An Anti-EGFR IgA That Displays Improved...
16
Therapeutics, Targets, and Chemical Biology An Anti-EGFR IgA That Displays Improved Pharmacokinetics and Myeloid Effector Cell Engagement In Vivo Stefan Lohse 1 , Saskia Meyer 2 , Laura A.P.M. Meulenbroek 2 , J.H. Marco Jansen 2 , Maaike Nederend 2 , Anna Kretschmer 1 , Katja Klausz 1 , Uwe M€ oginger 3,4 , Stefanie Derer 1 , Thies R € osner 1 , Christian Kellner 1 , Denis Schewe 5 , Peter Sondermann 6 , Sanjay Tiwari 7 , Daniel Kolarich 3 , Matthias Peipp 1 , Jeanette H.W. Leusen 2 , and Thomas Valerius 1 Abstract Antibodies of IgA isotype effectively engage myeloid effector cells for cancer immunotherapy. Here, we describe preclinical studies with an Fc engineered IgA2m(1) antibody containing the variable regions of the EGFR antibody cetuximab. Compared with wild-type IgA2m(1), the engineered molecule lacked two N- glycosylation sites (N166 and N337), two free cysteines (C311 and C472), and contained a stabilized heavy and light chain linkage (P221R mutation). This novel molecule displayed improved production rates and biochemical properties compared with wild-type IgA. In vitro, Fab- and Fc-mediated effector func- tions, such as inhibition of ligand binding, receptor modulation, and engagement of myeloid effector cells for antibody-dependent cell-mediated cytotoxicity, were similar between wild-type and engineered IgA2. The engineered antibody displayed lower levels of terminal galactosylation leading to reduced asialoglycoprotein- receptor binding and to improved pharmacokinetic properties. In a long-term in vivo model against EGFR-positive cancer cells, improved serum half-life translated into higher efficacy of the engineered molecule, which required myeloid cells expressing human FcaRI for its full efficacy. However, Fab-mediated effector functions contributed to the in vivo efficacy because the novel IgA antibody demonstrated therapeutic activity also in non- FcaRI transgenic mice. Together, these results demonstrate that engineering of an IgA antibody can significantly improve its pharmacokinetics and its therapeutic efficacy to inhibit tumor growth in vivo. Cancer Res; 76(2); 403–17. Ó2015 AACR. Introduction The EGFR constitutes an established tumor target antigen, which can be involved in tumorigenesis (1), and which is over- expressed or mutated in many common solid tumor types such as colorectal, head and neck, and lung cancer (2). Both tyrosine kinase inhibitors and monoclonal antibodies have been success- fully developed and approved for clinical applications (3, 4). The two approved monoclonal antibodies cetuximab (chimeric IgG1) and panitumumab (human IgG2) bind to overlapping epitopes in domain III of EGFR and are similar in Fab-mediated effector functions such as inhibition of ligand binding, tumor growth inhibition, and receptor modulation (5). However, both anti- bodies differ in their efficacy to recruit effector cells for antibody- dependent cell-mediated cytotoxicity (ADCC): although the IgG1 antibody cetuximab is particularly efficient with NK cells, the IgG2 antibody panitumumab effectively recruits myeloid cells (6). Interestingly, both antibodies proved to be similarly effective in the treatment of patients with chemotherapy-refractory metastatic colorectal cancer (7). Furthermore, there is increasing evidence that myeloid cells are important effector cells in cancer and cancer immunotherapy (8–10). In vitro, myeloid effector cell engage- ment is as effective with human IgG2 as with human IgG1 antibodies (6), but is significantly improved with antibodies of IgA isotypes (11–13). Enhanced myeloid cell recruitment by human IgA compared with IgG1 antibodies has been observed against different tumor target antigens such as CEA, CD20, HER- 2/neu, and EGFR. However, the immunotherapeutic potential of IgA antibodies has not been explored in humans. Natural IgA antibodies constitute the dominant isotype in mucosal immunology (14). By concerting multiple mechanisms of action, IgA antibodies control commensals and prevent the invasion of pathogens, or neutralize those that passed through mucosal barriers (14). Important defense mechanisms, such as phagocytosis, oxidative burst, cytokine release, and antigen 1 Division of Stem Cell Transplantation and Immunotherapy, 2 nd Department of Medicine, Christian-Albrechts-University, Kiel, Ger- many. 2 Laboratory for Translational Immunology, Immunotherapy, University Medical Center Utrecht, Utrecht, The Netherlands. 3 Depart- ment of Biomolecular Systems, Max Planck Institute for Colloids and Interfaces, Potsdam, Germany. 4 Institute for Chemistry and Biochem- istry, Freie Universit€ at Berlin, Berlin, Germany. 5 Department of General Pediatrics, Christian-Albrechts-University, Kiel, Germany. 6 SuppreMol GmbH, Martinsried, Germany. 7 Molecular Imaging North Competence Center, Christian-Albrechts-University, Kiel, Germany. Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). Current address for S. Lohse: Institute of Virology, Saarland University Medical Center, Homburg, Germany; and current address for S. Derer, Molecular Gas- troenterology/Medical Department 1, University Hospital Schleswig-Holstein, L€ ubeck, Germany. Corresponding Author: Thomas Valerius, Division of Stem Cell Transplantation and Immunotherapy, 2 nd Department of Medicine, Christian-Albrechts-Univer- sity, Schittenhelmstr 12, 24105 Kiel, Germany. Phone: 49-431-597-5802; Fax: 49- 431-597-5803; E-mail: [email protected] doi: 10.1158/0008-5472.CAN-15-1232 Ó2015 American Association for Cancer Research. Cancer Research www.aacrjournals.org 403 on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232
Transcript of An Anti-EGFR IgA That Displays Improved...

Therapeutics, Targets, and Chemical Biology
An Anti-EGFR IgA That Displays ImprovedPharmacokinetics and Myeloid EffectorCell Engagement In VivoStefan Lohse1, Saskia Meyer2, Laura A.P.M. Meulenbroek2, J.H. Marco Jansen2,Maaike Nederend2, Anna Kretschmer1, Katja Klausz1, Uwe M€oginger3,4, Stefanie Derer1,Thies R€osner1, Christian Kellner1, Denis Schewe5, Peter Sondermann6, Sanjay Tiwari7,Daniel Kolarich3, Matthias Peipp1, Jeanette H.W. Leusen2, and Thomas Valerius1
Abstract
Antibodies of IgA isotype effectively engage myeloid effectorcells for cancer immunotherapy. Here, we describe preclinicalstudies with an Fc engineered IgA2m(1) antibody containing thevariable regions of the EGFR antibody cetuximab. Comparedwithwild-type IgA2m(1), the engineered molecule lacked two N-glycosylation sites (N166 and N337), two free cysteines (C311and C472), and contained a stabilized heavy and light chainlinkage (P221R mutation). This novel molecule displayedimproved production rates and biochemical properties comparedwith wild-type IgA. In vitro, Fab- and Fc-mediated effector func-tions, such as inhibition of ligand binding, receptor modulation,and engagement ofmyeloid effector cells for antibody-dependentcell-mediated cytotoxicity, were similar between wild-type and
engineered IgA2. The engineered antibody displayed lower levelsof terminal galactosylation leading to reduced asialoglycoprotein-receptor binding and to improved pharmacokinetic properties.In a long-term in vivo model against EGFR-positive cancer cells,improved serum half-life translated into higher efficacy of theengineered molecule, which required myeloid cells expressinghuman FcaRI for its full efficacy. However, Fab-mediated effectorfunctions contributed to the in vivo efficacy because the novelIgA antibody demonstrated therapeutic activity also in non-FcaRI transgenic mice. Together, these results demonstrate thatengineering of an IgA antibody can significantly improve itspharmacokinetics and its therapeutic efficacy to inhibit tumorgrowth in vivo. Cancer Res; 76(2); 403–17. �2015 AACR.
IntroductionThe EGFR constitutes an established tumor target antigen,
which can be involved in tumorigenesis (1), and which is over-expressed ormutated inmany common solid tumor types such ascolorectal, head and neck, and lung cancer (2). Both tyrosinekinase inhibitors and monoclonal antibodies have been success-
fully developed and approved for clinical applications (3, 4). Thetwo approvedmonoclonal antibodies cetuximab (chimeric IgG1)and panitumumab (human IgG2) bind to overlapping epitopesin domain III of EGFR and are similar in Fab-mediated effectorfunctions such as inhibition of ligand binding, tumor growthinhibition, and receptor modulation (5). However, both anti-bodies differ in their efficacy to recruit effector cells for antibody-dependent cell-mediated cytotoxicity (ADCC): although the IgG1antibody cetuximab is particularly efficientwithNK cells, the IgG2antibody panitumumab effectively recruits myeloid cells (6).Interestingly, both antibodies proved to be similarly effective inthe treatment of patientswith chemotherapy-refractorymetastaticcolorectal cancer (7). Furthermore, there is increasing evidencethatmyeloid cells are important effector cells in cancer and cancerimmunotherapy (8–10). In vitro, myeloid effector cell engage-ment is as effective with human IgG2 as with human IgG1antibodies (6), but is significantly improved with antibodies ofIgA isotypes (11–13). Enhanced myeloid cell recruitment byhuman IgA compared with IgG1 antibodies has been observedagainst different tumor target antigens such as CEA, CD20, HER-2/neu, and EGFR. However, the immunotherapeutic potential ofIgA antibodies has not been explored in humans.
Natural IgA antibodies constitute the dominant isotype inmucosal immunology (14). By concerting multiple mechanismsof action, IgA antibodies control commensals and prevent theinvasion of pathogens, or neutralize those that passed throughmucosal barriers (14). Important defense mechanisms, suchas phagocytosis, oxidative burst, cytokine release, and antigen
1Division of Stem Cell Transplantation and Immunotherapy, 2nd
Department of Medicine, Christian-Albrechts-University, Kiel, Ger-many. 2Laboratory for Translational Immunology, Immunotherapy,University Medical Center Utrecht, Utrecht,TheNetherlands. 3Depart-ment of Biomolecular Systems, Max Planck Institute for Colloids andInterfaces, Potsdam, Germany. 4Institute for Chemistry and Biochem-istry, FreieUniversit€at Berlin, Berlin,Germany. 5Department ofGeneralPediatrics, Christian-Albrechts-University, Kiel, Germany. 6SuppreMolGmbH, Martinsried, Germany. 7Molecular Imaging North CompetenceCenter, Christian-Albrechts-University, Kiel, Germany.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Current address for S. Lohse: Institute of Virology, Saarland University MedicalCenter, Homburg, Germany; and current address for S. Derer, Molecular Gas-troenterology/Medical Department 1, University Hospital Schleswig-Holstein,L€ubeck, Germany.
Corresponding Author: Thomas Valerius, Division of Stem Cell Transplantationand Immunotherapy, 2nd Department of Medicine, Christian-Albrechts-Univer-sity, Schittenhelmstr 12, 24105 Kiel, Germany. Phone: 49-431-597-5802; Fax: 49-431-597-5803; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-15-1232
�2015 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 403
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

presentation, are mediated by the interaction of IgA antibodieswith the myeloid IgA Fc receptor, FcaRI (CD89), which isexpressed onmonocytes,macrophages, granulocytes, andKupffercells (14–16). There are important differences in the antibody-FcRinteraction between IgA and IgG antibodies (16): mutagenesisand X-ray crystal structure studies located the FcaRI interactionsite to the interface of the Ca2 and Ca3 domains of the Iga-chainand revealed a bivalent binding mode, although Fcg receptorsbind to IgG at the lower hinge in a 1:1 stoichiometry (16).Contribution of N-glycans to Fc receptor binding was shown forcarbohydrates of FcaRI but was not observed for those of IgAantibodies (17–19). This is in contrast to IgG1 where glycosyla-tion of both, FcgRIII and that of the antibody, does influenceinteractions with FcgRIII (20). Previous studies demonstrated theefficiency of IgA2 antibodies in activating myeloid effector cellsfor tumor cell lysis in ADCC assays in vitro (11–13). Thus,antibodies of IgA isotype may constitute interesting and potentmolecules for targeted cancer therapy.
