
An Electrochemical Immunosensor Based on Chemical Assembly of Vertically Aligned Carbon Nanotubes on...
8
An Electrochemical Immunosensor Based on Chemical Assembly of Vertically Aligned Carbon Nanotubes on Carbon Substrates for Direct Detection of the Pesticide Endosulfan in Environmental Water Guozhen Liu,* ,† Shuo Wang, ‡ Jingquan Liu, § and Dandan Song † † Key Laboratory of Pesticide and Chemical Biology of Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, P. R. China ‡ Tianjin Key Laboratory of Food Nutrition and Safety, Faculty of Food Engineering and Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, P. R. China § Laboratory of Fiber Materials and Modern Textile, School of Chemical and Environmental Engineering, Qingdao University, Qingdao 266071, Shandong, P. R. China ABSTRACT: A glassy carbon substrate was covalently modified with a mixed layer of 4-aminophenyl and phenyl via in situ electrografting of their aryldiazonium salts in acidic solutions. Single-walled carbon nanotubes (SWNTs) were covalently and vertically anchored on the electrode surface via the formation of amide bonds from the reaction between the amines located on the modified substrate and the carboxylic groups at the ends of the nanotubes. Ferrocenedimethylamine (FDMA) was subsequently attached to the ends of SWNTs through amide bonding followed by the attachment of an epitope, i.e., endosulfan hapten to which an antibody would bind. Association or dissociation of the antibody with the sensing interface causes a modulation of the ferrocene electrochemistry. Antibody-complexed electrodes were exposed to samples containing spiked endosulfan (unbound target analyte) in environment water and interrogated using the square wave voltammetry (SWV) technique. The modified sensing surfaces were characterized by atomic force microscopy, XPS, and electrochemistry. The fabricated electrochemical immunosensor can be successfully used for the detection of endosulfan over the range of 0.01-20 ppb by a displacement assay. The lowest detection limit of this immunosensor is 0.01 ppb endosulfan in 50 mM phosphate buffer at pH 7.0. E ndosulfan is a broad-spectrum pesticide widely used in agriculture to control insects and mites. 1-3 It is also used extensively in public health applications in developing countries. 4 Development of an immunobiosensor represents a challenging target, as Chinese Water Quality Guidelines set levels for endosulfan in drinking water of less than 0.01 ppb. Several studies have shown that endosulfan may exist in field water samples somewhat longer than in pure water by binding to sediments and soil particles. For assessment of the possible chronic health and environmental effects of long-term exposure to pesticides, extended monitoring of ground, surface, and drinking water, as well as analytical techniques with sufficiently low levels of detection, are essential. Endosulfan is generally analyzed by instrumental methods, such as gas chromatography with electron capture detection, 5 gas chromatography/mass spectrometry, 6 or high-performance liquid chromatography. 7 Each of these methods needs extraction, cleanup, and concentration of the sample. This is labor-intensive, time- consuming, and expensive, making it the rate-limiting step in environmental studies. Therefore, there is a need for a rapid, simple, and cost-effective method of analysis for endosulfan. Immunochemical techniques such as the immunoassay have lately gained a position as alternative and/or complementary methods for the analysis of agrochemicals because of their simplicity, cost-effectiveness, and high sample throughput. 8 Moreover, immunoassays are field-portable and can be performed on a simple and inexpensive instrument. Since Langone and Van Vunakis pioneered the work for the detection of cyclodienes by a radioimmunoassay, 9 several immunoassays capable of detecting endosulfan have been described in the literature. 10-13 However, those methods were of insufficient sensitivity for the analysis of water samples and can give false results due to the coexistence of the nontoxic metabolite, endosulfan diol. 14 Electrochemical immunoassays, in particular, are attractive for the detection of endosulfan because of the high specificity resulting from the introduction of the endosulfan monoclonal antibody to the sensing interface. Montoya and co-workers have developed an enzyme-linked immunosorbent assay based on monoclonal antibodies for the detection of organochlorine pesticides. 15 The authors claim that endosulfan can be determined in a competitive assay from 2 to 50 nM with a detection limit of 1 nM (2.46 ppb). The Received: October 18, 2011 Accepted: March 26, 2012 Published: March 26, 2012 Article pubs.acs.org/ac © 2012 American Chemical Society 3921 dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921-3928
Transcript of An Electrochemical Immunosensor Based on Chemical Assembly of Vertically Aligned Carbon Nanotubes on...

An Electrochemical Immunosensor Based on Chemical Assembly ofVertically Aligned Carbon Nanotubes on Carbon Substrates forDirect Detection of the Pesticide Endosulfan in Environmental WaterGuozhen Liu,*,† Shuo Wang,‡ Jingquan Liu,§ and Dandan Song†
†Key Laboratory of Pesticide and Chemical Biology of Ministry of Education, College of Chemistry, Central China NormalUniversity, Wuhan 430079, P. R. China‡Tianjin Key Laboratory of Food Nutrition and Safety, Faculty of Food Engineering and Biotechnology, Tianjin University of Scienceand Technology, Tianjin 300457, P. R. China§Laboratory of Fiber Materials and Modern Textile, School of Chemical and Environmental Engineering, Qingdao University,Qingdao 266071, Shandong, P. R. China
ABSTRACT: A glassy carbon substrate was covalentlymodified with a mixed layer of 4-aminophenyl and phenyl viain situ electrografting of their aryldiazonium salts in acidicsolutions. Single-walled carbon nanotubes (SWNTs) werecovalently and vertically anchored on the electrode surface viathe formation of amide bonds from the reaction between theamines located on the modified substrate and the carboxylicgroups at the ends of the nanotubes. Ferrocenedimethylamine(FDMA) was subsequently attached to the ends of SWNTsthrough amide bonding followed by the attachment of an epitope, i.e., endosulfan hapten to which an antibody would bind.Association or dissociation of the antibody with the sensing interface causes a modulation of the ferrocene electrochemistry.Antibody-complexed electrodes were exposed to samples containing spiked endosulfan (unbound target analyte) in environmentwater and interrogated using the square wave voltammetry (SWV) technique. The modified sensing surfaces were characterizedby atomic force microscopy, XPS, and electrochemistry. The fabricated electrochemical immunosensor can be successfully usedfor the detection of endosulfan over the range of 0.01−20 ppb by a displacement assay. The lowest detection limit of thisimmunosensor is 0.01 ppb endosulfan in 50 mM phosphate buffer at pH 7.0.
