAHs and related compounds, and in other experiments the bioavailability of the contaminants was studied by exposing earthworms to the soil [Paper II].
5.1 Characterization More than one hundred compounds were identified in the gasworks soil,
including PAHs, alkyl-PAHs, heterocyclic compounds and oxy-PAHs, as shown in Table 3 [Paper IV]. The concentrations of the compounds were similar to levels found at other gasworks sites [18,88], although concentrations in these soils vary considerably [5,19]. Large variations in the PAH-levels have also been recorded at the site at Husarviken (data not shown). Nevertheless, the quantified compounds, shown in Table 3, show that the PAHs with four, five, and six fused rings predominate in the soil, confirming that the contamination is old. Environmental processes, such as volatilization, leaching and biodegradation have changed the contaminant profile in the soil, so that the proportion of HMW PAHs has increased over time. Fresh coal tar contains larger relative amounts of the LMW PAHs [18,103], but these compounds are also more easily degraded and removed from the soil [6,101,104]. In Figure 14, a chromatogram obtained from an extract of the gasworks soil is compared against that obtained from an extract of fresh coal tar. Although this particular coal tar was not produced at the Husarviken site, this example indicates how the contaminant profile has changed over time, in favor of the HMW compounds (eluting late in the chromatogram). The predominance of HMW was seen not only among the PAHs; the heterocyclic compounds in the soil showed similar trends [Paper IV]. Furthermore, enrichment of alkylated PAHs probably also occurs over time, since the unsubstituted PAHs are generally more easily degraded than the alkylated PAHs [29,105]. However, this could not be verified in the soil from Husarviken.

The gasworks site at Husarviken
28
Table 3. PAHs and related compounds identified in the gasworks soil from Husarviken. Concentrations are given for the quantified compounds. PAHs and alkyl-PAHs Soil conc.
(ug/g, d.w.) PAHs and alkyl-PAHs (continued) Soil conc.
(ug/g, d.w.) Naphthalene 17 Dibenzo[b,k]fluoranthene 2-Methylnaphthalene 7.9 Naphtho[2,3-k]fluoranthene 1-Methylnaphthalene 5.2 Coronene 23 Biphenyl 2.6 Dibenzo[a,e]pyrene/Dibenzo[e,l]pyrene 2,6-Dimethylnaphthalene 7.0 C2-naphthalenes (4 peaks)
Naphtho[2,1-a]pyrene/ Benzo[b]perylene/ Dibenzo[2,3-a]pyrene/Dibenzo[a,i]pyrene
Acenaphthylene 29 Dibenzo[a,h]pyrene Acenaphthene 2.4 Methylbiphenyls (2 peaks) Soil conc. C3-naphthalenes (4 peaks) Heterocyclic PACs (ug/g, d.w.) 2,3,5-Trimethylnaphthalene 2.0 Benzothiophene 27 Fluorene 44 Dibenzofuran 42 C2-biphenyls (4 peaks) Methyldibenzofurans (3 peaks) 2-Methylfluorene Dibenzothiophene 28 1-Methylfluorene Benzoquinoline Methylfluorene Phenanthridine C3-Biphenyls (10 peaks) Carbazole 32 Phenanthrene 330 Methylacridine/phenantridine Anthracene 70 Methyldibenzothiophenes (2 peaks) 1-Phenylnaphthalene Methylcarbazoles (4 peaks) 3-Methylphenanthrene Azapyrenes/Azafluoranthenes (6 peaks) 2-Methylphenanthrene Phenanthro[4,5-bcd]thiophene 2-Methylanthracene Benzonaphthofurans (3 peaks) 4H-Cyclopenta[def]phenanthrene 29 Benzo[kl]xanthene 4-/9-Methylphenanthrene Benzo[def]carbazole 1-Methylphenanthrene 16 Benzo[b]naphtho[2,1-d]thiophene 2-Phenylnaphthalene Benzo[b]naphtho[1,2-d]thiophene C2-phenanthrenes/anthracenes (7 peaks) Benzo[b]naphtho[2,3-d]thiophene Fluoranthene 420 Benzoacridine/Azachrysene (2 peaks) Acephenanthrylene 25 Benzocarbazoles (3 peaks) Pyrene 290 Methylbenzoacridine Benzo[a]fluorene Dinaphthofurans (5 peaks) Benzo[b]fluorene Dibenzo[B,def]carbazole Methylpyrenes/fluoranthenes (4 peaks) Dibenzoacridines (3 peaks) 1-Methylpyrene Dinaphthothiophenes (5 peaks) Benzo[ghi]fluoranthene Dibenzocarbazole and isomers (7 peaks) Benzo[c]phenanthrene Cyclopenta[cd]pyrene 4.3 Soil conc. Benz[a]anthracene 190 Oxy-PAHs (ug/g, d.w.) Chrysene 180 1-Indanone 0.65 Methylbenz[a]anthracenes/chrysenes (9 peaks) 1-Acenaphthenone Binaphthalenes (3 peaks) 9-Fluorenone 26 C2-benz[a]anthracenes/chrysenes (4 peaks) Methyl-9-fluorenones (4 peaks) Benzo[b]fluoranthene 160 Acenaphthylene-1,2-dione Benzo[k]fluoranthene 130 4-hydroxy-9-fluorenone Benzo[a]fluoranthene Xanthone Benzo[e]pyrene 110 Anthracene-9,10-dione 24 Benzo[a]pyrene 120 Naphthalic anhydride Perylene 38 4H-Cyclopenta[def]phenathren-4-one Methyl-M252 (7 peaks) 1-Methylanthracenedione Indeno[7,1,2,3-cdef]chrysene 2-Methylanthracenedione 5.5 Dibenz[a,j]anthracene C2-anthracenedione Indeno[1,2,3-cd]pyrene 100 4-oxapyrene-5-one Dibenz[a,h]anthracene 28 Benzofluorenones (3 peaks) Pentaphene Methylbenzofluorenones (7 peaks) Benzo[b]chrysene 7H-Benz[de]anthracen-7-one 18 Picene 20 Benz[a]anthracene-7,12-dione 4.3 Benzo[ghi]perylene 84 Naphthacene-5,12-dione 4.5 Anthanthrene 19 Benzo[cd]pyrenones and isomers (4 peaks) Methyl-M278 (6 peaks) Chryseno[4,5-bcd]pyranone and isomers (2 peaks) Methyl-M276 (2 peaks) Dibenzofluorenones and isomers (6 peaks) Dibenzo[b,e]fluoranthene cyclopentaperylenone and isomers Naphtho[1,2-k]fluoranthene Indenopyrenone and isomers (4 peaks)

The gasworks site at Husarviken
29
Figure 14. Chromatograms of the PAHs in (a) a gasworks soil extract and (b) a coal tar extract (SRM 1597, NIST, MD USA), showing their different PAH-profile.
The compounds identified in the gasworks soil included a number of oxy-PAHs (Table 3) [Paper IV]. Ketones, such as 9-fluorenone, and quinones, such as anthracene-9,10-dione, were most common, but coumarins, such as xanthone and 4-oxapyren-5-one were also identified. Many of these oxy-PAHs have previously been found in other environmental samples, including, diesel exhaust [27,87,91], fly ash [61], airborne particulate matter [65,70,90], sewage sludge [63], sediments [68,106] and also some in contaminated soils [64,66,69]. The oxy-PAHs may have reached the soil at the same time as the PAHs, or may have been formed through oxidation of PAHs in the soil. Saturated carbons in the aromatic rings are especially susceptible to non-enzymatic oxidation [91], explaining the wide spectrum of ketones found in the soil. Accordingly, ketones of PAHs with saturated carbons in a five-membered ring, e.g. 9-fluorenone and 4H-cyclopenta[def]phenanthren-4-one, were found at approximately equal concentrations in the soil as the parent compounds. PAHs with a saturated carbon in a six-membered ring, e.g. 7H-benz[de]anthracene, are even more rapidly oxidized. Consequently, the corresponding ketones, but not the parent PAHs, were found in the soil. Furthermore, the quinones found in the soil have been formed from linear and other fairly reactive PAHs [107-109] (cf. PAH-reactivity, section 3.2). Large linear PAHs are especially reactive [108,109], explaining why napthacene-5,12-dione was detected in the soil, but not naphthacene. The smaller linear compounds anthracene and anthracene-9,10-dione were found at approximately equal concentrations in the soil.
5.2 Availability of the contaminants The soil at former gasworks sites is often highly weathered, and the
contaminants therein are strongly sorbed since they have been subject to aging. Besides the interactions with the natural organic matter, the contaminants are associated with particles of coal tar that are present in these soils [101,104].
R T: 5 .82 - 3 8 .6 2
1 0 15 20 25 30 3 5Tim e (m in )
0
5
1 0
1 5
2 0
2 5
3 0
3 5
4 0
4 5
5 0
5 5
6 0
6 5
7 0
7 5
8 0
8 5
9 0
9 5
1 00
Rel
ativ
e Ab
unda
nce
2 0 .1 8
20 .81
1 6 .6 7
24 .40
27 .40
2 8 .1 916 .79
23 .691 8 .2 3 2 1 .9 0 2 5 .6 6 2 8 .4 07 .52 1 1 .8 6 1 6 .3 1 3 0 .9 4
16 .119 .36 32 .00 35 .48 3 7 .3 8
N L :7 .20 E7TIC MS 2 01 110 12
R T: 5 .8 2 - 3 8 .6 2
1 0 1 5 2 0 2 5 3 0 3 5Tim e (m in )
0
5
1 0
1 5
2 0
2 5
3 0
3 5
4 0
4 5
5 0
5 5
6 0
6 5
7 0
7 5
8 0
8 5
9 0
9 5
1 0 0
Rel
ativ
e Ab
unda
nce
1 6 .6 8
1 1 .8 8
7 .5 2
2 0 .2 12 0 .8 2
1 6 .8 1
1 2 .3 4 1 3 .9 0
2 7 .4 22 4 .4 09 .3 6
1 8 .4 4
2 8 .1 91 6 .2 99 .6 5
2 1 .8 93 1 .5 51 4 .6 5 2 8 .4 02 6 .0 3 3 2 .0 0 3 5 .4 7 3 7 .2 1
N L :2 .1 3 E 8TIC MS 2 0 1 1 1 0 1 6
(a) (b)

