
Adsorption of amides at the mercury-solution interface
13
Electroanalytical Chemistry and Interfacial Eleclrochemistry, 47 (1973) 265-277 265 © ElsevierSequoiaS.A., Lausanne - Printed in The Netherlands ADSORPTION OF AMIDES AT THE MERCURY-SOLUTION INTERFACE RICHARD PAYNE Air Force Cambridge Research Laboratories, Hanscom Field, Bedford, Mass. 01730 (U.S.A.) (Received21st February 1973; in revisedform 26th April 1973) INTRODUCTION The aliphatic amides and their N-alkyl derivatives possess some unusual double layer properties which appear to be related indirectly to the dielectric constant 1. The fully substituted derivatives, for example dimethylformamide, are generally normal polar liquids 2. They have moderately high dielectric constants, attributable to the high dipole moment (~3.7 D*) associated with the amide group, and are unremarkable as solvents in double layer studies. The unsubstituted and the monosubstituted compounds, e.g. formamide and N-methylformamide, on the other hand have anomalously high dielectric constants indicative of strong association and show correspondingly unusual double layer behavior. In this type of solvent system the negative branch of the capacity-potential curve contains a broad hump, which is generally attributed to field reorientation of the solvent dipole in the inner region of the double layer. However, there is little direct experimental evidence to support this theory. Dutkiewicz and Parsons 3 attempted to determine the effective dielectric constant of the inner layer for formamide in the potential range of the hump by studying the adsorption of thiourea, but were unable to demonstrate that the dielectric constant passes through a maximum value as demanded by the theory. However, they were able to show that the adsorption of diethyl ether from formamide solutions is a maximum at the hump. This suggests that the solvent is least strongly adsorbed and therefore presumably has maximum freedom of orientation in this region of potential. Unfortunately experiments of this kind are usually ambiguous because the adsorbing species itself is dipolar and may als0 undergo specific interaction with either the electrode or the bulk solvent or both. Adsorption of the solvent may also be affected so that interpretation of the results becomes quite complex. An alternative approach which does not avoid the above ambiguities but which is, however, somewhat more direct is to study the adsorption of the amide itself from another solvent, e.g. water. In this way one might hope to gain some insight into the orientation of the dipole and its variation with the electrode potential. In the following paper the adsorption of formamide and its N-methyl derivatives from dilute aqueous solutions is examined. "1 D=3.3356 × 10 -30 Cm.
-
Upload
richard-payne -
Category
Documents
-
view
213 -
download
1
Transcript of Adsorption of amides at the mercury-solution interface

Electroanalytical Chemistry and Interfacial Eleclrochemistry, 47 (1973) 265-277 265 © Elsevier Sequoia S.A., Lausanne - Printed in The Netherlands
ADSORPTION OF AMIDES AT THE MERCURY-SOLUTION INTERFACE
RICHARD PAYNE Air Force Cambridge Research Laboratories, Hanscom Field, Bedford, Mass. 01730 (U.S.A.)
(Received 21st February 1973; in revised form 26th April 1973)
INTRODUCTION
The aliphatic amides and their N-alkyl derivatives possess some unusual double layer properties which appear to be related indirectly to the dielectric constant 1. The fully substituted derivatives, for example dimethylformamide, are generally normal polar liquids 2. They have moderately high dielectric constants, attributable to the high dipole moment (~3.7 D*) associated with the amide group, and are unremarkable as solvents in double layer studies. The unsubstituted and the monosubstituted compounds, e.g. formamide and N-methylformamide, on the other hand have anomalously high dielectric constants indicative of strong association and show correspondingly unusual double layer behavior. In this type of solvent system the negative branch of the capacity-potential curve contains a broad hump, which is generally attributed to field reorientation of the solvent dipole in the inner region of the double layer. However, there is little direct experimental evidence to support this theory. Dutkiewicz and Parsons 3 attempted to determine the effective dielectric constant of the inner layer for formamide in the potential range of the hump by studying the adsorption of thiourea, but were unable to demonstrate that the dielectric constant passes through a maximum value as demanded by the theory. However, they were able to show that the adsorption of diethyl ether from formamide solutions is a maximum at the hump. This suggests that the solvent is least strongly adsorbed and therefore presumably has maximum freedom of orientation in this region of potential. Unfortunately experiments of this kind are usually ambiguous because the adsorbing species itself is dipolar and may als0 undergo specific interaction with either the electrode or the bulk solvent or both. Adsorption of the solvent may also be affected so that interpretation of the results becomes quite complex.
An alternative approach which does not avoid the above ambiguities but which is, however, somewhat more direct is to study the adsorption of the amide itself from another solvent, e.g. water. In this way one might hope to gain some insight into the orientation of the dipole and its variation with the electrode potential. In the following paper the adsorption of formamide and its N-methyl derivatives from dilute aqueous solutions is examined.
"1 D=3.3356 × 1 0 - 3 0 Cm.

