
Accelerating the Prediction of Protein...
51
Accelerating the Prediction of Protein Interactions Alex Rodionov, Jonathan Rose, Elisabeth R.M. Tillier, Alexandr Bezginov October 21 2010
Transcript of Accelerating the Prediction of Protein...

Accelerating the Prediction of Protein Interactions
Alex Rodionov, Jonathan Rose, Elisabeth R.M. Tillier, Alexandr Bezginov
October 21 2010

Motivation● The human genome is sequenced, but we don't
know what all the genes do● Genes code for proteins● Genome function = protein interaction● We can learn about proteins and the
genome by studying protein-protein interactions

Motivation● Best known way of studying interactions is in
the lab (“high-throughput experiments”)● Lots of proteins, O(N2) possible pairs● We would like to be able to predict which
protein pairs interact before undertaking tedious and expensive lab experiments

Coevolution● One way to predict protein-protein interactions
is by using coevolution: two proteins that interact tend to evolve over time at similar rates.
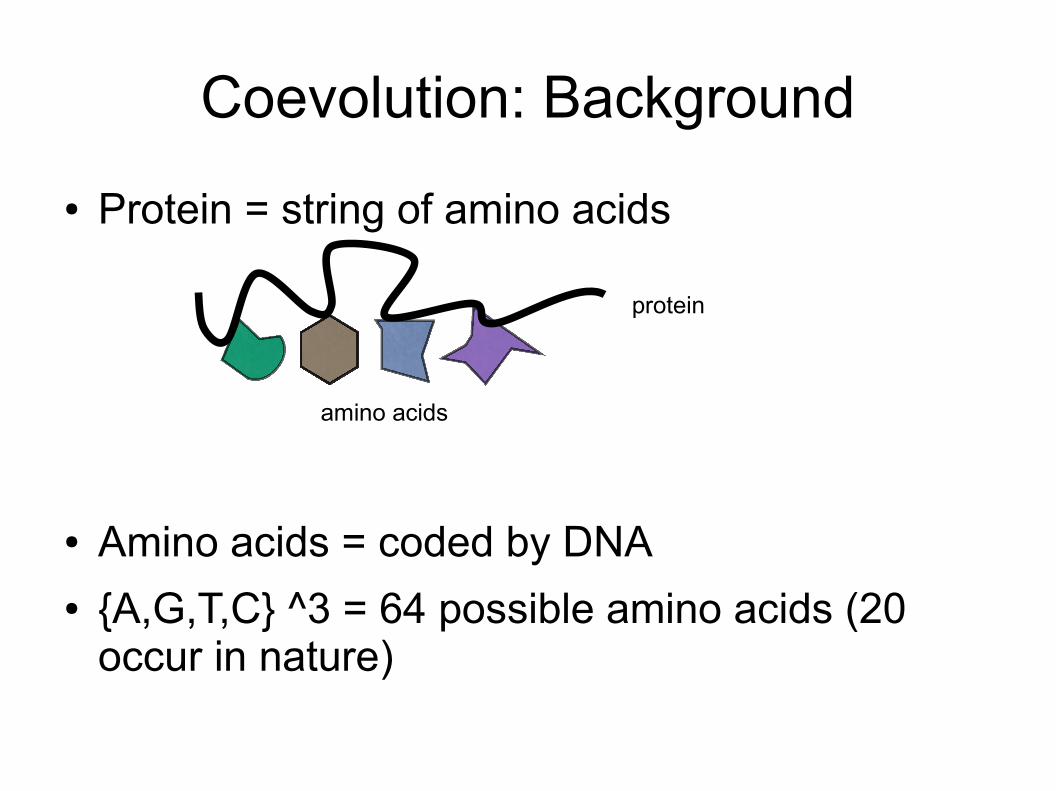
Coevolution: Background● Protein = string of amino acids
● Amino acids = coded by DNA● {A,G,T,C} ^3 = 64 possible amino acids (20
occur in nature)
protein
amino acids
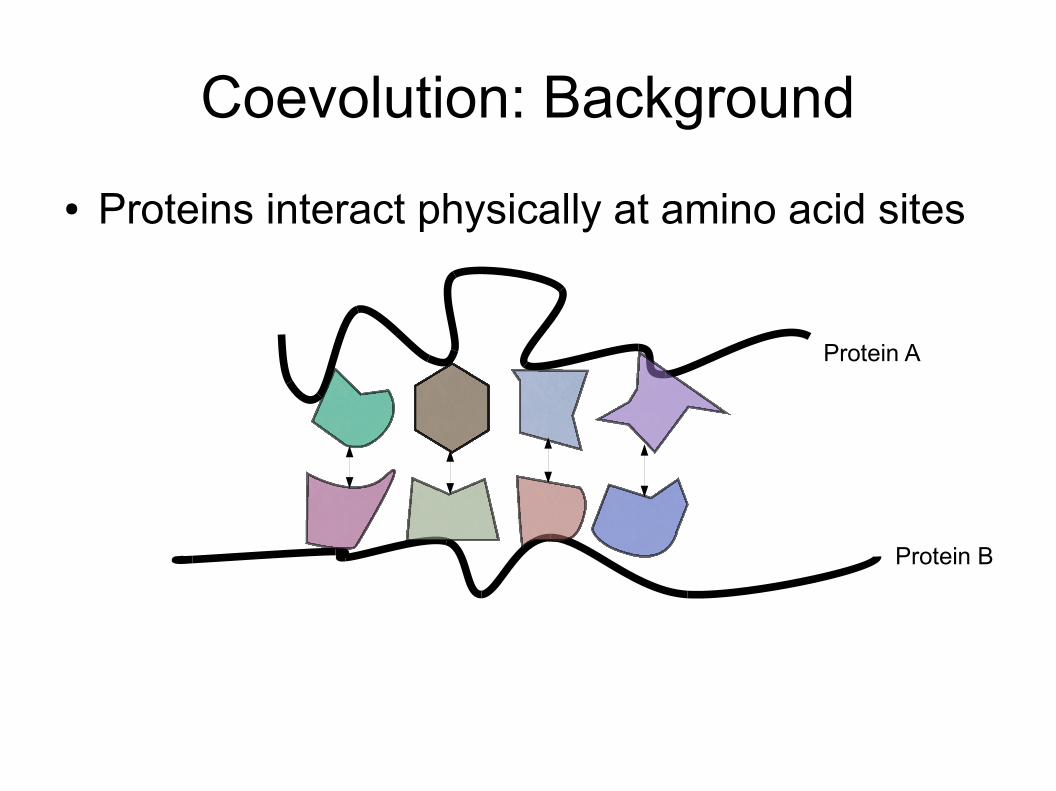
Coevolution: Background● Proteins interact physically at amino acid sites
Protein A
Protein B
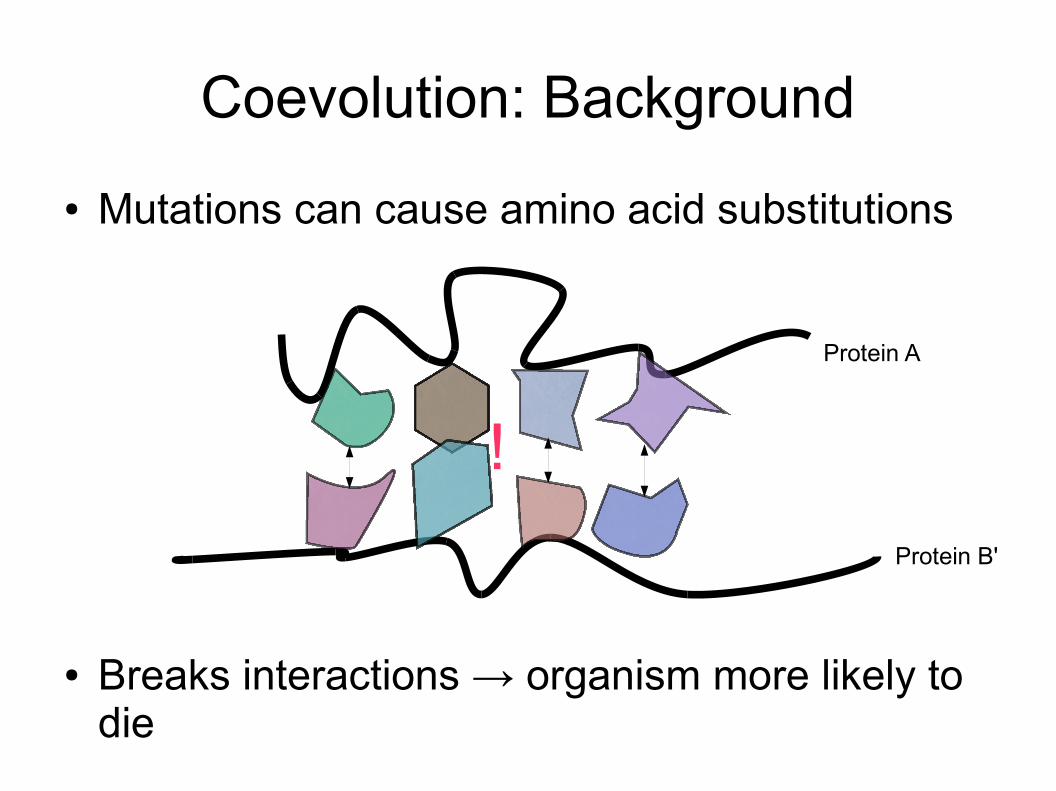
Coevolution: Background● Mutations can cause amino acid substitutions
● Breaks interactions → organism more likely to die
Protein A
Protein B'
!
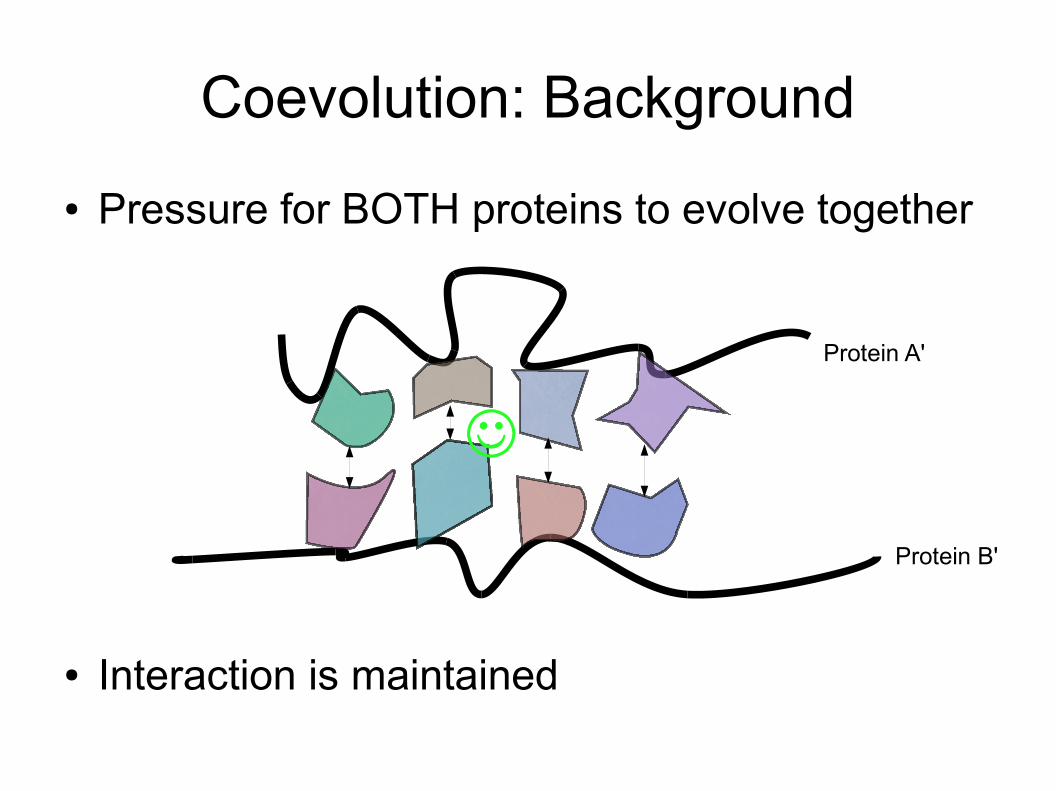
Coevolution: Background● Pressure for BOTH proteins to evolve together
● Interaction is maintained
Protein A'
Protein B'
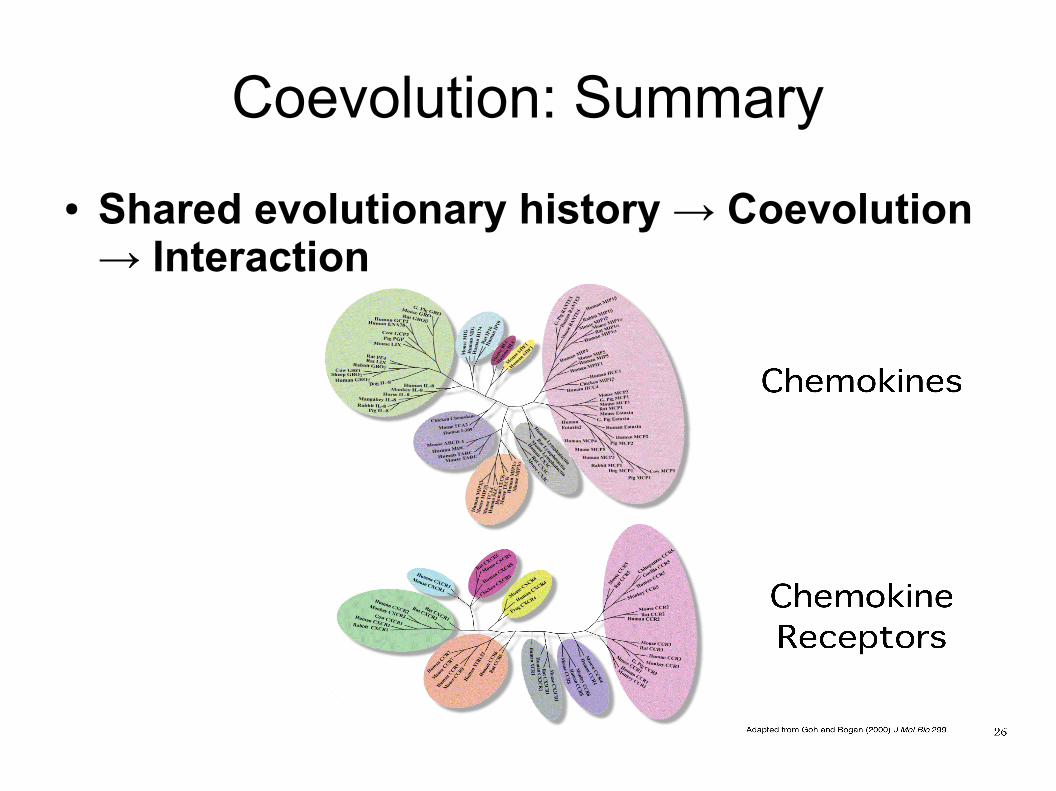
Coevolution: Summary● Shared evolutionary history → Coevolution
→ Interaction