The gasworks site at Husarviken
30
The contaminants therefore have low availability for degradation processes and for uptake by living organisms.
The availability of the contaminants in the soil from Husarviken was demonstrated in experiments reported in Paper II, where earthworms were exposed to the soil. The worms accumulated the PAHs to a lower extent than levels observed in earlier studies with earthworms exposed to PAHs in soil. This was probably because the PAHs in the gasworks soil were less bioavailable than the PAHs in the other soils, since earthworms mainly accumulate contaminants from the fraction dissolved in the interstitial pore-water, i.e. the bioavailable fraction [110,111]. This theory is supported by findings that the LMW compounds in the soil [Paper II] were taken up faster than the HMW compounds, and that the oxy-PAHs were taken up faster in the earthworms than their parent PAHs. Thus, the more water-soluble the contaminants are, the more readily they accumulate in the earthworms. The higher polarity of the oxy-PAHs implies that they generally are more water-soluble than their parent PAHs.
In Figure 15, the accumulation of benzo[a]anthracene and benzo[a]anthra-cene-7,12-dione in the earthworms [Paper II] is compared. It is evident that the biota to soil accumulation factor is much higher for the oxidation product than for the parent PAH. However, after the rapid increase, the concentration of benzo[a]anthracene-7,12-dione decreased in the worms. Similar results, yielding peak-shaped accumulation curves, were also observed for the other oxy-PAH, LMW PAHs, and heterocyclic compounds analyzed in Paper III, and have also been reported in previous studies [112,113]. It has been suggested [112] that the worms stimulate the microbial degradation of the compounds in the pore-water, thereby lowering the bioavailable fraction of the contaminants during the course of the exposure and eventually reducing the concentrations in the worms. Replenishment of contaminants from the soil particles is delayed by the very slow desorption processes.
The low availability of the contaminants in the gasworks soil was also observed during the degradation studies included in this thesis. The PAHs were thus difficult to degrade with either biological or chemical remediation methods [Papers IV and VI], as further discussed in chapter 6. The contaminants were also seen to desorb very slowly from the soil to the surrounding water in these studies [Papers IV and VI]. The PAH-levels in the water were, for instance, far lower than the aqueous saturation concentrations of the compounds, after the soil had been shaken with water for 24 hours at 25°C [Paper VI].

The gasworks site at Husarviken
31
Figure 15. Uptake of benzo[a]anthracene ( ) and benzo[a]anthracene-7,12-dione ( ) in earthworms from the gasworks soil [Paper II], plotted as the ratio of their concentrations in worm lipids (Cworms) to those in soil organic matter (Csoil) against time of exposure.
0
0.02
0.04
0.06
0 5 10 15 20 days
Cw
orm
s /C
soil

32
6. Degradation and formation in remedial processes
Although the natural degradation of PAHs in soil is slow, these processes could be enhanced and utilized for remedial purposes. Thus, both biological and chemical degradation methods have been used to treat PAH-contaminated soil [60,114]. Several methods have proven successful in reducing the PAH-levels in soil, but the fate of the contaminants has seldom been investigated. Consequently, it has rarely been clear whether the PAHs were completely mineralized or if persistent transformation products were accumulated during the process.
The degradation of PAHs and the formation of oxy-PAHs have been studied in three remedial processes: a bioslurry treatment [Paper IV], treatment with wood-rotting fungi [Paper III] and treatment with Fenton’s reagent [Paper VI]. All three techniques have been applied to the gasworks soil from Husarviken, but in Paper III only the results obtained from an artificially contaminated soil were included. However, in the following summary all the results are discussed.
6.1 Bioremediation The biological degradation processes in soil (cf. section 3.1) may be
enhanced by changing its chemical or physical conditions, such as texture, pH, moisture content, temperature and aeration, and nutrient status. The addition of specifically adapted microorganisms may also enhance the process [5,21,104,115]. Many different bioremediation methods have been applied or proposed, but they all involve stimulating the growth of microorganisms that degrade a certain contaminant. They may be classified in the following groups [5,104,116]:
• In situ methods: The contaminated soil is not excavated but is treated in situ by the addition of nutrients, oxygen (usually hydrogen peroxide), and sometimes specifically adapted microorganisms.
• Landfarming: The soil is excavated and spread in a thin layer across a large area. Water, nutrients and sometimes also microorganisms are added. The soil is aerated by mixing, often by standard agricultural equipment.
• Biopiles/Composting: The soil is excavated and placed in piles or vessels and may be mixed with organic bulk materials, such as straw or wood chips. The treatment bed is managed to enhance the degradation processes, by controlled addition of nutrients, water and oxygen, together with

Degradation and formation in remedial processes
33
temperature adjustment. The beds are also designed to minimize contaminant leakage.
• Bioreactors: This is the most controlled form of bioremediation. Usually, the soil is slurried with water and is treated in a specifically designed reactor, where the conditions for biodegradation are optimized.
Data summarized by Wilson & Jones 1993 [5] show that most PAHs in contaminated soil are poorly degraded with in situ methods. Landfarming and biopile/composting methods have also shown limited capacities to degrade PAHs in soil within a reasonable time, although LMW compounds may be degraded adequately with these methods. For the HMW PAHs, the degradation conditions need to be better optimized, and therefore bioreactors seem to be the only option. However, even this technology has to be carefully optimized to successfully manage this task, and even then some persistent PAHs are difficult to degrade [Paper IV]. The limiting factor is primarily the low aqueous solubility of PAHs and their strong adsorption to the soil material, i.e. their low bioavailability [117,118]. In order to be taken up and metabolized by a microorganism, the PAHs first need to be transferred from the soil to the water phase. This process is often the rate-limiting step in the biological degradation of organic contaminants in soil [14,115]. In bioreactors the mass transfer is accelerated by rigorous mixing of the soil and breaking up of the larger soil particles. The high water content in bioreactors also increases the dissolved fraction of PAHs. However, the bioavailable fraction of the HMW PAHs still appears to be limited in many cases [Paper IV].
The bioavailability of sorbed contaminants may be improved by the addition of surfactants, organic solvents or vegetable oil, which enhance the desorption and solubilisation of the contaminants [119-121]. However, the concentration of the additive used is important. High concentrations may inhibit microbial activity, or the additive may be preferentially degraded instead of the contaminant [5]. Furthermore, provision of high additive concentrations is expensive.
The problem of low bioavailability is perhaps less pronounced when wood-rooting fungi are used to degrade organic contaminants in soil [Paper III; 41]. The extracellular enzymes excreted by these fungi may have the ability to degrade even sorbed contaminants. However, one major problem that has to be solved before these fungi could be used in large-scale applications is their colonization of the soil. In order to efficiently degrade the soil contaminants the mycelium of these fungi has to penetrate all the soil material. This presents a challenge since wood-rotting fungi normally grow on dead wood [122].