Humans have two genes for IgA heavy chains (HC) encodingtwo isotypes—named IgA1 and IgA2—with three allelic forms ofIgA2, described as IgA2m(1), IgA2m(2), and IgA2n (21, 22). Eachof these IgA molecules has distinct structural characteristics. Forexample, the elongated hinge of IgA1 increases its flexibilitythereby possibly allowing binding of more distant epitopes(14), but the carbohydrates attached at the up to six O-glycosyl-ation sites are difficult to control during recombinant expression(23) and are critically involved in the pathogenesis of IgAnephropathy (24). This elongated hinge region of IgA1 is notpresent in human IgA2 allotypes, resulting in a more compactstructure (14, 25). IgA antibodies are stabilized by a variabledegree of interchain disulfide bridges between both HCs in theCa2 domain and between HC and light chains (LC; refs. 26, 27).However, LCs are not covalently connected to HCs in IgA2m(1),which is the most common allotype in Caucasians. The HCs ofboth IgA subclasses carry a C-terminal extension of 18 aminoacids, called tail piece,which is critically involved in the formationand transcytosis of dimeric and secretory IgA by the polymericimmunoglobulin receptor (pIgR) ontomucosal surfaces (14, 28).Notably, cysteines 471 and 311 inmonomeric IgA are required forcovalent linkage to the J-chain and the extracellular domain of thepIgR to form dimeric or secretory IgA, respectively (14). Incontrast to IgG1, IgA antibodies carry a more exposed and het-erogeneous N-glycosylation (14, 29); there are two conserved N-glycans in both IgA isotypes with two and three additional N-glycans for IgA2m(1) and IgA2m(2), respectively. Increasingevidence underlines the impact of glycosylation on pharmacoki-netic properties of IgA antibodies (30, 31). Naturally, IgA anti-bodies have a significantly shorter serum half-life compared withIgG in men and mice, which is partially explained by the lack ofbinding to the neonatal Fc receptor (FcRn; ref. 32). Additionally,recombinant IgA was previously reported to be rapidly cleared bythe asialoglycoprotein receptor (ASGPR), which is predominantlyexpressed in the liver and clears proteins with exposed terminalgalactose (30, 31, 33). Importantly, the serum half-life of IgGantibodies has been demonstrated to significantly impact on theirtherapeutic efficacy (34). Thus, glyco-engineering strategies toincrease the serum half-life of IgA antibodies may improve theimmunotherapeutic potential of this antibody isotype for clinicalapplications.
In the present study, we describe the rational design of anengineered IgA antibody directed against EGFR. We removed two
N-glycosylation sites (N166 and N337), stabilized HC and LClinkage by introduction of a P221Rmutation into the IgaHC andremoved two free cysteins (C311 and C472). Next, we investi-gated the effects of this engineering on the productivity, assembly,glycosylation, stability, and functionality. Importantly, the engi-neered IgA antibody demonstrated increased productivity andthermal stability. This novel IgA antibody had a significantlyimproved serum half-life compared with wild-type IgA2 and wasefficient in engaging myeloid effector cells in vitro and in vivo—thus constituting a promising antibody format for cancerimmunotherapy.
Materials and MethodsExperimentswithhumanmaterialwere approvedby theEthical
Committee of theChristian-Albrechts-University (Kiel,Germany)in accordance with theDeclaration ofHelsinki. All experiments inmice were approved by the Animal Ethical Committees of theUMC Utrecht or Christian-Albrechts-Universit€at in Kiel.
Animals and animal experimentsImmunodeficient SCID mice (CB17/Icr-Prkdcscid/IcrIcoCrl)
were used to evaluate the serum half-life in mice (four mice pergroup). Two hundred micrograms of the respective antibody wasinjected intravenously into the tail vein. Submandibular bloodsamples were collected daily during treatment, and antibodyconcentrations were evaluated using human IgA- or IgG-specificELISA as described earlier (30). The tumor model with A431-luc2in SCIDmice transgenic and nontransgenic for human FcaRI wasperformed similarly as previously described (30). Experimentswith B16F10-luc2-EGFR and BaF3-EGFR cells in C57BL/6 (BL6)and Balb/cmice transgenic (tg) or nontransgenic (ntg) for humanFcaRI were performed as described earlier (30).
For pharmacokinetic imaging antibodies were labeled andseparated from free dye using LICOR IRDye 800CW kit (LI-COR)according to the manufacturers' protocol. A total of 25 mg oflabeled antibodies were injected i.v. into tail veins of 5- to 6-week-old female mice (Athymic Nude-Foxn1nu, purchased from Har-lan Laboratories GmbH). For injection and imaging mice wereanaesthetized with i.p. injections of ketamine (80 mg/kg, AvecoPharmaceutical) and dorbene (0.5 mg/kg, Pfizer). Ventral viewswere imaged using a Peltier cooled charged-coupled device cam-era (NightOWL LB 983, Berthold Technologies). The excitationsource is a ring light used for epi-illumination, mounted 12 cmabove the mice. Filters of 740 and 790 nm were used to assessexcitation and emission signals, respectively. The exposure timewas set to 0.3 second. Fluorescent signal was quantified as countsper secondusing Indigo software (Berthold Technologies)with anautomated peak search function.
Cell linesThe human epidermoid carcinoma cell line A431 [German
Collection of Microorganisms and Cell Cultures (DSMZ),Braunschweig, Germany] and BHK-21 cells cotransfected withFcaRI (CD89) and the FcR g-chain (35) as well as the luciferase-transduced A431 cells (30) were cultured in RPMI 1640 contain-ing 10% heat-inactivated FBS, 100 U/mL penicillin, and 100mg/mL streptomycin (media and additives from Life Technolo-gies). Baby hamster kidney (BHK) transfectants were positivelyselected by addition of 1 mg/mL geneticin and 20 mmol/L meth-otrexate (Sigma). The human esophageal squamous carcinoma
Lohse et al.
Cancer Res; 76(2) January 15, 2016 Cancer Research404
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

cell line Kyse30 (DSMZ) was kept in 45% RPMI 1640 with 45%Ham's F12, 10% FBS, and 1% antibiotics (100 U/mL penicillin,100 mg/mL streptomycin). The human colon carcinoma cell lineDiFi and the human glioblastoma cell line A1207 (EuropeanCollection of Cell Culture, ECACC, Salisbury, UK) were main-tained in DMEM supplemented with 10% FBS, 100 U/mL pen-icillin, and 100 mg/mL streptomycin. A431-luc2, BaF3�EGFR,and B16F10-EGFR-luc2 [BaF3 (DSMZ) and B16F10 (ATCC)] cellswere kept in RPMI 1640 containing 10% heat-inactivated FBS,100 U/mL penicillin, and 100 mg/mL streptomycin (30). All celllines were obtained between 2011 and 2013.
IgA production, purification, and characterizationPurified human myeloma IgA2m(1) antibody was used as
control IgA2 (Meridian Life Science). Monomeric wild-type 225-IgA2m(1), named 225-IgA2-wt, and a wild-type human IgG1,named 225-IgG1, were produced from the variable regions of the225 antibody and human constant regions as previously described(13). The DNA-sequence encoding the mutated IgA2m(1) wasgenerated by Entelechon. The translated protein contains thefollowingmutations: asparagine to glycine at position 166, prolineto arginine at position 221, cysteine to serine at position 311,asparagine to threonine at position 337, isoleucine to leucine atposition 338, and threonine to serine at position 339. The nomen-clature of the new IgA2m(1) is therefore 225-IgA2m(1)-N166G-P221R-C331S-N337T-I338L-T339S-dC471-dY472 (further named225-IgA2.0). The coding sequences for the HCs of wild-typehuman IgA1, IgA2m(1), and the mutated IgA2.0 are aligned inFig. 1. Further cloning, production, purification, as wells as thedetermination of antibody concentrations, specific productionrates, gel electrophoresis,Western blotting, aswell as the functionalcharacterization were done as described earlier (13, 35).
GlycoprofilingThe releasedN-glycans were analyzed as described in detail in a
MIRAGE (36) compliant manner in the supplementary material(29, 37). Relative quantitation was performed using the QuantAnalysis tool (Bruker), which determines the area under the curveobtained from the individual extracted ion chromatograms frommultiple analyses (Supplementary Table S1).
Thermal shift assayThermal stability was analyzed in thermal shift assay using
SyproOrange (Life Technologies). A volumeof 7.5mL 300� SyproOrangewere dilutedwith 12.5 mL 1� PBS (Life Technologies) and5mLof 2.5mg/mLprotein and transferred on awhite 96-well thin-wall PCR plate and sealed with Optical-Quality Sealing Tape(both Roche). Plates were heated in a LightCycler 480 (Roche)from 37�C to 99�C with a heat-rate of 4�C/second and 1 minuteincubation at each degree. Fluorescence was recorded simulta-neously using 490 and 575 nm as excitation and emissionwavelengths, respectively.
Surface plasmon resonanceSurface plasmon resonance was investigated on a BIAcore
T100 system equipped with BIAcore evaluation software V1.2(both GE Healthcare). Recombinant human soluble FcgRIIIaand FcaRI (both R&D systems; 100 mg/mL) were coated usingrandom amine coupling kit in sodium acetate buffer with pH 4.5onto flow cell 1 (FcgRIIIa as reference) and flow cell 2 (FcaRI) ofCM5 chips (kit and chips from GE Healthcare). Different
Figure 1.Primary sequence and modeling of the IgA1/IgA2.0 hybrid antibody. A,alignment of primary sequences of the constant regions of hIgA1,IgA2m(1), and the new IgA1/IgA2m(1) hybrid (hIgA2.0). Residues arenumbered according to the myeloma IgA1 protein (Bur) scheme. Domainboundaries are indicated by vertical lines above the sequences. Thefollowing features are highlighted: light gray underlined residues areunique for IgA1, dark gray underlined asparagines are conservedN-glycosylation consensus sequences, and black underlined residues areunique for IgA2.0. B, the heavy chain of 225-IgA2.0 was modeled andillustrated in front and side view, with mutations marked in red. C, heavychains of wild-type and mutant IgA2 were modeled. The resultingalignment indicates a different orientation of C241 in the heavy chains ofIgA2-wt compared with IgA2.0, possibly due to the P221R mutation.D, focus on the tailpiece of 225-IgA2-wt (green, C471; red, Y 472) andIgA2.0 (red). Prediction and alignment of models were performed usingI-TASSER; models were modified in 3D-Mol Viewer.
Engineering IgA for Cancer Immunotherapy
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 405
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

concentrations (0–200 nmol/L) of 225-IgA2-wt and 225-IgA.0dialyzed three times against HPS-EPþ running buffer (GE Health-care)were injectedwith a constantflow rate of 30mL/min. Bindingto soluble FcgRIIIa and FcaRI was measured at 25�C. Sensogramswere recorded and kinetics were calculated using a bivalent ligandmodel.