Endosulfan is a broad-spectrum pesticide widely used inagriculture to control insects and mites.1−3 It is also used
extensively in public health applications in developingcountries.4 Development of an immunobiosensor represents achallenging target, as Chinese Water Quality Guidelines setlevels for endosulfan in drinking water of less than 0.01 ppb.Several studies have shown that endosulfan may exist in fieldwater samples somewhat longer than in pure water by bindingto sediments and soil particles. For assessment of the possiblechronic health and environmental effects of long-term exposureto pesticides, extended monitoring of ground, surface, anddrinking water, as well as analytical techniques with sufficientlylow levels of detection, are essential. Endosulfan is generallyanalyzed by instrumental methods, such as gas chromatographywith electron capture detection,5 gas chromatography/massspectrometry,6 or high-performance liquid chromatography.7
Each of these methods needs extraction, cleanup, andconcentration of the sample. This is labor-intensive, time-consuming, and expensive, making it the rate-limiting step inenvironmental studies. Therefore, there is a need for a rapid,simple, and cost-effective method of analysis for endosulfan.Immunochemical techniques such as the immunoassay have
lately gained a position as alternative and/or complementarymethods for the analysis of agrochemicals because of their
simplicity, cost-effectiveness, and high sample throughput.8
Moreover, immunoassays are field-portable and can beperformed on a simple and inexpensive instrument. SinceLangone and Van Vunakis pioneered the work for the detectionof cyclodienes by a radioimmunoassay,9 several immunoassayscapable of detecting endosulfan have been described in theliterature.10−13 However, those methods were of insufficientsensitivity for the analysis of water samples and can give falseresults due to the coexistence of the nontoxic metabolite,endosulfan diol.14 Electrochemical immunoassays, in particular,are attractive for the detection of endosulfan because of thehigh specificity resulting from the introduction of theendosulfan monoclonal antibody to the sensing interface.Montoya and co-workers have developed an enzyme-linkedimmunosorbent assay based on monoclonal antibodies for thedetection of organochlorine pesticides.15 The authors claim thatendosulfan can be determined in a competitive assay from 2 to50 nM with a detection limit of 1 nM (2.46 ppb). The
Received: October 18, 2011Accepted: March 26, 2012Published: March 26, 2012
Article
pubs.acs.org/ac
© 2012 American Chemical Society 3921 dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−3928

sensitivity has been greatly improved, but it still cannot meetthe requirement for monitoring endosulfan in water samples.Previously, a label-free immunosensor using oligo-
(phenylethynylene) molecular wires (MWs) for electrontransfer and poly(ethylene glycol) (PEG) for resistingunspecific protein adsorption was developed for the detectionof small molecule free biotin by a displacement assay.16 In thisdesign, MWs serve dual purposes in being rigid, therebyallowing access to the biotin by the binding antibodies withouthindrance from the surface, and being an efficient conduit forelectron transfer,17 which is necessary as the ferrocene islocated approximately 20 Å above the electrode surface. Theseattractive advantages of MWs, in that they are rigid and givewell-defined molecular architectures and efficient electrontransfer, were also observed by other researchers.18−21 TheMWs, however, suffer the disadvantages of being eitherunstable in air, difficult to synthesize in large quantities, orboth. Thus, it is crucial to find an alternative to these molecularwires, especially for the application of immunosensors for realanalytes. Vertical alignment of single-walled carbon nanotubes(SWNTs) on surfaces provides new materials with interestingproperties for future applications in electronic, optoelectronic,and sensing devices.22−24 Robust forests of SWNTs have beensuccessfully achieved via chemical assembly on carbon surfacesthat were first modified with amine-terminated aryldiazoniumsalt layers.25 Highly ordered covalent anchoring of carbonnanotubes through carbon−carbon bonds to electrode surfacesmodified with aryldiazonium salts has also been reported.26
Both structures show excellent stability over a wide potentialrange and are resistant to degradation from sonication in acids,bases, and organic solvents. An immunosensor based onSWNT-modified glassy carbon (GC) electrodes has beendeveloped for the detection of antibiotin IgG.27 It isdemonstrated that SWNTs are a good alternative to MWs.First, carbon nanotubes are small (as small as 1 nm indiameter), rigid, and simple to produce in large quantities.Their small size and conductivity means that they can beregarded as the smallest possible electrodes, with diameters ofless than 1 nm.28 Second, SWNTs are known to have a numberof carboxylic acid groups at each end of the tubes, which areimportant for further fabrication, after being cut in a 3:1 v/vmixture of concentrated H2SO4/HNO3.
29 Third, the assemblyof SWNTs increases the electronic communication between theelectrode and the environmental solutions.26 However, to thebest of our knowledge, no report of using SWNTs as the MWon a sensing interface for the detection of small molecules by adisplacement assay has been published to date.In this work, we aim to develop a label-free immunosensor
based on an SWNT−aryldiazonium salt-modified sensinginterface for the detection of endosulfan (Scheme 1). In thisdesign, four key components are introduced to the sensinginterface: (a) mixed layers of aryldiazonium salts, (b) SWNTs,(c) PEG molecules, and (d) endosulfan monoclonal antibodies.Due to the aryldiazonium salt chemistry, the SWNT assembliesare expected to have stability advantages over those based onSAMs of alkanethiols on gold and those where the anchoringlayer is simply chemisorbed to the surface.30 In addition, themixed layers of aryldiazonium salts aid the assembly of SWNTson the surface.26 Binding of the antibody to the surface-boundepitope immerses the redox probe ferrocene in a proteinmedium. A consequence of this change in the environment isthe attenuation of electron transfer to the ferrocene due to theinaccessibility of a counterion. The PEG antifouling layer
ensured that the antibody only interacted with the interfacewhen a specific biorecognition event occurred. Furthermore,the rigidity of SWNTs was essential to allow access of theantibody to the surface epitope without hindrance from thesurface.
■ EXPERIMENTAL SECTIONMaterials and Reagents. Sodium nitrite, potassium
ferricyanide, hydrochloric acid, 4-phenylenediamine, aniline,N,N′-dicyclohexylcarbodiimide (DCC), horseradish peroxidase(HRP), keyhole limpet hemocyanin (KLH), Freund’s adju-vants, bovine serum albumin (fraction V, BSA), and myoglobinwere purchased from Sigma-Aldrich. Endosulfan and otherendosulfan derivatives were purchased from Fluka. HybridomaFusion and Cloning Supplement (HFCS) was from RocheApplied Science (Indianapolis, IN). Peroxidase-labeled rabbitantimouse immunoglobulins and goat antimouse immunoglo-bulins were obtained from eBioscience (San Diego, CA).Culture media (high-glucose Dulbecco’s modified Eagle’smedium with Glutamax I and sodium pyruvate), fetal calfserum (Myoclone Super Plus), and supplements were fromGibco (HaoranBio, Shanghai). Culture plastic ware was fromBibby Sterilin Ltd. (Stone, UK). Flat-bottom polystyrene
Scheme 1. SWNT-Modified Sensing Interface for theDetection of Endosulfana
aThe structures for one isomer of endosulfan and its hapten areshown.
