
A TARGETABLE GATA2-IGF2 AXIS CONFERS … · Samuel J. Vidal Submitted in partial ... Stephen...
148
A TARGETABLE GATA2-IGF2 AXIS CONFERS AGGRESSIVENESS IN CHEMOTHERAPY RESISTANT PROSTATE CANCER Samuel J. Vidal Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2015
Transcript of A TARGETABLE GATA2-IGF2 AXIS CONFERS … · Samuel J. Vidal Submitted in partial ... Stephen...

A TARGETABLE GATA2-IGF2 AXIS CONFERS AGGRESSIVENESS IN
CHEMOTHERAPY RESISTANT PROSTATE CANCER
Samuel J. Vidal
Submitted in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy
under the Executive Committee
of the Graduate School of Arts and Sciences
COLUMBIA UNIVERSITY
2015

©2014
Samuel J. Vidal
All rights reserved

ABSTRACT
A Targetable GATA2-IGF2 Axis Confers Aggressiveness in
Chemotherapy Resistant Prostate Cancer
Samuel J. Vidal
Prostate cancer is a common malignancy with nearly one million annual diagnoses
worldwide. Among a subset of patients, primary disease eventually progresses to disseminated
castration resistant prostate cancer (CRPC). In recent years, treatment modalities that improve
survival in CRPC have emerged including taxane chemotherapy and second generation androgen
signaling inhibitors, among others. Indeed, today the first line chemotherapeutic docetaxel as
well as the second line agent cabazitaxel are mainstays of treatment. However, CRPC inexorably
progresses to a chemotherapy resistant state that ultimately precedes lethality. Elucidating the
molecular determinants of aggressiveness in chemotherapy resistant CRPC may therefore
stimulate new therapeutic strategies that improve clinical outcomes. We used laboratory models
and clinical databases to identify GATA2 as a regulator of chemotherapy resistance and
tumorigenicity in this context. Whole genome expression profiling, clinical validation and
genetic screening approaches revealed that GATA2 regulates a signature of cancer progression
associated genes. Mechanistically, direct upregulation of the growth hormone IGF2 emerged as a
significant mediator of the aggressive properties regulated by GATA2. IGF2 in turn activated

IGF1R and INSR as well as a downstream polykinase program. The characterization of this
regulatory axis prompted a combination strategy whereby dual IGF1R/INSR inhibition restored
the efficacy of chemotherapy and improved survival in preclinical models. These studies reveal a
GATA2-IGF2 aggressiveness axis in chemotherapy resistant prostate cancer and identify a
therapeutic opportunity in this challenging disease.

i
TABLE OF CONTENTS
LIST OF FIGURES iv
ACKNOWLEDGEMENTS vii
CHAPTER 1: Introduction 1
1.1: The natural history and management of prostate cancer 2
1.1.1: The development of metastatic prostate cancer 2
1.1.2: The changing therapeutic landscape of metastatic prostate cancer 6
1.1.3: The role of taxane chemotherapy in metastatic CRPC 9
1.1.4: Taxane resistance in metastatic CRPC 12
1.2: The role of GATA2 in physiology and disease 15
1.2.1: The role of GATA2 in mammalian physiology 15
1.2.2: The role of GATA2 in hematopoietic malignancy 20
1.2.3: The role of GATA2 in solid tumors 21
1.3: The role of IGF2 in physiology and disease 23
1.3.1: The role of IGF2 in mammalian physiology 23
1.3.2: The regulation of IGF2 in physiology and disease 27
1.3.3: The role of IGF2 in malignancy 29
1.3.4: IGF signaling as a therapeutic target 31
CHAPTER 2: GATA2 confers aggressiveness in CRPC 35
2.1: GATA2 is upregulated in chemotherapy resistant CRPC in laboratory models
and clinical databases 36
2.1.1: Generation of a docetaxel resistant ARCaPM subline 36
2.1.2: Characterization of cross resistance to cabazitaxel in docetaxel resistant cells 37

ii
2.1.3: GATA2 is upregulated in chemotherapy resistant CRPC in laboratory
models and clinical databases 39
2.2: GATA2 regulates chemotherapy resistance and tumorigenicity 41
2.3: GATA2 regulates a signature of cancer progression associated genes 43
2.3.1: Identification and characterization of GATA2 regulated genes 43
2.3.2: Genetic screen of a subset of clinically salient GATA2 regulated genes 45
2.3.3: The consensus signature is unrelated to AR biology 49
CHAPTER 3: IGF2 significantly mediates GATA2 biology in CRPC 52
3.1: Functional validation of IGF2 as a downstream effector of GATA2 53
3.2: GATA2 directly upregulates IGF2 59
3.2.1: GATA2 binds to and activates the IGF2 P4 promoter 59
3.2.2: GATA2 and IGF2 are co-expressed during prostate cancer progression 61
3.3: IGF2 activates a polykinase program downstream of GATA2 62
3.3.1: Characterization of dephosphorylation patterns in knockdown models 62
3.3.2: Chemical inhibition of the polykinase program is functionally significant 64
3.4: Dual IGF1R/INSR inhibition improves the efficacy of chemotherapy and
survival in preclinical models 67
3.4.1: Dual IGF1R/INSR inhibition improves the efficacy of chemotherapy
in vitro 67
3.4.2: OSI-906 improves the efficacy of chemotherapy in vivo 69
3.4.3: OSI-906 improves the efficacy of chemotherapy and survival in preclinical
models of disseminated CRPC without added general drug toxicity 71

iii
CHAPTER 4: Discussion 73
4.1: Chemotherapy resistant CRPC remains an intractable clinical entity 74
4.2: The emerging role of GATA2 in cancer biology 75
4.2.1: Previous characterization of GATA2 in prostate cancer and other
malignancies 75
4.2.2: GATA2 confers aggressiveness in CRPC through a consensus signature 77
4.2.3: The increasingly complex role of GATA2 in prostate cancer 81
4.3: A novel role for IGF2 in prostate cancer 83
4.4: Therapeutic implications of the GATA2-IGF2 axis 85
MATERIALS AND METHODS 89
REFERENCES 104

iv
LIST OF FIGURES
Figure 1: Characterization of the ARCaPM-DR subline 37
Figure 2: Docetaxel resistant sublines exhibit varying degrees of cross resistance to
cabazitaxel 38
Figure 3: GATA2 is upregulated in three models of chemotherapy resistant CRPC 39
Figure 4: GATA2 is upregulated in chemotherapy treated prostate cancer tissues from
multiple clinical databases 40
Figure 5: Characterization of two independent GATA2 shRNAs in chemotherapy
resistant CRPC sublines 41
Figure 6: GATA2 regulates chemotherapy resistance in CRPC cells 42
Figure 7: GATA2 regulates tumorigenicity in CRPC cells 43
Figure 8: GATA2 regulates a consensus 28 member signature of cancer associated
genes 44
Figure 9: The consensus signature is strongly expressed in clinical prostate cancer
tissues treated with chemotherapy 45
Figure 10: A subset of 7 genes is regulated by GATA2 in the chemotherapy resistant
sublines and exhibits a strongly complementary expression profile in clinical prostate
cancer tissues 46
Figure 11: Characterization of RNA interference reagents and expression constructs
for a focused genetic screen 47
Figure 12: Focused genetic screen of clinically salient GATA2 regulated genes 48

v
Figure 13: The consensus GATA2 regulated genes are unrelated to AR biology 49
Figure 14: GATA2 regulates a subset of AR targets that are not deregulated in
chemotherapy resistant CRPC cells or prostate cancer tissues 51
Figure 15: Characterization of two independent IGF2 shRNAs in chemotherapy
resistant CRPC sublines 53
Figure 16: IGF2 knockdown reduces the chemotherapy resistance and tumorigenicity
of CRPC cells 54
Figure 17: An IGF2 neutralizing antibody reduces the chemotherapy resistance and
tumorigenicity of CRPC cells 55
Figure 18: Characterization of an IGF2 vector in GATA2 shRNA expressing cells 56
Figure 19: IGF2 expression rescues the defects instigated by GATA2 knockdown on
chemotherapy resistance and tumorigenicity in CRPC cells 57
Figure 20: Exogenous IGF2 rescues the defects instigated by GATA2 knockdown on
chemotherapy resistance and tumorigenicity in CRPC cells 58
Figure 21: IGF2 protein levels are elevated and responsive to GATA2 knockdown in
chemotherapy resistant CRPC sublines 59
Figure 22: GATA2 occupies predicted binding elements in the IGF2 P4 promoter 60
Figure 23: GATA2 activates an IGF2 P4 luciferase reporter 60
Figure 24: IGF2 is co-expressed with GATA2 during prostate cancer progression 61
Figure 25: IGF2 activates a polykinase program through IGF1R and INSR downstream
of GATA2 63

vi
Figure 26: Chemicals inhibitors of the polykinase program reduce chemotherapy
resistance and soft agar growth 65
Figure 27: Alternative chemicals inhibitors of the polykinase program reduce
chemotherapy resistance and soft agar growth 66
Figure 28: Dual IGF1R/INSR inhibition improves the efficacy of chemotherapy in vitro 68
Figure 29: Monotherapy with OSI-906 does not significantly constrain the growth of
established CRPC xenografts 69
Figure 30: OSI-906 restores the efficacy of chemotherapy in vivo 70
Figure 31: OSI-906 improves the efficacy of chemotherapy and survival in preclinical
models of disseminated CRPC without added general drug toxicity 72

vii
ACKNOWLEDGEMENTS
As I conclude this chapter of my career and prepare for the next, I feel a profound sense
of gratitude and indebtedness to the extraordinary network of mentors, colleagues, family and
friends that has nurtured me throughout these last twenty eight years. It is beyond any doubt that
I would not be where I am today without their continuous support.
First and foremost, I wish to thank my thesis advisor Dr. Carlos Cordon-Cardo. From a
young age I dreamed of becoming a physician scientist and using the laboratory to contribute to
the field of medicine. In Carlos I found a mentor and role model deeply committed to this
endeavor and willing to take me on despite my limited knowledge and skills. The intellectual
freedom, enthusiasm for investigation, limitless resources and scientific mentorship afforded by
Carlos’ laboratory constituted an ideal environment for me to pursue translational research
projects. My time in Carlos’ laboratory was a period of immense personal and scientific growth
for which I will be everlastingly grateful.
I wish to thank the Columbia University faculty members who generously agreed to serve
on my thesis committee. I am particularly grateful to Dr. Cathy Mendelsohn, the chairwoman of
my committee, as well as Drs. Andrea Califano, Stephen Emerson and Ramon Parsons. These
faculty members patiently and insightfully guided my research projects during the last four years,
and I feel greatly honored to have stood on the shoulders of such a distinguished group of
scientists and mentors.
I wish to thank the members of the Cordon-Cardo laboratory. In particular, I thank Dr.
Josep Domingo-Domenech, whose highly skilled and passionate dedication to the improvement
of the oncology field has been a great source of inspiration and mentorship since my first days in

viii
Carlos’ laboratory. I thank Dennis Bonal, Janis de la Iglesia-Vicente, Nataliya Gladoun,
Elizabeth Charytonowicz, Mireia Castillo-Martin and Estrelania Williams for their scientific
support and camaraderie. I also wish to recognize all of the support staffers who worked behind
the scenes to make the scientific endeavor possible, including administrators, security officers,
housekeepers, and facility technicians. In particular, I thank Uncle Manu, Jorge Miranda,
Anthony Green, William Parkinson, Yu Zhou and Kenson Roberts.
I wish to thank the leaders of the MD/PhD program at Columbia University for their
institutional support, guidance and friendship over the years: Drs. Michael Shelanski, Ron Liem
and Patrice Spitalnik. Their generous invitation to the MD/PhD program has been one of the
most rewarding and exciting experiences in my life. I thank Stacy Warren, Zaia Sivo and Jeffrey
Brandt for their administrative support. I also thank the students of the MD/PhD program who
have been my peers, colleagues, mentors and friends during these last six years.
I thank my many scientific collaborators. I thank Dr. Veronica Rodriguez-Bravo from the
Memorial Sloan Kettering Comprehensive Cancer Center for assistance with
immunoprecipitation protocols. I thank Drs. Amaia Lumabio and Scott Lowe from the Memorial
Sloan Kettering Comprehensive Cancer Center for assistance with custom shRNA design and
cloning. I thank S. Aidan Quinn, Albert Lee and Dr. Raul Rabadan from Columbia University as
well as Xiaochen Sun, Ben Readhead, Xintong Chen and Drs. Joel Dudley and Yujin Hoshida
from the Mount Sinai Hospital for assistance with computational analyses. I thank Drs. Ruth
Rodriguez-Barrueco and Jose Silva from Columbia University for assistance with chromatin
immunoprecipitation and luciferase reporter assay protocols.

ix
Finally, I wish to thank my family and friends. I am everlastingly indebted to my mother
and father, Judy Stewart Vidal and Pierre-Paul Vidal, who in addition to making me from scratch
endowed me with a thirst for knowledge, a strong work ethic and a sense of commitment to the
uplift of humanity. I thank my brother William Vidal for his companionship and support over the
years. Among my friends, I am particularly grateful to Jason Long and Tsega Gebreyesus.
I feel grateful to all of these individuals. It is my fervent hope and expectation that in the
end my character and my contributions will prove worthy of their support.