MatrixMatchMaker● A method of protein coevolution detection● Developed by Elisabeth Tillier & Robert
Charlebois (Department of Molecular Biophysics)
● Has been shown to work better than other methods, at the expense of compute time

MMM: Big Picture● Looks at the evolutionary histories of two
proteins● Measures the similarity of the histories by
looking for common sections of those histories● Generates a numerical score indicating the
strength of evidence for coevolution

MMM: Homologous Proteins● There can exist many versions of one protein,
with slight variations, found in different species● These homologous protein variations form a
family. GGSSV
SSVNT
RYWWS
QNNEE
NNFFI
QPREP
SSEEY
RPQTT
LKLVV
VLVLL
TATAA
HumanMouseChickenRabbitFrog
amino acids

MMM: Homologous Proteins● The differences amongst the homologous
proteins provide an evolutionary context/history of the family
● How to quantify these differences and history?

MMM: Distance Matrices● Protein = sequence of amino acids = string● Can take two such strings and calculate a
number representing how different they are (=distance)
MADSTHRNMILEVNDEFHTMLEIMTHRNMILEVNRRFYY
MAD-STHRNMILEVNDEFHTMLEIMTHRNMILEVNRRFYY
0.4

MMM: Distance Matrices● Multiple Sequence Alignment takes all the
members of a protein family and generates all possible pairwise distances
● These distances form a distance matrix
p1 MAD-STHRNMILEVNDEFHTp2 MVDASTHRNMILEVNDEFTIp3 MID-MTHRNMILEVNDEFHTp4 MLEIMTHRNMILEVNRRFYY
p1 p2 p4
p1
p2
p4
0.1
p3
p3
0.30.1 0.6
0.7 0.2
0.5
0
0
0
0
0.1
0.3
0.6
0.7
0.2 0.5

MMM: Evolutionary History● Distance matrix implies evolutionary history of
protein● Submatrices represent histories of subsets of
the homologous variations
p1 p2 p4
p1
p2
p4
0.1
p3
p3
0.30.1 0.6
0.7 0.2
0.5
0
0
0
0
0.1
0.3
0.6
0.7
0.2 0.5p1 p2 p3 p4

MMM: Big Picture Revisited● MMM looks at two distance matrices A and B,
which represent the evolutionary histories of two proteins (two families of homologous proteins)
● Measures the similarity of the histories by looking for similar sub-matrices
● The size of the largest similar sub-matrices is output as a score, indicating the strength for the evidence of co-evolution

MMM: Sub-matrix Similarity● Distance within a matrix is relative to the family,
not absolute → submatrix equality isn't enough for similarity
● Use instead equality up to a scale factor (with some tolerance)
0 1.1 1.9
1.1 0 3.2
1.9 3.2 0
0 3.0 6.1
3.0 0 8.9
6.1 3.2 0
similar

MMM: Sub-matrix Similarity● Sub-matrices need to be at least size 3 (for
concept of similarity to make sense)
● Two similar sub-matrices of size K*K create K pairings of homologous proteins
0 x
x 0
0 y
y 0
always similar
0 1.1 1.9
1.1 0 3.2
1.9 3.2 0
0 3.0 6.1
3.0 0 8.9
6.1 3.2 0
a2 a8 a9
a2
a8
a9
b3 b5 b7
b3
b5
b7
Pairs:
a2 ↔ b3a8 ↔ b5a9 ↔ b7

MMM: Sub-matrix Similarity● Additional constraint: both proteins in a pairing
must belong to the same species● Size of largest similar submatrices = amount of
coevolution ● Homologous proteins paired up by submatrices
are useful for other purposes (downstream analysis tools)
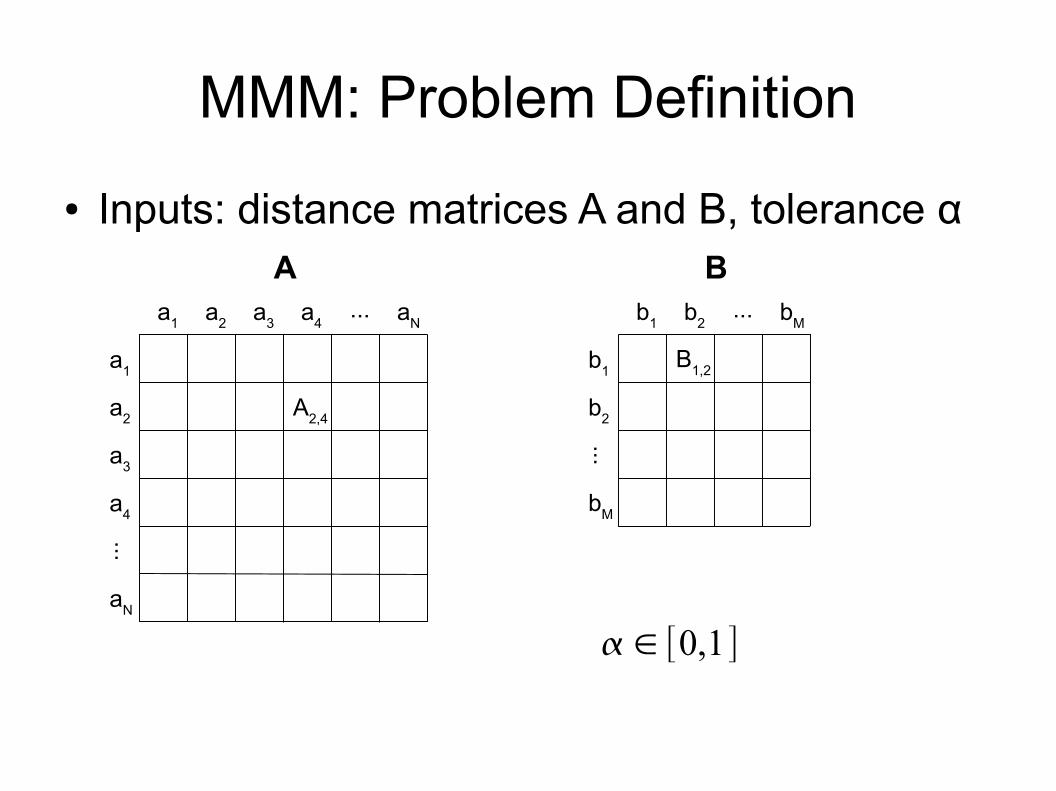
MMM: Problem Definition● Inputs: distance matrices A and B, tolerance α
a1 a2 a3 a4 aN
a1
a2
a3
a4
aN
...
...
A2,4
b1 b2 bM...
b1
b2
...
bM
B1,2
A B
∈ [0,1]

MMM: Problem Definition
● Submatrices A' of A and B' of B form a match M={(a'1,b'1), (a'2,b'2), … , (a'k,b'k)} iff:
a'1 a'2 a'k
a'1a'2
a'k
...
...
A'
0 A'1,2 A'1,k
A'2,1 0
0
0A'k,1
...
...
......
A'2,k
...
...A'k,2
b'1 b'2 b'k
b'1b'2
b'k
...
...
B'
0 B'1,2 B'1,k
B'2,1 0
0
0B'k,1
...
...
......
B'2,k
...
...B'k,2
1: 1−1
⋅Au ,v'
Bu ,v' ≤
Ai , j'
B i , j' ≤ 1
1−⋅Au , v'
Bu , v' ∀ i≠ j , u≠v
2: Ai , j' ≠0, Bi , j
' ≠0 ∀ i≠ j 3: species a i' =species bi
' ∀ i