Cell-based assaysGrowth inhibition of DiFi cells was analyzed using the 3-(3,5-
dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfop-henyl)-2H-tetrazolium inner salt (MTS) assay and performed asdescribed earlier (12). For the internalization assay, A431 cellswere seeded (2.5 � 104 cells) on 96-well plates (Sarstedt) andtreated with 5 mg/mL of 225-IgG1 or respective IgA2 antibodies. Ahuman kappa LC binding antibody fragment fused to a truncatedversion of Pseudomonas exotoxin A (a-kappa-ETA'; ref. 38) wasapplied in a dose-dependent manner. After 72 hours, MTS sub-stratewas added, and absorption at 490nmwasmeasured after 24hours to determine inhibition of cell growth as a measure forantibody internalization. All experimental points were set up intriplicates. Viable cell mass in the presence of control antibodyserved as reference (100% cell growth) to calculate growth inhi-bition by EGFR Abs according to the formula: absorption (EGFRAb)/absorption (control Ab) � 100. Apoptosis of DiFi cells wasinducedby incubating cellswith 20mg of the respective antibodiesusing a-kappa-ETA'. Twenty-four hours later cells were yielded bytrypsinization, stained with Annexin-FITC/PI kit (BD) and ana-lyzed by immunofluorescence analyses. Preparation and engage-ment of effector cells was analyzed in 51chromium release assaysas described earlier (13, 35). ASGPR-mediated uptake of anti-bodies was investigated using BHK cells, transfectedwith pCMV6-AC plasmid encoding ASGPR1 (OriGene) and Lipofectamine2000 (Life Technologies). Positive single clones were selected byFACS using ASGPR1-directed antibody (Affinity Bioreagents) andFITC-labeled goat-anti-mouse Ig Fab2 secondary antibody (Dia-nova). Cells (2.0 � 104 cells) were seeded in 96-well plates andincubated for 96 hours with respective antibodies at indicatedconcentrations with or without the supplement of ahu-kappa-ETA. Viable cellmass in the presence of 1� PBS served as reference(100% cell growth) to calculate growth inhibition by EGFR Absaccording to the formula: absorption (EGFR Ab)/absorption (1�PBS) � 100.
Data processing and statistical analyses and in silico modelingDataweregenerated fromat leastfive independent experiments.
Graphical and statistical analyseswereperformedusingGraphPadPrism 6.0 (GraphPad Software). Group data are reported as mean� SEM. Significancewasdeterminedby two-wayANOVA repeatedmeasures test with Bonferroni's post hoc correction. EC50 valueswere calculated from dose–response curves, reported as means �SEM and compared by paired Student t test to calculate significantdifferencesbetweendatagroups. SignificancewasacceptedwhenPvalues were �0.05. HCs of IgA2 antibodies were modeled on thebasis of the sequence available under the Genbank accessionnumber AY647979 and aligned using I-TASSER (39).
ResultsRational protein design and modeling of IgA HCs
Several strategies were combined for engineering an EGFR-directed IgA2m(1) antibody (Fig. 1A): first, the stability of the
molecule was increased by exchanging proline to arginine atposition 211 of the HC (P221R) to allow the covalent linkageof LCs and HCs. Second, the formation of dimeric aggregates andcomplex formation with serum proteins was reduced by deletingthe C-terminal cysteine 471 and by exchanging cysteine to serineat position 311 (C331S). Additionally, the number of glycosyl-ation siteswas reducedby exchanging the amino acids at positions166 and 337–339 to amino acids at corresponding positions inIgA1. The resulting monomeric molecule is therefore an Fc-engi-neered IgA2m(1) molecule with four instead of eight N-glycans.The three-dimensional structures of both IgA2 antibodies werepredicted using I-TASSER (Fig. 1B). Highlighting red space fillspheres indicate surface exposed residues. Consequently, theexposed nature of the residues at the amino acid positions 166and 337–339 indicates the accessibility of the respective N-gly-cosylation sites in wild-type IgA2m(1) antibodies. Next, HCs ofwild-type and mutated IgA2 were modeled and aligned. Intro-ducing the P221R mutation appears to allow a slight rotation ofthe HC, leading to a different orientation of the side chain ofcysteine 241 in wild-type and mutated IgA2.0, as describedpreviously (Fig. 1C; ref. 13). Furthermore, the tail piece wasmodeled differently for wild-type and mutated IgA2 (Fig. 1D).However, as IgA2.0 contains eight differentmutationswemaynotconclude that the predicted structural changes are the result of onespecific mutation.
Production and purification of 225-IgA2.0Both IgA antibodies were produced in CHO-K1 cells growing
serum-free under suspension culture conditions. Single clonesstably expressing the antibodies were generated by limiting dilu-tion cloning, and production rates of five 225-IgA2-wt and 225-IgA2.0 producing single clones were evaluated (Fig. 2A and B).Significantly higher production rates were detected for 225-IgA2.0producing clones. Next, antibodies were affinity-purified andsubjected to size exclusion chromatography to isolatemonomericIgA2 antibodies. Elution profiles of 225-IgA2-wt preparationsdisplayed a significant amount of polymeric aggregates, whereasthe preparations containing 225-IgA2.0 eluted mainly as a singlepeak (Fig. 2C). In both cases fractions containing monomericIgA2 antibodies were pooled and purity of those preparations wasassessed by size exclusion chromatography (Fig. 2D). Thus, higherproduction rates and lower formation of spontaneous aggregatessuggested improved biopharmaceutical properties of IgA2.0 com-pared with wild-type IgA2.
Biochemical characterizationMolecularmass and purity ofmonomeric IgA2 antibodies were
analyzed by capillary electrophoresis (Fig. 2E). Under reducingconditions 225-IgA2-wt displayed bands ranging from approxi-mately 75 to 110 kDa, whereas two distinct bands were detectedfor 225-IgA2.0 at approximately 75 and 80 kDa. On native PAGE,monomeric 225-IgA2-wt displayed a single band at themolecularmass of approximately 160 to 180 kDa, whereasmonomeric 225-IgA2.0 was detected at a molecular mass of approximately 150kDa (Fig. 2F). Next, affinity purified (using anti-human kappaLC directed beads) monomeric IgA2 antibodies were separatedby gel electrophoresis under denaturing nonreducing andreducing conditions, and silver staining was used for proteindetection (Fig. 2G). Again, the 225-IgA2.0 was detected at alower molecular mass under reducing (�50 and 60 kDa forHCs) and nonreducing conditions (�150 kDa for monomeric
Lohse et al.
Cancer Res; 76(2) January 15, 2016 Cancer Research406
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

Figure 2.Production and biochemical characterization of monomeric 225-IgA2-wt and 225-IgA2.0. A, antibody contents in supernatants of different 225-IgA2-wt and225-IgA2.0–producing single cloneswere analyzed by ELISA, and specific production rateswere calculated. B, median-specific production rates of 225-IgA2-wt and225-IgA2.0–producing single clones were calculated. �� , P � 0.01. C, antibody preparations purified by anti-human-kappa affinity columns (ahukappa) wereexposed to a Superdex 200 10� 300 size exclusion column to isolate monomeric antibodies. D, purified monomeric antibodies gained by pooling of fractions in Cwere separated on a Superdex 200 10 � 300 size exclusion column. Representative elution profiles are shown for 225-IgA2-wt (left) and 225-IgA2.0 (right)in C and D. E, concentration and purity of purified monomeric IgA2 antibodies were analyzed using capillary gel electrophoresis. F, purity of monomeric IgA2preparations was analyzed under native conditions using Native-PAGE. G, formation of aggregates was analyzed using denaturing SDS-PAGE stained withsilver nitrate. In H and I, proteins were transferred onto PVDF membranes and probed using a polyclonal POX-labeled antibody against human a- or k-chain,respectively. J, microheterogeneity of monomeric IgA preparations was analyzed by isoelectric focusing using pH 3–10 isoelectric focusing gels and silver staining.Lanes in A, B, and E: 1, 225-IgA2-wt; 2, 225-IgA2.0. Lanes in C and D: 1, anti-human-kappa purified 225-IgA2-wt; 2, anti-human kappa purified 225-IgA2.0; 3,monomeric 225-IgA2-wt; 4, monomeric 225-IgA2.0. Lanes in F: 1, control IgA2; 2, 225-IgA2-wt; 3, 225-IgA2.0. K, monomeric antibodies were incubated atincreasing temperature (37�C–99�C) in the presence of Sypro Orange. Thermal-induced fluorimetric shift was continuously measured in a LightCycler 480. L,monomeric antibodies were incubated for 5 minutes at different denaturing temperatures. Maintenance of functionality was analyzed in ADCC assays usingPMN as effector and A431 as target cells. Results of five independent experiments are presented as mean � SEM of "OD [575 nm]" in G and of "relative specificlysis [%]" in H. Significant differences between wild-type and mutant IgA2 are indicated by þ, P � 0.001.
Engineering IgA for Cancer Immunotherapy
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 407
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

antibody) than 225-IgA2-wt, which dissociates under nonreduc-ing but denaturing conditions in homodimers of LCs (�40 kDa)and HCs (�100 kDa) as previously described (13). Interestingly,polymeric aggregates were detected under nonreducing condi-tions in the affinity-purified 225-IgA2-wt but not in 225-IgA2.0.Western Blot analyses using peroxidase (POX)-labeled anti-human a-chain–specific antibody proved the existence of poly-meric aggregates in the affinity-purified wild-type but not in theIgA2.0 preparations (Fig. 2H). Western blot analyses usingk-chain–specific antibody confirmed the covalent linkage of LCsandHCs in 225-IgA2.0 but not in 225-IgA2-wt (Fig. 2I). Isoelectricfocusing was performed using pH 3–10 isoelectric focusing gelsunder native conditions to determine microheterogeneity (Fig.2J). Monomeric 225-IgA2.0 displayed a single band at pH 6,whereas 225-IgA2-wt displayed a remarkable heterogeneity witha pH from4.5 to 6.5. Next, thermal stability of bothwild-type andmutated IgA2 was analyzed in a thermal shift assay (Fig. 2K).Temperature (37�C–99�C) and fluorimetric shift of SyproOrangedue to protein unfolding was simultaneously measured in a RT-PCR cycler. The significantly increasedmelting temperature of themutated compared with the wt IgA2 antibody, which is similar tothat of 225-IgG1 (Table 1), indicated different kinetics in ther-mally induced protein unfolding between wild-type and engi-neered IgA2. Maintenance of functionality after incubation atdenaturing temperatures was tested in 51chromium release assaysusing polymorphonuclear (PMN) as effector and A431 as targetscells, respectively (Fig. 2L). Although 225-IgA2-wt was fully dena-tured under the employed conditions, functionality of 225-IgA2.0was at least partiallymaintained. Thus, after 5minutes at 96�C,1.7� 1.4%and58.9� 19.5% specific lysiswas detected for 225-IgA2-wt and 225-IgA2.0, respectively. Again, IgA2.0 displayed improv-ed biopharmaceutical properties compared with wild-type IgA2.