Degradation and formation in remedial processes
34
Bioslurry [Paper IV] As described in Paper IV, the gasworks soil was treated in a pilot-scale
bioslurry reactor for 29 days. Even though bioslurry treatment is considered an effective biological remediation method, the overall degradation result in this study was relatively poor (the total PAH-amount were reduced by approximately 40%). Most bi- and tri-cyclic PAHs were degraded, but of the PAHs with four fused rings half of the initial amount remained after the treatment. Furthermore, the PAHs with five and six fused rings were virtually unaffected by the treatment. These results are consistent with other bioslurry treatments of aged gasworks soil [19,88,123], although better degradation results have been obtained for less weathered soils [124]. It is generally accepted that the main reason for the poor degradation of weathered soils is the low bioavailability of the contaminants in such soils, due to aging effects [9,11,13]. The results in Paper IV support this theory since the dissolved fraction of most contaminants in the slurry decreased faster than the total concentration in the soil, indicating that the mass-transfer from the soil to the aqueous phase was the rate-limiting step for these compounds. On the other hand, for the PAHs with five and six fused rings the low initial concentrations in the water remained constant during the treatment, indicating that the poor degradation of these compounds was not due solely to their slow release from the soil, but other factors were also involved. One such factor might be the lack of microorganisms capable of degrading the HMW compounds.
No new oxy-PAHs, i.e. oxy-PAHs that were not present in the untreated soil, were detected in the soil following the bioslurry treatment. However, two of the initially present oxy-PAHs, 1-acenaphthenone and 4-oxapyrene-5-one, showed significantly higher concentrations at the end of the treatment than at the beginning (Figure 16). Consequently, these two compounds must have been formed to a greater extent than they were degraded. Further, as shown in Figure 16, the other oxy-PAHs in Paper IV were generally degraded more slowly than their parent compounds, indicating that these oxy-PAHs were also formed during the treatment, or alternatively that they were more persistent than the PAHs. However, since oxy-PAHs are generally more available to degradation than PAHs, as discussed in Chapter 5, they were probably formed in the bioslurry reactor. The temporary initial accumulation observed for some PAHs (Figure 16), provides further indications that these compounds really were formed during the treatment. However, the slow removal rates of the oxy-PAHs were not due solely to extensive formation. They also showed relatively high persistency. For example, naphthacene-5,12-dione and 7H-benz[de]anthracen-7-one, which were found at relatively constant
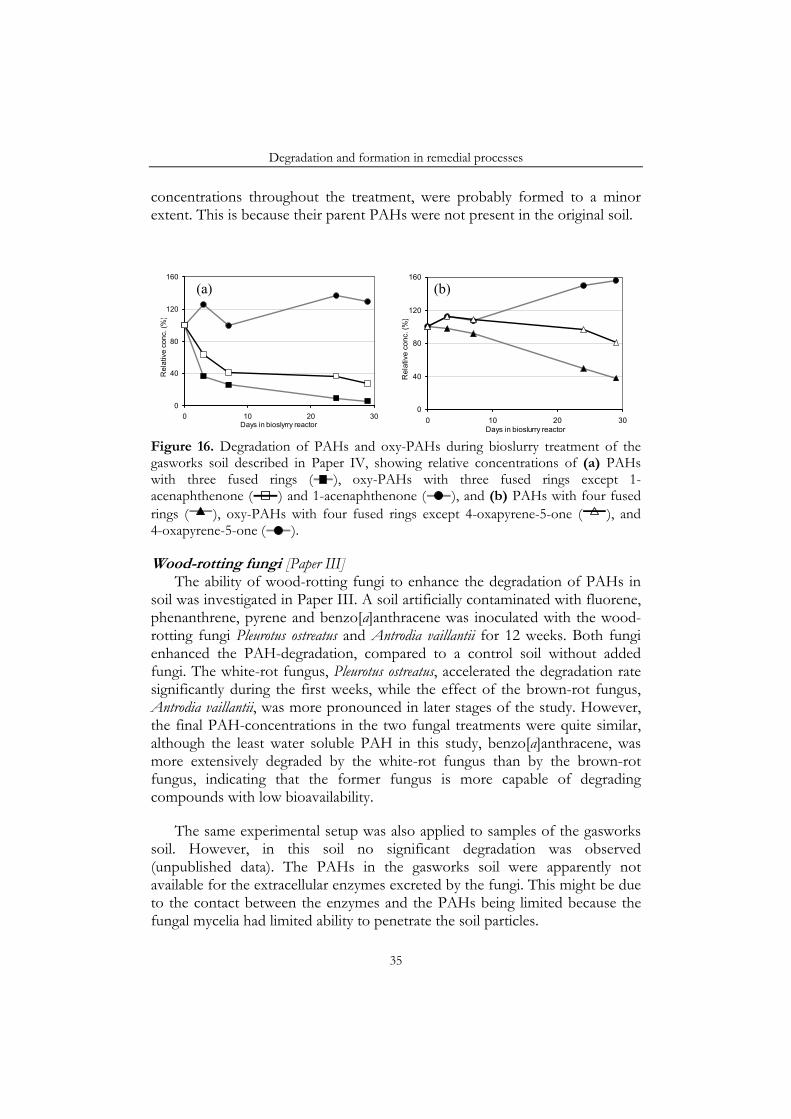
Degradation and formation in remedial processes
35
concentrations throughout the treatment, were probably formed to a minor extent. This is because their parent PAHs were not present in the original soil.
Figure 16. Degradation of PAHs and oxy-PAHs during bioslurry treatment of the gasworks soil described in Paper IV, showing relative concentrations of (a) PAHs with three fused rings ( ), oxy-PAHs with three fused rings except 1-acenaphthenone ( ) and 1-acenaphthenone ( ), and (b) PAHs with four fused rings ( ), oxy-PAHs with four fused rings except 4-oxapyrene-5-one ( ), and 4-oxapyrene-5-one ( ).
Wood-rotting fungi [Paper III] The ability of wood-rotting fungi to enhance the degradation of PAHs in
soil was investigated in Paper III. A soil artificially contaminated with fluorene, phenanthrene, pyrene and benzo[a]anthracene was inoculated with the wood- rotting fungi Pleurotus ostreatus and Antrodia vaillantii for 12 weeks. Both fungi enhanced the PAH-degradation, compared to a control soil without added fungi. The white-rot fungus, Pleurotus ostreatus, accelerated the degradation rate significantly during the first weeks, while the effect of the brown-rot fungus, Antrodia vaillantii, was more pronounced in later stages of the study. However, the final PAH-concentrations in the two fungal treatments were quite similar, although the least water soluble PAH in this study, benzo[a]anthracene, was more extensively degraded by the white-rot fungus than by the brown-rot fungus, indicating that the former fungus is more capable of degrading compounds with low bioavailability.
The same experimental setup was also applied to samples of the gasworks soil. However, in this soil no significant degradation was observed (unpublished data). The PAHs in the gasworks soil were apparently not available for the extracellular enzymes excreted by the fungi. This might be due to the contact between the enzymes and the PAHs being limited because the fungal mycelia had limited ability to penetrate the soil particles.
0
40
80
120
160
0 10 20 30Days in bioslyrry reactor
Rel
ativ
e co
nc. (
%)
0
40
80
120
160
0 10 20 30Days in bioslurry reactor
Rel
ativ
e co
nc. (
%)
(a) (b)
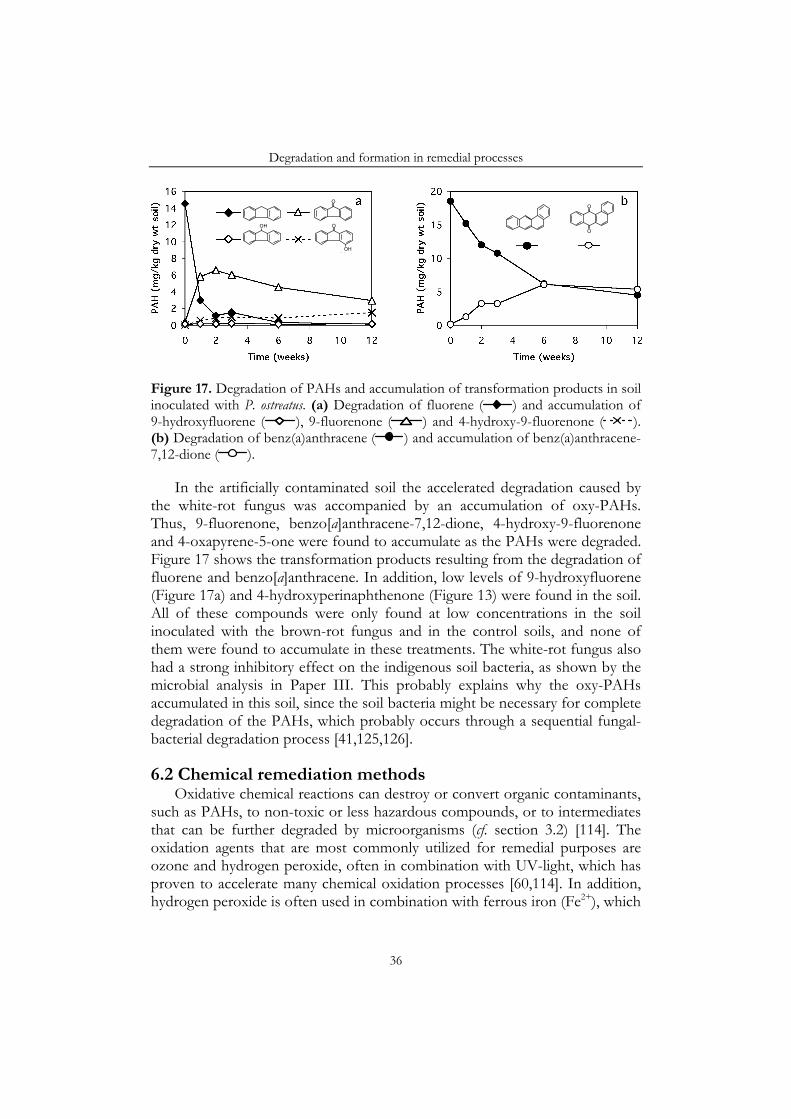
Degradation and formation in remedial processes
36
Figure 17. Degradation of PAHs and accumulation of transformation products in soil inoculated with P. ostreatus. (a) Degradation of fluorene ( ) and accumulation of 9-hydroxyfluorene ( ), 9-fluorenone ( ) and 4-hydroxy-9-fluorenone ( ). (b) Degradation of benz(a)anthracene ( ) and accumulation of benz(a)anthracene-7,12-dione ( ).
In the artificially contaminated soil the accelerated degradation caused by the white-rot fungus was accompanied by an accumulation of oxy-PAHs. Thus, 9-fluorenone, benzo[a]anthracene-7,12-dione, 4-hydroxy-9-fluorenone and 4-oxapyrene-5-one were found to accumulate as the PAHs were degraded. Figure 17 shows the transformation products resulting from the degradation of fluorene and benzo[a]anthracene. In addition, low levels of 9-hydroxyfluorene (Figure 17a) and 4-hydroxyperinaphthenone (Figure 13) were found in the soil. All of these compounds were only found at low concentrations in the soil inoculated with the brown-rot fungus and in the control soils, and none of them were found to accumulate in these treatments. The white-rot fungus also had a strong inhibitory effect on the indigenous soil bacteria, as shown by the microbial analysis in Paper III. This probably explains why the oxy-PAHs accumulated in this soil, since the soil bacteria might be necessary for complete degradation of the PAHs, which probably occurs through a sequential fungal-bacterial degradation process [41,125,126].
6.2 Chemical remediation methods Oxidative chemical reactions can destroy or convert organic contaminants,
such as PAHs, to non-toxic or less hazardous compounds, or to intermediates that can be further degraded by microorganisms (cf. section 3.2) [114]. The oxidation agents that are most commonly utilized for remedial purposes are ozone and hydrogen peroxide, often in combination with UV-light, which has proven to accelerate many chemical oxidation processes [60,114]. In addition, hydrogen peroxide is often used in combination with ferrous iron (Fe2+), which
O
OH O
OH
O
O

