
Addition reactionschem.yonsei.ac.kr/chem/upload/CHE4103-01/125837317354252.pdf · 5 10.3...
66
1 Chapter 10 Organic Reaction Mechanisms, Part 1: Reactions Involving Additions and/or Eliminations Addition reactions 10.2 Hydration of carbonyl structures R R RO OR acetal
Transcript of Addition reactionschem.yonsei.ac.kr/chem/upload/CHE4103-01/125837317354252.pdf · 5 10.3...

1
Chapter 10 Organic Reaction Mechanisms, Part 1: Reactions Involving Additions and/or Eliminations
Addition reactions10.2 Hydration of carbonyl structures
R RRO OR
acetal

2
10.2.1 Acid-base catalysis
See section 9.3

3
10.2.2 The thermodynamics of the formation of geminal diols and hemiacetals
1. Ketones and aryl aldehydes: less than unity, favoring the carbonyl group2. Aliphatic aldehydes, carbonyl structures with EWG, and carbonyls in strained rings:
greater than unity 3. Aldehydes are more hydrated than ketones because steric congestion in the geminal diol
is less.4. EWG destabilize the already electrophilic carbonyl, leading to greater hydration. 5. Strained rings such as cyclobutanone prefer the sp3 hybridization of a hemiacetal carbon over the sp2 hybridization of a carbonyl carbon. -> smaller bond angle of an sp3 center better matches the bond angles in small rings.

4
pyranose
0.003%
2-pyridone

5
10.3 Electrophilic addition of water to alkenes and alkynes: hydration
General acid catalysis: all forms of acid in the medium are reactive, and the protonation is rds
No scrambling of deuteriums -> the first step is not reversible

6
10.3.3 Regiochemistry
less stable product
H
H
more stable; less activation energy
enamine
vinyl ether
EDG: increase hydration rates EWG (Cl or CN): retard hydration rates
involvement of a localized carbenium ion on the more substituted carbon

7
10.3.4 Alkyne hydration
very similar to alkene hydrations

8
10.4 Electrophilic addition of hydrogen halides to alkenes and alkynes
rearrangement
The product ratio depends on neither [HCl] nor [added Cl-]. If Cl- and HOAc compete for addition to a transient carbenium ion, one would expect increasing chloride concentrations to divert the intermediate to the formation of alkyl chloride products, but this is not seen.
Cl- diffusion is slower than rearrangement -> added Cl- does not affect the product ratio.
Two cases A

9
The product ratio from the addition of HCl to cyclohexene in HOAc does depend on [added Cl-].
added Cl- ->Increase in this product
Second order reaction
Why? Consider stereochemistry of this reaction.
Added Cl- increases in this product ratio.
B