Fab-mediated effector mechanismsFunctional characterization of EGFR-directed IgA2 antibodies
was assessed by investigating their capability to induce Fab-mediated effector mechanisms. First, both antibodies were com-pared for binding to EGFR-expressing A431 cells (Fig. 3A). In
these experiments, 225-IgA2-wt and 225-IgA2.0 bound withsimilar avidity as confirmed by EC50 values (Table 1). Next, theirability to block binding of the ligand EGF was investigated byincubating A431 cells with FITC-labeled EGF and increasingconcentrations of both IgA2 antibodies (Fig. 3B). 225-IgA2.0preventedbinding of EGFwith similar efficacy as 225-IgA2-wt (forEC50 values see Table 1). Growth of EGFR-expressing DiFi coloncarcinoma cells was inhibited similarly effective by 225-IgA2.0and cetuximab, requiring lower concentrations than 225-IgA2-wt(Fig. 3C, EC50 Table 1). Internalization of EGFR upon antibodybinding was investigated using a-kappa-ETA' (Fig. 3D, Table 1).The toxic fusion protein induced growth inhibition of A431 cells,similarly when 5 mg/mL 225-IgA2-wt, 225-IgA2.0, or cetuximabwere initially bound to target cells. Apoptosis induction wasmeasured by incubating DiFi cells with EGFR-specific antibodiesfor 24 hours. Cells were then stained using Annexin V-FITC/PIand analyzed by flow cytometry. Results revealed all three anti-bodies to be similarly effective (Fig. 3E and F), indicating that Fab-mediated effector functionswere not affected by the antibodies' Fcregion.
Fc-mediated effector mechanismsThe ability of EGFR-directed IgA2 antibodies to bind to the IgA
Fc receptor, FcaRI, was analyzed first by surface plasmon reso-nance measurement (Fig. 4). Sensograms were derived frominjection of different concentrations of 225-IgA2-wt (Fig. 4A) or225-IgA2.0 (Fig. 4B) over randomly coupled FcaRI. Calculatingassociation and dissociation constants and the steady-state affin-ity using a bivalent ligandmodel revealed both antibodies to havesimilar kinetics and apparent affinities in binding to FcaRI (Fig.4C). These results are in line with previous reports describingFcaRI as a medium affinity receptor for monomeric IgA (17).Binding to human FcaRI was additionally measured by indirectimmunofluorescence analyses using FcaRI/FcRg-chain-cotrans-fected BHK cells. In these assays, 225-IgA2.0 demonstrated similarbinding as wild-type 225-IgA2-wt as confirmed by similar EC50
values (Table 1). Next, the potential of both IgA2 antibodies tomediate ADCC against EGFR-positive A431 cells was investigated
Table 1. Calculated EC50 and median values
Assay [Unit] Target cell line 225-IgA2-wt 225-IgA2.0 225-IgG1 Significance wt vs. 2.0
Thermal shift assay [�C] — 54.3 � 0.6 64.9 � 0.9 62.7 � 2.2 P � 0.001EGFR binding [mg/mL] A431 12.2 � 2.6 8.6 � 5.5 n.d. n.s.EGF blocking [mg/mL] A431 42.9 � 7.3 25.2 � 3.9 n.d. n.s.Growth inhibition [mg/mL] DiFi 4.1 � 1.2 1.0 � 0.2 0.99 � 0.27 P � 0.01Internalization [mg/mL] A431 0.02 � 0.01 0.03 � 0.01 0.02 � 0.007 n.s.FcaRI binding [mg/mL] BHK FcaRIþ 93.9 � 51.9 93.1 � 45.6 n.d. n.s.ADCC (wb) [mg/mL] A431 0.13 � 0.07 0.2 � 0.15 n.d. n.s.ADCC (wbþG-CSF) [mg/mL] A431 0.22 � 0.04 0.27 � 0.09 1.54 � 0.26 n.s.ADCC (PMN) [mg/mL] A431 0.22 � 0.16 0.75 � 0.54 n.d. n.s.ADCC (PMNþGM-CSF) [mg/mL] A431 0.12 � 0.09 0.14 � 0.07 0.47 � 0.27 n.s.ADCC (PMN) [mg/mL] DiFi 0.28 � 0.2 0.28 � 0.2 n.d. n.s.ADCC (PMNþGM-CSF) [mg/mL] DiFi 0.11 � 0.2 0.16 � 0.08 0.19 � 0.07 n.s.ADCC (PMN) [mg/mL] A1207 0.09 � 0.08 0.14 � 0.08 n.d. n.s.ADCC (PMNþGM-CSF) [mg/mL] A1207 0.01 � 0.001 0.008 � 0.005 0.41 � 0.27 n.s.ADCC (PMN) [mg/mL] Kyse30 0.25 � 0.09 0.27 � 0.07 n.d. n.s.ADCC (PMNþGM-CSF) [mg/mL] Kyse30 0.08 � 0.02 0.12 � 0.02 n.d. n.s.ADCC (monocytes) [mg/mL] A431 0.03 � 0.015 0.07 � 0.02 0.16 � 0.05 n.s.ADCC (macrophages) [mg/mL] A431 0.38 � 0.12 0.27 � 0.16 0.13 � 0.03 n.s.AUC of serum levels during treatment [ng/mL�d] tg mice 18.9 � 6.1 431.4 � 105.8 — P � 0.0001AUC of serum levels during treatment [ng/mL�d] ntg mice 15.7 � 0.3 1066.0 � 13.0 482101.0 � 4715.6 P � 0.0001
Abbreviations: wb, whole blood; PMN, polymorphonuclear cells; n.s., not significantly different; n.d., not detectable; AUC, area under the curve.
Lohse et al.
Cancer Res; 76(2) January 15, 2016 Cancer Research408
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232
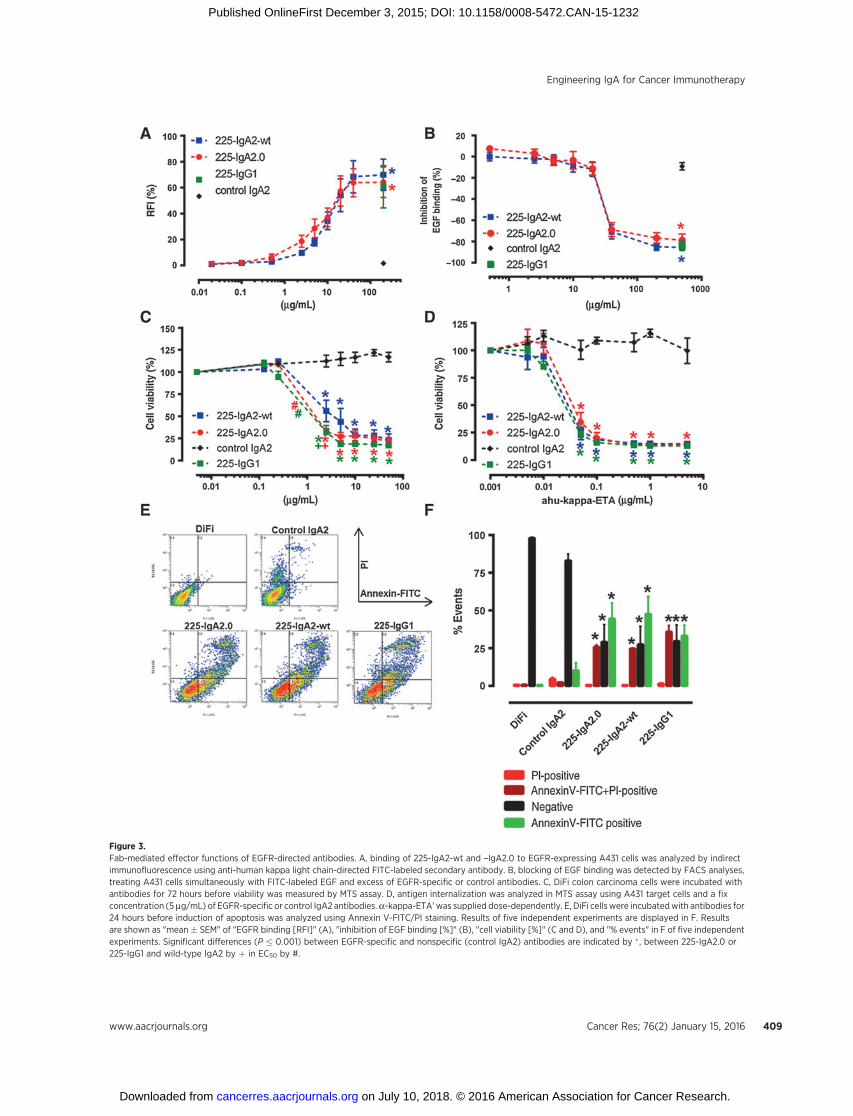
Figure 3.Fab-mediated effector functions of EGFR-directed antibodies. A, binding of 225-IgA2-wt and –IgA2.0 to EGFR-expressing A431 cells was analyzed by indirectimmunofluorescence using anti-human kappa light chain-directed FITC-labeled secondary antibody. B, blocking of EGF binding was detected by FACS analyses,treating A431 cells simultaneously with FITC-labeled EGF and excess of EGFR-specific or control antibodies. C, DiFi colon carcinoma cells were incubated withantibodies for 72 hours before viability was measured by MTS assay. D, antigen internalization was analyzed in MTS assay using A431 target cells and a fixconcentration (5 mg/mL) of EGFR-specific or control IgA2 antibodies.a-kappa-ETA' was supplied dose-dependently. E, DiFi cellswere incubatedwith antibodies for24 hours before induction of apoptosis was analyzed using Annexin V-FITC/PI staining. Results of five independent experiments are displayed in F. Resultsare shown as "mean� SEM" of "EGFR binding [RFI]" (A), "inhibition of EGF binding [%]" (B), "cell viability [%]" (C and D), and "% events" in F of five independentexperiments. Significant differences (P � 0.001) between EGFR-specific and nonspecific (control IgA2) antibodies are indicated by � , between 225-IgA2.0 or225-IgG1 and wild-type IgA2 by þ in EC50 by #.