1
CHAPTER 1:
Introduction

2
1.1: The natural history and management of prostate cancer
1.1.1: The development of metastatic prostate cancer
Prostate cancer is one of the most common malignant neoplasms in humans. Worldwide,
prostate cancer is the second leading cause of cancer incidence and the sixth leading cause of
cancer mortality in males, including more than 900,000 diagnoses and 250,000 fatalities in 2008
(Jemal et al., 2011). In the United States, prostate cancer is the leading cause of cancer incidence
and the second leading cause of cancer mortality in males, including more than 200,000
diagnoses and nearly 30,000 fatalities estimated in 2014 (Siegel et al., 2014).
Mortality from prostate cancer occurs as a result of complications associated with
metastatic disease. Accordingly, the disparity between prostate cancer incidence and mortality is
largely attributable to the fact that primary prostate cancer is highly heterogeneous with only a
subset patients progressing to metastatic disease within the adult lifetime. For example, in a
Swedish cohort of primary prostate cancer patients, 17.5% of initially untreated tumors
progressed to metastatic disease following a mean observation period of 21 years (Johansson et
al., 2004). In an American cohort of primary and locally advanced prostate cancer patients, 5.1%
progressed to metastatic disease within 15 years of radical prostatectomy (Pound et al., 1999).
This profound heterogeneity has stimulated intense interest in the classification and
treatment of patients with primary prostate cancer. Although there is considerable variability in
the management of primary prostate cancer today, decades of investigation have resulted in the
identification of several widely accepted practices. Classification is performed using
clinicopathological criteria, and treatment options include active surveillance, hormone therapy,

3
radiotherapy and surgery (Chang et al., 2014; Klotz and Emberton, 2014; Thompson et al.,
2007).
The salient clinicopathological criteria are tumor stage, Gleason grade and circulating
prostate specific antigen (PSA, encoded by the KLK3 gene) levels. Extensive clinical evidence
suggests that primary tumors with a definitive Gleason score ≤6 and a PSA <10ng/ml rarely
mestastasize and these cases are consequently termed low risk (Thompson et al., 2007). For
example, in a retrospective study of a large American radical prostatectomy cohort, only 22 of
14,123 patients with Gleason ≤6 disease exhibited lymph node metastasis at the time of surgery,
and 19 of these showed higher grade than originally assigned by pathologists upon secondary
review (Ross et al., 2012). Similarly, in a second retrospective study of a large American radical
prostatectomy cohort, only 3 of 9,557 patients with Gleason ≤6 disease experienced fatality from
prostate cancer over a mean of 15 years (Eggener et al., 2011).
These clinical observations cause many physicians to advocate active surveillance in
which patients undergo serial PSA and biopsy examinations as the appropriate management for
low risk disease (Klotz and Emberton, 2014). During active surveillance, local treatment is
deferred until the development of high risk disease. However, the excellent prognosis of low risk
disease is complicated by the fact that a substantial number of Gleason ≤6 diagnoses made by
needle biopsy harbor higher grade disease that is detectable post-operatively in prostatectomy
samples. For example, in the large Scandinavian Prostate Cancer Group Study Number 4 trial,
patients who underwent prostatectomy exhibited postoperative Gleason scores one step higher in
30% and two or more steps higher in 28% of cases (Bill-Axelson et al., 2011). Patient anxiety
may also limit the applicability of active surveillance (Klotz and Emberton, 2014).
Unfortunately, to date there are no published randomized trials comparing active surveillance

4
with radiotherapy or surgery, and as a result there is considerable variability in the management
of low risk primary prostate cancer.
Prostate cancer is termed high risk by the American Urological Association when it
exhibits spread to both lobes of the prostate, a Gleason score ≥8 or PSA >20 ng/ml (Thompson et
al., 2007). High risk patients exhibit a significant chance of local or metastatic progression as
well as prostate cancer related morbidity and mortality. For example, in a large French cohort
treated with prostatectomy and post-operative radiotherapy, 39.4% of high risk patients
experienced biochemical or clinical progression or death after a median follow up of 10.6 years
(Bolla et al., 2012). Similarly, in a retrospective study of a large German cohort, 43.8% of high
risk patients treated with prostatectomy experienced biochemical recurrence after 10 years
(Boorjian et al., 2011).
The literature on the management of high risk prostate cancer is limited as a result of
variations in definition, lack of randomized trials and underpowered studies (Chang et al., 2014).
Nonetheless, radiotherapy, hormone therapy and surgery are mainstays of management. Notably,
two large randomized trials demonstrated that a combination of radiotherapy and androgen
deprivation therapy (ADT) is superior to either alone (Bolla et al., 2002; Pilepich et al., 2005). In
contrast, the relative efficacy of radical prostatectomy versus a combination of radiotherapy and
ADT, as well as the benefits of more extensive surgeries and post-operative radiotherapy remain
unclear (Chang et al., 2014).
After local therapy both low and high risk patients are monitored by serial PSA testing.
Biochemical recurrence is heterogeneous and may signal either local recurrence or metastatic
disease (Pound et al., 1999). These outcomes are often distinguished by imaging studies and are

5
characterized by greatly differing prognoses. For example, in one American retrospective study
of patients who experienced biochemical recurrence following prostatectomy, prostate cancer
specific mortality occurred as rapidly as one year after recurrence but median overall survival for
the entire cohort was not reached after more than 15 years (Freedland et al., 2005). Although
accepted guidelines are limited, local recurrence is primarily treated with salvage radiotherapy,
ADT, surgery or a combination (Freedland and Moul, 2007).
Metastatic prostate cancer cells exhibit a strong tropism for bone and accordingly 90% of
advanced patients exhibit skeletal involvement (Gartrell and Saad, 2014). Although prostate
cancer may also colonize the liver, lungs and brain, its signature tropism for bone has garnered
the most scientific interest. The mechanisms of hematogenous dissemination in prostate cancer
include hallmarks described for other epithelial malignancies, including deregulation of cell
adhesion molecules, integrin and focal adhesion signaling and matrix metalloproteases, among
others (Jin et al., 2011). Prostate cancer tropism for bone is also multifactorial. For example,
bone marrow derived SDF1 may act as a chemoattractant for CXCR4 expressing prostate cancer
cells (Taichman et al., 2002). Moreover, prostate cancer cells may bind more avidly to bone
marrow endothelial cells than other endothelial cell types (Lehr and Pienta, 1998). In addition,
prostate cancer cells may commonly express integrins that facilitate adhesion to the bone marrow
extracellular matrix (Cooper et al., 2002). After prostate cancer cells enter the bone marrow, a
rich cellular and molecular microenvironment that includes osteoblasts and various growth
factors stimulates their growth into overt metastases. For example, co-culture with primary
mouse osteoblasts may increase the proliferation of bone metastatic prostate cancer cells (Fizazi
et al., 2003). In addition, bone marrow derived insulin-like growth factors may stimulate the
proliferation of prostate cancer cells (Ritchie et al., 1997).

6
1.1.2: The changing therapeutic landscape of metastatic prostate cancer
Although the management of metastastic prostate cancer is rapidly evolving, hormonal
manipulations remain first line therapy (Wong et al., 2014). Serum androgen levels in males are
primarily controlled by the hypothalamus–pituitary axis. Gonadotropin releasing hormone
(GnRH) secreted by the hypothalamus stimulates the anterior pituitary to secrete leutenizing
hormone (LH). LH in turn stimulates Leydig cells in the testes to secrete the vast majority of
circulating androgens. Finally, androgens regulate the development and maintenance of male
characteristics through binding to androgen receptor (AR).
Pioneering work published by Charles Huggins in 1941 demonstrated for the first time
that reduction of circulating androgen levels by surgical castration could induce significant
improvements in the progression of metastatic prostate cancer (Huggins et al., 1941). Moreover,
improved characterization of the hypothalamus–pituitary axis in the following decades resulted
in the discovery of medical castration with estrogens and GnRH agonists (Group, 1967; Schally
et al., 2000). Finally, discovery of AR led to the development of non-steroidal anti-androgens
that directly inhibit the receptor (Liao et al., 1974).
Today medical castration with GnRH agonists has largely replaced surgical castration in
developed countries, although medical and surgical castration are equally effective at reducing
circulating androgens to castrate levels and controlling the progression of metastatic disease
(Seidenfeld et al., 2000). Importantly, adrenal steroidogenesis results in residual circulating
androgens despite the 90-95% reductions achieved though castration (Gomella, 2009). Indeed,
this observation stimulated combination strategies with castration and anti-androgens to produce
combined androgen blockade (CAB). However, the modest survival benefit of CAB is mitigated

7
by added toxicity, and as a consequence CAB is only implemented in subsets of patients (Group,
2000).
ADT elicits clinically significant responses in 80-90% metastatic prostate cancer patients,
resulting in a median progression free survival benefit of 12-33 months (Hellerstedt and Pienta,
2002). However, ADT invariably precedes progression to CRPC, an aggressive disease
characterized by limited survival. The mechanisms of resistance to ADT have accordingly
attracted considerable interest and are generally classified into AR dependent and independent
categories (Zong and Goldstein, 2013).
For many years, the mechanisms of progression to CRPC were thought to be primarily
AR independent, a notion supported by considerable laboratory and clinical evidence. For
example, epithelial mesenchymal transition (EMT) may play a role in the emergence of CRPC.
Several groups reported that in vivo castration of well characterized androgen dependent
xenograft models results in the emergence of EMT markers including CDH2 and ZEB1, and
similar observations were made in clinical prostate cancer tissues managed with ADT
(Jennbacken et al., 2010; Sun et al., 2012; Tanaka et al., 2010). Altered apoptosis and survival
signaling may also promote CRPC independently of AR. For example, the anti-apoptotic factor
BCL2 is upregulated by castration in laboratory models and human prostate cancer tissues
(McDonnell et al., 1992). In addition, a number of groups reported that PTEN loss and AKT
activation contribute to castration resistant growth (Carver et al., 2011; Gao et al., 2006; Jiao et
al., 2007; Mulholland et al., 2011). Finally, several well characterized CRPC cell lines are AR
negative and form aggressive castration resistant tumors in vivo (van Bokhoven et al., 2003).

8
Nonetheless, a wealth of laboratory and clinical data suggest that AR dependent
mechanisms also contribute ADT resistance. Several groups initially reported that AR is both
necessary and sufficient for progression of androgen dependent cell lines to CRPC in vitro and in
vivo (Chen et al., 2004; Cheng et al., 2006; Snoek et al., 2009; Zegarra-Moro et al., 2002).
Moreover, a multitude of studies identified ligand dependent and independent mechanisms
whereby AR signaling remains active in CRPC. For example, intratumoral testosterone levels
may remain elevated during ADT through conversion of residual adrenal androgens or de novo
synthesis by prostate cancer cells (Chang et al., 2011; Locke et al., 2008; Montgomery et al.,
2008; Stanbrough et al., 2006). In addition, the AR gene is frequently amplified or mutated in
metastatic CRPC, and these alterations enable AR activation in the setting of reduced circulating
androgens during ADT (Grasso et al., 2012; Taylor et al., 2010; Visakorpi et al., 1995). Finally,
alternative splicing may result in the expression of transcripts that encode a constitutively active
AR (Dehm et al., 2008; Guo et al., 2009; Hu et al., 2009; Sun et al., 2010).
Perhaps the most unequivocal data that AR dependent mechanisms contribute to
progression to CRPC emerged from clinical trials in which second generation androgen signaling
inhibitors improved survival in patients with CRPC. 17-α-hydroxylase/17,20-lyase (encoded by
the CYP17A1 gene) is a critical enzyme required for androgen synthesis in the adrenal glands,
testes and prostate cancer. Abiraterone acetate is a CYP17A1 inhibitor with clinical activity in
CRPC both before and after chemotherapy (de Bono et al., 2011; Ryan et al., 2013). Moreover,
enzalutamide is a second generation anti-androgen with greater affinity for AR than bicalutamide
(Tran et al., 2009), and enzalutamide also exhibits clinical activity in CRPC both before and after
chemotherapy (Beer et al., 2014; Scher et al., 2012).