Degradation and formation in remedial processes
37
produces hydroxyl radicals (OH•) in a reaction commonly known as Fenton’s reaction [127; Paper VI]:
H2O2 + Fe2+ → OH• + OH- + Fe3+
Chemical remediation methods have a major advantage over biological methods in that they don’t need the contaminant to be taken up through a cell membrane prior to degradation. In addition, the chemical methods are faster and easier to control, since no biological factors have to be taken into consideration. However, pH and temperature must often be regulated to ensure that the reaction goes to completion [60]. The chemical oxidation methods are non-selective, which means that any oxidizable material will react with the oxidizing agent. Consequently, large amounts of the reagents may be consumed when soil is treated. For successful degradation, the oxidation agent must be thoroughly mixed with the contaminants, implying that slurry-reactors are best suited for chemical oxidation treatments [60]. However, in situ chemical oxidation methods have also been employed [128].
Even the chemical oxidation methods have limited capacity to degrade PAHs in highly weathered soils, where the contaminants have diffused deeply into the organic material [Paper VI; 129,130]. This is because such compounds are no longer available for the oxidation reactions, which mainly occur in the water phase. To be oxidized, the PAHs must be in close contact with the water, or preferably dissolved in it. The availability of the contaminants may, however, be enhanced in similar ways to the approaches used during bio-remediation. Thus, solvent [Paper VI; 131], vegetable oil [129] and surfactants [132] have been added to the soil prior to Fenton’s reagent to enhance the desorption of PAHs. Furthermore, large excesses of reagent have been used to disrupt the soil matrix and facilitate more rapid desorption [133].
Fenton’s reagent, [Paper VI] The gasworks soil was also treated with Fenton’s reagent. However, since
the oxidation of PAHs in highly weathered soils has been unsatisfactory in traditional Fenton reaction approaches, in which the soil is slurried in water at ambient temperature, some modifications were tested. For instance, the soil was pretreated with ethanol prior to addition of Fenton’s reagent [Paper VI]. This was done to enhance the desorption of the PAHs from the soil. In another (unpublished) study, elevated temperatures and aggressive oxidation conditions were used for the same purpose.
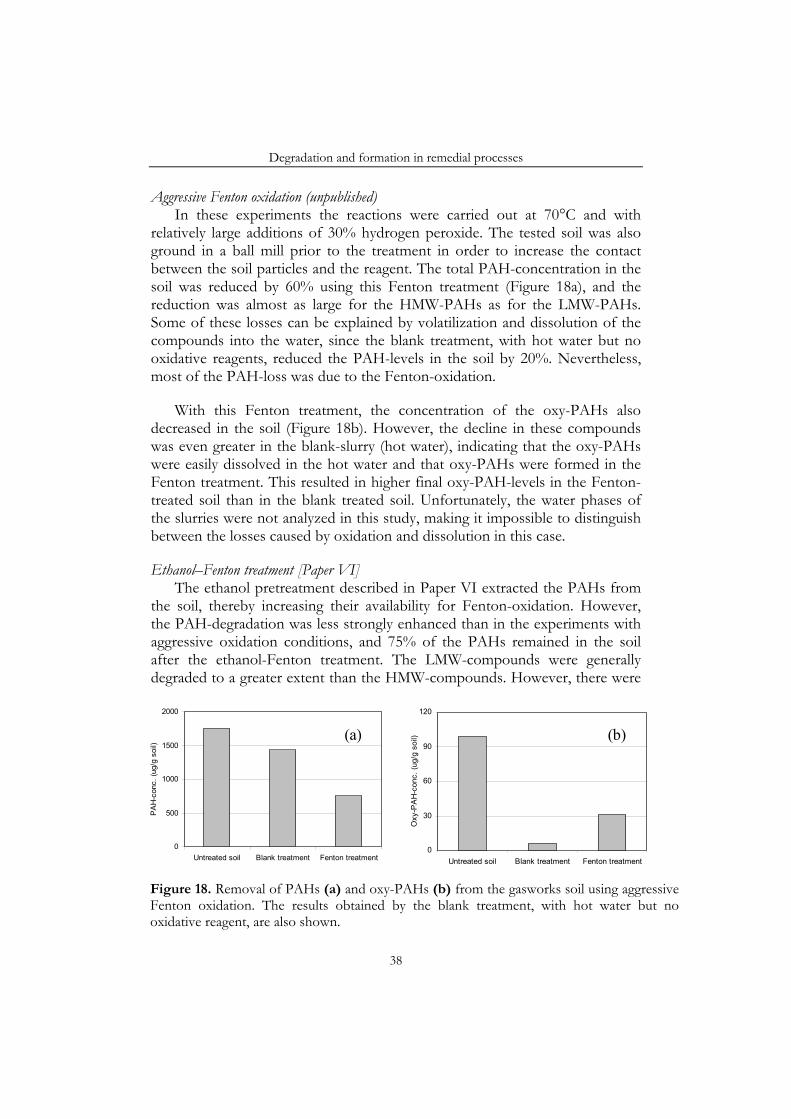
Degradation and formation in remedial processes
38
Aggressive Fenton oxidation (unpublished) In these experiments the reactions were carried out at 70°C and with
relatively large additions of 30% hydrogen peroxide. The tested soil was also ground in a ball mill prior to the treatment in order to increase the contact between the soil particles and the reagent. The total PAH-concentration in the soil was reduced by 60% using this Fenton treatment (Figure 18a), and the reduction was almost as large for the HMW-PAHs as for the LMW-PAHs. Some of these losses can be explained by volatilization and dissolution of the compounds into the water, since the blank treatment, with hot water but no oxidative reagents, reduced the PAH-levels in the soil by 20%. Nevertheless, most of the PAH-loss was due to the Fenton-oxidation.
With this Fenton treatment, the concentration of the oxy-PAHs also decreased in the soil (Figure 18b). However, the decline in these compounds was even greater in the blank-slurry (hot water), indicating that the oxy-PAHs were easily dissolved in the hot water and that oxy-PAHs were formed in the Fenton treatment. This resulted in higher final oxy-PAH-levels in the Fenton-treated soil than in the blank treated soil. Unfortunately, the water phases of the slurries were not analyzed in this study, making it impossible to distinguish between the losses caused by oxidation and dissolution in this case.
Ethanol–Fenton treatment [Paper VI] The ethanol pretreatment described in Paper VI extracted the PAHs from
the soil, thereby increasing their availability for Fenton-oxidation. However, the PAH-degradation was less strongly enhanced than in the experiments with aggressive oxidation conditions, and 75% of the PAHs remained in the soil after the ethanol-Fenton treatment. The LMW-compounds were generally degraded to a greater extent than the HMW-compounds. However, there were
Figure 18. Removal of PAHs (a) and oxy-PAHs (b) from the gasworks soil using aggressive Fenton oxidation. The results obtained by the blank treatment, with hot water but no oxidative reagent, are also shown.
0
500
1000
1500
2000
Untreated soil Blank treatment Fenton treatment
PA
H-c
onc.
(ug/
g so
il)
0
30
60
90
120
Untreated soil Blank treatment Fenton treatment
Oxy
-PA
H-c
onc.
(ug/
g so
il)(a) (b)

Degradation and formation in remedial processes
39
notable exceptions to this general rule. For instance, anthracene, benzo[a]pyrene and perylene were much more efficiently removed form the soil than other PAHs with equal numbers of fused rings. This is probably because PAHs differ in their chemical reactivity, as discussed in section 3.2. The result is in agreement with previous studies [50,134], which have found Fenton’s reagent to react more efficiently with some PAHs than with others. However, when comparing the results of these studies it appears as if the reaction rates of individual PAHs are influenced to different degrees by the presence of ethanol. As shown in Paper VI, anthracene and the methyl anthracenes were among the most efficiently oxidized compounds, while phenanthrene and the methyl phenanthrenes were much less oxidized. This is consistent with the higher reactivity of anthracene, but on the other hand phenanthrene has higher aqueous solubility and should therefore be more readily available to the degradation processes. The latter fact explains why phenanthrene is normally more easily biodegraded than anthracene [Paper IV; 134].
The enhanced degradation observed in the ethanol-pretreated soil was accompanied by increasing concentrations of some oxy-PAHs. For instance, anthracene-9,10-dione, 1-methylanthracenedione, 2-methylanthracenedione, benzo[a]anthracene-7,12-dione and 4-hydroxy-9-fluorenone were found at higher concentrations in the soil after the treatment than before the treatment. These compounds were also found in considerable amounts in the liquid phase after the treatment. Analysis of the liquid phase also revealed that the total amounts of 1-indanone and 1,8-naphthalic anhydride had increased during the treatment, and that a second isomer of hydroxy-9-fluorenone had been formed. The increased concentrations of the anthracenediones are consistent with the high removal rates of the anthracenes. Similarly, benzo[a]anthracene-7,12-dione, 1,8-naphthalic anhydride and the hydroxy-9-fluorenones were probably formed through the oxidation of benzo[a]anthracene, acenaphthene/ acenaphthylene and fluorene, respectively. Previous studies have shown that quinones are important intermediates during Fenton-oxidation of PAHs [50]. The studies presented here verify these findings since quinones were found to be the main compounds to accumulate.
6.3 Oxy-PAHs in remedial processes The potential accumulation of oxy-PAHs and other oxidation products of
PAHs are often ignored during soil remediation programs. Although many studies have shown that these compounds are formed during the biological and chemical degradation of PAHs (as shown in Chapter 3), there have been few studies on their possible accumulation during soil remediation processes