Engineering IgA for Cancer Immunotherapy
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 409
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232
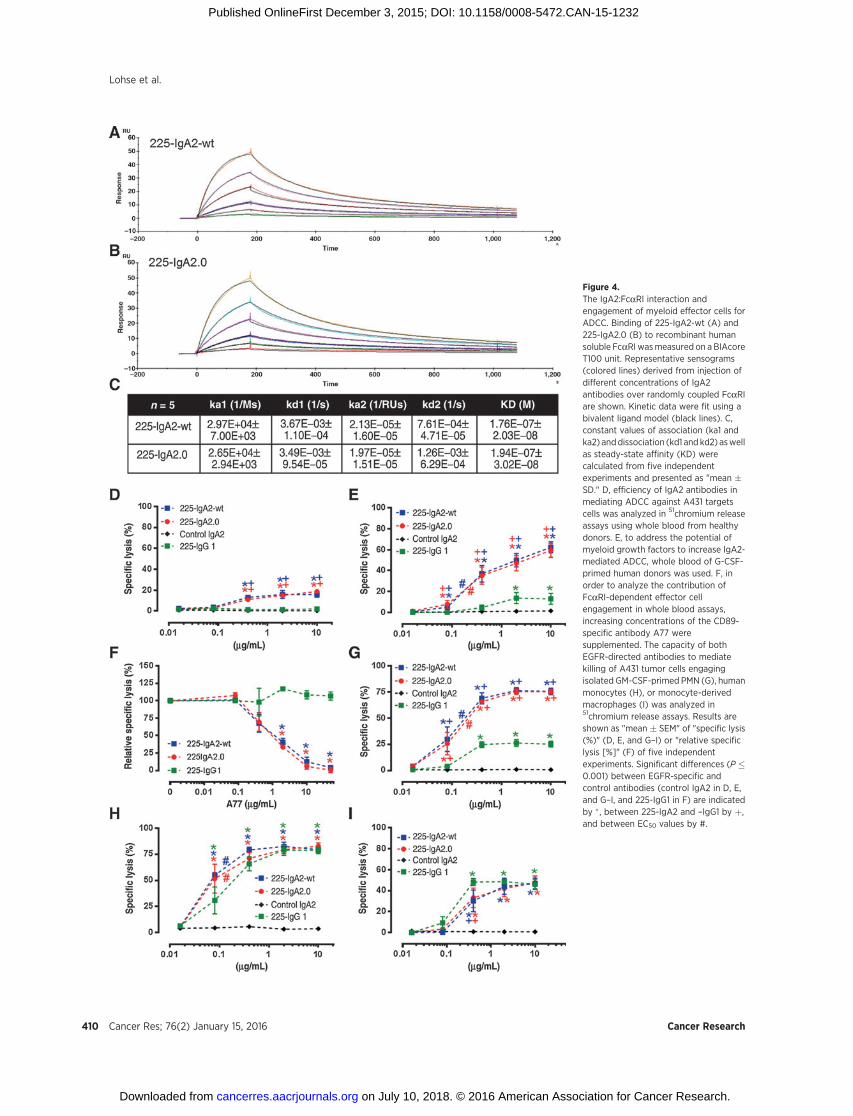
Figure 4.The IgA2:FcaRI interaction andengagement of myeloid effector cells forADCC. Binding of 225-IgA2-wt (A) and225-IgA2.0 (B) to recombinant humansoluble FcaRI wasmeasured on a BIAcoreT100 unit. Representative sensograms(colored lines) derived from injection ofdifferent concentrations of IgA2antibodies over randomly coupled FcaRIare shown. Kinetic data were fit using abivalent ligand model (black lines). C,constant values of association (ka1 andka2) anddissociation (kd1 and kd2) aswellas steady-state affinity (KD) werecalculated from five independentexperiments and presented as "mean �SD." D, efficiency of IgA2 antibodies inmediating ADCC against A431 targetscells was analyzed in 51chromium releaseassays using whole blood from healthydonors. E, to address the potential ofmyeloid growth factors to increase IgA2-mediated ADCC, whole blood of G-CSF-primed human donors was used. F, inorder to analyze the contribution ofFcaRI-dependent effector cellengagement in whole blood assays,increasing concentrations of the CD89-specific antibody A77 weresupplemented. The capacity of bothEGFR-directed antibodies to mediatekilling of A431 tumor cells engagingisolated GM-CSF-primed PMN (G), humanmonocytes (H), or monocyte-derivedmacrophages (I) was analyzed in51chromium release assays. Results areshown as "mean � SEM" of "specific lysis(%)" (D, E, and G–I) or "relative specificlysis [%]" (F) of five independentexperiments. Significant differences (P �0.001) between EGFR-specific andcontrol antibodies (control IgA2 in D, E,and G–I, and 225-IgG1 in F) are indicatedby � , between 225-IgA2 and –IgG1 by þ,and between EC50 values by #.
Lohse et al.
Cancer Res; 76(2) January 15, 2016 Cancer Research410
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

in 51chromium release assays using increasing antibody concen-trations and whole blood as a source of effector cells (Fig. 4D). Inthese assays, 225-IgA2.0 and 225-IgA2-wt were similarly effectivein tumor cell killing, whereas cetuximab was not effective(Table 1). To address the potential of myeloid growth factors toenhance IgA2-mediated ADCC, whole blood of G-CSF-primeddonors was used in ADCC assays (Fig. 4E). Both EGFR-directedIgA2 antibodiesmediated significant lysis of A431 tumor cells andwere similarly effective. The contribution of effector cell recruit-ment in whole blood ADCC assays was investigated by supple-menting the FcaRI-specific murine antibody A77. ADCC medi-ated by both EGFR-specific IgA2 antibodies was significantlyinhibited using increasing concentrations of A77, whereasIgG1-mediated lysis was not affected (Fig. 4F). Subsequently, weisolated FcaRI-expressingmyeloid effector cells from the blood ofhealthy donors. In ADCC assays using A431 target cells and PMN(Table 1) and GM-CSF-primed PMN (Fig. 4G), monomeric 225-IgA2-wt and 225-IgA2.0 triggered similarly effective ADCC. InADCC assays employing the colon carcinoma cell line DiFi, theglioblastoma cell line A1207, or the esophageal squamous car-cinoma cell line Kyse30 both EGFR-specific IgA2 antibodieswere similarly effective. In contrast, cetuximab did not engagegranulocytes for ADCC against any of these target cells (Table 1).Both IgA2 antibodies were even more effective using GM-CSFprimed compared with nonprimed PMN, as confirmed by EC50
values (Table 1). Next, monocytes, which were isolated usingCD14-specific magnetic beads (Fig. 4H), and human monocyte–derived macrophages (Fig. 4I) were analyzed as effector cells inovernight ADCC assays. In these experiments, both 225-IgA2antibodies were similarly effective in mediating ADCC againstA431 target cells by activating monocytes or macrophages fortumor cell killing (Table 1).
In vivo efficacy and pharmacokinetic properties of EGFR-directed IgA2 antibodies
The efficacy of EGFR-directed antibodies was first evaluatedin a syngeneic short-term i.p. model employing equal amountsof wild-type or engineered IgA2. One group of mice waspretreated with Ly6G-specific antibody to deplete granulocytesand monocytes. Briefly, BaF3 cells negative or positive for hu-man EGFR (Supplementary Fig. S1) were labeled with 0.125and 1 mmol/L carboxyfluorescein diacetate succinimidyl ester(CFSE; ref. 30), respectively, and were then injected i.p. intoBalb/c mice ntg or tg for human FcaRI (Fig. 5A). After 16 hours,cells were recovered by peritoneal lavage and the relativeamount of BaF3 cells was counted by calibrated flow cytometry,and the ratio of EGFR-negative to EGFR-positive cells wasdetermined. Both IgA2 antibodies significantly decreased theamount of EGFR-expressing BaF3 cells in ntg mice with asignificant increment in efficacy if human FcaRI was presentin tg mice (Fig. 5B). Pretreatment with Ly6G-specific antibody 2days before injection of tumor cells had no effect on the efficacyof 225-IgA2.0, although the treatment did efficiently deplete allneutrophils and most monocytes (data not shown). Impor-tantly, this was in line with previous data demonstrating thatperitoneal macrophages but neither granulocytes nor mono-cytes are responsible for the efficacy of IgA antibodies in thismodel (30). Next, a long-term peritoneal tumor model in SCIDmice, tg or ntg for human FcaRI, was employed to furtherevaluate the in vivo efficacy of the novel IgA antibody (Fig. 5C).A431-luc2 tumor cells were administered at day 1 and allowed
to grow until day 7 when EGFR-directed antibodies wereapplied (1 � 50 mg of 225-IgG1) or daily from day 7 to day16 (10� 50 mg of IgA antibodies) to compensate for the shorterserum half-life of IgA antibodies compared with IgG1. Growthof tumor cells was detected using BLI at indicated time points.Previous studies demonstrated that 225-IgG1 was similarlyeffective in FcaRI-tg and ntg mice. In ntg mice, 225-IgA2.0 butnot the 225-IgA2-wt antibody blocked tumor outgrowth sig-nificantly, but the 225-IgG1 antibody was more effective thanboth IgA2 antibodies (Fig. 5D, left). However, in human FcaRItg mice the efficacy of the engineered 225-IgA2.0 was signifi-cantly enhanced (Fig. 5D, right), indicating that FcaRI-positivemyeloid effector cells contribute to the therapeutic efficacy of225-IgA2.0 in transgenic animals. However, 225-IgA2-wt wasnot effective neither in tg nor in ntg mice (Fig. 5D). In Fig. 5E,representative BLI images are shown. Furthermore, ntg as wellas tg mice treated with 225-IgA2.0 survived significantly longerthan those treated with 225-IgA2-wt (Fig. 5F). In order tofurther investigate the immunotherapeutic activity of IgA2.0in immune competent mice, we evaluated if the engineeredIgA antibody was able to prevent tumor engraftment in asyngeneic long-term tumor model in BL6 mice (Fig. 5G). Forthis purpose, B16F10 cells, transfected to express luciferase andhuman EGFR (Supplementary Fig. S1), were injected i.v. intothe tail veins of FcaRI-tg BL6 mice. Mice were then treated withPBS or 225-IgA2.0 (50 mg/day) from days 0 to 9. Growth oftumor cells was imaged using BLI at indicated time points(Fig. 5H). Engraftment of tumor cells was significantly delayedin FcaRI-tg mice treated with the engineered 225-IgA2.0,whereas in PBS-treated control mice tumors grew out morerapidly (Fig. 5H and I). Mice were sacrificed at day 26 to deter-mine lung scores by visually scoring the number and size oflung metastases. In line with the BLI data, mice treated with225-IgA2.0 displayed a significantly decreased number and sizeof lung metastases (Fig. 5J). Two representative lungs aredisplayed in Fig. 5K. Together, IgA2.0 demonstrated therapeuticactivity in three in vivo models against tumor cells expressingdifferent levels of EGFR. Importantly, the engineered IgA anti-body was more effective than wt IgA in the long-term i.v. butnot in the short-term i.p. model, suggesting that phamacoki-netic differences may contribute to its enhanced activity.