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283922

ELISA high binding plates were from Costar (Cambridge, MA).SWNTs prepared by the HiPco process were purchased fromCarbon Nanotechnologies Incorporated. Cut SWNTs wereprepared as reported previously.31 The aryldiazonium cationsfor 2-(2-(2-(4-aminophenoxy)ethoxy)ethoxy)ethanol (PEG)was custom synthesized by following the modified proceduresof Bahr et al.17,32 Ferrocenedimethylamine (FDMA) wassynthesized by the literature method.33 The synthesis ofendosulfan haptens was accomplished by following theprocedures of Lee et al.10 N-Hydroxysuccinimide activatedesters of haptens were conjugated to HRP and KLH using themethod described earlier.34 All other reagents were used asreceived. Aqueous solutions were prepared using Mill-Q water(>18 MΩ cm). Phosphate-buffered saline (PBS) solutions were0.15 M NaCl and 0.1 M phosphate buffer, pH 7.3. Phosphatebuffer solution for electrochemistry was prepared using 0.1 Mbuffer with added 0.05 M KCl (pH 7.0).Apparatus. All electrochemical experiments were con-
ducted using a GaossUnion EC510 potentiostat (GaossUnion,China). GC electrodes were 3 mm disks embedded in epoxyresin (GaossUnion). All experiments utilized a Pt secondaryelectrode and a Ag/AgCl (3.0 M NaCl) reference electrode. Allvoltammetric measurements were obtained with a scan rate of100 mV s−1. X-ray photoelectron spectra (XPS) were collectedfrom GC plates on a VG multilab 2000 spectrometer with amonochromated Al Kα source (1486.6 eV), hemisphericalanalyzer, and multichannel detector. The spectra werecalibrated on the C1s peak (285.0 eV). Spectra were analyzedusing XPSPEAK41 software. The quoted percentage coveragefor the different elements and subgroups were estimated fromthe corresponding fitted areas and associated sensitivity factors.Atomic force microscopy (AFM) images were taken on GCplates using a Digital Instruments Dimension 3100 scanningprobe microscope. All images were acquired in tapping modeusing commercial Si cantilevers/tips (Olympus) at theirfundamental resonance frequencies, which typically variedbetween 275 and 320 kHz.Preparation of Endosulfan Monoclonal Antibodies
IgG. BALB/c female mice were used for the production ofendosulfan monoclonal antibodies according to the methoddescribed by Manclus et al.15 Antibodies were raised byintraperitoneal injections of endosulfan hapten conjugated toKLH. The immunogens (100 μg of conjugate) were diluted inPBS and emulsified in Freund’s complete (first immunization)or incomplete adjuvant (subsequent immunizations). Afterthree initial fortnightly intervals, booster injections were givenmonthly. Blood was collected from mouse tail one week aftereach monthly booster injection, and the antiserum was testedfor antihapten antibody titer by indirect ELISA and for analyterecognition properties by competitive indirect ELISA. After 4weeks from the last injection in adjuvant, mice selected to bespleen donors for hybridoma production received a final solubleintraperitoneal injection of 100 μg of conjugate in PBS fourdays prior to cell fusion. HB-10744 SH-3 marine myeloma cells(ATCC, Xiangf bio, Shanghai) were cultured in high-glucoseculture media supplemented with 2 mM L-glutamine, 1 mMnonessential amino acids, 25 μg mL−1 gentamicin, and 15%fetal bovine serum. Cell fusion procedures were carried out asdescribed by Nowinski et al.35 Ten days after cell fusion, culturesupernatants were screened for the presence of antibodies thatrecognized the analyte. Selected hybridomas were cloned bylimiting dilution using HT (hypoxanthine−thymidine) mediumsupplemented with 2% HFCS (v/v). Stable antibody-producing
clones were expanded and cryopreserved in liquid nitrogen.The achieved antibodies were classified as antiendosulfan IgGwhich were purified on a small scale directly from latestationary phase culture supernatants by saline precipitationwith saturated ammonium sulfate followed by affinitychromatography.36
Preparation of the Immunosensor Interface forDetection of Endosulfan. Prior to modification, GCelectrodes were polished successively with 1.0, 0.3, and 0.05μm alumina slurries (alumina: Buehler, Lake Bluff, IL) onmicrocloth pads (Buehler). The electrodes were thoroughlyrinsed and sonicated in Milli-Q water for 1 min betweenpolishing steps. Before modification, the electrodes were driedwith a stream of argon. The derivatization of the clean GCelectrode with mixed layers of 4-aminophenyl and phenyl wasconducted with in situ-generated aryldiazonium cations thatinvolved the electrochemical reduction of the correspondinganilines in acidic media to achieve 4-aminophenyl/phenyl-modified surfaces (GC-Ph-NH2).
37,38 Specifically, 0.5 mM p-phenylenediamine and 0.5 mM aniline were solubilized in 0.5M aqueous HCl, and 1 mM sodium nitrite was added togenerate the aryldiazonium salts in the electrochemical cell (insitu). The mixture was degassed and left to react for about 10min at 0 °C. The electrochemical reductive modification of theGC surface with an in situ-generated mixture of 4-amino-phenyl- and phenyldiazonium salts was carried out by applyinga potential to the electrode between 1.0 V and −1.0 V for twocycles at a scan rate of 100 mV s−1. After surface derivatization,the electrodes were rinsed with copious amounts of Milli-Qwater and dried under a stream of argon prior to the next step.Modification of SWNTs with carboxylic acid-terminated groupswas carried out by immersing GC-Ph-NH2-modified GCelectrodes in the ethanol solution of cut SWNTs (0.2 mgmL−1) in the presence of 0.5 mg mL−1 DCC. For resisting thenonspecific protein adsorption, the SWNT-modified GCsurfaces (GC-Ph-NH2/SWNT) were modified with PEGmolecules through aryldiazonium treatment of SWNTsreported by Tour et al.32 For coupling FDMA to the openends of the SWNTs assembled on GC substrates, SWNT- andPEG-modified GC surfaces (GC-Ph-NH2/SWNT/PEG) wereincubated in an absolute ethanol solution containing 40 mMDCC and 5 mM FDMA for 6 h at room temperature to achievethe surface of GC-Ph-NH2/SWNT/PEG/FDMA. Endosulfanhapten was coupled to the ferrocene by an amide couplingreaction with 1 mg mL−1 hapten in 0.1 M PBS for 2 h at 4 °C.The endosulfan hapten-modified electrodes were rinsed withcopious amounts of water and PBS before immersion into aPBS solution of endosulfan monoclonal IgG for 30 min at 4 °C.The electrodes were transferred to phosphate buffer solutionfor measurement using cyclic voltammetry (CV) and squarewave voltammetry (SWV).
