
Achromatopsia
37
Achromatopsia By: Yousuf Kamran BS-Biotechnology (2010-14) IMBB, BZU Multan
-
Upload
yousuf-kamran -
Category
Healthcare
-
view
264 -
download
0
description
Introduction, types, causes, and molecular genetics of achromatopsia.
Transcript of Achromatopsia

AchromatopsiaBy:
Yousuf Kamran
BS-Biotechnology (2010-14)
IMBB, BZU Multan

1.Introduction2.Signs & Symptoms
3.Diagnosis & Testing
4.Types 5.Causes6.Differential
Diagnosis
7.Treatments/Management
8.Molecular Genetics
9.Molecular Genetic Testing
10.Molecular genetic
Pathogenesis
11.Achromatopsia in Pakistani Families
CONTENTS

What is Achromatopsia?
Achromatopsia simply means “loss of color vision”
We can define it as “total inability to see colors”
A rare, non-progressive inability to distinguish any colors as a result of absent or nonfunctioning retinal cones
Persons with achromatopsia see everything in black, white and shades of grey
They have little or no cone vision
In normal eyes there are 6 million cone cells and 100 million rod cells
Patients have to rely just on rods which do not provide color vision and “saturate” at higher levels of illumination


So an achromat (person having achromatopsia) has impaired color discrimination and is either totally colorblind or almost totally colorblind
The severity varies between individuals
Achromatopsia is rather a syndrome because it exhibits symptoms of five different related disorders
(1) Hemeralopia (2) Amblyopia (3) Photophobia
(4) Nystagmus (5) Reduced visual acuity
Other names of achromatopsia are ACHM, Complete or incomplete color blindness, Rod monochromacy, Total color blindness etc
Usually autosomal recessive
Prevalence is 1 in 30,000 people (0.0033%)

SIGNS AND SYMPTOMS
Little or no color perception
Hemeralopia: severe day blindness
Amblyopia: reduced visual acuity
Nystagmus: uncontrolled oscillatory movement of eyes
Photophobia: aversion from light

Diagnosis and Testing
Color Vision Test The color perception of individuals
with achromatopsia (achromats) is unreliable
In general, all achromats have anomalous (impaired) color discrimination along all three axes of color vision corresponding to the three cone classes
(1) the protan or long-wavelength-sensitive cone axis (red)
(2) the deutan or middle-wavelength-sensitive cone axis (green)
(3) the tritan or short-wavelength-sensitive cone axis (blue)

Electrophysiology In the single-flash electroretinogram (ERG), the photopic
(high light levels) response is absent or markedly diminished while the scotopic (low light levels) response is normal or mildly abnormal.
Visual Fields Small central scotomas can be demonstrated in some
individuals by careful testing
Fundus Appearance Many affected individuals have a normal-appearing
fundus

Complete inability to see colors
Lack of function of all three types of cone (or photopic) photoreceptors of the eye
Characterized with:
greatly reduced visual acuity
Hemeralopia
Nystagmus
Severe photophobia
Ability to see colors partially
one or more cone types may be partially functioning
Symptoms are less severe than complete achromatopsia
Reduced visual acuity with or without nystagmus or photophobia
Also called dyschromatopsia
CompleteAchromatopsia
IncompleteAchromatopsia
Types
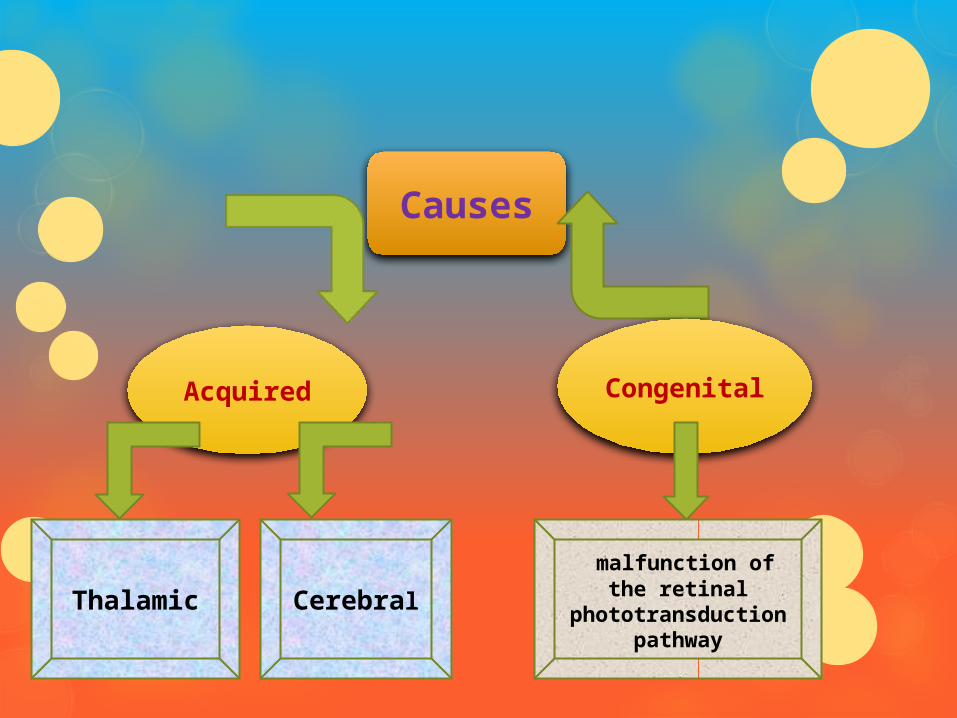
Causes
AcquiredCongenita
l
Thalamic Cerebral
malfunction of the retinal
phototransduction pathway