9
Apart from second generation androgen signaling inhibitors and chemotherapy, two other
treatments exhibit clinical activity in CRPC. Sipuleucel-T is a dendritic cell vaccine prepared
from peripheral blood mononuclear cells obtained by leukapheresis and exposed ex vivo to a
recombinant immunogenic protein. Sipuleucel-T improves survival in CRPC patients with
minimally symptomatic metastatic disease, although its clinical activity is impossible to monitor
with imaging or PSA (Kantoff et al., 2010). This limitation in clinical management as well as the
cost of Sipuleucel-T have greatly restricted its widespread application (Bishr and Saad, 2013).
Moreover, radium-223 is an alpha particle emitting radiopharmaceutical with tropism for bone.
Radium-223 also improves survival in CRPC and reduces skeletal events, although its use is
limited to patients lacking symptomatic visceral metastases (Parker et al., 2013).
1.1.3: The role of taxane chemotherapy in metastatic CRPC
Until recently metastatic CRPC was considered a chemotherapy resistant disease (Schutz
et al., 2014). Prior to the turn of the century, a large number of chemotherapy agents including
anthracyclines, alkylating agents, antimetabolites, platinums and topoisomerase inhibitors had
been evaluated in phase II clinical trials. Remarkably, a review of 26 trials conducted between
1987 and 1991 found that the average response rate to cytotoxic chemotherapy was less than
10% (Yagoda and Petrylak, 1993). In the following decade, the topoisomerase inhibitor
mitoxantrone was found in several large trials to provide palliative benefit in CRPC (Berry et al.,
2002; Kantoff et al., 1999; Tannock et al., 1996). Although mitoxantrone conferred no survival
benefit in these trials, the grim therapeutic landscape of CRPC resulted in the adoption of this
agent as standard chemotherapy (Schutz et al., 2014).

10
The taxane family of cytotoxic chemotherapy agents consists of paclitaxel (also called
taxol), docetaxel (also called taxotere) and cabazitaxel. The United States Congress created the
National Cancer Chemotherapy Service Center at the National Cancer Institute in 1955, and
under its auspices paclitaxel was discovered as a natural isolate from the bark of the Pacific Yew
tree in the 1960s (Chabner and Roberts, 2005). Docetaxel and cabazitaxel were later developed
by the pharmaceutical company Sanofi-Aventis as semi-synthetic derivatives of paclitaxel
(Schutz et al., 2014). The taxane chemotherapeutics bind to polymerized tubulin dimers and
function as microtubule stabilizing agents (Dumontet and Sikic, 1999). Although the
mechanisms are incompletely characterized, microtubule stabilization interferes with mitosis and
intracellular transport and activates apoptosis through the mitochondrial pathway. Some of the
molecular events that accompany this process include G2/M arrest (Fabbri et al., 2006),
activation of the spindle assembly checkpoint (Sudo et al., 2004) and inactivation of BCL2 by
phosphorylation (Haldar et al., 1996; Wang et al., 1999).
Following the clinical introduction of paclitaxel in the 1980s and docetaxel in the 1990s,
results from phase II trials began to emerge on their activity in metastatic CRPC. While
paclitaxel showed modest activity (Roth et al., 1993), two phase II trials suggested docetaxel
may be more effective (Beer et al., 2001; Berry et al., 2001). On the basis of these results, two
randomized phase III studies were initiated, TAX-327 and SWOG-9916, which included more
than 1,500 CRPC patients (Petrylak et al., 2004; Tannock et al., 2004). In both trials docetaxel
instigated PSA responses of approximately 50% and conferred an overall survival benefit.
Moreover, the results from the TAX-327 trial were later reaffirmed after longer follow up
(Berthold et al., 2008). Remarkably, in 2004 single agent docetaxel thereby became the first

11
known therapeutic intervention to prolong survival in CRPC and presently remains a mainstay of
treatment (Bishr and Saad, 2013).
Docetaxel remained the only approved treatment for CRPC for several years until the
introduction of cabazitaxel in 2010. Cabazitaxel showed activity in preclinical models of
docetaxel resistant cells and the ability to cross the blood-brain barrier, an attractive property for
the management of patients with brain metastasis (Cisternino et al., 2003; Schutz et al., 2014).
Moreover, promising results from a phase II clinical trial (Pivot et al., 2008) laid the groundwork
for the randomized phase III TROPIC study (de Bono et al., 2010). The TROPIC study showed
that cabazitaxel confers a survival benefit in patients who progress with docetaxel, and
cabazitaxel was subsequently widely approved as a second line chemotherapy agent for patients
with CRPC (Bishr and Saad, 2013).
Finally, although ADT has historically been the first line therapy for patients with
metastatic prostate cancer, recent evidence suggests that taxane chemotherapy may soon play a
major role in this context. An initial phase III trial showed that ADT combined with docetaxel
confers a measurable but not statistically significant survival benefit in men with untreated
metastatic disease (Gravis et al., 2013). However, the overwhelming majority (>75%) of patients
in this trial exhibited low or intermediate risk disease. In contrast, the larger phase III
CHAARTED trial enrolled a majority (>60%) of high risk patients, and an interim analysis
revealed an unprecedented survival benefit of 17 months in this subgroup (Sweeney, 2014).
While continued follow up is required to provide definitive results from the CHAARTED trial,
this preliminary analysis suggests that taxane chemotherapy may soon play a significant role in
the setting of untreated metastatic prostate cancer.

12
1.1.4: Taxane resistance in metastatic CRPC
Despite the advances of the last decade—first and second line chemotherapy,
immunotherapy, second generation androgen signaling inhibitors, a bone targeting
radiopharmaceutical and combination strategies—CRPC remains a uniformly lethal diagnosis
(Bishr and Saad, 2013). This harsh reality is due in part to the fact that CRPC exhibits
mechanisms of resistance to currently available therapeutic modalities. For example, although
taxanes instigate high response rates, treatment is never curative because patients universally
relapse with chemotherapy resistant disease (Seruga et al., 2011). The mechanisms of resistance
to taxane chemotherapy in metastatic CRPC are therefore of considerable clinical significance.
Mechanisms of resistance to chemotherapy are generally divided into distinct
pharmacokinetic and cellular categories (Holohan et al., 2013). Pharmacokinetic mechanisms
include systemic distribution, metabolism and elimination and are primarily determined by
chemical structure. Consequently, pharmacokinetic considerations figure primarily in drug
development and medicinal chemistry rather than the study of drug resistance. Indeed, the best
characterized mechanisms of taxane resistance in CRPC are cellular in nature (Chien and
Moasser, 2008; Murray et al., 2012; Seruga et al., 2011).
Perhaps the most clinically significant mechanism of docetaxel resistance in CRPC to
date is the molecular chaperone clusterin (Zoubeidi et al., 2010). Although the precise
mechanisms are poorly characterized, clusterin is thought to inhibit stress induced protein
aggregation and apoptosis (Yerbury et al., 2007). Interestingly, an initial study reported that
clusterin suppression with an antisense oligonucleotide could improve the efficacy of docetaxel
in CRPC cell line xenograft models (Springate et al., 2005). Although poorly characterized, the

13
increased efficacy of docetaxel in this context appears to be reduced resistance to apoptosis
(Sallman et al., 2007), a classical mechanism of chemotherapy resistance (Holohan et al., 2013).
Moreover, the second generation clusterin antisense oligonucleotide OGX-011 was well
tolerated in a phase I clinical trial in combination with docetaxel (Chi et al., 2008). Notably, two
subsequent randomized phase II trials showed that addition of OGX-011 to docetaxel may confer
a substantial survival benefit, and phase III trials are ongoing (Chi et al., 2010a; Saad et al.,
2011).
A number of studies have linked other apoptosis and survival signaling pathways to
docetaxel resistance in CRPC (Seruga et al., 2011). For example, chemical inhibition of the
PI3K/AKT (Domingo-Domenech et al., 2012; Qian et al., 2010; Wu et al., 2005), NFκB/IL6
(Domingo-Domenech et al., 2006) and EGFR (Mimeault et al., 2007) survival pathways
improves the efficacy of docetaxel in CRPC cells. Likewise, expression of the antiapoptotic
molecules SPHK1 (Pchejetski et al., 2008) and BCL2 (Domingo-Domenech et al., 2012) reduces
the efficacy of docetaxel in CRPC cells.
Alteration of drug targets is another classical mechanism of chemotherapy resistance
(Holohan et al., 2013). Microtubules are composed of tubulin subunits that belong to six distinct
classes based on their C-terminal sequence variability, and purified preparations of these distinct
classes exhibit differing dynamics (Chien and Moasser, 2008; Sullivan and Cleveland, 1986).
Interestingly, a widely reported mechanism of resistance to paclitaxel is increased composition
of class III beta tubulin (TUBB3) in microtubules. Microtubules composed of class III tubulin
are less sensitive to the effects of paclitaxel on microtubule dynamics (Derry et al., 1997),
possibly as a result of the increased microtubule instability (Banerjee et al., 1990; Panda et al.,
1994). In one study, genetic manipulation of TUBB3 in CRPC cell lines modestly regulated

14
docetaxel resistance, and elevated TUBB3 protein levels predicted poor survival in metastatic
CRPC patients receiving docetaxel (Ploussard et al., 2010). However, the reproducibility and
clinical significance of these findings remain unclear, and evidence implicating TUBB3 in
docetaxel resistance in CRPC is therefore limited at this time.
Several other mechanisms of taxane resistance have been widely reported, although their
relevance to docetaxel and CRPC are unclear. Perhaps foremost among these mechanisms is the
family of ATP-binding cassette (ABC) proteins that are energy dependent transporters for a wide
range of substrates (Holohan et al., 2013). Paclitaxel and docetaxel are known substrates for
ABCB1 (also known as MDR1 or P-gp), the best characterized ABC transporter (Gottesman et
al., 2002). However, data from cell lines and in particular from cancer tissues are conflicting,
with a substantial number of studies finding no association between taxane resistance and
ABCB1 (Chien and Moasser, 2008; Murray et al., 2012). Moreover, the failure of three
generations of ABCB1 inhibitors over a period of two decades to improve the efficacy of
chemotherapy in clinical trials has further cast doubt on its widespread significance (Tamaki et
al., 2011). In prostate cancer, one study reported that a docetaxel resistant C4-2B subline
exhibited increased ABCB1 expression and could be sensitized by genetic or pharmacological
inhibition of that transporter (Zhu et al., 2013). However, the reproducibility and clinical
significance of this finding remains unknown.
Other possible mechanisms of taxane resistance include tubulin mutations (Giannakakou
et al., 1997; Gonzalez-Garay et al., 1999), microtubule associations with other cytoskeletal
elements (Verrills et al., 2006) and limited tissue penetration (Kyle et al., 2007). However, the
evidence for these mechanisms of resistance is limited, and their relevance to docetaxel
resistance in CRPC is unknown (Chien and Moasser, 2008; Murray et al., 2012). Therefore, to