10

11
10.4.3 Addition to alkynes
vinyl carbocation-> lower stability than trigonal sp2 carbenium ions-> addition of HX to alkynes is slower than with alkenes
Cl+
major product
Because of the lower stability of vinyl cations relative to alkyl carbenium ions, concerted reactions occur.
Anti addition product; predominantly
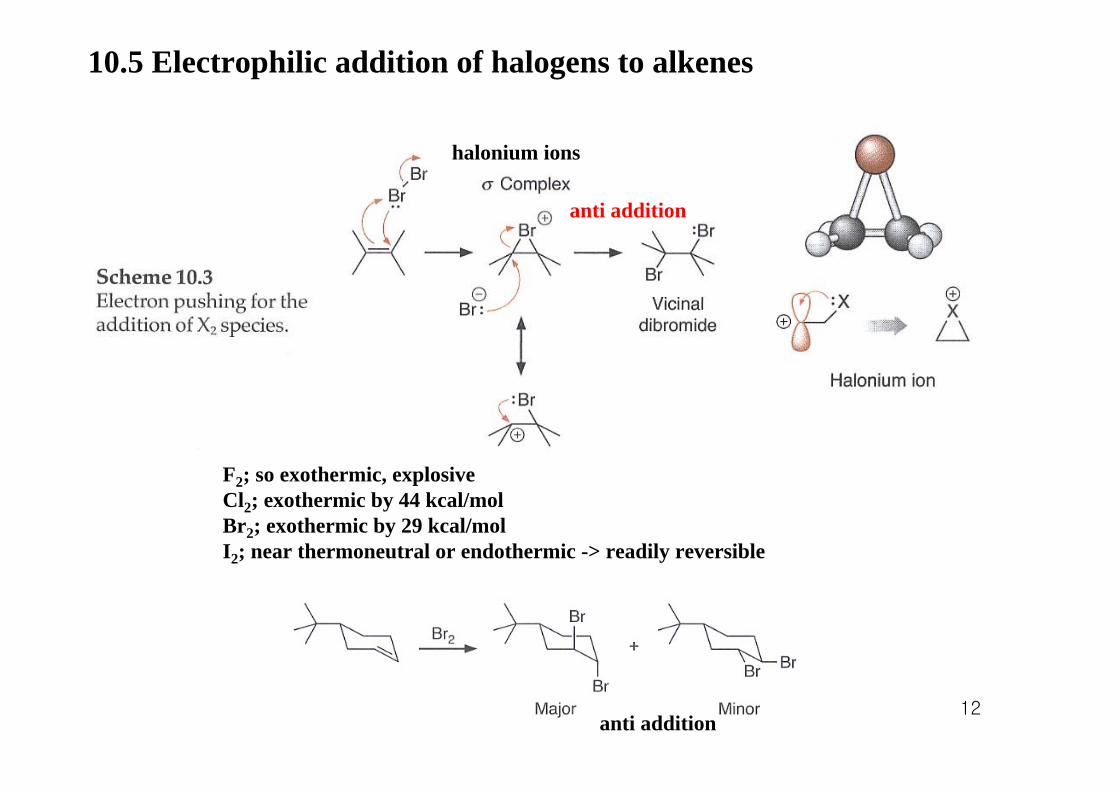
12
10.5 Electrophilic addition of halogens to alkenes
halonium ions
F2; so exothermic, explosiveCl2; exothermic by 44 kcal/molBr2; exothermic by 29 kcal/molI2; near thermoneutral or endothermic -> readily reversible
anti addition
anti addition

13
10.5.3 Other evidence supporting a σ complex
1. kinetics
hydration rates
If a non-bridging carbenium ion were formed, one would expect substituent effects similar to that seen for alkene hydration. -> but this is not the case. -> σ complex formation