■ RESULTS AND DISCUSSIONCharacterization of the SWNT-Modified GC Surfaces
by AFM. SWNTs were assembled on the mixed layers of 4-aminophenyl/phenyl-modified GC surface by reacting thesubstrate with SWNTs for 24 h in the ethanol solution of cutSWNTs (0.2 mg mL−1) in the presence of 0.5 mg mL−1 DCC.Bond formation involving the multiple carboxylic acid groups atthe end of each SWNT is expected to result in verticalalignment of SWNTs, as demonstrated in studies using otheramine-functionalized substrates.29,39,40 The so-fabricated surfa-ces are referred to as GC-Ph-NH2/SWNTs and were imaged by
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283923
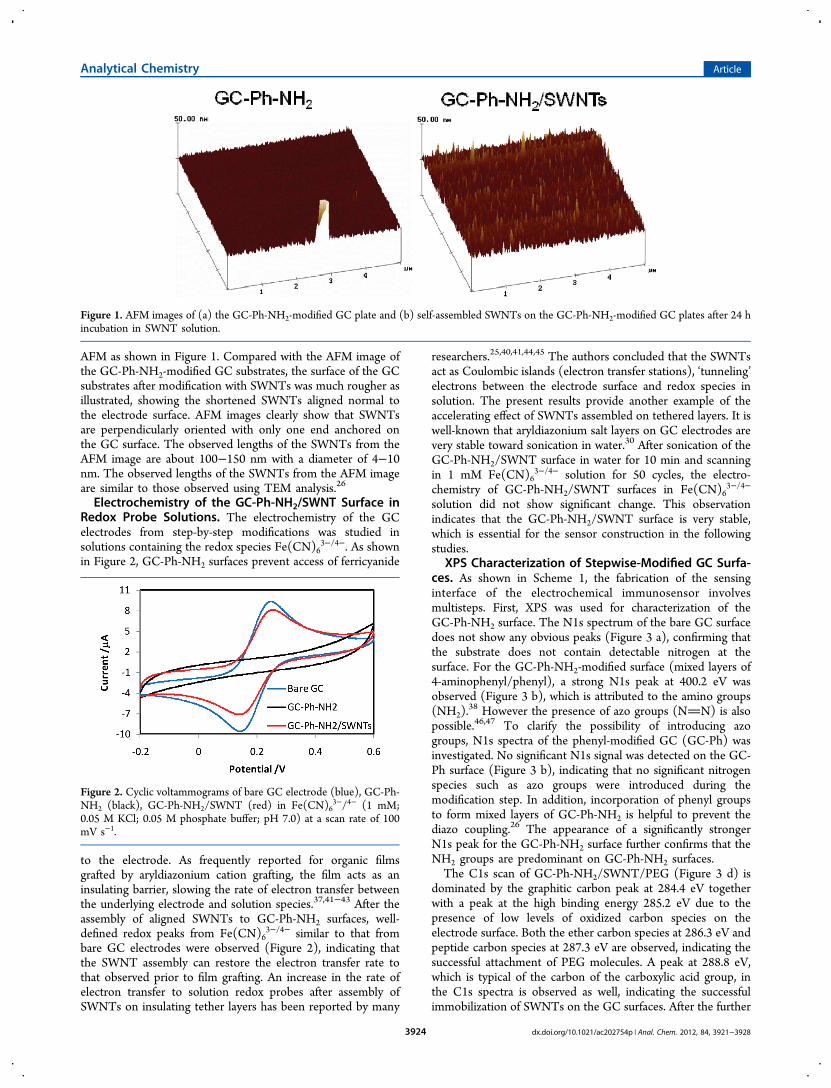
AFM as shown in Figure 1. Compared with the AFM image ofthe GC-Ph-NH2-modified GC substrates, the surface of the GCsubstrates after modification with SWNTs was much rougher asillustrated, showing the shortened SWNTs aligned normal tothe electrode surface. AFM images clearly show that SWNTsare perpendicularly oriented with only one end anchored onthe GC surface. The observed lengths of the SWNTs from theAFM image are about 100−150 nm with a diameter of 4−10nm. The observed lengths of the SWNTs from the AFM imageare similar to those observed using TEM analysis.26
Electrochemistry of the GC-Ph-NH2/SWNT Surface inRedox Probe Solutions. The electrochemistry of the GCelectrodes from step-by-step modifications was studied insolutions containing the redox species Fe(CN)6
3−/4−. As shownin Figure 2, GC-Ph-NH2 surfaces prevent access of ferricyanide
to the electrode. As frequently reported for organic filmsgrafted by aryldiazonium cation grafting, the film acts as aninsulating barrier, slowing the rate of electron transfer betweenthe underlying electrode and solution species.37,41−43 After theassembly of aligned SWNTs to GC-Ph-NH2 surfaces, well-defined redox peaks from Fe(CN)6
3−/4− similar to that frombare GC electrodes were observed (Figure 2), indicating thatthe SWNT assembly can restore the electron transfer rate tothat observed prior to film grafting. An increase in the rate ofelectron transfer to solution redox probes after assembly ofSWNTs on insulating tether layers has been reported by many
researchers.25,40,41,44,45 The authors concluded that the SWNTsact as Coulombic islands (electron transfer stations), ‘tunneling’electrons between the electrode surface and redox species insolution. The present results provide another example of theaccelerating effect of SWNTs assembled on tethered layers. It iswell-known that aryldiazonium salt layers on GC electrodes arevery stable toward sonication in water.30 After sonication of theGC-Ph-NH2/SWNT surface in water for 10 min and scanningin 1 mM Fe(CN)6
3−/4− solution for 50 cycles, the electro-chemistry of GC-Ph-NH2/SWNT surfaces in Fe(CN)6
3−/4−
solution did not show significant change. This observationindicates that the GC-Ph-NH2/SWNT surface is very stable,which is essential for the sensor construction in the followingstudies.
XPS Characterization of Stepwise-Modified GC Surfa-ces. As shown in Scheme 1, the fabrication of the sensinginterface of the electrochemical immunosensor involvesmultisteps. First, XPS was used for characterization of theGC-Ph-NH2 surface. The N1s spectrum of the bare GC surfacedoes not show any obvious peaks (Figure 3 a), confirming thatthe substrate does not contain detectable nitrogen at thesurface. For the GC-Ph-NH2-modified surface (mixed layers of4-aminophenyl/phenyl), a strong N1s peak at 400.2 eV wasobserved (Figure 3 b), which is attributed to the amino groups(NH2).
38 However the presence of azo groups (NN) is alsopossible.46,47 To clarify the possibility of introducing azogroups, N1s spectra of the phenyl-modified GC (GC-Ph) wasinvestigated. No significant N1s signal was detected on the GC-Ph surface (Figure 3 b), indicating that no significant nitrogenspecies such as azo groups were introduced during themodification step. In addition, incorporation of phenyl groupsto form mixed layers of GC-Ph-NH2 is helpful to prevent thediazo coupling.26 The appearance of a significantly strongerN1s peak for the GC-Ph-NH2 surface further confirms that theNH2 groups are predominant on GC-Ph-NH2 surfaces.The C1s scan of GC-Ph-NH2/SWNT/PEG (Figure 3 d) is
dominated by the graphitic carbon peak at 284.4 eV togetherwith a peak at the high binding energy 285.2 eV due to thepresence of low levels of oxidized carbon species on theelectrode surface. Both the ether carbon species at 286.3 eV andpeptide carbon species at 287.3 eV are observed, indicating thesuccessful attachment of PEG molecules. A peak at 288.8 eV,which is typical of the carbon of the carboxylic acid group, inthe C1s spectra is observed as well, indicating the successfulimmobilization of SWNTs on the GC surfaces. After the further
Figure 1. AFM images of (a) the GC-Ph-NH2-modified GC plate and (b) self-assembled SWNTs on the GC-Ph-NH2-modified GC plates after 24 hincubation in SWNT solution.
Figure 2. Cyclic voltammograms of bare GC electrode (blue), GC-Ph-NH2 (black), GC-Ph-NH2/SWNT (red) in Fe(CN)6
3−/4− (1 mM;0.05 M KCl; 0.05 M phosphate buffer; pH 7.0) at a scan rate of 100mV s−1.