Glycosylation and pharmacokinetic properties of EGFR-directed antibodies
Previous studies indicated that the serum half-life of recombi-nant IgA antibodies is affected by their glycosylation and by rapidASGPR-dependent hepatic clearance. Thus, we investigated if thereduced number of N-glycosylation sites of the 225-IgA2.0 lead toan altered global glycosylation profile. For this purpose, wild-typeand engineered IgA2 were separated on SDS-PAGE, transferredonto polyvinylidene difluoride (PVDF) membranes, and stainedwith direct blue 71 (Supplementary Fig. S2). The respective IgA-bands were cut out, N-glycans were enzymatically released usingPNGase F, reduced, and subsequently analyzed in-depth by PGCnano-Liquid Chromatography-ESI MS/MS glycoprofiling (Fig. 6Aand B). The systematic removal of two N-glycosylation sites in225-IgA2.0 resulted inmajor glycosylation differences in terms ofsialylation (and subsequently terminal galactoses) andoligoman-nosidic structures (Fig. 6A). In total, 86 different N-glycan struc-tures (present in 50 different compositions) were identified(Supplementary Tables S2 and S3). 225-IgA2.0 displayed an
Engineering IgA for Cancer Immunotherapy
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 411
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232
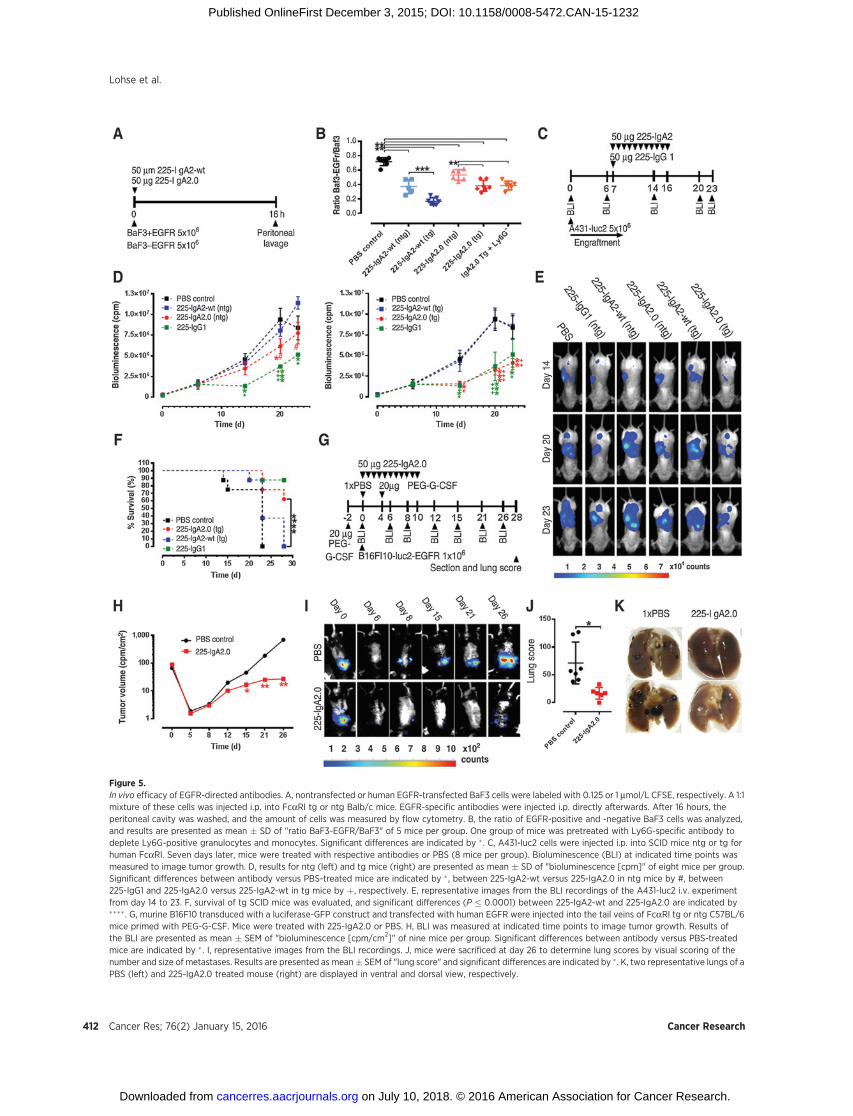
Figure 5.In vivo efficacy of EGFR-directed antibodies. A, nontransfected or human EGFR-transfected BaF3 cells were labeled with 0.125 or 1 mmol/L CFSE, respectively. A 1:1mixture of these cells was injected i.p. into FcaRI tg or ntg Balb/c mice. EGFR-specific antibodies were injected i.p. directly afterwards. After 16 hours, theperitoneal cavity was washed, and the amount of cells was measured by flow cytometry. B, the ratio of EGFR-positive and -negative BaF3 cells was analyzed,and results are presented as mean � SD of "ratio BaF3-EGFR/BaF3" of 5 mice per group. One group of mice was pretreated with Ly6G-specific antibody todeplete Ly6G-positive granulocytes and monocytes. Significant differences are indicated by � . C, A431-luc2 cells were injected i.p. into SCID mice ntg or tg forhuman FcaRI. Seven days later, mice were treated with respective antibodies or PBS (8 mice per group). Bioluminescence (BLI) at indicated time points wasmeasured to image tumor growth. D, results for ntg (left) and tg mice (right) are presented as mean � SD of "bioluminescence [cpm]" of eight mice per group.Significant differences between antibody versus PBS-treated mice are indicated by � , between 225-IgA2-wt versus 225-IgA2.0 in ntg mice by #, between225-IgG1 and 225-IgA2.0 versus 225-IgA2-wt in tg mice by þ, respectively. E, representative images from the BLI recordings of the A431-luc2 i.v. experimentfrom day 14 to 23. F, survival of tg SCID mice was evaluated, and significant differences (P � 0.0001) between 225-IgA2-wt and 225-IgA2.0 are indicated by���� . G, murine B16F10 transduced with a luciferase-GFP construct and transfected with human EGFR were injected into the tail veins of FcaRI tg or ntg C57BL/6mice primed with PEG-G-CSF. Mice were treated with 225-IgA2.0 or PBS. H, BLI was measured at indicated time points to image tumor growth. Results ofthe BLI are presented as mean � SEM of "bioluminescence [cpm/cm2]" of nine mice per group. Significant differences between antibody versus PBS-treatedmice are indicated by � . I, representative images from the BLI recordings. J, mice were sacrificed at day 26 to determine lung scores by visual scoring of thenumber and size of metastases. Results are presented as mean� SEM of "lung score" and significant differences are indicated by � . K, two representative lungs of aPBS (left) and 225-IgA2.0 treated mouse (right) are displayed in ventral and dorsal view, respectively.
Lohse et al.
Cancer Res; 76(2) January 15, 2016 Cancer Research412
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232
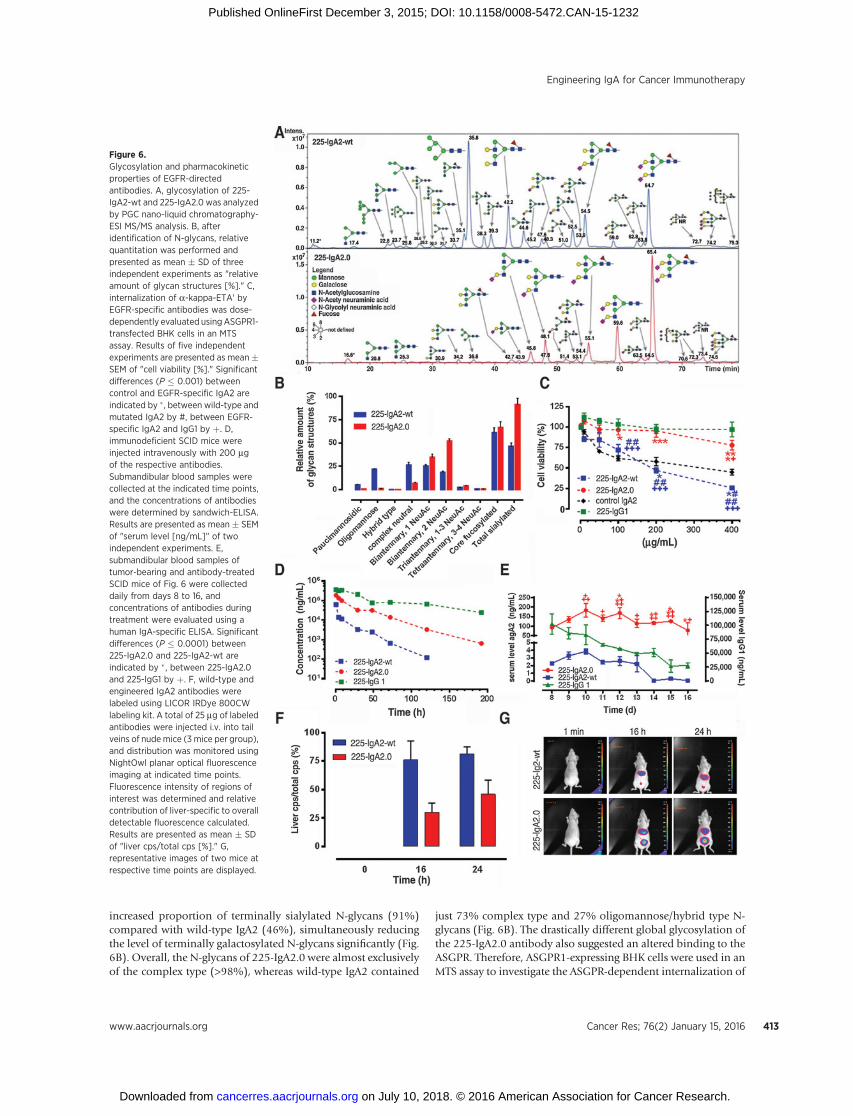
increased proportion of terminally sialylated N-glycans (91%)compared with wild-type IgA2 (46%), simultaneously reducingthe level of terminally galactosylated N-glycans significantly (Fig.6B). Overall, the N-glycans of 225-IgA2.0 were almost exclusivelyof the complex type (>98%), whereas wild-type IgA2 contained
just 73% complex type and 27% oligomannose/hybrid type N-glycans (Fig. 6B). The drastically different global glycosylation ofthe 225-IgA2.0 antibody also suggested an altered binding to theASGPR. Therefore, ASGPR1-expressing BHK cells were used in anMTS assay to investigate the ASGPR-dependent internalization of
Figure 6.Glycosylation and pharmacokineticproperties of EGFR-directedantibodies. A, glycosylation of 225-IgA2-wt and 225-IgA2.0 was analyzedby PGC nano-liquid chromatography-ESI MS/MS analysis. B, afteridentification of N-glycans, relativequantitation was performed andpresented as mean � SD of threeindependent experiments as "relativeamount of glycan structures [%]." C,internalization of a-kappa-ETA' byEGFR-specific antibodies was dose-dependently evaluated usingASGPR1-transfected BHK cells in an MTSassay. Results of five independentexperiments are presented as mean�SEM of "cell viability [%]." Significantdifferences (P � 0.001) betweencontrol and EGFR-specific IgA2 areindicated by � , between wild-type andmutated IgA2 by #, between EGFR-specific IgA2 and IgG1 by þ. D,immunodeficient SCID mice wereinjected intravenously with 200 mgof the respective antibodies.Submandibular blood samples werecollected at the indicated time points,and the concentrations of antibodieswere determined by sandwich-ELISA.Results are presented as mean � SEMof "serum level [ng/mL]" of twoindependent experiments. E,submandibular blood samples oftumor-bearing and antibody-treatedSCID mice of Fig. 6 were collecteddaily from days 8 to 16, andconcentrations of antibodies duringtreatment were evaluated using ahuman IgA-specific ELISA. Significantdifferences (P � 0.0001) between225-IgA2.0 and 225-IgA2-wt areindicated by � , between 225-IgA2.0and 225-IgG1 by þ. F, wild-type andengineered IgA2 antibodies werelabeled using LICOR IRDye 800CWlabeling kit. A total of 25 mg of labeledantibodies were injected i.v. into tailveins of nudemice (3 mice per group),and distribution was monitored usingNightOwl planar optical fluorescenceimaging at indicated time points.Fluorescence intensity of regions ofinterest was determined and relativecontribution of liver-specific to overalldetectable fluorescence calculated.Results are presented as mean � SDof "liver cps/total cps [%]." G,representative images of two mice atrespective time points are displayed.