14
2. Kinetic isotope effects
kH/kD = 0.53 (large inverse KIE -> significant rehybridization of both alkene carbons in rds)sp2 -> sp3
3. Addition of other nucleophiles other than bromide
BrBr
δ+ δ+
MeOH
4. Isolation of bromonium ionsSteric hindrance impedes nucleophilic attack
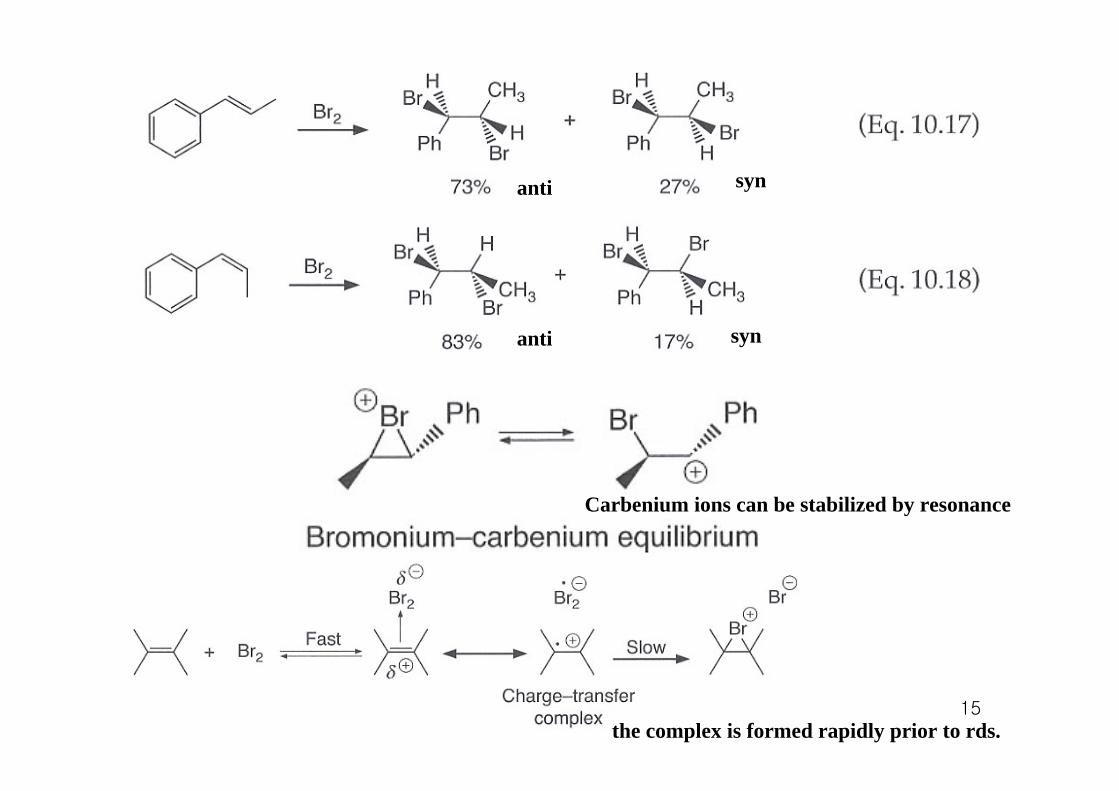
15
anti syn
anti syn
Carbenium ions can be stabilized by resonance
the complex is formed rapidly prior to rds.

16
10.5.4 Mechanistic variantsat low concentrations of bromine or in water and alcohol
In solvents of lower polarity, even acetic acid, the reaction is second order in bromine.
F2 addition -> syn addition
Carbocation rapidly combines with F- before dissociation of the ion pair.

17
10.5.5 Addition to alkynesAlkyl-substituted alkynes show anti addition products with bromine, again supporting a brominium ion intermediate. However, alkynes generally react 103 to 107 times slower than alkenes.
ring strain and positive charge on sp2 orbital
Aryl alkynes
ρ = -5.17 (large negative value) -> EDG 반응속도증가 -> vinyl cation intermediate

18
10.6 Hydroboration
diborane borane
THF
syn addition
H2O2, NaOH
H OH
Addition; anti-Markovnikov product
Addition of boron to a less hindered carbon

19
10.7 EpoxidationmCPBA (m-chloroperbenzoic acid)
syn addition
The more electron rich the double bond, the faster it will react with the peracid.Sterics are the primary factor directing the epoxidation stereochemistry. The least hindered face of a double bond is predominantly epoxidized.
carbocation charater가크다; sp2 -> sp2
sp2 -> sp3
Almost no secondary KIE
A relatively large inverse KIE

20
10.8 Nucleophilic additions to carbonyl compounds
Cyanohydrin formation
cyanohydrin
Aldehydes are more reactive than ketonesbecause steric congestion in the cyanohydrinis less.
rate = k[CN-][C=O]
CN- is a very good nucleophile-> protonation is not required prior to or at rds-> no dependence on acid

21
Grignard additionExtremely fast even at -85 oC
two electron nucleophile
For some carbonyl compounds
electron transfer mechanism:carbonyl structures that lead to stabilized ketyl anions will favor this mechanism, such as conjugate enones, phenyl ketones, and phenyl aldehydes.
-> evidence; hydrogen abstraction products (B 첫번째)or radical coupling products (B 두번째) are formed.