266 R. PAYNE
EXPERIMENTAL
The methods described previously were used to measure the capacity at a growing mercury drop by a bridge method 4 and the interfacial tension using the capillary electrometer 5. Potential of zero charge (p.z.c.) was measured for each solution by the method of the streaming mercury electrode 6. A 0.1 M NaCI calomel electrode was used as a reference in a cell of the type,
amide solution 0.1 M aq. NaF i0.1 M aq. NaC11Hg2C12 Hg Hg in 0.1 M aq. NaF
All liquid junctions were left undisturbed during a series of measurements with the exception of that between the test solution and 0.1 M NaF. The stability of the liquid junction potentials and the reference electrode were checked by frequent measurement of the p.z.c. An apparent negative shift of ~ 4 mV in the p.z.c, for the base electrolyte over a two-week period was traced to the reference electrode. 'The proper value could be restored by replacing the NaCI electrolyte in the reference electrode, and this was therefore done routinely at intervals. All solutions were deaerated with pre-saturated oxygen-free nitrogen. The electrocapillary measure- ments were conducted in an air thermostat controlled to ___ 0.5 °C.
Solutions were prepared from A.R. grade salts further purified by recrystalli- zation from permanganate-distilled water. The amides were purified by triple distillation under reduced pressure. Mercury was purified by chemical pre- treatment followed by triple distillation in vacuo. Capillaries for the capacity measurements were prepared from pyrex capillary stock and treated with dimethyl- dichlorosilane to prevent solution-creep problems as described previously 7.
The capacity was normally measured at a frequency of 1 kHz with bridge balance achieved 8.20 s after the birth of the drop. A few measurements were made in the frequency range 0 .2-4 kHz in order to check the frequency dependence of the results. The 1 kHz measurements were shown to be within 1~o of the zero frequency value for DMF solutions at the desorption peaks. Elsewhere the frequency dependence below 1 kHz was generally negligible ( ~ 0.1~o).
The capillary electrometer was calibrated by measurements with pure water using Gouy's value s of 426.7 erg cm -2 at 18°C for the interracial tension at the electrocapillary maximum.
RESULTS AND DISCUSSION
(1) . Capacity curves The capacity and the interfacial tension were measured as a function of
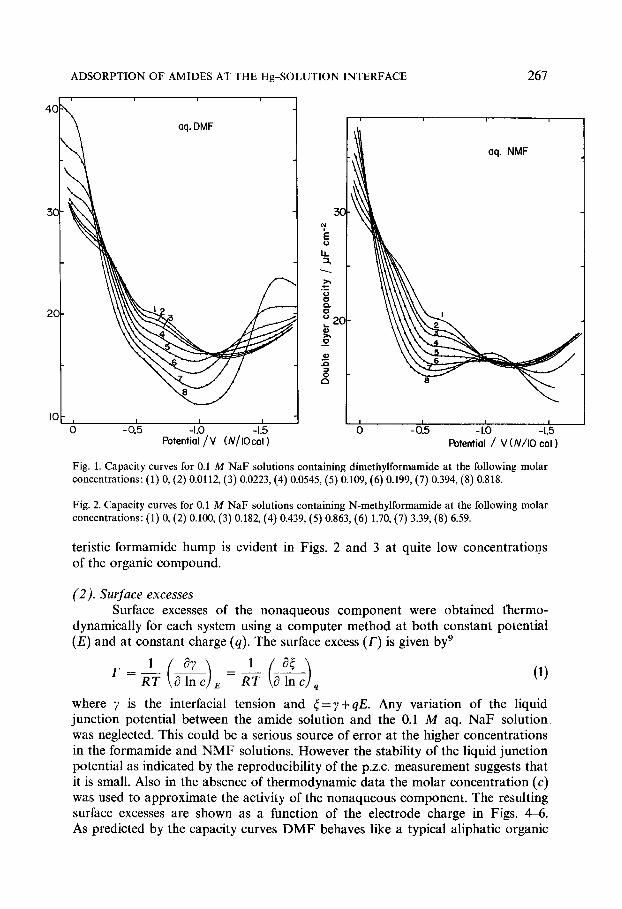
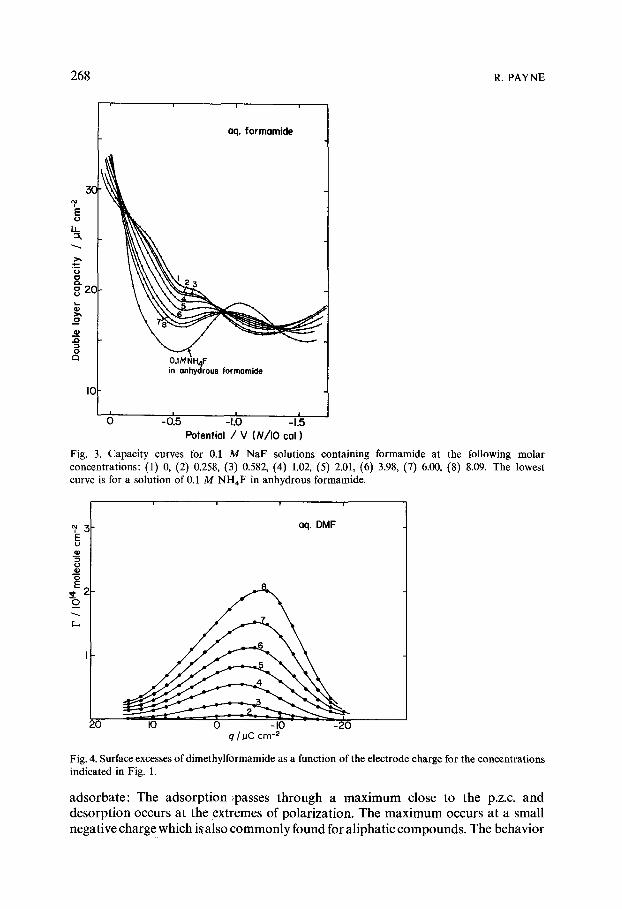