15
date the mechanisms of resistance to chemotherapy in CRPC remain unclear. Moreover, the
limited understanding of these mechanisms has failed to translate to therapeutic strategies with
clinical benefit in a phase III clinical trial. The continued investigation of docetaxel resistance in
CRPC is thus both biologically and clinically meritorious.
1.2: The role of GATA2 in physiology and disease
1.2.1: The role of GATA2 in mammalian physiology
The GATA genes are a family of evolutionarily conserved transcription factors that
regulate development and differentiation in eukaryotic organisms (Vicente et al., 2012a). In
mammals, the GATA family contains six members termed GATA1-6 and is a prototype of
lineage restricted transcription factors that regulate cell fate determination. GATA1 is the
founding member of the GATA family and was initially discovered as an erythroid nuclear
protein involved in the regulation of globin genes (Evans and Felsenfeld, 1989; Tsai et al., 1989).
Although the gene family owes its name to the consensus DNA binding motif
(A/T)GATA(A/G), GATA binding elements are now known to vary considerably in their
sequences (Kobayashi-Osaki et al., 2005; May et al., 2013; Merika and Orkin, 1993).
GATA1 is extensively characterized in the hematopoietic system where it regulates the
maturation of erythroid (Fujiwara et al., 1996) and megakaryocytic (Shivdasani et al., 1997)
lineages, among others. GATA3 regulates brain (Pandolfi et al., 1995), breast (Kouros-Mehr et
al., 2006) and kidney (Grote et al., 2008) development, among others. Interestingly, GATA3 is
also a luminal differentiation marker in breast cancer that inhibits progression and signals
favorable prognosis (Dydensborg et al., 2009; Kouros-Mehr et al., 2008; Mehra et al., 2005).
GATA4, GATA5 and GATA6 are less well characterized but known to play important roles in

16
the development of mesodermal and endodermal lineages including heart, liver, lung, gut and
gonads (Molkentin, 2000). Notably, null mutations in all GATA family genes with the exception
of GATA5 are embryonic lethal in mice (Vicente et al., 2012a).
The vertebrate GATA factors contain a conserved DNA binding domain that is
comprised of two multifunctional zinc finger domains (Trainor et al., 2000). The N-terminal and
C-terminal zinc finger domains of GATA1 are essential for its protein-DNA and protein-protein
interactions and have been studied in considerable detail. However, little is known about the
structural properties of GATA2 (Vicente et al., 2012a). Like the other GATA family members,
the two zinc finger domains of GATA2 are required for protein-DNA interactions (Evans and
Felsenfeld, 1989). In addition, the GATA2 zinc finger domains are known to be required for
protein-protein interactions. For example, the zinc finger domains are required for binding to the
transcriptional co-activator FOG1 (Chang et al., 2002), the transcription factor PU.1 (Zhang et
al., 1999) and the chromatin modifying enzyme HDAC3 (Ozawa et al., 2001). Finally, GATA2
contains a number of other poorly characterized domains, including two transactivation domains,
a nuclear localization signal and a negative regulatory domain (Vicente et al., 2012a).
The GATA2 protein undergoes a number of post-translational modifications. For
example, phosphorylation of GATA2 by AKT reduces its localization to the nucleus and its
DNA binding activity (Menghini et al., 2005), while phosphorylation by cyclin dependent
kinases regulates its protein levels during the cell cycle (Koga et al., 2007). In addition,
acetylation of at least six lysine residues by EP300 results in increased transcriptional activity
(Hayakawa et al., 2004). Moreover, sumoylation of GATA2 reduces its transcriptional activity at
the ET1 promoter of endothelial cells (Chun et al., 2003). Finally, three ubiquination sites
regulate GATA2 protein levels through the proteasome pathway (Minegishi et al., 2005).

17
GATA2 developmental biology is mostly characterized in the hematopoietic system,
although a role in the vascular system is also emerging (Vicente et al., 2012a). Hematopoietic
and vascular development are complex processes that involve multiple tissues, and efficient
blood circulation commences at E8.5 in the mouse (McGrath et al., 2003). As the site of
primitive hematopoiesis, the extraembryonic yolk sac produces hematopoietic cells beginning on
E7.5 (Ueno and Weissman, 2010). Definitive hematopoietic stem cells (HSCs) that will give rise
to all the hematopoietic cells of the adult emerge primarily from the hemogenic endothelium of
the large vessels of the intraembryonic aorta-gonad-mesonephros (AGM) region beginning on
E10.5. Interestingly, although vascular development is less well characterized, primitive
vasculogenesis also occurs in the yolk sac (Udan et al., 2013).
The first strong evidence that GATA2 regulates the development of the hematopoietic
system emerged from genetically engineered mice bearing null mutations at the Gata2 locus
(Tsai et al., 1994). Gata2-/- embryos succumb to severe anemia at E10.5, presumably as a result
of defective primitive hematopoiesis. Moreover, the profound loss or absence of Gata2-/- cells in
every hematopoietic compartment in Gata2+/+/Gata2-/- chimeric embryos after E13.5 and adults
indicates a defect in the ontogeny definitive HSCs. Indeed, later studies confirmed that GATA2
loss causes a dramatic reduction in the ontogeny of HSCs from both cultured embryonic stem
cells (Tsai and Orkin, 1997) and the AGM (de Pater et al., 2013; Gao et al., 2013; Ling et al.,
2004). Interestingly, the emergence of HSCs from the AGM is dependent on several
transcriptional activators of GATA2, including NOTCH1 (Robert-Moreno et al., 2005) and EVI1
(Yuasa et al., 2005), among others.
In addition to its widely reported role in hematopoietic development, several studies
suggest that GATA2 regulates vascular development. GATA2 mRNA and protein are strongly

18
expressed in the lymphatic vasculature of developing and adult mice, and GATA2 regulates
genes associated with vascular development in cultured primary lymphatic cells isolated from
mouse embryos (Kazenwadel et al., 2012; Lim et al., 2012). Moreover, second generation
genetically engineered mice that survive several days longer than the originally described null
mutants provide strong in vivo evidence that GATA2 regulates vascular development in the
developing embryo (Johnson et al., 2012; Lim et al., 2012). Mechanistically, GATA2 may
regulate the migration and sprouting of the developing vasculature through the VEGF co-
receptor NRP2 (Coma et al., 2013).
The severe hematopoietic defects and consequent embryonic lethality of null mice were
later found to unexpectedly mask a second facet of GATA2 developmental biology. In one study,
a yeast artificial chromosome (YAC) containing the Gata2 locus as well as >150kbp of 5’
sequence and >80kbp of 3’ sequence was found to completely rescue Gata2-/- embryos,
suggesting that all the regulatory elements required for GATA2 expression in the hematopoietic
system are contained in this genomic area (Zhou et al., 1998). Importantly, however, all the
resulting newborns rapidly succumbed to severe hydronephrosis. More detailed analysis revealed
that GATA2 is widely expressed in embryonic structures that give rise to the urogenital system,
including the uretic bud, Wolffian duct and urogenital sinus. This embryonic expression pattern
is linked to numerous postnatal abnormalities, including defects in the ureters, seminal vesicles
and vas deferens, among others (Zhou et al., 1998). Intriguingly, the authors did not comment on
the development of the prostate, a structure which emerges from the GATA2 expressing cells
that line the urogenital sinus. Nonetheless, these studies with a Gata2 YAC in Gata2-/- embryos
revealed a significant role for GATA2 in urogenital development. Moreover, a later study found

19
that GATA2 is expressed in the developing mouse prostate as well as the adult mouse and human
prostate (Perez-Stable et al., 2000).
Similar to its role in embryonic development, the role of GATA2 in adult physiology is
best characterized in the hematopoietic system (Vicente et al., 2012a). For example, GATA2 is
required for the survival and proliferation of adult HSCs. First, GATA2 is highly expressed in
adult HSCs (Orlic et al., 1995; Suzuki et al., 2006). Moreover, conditional homozygous
inactivation of Gata2 in adult mice results in a profound reduction in the number of bone marrow
HSCs (Lim et al., 2012). Likewise, adult Gata2+/- bone marrow HSCs perform poorly in
competitive transplantation experiments and exhibit a proliferation defect following cytotoxic
insult with 5-fluorouracil in vivo (Ling et al., 2004; Rodrigues et al., 2005).
In addition to its role in adult HSCs, GATA2 plays a complex role in adult hematopoiesis
(Vicente et al., 2012a). A dynamic reciprocal regulation pattern at several genomic loci called
GATA factor switching primarily involves GATA1 and GATA2 and is essential for erythroid
and myeloid differentiation. For example, disruption of GATA2 binding to its own promoter and
subsequent silencing by GATA1 is required for erythroid differentiation (Grass et al., 2003;
Ikonomi et al., 2000). Likewise, GATA factor switching involving GATA2 also regulates mast
cell differentiation (Cantor et al., 2008). In addition, GATA2 is highly expressed during
megakaryocyte differentiation, promotes megakaryocytic progenitor cell proliferation and drives
megakaryocytic lineage gene expression (Huang et al., 2009; Terui et al., 2000).
In recent years the laboratory findings implicating GATA2 in hematopoietic and vascular
biology have been complemented by a growing number of genetic studies in humans exhibiting
rare heritable diseases in which these tissues are dysfunctional. For example, two closely related

20
immunodeficiency syndromes known as monocytopenia with Mycobacterium avium complex
(MonoMAC) and dendritic cell, monocyte, B and NK lymphoid (DCML) deficiency are
characterized by mutations in GATA2 (Dickinson et al., 2011; Hsu et al., 2011). Interestingly,
patients with MonoMAC/DCML and Emberger disease, another heritable immunodeficiency
syndrome associated with GATA2 mutations, may exhibit primary lymphedema (Kazenwadel et
al., 2012; Ostergaard et al., 2011), complementing the laboratory studies linking GATA2 to
vascular biology. Finally, recent large retrospective studies have confirmed the association
between GATA2 mutation and hematological and vascular defects in human adults (Dickinson et
al., 2014; Spinner et al., 2014).
1.2.2: The role of GATA2 in hematopoietic malignancy
The heritable immunodeficiency syndromes characterized by GATA2 mutation are also
associated with malignant transformation. For example, MonoMAC/DCML deficiency and
Emberger disease are both associated with myelodysplastic syndrome (MDS) and AML
(Dickinson et al., 2011; Hsu et al., 2011; Ostergaard et al., 2011). In addition, familial GATA2
mutations may result in MDS/AML without previous hematological or vascular symptoms
(Hahn et al., 2011). Although the majority of the GATA2 mutations occurring these families have
not been characterized mechanistically, at least some of them are known to exhibit loss-of-
function and possibly dominant negative properties (Hahn et al., 2011). These observations have
led to speculation that MDS arises in these patients as a consequence of stem cell exhaustion and
bone marrow stress (Migliaccio and Bieker, 2011). In contrast, high GATA2 mRNA expression
is associated with poor prognosis in sporadic MDS (Fadilah et al., 2002) and AML (Luesink et
al., 2012; Vicente et al., 2012b), although the mechanistic role of GATA2 in this context is
unknown.