22
Lithium aluminum hydride reduction
A lithium cation is involved in the reaction.
A lithium-specific cryptand

23
10.8.6 Conformational effects in additions to carbonyl compoundsThe addition of nucleophiles to carbonyl compounds is often found to occur faster with six-memberedring cyclic ketones than with acyclic ketones or cyclopentanones.
Why? The dihedral angle between the Heq and the carbonyl oxygen is only 4o. This near eclipsing interaction produces a conformational strain of around 4 kcal/mol that raises the ground state energy of cyclohexanones relative to acyclic systems. Upon nucleophilic attack, this near eclipsing interaction is relieved, but we introduce a 1,3-diaxial interaction with an oxygen anion. However, the diaxialinteraction is estimated to be only 0.7 kcal/mol destabilizing (the A value for an OH), and so the net effect is that a significant amount of strain has been released in this reaction.
near eclipsing interaction
Release of eclipsing interaction (4 kcal/mol)but increase in 1,3 diaxial interaction (0.7 kcal/mol) -> a significant amount of strain has been released

24
10.8.7 Stereochemistry of nucleophilic additions
Cram’s model
Felkin-Ahn model
Karabatsos’ model
Rl
O
R
RmRs
Rl
OH
R
RmRs
NuRl
OH
Nu
RmRs
R+
major minor
S기쪽으로공격
S기쪽으로공격

25
Cram’s model
Felkin-Ahn model

26
Cyclic carbonyl structures
LAH reduction; trans -> major
Due to these strains, H- attacks more stericallyhindered face of the ketone
1. 3번위치에 H 이외의치환기가있으면 hydride 반응시 cis가major2. Larger Nu -> cis product as a major
due to steric hindrance

27
Meerwein-Pondorrf-Verley reduction (the reverse reaction is called the Oppennauer ocidation)
equilibrium
More stable
less stable

28
FAD: one electron and two electron reactionsNADH(P): two electron (or hydride) reactions

29
10.9 Nucleophilic additions to alkenesMuch less favorable than a carbonyl. But when strongly EWG are placed on an alkene, nucleophilic addition can occur.
Michael addition (1,4 addition)

30
10.9.4 Baldwin’s rule
These rules allow chemists to predict the ease of ring closure reactions.Three factors are considered: 1. ring size, 2. hybridization of the carbon undergoing attack,3. whether the bond undergoing attack will be endocyclic or exocyclic to the forming ring in the product.
The ease of intramolecular formation of a particular ring size generally followed the trend, 5> 6 > 3 > 7 > 4 > 8-10. This holds for intramolecular nucleophilic, as well as radical and cationic ring closures.
sp = digonal, sp2 = trigonal, sp3 = tetragonal

