Amino Acidss & Aminoacidopathies
55
Amino Acids & Aminoacidopathies 16/5/2015
-
Upload
mustafa-khandagawi -
Category
Documents
-
view
6 -
download
0
description
Amino Acidss & Aminoacidopathies
Transcript of Amino Acidss & Aminoacidopathies

Amino Acids & Aminoacidopathies
16/5/2015

• Amino acids are the building blocks of proteins, which have structural and functional properties.
• Proteins are polymers of amino acids covalently bonded in specific sequences.
• When amino acids are covalently linked to one another, this chain can twist and fold to form a unique three-dimensional structure that has a specific function.

• There are 20 commonly occurring amino acids that make up proteins, and the order of amino acids in proteins determines its structure and biological function.

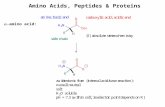
Amino Acid Structure
Each amino acid (except for proline) has:
1. A carboxyl group (-COO-) .
2. Protonated amino group (-NH3+) .
3. Side chain ("R-group") bonded to the α-carbon atom.
These carboxyl and amino groups are combined in peptide linkage.

• The carbon is also bonded to a hydrogen atom and a larger side chain. The side chain is unique for each amino acid

Amino Acids have two enantiomers
The L-amino acids are the building blocks for proteins. Some D-amino acids occur in nature, but not in proteins.

The 20 Amino acids are differ by their R group

organic compounds represented in the structures of different amino acids
1. Alkanes
2. Aromatics
3. Thioethers
4. Alcohols
5. Phenols
6. Thiols
7. Amides
8. Carboxylic acids
9. Amines

Essential amino acids
• There are 10 amino acids that are essential amino acids because they cannot be synthesized in the human body and must be obtained in the diet.
• Two of these amino acids, arginine and histidine, are essential in children, but not adults.

Essential amino acids – Arginine (Arg) : amine , cell division , wound healing– Histidine (His) : basic (imidazole), – Isoleucine (Ile): branched-chain, healing muscle cells– Leucine (Leu) : branched-chain, healing muscle cells– Lysine (Lys)– Methionine (Met): initiatetranslation of messenger
RNA– Phenylalanine (Phe): non polar , used by brain – Threonine (Thr)– Tryptophan (Trp)– Valine (Val): branched-chain, healing muscle cells

Metabolism of amino acids
• Essential amino acids should be provided by diet
• Other amino acid synthesized by the body or from essential amino acids
• Tyrosine produced from phenylalanine• Dietary proteins are completely digested into
amino acids by proteolytic enzymes

Overview of amino acids metabolism

Metabolism of amino acids
• Amino acids are then rapidly absorbed from the intestine into the blood and subsequently become part of the body’s pool of amino acids
• Amino acids are also released by the normal breakdown of body proteins
• The primary purpose of amino acids is for the synthesis of body proteins
• synthesis of nonprotein nitrogen-containing compounds such as purines, pyrimidines, porphyrins, creatine

Classification of amino acids
• based on the side chains:
1. Nonpolar amino acids
2. Polar amino acids, which are further divided into:• Neutral amino acids• Acidic amino acids• Basic amino acids

Non-polar amino acids
• Side chains consists entirely of carbon and hydrogen.
• Compounds composed of only carbon and hydrogen are nonpolar and hydrophobic.

Polar amino acids
• Contain functional groups, such as hydroxyl (–OH) and amide (–CONH2).
• Side chains can form hydrogen bonds with water.
• Hydrophilic (An exception is cysteine, which does not form hydrogen bonds)


Protein formation
• When two amino acids condense, a dipeptide is formed.
• The carboxylate ion (–COO-) of one amino acid reacts with the protonated amine (–NH3
+) of a second amino acid.
• A water molecule is lost and an amide functional group is formed. An amide bond is formed between the two amino acids.

Chapter 9 19© 2011 Pearson Education, Inc.
When amino acids combine in a condensation reaction, the amide bond that is formed between them is called a peptide bond.