21
Chronic myeloid leukemia (CML) is characterized by an indolent chronic phase (CP) that
often progresses to a lethal blast crisis (BC) (Goldman and Melo, 2003). Although tyrosine
kinase inhibitors have dramatically altered the clinical progression of CML, a subset of patients
develops resistance and succumbs to BC (Cortes et al., 2011). CP is associated with an expansion
of differentiated myeloid cells, while BC is characterized by a reduction in differentiation and
the appearance of myeloblasts as well as lymphoblasts. The transcription factor PU.1 is a critical
regulator of myeloid differentiation (Kastner and Chan, 2008) that is repressed though a protein-
protein interaction with the C-terminal zinc finger domain of GATA2 (Zhang et al., 1999).
Interestingly, in one study 8/85 blast crisis patients exhibited a single gain-of-function mutation
in the C-terminal zinc finger domain of GATA2 (Zhang et al., 2008). The mutation increased the
transactivation potential of GATA2, accentuated repressive association of GATA2 with PU.1
and reduced the differentiation potential of human leukemia cells. Thus, gain-of-function
mutations in GATA2 may contribute to the development of BC in CML.
1.2.3: The role of GATA2 in solid tumors
Lung cancer is the most common cause of cancer incidence and mortality worldwide, and
NSCLC is the most common histological category (Ferlay et al., 2010). Although the RAS
family of GTPases and upstream receptor tyrosine kinases are among the most commonly
mutated genes in NSCLC, decades of investigation have failed to produce RAS pathway targeted
agents that provoke durable responses (Vasan et al., 2014). Consequently, synthetic lethal
interactions with RAS pathway mutation in NSCLC have attracted considerable laboratory
investigation. Notably, in one study an RNA interference screen identified GATA2 as a synthetic
lethal partner with RAS pathway mutation in NSCLC (Kumar et al., 2012). GATA2 knockdown
in cultured RAS pathway mutated NSCLC cells reduced their viability and tumorigenicity, and

22
Gata2 knockout in genetically engineered mouse models reduced lung tumorigenesis and
provoked regression of established tumors. Moreover, integrated transcriptome and genome
occupancy analyses revealed that GATA2 transcriptionally activates proteasome, IL1/NFκB and
RHO signaling, leading to a novel preclinical combination strategy. Surprisingly, this extensive
in vitro and in vivo characterization was recently challenged by a report that GATA2 expression
is strongly reduced in lung cancer versus normal tissues and that GATA2 knockdown does not
reduce the viability of cultured RAS pathway mutated NSCLC cells (Tessema et al., 2014).
Therefore, further investigation is needed to clarify the biology and therapeutic significance of
GATA2 in NSCLC.
In addition to its possible role in NSCLC, GATA2 has been studied by several groups in
prostate cancer. These studies have predominantly focused on the role of GATA2 in castration
naive disease, in particular the widely characterized AR expressing and androgen dependent
LNCaP cell line. First and foremost, GATA2 has emerged as a pioneer factor for AR.
An initial study exploring the role of GATA family proteins in the regulation of prostate
specific genes revealed that multiple GATA2 binding elements (GBEs) flank an AR responsive
site in an enhancer of KLK3 (Perez-Stable et al., 2000). Moreover, the GBEs were required for
androgen stimulated induction of PSA. A second study showed that GATA2 regulates androgen
stimulated induction of TMPRSS2, another canonical AR target, again through GBEs in an AR
regulated enhancer (Wang et al., 2007). Furthermore, the latter study showed that GATA2
binding to the KLK3 and TMPRSS2 enhancers is required for AR recruitment to these sites,
revealing a regulatory hierarchy whereby GATA2 is a pioneer factor for AR at these loci. This
regulatory hierarchy has been confirmed at several other AR regulated loci, including UBE2C
(Wang et al., 2009) as well as ABCC4 and ADPGK (Wu et al., 2014). Moreover, high

23
throughput data from both LNCaP cells (Wu et al., 2014) and genetically engineered mouse
models (Chen et al., 2013) suggest that this hierarchy operates at a genome wide level.
Finally, a recent study suggests that GATA2 may promote metastasis in the hormone
dependent setting. GATA2 knockdown in the LNCaP cell line resulted in reduced wound healing
and matrigel invasion (Chiang et al., 2014). Moreover, mechanistic data suggested that GATA2
regulates migration and invasion through focal adhesion disassembly. Therefore, the current
literature related to GATA2 in prostate cancer is limited to the castration naïve context,
advocates an important function as a pioneer factor for AR regulated genes and supports a
possible role in metastatic progression.
1.3: The role of IGF2 in physiology and disease
1.3.1: The role of IGF2 in mammalian physiology
More than half a century ago, the observation that growth hormone (GH) fails to directly
stimulate uptake of sulphate into cartilage led to the proposed existence of an intermediate
sulphation factor (SF) (Salmon and Daughaday, 1957). Shortly afterward an independent group
described a serum factor that exhibited insulin-like activities but could not be neutralized by an
insulin antibody and was accordingly termed non-suppressible insulin-like activity (NSILA)
(Froesch et al., 1963). Interestingly, SF and NSILA were later shown to regulate similar
properties, suggesting that they may in fact be the same factor (Underwood et al., 1972; Zingg
and Froesch, 1973). This factor, which had been renamed somatomedin by investigators in the
SF field (Daughaday et al., 1972), was later shown by investigators in the NSILA field to in fact
consist of two peptides (Rinderknecht and Humbel, 1976b). These peptides were soon found to
exhibit amino acid sequence homology with insulin and were consequently renamed IGF1 and

24
IGF2 (Rinderknecht and Humbel, 1976a). These early findings laid the groundwork for decades
of research that have predominantly focused on the biology of IGF1.
The IGFs bind to a family of homologous receptor tyrosine kinases that includes IGF1R
and insulin receptor (INSR) and is thought to have evolved from a common ancestral gene
(Pollak, 2008). Both IGF1 and IGF2 bind to IGF1R with very high affinity that is comparable to
that of insulin binding to INSR (Pandini et al., 2002). The INSR gene encodes two isoforms
termed A and B, and the A isoform is predominantly expressed in embryonic tissues (Giddings
and Carnaghi, 1992). In addition, the INSR A isoform is widely expressed by cancer cells and
regulates a range of malignant properties (Belfiore and Malaguarnera, 2011). Importantly, IGF2
binds with high affinity to the A isoform (Frasca et al., 1999). Moreover, IGF2 binds with high
affinity to IGF2R, which is structurally unrelated to IGF1R and INSR, lacks tyrosine kinase
activity and is thought to primarily function by sequestering IGF2 (Brown et al., 2009). In
contrast, IGF1 binds weakly to both INSR and IGF2R (Pollak, 2008).
The human IGF2 gene contains at least four promoters arranged among several exons of
which the last three are protein coding (Bergman et al., 2013; Nordin et al., 2014). The
promoters give rise to differing transcripts in a context dependent manner, although the protein
coding segment is always the same. This segment is initially translated as a 180 amino acid pre-
propeptide that contains five domains as well as a signal sequence (O'Dell and Day, 1998). The
signal sequence is cleaved to yield the 156 amino acid propeptide, which itself undergoes
sequential proteolysis to yield a mature 67 amino acid peptide that lacks the large E domain.
A wealth of evidence implicates IGF2 as a key regulator of fetal growth (Harris and
Westwood, 2012). Early RNA in situ hybridization studies revealed that IGF2 is widely

25
expressed in both human (Han et al., 1987) and rodent (Stylianopoulou et al., 1988) embryos.
However, perhaps the most decisive data originated from genetically engineered mouse models.
In an initial study, germline transmission of a null allele from male chimeras resulted in
heterozygous progeny that were approximately half the size but otherwise phenotypically
indistinguishable relative to their wild type littermates (DeChiara et al., 1990; DeChiara et al.,
1991). Reciprocally, null mutations in Igf2r result in fetal overgrowth (Lau et al., 1994; Wang et
al., 1994). In addition, IGF2 increases fetal weight when directly infused into the circulation of
pregnant guinea pigs (Sferruzzi-Perri et al., 2006). Finally, evidence from human disease also
supports a role of IGF2 in fetal development. For example, Beckwith-Wiedemann syndrome
(BWS) is characterized by fetal overgrowth and cancer predisposition and specifically involves
alterations in a genomic locus that emcompasses the IGF2 gene (Cooper et al., 2005).
At the tissue level, both Igf2 null mice and mice lacking Igf2 specifically in placental
tissues exhibit reduced placental and fetal growth (Constancia et al., 2002). Indeed, tissue
explant experiments showed that IGF2 stimulates the survival and proliferation of the
cytotrophoblast progenitors that give rise to the syncytiotrophoblast layer (Forbes et al., 2008).
Interestingly, IGF2 is also highly expressed in the trophoblastic columns that invade the maternal
endometrium (Han et al., 1996), and IGF2 regulates the migration (Irving and Lala, 1995) and
invasion (Hamilton et al., 1998) properties of these cells. IGF2 may also promote fetal
development by regulating the glucose and amino acid transport of nutrients through placental
trophoblasts (Karl, 1995; Kniss et al., 1994). Finally, IGF2 is expressed in the embryonic mouse
epicardium during midgestation heart development, and Igf2 null mice exhibit reduced
ventricular wall proliferation and ventricular wall hypoplasia (Li et al., 2011).

26
IGF2 expression in rodents declines sharply after birth with the exception of the brain
(Murphy et al., 1987; Soares et al., 1985). In postnatal humans, IGF2 is expressed form the liver
specific P1 promoter and is secreted abundantly into the circulation (Blum et al., 1988;
Sussenbach et al., 1992; Zapf et al., 1981). However, little is known about the function of this
phenomenon in adult physiology, and in recent years the biology of IGF2 has been investigated
almost exclusively in the context of malignancy (Livingstone, 2013). Nonetheless, the potential
consequences of IGF2 expression in postnatal life have been studied in several genetically
engineered mouse models (Wolf et al., 1998).
Mirroring the role of IGF2 in fetal development, overexpression also causes the
overgrowth of several tissues during adult life. In an initial study, IGF2 expression driven by a
bovine keratin 10 promoter resulted in overgrowth of the skin as well as the colon, appendix and
uterus (Ward et al., 1994). In another study, IGF2 expression driven by a rat
phosphoenolpyruvate carboxykinase promoter resulted in increased size of the kidneys, adrenal
glands and testes (Wolf et al., 1994). In a third study, IGF2 expression driven by a mouse H-2K
promoter caused increased weight of the thymus as a result of increased numbers of CD4 T cells
(Kooijman et al., 1995; van Buul-Offers et al., 1995).
Some transgenic mouse lines expressing elevated IGF2 also exhibit metabolic alterations.
In one study, IGF2 expression driven by a mouse major urinary protein promoter resulted in
animals with hypoglycemia and hypoinsulinemia, suggesting IGF2 may exert insulin-like effects
(Rogler et al., 1994). Indeed, in another study the authors confirmed that IGF2 overexpressing
animals exhibit physiological properties consistent with markedly increased peripheral insulin
action, primarily in skeletal muscle (Rossetti et al., 1996). Interestingly, both the bovine kertain
10 and mouse major urinary protein driven models exhibit markedly reduced levels body fat,

27
likely as a result of increased dietary lipid oxidation and reduced lipid incorporation into body fat
(Da Costa et al., 1994). Therefore, elevated serum IGF2 levels in human adults may regulate
tissue growth as well as glucose and lipid metabolism.
1.3.2: The regulation of IGF2 in physiology and disease
Genomic imprinting is a complex epigenetic phenomenon whereby genes are expressed
from a single allele according to parental origin (Ferguson-Smith, 2011). Although originally
described in plants four decades ago, imprinting is now known to broadly regulate development,
adult physiology and disease in mammals (Peters, 2014). Notably, the first three genes shown to
be imprinted in mice were the IGF2 receptor Igf2r (Barlow et al., 1991), Igf2 itself (DeChiara et
al., 1991) and the neighboring H19 gene (Bartolomei et al., 1991). Moreover, IGF2 was soon
confirmed as an imprinted gene in humans (Giannoukakis et al., 1993; Ogawa et al., 1993;
Ohlsson et al., 1993; Rainier et al., 1993).
Prior to the decisive data showing that Igf2 is an imprinted gene in mice, an initial clue
was the observation that the heterozygous progeny of male germline chimeras exhibited IGF2
mRNA levels ten times lower than their wild type littermates (DeChiara et al., 1990). Moreover,
the authors soon demonstrated that the growth phenotype observed in heterozygotes was never
observed in the progeny of wild type males and that the heterozygous progeny of mutant males
were phenotypically indistinguishable from homozygous mutants (DeChiara et al., 1991).
Moreover, RNAase protection assays provided definitive molecular evidence that only the
paternal Igf2 allele is expressed (DeChiara et al., 1991). The mechanism of maternal IGF2 allele
silencing in mice and humans has since attracted considerable interest and is now known to

28
involve well characterized differentially methylated regions (also called imprinting control
regions) located in and around the IGF2 locus (Nordin et al., 2014).
The finding that the maternal IGF2 allele is silenced through imprinting was quickly
followed by the discovery that LOI occurs in human cancer. Two landmark studies showed that
the maternal IGF2 allele may become reactivated in Wilms tumors (Ogawa et al., 1993; Rainier
et al., 1993). In these studies, IGF2 was shown to be exclusively expressed form the paternal
allele in embryonic and normal adult kidneys, while a subset of tumors exhibited biallelic
expression. Subsequent reports showed that maternal allele reactivation is associated with
methylation changes in the imprinting control regions (Moulton et al., 1994; Steenman et al.,
1994; Sullivan et al., 1999; Taniguchi et al., 1995) and results in substantially increased mRNA
levels of IGF2 (Ravenel et al., 2001).
The discovery of IGF2 LOI in Wilms tumors stimulated numerous complementary
studies in other tumor types. Notably, these studies have repeatedly shown that IGF2 LOI is
widespread and characterizes a large subset of patients in a vast array of malignancies (Cui,
2007). For example, an initial study in prostate cancer reported biallelic IGF2 expression in a
small fraction (2/11) of benign prostatic hyperplasia samples and in a large fraction (8/10) of
primary prostate cancers (Jarrard et al., 1995). Interestingly, further studies showed that IGF2
LOI is a consistent feature of aging prostates in both mice and humans, and in mice LOI is
associated with alterations in the well characterized differentially methylated regions that
regulate IGF2 imprinting (Fu et al., 2008).
IGF2 overexpression is extensively documented in human cancer, and a variety of
mechanisms in addition to LOI are known to underlie this phenomenon (Livingstone, 2013).