Engineering IgA for Cancer Immunotherapy
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 413
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

EGFR-specific antibodies (Fig. 6C). Cells were incubated withdilutions of respective antibodies in the presence ofa-kappa-ETA'.Significant growth inhibitionwasmediatedbybothwild-type andcontrol IgA2 antibodies in a dose-dependentmanner, whereas the225-IgA2.0 and -IgG1 did not mediate target cell killing underthese conditions. Subsequently, the serum half-life of IgA anti-bodies was investigated by injecting 200 mg of 225-IgA2-wt, 225-IgA2.0, and 225-IgG1 i.v. into tail veins of SCID mice. Blood wascollected at different time points, and serum concentrations weremeasured by ELISA (Fig. 6D). These pharmacokinetic studiesrevealed a significantly increased serum half-life of 225-IgA2.0compared with 225-IgA2-wt. Actually, the rapid eliminationwithin the first 24 hours as well as the second elimination phaseduring the following days was significantly decelerated for 225-IgA2.0 compared with 225-IgA2-wt. Although the serum half-life of 225-IgA2.0 was significantly improved, there was still adifference to 225-IgG1, of which prolonged serum levels werelikely maintained by FcRn-mediated recycling. Next, we ana-lyzed the serum levels of EGFR-directed antibodies duringtreatment in A431-luc2 tumor-bearing SCID mice of Fig. 5.For this purpose, submandibular blood samples were collecteddaily during treatment, and serum levels of therapeutic anti-bodies were evaluated from day 8 to day 16 using human Ig-specific Sandwich ELISA (Fig. 6E). Results demonstrated asustained higher concentration of 225-IgA2.0 during treatmentcompared with 225-IgA2-wt in transgenic and in nontransgenicmice (Fig. 6E, Table 1). Nevertheless, the serum levels of therespective 225-IgG1 were significantly higher than those ofboth 225-IgA2-wt and 225-IgA2.0 (Fig. 6E, Table 1). However,there was a significant decline in antibody concentrationsdetectable for 225-IgA2-wt and 225-IgG1, whereas the engi-neered 225-IgA2.0 demonstrated sustained high serum levels(Fig. 6E, Table 1). Next, fluorescently labeled IgA antibodieswere injected into nude mice. Planar optical whole body andsingle organ fluorescence imaging indicated enhanced hepaticfluorescence for wild-type IgA2 compared with IgA2.0-treatedmice (Fig. 6F and G; ref. 40). Thus, the significantly lower levelsof terminal galactoyslation in IgA2.0 compared with wt IgAreduced hepatic up-take via the ASGPR, leading to the extendedserum half-life of the engineered IgA antibody.
DiscussionAntibody isotypes for tumor immunotherapy
Human IgG1 is by far the most commonly selected antibodyisotype in tumor immunotherapy (41). This decision is basedon the well documented activity of human IgG1 to mediatecomplement-dependent cytotoxicity and to activate NK cellsand monocytes/macrophages for ADCC (42) as well as itsprolonged FcRn-mediated serum half-life (32). Furthermore,production and purification technologies are well establishedfor human IgG1 antibodies (43). However, in vitro studiesdocumented that myeloid effector cells—and PMN in particu-lar—were more effective in ADCC by IgA than by IgG1 anti-bodies (11–13). Human IgA antibodies demonstrated signifi-cant antitumor activity in huFcaRI-transgenic mice, which arerequired to investigate Fc-mediated effector functions of thisantibody isotype in vivo, because mice do not express a func-tional FcaRI orthologue (30, 44, 45). However, the serum half-life of recombinant human IgA antibodies in mice was unex-pectedly short—probably due to rapid clearance by the ASGPR
(30, 31) and the lack of binding to FcRn. This hepaticallyexpressed receptor is known to mediate internalization anddegradation of proteins with terminally exposed galactose (33),which was present on the majority of our previous IgA pre-parations (30). These results suggested engineering of an IgAantibody with the aim to improve its pharmacokinetic prop-erties and to evaluate its therapeutic activity.
Engineering of an EGFR-directed IgA2m(1) antibodyWhen we started to engineer IgA antibodies for tumor immu-
notherapy, we decided to improve an IgA2m(1) antibody, whichis the most common Caucasian IgA2 allotype (22). The decisionto use IgA2 rather than IgA1 is based on a number of reasons: IgA2lacks the elongated hinge of IgA1, which is associated withpotential disadvantages. For example, the IgA1 hinge region istargeted by bacterial proteases and contains difficult to control O-glycans, which are involved in the pathogenesis of IgA nephrop-athy (14). Furthermore, IgA2 proved significantly more effectivein ADCC than IgA1 andhas a prolonged serumhalf-life comparedwith IgA1 (11, 30). First,we introduced aP221Rmutation into theIga HC, which enabled a covalent linkage between LCs and HCs(13). Additionally, this mutation increased the thermal and long-term stability of the antibody. Next, the removal of the free andaccessible thiol group in the tail piece further enhanced thestability, as it prevented the formation of dimeric aggregates andpotential complexes with other serum proteins (35). The addi-tional mutations described in this manuscript increased produc-tivity and thermal stability as relevant pharmaceutical properties.Except for an yet unexplained difference in growth inhibition ofDiFi cells (Fig. 3C), Fab- or Fc-mediated effector functions in vitrowere not different betweenwild-type and engineered IgA2 (Figs. 3and 4). Importantly, however, both molecules differed signifi-cantly in their glycosylation patterns.
The glycosylation of IgA antibodies is closely related to theirpharmacokinetic properties (30), suggesting that glyco-engineer-ing strategies may improve their therapeutic efficacy. By compar-ing sequences and structures of different IgA iso- and allotypes,deletion of the N-glycans at positions 166 and 337 appeared as arational approach to reduce the overall level of glycosylation,because these sites are not conserved in IgA1. Interestingly, thesemutations resulted in an altered global glycosylation profile. Itwill require further in-depth studies to elucidate whether thiseffect is caused by minor protein structure alterations making thesites of glycosylation better accessible to the glycan-modifyingenzymes of the endoplasmatic reticulum and Golgi network, orwhether the oligomannose and less sialylated N-glycans arespecific for glycosylation sites Asn166 and Asn337 in the wild-type IgA2 protein expressed in CHO cells. Whether these altera-tions in glycosylation also affected antibody productivity orsecretion would require additional studies. Potentially, thereduced number of N-glycans led to an accelerated posttransla-tional processing and thereby enhanced production.However, wecannot exclude that transfection or clone effects (higher gene copynumber, gene amplification, and genomic loci of insertion) mayhave altered the production rates of the antibodies. Importantly,our data indicate that the increased terminal sialylation (whichmasks ASGPR recognized glyco-epitopes) of the engineered 225-IgA2.0 considerably delayed the clearance of IgA antibodies inmice—thereby increasing the otherwise short half-life of recom-binant IgA compared with IgG antibodies (30). Hence, this reportfurther confirms the important role of glycosylation and hepatic
Cancer Res; 76(2) January 15, 2016 Cancer Research414
Lohse et al.
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

clearance by the ASGPR for the pharmacokinetics of IgA anti-bodies (30, 31). Although the engineering strategy describedabove resulted in altered glycosylation and enhanced pharmaco-kinetic properties of the IgA2.0 antibody, additional improve-ments appear reasonable. For example, C-terminal fusion ofhuman albumin or FcRn-binding motifs to IgA antibodies, orthe construction of IgG/A hybrid antibodies (46). Also, furtherglyco-engineering strategies or the combination of these novelapproaches may further enhance the serum half-life and thera-peutic efficacy of IgA antibodies (47).
Engagement of myeloid effector cellsMyeloid effector cells are considered as important effector cells
in cancer and in cancer immunotherapy. Depending on themicroenvironment, tumor-associated myeloid cells switch fromtumor-promoting to tumor-preventing cells (8–10, 48, 49). Thus,myeloid cells constitute a numerous population of powerfuleffector cells, which are present at many tumor sites and whichcould possibly be recruited by tumor directed antibodies to killmalignant cells (13, 30, 35). Several studies have previouslyreported that FcaRI (CD89) is a potent triggermolecule to activatethese effector cells in vitro and also in vivo using FcaRI-transgenicmice expressing FcaRI on myeloid cells (11–13, 30, 35). Further-more, especially IgA2 antibodies were strong activators to inducemyeloid effector cell-mediated tumor cell lysis, and co- or pre-stimulation with GM-CSF and G-CSF, respectively, could fur-ther enhance their efficacy (11). In this manuscript we describethe development of an engineered IgA2 antibody, which was aseffective as wild-type IgA2 in engaging myeloid effector cells forADCC in vitro against cell lines derived from different tumorentities. In a long-term xenogeneic model, the engineered 225-IgA2.0 was effective employing FcaRI-transgenic or nontrans-genic mice, indicating recruitment of both Fc- and Fab-medi-ated effector functions (Fig. 5D–F). However, there was a clearincrement in its efficacy if myeloid effector cell engagement viaFcaRI-interaction was enabled in human FcaRI transgenicmice. In addition, the application of the engineered 225-IgA2.0prevented engraftment and reduced the number of syngeneictumor cells in FcaRI transgenic immune competent C57BL/6and Balb/c mice, respectively. Thus, the engineered IgA anti-body demonstrated significant in vivo efficacy against differenttarget cells. Furthermore, the efficacy of IgA antibodies in wild-type mice lacking FcaRI in the short-term model supports theirefficacy in Fab-mediated killing against low EGFR-expressingtumor cells.
Because there was no difference in Fab- or Fc-mediated effectormechanisms between wild-type and engineered IgA2 in vitro, theincreased efficacy of the mutated IgA2 compared with wild-typeIgA2 in the long-term tumor model in vivo could be explained bythe sustained higher concentrations during treatment in trans-genic as well as nontransgenic mice, whereas wild-type IgA2 wasrapidly cleared. According to a previous report, where in vivomechanisms of EGFR-directed antibodies were shown to dependon local antibody concentrations (50), we may argue that thehigher serum levels of 225-IgA2.0 compared with wild-type IgA2may constitute more appropriate conditions for the engineeredantibody to induce growth inhibition or signaling abrogation innontransgenic mice and ADCC in FcaRI transgenic mice, respec-tively. Thus, the prolonged serum half-life of IgA2.0 led to atherapeutic benefit in a long-term, but not in short-term treatmentmodels, in which pharmacokinetic properties apparently did not
contribute to therapeutic efficacy. Nevertheless, serum levels ofIgG were multiple times higher than those of the respectiveengineered IgA2 antibody although higher amounts of the latterwere applied, although both demonstrated similar in vivo activityunder these conditions. Further studies are required to evaluatethe relation between the serum half-life of recombinant IgAantibodies and local antibody concentrations, which are sufficientfor preventing tumor growth in vivo. Noteworthy, antibody iso-type comparisons in mice are difficult to translate into humansbecause, e.g., FcRn binding, Fc receptor biology, and effector cellfunctions are all very different between men and mice.
In conclusion, an engineered IgA antibody with improvedpharmacokinetic properties demonstrated enhanced efficacy invivo by employing both Fc- and Fab-mediated effector func-tions. Engagement of myeloid effector cells via FcaRI consti-tutes a promising approach for immunotherapy against EGFR-expressing tumors. Furthermore, our results demonstrate thatthe presented molecule is a novel and innovative antibodyformat that overcomes some of the limitations (production,stability, glycosylation, serum half-life, and in vivo efficacy)of previous IgA antibodies. Thus, these results further supportthe concept of introducing IgA antibodies into clinicaldevelopment.
Disclosure of Potential Conflicts of InterestP. Sondermann is a CSO at SuppreMol GmbH. No potential conflicts of
interest were disclosed by the other authors.
Authors' ContributionsConception and design: S. Lohse, L.A.P.M. Meulenbroek, S. Derer, T. ValeriusDevelopment of methodology: S. Lohse, U. M€oginger, D. Kolarich, T. ValeriusAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): S. Lohse, S. Meyer, L.A.P.M. Meulenbroek,J.H.M. Jansen, A. Kretschmer, U. M€oginger, S. Tiwari, D. KolarichAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): S. Lohse,U.M€oginger, D. Schewe,D. Kolarich, J.H.W.Leusen, T. ValeriusWriting, review, and/or revision of the manuscript: S. Lohse, J.H.M. Jansen,K. Klausz, U. M€oginger, S. Derer, T. R€osner, C. Kellner, D. Schewe, P. Sonder-mann, D. Kolarich, M. Peipp, J.H.W. Leusen, T. ValeriusAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): S. Lohse, M. Nederend, T. ValeriusStudy supervision: S. Lohse, S. Derer, T. ValeriusOther (support in engineering the IgA molecule): P. Sondermann
AcknowledgmentsThe authors thank the excellent technical assistance from Kathinka T€uxen,
Christyn Wildgrube, Yasmin Brodtmann, the group of Prof. Dr. Axel Scheidig(Section of Structural Biology, Centre of Molecular Biosciences, Christian-Albrechts-University, Kiel), and the group of Prof. Dr. Susanne Sebens (Depart-ment of InternalMedicine I, Inflammatory Carcinogenesis, Christian-Albrechts-University, Kiel) for their help with BIAcore and Real Time PCR instruments,respectively.