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283924
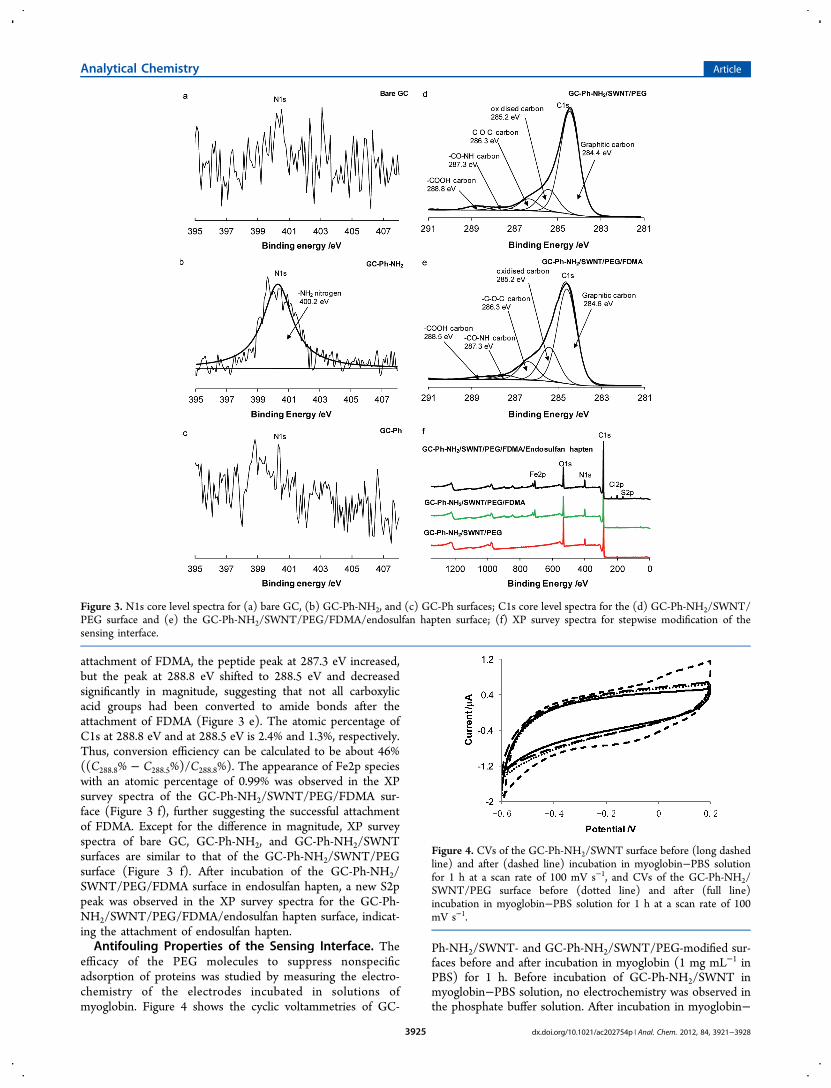
attachment of FDMA, the peptide peak at 287.3 eV increased,but the peak at 288.8 eV shifted to 288.5 eV and decreasedsignificantly in magnitude, suggesting that not all carboxylicacid groups had been converted to amide bonds after theattachment of FDMA (Figure 3 e). The atomic percentage ofC1s at 288.8 eV and at 288.5 eV is 2.4% and 1.3%, respectively.Thus, conversion efficiency can be calculated to be about 46%((C288.8% − C288.5%)/C288.8%). The appearance of Fe2p specieswith an atomic percentage of 0.99% was observed in the XPsurvey spectra of the GC-Ph-NH2/SWNT/PEG/FDMA sur-face (Figure 3 f), further suggesting the successful attachmentof FDMA. Except for the difference in magnitude, XP surveyspectra of bare GC, GC-Ph-NH2, and GC-Ph-NH2/SWNTsurfaces are similar to that of the GC-Ph-NH2/SWNT/PEGsurface (Figure 3 f). After incubation of the GC-Ph-NH2/SWNT/PEG/FDMA surface in endosulfan hapten, a new S2ppeak was observed in the XP survey spectra for the GC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten surface, indicat-ing the attachment of endosulfan hapten.Antifouling Properties of the Sensing Interface. The
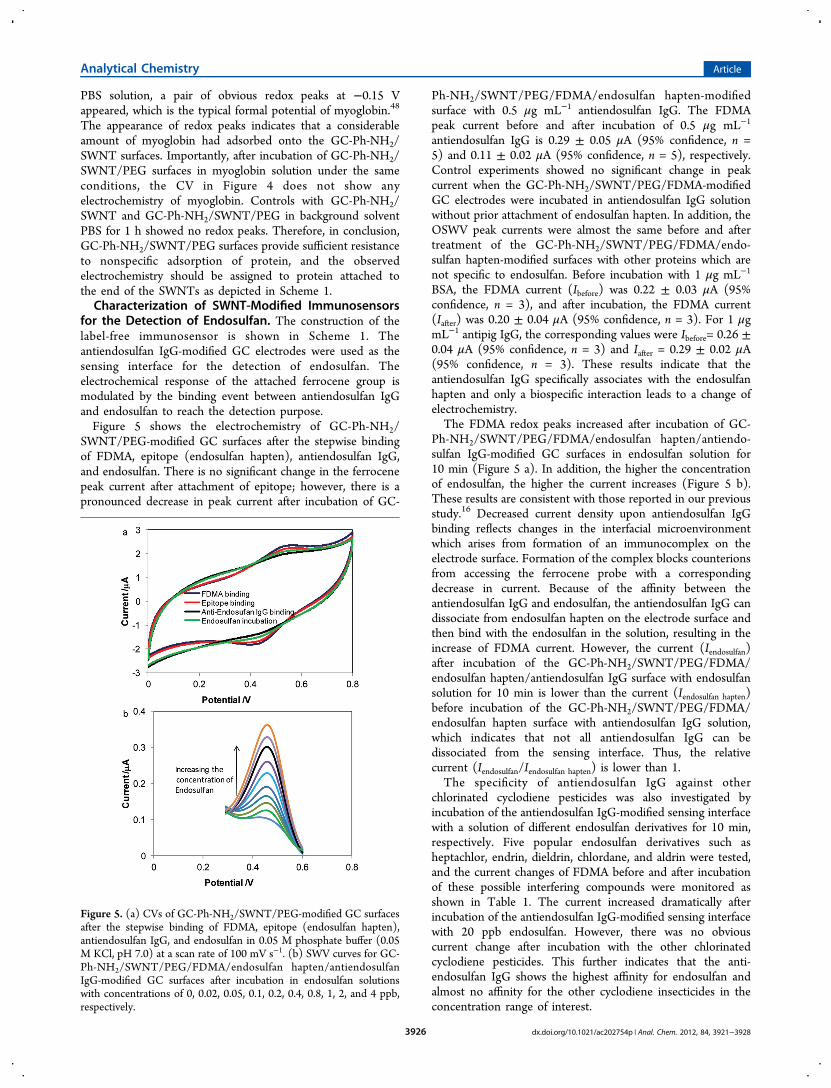
efficacy of the PEG molecules to suppress nonspecificadsorption of proteins was studied by measuring the electro-chemistry of the electrodes incubated in solutions ofmyoglobin. Figure 4 shows the cyclic voltammetries of GC-
Ph-NH2/SWNT- and GC-Ph-NH2/SWNT/PEG-modified sur-faces before and after incubation in myoglobin (1 mg mL−1 inPBS) for 1 h. Before incubation of GC-Ph-NH2/SWNT inmyoglobin−PBS solution, no electrochemistry was observed inthe phosphate buffer solution. After incubation in myoglobin−
Figure 3. N1s core level spectra for (a) bare GC, (b) GC-Ph-NH2, and (c) GC-Ph surfaces; C1s core level spectra for the (d) GC-Ph-NH2/SWNT/PEG surface and (e) the GC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten surface; (f) XP survey spectra for stepwise modification of thesensing interface.
Figure 4. CVs of the GC-Ph-NH2/SWNT surface before (long dashedline) and after (dashed line) incubation in myoglobin−PBS solutionfor 1 h at a scan rate of 100 mV s−1, and CVs of the GC-Ph-NH2/SWNT/PEG surface before (dotted line) and after (full line)incubation in myoglobin−PBS solution for 1 h at a scan rate of 100mV s−1.