29
Indeed, early studies commented on the complexity of the regulatory sequences that comprise the
various IGF2 promoters (Holthuizen et al., 1993). For example, the transcription factors C/EBP
(van Dijk et al., 1992), EGR1 (Bae et al., 1999) and SP1 (Lee et al., 1998) were found to bind to
and activate the P1, P3 and P4 promoters, respectively, in human liver cells. Notably, the well
characterized tumor suppressor WT1 was also found to bind to the P3 promoter in human cells
and repress the expression of IGF2 (Drummond et al., 1992).
More recently, a number of additional transcription factors and a microRNA have been
found to promote IGF2 expression in cancer. For example, the transcription factor ID1 promotes
the growth and metastasis of esophageal cancer cells through upregulation of IGF2 (Li et al.,
2014), and the transcription factor ZFP57 promotes the tumorigenicity of fibrosarcoma cells
through upregulation of IGF2 (Tada et al., 2014). In addition, downregulation of the microRNA
miR-100 in breast cancer promotes tumor growth through derepression of IGF2 (Gebeshuber and
Martinez, 2013). Finally, transcription factors may drive IGF2 expression in prostate cancer.
Indeed, the transcription factor E2F3 binds to and activates the Igf2 P2 promoter in mouse
tissues, and the genes are co-expressed in clinical prostate cancer samples (Lui and Baron, 2013).
1.3.3: The role of IGF2 in malignancy
Although IGF1 has attracted considerably more interest over the last four decades, the
role of IGF2 in cancer has also been the subject of investigation (Livingstone, 2013). IGF2 binds
to canonical multimeric receptor tyrosine kinase complexes composed of IGF1R and INSR alone
or in combination as hydrid receptors (Belfiore et al., 2009). Extracellular ligand binding results
in autophosphorylation of the intracellular tyrosine kinase domains. These activated domains in
turn phosphorylate scaffolding intermediaries from the phosphoinositide-3-kinase (PI3K) and

30
mitogen activated protein kinase (MAPK) signaling pathways, primarily members of the insulin
receptor substrate (IRS) and Src homology 2 domain containing (SHC) protein families,
respectively. The activated IRS intermediaries principally activate AKT, while the SHC
intermediaries may activate JNK, ERK and P38 alone or in combination. These effector kinases
in turn regulate canonical cancer properties including survival, proliferation and metastasis in a
context dependent manner (Weroha and Haluska, 2012).
Observations from genetically engineered mouse models suggest that IGF2 may play an
important role in the pathogenesis of a number of tumor types (Livingstone, 2013). In an initial
study, expression of IGF2 in mammary tissue with a sheep beta-lactoglobulin promoter resulted
in transgenic animals that developed breast tumors beginning at five months of age (Bates et al.,
1995), and in a second study IGF2 overexpression was sufficient to initiate metastatic breast
tumors (Pravtcheva and Wise, 1998). In addition, after longer latencies IGF2 is sufficient to
initiate lung cancer (Moorehead et al., 2003) as well as liver, lymphoid, skin, soft tissue and
thyroid cancer (Rogler et al., 1994). Similarly, IGF2 modulates the penetrance of large T antigen
induced islet cell tumors (Christofori et al., 1994) and PTEN deficient breast tumors (Church et
al., 2012), and IGF2 is indispensable for the formation of PTCH deficient medulloblastoma and
rhabdomyosarcoma (Hahn et al., 2000).
In addition to this role in tumorigenesis, IGF2 may also regulate several aspects of tumor
progression. Interestingly, hypoxia instigates a rapid upregulation IGF2 in human liver cancer
cells in an EGR1 dependent manner (Bae et al., 1999). Moreover, IGF2 stimulates the secretion
of VEGF in this context, suggesting that IGF2 may regulate angiogenesis (Kim et al., 1998; Yao
et al., 2012). Furthermore, as alluded to earlier IGF2 stimulates the invasion and migration of
human placental trophoblasts (Hamilton et al., 1998; Irving and Lala, 1995), and IGF2

31
expression is sufficient to initiate metastatic breast tumors in transgenic mice (Pravtcheva and
Wise, 1998). Indeed, IGF2 confers invasive properties in leiomyosarcoma (Sciacca et al., 2002)
and choriocarcinoma (Diaz et al., 2007) cells, supporting a role for IGF2 in metastatic
progression. Finally, consistent with a large literature that modifiers of apoptosis and survival
signaling regulate the efficacy of chemotherapy (Pommier et al., 2004), IGF2 may regulate
cisplatin resistance in head and neck cancer (Ogawa et al., 2010).
Today knowledge about the role of IGF2 in prostate cancer is exceedingly scarce. As
alluded to earlier, LOI may contribute to increased IGF2 expression in primary prostate cancer
(Jarrard et al., 1995). Moreover, in one study exposure of LNCaP cells to IGF2 increased the
expression of steroidogenic enzymes, the production and secretion of androgens and the
expression of AR regulated genes (Lubik et al., 2013). Notably, the authors also reported that
IGF2 expression increases in patients undergoing ADT, suggesting that IGF2 may regulate
steroidogenesis and castration resistance in prostate cancer cells.
1.3.4: IGF signaling as a therapeutic target
The success of trastuzumab and the extensive laboratory research implicating IGF1 and
IGF1R in cancer biology stimulated the development of multiple IGF1R monoclonal antibodies
(Pollak, 2008). Unfortunately, these antibodies uniformly yielded disappointing results in large
randomized clinical trials. For example, CP-751,871 (also called figitumumab) failed to confer a
survival benefit when combined with paclitatexl and carboplatin for lung cancer in a phase III
trial (Langer et al., 2014). Similarly, R1507 is a monoclonal antibody that failed to improve
survival in combination with erlotinib for lung cancer in a large randomized phase II trial
(Ramalingam et al., 2011). Likewise, AMG479 (also called ganitumab) failed to improve

32
survival in large randomized phase II trials in combination with endocrine therapy for breast
cancer (Robertson et al., 2013) and gemcitabine for pancreatic cancer (Cohn et al., 2013).
Finally, figitumumab failed to improve survival in combination with docetaxel for CRPC in a
large randomized phase II study (de Bono et al., 2014).
These results raised considerable interest in understanding why IGF1R blockade fails to
confer clinically meaningful results, and several possible explanations have emerged (Pollak,
2012; Yee, 2012). The hypothalamus stimulates GH secretion by the anterior pituitary, which in
turn causes the liver to produce IGF1. Importantly, this endocrine system is regulated by
negative feedback at the level of both the hypothalamus and pituitary. IGF1R blockade therefore
predictably causes large compensatory increases in GH and IGF1 in humans (Atzori et al., 2011;
Haluska et al., 2007; Tolcher et al., 2009). In the setting of supraphysiological IGF1, weak
binding of that ligand to INSR may activate downstream signaling in spite of IGF1R blockade.
Moreover, GH excess is known in the endocrinology field to promote insulin resistance (Moller
and Jorgensen, 2009). Indeed, hyperglycemia and compensatory hyperinsulinemia are well
documented consequences of IGF1R blockade (Atzori et al., 2011; Haluska et al., 2007; Tolcher
et al., 2009), and hyperinsulinemia would be expected to activate INSR expressed by tumor cells.
The finding that IGF1R knockdown causes INSR hypersensitivity to insulin is particularly
relevant in this context (Dinchuk et al., 2010). In addition, the high serum IGF2 levels
physiologically present in humans would be expected to efficiently activate INSR and
downstream signaling irrespective of IGF1R blockade. Finally, laboratory models suggest that
IGF1R antibodies may cause compensatory INSR activation in the absence of changes in ligand
concentrations (Buck et al., 2010). Therefore, a number of mechanisms are thought to limit the
efficacy of IGF1R blockade, and these mechanisms generally involve activation of INSR.

33
The data emphasizing the likely significant role of INSR in IGF1R blockade resistance is
particularly relevant to the recent development of dual IGF1R/INSR tyrosine kinase inhibitors
and anti-ligand therapeutics. For example, OSI-906 is a potent, selective and orally bioavailable
small molecule inhibitor of IGF1R and INSR (Mulvihill et al., 2009). OSI-906 shows therapeutic
activity in an array of in vivo models (Flanigan et al., 2010; Flanigan et al., 2013; Fox et al.,
2011; Kuhn et al., 2012; Mulvihill et al., 2009; Zeng et al., 2012; Zhao et al., 2012; Zinn et al.,
2013), with some studies reporting abrogation of IGF1R induced activation of INSR (Buck et al.,
2010; Zhao et al., 2012). Moreover, OSI-906 was well tolerated in phase I clinical trials (Jones et
al., 2014; Puzanov et al., 2014), and a phase III study is underway in the United States for
adrenocortical carcinoma (NCT00924989). Like OSI-906, BMS-754807 is a potent, selective
and orally bioavailable small molecule inhibitor of IGF1R and INSR (Carboni et al., 2009;
Wittman et al., 2009). BMS-754807 also exhibits therapeutic activity in animal models and is
currently in clinical development (Awasthi et al., 2012; Heilmann et al., 2014; Hou et al., 2011;
Litzenburger et al., 2011; Tognon et al., 2011).
In addition to dual IGF1R/INSR small molecule inhibitors, antibodies that target the IGF
ligands have been developed. For example, MEDI-573 binds to IGF1 and IGF2 with high
affinity and reverses IGF ligand induced signaling (Gao et al., 2011). Moreover, MEDI-573
exhibits therapeutic activity in vivo (Gao et al., 2011; Zhong et al., 2014) and was well tolerated
in a phase I clinical trial (Haluska et al., 2014). Similarly, BI-836845 binds to IGF1 and IGF2
with high affinity, reverses IGF ligand induced signaling, exhibits therapeutic activity in vivo
and is currently in clinical development (Friedbichler et al., 2014). Notably, both MEDI-573 and
BI-836845 inhibit signaling downstream of IGF2 and INSR. Therefore, disappointing results
from the first generation of IGF1R antibodies have given rise to a second generation of small

34
molecule inhibitors and antibodies. These second generation therapeutics are more aligned with
the molecular pathophysiology of IGF signaling in cancer and therefore may instigate more
favorable outcomes (Pollak, 2012; Yee, 2012).

35
CHAPTER 2:
GATA2 confers aggressiveness in CRPC

36
2.1: GATA2 is upregulated in chemotherapy resistant CRPC in laboratory models and clinical
databases
2.1.1: Generation of a docetaxel resistant ARCaPM subline
We previously generated two in vitro models of docetaxel resistance using the well
characterized CRPC cell lines DU145 and 22Rv1 (Domingo-Domenech et al., 2012). The
resulting docetaxel resistant sublines were termed DU145-DR and 22Rv1-DR. In addition to
docetaxel resistance, DU145-DR and 22Rv1-DR were characterized by increased tumorigenicity
in immunocompromised mice.
To further investigate the mechanisms underlying docetaxel resistance in CRPC, we
generated a third model using the CRPC cell line ARCaPM. ARCaPM is a derivative of a cell
line generated from the ascites fluid of a patient with metastatic CRPC (Zhau et al., 1996).
ARCaPM cells were grown to 50% confluence, treated with 50nM docetaxel for 72 hours and
the emergence of docetaxel resistant clones was monitored microscopically over a period of 10-
20 days. This selection process was repeated serially over a period of six months with gradually
increasing concentrations of docetaxel until a dose of 500nM was reached. The resulting subline
was termed ARCaPM-DR and exhibited no reduction in two dimensional colony formation
capacity following 72 hour exposure to 125nM docetaxel, a concentration uniformly lethal in the
parental cell line (Figure 1(a)). Moreover, immunoblot studies confirmed that there was no
increase in the induction of apoptosis following 72 hour exposure to 125nM docetaxel (Figure
1(b)). Finally, consistent with our previous characterization of DU145-DR and 22Rv1-DR, we
observed that ARCaPM-DR cells are more tumorigenic than their parental counterparts
following subcutaneous implantation in immunocompromised mice (Figure 1(c)).