concentration in the range 0-0.8 tool 1-1 (DMF) 0-7 tool 1-1 (formamide) and 0-8 mol 1-1 (NMF). The capacity curves are shown in Figs. 1-3. The behavior of the DMF system (Fig. 1) is fairly typical for aliphatic compounds: lowering of the capacity corresponding to maximum adsorption occurs close to the p.z.c., and desorption occurs at both extremes of polarization. However desorption is in- complete. The curves for formamide (Fig. 2) and NMF (Fig. 3) in contrast are atypical. The usual desorption peaks are absent. It appears therefore that although formamide and NMF are generally less strongly adsorbed than DMF they tend to remain adsorbed at the extremes of polarization. The formation of the charac-
ADSORPTION OF AMIDES AT THE Hg-SOLUTION INTERFACE 267
IC
oq. DMF
I I I I
-0.5 -I.0 -I.5 0 -0.5 Potential/V (N/lOcal)
i i
aq. NMF
- 1.0 -1.5
Potential / V(N/IO col )
Fig. 1. Capacity curves for 0.1 M NaF solutions containing dimethylformamide at the following molar concentrations: (1) 0, (2) 0.0112, (3) 0.0223, (4) 0.0545, (5) 0.109, (6) 0.199, (7) 0.394, (8) 0.818.
Fig. 2. Capacity curves for 0.1 M NaF solutions containing N-methylformamide at the following molar concentrations: (1) 0, (2) 0.100, (3) 0.182, (4) 0.439, (5) 0.863, (6) 1.70, (7) 3.39, (8) 6.59.
teristic formamide hump is evident in Figs. 2 and 3 at quite low concentrations of the organic compound.
(2). Surface excesses Surface excesses of the nonaqueous component were obtained thermo-
dynamically for each system using a computer method at both constant potential (E) and at constant charge (q). The surface excess (F) is given by 9
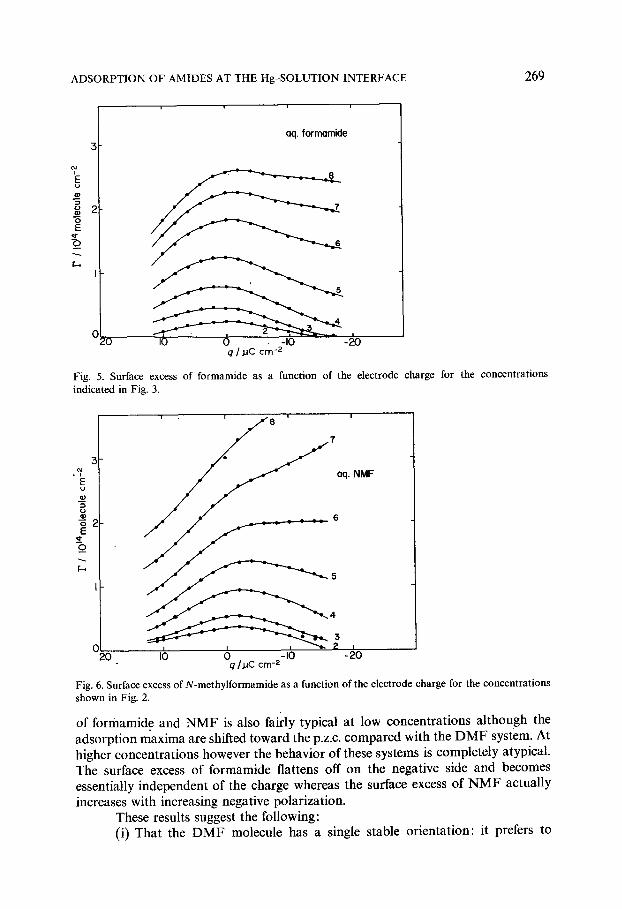
R ~ RT (1) = (0- n c), where 7 is the interracial tension and ~ =7+qE. Any variation of the liquid junction potential between the amide solution and the 0.1 M aq. NaF solution~ was neglected. This could be a serious source of error at the higher concentrations in the formamide and N M F solutions. However the stability of the liquid junction potential as indicated by the reproducibility of the p.z.c, measurement suggests that it is small. Also in the absence of thermodynamic data the molar concentration (c) was used to approximate the activity of the nonaqueous component. The resulting surface excesses are shown as a function of the electrode charge in Figs. 4-6. As predicted by the capacity curves D M F behaves like a typical aliphatic organic
268 R. PAYNE
i
oq. formomide
E ¢J
LL
._-, ¢.~ o
o u
in anhydrous formamide
I , ~ ) I I I I - 0 . 5 - I .0 - I .5
Potentiul / V (N/ IO col )
Fig. 3. Capacity curves for 0.1 M NaF solutions containing formamide at the following molar concentrations: (1) 0, (2) 0.258, (3) 0.582, (4) 1.02, (5) 2.01, (6) 3.98, (7) 6.00, (8) 8.09. The lowest curve is for a solution of 0.1 M NH4F in anhydrous formamide.