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283925
PBS solution, a pair of obvious redox peaks at −0.15 Vappeared, which is the typical formal potential of myoglobin.48
The appearance of redox peaks indicates that a considerableamount of myoglobin had adsorbed onto the GC-Ph-NH2/SWNT surfaces. Importantly, after incubation of GC-Ph-NH2/SWNT/PEG surfaces in myoglobin solution under the sameconditions, the CV in Figure 4 does not show anyelectrochemistry of myoglobin. Controls with GC-Ph-NH2/SWNT and GC-Ph-NH2/SWNT/PEG in background solventPBS for 1 h showed no redox peaks. Therefore, in conclusion,GC-Ph-NH2/SWNT/PEG surfaces provide sufficient resistanceto nonspecific adsorption of protein, and the observedelectrochemistry should be assigned to protein attached tothe end of the SWNTs as depicted in Scheme 1.Characterization of SWNT-Modified Immunosensors
for the Detection of Endosulfan. The construction of thelabel-free immunosensor is shown in Scheme 1. Theantiendosulfan IgG-modified GC electrodes were used as thesensing interface for the detection of endosulfan. Theelectrochemical response of the attached ferrocene group ismodulated by the binding event between antiendosulfan IgGand endosulfan to reach the detection purpose.Figure 5 shows the electrochemistry of GC-Ph-NH2/
SWNT/PEG-modified GC surfaces after the stepwise bindingof FDMA, epitope (endosulfan hapten), antiendosulfan IgG,and endosulfan. There is no significant change in the ferrocenepeak current after attachment of epitope; however, there is apronounced decrease in peak current after incubation of GC-
Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten-modifiedsurface with 0.5 μg mL−1 antiendosulfan IgG. The FDMApeak current before and after incubation of 0.5 μg mL−1
antiendosulfan IgG is 0.29 ± 0.05 μA (95% confidence, n =5) and 0.11 ± 0.02 μA (95% confidence, n = 5), respectively.Control experiments showed no significant change in peakcurrent when the GC-Ph-NH2/SWNT/PEG/FDMA-modifiedGC electrodes were incubated in antiendosulfan IgG solutionwithout prior attachment of endosulfan hapten. In addition, theOSWV peak currents were almost the same before and aftertreatment of the GC-Ph-NH2/SWNT/PEG/FDMA/endo-sulfan hapten-modified surfaces with other proteins which arenot specific to endosulfan. Before incubation with 1 μg mL−1
BSA, the FDMA current (Ibefore) was 0.22 ± 0.03 μA (95%confidence, n = 3), and after incubation, the FDMA current(Iafter) was 0.20 ± 0.04 μA (95% confidence, n = 3). For 1 μgmL−1 antipig IgG, the corresponding values were Ibefore= 0.26 ±0.04 μA (95% confidence, n = 3) and Iafter = 0.29 ± 0.02 μA(95% confidence, n = 3). These results indicate that theantiendosulfan IgG specifically associates with the endosulfanhapten and only a biospecific interaction leads to a change ofelectrochemistry.The FDMA redox peaks increased after incubation of GC-
Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten/antiendo-sulfan IgG-modified GC surfaces in endosulfan solution for10 min (Figure 5 a). In addition, the higher the concentrationof endosulfan, the higher the current increases (Figure 5 b).These results are consistent with those reported in our previousstudy.16 Decreased current density upon antiendosulfan IgGbinding reflects changes in the interfacial microenvironmentwhich arises from formation of an immunocomplex on theelectrode surface. Formation of the complex blocks counterionsfrom accessing the ferrocene probe with a correspondingdecrease in current. Because of the affinity between theantiendosulfan IgG and endosulfan, the antiendosulfan IgG candissociate from endosulfan hapten on the electrode surface andthen bind with the endosulfan in the solution, resulting in theincrease of FDMA current. However, the current (Iendosulfan)after incubation of the GC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten/antiendosulfan IgG surface with endosulfansolution for 10 min is lower than the current (Iendosulfan hapten)before incubation of the GC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten surface with antiendosulfan IgG solution,which indicates that not all antiendosulfan IgG can bedissociated from the sensing interface. Thus, the relativecurrent (Iendosulfan/Iendosulfan hapten) is lower than 1.The specificity of antiendosulfan IgG against other
chlorinated cyclodiene pesticides was also investigated byincubation of the antiendosulfan IgG-modified sensing interfacewith a solution of different endosulfan derivatives for 10 min,respectively. Five popular endosulfan derivatives such asheptachlor, endrin, dieldrin, chlordane, and aldrin were tested,and the current changes of FDMA before and after incubationof these possible interfering compounds were monitored asshown in Table 1. The current increased dramatically afterincubation of the antiendosulfan IgG-modified sensing interfacewith 20 ppb endosulfan. However, there was no obviouscurrent change after incubation with the other chlorinatedcyclodiene pesticides. This further indicates that the anti-endosulfan IgG shows the highest affinity for endosulfan andalmost no affinity for the other cyclodiene insecticides in theconcentration range of interest.
Figure 5. (a) CVs of GC-Ph-NH2/SWNT/PEG-modified GC surfacesafter the stepwise binding of FDMA, epitope (endosulfan hapten),antiendosulfan IgG, and endosulfan in 0.05 M phosphate buffer (0.05M KCl, pH 7.0) at a scan rate of 100 mV s−1. (b) SWV curves for GC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten/antiendosulfanIgG-modified GC surfaces after incubation in endosulfan solutionswith concentrations of 0, 0.02, 0.05, 0.1, 0.2, 0.4, 0.8, 1, 2, and 4 ppb,respectively.