• In this dipeptide, alanine is called the N-terminus because it has an unreacted -amino group.
• Valine is called the C-terminus because it has an unreacted -carboxylate group.

Aminoacidopathies• class of rare inherited errors of metabolism in
which there is an enzyme defect that inhibits the body’s ability to metabolize certain amino acids
• The abnormalities may involve– Activity of specific enzymes– Metabolic pathway– Or membrane transport system for amino acids

Aminoacidopathies
• These defects leads to accumulation of amino acids , its precursors or by products
• Excessive accumulation in blood or tissues leads to physical symptoms of the disease

Clinical Manifestation • Aminoaciduria • CNS dysfunction:
– Mental retardation– Seizure disorders– Behavioral disturbances
• Metabolic ketoacidosis• Occasionally:
– Liver disorders– Renal failure– Ocular lesions

Aminoacidopathies
Neurological • PKU• Homocyctinuria• Hypertyrosinemia• Maple-syrup urine disease• Hyperglycenemia• Methyle melonic aciduria• Citrullinemia• Carnosinemia
Hypopigmentation• Brown –syndrome• Albinism• Chediac Higashi syndrome

Phenyl Ketonuria (PKU)


Phenyleketoneuria• autosomal recessive disorder• Incidence rate 1 in 14000• Common compared to other
aminoacidopathy• Deficiency of phenylanaline
hydroxylase• Phenylalanine is metabolized via
alterative pathway leading to accumulation phenyle pyruvic acid (deamination)
• Accumulate in blood and urine and gives musty odor to it

Phenyleketoneuria
• Phenyl pyruvic acid is neurotoxic and in undiagnosed newborn can cause severe mental retardation due to brain damage, which starts in 2nd and 3rd week post birth.
• Early detection allows diet control therapy low in phenylalanine up to the age of 5-6yrs until normal metabolism develops, however low IQ levels have been reported after termination of special diet.
• Phenylalanine > 1200 µmole/l (upper limit 120 µmole/l)

Tests of PKU• Detection of phe (Screening ):
– Ferric chloride test– Guthrie test : bacterial inhibition assay for
phenylalanine that uses the ability of phenylalanine to facilitate bacterial growth in a culture medium with an inhibitor (Bacillus subtilis and -2-thienylalanine).
– Automated methods• Plasma level estimation (Confirmatory or
reference method)– Chromatography– Fluoremetric methods

Tests of PKU• principle of Guthrie test :
– Spores of organism Bacillus subtilisare incorporated into an agar plate containing β-2-thienylalanine (a metabolic antagonist to B. Subtilis).
– A filter paper disk, impregnated with dried blood sample is placed onto the agar
– If the blood phenylalanine level is higher than 2.5 mg/dl, the phenylalanine counteracts the antagonist and the bacteria grows.
– The sample should be collected prior to administration of antibiotics or transfusion of blood or blood products.

Tests of PKU• principle of Guthrie test :

Tests of PKU
• Genetic pre-natal diagnosis of PKU: – detection of carrier status in families with PKU
using DNA analysis– PKU is a complex polygenic disorder and results in
multiple independent mutations at the PAH locus– Screening for mutations using PAH gene probes

2. ALCAPTONURIA• AKU
• It is rare autosomal recessive inherited genetic disorder of phenylalanine and tyrosine metabolism
• defect in the HGD gene (Homogentisic acid oxidase deficiency)
• the body unable to properly break down certain amino acids (tyrosine and phenylalanine)
• homogentisic acid accumulates in the skin and other body tissues
– BLACK URINE– BLACK NAILS (OCHRONOSIS), SKIN– BLACK JOINT CARTILAGE (SEVERE ARTHRITIS)

Defect here causes Type I Tyrosinemia
Defect here causes alkaptonuria
Catabolic pathway for phenylalanine and tyrosine
Homogentisate dioxygenase
Fumarylacetoacetate hydrolase



Albinism
• Autosomal recessive • Result from loss of TYROSINASE enzyme in
skin which converts tyr to DOPA and DOPA to melanin pigments