37
Figure 1: Characterization of the ARCaPM-DR subline. (a) Representative colony formation assays and quantifications of ARCaPM parental and ARCaPM-DR cells following indicated docetaxel treatment for 72 hours. Data represent the mean ± SD. (b) Representative immunoblots of cleaved PARP protein levels in ARCaPM parental and ARCaPM-DR cells following treatment with DMSO or docetaxel (125nM) for 72 hours. (c) Tumor incidence and latency of ARCaPM parental and ARCaPM-DR cells following subcutaneous implantation in immunocompromised mice. Data represent the mean ± SD. *P<0.05.
2.1.2: Characterization of cross resistance to cabazitaxel in docetaxel resistant cells
Cabazitaxel is a second generation semi-synthetic taxane agent that improves survival in
CRPC patients who progress with docetaxel (de Bono et al., 2010). However, while docetaxel
elicits responses of approximately 50% (Petrylak et al., 2004; Tannock et al., 2004), cabazitaxel
does so only in a small subset of docetaxel resistant patients (de Bono et al., 2010). Interestingly,
cell viability assays showed that the three docetaxel resistant sublines exhibit varying degrees of
cross resistance to cabazitaxel (Figure 2(a)). Colony formation assays confirmed the relative
resistance of the docetaxel resistant sublines to cabazitaxel (Figure 2(b)). Moreover, immunblot
(Figure 2(c)) and flow cytometry (Figure 2(d)) studies showed that cabazitaxel efficiently

38
induces apoptosis in the parental cell lines, but that this effect is abolished in the docetaxel
resistant sublines. These results suggest that the molecular landscape of the docetaxel resistant
models confers varying degrees of resistance to the second line agent cabazitaxel.
Figure 2: Docetaxel resistant sublines exhibit varying degrees of cross resistance to cabazitaxel. (a) MTS assays of parental and docetaxel resistant cells treated for 72 hours with DMSO or indicated cabazitaxel concentrations. Data represent the mean ± SD. *P<0.05. (b) Representative colony formation assays and quantifications of parental and docetaxel resistant cells following indicated treatments for 72 hours. Data represent the mean ± SD. (c) Representative immunoblots of cleaved PARP protein levels in the cells and 72 hour treatments described in (b). (d) Flow cytometry detection of Annexin V and propidium iodide (PI) in the cells and 72 hour treatments described in (b). Data represent the mean ± SD. *P<0.05.

39
2.1.3: GATA2 is upregulated in chemotherapy resistant CRPC in laboratory models and clinical
databases
To investigate whether GATA2 contributes to the properties of the chemotherapy
resistant sublines, we first evaluated whether its expression levels vary between the sublines and
their parental counterparts. Indeed, we observed that GATA2 mRNA and protein are
substantially elevated in the previously characterized DU145-DR and 22Rv1-DR models (Figure
3(a)). Moreover, we observed that GATA2 mRNA and protein levels are also increased in the
third independent ARCaPM-DR model (Figure 3(b)).
Figure 3: GATA2 is upregulated in three models of chemotherapy resistant CRPC. (a) qRT-PCR and immunoblot analyses of GATA2 mRNA and protein levels, respectively, in DU145-DR and 22Rv1-DR cells relative to their parental counterparts. Data represent the mean ± SD. *P<0.05. (b) qRT-PCR and immunoblot analyses of GATA2 mRNA and protein levels, respectively, in the ARCAPM-DR subline relative to its parental counterpart. Data represent the mean ± SD. *P<0.05.
To assess the clinical relevance of these findings, we interrogated published gene
expression datasets derived from clinical prostate cancer tissues. Specifically, we investigated
whether GATA2 is significantly deregulated during the progression from primary disease to both
heavily treated lethal prostate cancer in the Grasso et al. study (Grasso et al., 2012) and
disseminated chemotherapy treated disease in the Taylor et al. study (Taylor et al., 2010).

40
Indeed, we observed that GATA2 mRNA increases during the progression to chemotherapy
treated disease in both published datasets (Figure 4(a)).
To further characterize the expression of GATA2 in clinical prostate cancer, we
performed an immunohistochemistry analysis in a series of 124 human paraffin embedded
tissues. We observed that GATA2 protein levels increase during the progression from primary
prostate cancer to disseminated CRPC, while the subset of patients treated with taxane
chemotherapy exhibit the highest levels (Figure 4(b)).
Figure 4: GATA2 is upregulated in chemotherapy treated prostate cancer tissues from multiple clinical databases. (a) Box plots of GATA2 mRNA levels during disease progression in the indicated clinical transcriptome datasets. Line, median; box, 25th to 75th percentiles; bars, 1.5 times interquartile range (IQR); dots, outliers. *P<0.05. (b) Representative immunohistochemistry images and quantifications of GATA2 protein levels during disease progression in a series of human paraffin embedded clinical prostate cancer tissues. Line, median; box, 25th to 75th percentiles; bars, 1.5 times IQR; dots, outliers. n=124. Bar, 100µm. *P<0.05.

41
2.2: GATA2 regulates chemotherapy resistance and tumorigenicity
Our data from laboratory models and clinical databases supported a possible association
between GATA2, chemotherapy resistance and tumorigenicity in CRPC. To further investigate
this association, we screened a panel of custom short hairpin RNAs (shRNAs) and characterized
two that efficiently reduced the mRNA (Figure 5(a)) and protein (Figure 5(b)) levels of GATA2
in the three chemotherapy resistant sublines.
Figure 5: Characterization of two independent GATA2 shRNAs in chemotherapy resistant CRPC sublines. (a) qRT-PCR of GATA2 mRNA levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing two independent GATA2 shRNAs or a control shRNA. Data represent the mean ± SD. (b) Representative immunoblots of GATA2 protein levels in cells from (a).
Colony formation assays revealed that GATA2 knockdown potently sensitizes the three
chemotherapy resistant sublines to docetaxel and cabazitaxel (Figure 6(a)). Moreover,
immunoblot (Figure 6(b)) and flow cytometry (Figure 6(c)) experiments showed that
sensitization is accompanied by increased induction of an apoptotic response.

42
Figure 6: GATA2 regulates chemotherapy resistance in CRPC cells. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs and following 72 hour treatment with DMSO, docetaxel (125nM) or cabazitaxel (DU145-DR and 22Rv1-DR, 25nM; ARCaPM-DR, 5nM). Data represent the mean ± SD. (b) Representative immunoblots of cleaved PARP protein levels in the cells and 72 hour treatments described in (b). (c) Flow cytometry detection of Annexin V and PI in the cells and 72 hour treatments described in (b). Data represent the mean ± SD. *P<0.05.
In addition, detailed transplantation studies showed that GATA2 knockdown reduces the
capacity of CRPC cells to form tumors in immunocompromised mice (Figure 7(a)). For example,
following implantation of 100 cells GATA2 knockdown completely abrogated the
tumorigenicity of the chemotherapy resistant sublines. Moreover, while infrequent tumors
formed at larger inoculums, they did so after long latencies and likely as a result of escape from
GATA2 knockdown (Figure 7(b)).

43
Figure 7: GATA2 regulates tumorigenicity in CRPC cells. (a) Tumor incidence and latency of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs. Data represent the mean ± SD. *P<0.05. (b) qRT-PCR of GATA2 mRNA levels in preimplanted DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs as well as infrequent explanted tumors (103 cell inoculum). Adjoining representative immunohistochemistry images of GATA2 protein levels in the explanted tumors. Data represent the mean ± SD. Bar, 100µm.
2.3: GATA2 regulates a signature of cancer progression associated genes
2.3.1: Identification and characterization of GATA2 regulated genes
To elucidate mechanisms whereby GATA2 regulates chemotherapy resistance and
tumorigenicity, we performed expression profiling and knowledge based computational studies
comparing control and GATA2 knockdown conditions in the chemotherapy resistant sublines.
Prompted by the observation that GATA2 regulates similar biological properties in DU145-DR,

44
22Rv1-DR and ARCaPM-DR, we focused an initial series of studies on mechanisms overlapping
among the three models. RNA sequencing followed by differential expression analysis
(Padj<0.05, fold change (FC)≥1.5) resulted in the identification of a consensus 28 member
signature of GATA2 regulated genes (Figures 8(a) and 8(b)). In addition, gene ontology analysis
showed that ‘Cancer’, ‘Cell Death and Survival’ and ‘Cellular Growth and Proliferation’ are the
most commonly deregulated biological categories (Figure 8(c)).
Figure 8: GATA2 regulates a consensus 28 member signature of cancer associated genes. (a) Venn diagram of differentially expressed genes (Padj<0.05 and FC≥1.5) identified by RNA sequencing after shRNA mediated GATA2 knockdown in DU145-DR, 22Rv1-DR and ARCaPM-DR cells. (b) Heatmap of the consensus 28 member signature of GATA2 regulated genes from (a). (c) Ingenuity knowledge based molecular and cellular functions analysis of GATA2 regulated genes in DU145-DR, 22Rv1-DR and ARCaPM-DR cells from (a).
To assess the clinical relevance of the consensus signature, we interrogated its
representation in patient derived datasets. Specifically, we performed Pearson correlation
coefficient calculations comparing the average expression of the signature and the expression
data of patients as previously described (Liu et al., 2007). These calculations showed that the 28

45
gene signature is remarkably enriched in the heavily treated lethal prostate cancer population
from the large Grasso et al. study (Figure 9). Similarly, the signature is also significantly
enriched in the disseminated chemotherapy treated population from the independent Taylor et al.
study (Figure 9). Therefore, the collective expression pattern of the consensus signature genes
during prostate cancer progression is strongly consistent with regulation by GATA2.
Figure 9: The consensus signature is strongly expressed in clinical prostate cancer tissues treated with chemotherapy. Box plots of the Pearson coefficients for the correlation between the expression data of each patient in the indicated clinical transcriptome datasets and the average expression of the consensus GATA2 signature during prostate cancer progression. Line, median; box, 25th to 75th percentiles; bars, 1.5 times IQR; dots, outliers.
2.3.2: Genetic screen of a subset of clinically salient GATA2 regulated genes
To further distill the consensus signature to its clinically salient components, we focused
on genes that exhibited the most robust differential expression pattern consistent with regulation
by GATA2 in patient derived datasets. We thereby identified a core subset of 7 genes that
demonstrate both uniform deregulation by GATA2 knockdown in the three chemotherapy
resistant sublines (Figure 10(a)) as well as a strongly complementary (FDR<0.1) expression
pattern during disease progression in the Grasso et al. and Taylor et al. studies (Figure 10(b)).