MMM: Problem Definition● α → 0 : more strict matching● α → 1 : more lenient matching● Goal: Find the set of matches of largest size● Outputs: Largest match size, protein pairs for
each match

Initial Algorithm● Tillier et al. already had a first try at an
algorithm● It took 6 days to process ~6 million matrix pairs

Initial Algorithm● For all protein triplets (a,b,c) from A
● For all protein triplets (w,x,y) from B– If {(a,w), (b,x), (c,y)} is a match then– * For remaining proteins d from A
● ! For remaining proteins z from B– If current match plus (d,z) is also a match, add (d,z), goto *– Else record current match
– Remove latest pair from match, goto ! to resume loop ● Keep largest matches, clear list when larger
example found, report match list at the end● Slow, exhaustive, no pruning of recursion tree

New Algorithm● Our (ECE) work begins here● Make a faster algorithm, maintain correctness● Big picture: recast MMM problem as a graph
problem, use well-known and efficient algorithms to solve sub-problems
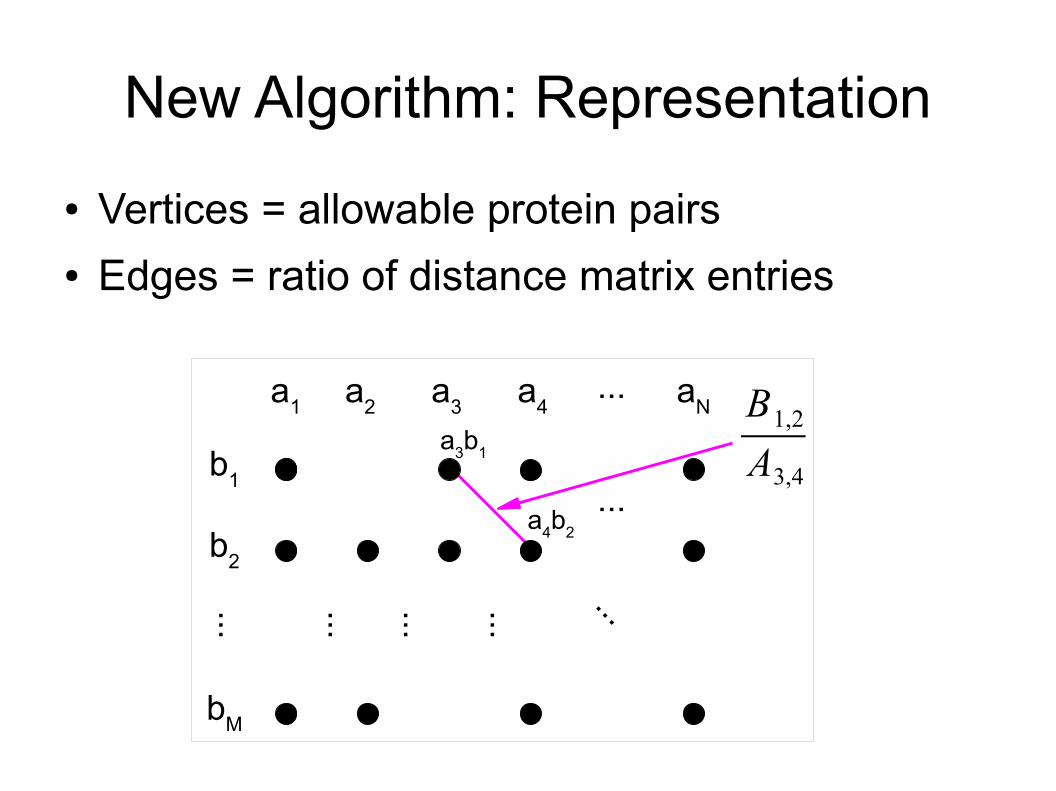
New Algorithm: Representation● Vertices = allowable protein pairs● Edges = ratio of distance matrix entries
a1 a2 a3 a4 aN...
b1
b2
...
bM
a3b1
a4b2
B1,2
A3,4......... ... ...
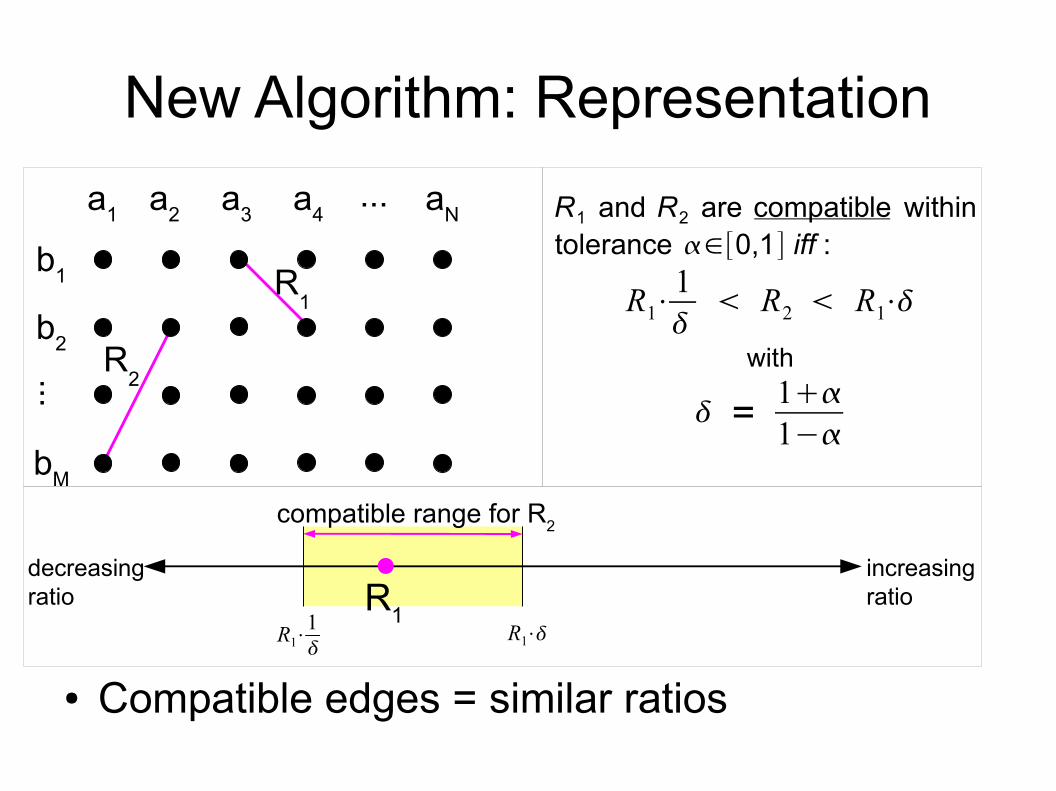
New Algorithm: Representation
● Compatible edges = similar ratios
a1 a2 a3 a4 aN...
b1
b2
...
bM
R1
R2
R1 and R2 are compatible within tolerance ∈[0,1] iff :
R1⋅1
R2 R1⋅
with
= 11−
decreasingratio
increasingratioR1
R1⋅1
R1⋅
compatible range for R2
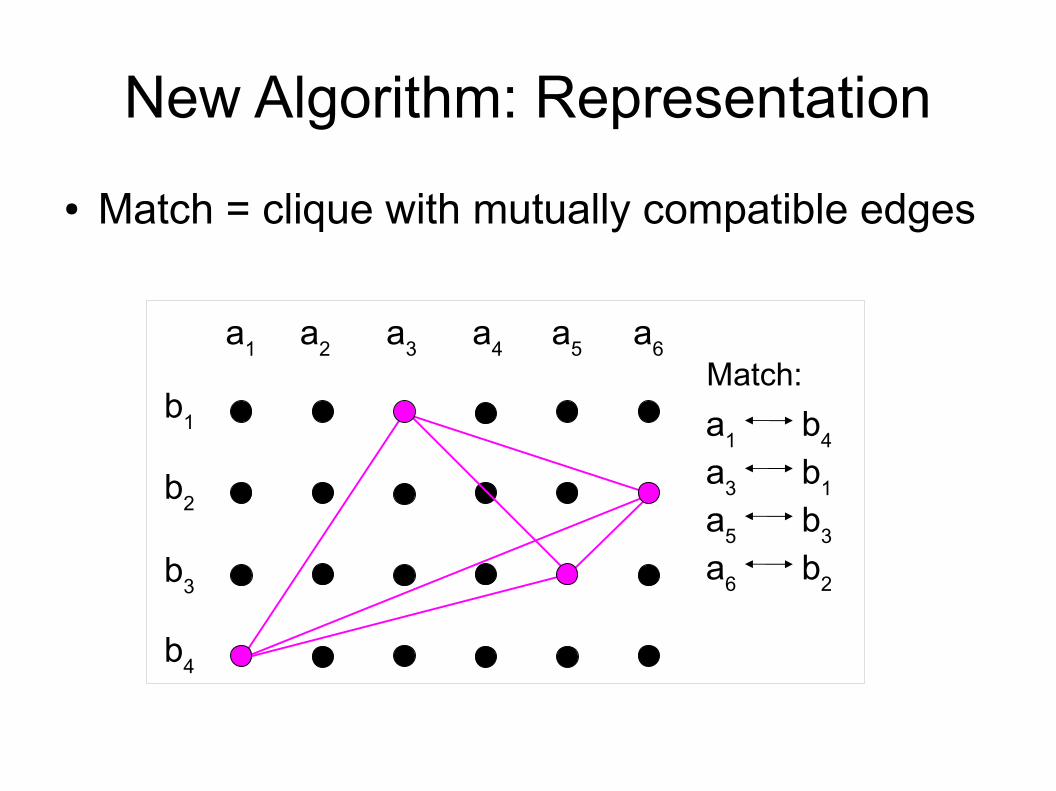
New Algorithm: Representation● Match = clique with mutually compatible edges
a1
a3
a5
a6
b4
b1
b3
b2
Match:a1 a2 a3 a4 a6
b1
b2
b4
a5
b3