5 ¢D -5
o
f.~
i i
oq. DMF
2 0 10 0 - I0 - 2 0 q / j J C cm -2
Fig. 4. Surface excesses of dimethylformamide as a function of the electrode charge for the concentrations indicated in Fig. 1.
adsorbate: The adsorption ~passes through a maximum close to the p.z.c, and desorption occurs at the extremes of polarization. The maximum occurs at a small negative charge which isalso commonly found for aliphatic compounds. The behavior
ADSORPTION OF AMIDES AT THE Hg-SOLUTION INTERFACE 269
,?
¢,
o E
r--,
aq. fo rmamide
z I ~ ) 0 -IO -20
q/.uC c m -2
Fig. 5. Surface excess of formamide as a function of the electrode charge for the concentrations indicated in Fig. 3.
E u
m
O
E
o
1 • I / 8 I I
7
3 • 7
~ q. N M F
/ f / t / ~ 3
0 I I ~ I 2 0 I0 0 - I 0 - 2 0
q/JJC cm -2
Fig. 6. Surface excess of N-methylformamide as a function of the electrode charge for the concentrations shown in Fig. 2.
of formamide and NMF is also fairly typical at low concentrations although the adsorption maxima are shifted toward the p.z.c, compared with the DMF system. At higher concentrations however the behavior of these systems is completely atypical. The surface excess of formamide flattens off on the negative side and becomes essentially independent of the charge whereas the surface excess of NMF actually increases with increasing negative polarization.
These results suggest the following: (i) That the DMF molecule has a single stable orientation: it prefers to

270 R. PAYNE
desorb rather than to reorientate at the extremes of polarization. (ii) At sufficiently negative potentials both formamide and NMF undergo
some kind of reorientation to a stable configuration which can compete favorably with the water dipole at high field strengths. The new orientation presumably must have at least a component of the dipole moment positively oriented toward the negatively charged electrode.
(iii) The reorientation effect is only apparent at high concentrations which suggests that the new orientation is stabilized by the presence of the amide in the bulk of the solution. This could occur for example through a hydrogen-bonding type of interaction which, significantly, is not possible in the case of DMF where the amide group is fully substituted.
These tentative conclusions are confirmed by a more detailed examination of the results.
(3). Shift of the potential Shift of the potential at constant electrode charge can be produced by changes
in thickness and dielectric constant of the double layer, adsorption of oriented dipoles or displacement of oriented solvent dipoles. Assuming for simplicity that one water molecule is replaced by one organic molecule one can write the potential difference across the inner region of the double layer as
c~m_ 2 = __4n [qx2 + NsOftor, + Ns(1-0)~w] (2)
where e is the mean dielectric constant, x 2 the thickness, Ns the number of adsorption sites, 0 the fractional coverage of the organic molecules, f~rg and ftw the normal components of the dipole for the organic molecules and the water molecules respectively. The effect of the various terms in this expression can now be considered.
(a). Variation of the thickness, x2. The thickness of the inner layer is expected to increase as solvent molecules are replaced by the larger organic molecules. The potential shift produced by the adsorption therefore will be in the positive direction for a positively charged electrode and in the negative direction for a negatively charged electrode. At the p.z.c, the variation of x2 has no effect on the potential. If the thickness (strictly the ratio x2/e) is linearly dependent on the amount adsorbed the potential shift will also be linear in 0.
(b). Variation of the dipole contributions. Adsorption of a dipole with the positive end facing the electrode will produce a potential shift in the positive direction and vice versa.
If the adsorbing molecule has no dipole moment (or the normal com- ponent of the dipole moment is zero) the shift of potential is entirely due to replacement of oriented solvent dipoles which are expected to be positively oriented on a negatively charged electrode and vice versa. Consequently replacement of solvent dipoles should generally cause a positive shift of potential for a positively charged electrode and vice versa. This is in the same sense as the thickness variation, but unlike the thickness effect it need not vanish at the p.z.c, since the solvent molecules may be preferentially oriented on a uncharged electrode.