Degradation and formation in remedial processes
40
[41,66,88,135-138]. In the present investigation, the oxy-PAHs found after the treatments were primarily those that were detected in the untreated gasworks soil. The exceptions were the 9-hydroxyfluoren and 4-hydroxyperinaphthenone found in small amounts in the PAH-spiked soil after the fungal treatment [Paper III], and the hydroxy-9-fluorenone found (again in small quantities) in the slurry liquid of the gasworks soil after the ethanol-Fenton treatment [Paper VI].
Many of the oxy-PAHs that were seen to accumulate were probably temporary intermediates, and their abundance in the soil might decrease if a longer process time was applied, or larger amounts of reagents. However, since most of them were present in the untreated gasworks soil and are also found in other environmental samples (Table 4) they are probably relatively persistent and may remain in the soil after a remedial treatment. Similar compounds have also been found to accumulate in other studies where PAH-contaminated soils have been treated in different remedial processes (Table 4). It might therefore be concluded that most of the oxy-PAHs that are frequently found in environmental samples, could potentially accumulate during the remediation of PAH-contaminated soil.
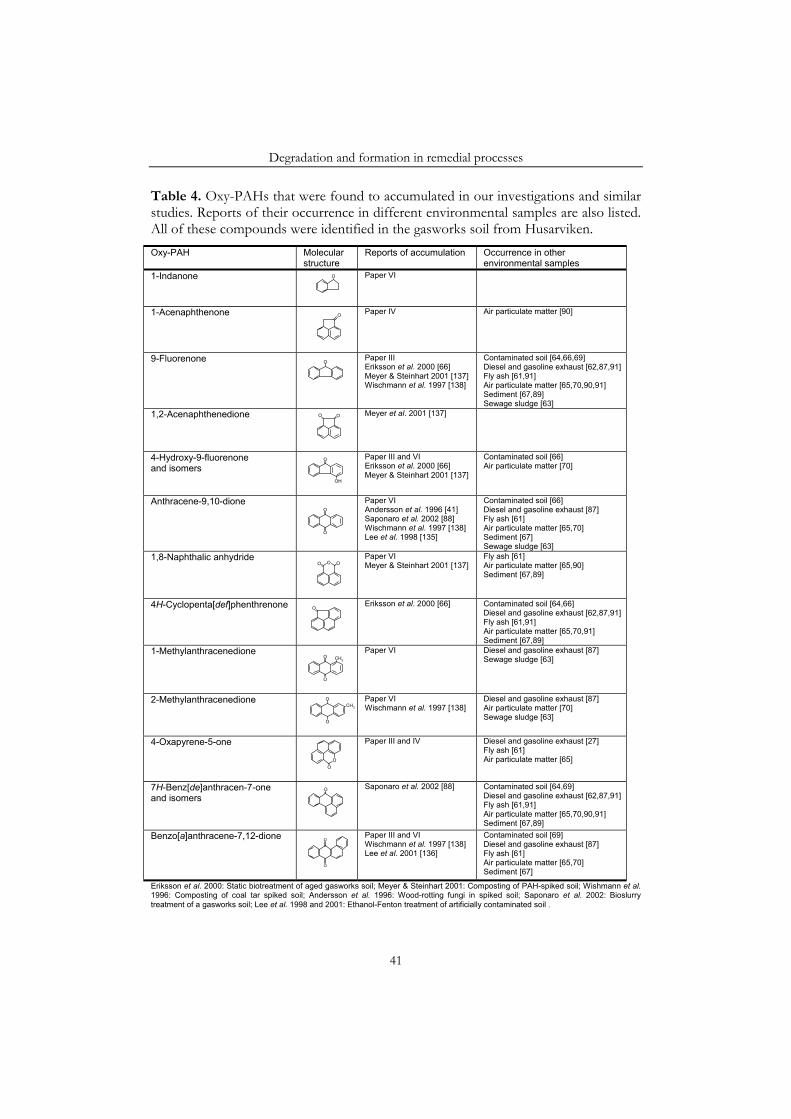
Degradation and formation in remedial processes
41
Table 4. Oxy-PAHs that were found to accumulated in our investigations and similar studies. Reports of their occurrence in different environmental samples are also listed. All of these compounds were identified in the gasworks soil from Husarviken. Oxy-PAH Molecular
structure Reports of accumulation Occurrence in other
environmental samples 1-Indanone
Paper VI
1-Acenaphthenone
Paper IV Air particulate matter [90]
9-Fluorenone
Paper III Eriksson et al. 2000 [66] Meyer & Steinhart 2001 [137] Wischmann et al. 1997 [138]
Contaminated soil [64,66,69] Diesel and gasoline exhaust [62,87,91] Fly ash [61,91] Air particulate matter [65,70,90,91] Sediment [67,89] Sewage sludge [63]
1,2-Acenaphthenedione
Meyer et al. 2001 [137]
4-Hydroxy-9-fluorenone and isomers
Paper III and VI Eriksson et al. 2000 [66] Meyer & Steinhart 2001 [137]
Contaminated soil [66] Air particulate matter [70]
Anthracene-9,10-dione Paper VI Andersson et al. 1996 [41] Saponaro et al. 2002 [88] Wischmann et al. 1997 [138] Lee et al. 1998 [135]
Contaminated soil [66] Diesel and gasoline exhaust [87] Fly ash [61] Air particulate matter [65,70] Sediment [67] Sewage sludge [63]
1,8-Naphthalic anhydride
Paper VI Meyer & Steinhart 2001 [137]
Fly ash [61] Air particulate matter [65,90] Sediment [67,89]
4H-Cyclopenta[def]phenthrenone
Eriksson et al. 2000 [66]
Contaminated soil [64,66] Diesel and gasoline exhaust [62,87,91] Fly ash [61,91] Air particulate matter [65,70,91] Sediment [67,89]
1-Methylanthracenedione
Paper VI Diesel and gasoline exhaust [87] Sewage sludge [63]
2-Methylanthracenedione
Paper VI Wischmann et al. 1997 [138]
Diesel and gasoline exhaust [87] Air particulate matter [70] Sewage sludge [63]
4-Oxapyrene-5-one
Paper III and IV Diesel and gasoline exhaust [27] Fly ash [61] Air particulate matter [65]
7H-Benz[de]anthracen-7-one and isomers
Saponaro et al. 2002 [88]
Contaminated soil [64,69] Diesel and gasoline exhaust [62,87,91] Fly ash [61,91] Air particulate matter [65,70,90,91] Sediment [67,89]
Benzo[a]anthracene-7,12-dione
Paper III and VI Wischmann et al. 1997 [138] Lee et al. 2001 [136]
Contaminated soil [69] Diesel and gasoline exhaust [87] Fly ash [61] Air particulate matter [65,70] Sediment [67]
Eriksson et al. 2000: Static biotreatment of aged gasworks soil; Meyer & Steinhart 2001: Composting of PAH-spiked soil; Wishmann et al. 1996: Composting of coal tar spiked soil; Andersson et al. 1996: Wood-rotting fungi in spiked soil; Saponaro et al. 2002: Bioslurry treatment of a gasworks soil; Lee et al. 1998 and 2001: Ethanol-Fenton treatment of artificially contaminated soil .
O
O
O
OO
O
O
O
O
CH3
O
O
CH3
O
O OO
O
OH
OO
O
O
O

42
7. Concluding remarks and future perspectives
In this thesis it is shown that not only PAHs but also oxy-PAHs should be taken into account during risk assessment and remediation of PAH-contaminated soils. This is because as well as being toxic and mutagenic, oxy-PAHs are also relatively persistent in the environment [Paper IV]. The oxy-PAHs may also be formed during degradation of PAHs, and they could therefore potentially accumulate during remedial processes [Papers III, IV and VI]. Furthermore, exposure to the oxy-PAHs may pose a greater risk to health and the environment than exposure to the PAHs, since the former compounds are more available for uptake in living organisms [Paper II].
New extraction and fractionation methods have been developed to facilitate the concurrent analysis of PAHs and oxy-PAHs in soil. The PLE-technique was investigated for its reliability and efficiency to extract these compounds from soil [Paper I], and it was shown to be rapid and efficient. A selective PLE-method was also developed, exploiting the functional similarity between the PLE-cell and a chromatographic column [Paper V]. With chromatographic material packed in the extraction cell it was possible to selectively extract the PAHs and oxy-PAHs into two different fractions. This method is much faster than a traditional method, and it uses considerably less solvent.
The persistency of oxy-PAHs was indicated by their presence in the gasworks soil analyzed in most of the studies [Paper IV]. This soil was highly weathered and had been subject to various environmental degradation processes, which had changed the contaminant profile in the soil in favor of the more recalcitrant compounds. Nevertheless, oxy-PAHs were relatively abundant. This is noteworthy since oxy-PAHs have higher aqueous solubility and are therefore generally believed to be more available to degradation and leaching than the PAHs.
The assumption that oxy-PAHs could accumulate during soil remediation was supported by the findings from tests of three different remediation methods. In a bioslurry treatment the formation of oxy-PAHs lead to increased amounts of two oxy-PAHs (1-acenaphthenone and 4-oxapyrene-5-one) and an unexpectedly slow removal of other oxy-PAHs [Paper IV]. In soil inoculated with white-rot fungus the enhanced PAH-degradation was accompanied by the accumulation of 9-fluorenone, benzo[a]anthracene-7,12-dione, 4-hydroxy-9-fluorenone and 4-oxapyrene-5-one [Paper III]. In the ethanol-Fenton treatment, the extensive oxidation of some specific PAHs, e.g. anthracene, led

Concluding remarks and future perspectives
43
to increased amounts of oxy-PAHs, which mainly originated from these PAHs [Paper VI].
The gasworks soil examined in the present studies was highly weathered and the contaminants therein had been subject to prolonged aging. This explains why the availability of the PAHs to the biological and chemical degradation processes was poor [Papers IV & VI], resulting in insufficient degradation, and why the uptake in earthworms was low [Paper II]. Consequently, to remediate a site such as Husarviken, where this soil was collected, a robust remediation method will be needed.
Future perspectives At present, it is difficult to predict when the formation and accumulation
of specific oxy-PAHs will occur. To avoid increasing levels of these compounds during soil remediation the conditions that promote their formation must be further elucidated.
It may be necessary to ensure that the amounts of oxy-PAHs do not increase in the soil. Therefore, it is suggested that PAH-contaminated soil should be screened for oxy-PAHs as well as PAHs during the course of remedial monitoring programs. The selective PLE-method could be used to simplify the inclusion of oxy-PAHs in such programs.
Monitoring oxy-PAHs would be much easier if adequate marker compounds were available. Such marker compounds should be easy to analyze together with the PAHs, and should give information about the status of all other oxy-PAHs in the soil. However, a justified selection of marker compounds cannot be made until sufficient knowledge about the formation of oxy-PAHs in different remedial processes has been obtained.
If biological or chemical remediation methods are to be used at Husarviken, pretreatment of the soil will be needed to facilitate desorption and degradation. Probably, a better option for this particular soil would be to use an extraction method that separates the contaminants from the soil into an organic solvent, e.g. ethanol, or hot pressurized water, before subjecting them to destructive treatments.