New Algorithm: Method● Every match has edge of minimum ratio● Edges in match are mutually compatible iff they
are forward-compatible with the edge of minimum ratio
R1
R3= R
min
R2
R4
R6
R5
decreasingratio
increasingratio
RminRmin⋅1
Rmin⋅
forward-compatible range for Rmin
R2
R4R
5R
1R
6

New Algorithm: Method● Go through every edge e in the graph● Assume e is the edge of minimum ratio of some
match(es)● Work backwards to find the largest of those
matches● After picking e, this just means finding the
maximum cliques on a subgraph H:● V(H) = vertices adjacent to e● E(H) = edges forward-compatible with e
● Repeat for all e → Find all largest matches
New Algorithm: Method
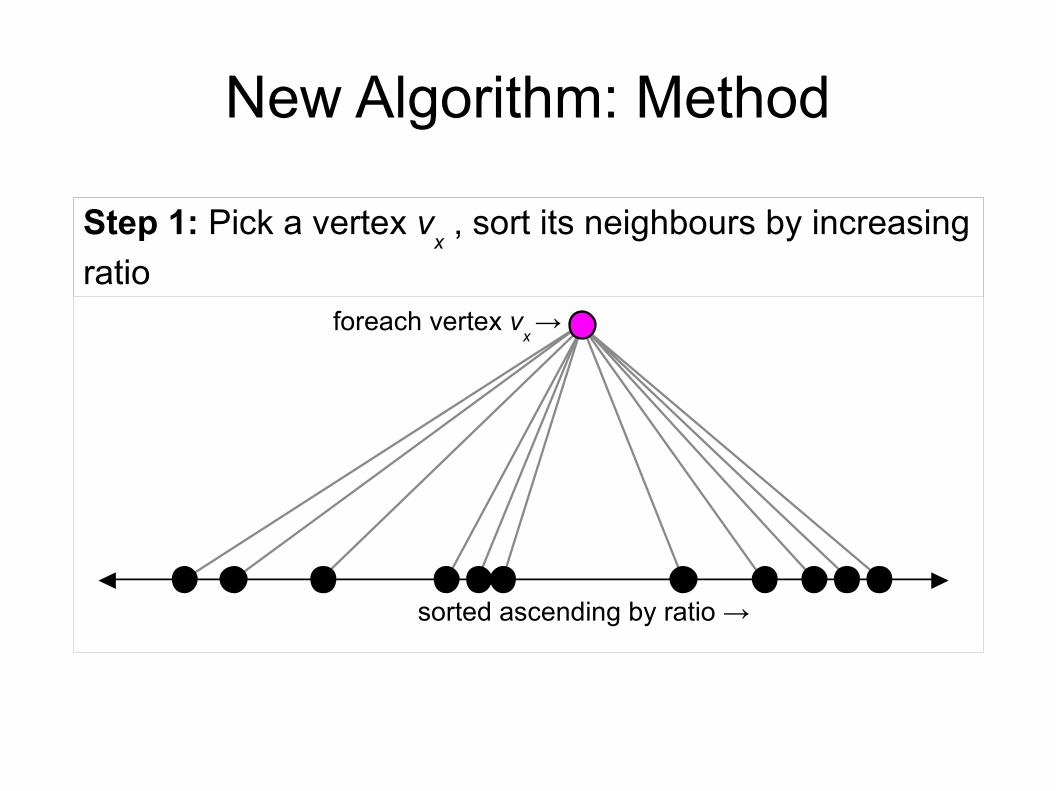
foreach vertex vx →
sorted ascending by ratio →