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283926
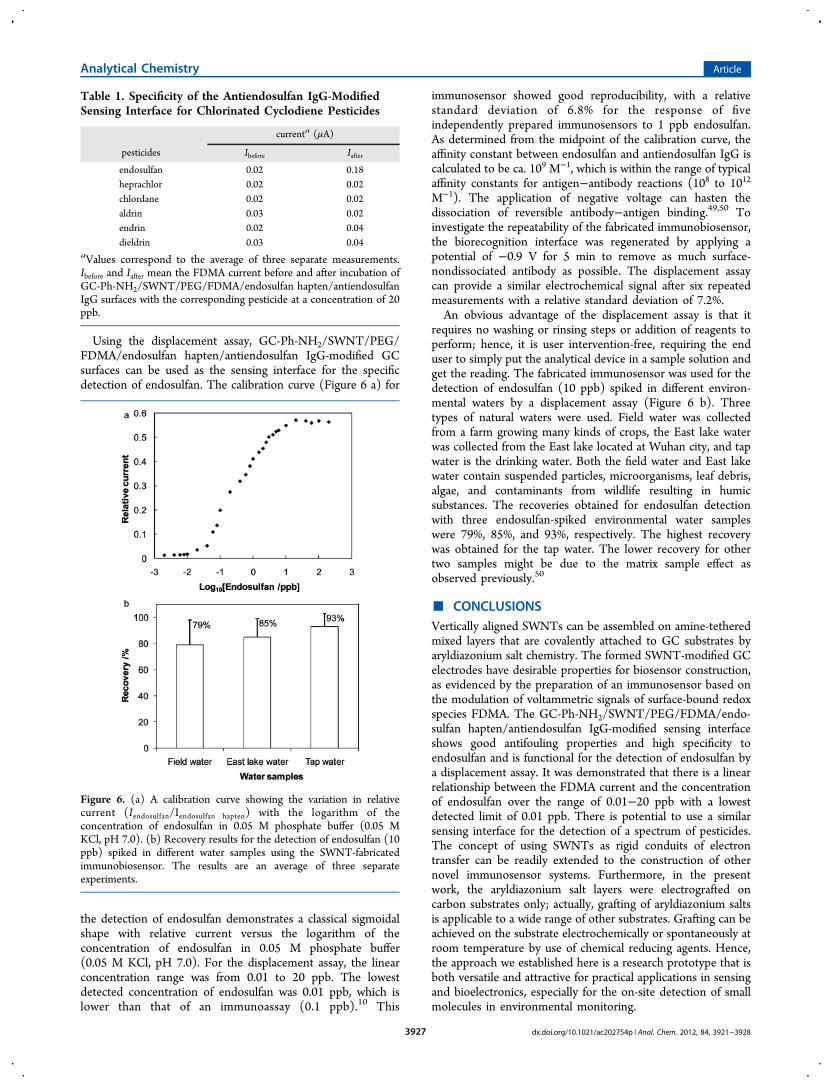
Using the displacement assay, GC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten/antiendosulfan IgG-modified GCsurfaces can be used as the sensing interface for the specificdetection of endosulfan. The calibration curve (Figure 6 a) for
the detection of endosulfan demonstrates a classical sigmoidalshape with relative current versus the logarithm of theconcentration of endosulfan in 0.05 M phosphate buffer(0.05 M KCl, pH 7.0). For the displacement assay, the linearconcentration range was from 0.01 to 20 ppb. The lowestdetected concentration of endosulfan was 0.01 ppb, which islower than that of an immunoassay (0.1 ppb).10 This
immunosensor showed good reproducibility, with a relativestandard deviation of 6.8% for the response of fiveindependently prepared immunosensors to 1 ppb endosulfan.As determined from the midpoint of the calibration curve, theaffinity constant between endosulfan and antiendosulfan IgG iscalculated to be ca. 109 M−1, which is within the range of typicalaffinity constants for antigen−antibody reactions (108 to 1012
M−1). The application of negative voltage can hasten thedissociation of reversible antibody−antigen binding.49,50 Toinvestigate the repeatability of the fabricated immunobiosensor,the biorecognition interface was regenerated by applying apotential of −0.9 V for 5 min to remove as much surface-nondissociated antibody as possible. The displacement assaycan provide a similar electrochemical signal after six repeatedmeasurements with a relative standard deviation of 7.2%.An obvious advantage of the displacement assay is that it
requires no washing or rinsing steps or addition of reagents toperform; hence, it is user intervention-free, requiring the enduser to simply put the analytical device in a sample solution andget the reading. The fabricated immunosensor was used for thedetection of endosulfan (10 ppb) spiked in different environ-mental waters by a displacement assay (Figure 6 b). Threetypes of natural waters were used. Field water was collectedfrom a farm growing many kinds of crops, the East lake waterwas collected from the East lake located at Wuhan city, and tapwater is the drinking water. Both the field water and East lakewater contain suspended particles, microorganisms, leaf debris,algae, and contaminants from wildlife resulting in humicsubstances. The recoveries obtained for endosulfan detectionwith three endosulfan-spiked environmental water sampleswere 79%, 85%, and 93%, respectively. The highest recoverywas obtained for the tap water. The lower recovery for othertwo samples might be due to the matrix sample effect asobserved previously.50
■ CONCLUSIONSVertically aligned SWNTs can be assembled on amine-tetheredmixed layers that are covalently attached to GC substrates byaryldiazonium salt chemistry. The formed SWNT-modified GCelectrodes have desirable properties for biosensor construction,as evidenced by the preparation of an immunosensor based onthe modulation of voltammetric signals of surface-bound redoxspecies FDMA. The GC-Ph-NH2/SWNT/PEG/FDMA/endo-sulfan hapten/antiendosulfan IgG-modified sensing interfaceshows good antifouling properties and high specificity toendosulfan and is functional for the detection of endosulfan bya displacement assay. It was demonstrated that there is a linearrelationship between the FDMA current and the concentrationof endosulfan over the range of 0.01−20 ppb with a lowestdetected limit of 0.01 ppb. There is potential to use a similarsensing interface for the detection of a spectrum of pesticides.The concept of using SWNTs as rigid conduits of electrontransfer can be readily extended to the construction of othernovel immunosensor systems. Furthermore, in the presentwork, the aryldiazonium salt layers were electrografted oncarbon substrates only; actually, grafting of aryldiazonium saltsis applicable to a wide range of other substrates. Grafting can beachieved on the substrate electrochemically or spontaneously atroom temperature by use of chemical reducing agents. Hence,the approach we established here is a research prototype that isboth versatile and attractive for practical applications in sensingand bioelectronics, especially for the on-site detection of smallmolecules in environmental monitoring.
Table 1. Specificity of the Antiendosulfan IgG-ModifiedSensing Interface for Chlorinated Cyclodiene Pesticides
currenta (μA)
pesticides Ibefore Iafter
endosulfan 0.02 0.18heprachlor 0.02 0.02chlordane 0.02 0.02aldrin 0.03 0.02endrin 0.02 0.04dieldrin 0.03 0.04
aValues correspond to the average of three separate measurements.Ibefore and Iafter mean the FDMA current before and after incubation ofGC-Ph-NH2/SWNT/PEG/FDMA/endosulfan hapten/antiendosulfanIgG surfaces with the corresponding pesticide at a concentration of 20ppb.
Figure 6. (a) A calibration curve showing the variation in relativecurrent (Iendosulfan/Iendosulfan hapten) with the logarithm of theconcentration of endosulfan in 0.05 M phosphate buffer (0.05 MKCl, pH 7.0). (b) Recovery results for the detection of endosulfan (10ppb) spiked in different water samples using the SWNT-fabricatedimmunobiosensor. The results are an average of three separateexperiments.
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283927

■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected]. Tel: +86-27-6786 7535.
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work was financially supported by the National NaturalScience Foundation of China (grant 209237057).