44
8. Acknowledgement
This study was performed within the Swedish national research program “Soil Remediation in a Cold Climate” (COLDREM), within financial support from the Foundation for Strategic Environmental Research (MISTRA) for which I am very grateful. Further, thanks for the collaboration and support form the North Sweden Soil Remediation Center (MCN), which is financed by the EU Structural Funds and New Objectives 1.
I would also like to thank all my fellow co-authors of the papers, EkoTec AB in Skelleftehamn for the collaboration resulting in Paper IV, and John Blackwell for proofreading the papers and the summary in this thesis.
Tack Lars för handledning, psykologisk stöd och uppmuntran under arbetet. Det blev en rätt tuff avslutning på det hela, men det var ju många roliga år före det. Nu får du kanske mer tid att tänka på dig själv. Tack också för det lånet av skidpulkan, det kanske är dags att sälja….? Och så vill jag tacka mina biträdande handledare, Peter och Mats, för all hjälp ni gav och för att ni baxade mig i mål på slutet.
Tack hela gänget på Miljökemi. Jag har trivts mycket bra att jobba med er alla och kommer så säkerligen att fortsätta. Lita på att jag också kommer att synas mer i fikarummet framöver än jag har gjort den sista tiden. Det blir nog fler luncher på Universum också, matlådan får vila därhemma ett tag. Så när det knackar på rutan kl. 11.13 (inga namn) då är jag redo. Med ny glöd kommer jag även att äntra innebandyplan, det har ju blivit ett litet avbrott där också. Men det är väl så att alla stjärnor ska lägga av ett tag för att sedan kunna göra comeback.
Ett tack till COLDREM gänget, det har varit många roliga samarbeten, symposium och inte minst internat. Måste säga att Vålådalen och Duved är särskilt stimulerande ”forskningsmiljöer”. Men vad blev det av Hummerfisket på västkusten? Ett extra tack till Erik Andersson för många givande samarbeten och diskussioner, och inte minst för mat och logi i Lunds studentgetto. Nästa gång vi hörs på telefon är jag möjligen något mindre disträ.
Tack alla vänner utanför Miljökemi för att ni finns och för att ni har hjälpt mig att tänka på annat ibland. Det har varit extra viktigt nu under den mer intensiva skrivarperioden. Stefan, vi får ta nya tag i sanden nästa säsong, kanske är det då dags att börja vinna lite?
Jag vill också rikta ett tack till familjen hemma i Gävle för all er uppmuntran, det har betytt mer än ni kanske tror. Sist men definitivt inte minst, Stina och Hugo, det har varit en intensiv period för er också de senaste månaderna. Tack för att ni har förstått och ställt upp. Ni har funnits och finns i mina tankar hela tiden.

45
9. References
1. Howsam M Jones KC (1998) Sources of PAHs in the environment. In: Neilson AH (ed). Anthropogenic compounds. PAHs and related compounds. Springer, Berlin, Germany, pp. 137-174.
2. Blumer M (2003) Polycyclic aromatic compounds in nature. Scientific American 35-45.
3. Mackay D, Shiu WY Ma KC (1992) Illustrated handbook of physical-chemical properties and environmental fate for organic chemicals: Polynuclear aromatic hydrocarbons, polychlorinated dioxins and dibenzofurans. Lewis Publishers, Chelsea, Michigan, USA.
4. Wild SR, Jones KC (1995) Polynuclear aromatic hydrocarbons in the United-Kingdom environment – a preliminary source inventory and budget. Environ.Pollut. 88: 91-108.
5. Wilson SC, Jones KC (1993) Bioremediation of soil contaminated with polynuclear aromatic hydrocarbons (PAHs) - a review. Environ.Pollut. 81: 229-249.
6. Johnston N, Sadler R, Shaw GR, Connell DW (1993) Environmental modification of PAH composition in coal tar containing samples. Chemosphere 27: 1151-1158.
7. Bossert IP, Bartha R (1986) Structure-biodegradability relationsships of polycyclic aromatic hydrocarbons in soil. Bull.Environ.Contam.Toxicol. 37: 490-495.
8. Sims RC, Overcash MR (1983) Fate of polynuclear aromatic compounds (PNAs) in soil-plant systems. Res.Rev. 88: 1-67.
9. Hatzinger PB, Alexander M (1995) Effect of aging of chemicals in soil on their biodegradability and extractability. Environ.Sci.Technol. 29: 537-545.
10. Loehr RC, Webster MT (1996) Behavior of fresh vs aged chemicals in soil. J Soil Contam. 5: 361-383.
11. Weissenfels WD, Klewer HJ, Langhoff J (1992) Adsorption of polycyclic aromatic hydrocarbons (PAHs) by soil particles - influence on biodegradability and biotoxicity. Appl.Microbiol.Biotechnol. 36: 689-696.
12. Allard AS, Remberger M, Neilson AH (2000) The negative impact of aging on the loss of PAH components in a creosote-contaminated soil. International Biodeterioration & Biodegradation 46: 43-49.
13. Alexander M (1995) How toxic are toxic chemicals in soil? Environ.Sci.Technol. 29: 2713-2717.

References
46
14. Pignatello JJ, Xing BS (1996) Mechanisms of slow sorption of organic chemicals to natural particles. Environ.Sci.Technol. 30: 1-11.
15. Scow KM Johnson CR (1997) Effect of sorption on biodegradation of soil pollutants. In: Sparks DL (ed). Advances in Agronomy, Vol 58. Academic Press Inc, San Diego, pp. 1-56.
16. Jones KC, Stratford JA, Waterhouse KS, Vogt NB (1989) Organic contaminants in welsh soils - polynuclear aromatic hydrocarbons. Environ.Sci.Technol. 23: 540-550.
17. Johnston WR, Harrison RM (1984) Deposition of metallic and organic pollutants alongside the M6 motorway. Sci.Total Environ. 33: 119-127.
18. Haeseler F, Blanchet D, Druelle V, Werner P, Vandecasteele JP (1999) Analytical characterization of contaminated soils from former manufactured gas plants. Environ.Sci.Technol. 33: 825-830.
19. Kilbane JJ (1998) Extractability and subsequent biodegradation of PAHs from contaminated soil. Water Air Soil Pollut. 104: 285-304.
20. IARC (1984) Monographs on the evalutaion of the carcinogenic risk of chemicals to humans; Polynuclear Aromatic Compounds, Part 3, Industrial exposures in aluminium production, coal gasification, coke production and iron and steel founding. Vol. 34, IARC, Lyon, Fracnce.
21. Mueller JG, Chapman PJ, Pritchard PH (1989) Creosote-contaminated sites. Their potential for bioremediation. Environ.Sci.Technol. 23: 1197-1201.
22. IARC (1985) Monographs on the evalutaion of the carcinogenic risk of chemicals to humans; Polynuclear Aromatic Compounds, Part 4, Bitumens, coal-tars and derived products, shale oils and soots, Vol. 35, IARC, Lyon, France.
23. Delistraty D (1997) Toxic equivalency factor approach for risk assessment of polycyclic aromatic hydrocarbons. Toxicol.Environ.Chem. 64: 81-108.
24. Pickering RW (1999) A toxicological review of polycyclic aromatic hydrocarbons. J.Toxicol.Cutan.Ocul.Toxicol. 18: 101-135.
25. IARC (1983) Monographs on the evalutaion of the carcinogenic risk of chemicals to humans; Polynuclear Aromatic Compounds, Part 1, Chemical, environmental and experimental data. Vol. 32, IARC, Lyon, France.
26. Moller M, Hagen I, Ramdahl T (1985) Mutagenicity of polycyclic aromatic compounds (PAC) identified in source emissions and ambient air. Mutation Research 149-156.

References
47
27. Pitts JN, Lokensgard DM, Harger W, Fisher TS, Mejia V, Schuler JJ, Scorziell GM, Katzenstein YA (1982) Mutagens in diesel exhaust particulate. Identification and direct activities of 6-nitrobenzo[a]pyrene, 9-nitroanthracene, 1-nitropyrene and 5H-phenanthro[4,5-bcd]pyran-5-one. Mutation Research 103: 241-249.
28. Cerniglia CE (1984) Microbial metabolism of polycyclic aromatic hydrocarbons. Advances in Applied Microbiology 30: 31-71.
29. Cerniglia CE (1992) Biodegradation of polycyclic aromatic hydrocarbons. Biodegradation 3: 351-368.
30. Gibson DT (1993) Biodegradation, biotransformation and the Belmont. J.Ind.Microbiol. 12: 1-12.
31. Heitkamp MA, Cerniglia CE (1988) Mineralization of polycyclic aromatic-hydrocarbons by a bacterium isolated from sediment below an oil-field. Appl.Environ.Microbiol. 54: 1612-1614.
32. Kanaly RA, Harayama S (2000) Biodegradation of high-molecular-weight polycyclic aromatic hydrocarbons by bacteria. J.Bacteriol. 182: 2059-2067.
33. Cerniglia CE (1997) Fungal metabolism of polycyclic aromatic hydrocarbons: Past, present and future applications in bioremediation. J Ind.Microbiol.Biotechnol. 19: 324-333.
34. Sutherland JB, Rafii F, Khan AA Cerniglia CE (1995) Mechanisms of polycyclic aromatic hydrocarbon degradation. In: Young LY Cerniglia CE (ed), Microbial Transformation and Degradation of Toxic Organic Chemicals, Wiley-Liss, Inc, New York, pp. 269-306.
35. Wunder T, Marr J, Kremer S, Sterner O, Anke H (1997) 1-methoxypyrene and 1,6-dimethoxypyrene: Two novel metabolites in fungal metabolism of polycyclic aromatic hydrocarbons. Arch.Microbiol. 167: 310-316.
36. Casellas M, Grifoll M, Bayona JM, Solanas AM (1997) New metabolites in the degradation of fluorene by Arthrobacter sp strain F101. Appl.Environ.Microbiol. 63: 819-826.
37. Kelley I, Freeman JP, Evans FE, Cerniglia CE (1993) Identification of metabolites from the degradation of fluoranthene by Mycobacterium sp strain Pyr-1. Appl.Environ.Microbiol. 59: 800-806.
38. Bumpus JA, Tien M, Wright D, Aust SD (1985) Oxidation of persistent environmental pollutants by a white rot fungus. Science 228: 1434-1436.
39. Bezalel L, Hadar Y, Fu PP, Freeman JP, Cerniglia CE (1996) Metabolism of phenanthrene by the white rot fungus Pleurotus ostreatus. Appl.Environ.Microbiol. 62: 2547-2553.