Step 1: Pick a vertex vx , sort its neighbours by increasing
ratio
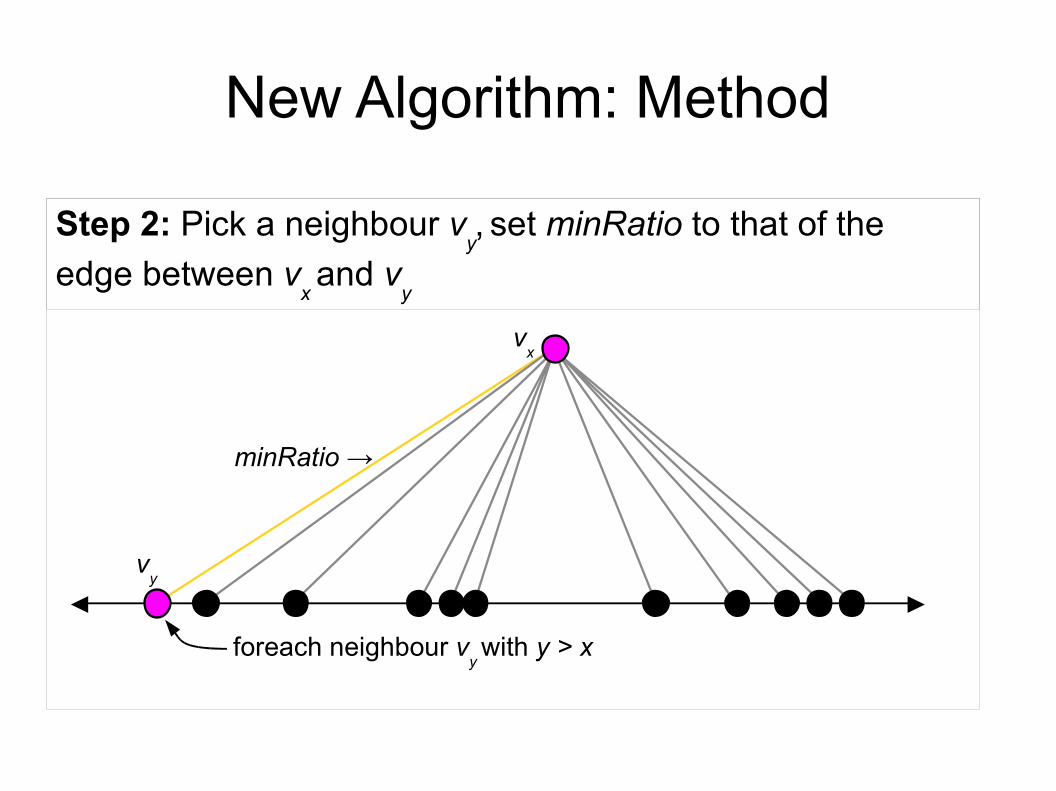
New Algorithm: Method
vx
Step 2: Pick a neighbour vy, set minRatio to that of the
edge between vx and v
y
vy
foreach neighbour vy with y > x
minRatio →
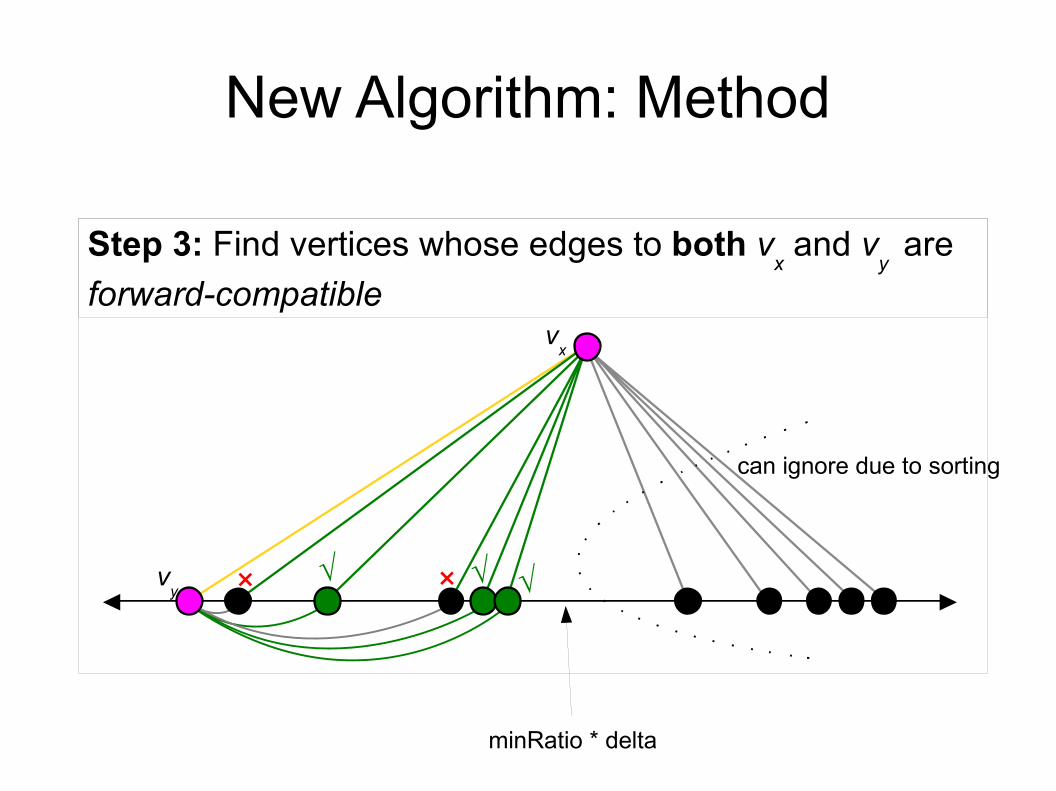
New Algorithm: Method
vx
Step 3: Find vertices whose edges to both vx and v
y are
forward-compatible
vy × ×√ √ √
can ignore due to sorting
minRatio * delta
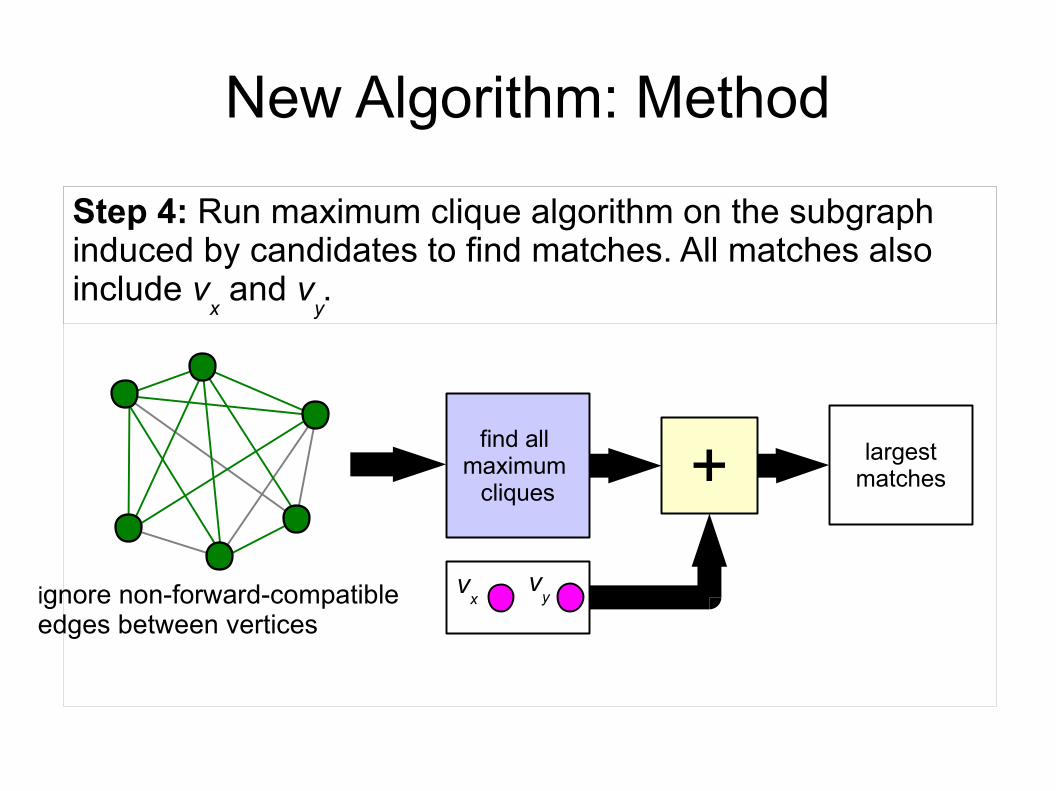
New Algorithm: Method
find all maximum
cliques
Step 4: Run maximum clique algorithm on the subgraph induced by candidates to find matches. All matches also include v
x and v
y.
ignore non-forward-compatibleedges between vertices
vx
vy
+ largestmatches