31
trigonal
digonal
Nu
Nu
Nu
Nu
Nu
large distortion -> unfavorable
favorable
32
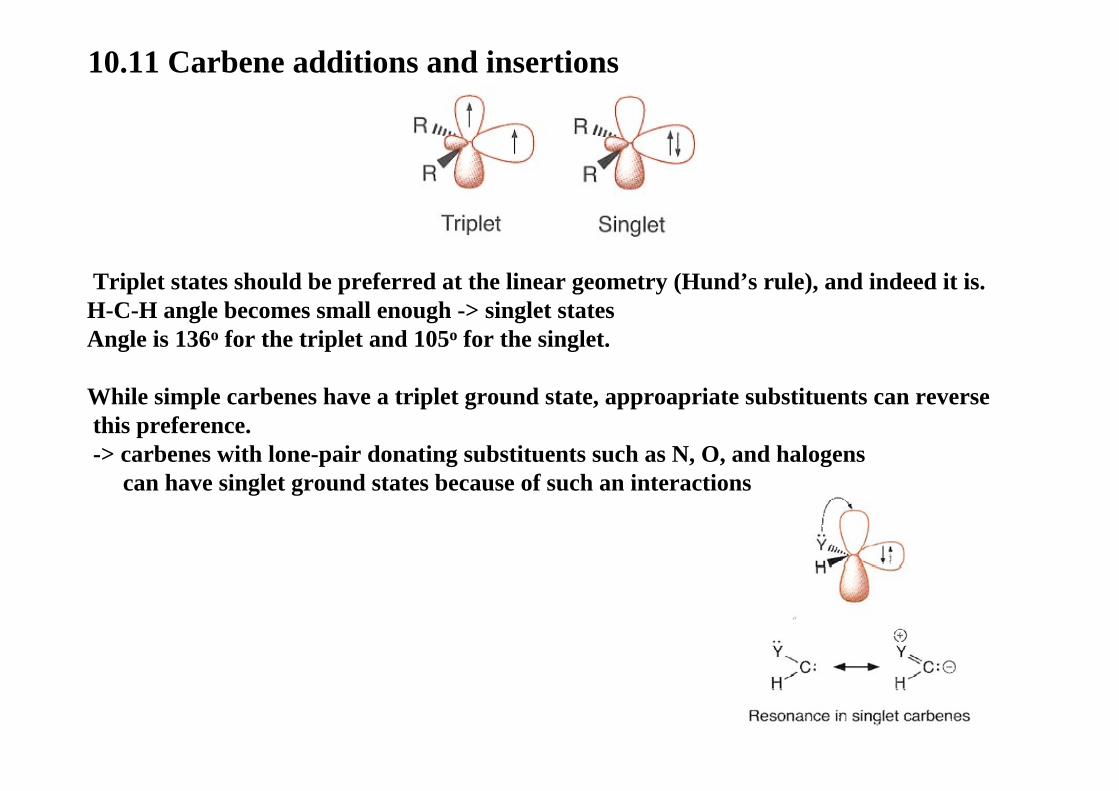
10.11 Carbene additions and insertions
Triplet states should be preferred at the linear geometry (Hund’s rule), and indeed it is.H-C-H angle becomes small enough -> singlet statesAngle is 136o for the triplet and 105o for the singlet.
While simple carbenes have a triplet ground state, approapriate substituents can reverse this preference.-> carbenes with lone-pair donating substituents such as N, O, and halogens
can have singlet ground states because of such an interactions

33
Singlet carbene addition to alkenes Triplet carbene addition to alkenes
Singlet carbene insertion into a C-H bond Triplet carbene insertion goes via radical abstraction followed by recombination

34
Carbene generation Thermal decomposition of diazoalkanes
N-nitrosourea
Base-induced eleimination of nitrosourea
Base-induced eleiminationof tosylhydrazone
Decomposition of diazirine
Base-induced alpha eleimination of haloform
All the thermal protocols described above initially form singlet carbenes, as do the photolysis of diazo and diazirine compounds.

35
The Simmons-Smith reagent (ICH2ZnI) also act as a carbene source. However, the reaction between CH2I2 and Zn does not generate a full-fledged free carbene, but instead a carbenoid.
Carbenoid is a carbene that is stabilized by complexation to a metal.

36
MechanismSinglet carbenes give 100% stereospecific reactions (syn addition).Triplet carbenes give mixtures.
Triplet carbenes
triplet biradical
spin flip
spin flip
- Stereochemistry of the alkene is typically not completely lost in the product, which indicates that the spin inversion and bond rotation rates must be comparable.
- The more electron rich the alkene, the faster the carbene addition.- The dialkylcarbenes (more unstable) are less selective than dihalocarbenes or carbenes
with neighboring N, O atoms. (more stable carbenes are more selective)- Carbenes are highly reactive species, and if an olefin or other addition partner is not available,
carbenes will indiscriminately insert into C-H bonds.