References
48
40. Hammel KE, Gai WZ, Green B, Moen MA (1992) Oxidative degradation of phenanthrene by the ligninolytic fungus Phanerochaete chrysosporium. Appl.Environ.Microbiol. 58: 1832-1838.
41. Andersson BE, Henrysson T (1996) Accumulation and degradation of dead-end metabolites during treatment of soil contaminated with polycyclic aromatic hydrocarbons with five strains of white-rot fungi. Appl.Microbiol.Biotechnol. 46: 647-652.
42. Kochany J, Maguire RJ (1994) Abiotic transformations of polynuclear aromatic hydrocarbons and polynuclear aromatic nitrogen heterocycles in aquatic environments. Sci.Total Environ. 144: 17-31.
43. Neilson AH (1994) Organic chemicals in the aquatic environment; Distribution, persistency and toxicity. Lewis Publishers, Boca Raton, Florida, USA.
44. Berg Mvd, Meent Dvd, Peijnenburg WJGM, Sijm DTHM, Struijs J Tas JW (1995) Transport, accumulation and transformation processes. In: C.J. van Leeuwen and J.L.M. Hermens (ed.) Risk assessment of chemicals: An introduction,. Kluwer Academic Publishers, Dordrecht, The Netherlands.
45. Haag WR, Yao CCD (1992) Rate constants for reaction of hydroxyl radicals with several drinking water contaminants. Environ.Sci.Technol. 26: 1005-1013.
46. Legrini O, Oliveros E, Braun AM (1993) Photochemical processes for water-treatment. Chemical Reviews 93: 671-698.
47. Gurol MD, Singer PC (1982) Kinetics of ozone decomposition: A dynamic approach. Environ.Sci.Technol. 16: 377-383.
48. Yao JJ, Huang ZH, Masten SJ (1998) The ozonation of pyrene: Pathway and product identification. Water Res. 32: 3001-3012.
49. Yao JJ, Huang ZH, Masten SJ (1998) The ozonation of benz[a]anthracene: Pathway and product identification. Water Res. 32: 3235-3244.
50. Lee BD, Iso M, Hosomi M (2001) Prediction of Fenton oxidation positions in polycyclic aromatic hydrocarbons by Frontier electron density. Chemosphere 42: 431-435.
51. Rivas FJ, Beltran FJ, Acedo B (2000) Chemical and photochemical degradation of acenaphthylene. Intermediate identification. Journal of Hazardous Materials 75: 89-98.
52. Reisen F, Arey J (2002) Reactions of hydroxyl radicals and ozone with acenaphthene and acenaphthylene. Environ.Sci.Technol. 36: 4302-4311.

References
49
53. Koeber R, Bayona JM, Niessner R (1997) Analysis of ozonolysis products of benzo[a]pyrene with capillary gas chromatography mass spectrometry and liquid chromatography mass spectrometry. International Journal of Environmental Analytical Chemistry 66: 313-325.
54. Barbas JT, Sigman ME, Dabestani R (1996) Photochemical oxidation of phenanthrene sorbed on silica gel. Environ.Sci.Technol. 30: 1776-1780.
55. David B, Boule P (1993) Phototransformation of hydrophobic pollutants in aqueous medium .1. PAHs adsorbed on silica. Chemosphere 26: 1617-1630.
56. Mallakin A, Dixon DG, Greenberg BM (2000) Pathway of anthracene modification under simulated solar radiation. Chemosphere 40: 1435-1441.
57. Wheland GW (1942) A quantum-mecahnical investigation of the orientation of substituents in aromatic molecules. J.Amer.Chem.Soc. 64: 900-908.
58. Dewar MJS (1952) A molecular-orbital theory of organic chemistry. VI. Aromatic substitution and addition. J.Amer.Chem.Soc. 74: 3357-3363.
59. Zander M (1979) Z.Naturforsch 34a: 521
60. Freeman HM, Harris EF (1995) Hazardous Waste Remediation. Innovative treatment technologies, Technomic Publishing Company, Lancaster, Pennsylvania, USA.
61. Akimoto Y, Aoki T, Nito S, Inouye Y (1997) Oxygenated polycyclic aromatic hydrocarbons from MSW incinerator fly ash. Chemosphere 34: 263-273.
62. Alsberg T, Stenberg U, Westerholm R, Strandell M, Rannug U, Sundvall A, Romert L, Bernson V, Pettersson B, Toftgard R, Franzen B, Jansson M, Gustafsson JA, Egeback KE, Tejle G (1985) Chemical and biological characterization of organic material from gasoline exhaust particles. Environ.Sci.Technol. 19: 43-50.
63. Bodzek D, Janoszka B, Dobosz C, Warzecha L, Bodzek M (1997) Determination of polycyclic aromatic compounds and heavy metals in sludges from biological sewage treatment plants. J.Chromatogr.A 774: 177-192.
64. Brooks LR, Hughes TJ, Claxton LD, Austern B, Brenner R, Kremer F (1998) Bioassay-directed fractionation and chemical identification of mutagens in bioremediated soils. Environ.Health Perspect. 106: 1435-1440.
65. Casellas M, Fernandez P, Bayona JM, Solanas AM (1995) Bioassay-directed chemical-analysis of genotoxic components in urban airborne particulate matter from Barcelona (Spain). Chemosphere 30: 725-740.

References
50
66. Eriksson M, Dalhammar G, Borg-Karlsson AK (2000) Biological degradation of selected hydrocarbons in an old PAH/creosote contaminated soil from a gasworks site. Appl.Microbiol.Biotechnol. 53: 619-626.
67. Fernandez P, Bayona JM (1992) Use of off-line gel-permeation chromatography normal-phase liquid-chromatography for the determination of polycyclic aromatic-compounds in environmental-samples and standard reference materials (air particulate matter and marine sediment). Journal of Chromatography 625: 141-149.
68. Fernandez P, Grifoll M, Solanas AM, Bayona JM, Albaiges J (1992) Bioassay-directed chemical analysis of genotoxic components in coastal sediments. Environ.Sci.Technol. 26: 817-829.
69. Meyer S, Cartellieri S, Steinhart H (1999) Simultaneous determination of PAHs, hetero-PAHs (N, S, O), and their degradation products in creosote-contaminated soils. Method development, validation, and application to hazardous waste sites. Anal.Chem. 71: 4023-4029.
70. Moyano E, Galceran MT (1997) Determination of oxy-, nitro- and hydroxy- polycyclic aromatic hydrocarbons in atmospheric aerosol samples. Quim.Anal.(Barcelona) 16: 159-164.
71. Huang XD, Dixon DG, Greenberg BM (1993) Impacts of UV-radiation and photomodification on the toxicity of PAHs to the higher plant Lemna gibba (Duckweed). Environ.Toxicol.Chem. 12: 1067-1077.
72. Mallakin A, McConkey BJ, Miao GB, McKibben B, Snieckus V, Dixon DG, Greenberg BM (1999) Impacts of structural photomodification on the toxicity of environmental contaminants: Anthracene photooxidation products. Ecotoxicol.Environ Safety 43: 204-212.
73. McConkey BJ, Duxbury CL, Dixon DG, Greenberg BM (1997) Toxicity of a PAH photooxidation product to the bacteria Photobacterium phosphoreum and the duckweed Lemna gibba: Effects of phenanthrene and its primary photoproduct, phenanthrenequinone. Environ.Toxicol.Chem. 16: 892-899.
74. Ramdahl T (1985) Characterization of polarcompounds such as polycyclic aromatic ketones in air pollution including wood smoke. Enironment International 11: 197-203.
75. Chesis PL, Levin DE, Smith MT, Ernster L, Ames BN (1983) Mutagenicity of quinones: Pathways of metabolic activation and detoxification. Proc.Natl.Acad.Sci.USA 81: 1696-1700.
76. Wischmann H, Steinhart H, Hupe K, Montresori G, Stegmann R (1996) Degradation of selected PAHs in soil/compost and identification of intermediates. Int.J.Environ.Anal.Chem. 64: 247-255.

References
51
77. Bergknut M, Kitti A, Lundstedt S, Tysklind M, Haglund P (2003) Assessment of the availability of PAHs from gasworks soil using different extraction solvents and techniques. Manuscript
78. Richter BE, Jones BA, Ezzell JL, Porter NL, Avdalovic N, Pohl C (1996) Accelerated solvent extraction: A technique for sample preparation. Anal.Chem. 68: 1033-1039.
79. Bjorklund E, Nilsson T, Bowadt S (2000) Pressurised liquid extraction of persistent organic pollutants in environmental analysis. Trac-Trends in Analytical Chemistry 19: 434-445.
80. Fisher JA, Scarlett MJ, Stott AD (1997) Accelerated solvent extraction: An evaluation for screening of soils for selected US EPA semivolatile organic priority pollutants. Environ.Sci.Technol. 31: 1120-1127.
81. Hubert A, Wenzel KD, Manz M, Weissflog L, Engewald W, Schuurmann G (2000) High extraction efficiency for POPs in real contaminated soil samples using accelerated solvent extraction. Anal.Chem. 72: 1294-1300.
82. Box GEP, Hunter WG Hunter JS (1978) Statistics for experimenters: An introduction to design, data analysis and model building. John Wiley & Sons, New York, USA.
83. Popp P, Keil P, M”der M, Paschke A, Thuss U (1997) Application of accelerated solvent extraction followed by gas chromatography, high-performance liquid chromatography and gas chromatography mass spectrometry for the determination of polycyclic aromatic hydrocarbons, chlorinated pesticides and polychlorinated dibenzo-p-dioxins and dibenzofurans in solid wastes. J.Chromatogr.A 774: 203-211.
84. Saim N, Dean JR, Abdullah MP, Zakaria Z (1998) An experimental design approach for the determination of polycyclic aromatic hydrocarbons from highly contaminated soil using accelerated solvent extraction. Anal.Chem. 70: 420-424.
85. EPA Method 3545, Pressurised Fluid Extraction, Test Methods for evaluating solid waste, 3rd ed., Update III; EPA SW-846. U.S. GPO, Washington, DC, July 1995.
86. Zdrahal Z, Karasek P, Lojkova L, Buckova M, Vecera Z, Vejrosta J (2000) Pressurised liquid extraction of ketones of polycyclic aromatic hydrocarbons from soil. J.Chromatogr.A 893: 201-206.
87. Choudhury DR (1982) Characterization of polycyclic ketones and quinones in diesel emission particulates by gas-chromatography mass-spectrometry. Environ.Sci.Technol. 16: 102-106.
88. Saponaro S, Bonomo L, Petruzzelli G, Romele L, Barbafieri M (2002) Polycyclic aromatic hydrocarbons (PAHs) slurry phase bioremediation of a manufacturing gas plant (MGP) site aged soil. Water Air and Soil Pollution 135: 219-236.