New Algorithm: Method● Repeat● Every choice of v
x and v
y creates a max clique
problem● Keep list of largest matches, and the largest
match size

Max Clique Algorithm● Using Ostergard (2002) algorithm● Recursive, branch and bound algorithm:
0 1 2 3 4 5 6
0 1 2 3 4 5 6
0 1 2 3 4 5 6
0 1 2 3 4 5 6
clique: 0,3,5 (new max)
0 1 2 3 4 5 6
clique: 0,5 (less than max, no report)
0 1 2 3 4 5 6
clique: 0,6 (less than max, no report)
0 1 2 3 4 5 6
back out: can't match or beat max of 3
0 1 2 3 4 5 6
etc..
0 1 2 3 4 5 60 1 2 3 4 5 6

Max Clique Algorithm● Outer loop:
0 1 2 3 4 5 60 1 2 3 4 5 6
perform B&B algorithm on last vertex to the end
1
record max clique size in MSPV
0 1 2 3 4 5 60 1 2 3 4 5 6 2 1
work backwards...
0 1 2 3 4 5 60 1 2 3 4 5 6 3 3 3 2 2 1
new bound condition: if current size + MSPV[v] < max size then leave early
MSPV[ ]

Max Clique Algorithm● Ordering of vertices important for performance● Recommended ordering (Ostergard):
● Perform greedy vertex coloring● Sort vertices by decreasing color class● Sort by decreasing degree within color class

New Algorithm: Results● Data set: ~3500 matrices, ~6 million matrix
pairs● Matrices represent proteins with at least 1
human variant● Tolerance (alpha) set to 0.1● Compared total and per-problem runtime of
new vs. old algorithms
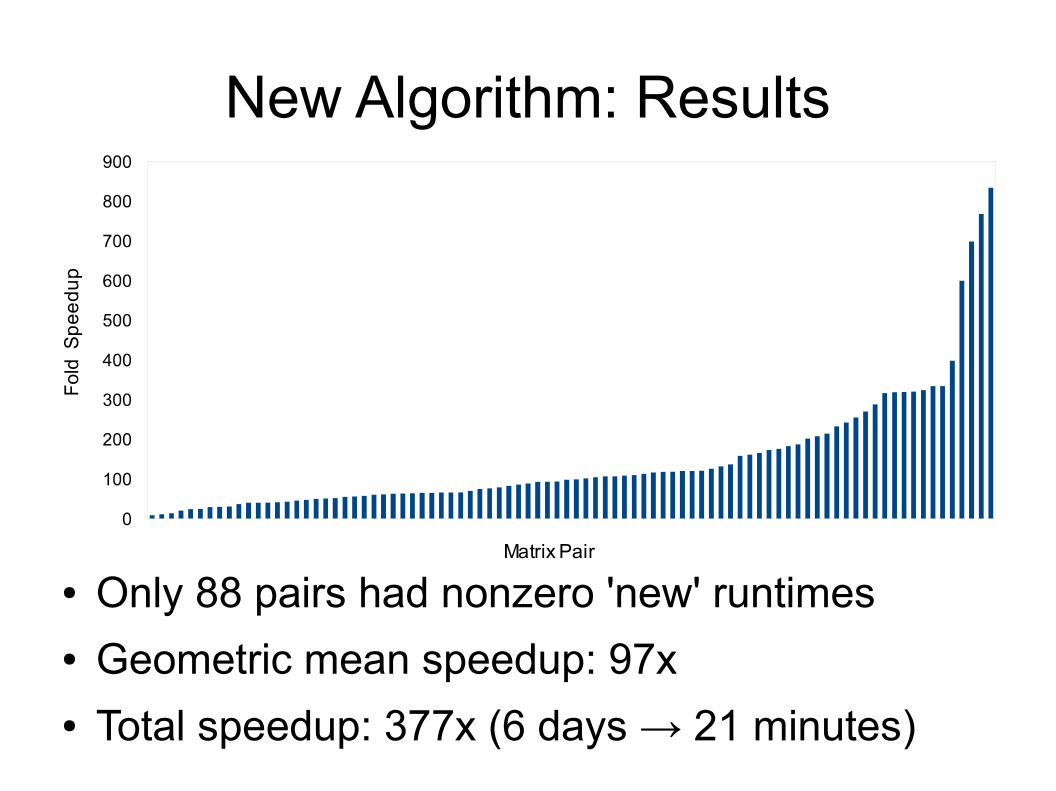
New Algorithm: Results
● Only 88 pairs had nonzero 'new' runtimes● Geometric mean speedup: 97x● Total speedup: 377x (6 days → 21 minutes)

Hardware Acceleration● FPGA-related part of this talk● Max clique is NP-complete● Setup portion of algorithm is O(n^4)● Max clique time → 100% for larger problems● Setup tasks include things like sorting → best
leave this on the CPU● Max clique algorithm is mostly serial, but there
are LOTS of max clique problems to solve!

Max Clique: FPGA vs GPU● Recursive, depth-first● Not data parallel at all● Input data = adjacency matrix = can be
represented as array of bits

Hardware Platform
● Terasic DE3● Stratix III L340● USB 2.0● 1GB DDR2 memory
● Hopefully porting to DE4 → PCI Express!