For many aliphatic compounds adsorbed from aqueous solutions the potential
ADSORPTION OF AMIDES AT THE Hg-SOLUTION INTERFACE 271
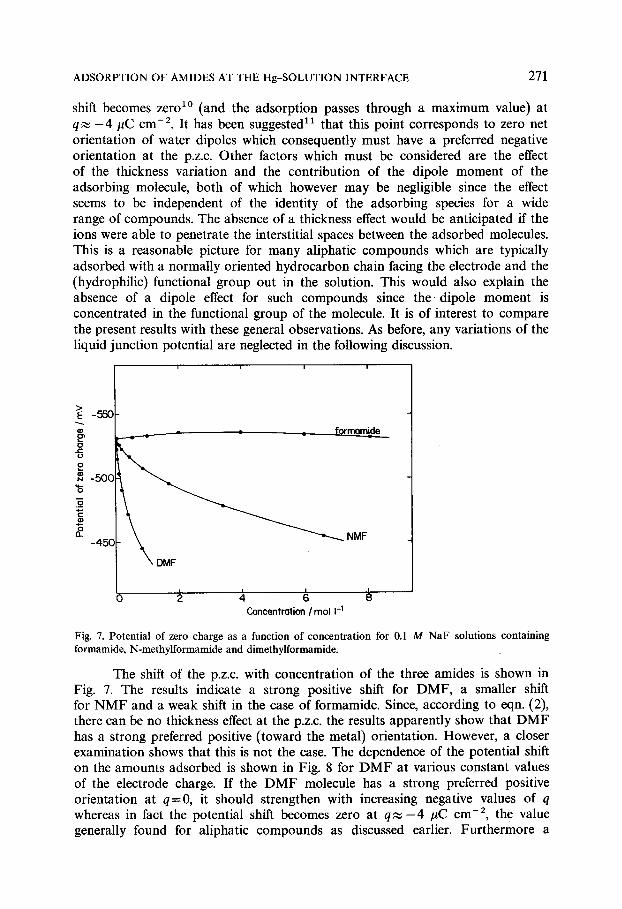
shift becomes zero 1° (and the adsorption passes through a maximum value) at q , ,~ -4 #C cm -2. It has been suggested 11 that this point corresponds to zero net orientation of water dipoles which consequently must have a preferred negative orientation at the p.z.c. Other factors which must be considered are the effect of the thickness variation and the contribution of the dipole moment of the adsorbing molecule, both of which however may be negligible since the effect seems to be independent of the identity of the adsorbing species for a wide range of compounds. The absence of a thickness effect would be anticipated if the ions were able to penetrate the interstitial spaces between the adsorbed molecules. This is a reasonable picture for many aliphatic compounds which are typically adsorbed with a normally oriented hydrocarbon chain facing the electrode and the (hydrophilic) functional group out in the solution. This would also explain the absence of a dipole effect for such compounds since t h e dipole moment is concentrated in the functional group of the molecule. It is of interest to compare the present results with these general observations. As before, any variations of the liquid junction potential are neglected in the following discussion.
i l I i
~, • • , forrnami, d e _
=o o
-500 "6 -6
n° ~ NMF
-450 ~ DMF
o Concentration /tool 1-1
Fig. 7. Potential of zero charge as a function of concentration for 0.1 M NaF solutions containing formamide, N-methylformamide and dimethylformamide.
The shift of the p.z.c, with concentration of the three amides is shown in Fig. 7. The results indicate a strong positive shift for DMF, a smaller shift for N M F and a weak shift in the case of formamide. Since, according to eqn. (2), there can be no thickness effect at the p.z.c, the results apparently show that D M F has a strong preferred positive (toward the metal) orientation. However, a closer examination shows that this is not the case. The dependence of the potential shift on the amounts adsorbed is shown in Fig. 8 for D M F at various constant values of the electrode charge. If the D M F molecule has a strong preferred positive orientation at q=0, it should strengthen with increasing negative values of q whereas in fact the potential shift becomes zero at q ~ - 4 #C cm -2, the value generally found for aliphatic compounds as discussed earlier. Furthermore a
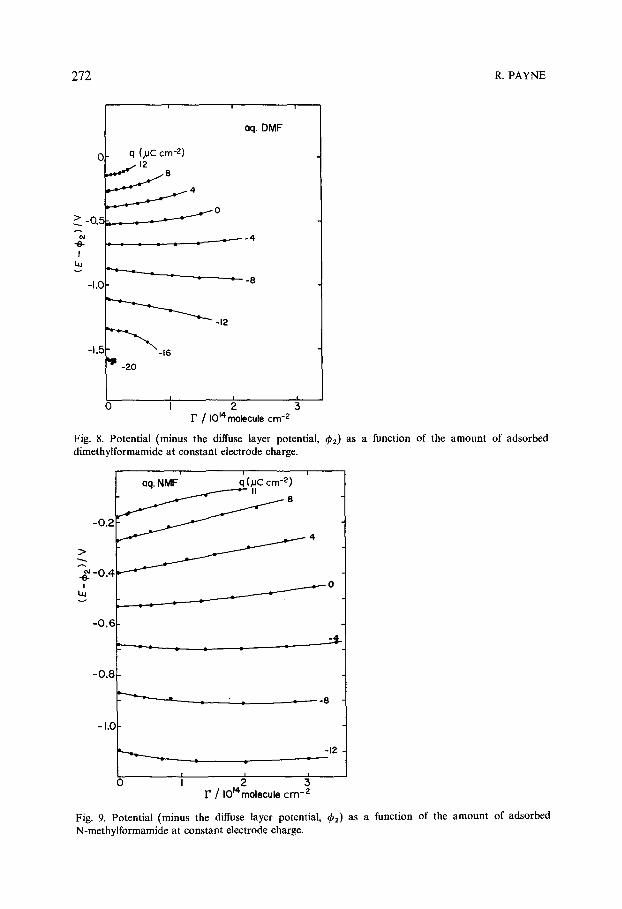
272 R. PAYNE
> _ 0 =
.g I
i
q (pC cm -2)
. _ . . _ . . . . . 4 - - - , - - - 4
i
oq. DMF
Fig. 8. Potential (minus the diffuse layer potential, (~2) as a function of the amount of adsorbed dimethylformamide at constant electrode charge.