References
52
89. Grifoll M, Solanas AM, Bayona JM (1990) Characterization of Genotoxic Components in Sediments by Mass- Spectrometric Techniques Combined with Salmonella Microsome Test. Archives of Environmental Contamination and Toxicology 19: 175-184.
90. Allen JO, Dookeran NM, Taghizadeh K, Lafleur AL, Smith KA, Sarofim AF (1997) Measurement of oxygenated polycyclic aromatic hydrocarbons associated with a size-segregated urban aerosol. Environ.Sci.Technol. 31: 2064-2070.
91. Ramdahl T (1983) Polycyclic aromatic ketones in environmental samples. Environ.Sci.Technol. 17: 666-670.
92. Hale RC, Aneiro KM (1997) Determination of coal tar and creosote constituents in the aquatic environment. J.Chromatogr.A 774: 79-95.
93. Muller A, Bjorklund E, von Holst C (2001) On-line clean-up of pressurized liquid extracts for the determination of polychlorinated biphenyls in feedingstuffs and food matrices using gas chromatography-mass spectrometry. J.Chromatogr.A 925: 197-205.
94. Bjorklund E, Muller A, von Holst C (2001) Comparison of fat retainers in accelerated solvent extraction for the selective extraction of PCBs from fat-containing samples. Anal.Chem. 73: 4050-4053.
95. Gomez-Ariza JL, Bujalance M, Giraldez I, Velasco A, Morales E (2002) Determination of polychlorinated biphenyls in biota samples using simultaneous pressurized liquid extraction and purification. J.Chromatogr.A 946: 209-219.
96. Dionex Corporation (2002) Selective Extraction of PCBs from fish tissue using accelerated solvent extraction (ASE). Application Note 313, Dionex, Sunnyvale, CA, USA.
97. Lee ML, Vassilaros DL, White CM, Novotny M (1979) Retention indexes for programmed-temperature capillary-column gas-chromatography of polycyclic aromatic-hydrocarbons. Anal.Chem. 51: 768-774.
98. McLafferty FW Turecek F (1993) Interpretation of mass spectra, Fourth edition, University Science Books, Mill Valley, California, USA
99. Poole CF Poole SK (1991) Chromatography today. First edition, Elsevier Science Publishers, Amsterdam, The Netherlands.
100. Drozd J (1981) Chemical derivatization in gas chromatography. First edition, Elsevier Scientific Publishing Company, Amsterdam, The Netherlands
101. Luthy RG, Dzombak DA, Peters CA, Roy SB, Ramaswami A, Nakles DV, Nott BR (1994) Remediating Tar-Contaminated Soils at Manufactured Gas Plant Sites. Environ.Sci.Technol. 28: A266-A276

References
53
102. Thomas AO, Lester JN (1994) The Reclamation of Disused Gasworks Sites - New Solutions to An Old Problem. Sci.Total Environ. 152: 239-260.
103. Peters CA, Luthy RG (1993) Coal Tar Dissolution in Water-Miscible Solvents - Experimental Evaluation. Environ.Sci.Technol. 27: 2831-2843.
104. Thomas AO, Lester JN (1993) The microbial remediation of former gasworks sites - a review. Environ.Technol. 14: 1-24.
105. Douglas GS, Bence AE, Prince RC, McMillen SJ, Butler EL (1996) Environmental stability of selected petroleum hydrocarbon source and weathering ratios. Environ.Sci.Technol. 30: 2332-2339.
106. Mosi AA, Reimer KJ, Eigendorf GK (1997) Analysis of polyaromatic quinones in a complex environmental matrix using gas chromatography ion trap tandem mass spectrometry. Talanta 44: 985-1001.
107. Behymer TD, Hites RA (1988) Photolysis of Polycyclic Aromatic-Hydrocarbons Adsorbed on Fly- Ash. Environ.Sci.Technol. 22: 1311-1319.
108. Aihara J (1999) Reduced HOMO-LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J.Phys.Chem.A 103: 7487-7495.
109. Zander M (1983) Physical and chemical properties of polycyclic aromatic hydrocarbons. In: Bjorseth A (ed). Handbook of polycyclic aromatic hydrocarbons. Marcel Dekker, New York, pp. 1-25.
110. Jager T (1998) Mechanistic approach for estimating bioconcentration of organic chemicals in earthworms (Oligochaeta). Environ.Toxicol.Chem. 17: 2080-2090.
111. Ma WC, Vankleunen A, Immerzeel J, DeMaagd PGJ (1998) Bioaccumulation of polycyclic aromatic hydrocarbons by earthworms: Assessment of equilibrium partitioning theory in in situ studies and water experiments. Environ.Toxicol.Chem. 17: 1730-1737.
112. Jager T, Sanchez FAA, Muijs B, Vandervelde EG, Posthuma L (2000) Toxicokinetics of polycyclic aromatic hydrocarbons in Eisenia andrei (Oligochaeta) using spiked soil. Environ.Toxicol.Chem. 19: 953-961.
113. Ma WC, Immerzeel J, Bodt J (1995) Earthworm and food interactions on bioaccumulation and disappearance in soil of polycyclic aromatic hydrocarbons: Studies on phenanthrene and fluo ranthene. Ecotoxicol.Environ.Safety 32: 226-232.
114. Hamby DM (1996) Site remediation techniques supporting environmental restoration activities - A review. Sci.Total Environ. 191: 203-224.
115. Boopathy R (2000) Factors limiting bioremediation technologies. Bioresource Technol. 74: 63-67.

References
54
116. Pollard SJT, Hrudey SE, Fedorak PM (1994) Bioremediation of Petroleum - and Creosote-Contaminated Soils - A Review of Constraints. Waste Management & Research 12: 173-194.
117. Olsen RL, Davis A (1990) Predicting the fate of and transport of organic compounds in groundwater. Hazard.Mater.Control 3: 38-63.
118. Rao PSC (1990) Sorption of Organic Contaminants. Water Science and Technology 22: 1-6.
119. Bonten LTC, Grotenhuis TC, Rulkens WH (1999) Enhancement of PAH biodegradation in soil by physicochemical pretreatment. Chemosphere 38: 3627-3636.
120. Yeom IT, Ghosh MM (1998) Mass transfer limitation in PAH-contaminated soil remediation. Water.Sci.Technol. 37: 111-118.
121. Bogan BW, Lamar RT (1999) Surfactants enhancement of white-rot fungal PAH soil remediation. In: Leeson A, Alleman BC (ed.) Bioremediation technologies for polycyclic aromatic hydrocarbon compounds. Battelle Press, Coulumbus, pp 81-86.
122. Andersson E (2001) Colonisation and PAH degradation by wood-rotting fungi in contaminated soil. PhD-Thesis, Department of Biotechnology, Lund University.
123. Pradhan SP, Paterek JR, Liu BY, Conrad JR, Srivastava VJ (1997) Pilot-scale bioremediation of PAH-contaminated soils. Appl.Biochem.Biotechnol. 63-65: 759-773.
124. Pinelli D, Fava F, Nocentini M, Pasquali G (1997) Bioremediation of a polycyclic aromatic hydrocarbon-contaminated soil by using different aerobic batch bioreactor systems. J.Soil Contam. 6: 243-256.
125. In der Wiesche C, Martens R, Zadrazil F (1996) Two-step degradation of pyrene by white-rot fungi and soil microorganisms. Appl.Microbiol.Biotechnol. 46: 653-659.
126. Kotterman MJJ, Vis EH, Field JA (1998) Successive mineralization and detoxification of benzo[a]pyrene by the white rot fungus Bjerkandera sp. strain BOS55 and indigenous microflora. Appl.Environ.Microbiol. 64: 2853-2858.
127. Walling C (1975) Fenton's reagent revisited. Acc.Chem.Res. 8: 125-131.
128. Ho CL, Shebl MAA, Watts RJ (1995) Development of An Injection System for In-Situ Catalyzed Peroxide Remediation of Contaminated Soil. Hazardous Waste & Hazardous Materials 12: 15-25.
129. Bogan BW, Trbovic V, Paterek JR (2003) Inclusion of vegetable oils in Fenton's chemistry for remediation of PAH-contaminated soils. Chemosphere 50: 15-21.
130. Sedlak DL, Andren AW (1994) The Effect of Sorption on the Oxidation of Polychlorinated Biphenyls (PCBs) by Hydroxyl Radical. Water Res. 28: 1207-1215.

References
55
131. Lee BD, Nakai S, Hosomi M (2002) Application of Fenton oxidation to remediate polycyclic aromatic hydrocarbons-contaminated soil. Journal of Chemical Engineering of Japan 35: 582-586.
132. Martens DA, Frankenberger JrWT (1995) Enhanced degradation of polycyclic aromatic hydrocarbons in soil treated with an advanced oxidation process - Fenton's Reagent. Journal of soil contamination 4: 175-190.
133. Watts RJ, Stanton PC, Howsawkeng J, Teel AL (2002) Mineralization of a sorbed polycyclic aromatic hydrocarbon in two soils using catalyzed hydrogen peroxide. Water Res. 36: 4283-4292.
134. Nam K, Rodriguez W, Kukor JJ (2001) Enhanced degradation of polycyclic aromatic hydrocarbons by biodegradation combined with a modified Fenton reaction. Chemosphere 45: 11-20.
135. Lee BD, Hosomi M, Murakami A (1998) Fenton oxidation with ethanol to degrade anthracene into biodegradable 9,10-anthraquinon: A pretreatment method for anthracene-contaminated soil. Water Science and Technology 38: 91-97.
136. Lee BD, Hosomi M (2001) A hybrid Fenton oxidation-microbial treatment for soil highly contaminated with benz[a]anthracene. Chemosphere 43: 1127-1132.
137. Meyer S, Steinhart H (2001) Fate of PAHs and hetero-PAHs during biodegradation in a model soil/compost-system: Formation of extractable metabolites. Water Air and Soil Pollution 132: 215-231.
138. Wischmann H, Steinhart H (1997) The formation of PAH oxidation products in soils and soil/compost mixtures. Chemosphere 35: 1681-1698.

56


