HW ↔ SW Interface● 'Ports' package over USB 2.0● Looks like open/read/write C calls to SW● Data sent via TCP/IP to computer hosting the
DE3● Auto-generated hardware block feeding desired
signals + handshaking to design● Work in progress itself
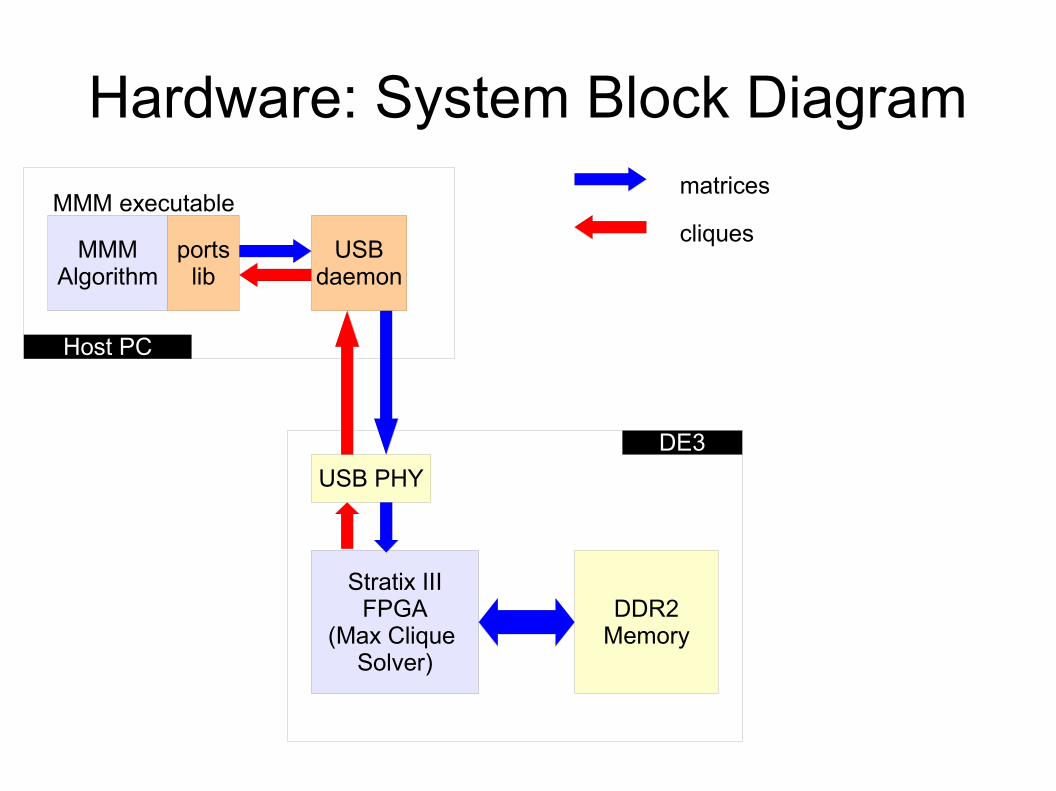
Hardware: System Block Diagram
MMMAlgorithm
portslib
USBdaemon
MMM executable
USB PHY
Stratix IIIFPGA
(Max Clique Solver)
DDR2Memory
Host PC
DE3
matrices
cliques
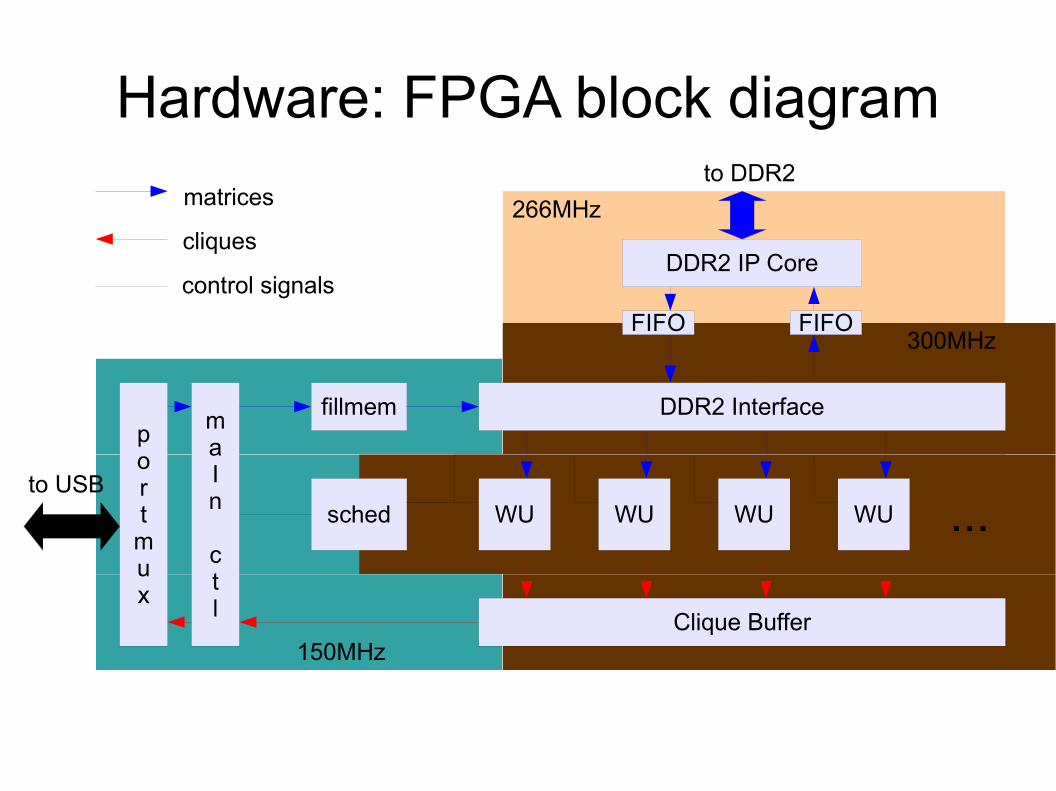
Hardware: FPGA block diagram
DDR2 Interface
WU
Clique Buffer
fillmem
sched WU WU WU
maIn
ctl
...
DDR2 IP Core
port
mux
to DDR2
to USB
matrices
cliques
control signalsFIFO FIFO
266MHz
300MHz
150MHz
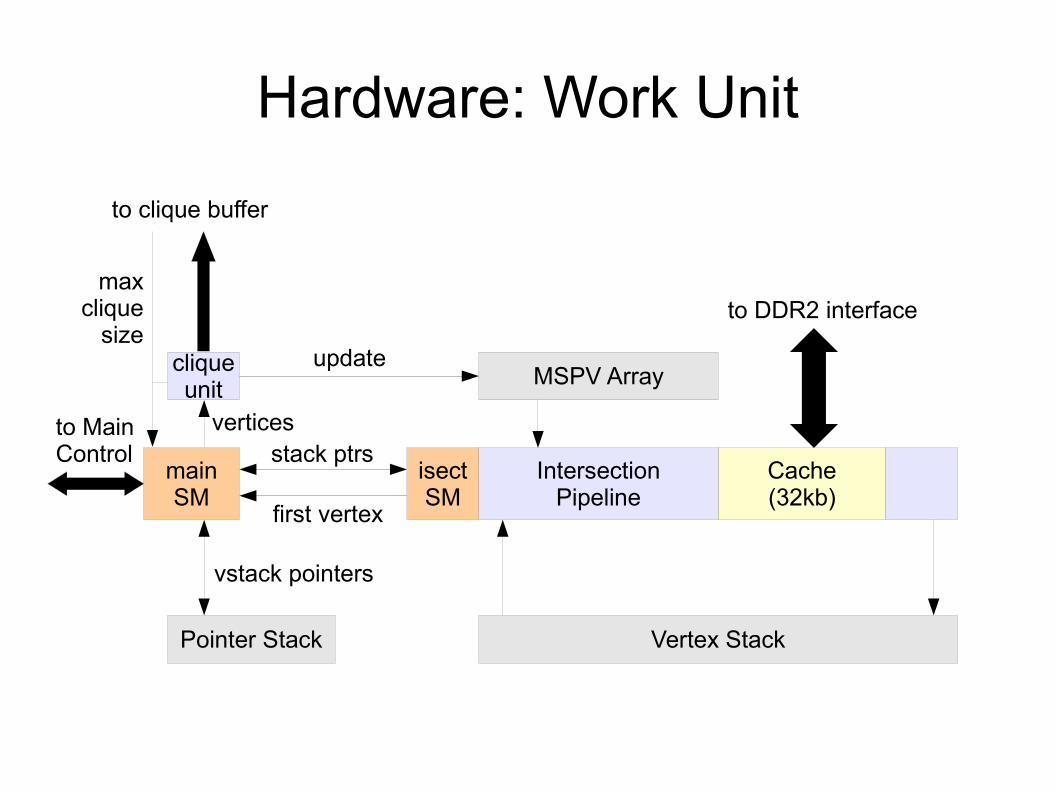
Hardware: Work Unit
Cache(32kb)
IntersectionPipeline
Vertex Stack
isectSM
mainSM
to DDR2 interface
MSPV Array
to MainControl stack ptrs
first vertex
cliqueunit
update
vertices
Pointer Stack
vstack pointers
to clique buffer
maxclique
size

Hardware: Progress and Future Work
● No speed results yet● Version 1: One WU, no DDR2, no MMM
support● Used to verify WU operation
● Version 2: One WU, DDR2● Hardware ready, software support coming
● Version 3: Multiple WU, DDR2● Problem: USB 2.0 bandwidth → need PCIe

Conclusion● Interesting biological problem● Interesting mathematical problem● Great algorithmic/software speedup achieved● Hardware work in progress

Done
Questions?


