oq. NMF' q (pC cm -2) '
-0.2 ~ s
>
.~-0.4 I Lu _.....,,..___.._._._,........~ 0
-0.6
--.---.-,-..,_ _. : : ~
-0.8
" " ~ : , ~,------- -e
-I.C
-12
o i ~ P / 10~4molecule cm -2
Fig. 9. Potential (minus the diffuse layer potential, (k2) as a function of the amount of adsorbed N-methylformamide at constant electrode charge.
- I . 0 ~ - - - - ' * - - -8
_ 1 . 5 ~ _ 1 6 ~ -12
~ 0
0 I 2 1" / 10=4molecule cm -2

ADSORPTION OF AMIDES AT THE Hg-SOLUTION INTERFACE 273
positively oriented dipole should become less strongly oriented with increasing positive charge and perhaps eventually reverse its orientation. There is no evidence for this in the experimental results. It must be concluded therefore that DMF does not have a strong preferred positive orientation and that the positive shift of the p.z.c, is due to replacement of negatively oriented solvent molecules. The increasing magnitude of this effect (Fig. 7) in the series formamide < NMF < DMF can be attributed to increasing size of the molecule resulting in the displacement of more solvent molecules. Since there is apparently little or no contribution from the DMF dipole it must be further concluded that the organic molecules are adsorbed either in a flat orientation (no normal component of the dipole moment) or with the polar part of the molecule in the diffuse layer where its effect would be screened out by the ions. The size of the water molecule controls the effective thickness of the inner layer at low coverages of the adsorbate. Molecular models indicate that the polar group of even the smallest molecule in this group (formamide) can be outside the inner layer under these conditions. However at high surface concentra- tions the mean thickness is set by the organic molecule, and the polar group is now in a region of high field strength.
The shift of the potential at Constant charge is shown for the NMF system in Fig. 9. As expected from the predicted increase of thickness and replacement of oriented water molecules, the potential shift is in the positive direction when q is positive. As in the case of DMF the shift is small at small negative values of q. At more negative values of q the initial potential shift is negative, consistent again with increase, of thickness or replacement of oriented water. However, at higher concentrations the shift becomes positive. In terms of the simple model under discussion this effect could only be due to adsorption of a positNely oriented dipole. The behavior of the formamide system is similar.
There appear to be two possible explanations of these results: (i) That the amide dipoles are always adsorbed with a more or less normal
orientation, positive side toward the electrode, screened by diffuse layer ions at low surface concentrations, where the thickness of the inner layer is controlled by the water molecules, but contributing to the potential at high surface concentrations where the organic molecules control the thickness.
(ii) That the amide molecules are adsorbed in a more or less flat orientation at the p.z.c, and undergo field reorientation at sufficiently negative charges and high concentrations.