37
Eliminations10.12 Eliminations to form carbonyls or carbonyl-like intermediates
acetal
Correct; 1. stereocenter in R -> retention2. -18OR -> release in solution after reaction

38
10.12.3 Catalysis of the hydrolysis of acetals
Specific-acid catalyzed pathway: OR -> poor leaving groups
Not plausible pathway
General-acid catalyzed pathway: OR -> good leaving groups

39
10.12.4 Stereoelectronic effects
two antiperiplanar

40
10.12.5 CrO3 oxidation – The Jones reagent
R OH(ClCO)2, DMSO
pyridine R O
H
Swern oxidation

41
10.14 Elimination reactions for aliphatic systems–formation of alkenesE1 and E2 reactions1,2-elimination (β-elimination) and 1,4-elimination
1,2-elimination or β-elimination
1,4-elimination
l
4l
4Acid-catalyzed 1,4-elimination
E1:
E2:

42
Other types of elimination
elimination of 1,2-dihaloalkanes
elimination of 1,4-dihaloalkanes
oxidative addition of Zn to C-X bond
Reverse aldol reaction
Reverse Michael reaction

43
10.13.3 Contrasting elimination and substitution
1. Elimination will dominate if the carbon with the leaving group (LG) is not susceptible to nucleophilicattack, such as a tertiary R group.2. E1 involves carbenium ion intermediates, and thus are facilitated by all the factors that stabilize carbeniumions. These are the same factors that facilitate SN1reactions.3. In highly ionizing solvents and with R groups that readily form carbenium ions, the ratio of substitution to elimination products is typically independent of the LG.4. In solents of lower ionizing power, the ratio of substitution to elimination products does depend on the LG.

44
10.13.4 E1cB
Any elimination that first form the conjugate base of the reactant is referred to as E1cB (elimination, unimolecular, conjugate base).
α-CH

45
10.13.7 Regiochemistry of elimination
- Saytzeff’s rule: the more substituted double bond will dominate, a common observation for both E2 and E1 reactions. -> Saytzeff elimination
- Hofmann elimination: the product with the less substituted double bond is formed.
E1E2
- Saytzeff’s rule: the more substituted double bond will dominate, a common observation for both E2 and E1 reactions. -> Saytzeff elimination
- Hofmann elimination: the product with the less substituted double bond is formed.- With E1 reaction, Saytzeff elimination dominates because the transition state for proton removal
from the carbenium ion has double bond character.- The rationalization for Sayzeff elimination in E2 reactions is similar to the reasoning for E1 reactions.

46
Elimination reactions with quaternary ammonium and sulfonium LG give preferential Hofmann elimination.
1. Steric hindrance by bulk quaternary ammonium and sulfonium LG, 2. The number of hydrogens to be removed3. Electronic effects; a strongly electron withdrawing cationic LG creates a significant amount of
positive charge on the neighboring hydrogens.However, electron donating alkyl group diminish this charge on the neighboring hydrogens, and thus the most positive hydrogens are those on the less substituted carbon -> preferential deprotonation of the less substituted carbon.
If there are severe steric factors that make the hydrogen on the more substituted carbon inaccessible, Hofmann elimination will dominate the product mixture.
H H
1H3H
3H2H

47
10.13.8 Stereochemistry of eliminations-orbital considerations
E1; in the low-ionizing solvent such as nitromethane-> gives only elimination products via a syn pathways; a contact ion pair is formed and
the tosylate is the base that removes the proton.in more ionizing solvents such as aqueous ethanol, all four possible products are formed.

48
E2; anti elimination is preferred1. conformational preferences, 2. orbital effect
eclipsed
(Bs = SO2C6H4Br)

49
E2; syn elimination can occur when one or more of the following circumstances occurs1. a synperiplanar arrangement can be achieved but an an antiperiplananr one cannot
120o
2. The counterion of the base is ion paired with the base and the leaving group.
3. Strong steric factors favor the syn pathway.
13%Addition of 18-crown-6 0%

50Two products are observed
one product is observed
Antiperiplanar elimination

51
10.13.10 Thermal elimination (pyrolysis)
Cope elimination
Chugaev elimination
heat
10.13.9 Dehydration
Only syn elimination
N-oxides
Xanthate esters
Esters (400-450 oC)

52

53
Combining addition and elimination reactions(substitutions at sp2 centers)

54
10.15 The addition of nitrogen nucleophiles to carbonyl structures, followed by elimination
Schiff base; unstable to be isolated.However, when aromatic groups are placedon either C or N, imines are stable to be often isolated. In addition, oximes (R’ = OH),semizarbazones (R’ = NHCONH2) and hydrazone (R’ = NHR) are very stable.Schiff base

55
10.15.2 Acid-base catalysis
Commonly bell-shaped pH versus rate profiles for imine and enamine formation.