■ REFERENCES(1) Lee, N. J.; Beasley, H. L.; Kimber, S. W. L.; Silburn, M.; Woods,N.; Skerritt, J. H.; Kennedy, I. R. J. Agric. Food Chem. 1997, 45, 4147.(2) Goebel, H.; Gorbath, S. G.; Knauf, W.; Rimpau, R. H.;Huttenbach, H. Residue Rev. 1982, 83, 1.(3) Peterson, S. M.; Batley, G. E. Environ. Pollut. 1993, 82, 143.(4) Mattiessen, P.; Fox, P. J.; Wood, A. B. Pestic. Sci. 1982, 13, 39.(5) Jimenez, J. J.; Bernal, J. L.; del Nozal, M. J.; Rivera, J. M. J.Chromatogr., A 1997, 778, 289.(6) Wilkes, P. S. J. Assoc. Off. Anal. Chem. 1981, 64, 1208.(7) Galeano, T.; Guiberteani, A.; Salinas, F. Anal. Lett. 1992, 25,1797.(8) Gabaldon, J. A.; Cascales, J. M.; Morais, S.; Maquieira, A.;Puchades, R. Food Addit. Contam. 2003, 20, 707.(9) Langone, J. J.; Van Vunakis, H. Res. Commun. Chem. Pathol.Pharmacol. 1975, 10, 163.(10) Lee, N. J.; Skerrit, J. H.; Mcadam, D. P. J. Agric. Food Chem.1995, 43, 1730.(11) Wang, S.; Zhang, J.; Yang, Z. Y.; Wang, J. P.; Zhang, Y. J. Agric.Food Chem. 2005, 53, 7277.(12) Zhang, C.; Zhang, Y.; Wang, S. J. Agric. Food Chem. 2006, 54,2502.(13) Rani, B. E. A.; Roshni, K. R.; Pasha, A.; Karanth, N. G. K. FoodAgric. Immunol. 2003, 15, 105.(14) Dreher, R. M.; Podratzki, B. J. Agric. Food Chem. 1988, 36, 1072.(15) Manclus, J. J.; Abad, A.; Lebedev, M. Y.; Mojarrad, F.; Mickova,B.; Mercader, J. V.; Promo, J.; Miranda, M. A.; Montoya, A. J. Agric.Food Chem. 2004, 52, 2776.(16) Liu, G. Z.; Paddon-Row, M. N.; Gooding, J. J. Chem. Commun.2008, 3870.(17) Liu, G. Z.; Gooding, J. J. Langmuir 2006, 22, 7421.(18) Creager, S.; Yu, C. J.; Bamdad, C.; O’Connor, S.; MacLean, T.;Lam, E.; Chong, Y.; Olsen, G. T.; Luo, J.; Gozin, M.; Kayyem, J. F. J.Am. Chem. Soc. 1999, 121, 1059.(19) Fan, F. R. F.; Yang, J. P.; Cai, L. T.; Price, D. W.; Dirk, S. M.;Kosynkin, D. V.; Yao, Y. X.; Rawlett, A. M.; Tour, J. M.; Bard, A. J. J.Am. Chem. Soc. 2002, 124, 5550.(20) Hess, C. R.; Juda, G. A.; Dooley, D. M.; Amii, R. N.; Hill, M. G.;Winkler, J. R.; Gray, H. B. J. Am. Chem. Soc. 2003, 125, 7156.(21) Umek, R. M.; Lin, S. W.; Vielmetter, J.; Terbrueggen, R. H.;Irvine, B.; Yu, C. J.; Kayyem, J. F.; Yowanto, H.; Blackburn, G. F.;Farkas, D. H.; Chen, Y.-P. J. Mol. Diagn. 2001, 3, 74.(22) Choi, W. B.; Bae, E.; Kang, D.; Chae, S.; Cheong, B. H.; Ko, J.H.; Lee, E. M.; Park, W. Nanotechnology 2004, 15, S512.(23) Dai, L. M. Smart Mater. Struct. 2002, 11, 645.(24) Li, J.; Stevens, R.; Delzeit, L.; Ng, H. T.; Cassell, A.; Han, J.;Meyyappan, M. Appl. Phys. Lett. 2002, 81, 910.(25) Garrett, D. J.; Flavel, B. S.; Shapter, J. G.; Baronian, K. H. R.;Downard, A. J. Langmuir 2010, 26, 1848.(26) de Fuentes, O. A.; Ferri, T.; Frasconi, M.; Paolini, V.; Santucci,R. Angew. Chem., Int. Ed. 2011, 50, 3457.(27) Liu, G. Z.; Gooding, J. J. Electrochem. Commun. 2009, 11, 1982.(28) Zhao, Q.; Gan, Z. H.; Zhuang, Q. K. Electroanalysis 2002, 14,1609.(29) Gooding, J. J.; Wibowo, R.; Liu, J.; Yang, W.; Losic, D.; Orbons,S.; Mearns, F. J.; Shapter, J. G.; Hibbert, D. B. J. Am. Chem. Soc. 2003,125, 9006.
(30) Liu, G. Z.; Bocking, T.; Gooding, J. J. J. Electroanal. Chem. 2007,600, 335.(31) Liu, J.; Rinzler, A. G.; Dai, H. J.; Hafner, J. H.; Bradley, R. K.;Boul, P. J.; Lu, A.; Iverson, T.; Shelimov, K.; Huffman, C. B.;Rodriguez-Macias, F.; Shon, Y. S.; Lee, T. R.; Colbert, D. T.; Smalley,R. E. Science 1998, 280, 1253.(32) Bahr, J. L.; Yang, J.; Kosynkin, D. V.; Bronikowski, M. J.;Smalley, R. E.; Tour, J. M. J. Am. Chem. Soc. 2001, 123, 6536.(33) Ossola, F.; Tomasin, P.; Benetollo, F.; Foresti, E.; Vigato, P. A.Inorg. Chim. Acta 2003, 353, 292.(34) Wang, S.; Allan, R. D.; Hill, A. S.; Kennedy, I. R. J. Environ. Sci.Health 2002, B37, 521.(35) Nowinski, R. C.; Lostrom, M. E.; Tam, M. R.; Stone, M. R.;Burnette, W. N. Virology 1979, 93, 111.(36) Goding, J. W. J. Immunol. Methods 1978, 13, 215.(37) Lyskawa, J.; Belanger, D. Chem. Mater. 2006, 18, 4755.(38) Liu, G. Z.; Chockalingham, M.; Khor, S. M.; Gui, A. L.;Gooding, J. J. Electroanalysis 2010, 22, 918.(39) Cai, L.; Bahr, J. L.; Yao, Y.; Tour, J. M. Chem. Mater. 2002, 14,4235.(40) Diao, P.; Liu, Z. F. J. Phys. Chem. B 2005, 109, 20906.(41) Chou, A.; Eggers, P. K.; Paddon-Row, M. N.; Gooding, J. J. J.Phys. Chem. C 2009, 113, 3203.(42) D’Amours, M.; Belanger, D. J. Phys. Chem. B 2003, 107, 4811.(43) Downard, A. J.; Prince, M. J. Langmuir 2001, 17, 5581.(44) Hauquier, F.; Pastorin, G.; Hapiot, P.; Prato, M.; Bianco, A.;Fabre, B. Chem. Commun. 2006, 4536.(45) Yu, J. X.; Shapter, J. G.; Quinton, J. S.; Johnston, M. R.; Beattie,D. A. Phys. Chem. Chem. Phys. 2007, 9, 510.(46) Toupin, M.; Belanger, D. J. Phys. Chem. C 2007, 111, 5394.(47) Doppelt, P.; Hallais, G. R.; Pinson, J.; Podvorica, F.; Verneyre, S.Chem. Mater. 2007, 19, 4570.(48) Yu, X.; Chattopadhyay, D.; Galeska, I.; Papadimitrakopoulos, F.;Rusling, J. F. Electrochem. Commun. 2003, 5, 408.(49) Asanov, A. N.; Wilson, W. W.; Oldham, P. B. Anal. Chem. 1998,70, 1156.(50) Khor, S. M.; Liu, G. Z.; Peterson, J. R.; Iyengar, S. G.; Gooding,J. J. Electroanalysis 2011, 23, 1797.
Analytical Chemistry Article
dx.doi.org/10.1021/ac202754p | Anal. Chem. 2012, 84, 3921−39283928


