Tyrosinemia • Tyrosine is an amino acid which is found in most
animal and plant proteins. The metabolism of tyrosine in humans takes place primarily in the liver
• Hereditary tyrosinemia is a genetic inborn error of metabolism associated with severe liver disease in infancy. The disease is inherited in an autosomal recessive fashion.
• Tyrosine catabolism defect• characterized by the excretion of tyrosine and
tyrosine catabolites in urine•

Tyrosinemia • Type I tyrosinemia : caused by an absence of the
enzyme fumarylacetoacetate hydrolase (FAH)– most severe form of this aminoacidopathy
– found in about 1 in 100,000 births
– lead to liver and kidney failure
• Type II tyrosinemia: – deficiency of the enzyme tyrosine
aminotransferase– occurs in fewer than 1 in 250,000 births– mentally retarded ,excessive tearing and
photophobia

Tyrosinemia • Diagnosis :
– Elevation of tyrosine in plasma and urine• MS/MS chromatography
• Treatment : – Low protein diet – Drugs that inhibits maleylacetoacetic acid and
fumarylacetoacetic acid formation


• results from an absence or greatly reduced activity of the enzyme branched-chain -ketoacid decarboxylase
• Normal metabolism of 3 amino acids inhibited• MSUD is an autosomal recessive genetic
inherited disorder• 1 in 150,000 births in the general population

• characteristic maple syrup or burnt sugar odor of the urine, breath, and skin
• accumulation of the branched-chain amino acids and their corresponding ketoacids in the blood, urine, and cerebrospinal fluid (CSF).
• Within a week, develop lethargy, vomiting, lack of appetite, and signs of failure to thrive
• severe mental retardation, seizures, acidosis, and hypoglycemia
• Can lead to death

• Detection and screening:– modified Guthrie test
• The metabolic inhibitor to B. subtilis, includedin the growth media, is 4-azaleucine
– Microfluorometric assay for the three branched-chain amino

Homocystinuria
• Autosomal recessive disorder• Incidence : 1 in 200,000 birth• Deficiency y of the enzyme cystathionine-
synthetase, necessary for the metabolism of the methionine amino acid, that results in elevated plasma and urine levels of methionine and of the precursor homocysteine
• Urinary exertion of homocystein is > 300 mg/24 hrs

Homocystin

Homocystinuria
• Signs and complications:– Increased risk of blood clot (thrombosis)– Dislocation of eye lenses– Skeletal abnormality– Developmental delay– osteoporosis

Laboratory tests for homocysteinuria
• Guthrie test (blood) : using L-methionine sulfoximine as the metabolic inhibitor
• Plasma methionine level – HPLC – GC& MS
• Urinary homocysteine:– HPLC

Laboratory tests for homocysteinuria• Detection in urine : Cyanide-nitroprusside spot
test– cyanide-nitroprusside is added to urine sample– High levels of homocysteine will be detected by
the development of purple-red colour – urinary cysteine might interfere in this test to give
false +ve. – Confirmation by adding silver, homocysteine is
reduced but not cysteine thus allowing homocysteine to react with nitro-prusside to produce reddish color.


Cystinuria
• Autosomal recessive defect that is caused by a defect in the amino acid transport system rather than a metabolic enzyme deficiency
• inadequate reabsorption of cystine during the filtering process in the kidneys
• Cystine
• precipitates out of the urine and forms stones in the kidneys,ureters, or bladder

Cystinuria
• diagnosed by testing the urine for cystine using cyanide nitroprusside, which produces a red-purple color on reaction with sulfhydryl groups

Amino Acid analysis• Samples
– Plasma , urine and amnoitic fluid• Fasting sample 6 to 8 hrs • Heparin • Separate plasma as soon as possible• Contamination with platelets and WBCs should be
prevented• Heamolysis affect the results• Immediately analysed or sample should be frozen -20• Deproteinization within 30 mins

Amino Acid analysis
• Urine samples : – Random for screening– 24 hr with thymol preservative for quantitative
analysis

Amino Acid analysis
• Methods – TLC for screening– HPLC– MS/MS


