The reorientation explanation seems the more plausible for the following reason. As we have seen, the p.z.c, is essentially independent of the amount adsorbed in the case of formamide. If water has a small negative orientation at the p.z.c, then it follows that formamide must have a similar negative orientation. Hence the polar group must be close to the electrode surface and the first explanation therefore cannot be correct.
(4). The adsorption isotherm It is of interest to examine the form and constants of the adsorption iso-
therms which should be consistent with the conclusions drawn from the potential shift. In the formamide and NMF systems the concentration range is too high for the surface concentration to be approximated by the surface excess. The following
274 R. PAYNE
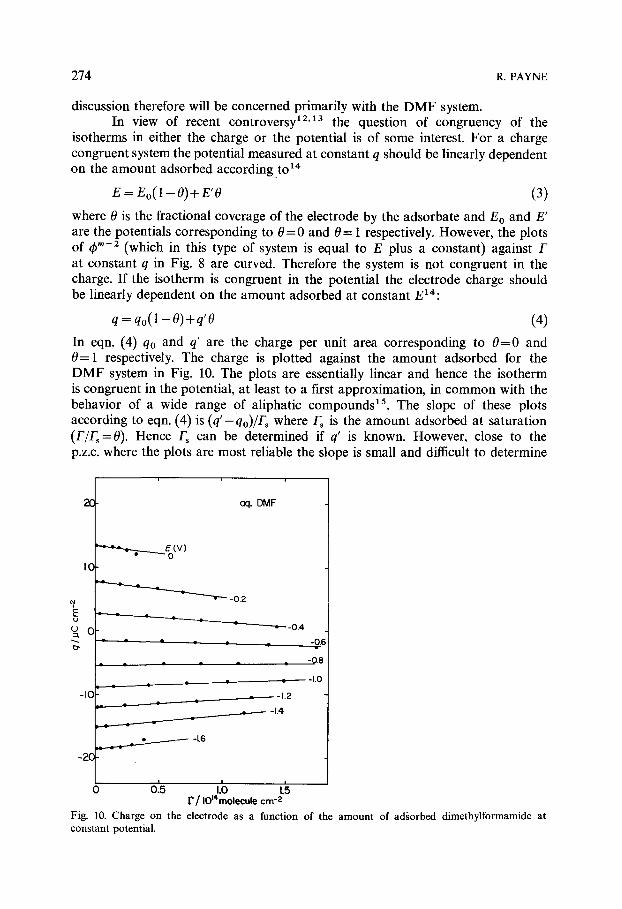
discussion therefore will be concerned primarily with the D M F system. In view of recent controversy 12'13 the question of congruency of the
isotherms in either the charge or the potential is of some interest. For a charge congruent system the potential measured at constant q should be linearly dependent on the amount adsorbed according to TM
E = E o ( 1 - O)+E'O (3)
where 0 is the fractional coverage of the electrode by the adsorbate and E 0 and E' are the potentials corresponding to 0 = 0 and 0 = 1 respectively, However, the plots of ~b m- 2 (which in this type of system is equal to E plus a constant) against F at constant q in Fig. 8 are curved. Therefore the system is not congruent in the charge. If the isotherm is congruent in the potential the electrode charge should be linearly dependent on the amount adsorbed at constant E14:
q=qo(1-O)+q'O (4) In eqn. (4) qo and q' are the charge per unit area corresponding to 0 = 0 and 0= 1 respectively. The charge is plotted against the amount adsorbed for the D M F system in Fig. 10. The plots are essentially linear and hence the isotherm is congruent in the potential, at least to a first approximation, in common with the behavior of a wide range of aliphatic compounds 15. The slope of these plots according to eqn. (4) is (q'-'qo)/Fs where Fs is the amount adsorbed at saturation (F/Fs= 0). Hence Fs can be determined if q' is known. However, close to the p.z.c, where the plots are most reliable the slope is small and difficult to determine
i i
E aq. DMF
IC
~ -0.2
E u ~ , - o - . ~ _ ~ _ _ . ~ _ . -0.4
""-- ; "- . -0 t
. - -0.8
f . - -LO - I C : - " " " - I .2
-I.4
._~r__~..~.------- -L6
- 2 £
0 0 5 1.0 1.5 1"1 / 1014molecule c m - 2
Fig. 10. Charge on the electrode as a function of the amount of adsorbed dimethylformamide at constant potential.
ADSORPTION OF AMIDES AT THE Hg-SOLUTION INTERFACE 275
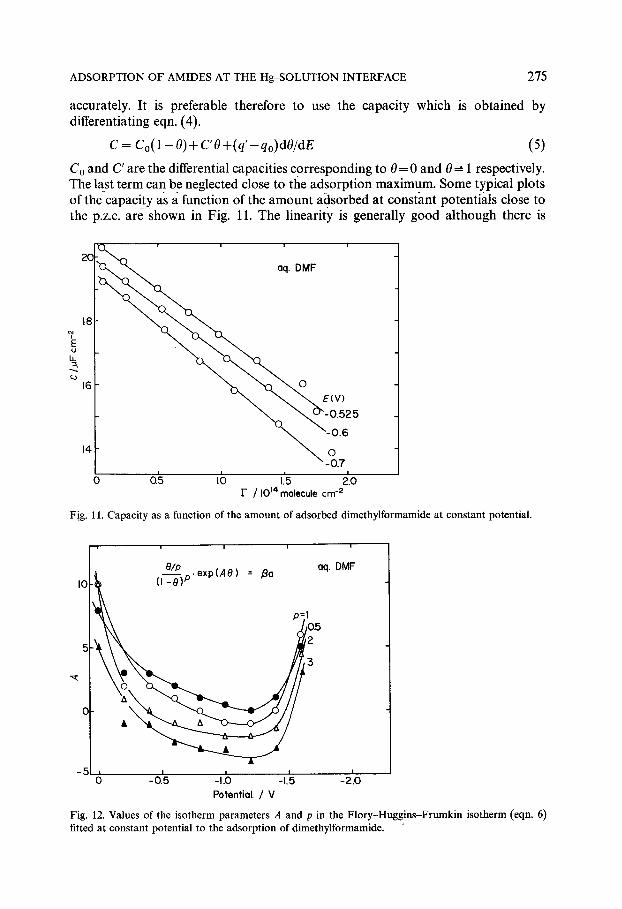
accurately. It is preferable therefore to use the capaci ty which is obtained by differentiating eqn. (4).
C = C0(1 - 0) + C'O +(q'- qo)dO/dE (5)
Co and C' are the differential capacities cor responding to 0 = 0 and 0--- 1 respectively. The last term can be neglected close to the adsorpt ion maximum. Some typical plots of the capaci ty as a function of the a m o u n t adsorbed at constant potentials close to the p.z.c, are shown in Fig. 11. The linearity is generally g o o d a l though there is
E u
L L
2C
18
16
14-
0
i i i i
(v)
,.6 ,;s r / I014 molecule cm "2
Fig. 11. Capacity as a function of the amount of adsorbed dimethylformamide at constant potential.
I0
-5
i i i !