56
Strongly nucleophilic amines (hydroxylamines and alkylamines): A -> amines add directly at all pHs, but below pHs around 4 this direct addition becomes rds. This is because there is a low concentration of unprotonated amine present at low pHs. At the high pHs where the carbinolamine breakdown is rds, kobs decreases as the pH is increased.The rate has a maximum where the amine is present in high enough concentrations as the free base form to react with a reasonable rate, but there is also enough acid present to catalyze the elimination of water from the carbinolamine, hence the bell-shaped pH-rate profile.weakly nucleophilic amines (aryl amines): B -> the amines are not nucleophilic enough to directly add to the carbonyl, and general-acid catalysis is found for this step.The amines should be in its free base form, and therefore the rate still increases with increasing pH. At higher pHs, the dehydration becomes rds (step 3), and it involves general-acid catalysis. Therefore, in this reaction both the addition and elimination steps are general-acid catalyzed, but enough free base form of the amine still needs to be present to produce a reasonable rate.
carbinolamine

57
10.16 The addition of carbon nucleophiles, followed by elimination-the Wittig reaction
ylide
P=C or P=O can be acceptable, but the d orbitals on P are too high in energy to participate in a significant manner in the bonding to phosphorus. Thus, the zwitterion forms (ylide) are more representative of the true chemical structure.

58
10.17 Acyl transfers
10.17.1 General electron-pushing schemes
tetrahedral intermediate

59
Other possible pathways, however, the most common pathway is addition-elimination via a tetrahedral intermediate.
highly acidic conditions

60
10.17.2 Isotope scrambling
Apparently all the carboxylic acid derivatives (esters, acyl halides, anhydrides and amides) can proceed through tetrahedral intermediates during acyl transfers.
R
O
OR' R
HO
OR'
18OHH2O18
R
O18
OR'+
R
O
OR'
Caution: observation of isotope exchange -> a good evidence for a tetrahedral intermediatesthe lack of isotope exchange -> we do not know which is correct because if nucleophilicattack is rds (k2 >> k-1), little exchange into the starting material will be seen. Amides display such behavior under acidic conditions.
rds little exchange

61
10.17.4 Catalysis
Reaction of an alcohol or water with an acid halide in the presence of trialkyl amine -> not only base neutralizes the HX, but also significantly enhances the rate of the reaction.
Other species such as anhydrides and esters are also susceptible to this form of catalysis.
a better nucleophile than ROH or H2O
a highly reactive carbonyl
Amide hydrolysis under basic conditions
base-initiated reaction, not base-catalyzed reaction
rds
very bad LG
very rapid 18O scrambling is observed.
Base-mediated reaction is not very effective for amide hydrolysis.
not very effective

62
Amide hydrolysis under acidic conditions
Since amides are so unreactive toward nucleophilic attack, specific-acid catalysis is most commonlyobserved.
Little to no 18O scrambling into reactants is observed, but still tetrahedral intermediate exists.
extremely acidic conditions
acidic conditions
effective

63
Ester hydrolysis under basic conditions very effective
k2 ~ k-1 (the rate of departure of -OH ~ the rate of departure of -OR)However, a good LG departs much faster.
base-initiated reaction, not base-catalyzed reaction
Ester hydrolysis under acidic conditions very effective
An addition-elimination process

64
Catalytic triad
Serine proteases: the catalytic triadeg) chymotrypsin

65
Metalloproteases: Zn(II) catalysiseg) carboxypeptidase A

66
Peptide synthesis


