Grant SupportThis work was supported by the German Research Organization (Lo 1853/1-
1, Va 124/7-2), the Wilhelm Sander-Foundation (2009.098.1 and 2), the MaxPlanck Society, European Union Seventh Framework Program (grant numberPCIG09-GA-2011-293847), and by intramural funding from the Christian-Albrechts-University.
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received May 6, 2015; revised August 17, 2015; accepted September 7, 2015;published OnlineFirst December 3, 2015.
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 415
Engineering IgA for Cancer Immunotherapy
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

References1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell
2011;145:646–74.2. Mendelsohn J, Baselga J. Status of epidermal growth factor receptor
antagonists in the biology and treatment of cancer. J Clin Oncol2003;21:2787–99.
3. Pao W, Miller VA. Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowl-edge and future directions. J Clin Oncol 2005;2:e73.
4. Baselga J. Targeting tyrosine kinases in cancer: the second wave. Science2006;312:1175–8.
5. Schmitz KR, Ferguson KM. Interaction of antibodies with ErbB receptorextracellular regions. Exp Cell Res 2009;315:659–70.
6. Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van BerkelPH, Derer S, et al. Human IgG2 antibodies against epidermal growth factorreceptor effectively trigger antibody-dependent cellular cytotoxicity but, incontrast to IgG1, only by cells of myeloid lineage. J Immunol 2010;184:512–20.
7. Price TJ, Peeters M, Kim TW, Cascinu S, Ruff P, Suresh AS, et al. Panitu-mumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised,multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol2014;15:569–79.
8. Uchida J, Hamaguchi Y, Oliver JA, Ravetch JV, Poe JC, Haas KM, et al. Theinnate mononuclear phagocyte network depletes B lymphocytes throughFc receptor- dependent mechanisms during anti-CD20 antibody immu-notherapy. J Exp Med 2004;199:1659–69.
9. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms totherapy. Immunity 2014;41:49–61.
10. Albanesi M, Mancardi DA, J€onsson F, Iannascoli B, Fiette L, Di Santo JP,et al. Neutrophils mediate antibody-induced antitumor effects in mice.Blood 2013;122:3160–4.
11. Dechant M, Beyer T, Schneider-Merck T, Weisner W, Peipp M, van deWinkel JGJ, et al. Effector mechanisms of recombinant IgA antibodiesagainst EGFR. J Immunol 2007;179:2936–43.
12. Huls G, Heijnen IA, Cuomo E, van der Linden J, Boel E, van de Winkel JG,et al. Antitumor immune effector mechanisms recruited by phage display-derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res1999;59:5778–84.
13. Lohse S, Brunke C, Derer S, Peipp M, Boross P, Kellner C, et al. Charac-terization of a mutated IgA2 antibody of the m(1) allotype against theepidermal growth factor receptor as novel cytotoxic triggermolecule for therecruitment of monocytes and macrophages. J Biol Chem 2012;287:25139–50.
14. Woof JM, RussellMW. Structure and function relationships in IgA.MucosalImmunol 2011;4:590–7.
15. Monteiro RC, van de Winkel JG. IgA Fc receptors. Annu Rev Immunol2003;21:177–204.
16. Woof JM, Burton DR. Human antibody-Fc receptor interactions illumi-nated by crystal structures. Nat Rev Immunol 2004;4:89–99.
17. Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, et al.The glycosylation and structure of human serum IgA1, Fab, and Fc regionsand the role of N-glycosylation on Fca receptor interactions. J Biol Chem1998;273:2260–72.
18. GomesMM,Wall SB, Takahashi K, Novak J, RenfrowMB,Herr AB. Analysisof IgA1 N-glycosylation and its contribution to FcaRI binding. Biochem-istry 2008;47:11285–99.
19. Xue J, Zhao Q, Zhu L, Zhang W. Deglycosylation of FcaR at N58 increasesit's binding to IgA. Glycobiology 2010;20:905–15.
20. Ferrara C, Stuart F, Sondermann P, Br€unker P, Uma~na P. The carbohydrateat FcgRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J Biol Chem 2006;281:5032–6.
21. Kawamura S, Saitou N, Ueda S. Concerted evolution of the primateimmunoglobulin alpha-gene through gene conversion. J Biol Chem1992;267:7359–67.
22. Chintalacharuvu KR, Raines M, Morrison SL. Divergence of human alpha-chain constant region sequences. A novel recombinant alpha 2 gene. JImmunol 1994;152:5299–304.
23. Wurm FM. Production of recombinant protein therapeutics in cultivatedmammalian cells. Nat Biotechnol 2004;22:1393–8.
24. Kiryluk K,Novak J. The genetics and immunobiology of IgA nephropathy. JClin Invest 2014;124:2325–32.
25. Furtado PB, Whitty PW, Robertson A, Eaton JT, Almogren A, Kerr MA, et al.Solution structure determination of monomeric human IgA2 by X-ray andneutron scattering, analytical ultracentrifugation and constrained model-ing: a comparison with monomeric human IgA1. J Mol Biol 2004;338:921–41.
26. Chintalacharuvu KR, Yu LJ, BholaN, Kobayashi K, FernandezCZ,MorrisonSL. Cysteine residues required for the attachment of the light chain inhuman IgA2. J Immunol 2002;169:5072–7.
27. Tsuzukida Y, Wang CC, Putnam FW. Structure of the A2m(1) allotype ofhuman IgA - a recombinant molecule. Proc Natl Acad Sci U S A 1979;76:1104–8.
28. Johansen FE, Braathen R, Brandtzaeg P. The J chain is essential forpolymeric Ig receptor-mediated epithelial transport of IgA. J Immunol2001;167:5185–92.
29. Deshpande N, Jensen PH, Packer NH, Kolarich D. GlycoSpectrumScan:fishing glycopeptides from MS spectra of protease digests of humancolostrum sIgA. J Proteome Res 2010;9:1063–75.
30. Boross P, Lohse S,NederendM, Jansen JH, van TeteringG,DechantM, et al.IgA EGFR antibodies mediate tumor killing in vivo. EMBO Mol Med2013;5:1213–26.
31. Rifai A, Fadden K, Morrison SL, Chintalacharuvu KR. The N-glycansdetermine the differential blood clearance and hepatic uptake of humanimmunoglobulin IgA1 and IgA2 isotypes. J Exp Med 2000;191:2171–82.
32. Ghetie V, Ward ES. Multiple roles for the major histocompatibilitycomplex class I- related receptor FcRn. Annu Rev Immunol 2000;18:739–66.
33. Seested T, Nielsen HM, Christensen EI, Appa RS. The unsialylated sub-population of recombinant activated factor VII binds to the asialo-glyvo-protein receptor (ASGPR) on primary rat hepatocytes. Thromb Haemost2010;104:1166–73.
34. Ko SY, Pegu A, Rudicell RS, Yang ZY, Joyce MG, Chen X, et al. Enhancedneonatal Fc receptor function improves protection against primate SHIVinfection. Nature 2014;514:642–5.
35. Brunke C, Lohse S, Derer S, Peipp M, Boross P, Kellner C, et al. Impact of atail piece cysteine deletion on biochemical and functional properties of anepidermal growth factor receptor-directed IgA2m(1) antibody. mAbs2013;5:936–45.
36. Kolarich D, Rapp E, Struwe WB, Haslam SM, Zaja J, McBride R, et al. Theminimum information required for a glycomics experiment (MIRAGE)project: improving the standards for reporting mass-spectrometry-basedglycoanalytic data. Mol Cell Proteomics 2013;12:991–5.
37. Jensen PH, Karlsson NG, Kolarich D, Packer NH. Structural analysisof N- and O-glycans released from glycoproteins. Nat Protoc 2012;7:1299–310.
38. Kellner C, Bleeker WK, Lammerts van Bueren JJ, Staudinger M, Klausz K,Derer S, et al. Human kappa light chain targeted Pseudomonas exotoxin Aidentifying human antibodies and Fab fragments with favorable charac-teristics of antibody-drug conjugate development. J Immunol Methods2011;371:122–33.
39. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for auto-mated protein structure and function prediction. Nat Protoc 2010;5:725–38.
40. Filipe V, Que I, Carpenter JF, L€owik C, Jiskoot W. In vivo imaging ofaggregates after subcutaneous and intravenous injection in mice. PharmRes 2014;31:216–27.
41. Reichert JM. Antibodies to watch in 2014. MAbs 2014;6:5–14.42. Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis
R, et al. Comparison of the effector functions of human immunoglo-bulins using a matched set of chimeric antibodies. J Exp Med 1987;166:1351–61.
43. Liu HF, Ma J, Winter C, Bayer R. Recovery and purification processdevelopment for monoclonal antibody. mAbs 2010;2:480–99.
44. Pascal V, Laffleur B, Debin A, Cuvillier A, van EgmondM,Drocourt D, et al.Anti-CD20 IgA can protect mice against lymphoma development: evalu-ationof the direct impact of IgA and cytotoxic effector recruitment onCD20target cells. Haematologica 2012;97:1686–94.
Cancer Res; 76(2) January 15, 2016 Cancer Research416
Lohse et al.
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

45. Marouka T, Nagata T, Kasahara M. Identification of the rat IgA Fc receptorencoded in the leukocyte receptor complex. Immunogenetics 2004;55:712–6.
46. KeltonW,MehtaN, CharabW, Lee J, Lee CH, Kojima T, et al. IgGA: a "cross-isotype" engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol 2014;21:1603–9.
47. Sand KM, Dalhus B, Christianson GJ, Bern M, Foss S, Cameron J, et al.Dissection of the neonatal Fc receptor (FcRn)-albumin interface usingmutagenesis and anti-FcRn albumin-blocking antibodies. J Biol Chem2014;289:17228–39.
48. Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes inthe tumor microenvironment: mechanisms and consequences for tumorprogression. Semin Cancer Biol 2013;23:141–8.
49. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognosticsignificance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 2012;7:e50946.
50. BleekerWK, Lammerts van Bueren JJ, vanOjikHH,Gerristen AF, PluyterM,Houtkamp M, et al. Dual mode of action of a human anti-epidermalgrowth factor receptormonoclonal antibody for cancer therapy. J Immunol2004;173:4699–707.
www.aacrjournals.org Cancer Res; 76(2) January 15, 2016 417
Engineering IgA for Cancer Immunotherapy
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232

2016;76:403-417. Published OnlineFirst December 3, 2015.Cancer Res Stefan Lohse, Saskia Meyer, Laura A.P.M. Meulenbroek, et al.
In VivoMyeloid Effector Cell Engagement An Anti-EGFR IgA That Displays Improved Pharmacokinetics and
Updated version
10.1158/0008-5472.CAN-15-1232doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2015/12/03/0008-5472.CAN-15-1232.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/76/2/403.full#ref-list-1
This article cites 50 articles, 22 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/76/2/403.full#related-urls
This article has been cited by 1 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/76/2/403To request permission to re-use all or part of this article, use this link
on July 10, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst December 3, 2015; DOI: 10.1158/0008-5472.CAN-15-1232