Causes
Acquired Thalamic Achromatopsia: caused by damage to
the thalamus
Most frequently caused by tumor growth
Cerebral Achromatopsia: caused by damage to the cerebral cortex
Most frequently caused by physical trauma, hemorrhage or tumor tissue growth

Causes
Congenital Congenital forms of achromatopsia are due to
malfunction of the retinal photo transduction pathway
Result from the inability of cone cells to properly respond to light input by hyperpolarizing
Known genetic causes of this are mutations in the cone cell cyclic nucleotide-gated ion channels CNGA3 (ACHM2) and CNGB3 (ACHM3) as well as the cone cell transducin, GNAT2 (ACHM4)

Differential Diagnosis
As achromatopsia is characterized with severely reduced visual acuity, pendular nystagmus, increased sensitivity to light, and reduced or complete loss of color discrimination and other psychophysical and electroretinographic findings
The following retinopathies may be confused with achromatopsia

Differential Diagnosis
Blue cone monochromatism Also called S-cone monochromacy or X-chromosome-linked
achromatopsia
Like achromatopsia, it is also characterized with reduced visual acuity, nystagmus, normal fundus and poor or no color discrimination
BUT in patient with BCM the peak of the photopic luminosity function is near 440 nm not 507nm
Actually 440nm is the peak sensitivity of the S cones and 507nm is the peak sensitivity of rods
Which means that S cones are also functioning alongwith rods
A special four-color plate test or a two-color filter test can clinically distinguish blue-cone monochromats from achromats (rod monochromats)

Differential Diagnosis
Cone Monochromatism Complete achromatopsia with normal visual acuity
Achromatopsia is less often confused with two other extremely rare forms of cone monochromatism which are L or red cone monochromacy and M or green cone monochromacy
In these nystagmus and light aversions are not present and the visual acuity and the cone ERG are normal:
Red cone monochromacy: in this only red cones may be functioning in addition to rods
Green cone monochromacy: in this only green cones may be functioning along with rods

Differential Diagnosis
Cone Dystrophies In cone dystrophy, cone function is normal at birth and symptoms
appear later
These include reduced visual acuity, photophobia, increased sensitivity to glare, and abnormal color vision
The age of onset of vision loss may be as early as childhood or as late as the seventh decade
Differentiating between achromatopsia and cone dystrophy can be difficult, particularly in individuals with onset in early childhood
But best clinical discriminator between achromatopsia & cone dystrophy is progression
Cone dystrophy is progressive in nature while achromatopsia is not

Treatments/Management
Generally there is no as such treatment to cure achromatopsia
But to cope with achromatosia there are
Dark or special filter glasses
Red tinted contact lenses
Eyeborg: it is a device that help people to percieve colors through sound waves