( l~-~)p.exp(AO) = .8o P:
! 6 -,b -,;
Potentia[ / V
i
aq. DMF
I ~35 2
3
I
-2.0
Fig. 12. Values of the isotherm parameters A and p in the Flory-Huggins-Frumkin isotherm (eqn. 6) fitted at constant potential to the adsorption of dimethylformamide.

276 R. PAYNE
some variation in the slope. According to eqn. 5 the slope is equal to (C'-Co)/Fs from which F~ can be calculated. Using values of C' estimated from previously published measurements I for 0.1 M KPF 6 in anhydrous DMF, values of Fs in the range 2.2 to 2.4 x 10 t4 were obtained, corresponding to area/molecule values of 42-46/~2. Similar calculations for formamide and NMF although less reliable give substantially lower values of the saturation area as would be expected. The value obtained for DMF is consistent with the size of the molecule estimated from molecular models.
The linear plots in Fig. 10 indicate that the isotherm is congruent in the potential. It should be possible therefore to fit the amounts adsorbed to a theoretical isotherm at constant potential with potential independent parameters. An attempt was made to fit the data to the Flory-Huggins modification of Frumkin's isothermt 5, ~6
O/p exp (AO) = tic (6) (1-0) p
in which A is a lateral interaction parameter, /~ is the adsorption equilibrium constant and p the number of solvent molecules displaced by one adsorbed DMF molecule. A value for p of 3 or 4 would be expected. A reasonable fit of the data to eqn. (6) could be obtained for a range of parameters as shown in Fig. 12. However, the best fit was obtained for p = 1. Also for a given value of p, the results could only be fitted to the isotherm by allowing A to vary with the potential. According to Fig. 12 the interaction parameter is always positive, corresponding to repulsive interaction, and increases strongly at the extremes of polarization. Consequently the congruency condition is only approximately met since at least one of the isotherm parameters is potential dependent. This is confirmed by the deviation from linearity of the charge plots in Fig. 10 at the extremes of potential. This is to be expected since high field strengths will in general distort and compress the molecules in the inner layer and produce the kind of deviations from congruency observed.
ACKNOWLEDGEMENT
The experimental part of this work was performed at the University of Bristol during the tenure of a Senior Research Fellowship of the Science Research Council (U.K.).
SUMMARY
The adsorption of formamide, N-methylformamide and dimethylformamide from 0.1 M NaF solutions in water has been studied at 25°C. The behavior Of dimethylformamide is typical of simple aliphatic compounds. The adsorption isotherm is approximately congruent in the potential, and the shift of potential at constant charge resulting fro m the adsorption is primarily due to replacement of oriented water dipoles. Formamide and N-methylformamide on the other hand are quite atypical. Their adsorption isotherms are congruent in neither the potential nor the charge. The shift of the potential at constant electrode charge in these systems

ADSORPTION OF AMIDES AT THE Hg-SOLUTION INTERFACE 277
is cons i s t en t w i t h a pa ra l l e l o r i e n t a t i o n o f the d i p o l e a t the p o i n t o f z e r o charge ,
w i t h r e o r i e n t a t i o n o c c u r r i n g o n a nega t i ve ly c h a r g e d e lec t rode . Th i s c o n f i r m s the
d i p o l e r e o r i e n t a t i o n t h e o r y o f the o r ig in o f the c a p a c i t y h u m p f o u n d in the a n h y d r o u s so lvent .
REFERENCES
1 R. Payne, J. Phys. Chem., 73 (1969) 3598. 2 S. J. Bass, W. I. Nathan, R. M. Meighan and R. H. Cole, J. Phys. Chem., 68 (1964) 509. 3 E. Dutkiewicz and R. Parsons, J. Electroanal. Chem., 11 (1966) 196. 4 G. J. Hills and R. Payne, Trans. Faraday Soc., 61 (1965) 316. 5 K. M. Joshi and R. Parsons, Electrochim. Acta, 4 (1"961) 129. 6 D. C. Grahame, E. M. Coffin, J. I. Cummings and M. A. Poth, J. Amer. Chem. Soc., 74 (1952) 1207. 7 R. Payne, J. Electroanal. Chem., 7 (1964) 343. 8 G. Gouy, Ann. Phys., 6 (1916) 5. 9 R. Parsons, Trans. Faraday Soc., 51 (1955) 1518.
10 E. Blomgren, J.O'M. Bockris and C. Jesch, J. Phys. Chem., 65 (1961) 2000. 11 J. O'M. Bockris, M. A. V. Devanathan and K. Miiller, Proc. Roy. Soc., A274 (1963) 55. 12 A. N. Frumkin, B. B. Damaskin and A. A. Survila, J. Electroanal. Chem., 16 (1968) 493. 13 E. Dutkiewicz, J. D. Garnish and R. Parsons, J. Electroanal. Chem., 16 (1968) 505. 14 R. Parsons, Trans. Faraday Soc., 55 (1959) 999. 15 B. B. Damaskin, A. N. Frumkin and A. Chizhov, J. Electroanal. Chem., 28 (1970) 93. 16 S. Levine, G. M. Bell and D. Calvert, Can. J. Chem., 40 (1962) 518. 17 R. Parsons, Rev. Pure Appl. Chem., 18 (1968) 91.


