Molecular GeneticsLocus name
Gene Symbol
Chromosomal locus
Protein Name
ACHM2 CNGA3 2q11.2 Cyclic nucleotide-gated cation channel alpha 3
ACHM3 CNGB3 8q21.3 Cyclic nucleotide-gated cation channel beta 3
ACHM4 GNAT2 1p13.3 Guanine nucleotide binding protein G subunit alpha 2
ACHM5 PDE6C 10q23.33 Cone cGMP specific 3’,5’ cyclic phosphdiesterase subunit alpha
ACHM6 PDE6H 12p12.3 Retinal cone rhodopsin sensitive cGMP 3’,5’ –cyclic phosphodiesterase subunit gamma

Molecular Genetic TestingGenes Proportion of
Achromatopsia Attributed to Mutations in This Gene
Test Method Mutations detected
Test availability
CNGB3 ~40-50% Targeted mutation analysis
c.1148delC Clinical
Sequence analysis
Sequence variants
Deletion/duplication analysis
Unknown
CNGA3 ~25% Sequence analysis
Sequence variants Clinical
Deletion/duplication analysis
unknown
GNAT2 <2% Sequence analysis
Sequence variants Clinical
Deletion/duplication analysis
Exonic or whole-gene deletions
PDE6C <2% Sequence analysis
Sequence variants Clinical
PDE6H ~0.3% Sequence analysis
Sequence variants Clinical

Molecular Genetic Pathogenesis
Molecular pathomechanism of ACHM is either the inability to properly control or respond to altered levels of cGMP
Levels of cGMp controls the opening of cyclic nucleotide-gated ion channels (CNGs)
Mutation in any of the described genes disturb this pathomechanism
In normal eye this pathway is as follows:

Light excites cone visual pigment molecules
GDP is converted into GTP at the guanosine binding site of the transducin alpha subunit (GNAT2)
This activated GTP transducin is released from the inhibitory beta/gamma subunits binds and activates the alpha’-subunit of the cone phosphodiesterase (PDE6C) by retracting the inhibitory gamma-subunit (PDE6H)
PDE hydrolyzes cGMP and effectively reduces its intracellular concentration
This results in the closure of the hetero-tetrameric cGMP-gated cation channels (CNGA3/CNGB3) and, subsequently, membrane hyperpolarization

Molecular Genetic Pathogenesis
CNGA3
Normal allelic variants: CNGA3 consists of eight coding exons
Only a few normal allelic variants
Mostly occurring in non coding region and do not result in amino acid substitution
Pathologic allelic variants: More than 80 different mutations have been reported
Majority of mutations are missense (<80%)
Few nonsense mutations, insertions, and deletions have been observed.

Nucleotide mutation
Protein mutation
Functional? (known or predicted)
Effect
c.C67T p.R23X NO
c.A542G p.Y181C NO Does not properly traffic out of the endoplasmic reticulum
c.C1106G p.T369S Yes Increased calcium influx
c.G830A p.R277H
c.C556T p. L186F No Does not properly traffic out of the endoplasmic reticulum
c.T1565C p. I522T
c.G580A p.E194K NO Does not properly traffic out of the endoplasmic reticulum
c.A485T p.D162V
c.934_936del p. 312delI

Molecular Genetic Pathogenesis
GNAT2 Normal allelic variants: GNAT2 consists of eight
coding exons
Only a few polymorphisms and rare variants are observed; most occur within non-coding regions or do not result in an amino acid substitution.
Pathogenic allelic variants: Only 10 different disease-associated mutations
1 nonsense mutation, 7 deletions/insertions, one large deletion of exon 4, and a mutation c.461+24G>A activating a cryptic splice site and resulting in frame-shift

Nucleotide mutaion protein Functional? (predicted)
c.C235T p.Q79X No?
c.285_291del p.Y95fsX61 No?
IVS3+365_IVS4+974del
p.A101fsX12 No?
c.503_504insT p. L168fsX3 No?
c.802_803insTCAA p. L268fsX9 No?
c.955del p. I319SfsX5 No?

Molecular Genetic Pathogenesis
CNGB3 Normal allelic variants: CNGB3 consists of 18 coding
exons
Only a few polymorphisms and rare variants are observed; most occur within non-coding regions or do not result in an amino acid substitution
Pathologic allelic variants: More than 40 different mutations have been reported
Mostly are non-sense mutations, frame shift deletions and insertions and putative splice site mtations
Mis-sense mutations are ~10 %

Nucleotide mutation
Amino acid mutation
Functional? (known or predicted)
Effects
c.1148delC p.T383IfsX12 NO? Does not traffic to the surface
c.G1006T p.E336X NO?
c.G1208A p.R403Q Yes Increased outward rectification, increased cGMP affinity
c.819_826del p.P273fsX13 NO?
c.29_30insA p.K10fsX9 NO?
c.595delG p.E199SfsX2 NO?
c.1573_1574delinsTT
p.F525N Yes Increased surface expression in oocytes, decreased outward rectification, increased cGMP and cAMP affinity
c.C391T p.Q131X NO?
c.T991-3G Splicing NO?
c.T1635A p.Y545X NO?
c.C926T p.P309L
p.P309L p.R216X NO?

PDE6C Normal allelic variants: PDE6C consists of 22 coding
exons
Several polymorphisms and rare variants are observed; most occur within non-coding regions or do not result in an amino acid substitution
Pathologic allelic variants: To date sixteen different mutations in PDE6C in eight independent families have been described
seven missense and two nonsense mutations, three small indels, and four mutations affecting splicing

PDE6H Normal allelic variants: PDE6CH consists of only three
coding exons
Only few polymorphisms and rare variants are observed
Pathologic allelic variants: To date only a single homozygous nonsense mutation c.35C>G in PDE6H in three affected individuals from two independent families originating from Belgium and the Netherlands have been described

Genetics: pattern of inheritance
2 major types of achromatopsia (rod monochromacy & blue cone monochromatism) have different pattern of inheritance
Rod Monochromacy
Autosomal recessive
Usually this form of achromatopsia occurs
Males and females are equally affected
Persons with this vision disorder have inherited 2 faulty genes, 1 from each parent
Usually found in one generation only
BCM
X-linked recessive
Rare form of achromatopsia
In BCM, cone cells develop normally but the retina is unable to fill them with red or green pigment, thus leaving only blue cones
Almost always, only males are affected by BCM



Genetic analysis of 2 Pakistani families with achromatopsia
Family RP26 Family RP44
CNGA3 Nucleotidemutation
protein type
No mutation detectedc.822G>T p.R274S missense
CNGB3
No mutation detected
Nucleotidemutation
protein type
c.1825delG p.V609WfsX9
frameshift

Genetic analysis of four Pakistani families with achromatopsia
Family 50 Family 55
Family 70
Family 74
CNGA3 nucleotide
protein type No mutation detected
No mutation detected
No mutation detected
c.827A>G p.N276S substitution
missense
CNGB3 No mutation detected No mutation detected
No mutation detected
No mutation detected*

The Island of Colorblinds
The Pingelap Atoll is an island consisting of a circular coral reef surrounding a lagoon in the Pacific Ocean.
In 1775 a huge storm called Typhoon Liengkieki ravaged the island, leaving only 20 inhabitants (90% were killed, meaning the original population was around 200)
One of the survivor was carrier for achromatopsia
Inbreeding was necessary to replenish the population and in the 4th generation of inbreeding Achromatopsia appeared
Today, it has roughly 200-250 residents, roughly 10% of which are affected by total color blindness - known as Achromatopsia and 30% are carriers

References
Azam, M., et al. (2010). "Novel CNGA3 and CNGB3 mutations in two Pakistani families with achromatopsia." Molecular Vision 16: 774.
Sacks, O. (1997). The island of the colour-blind: and, Cycad Island, Pan Macmillan.
Futterman, F. (1998). Understanding and Coping with Achromatopsia, F. Futterman.
Kohl, S., et al. (2004). "CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia." European Journal of Human Genetics 13(3): 302-308.
Achromatopsia GeneReviews™ [Internet] by Kohl S, Jägle H, Wissinger B. 2004 Jun 24 [Updated 2013 Jun 27]
Genetic analysis of four Pakistani families with achromatopsia and a novel S4 motif mutation of CNGA3 by Muhammad Arif Nadeem Saqib, Bilal Malik Awan, Mehwish Sarfraz, Muhammad Nasim Khan, Sajid Rashid, Muhammad Ansar