46
Figure 10: A subset of 7 genes is regulated by GATA2 in the chemotherapy resistant sublines and exhibits a strongly complementary expression profile in clinical prostate cancer tissues. (a) qRT-PCR of mRNA levels of a subset of cancer progression associated genes in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs. Data represent the mean ± SD. (b) Z scores of mRNA levels of genes from (a) in the indicated clinical transcriptome datasets. Line, median; box, 25th to 75th percentiles; bars, 1.5 times IQR. (c) qRT-PCR of mRNA levels of genes from (a) and (b) in DU145-DR, 22Rv1-DR and ARCaPM-DR cells relative to their parental counterparts. Data represent the mean ± SD. *P<0.05.
Interestingly, all 7 genes also exhibited corresponding expression patterns in the
chemotherapy resistant cultures relative to their parental counterparts (Figure 10(c)), suggesting
possible roles in chemotherapy resistance, tumorigenicity or both. Moreover, complementing the
knowledge based identification of ‘Cancer’, ‘Death and Survival’ and ‘Growth and Proliferation’
as relevant GATA2 regulated biological categories (Figure 8(c)), this core subset was rich in
genes known to regulate malignant properties. For example, known cancer progression
promoting genes (e.g. IGF2, PAK4, FOXM1) were repressed by GATA2 knockdown and
upregulated during prostate cancer progression, while known cancer progression inhibiting genes

47
(e.g. GALNT7, ARRDC3) were derepressed by GATA2 knockdown and downregulated during
prostate cancer progression (Figures 10(a) and 10(b)).
Figure 11: Characterization of RNA interference reagents and expression constructs for a focused genetic screen. qRT-PCR of mRNA levels of a subset of clinically salient GATA2 regulated genes in DU145-DR, 22Rv1-DR and ARCaPM-DR following the indicated genetic manipulations. Data represent the mean ± SD.
In order to functionally characterize the 7 gene signature, we performed a genetic screen.
Specifically, we performed RNA interference (genes repressed by GATA2 knockdown) and
overexpression (genes derepressed by GATA2 knockdown) studies using the three chemotherapy
resistant sublines (Figure 2.11). We monitored both chemotherapy resistance and soft agar
colony formation as a surrogate for in vivo tumorigenicity. Notably, 5 of the 7 genes consistently
regulated chemotherapy resistance (Figure 12(a)), soft agar growth (Figure 12(b)) or both,
revealing a discrete network of functionally validated genes. For example, IGF2, PAK4 and
FOXM1 suppression diminished chemotherapy resistance and soft agar growth, while re-
expression of ARRDC3 primarily impinged on soft agar growth. These results suggest that
GATA2 regulates a core subset of clinically and biologically significant genes during the
progression to chemotherapy resistant CRPC.

48
Figure 12. Focused genetic screen of clinically salient GATA2 regulated genes. (a) Colony formation assay quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells transfected with indicated siRNAs or stable expression vectors and following 72 hour treatment with DMSO, docetaxel (125nM) or cabazitaxel (DU145-DR and 22Rv1-DR, 25nM; ARCaPM-DR, 5nM). Data represent the mean ± SD. *P<0.05. (c) Soft agar colony formation assay quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR transfected with indicated siRNAs or stable expression vectors. Data represent the mean ± SD. *P<0.05.
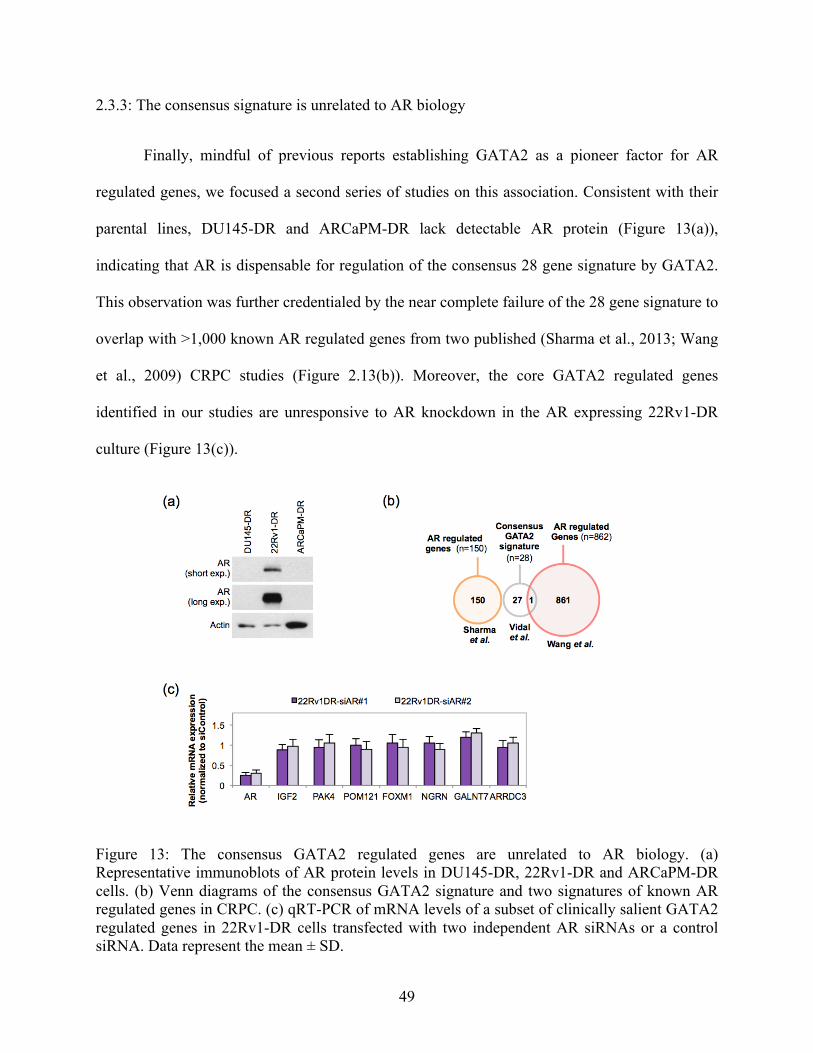
49
2.3.3: The consensus signature is unrelated to AR biology
Finally, mindful of previous reports establishing GATA2 as a pioneer factor for AR
regulated genes, we focused a second series of studies on this association. Consistent with their
parental lines, DU145-DR and ARCaPM-DR lack detectable AR protein (Figure 13(a)),
indicating that AR is dispensable for regulation of the consensus 28 gene signature by GATA2.
This observation was further credentialed by the near complete failure of the 28 gene signature to
overlap with >1,000 known AR regulated genes from two published (Sharma et al., 2013; Wang
et al., 2009) CRPC studies (Figure 2.13(b)). Moreover, the core GATA2 regulated genes
identified in our studies are unresponsive to AR knockdown in the AR expressing 22Rv1-DR
culture (Figure 13(c)).
Figure 13: The consensus GATA2 regulated genes are unrelated to AR biology. (a) Representative immunoblots of AR protein levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells. (b) Venn diagrams of the consensus GATA2 signature and two signatures of known AR regulated genes in CRPC. (c) qRT-PCR of mRNA levels of a subset of clinically salient GATA2 regulated genes in 22Rv1-DR cells transfected with two independent AR siRNAs or a control siRNA. Data represent the mean ± SD.

50
Our data suggests that the consensus GATA2 signature is unrelated to AR biology.
Nonetheless, we observed that subsets of AR regulated genes from the Sharma et al. and Wang et
al. studies are significantly deregulated by GATA2 knockdown in the AR expressing 22Rv1-DR
culture (Figure 14(a)). Notably, these GATA2 and AR regulated genes include the canonical AR
targets KLK3 and TMPRSS2 (Figure 14(b)), corroborating previous reports (Perez-Stable et al.,
2000; Wang et al., 2007) that GATA2 functions as a pioneer factor for AR at these loci.
However, the GATA2 and AR regulated signatures are not consistently deregulated in 22Rv1-
DR relative to its parental line (Figure 14(c)), suggesting that they do not contribute to
chemotherapy resistance or tumorigenicity. Complementing this hypothesis, the GATA2 and AR
regulated signatures are poorly correlated with progression to heavily treated disease in clinical
datasets (Figure 14(d)). Therefore, our data collectively suggest that in addition to its established
pioneer function at androgen regulated sites, GATA2 regulates clinically relevant AR
independent genes that confer chemotherapy resistant and tumorigenic properties in CRPC.

51
Figure 14: GATA2 regulates a subset of AR targets that are not deregulated in chemotherapy resistant CRPC cells or prostate cancer tissues. (a) Venn diagrams of GATA2 regulated genes in the AR expressing 22Rv1-DR subline and two independent signatures of known AR regulated genes in CRPC. (b) qRT-PCR of mRNA levels of two canonical AR regulated genes in 22Rv1-DR cells stably expressing control or GATA2 shRNAs. Data represent the mean ± SD. *P<0.05. (c) Heat maps of mRNA levels of GATA2 and AR regulated genes in 22Rv1 parental versus 22Rv1-DR cells. (d) Box plots of the Pearson coefficients for the correlation between the expression data of each patient in the indicated clinical transcriptome datasets and the average expression of the overlapping genes from (a) during prostate cancer progression. Line, median; box, 25th to 75th percentiles; bars, 1.5 times IQR; dots, outliers.

52
CHAPTER 3:
IGF2 significantly mediates GATA2 biology in CRPC

53
3.1: Functional validation of IGF2 as a downstream effector of GATA2
We next investigated whether GATA2 regulates actionable targets with the potential to
confer therapeutic benefit. Among the functionally validated GATA2 regulated genes (Figure
12), IGF2 was distinctive through its strong and consistent regulation of both chemotherapy
resistance and soft agar colony formation. Indeed, as a canonical activator of IGF signaling,
IGF2 promotes aggressive properties in a multitude of malignancies (Livingstone, 2013). In
addition, we noted that several IGF pathway inhibitors are currently in advanced clinical
development, highlighting a possible therapeutic opportunity. Together, these observations
credentialed IGF2 as a biologically and therapeutically meritorious candidate downstream of
GATA2, and we therefore selected it for more detailed analysis.
Figure 15: Characterization of two independent IGF2 shRNAs in chemotherapy resistant CRPC sublines. (a) qRT-PCR of IGF2 mRNA levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing two independent IGF2 shRNAs or a control shRNA. Data represent the mean ± SD. (b) Representative immunoblots of cells from (a).
We first performed a series of studies to clearly establish IGF2 as a functional mediator
of GATA2 biology. Two independent shRNAs efficiently reduced the mRNA and protein levels
of IGF2 in the three chemotherapy resistant sublines (Figure 15). Notably, colony formation

54
(Figure 16(a)) and xenotransplantation (Figure 16(b)) studies showed that IGF2 knockdown
reduces the chemotherapy resistant and tumorigenic properties of these cells, respectively.
Figure 16: IGF2 knockdown reduces the chemotherapy resistance and tumorigenicity of CRPC cells. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or IGF2 shRNAs and following 72 hour treatment with DMSO, docetaxel or cabazitaxel. Data represent the mean ± SD. (b) Tumor incidence and latency of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or IGF2 shRNAs. Data represent the mean ± SD. *P<0.05.

55
Moreover, we observed that IGF2 is abundantly secreted into the culture medium of the
chemotherapy resistant sublines (Figure 17(a)), and addition of a neutralizing IGF2 antibody
reduces their chemotherapy resistance (Figure 17(b)) and growth in soft agar (Figure 17(c)).
Figure 17: An IGF2 neutralizing antibody reduces the chemotherapy resistance and tumorigenicity of CRPC cells. (a) Enzyme-linked immunosorbent assay (ELISA) of IGF2 protein levels in the culture medium of DU145-DR, 22Rv1-DR and ARCaPM-DR cells relative to their parental cell lines. Data represent the mean ± SD. *P<0.05. (b) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells following 72 hour treatment with isotype control or anti-IGF2 immunoglobulin (10µg/ml) as well as DMSO, docetaxel or cabazitaxel. Data represent the mean ± SD. *P<0.05. (c) Representative soft agar colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells treated continuously with isotype control or anti-IGF2 immunoglobulin (10µg/ml). Data represent the mean ± SD. *P<0.05.

56
Next, we transduced control and GATA2 shRNA expressing chemotherapy resistant cells
with a control or IGF2 expression vector (Figure 18). Notably, colony formation (Figure 19(a))
and xenotransplantation (Figure 19(b)) studies showed that IGF2 expression significantly rescues
the biological defects instigated by GATA2 knockdown in these models.
Figure 18: Characterization of an IGF2 vector in GATA2 shRNA expressing cells. (a) qRT-PCR of IGF2 mRNA levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing control or GATA2 shRNAs as well as a control or IGF2 expression vector. Data represents the mean ± SD. (b) Representative immunoblots of cells from (a).
Moreover, we observed that addition of recombinant IGF2 (rIGF2) to the culture medium
of the chemotherapy resistant sublines rescues the defects instigated by GATA2 knockdown on
taxane resistance (Figure 20(a)) and soft agar growth (Figure 20(b)). Collectively, these results
suggest that IGF2 significantly mediates chemotherapy resistant and tumorigenic properties of
CRPC cells downstream of GATA2.

57
Figure 19: IGF2 expression rescues the defects instigated by GATA2 knockdown on chemotherapy resistance and tumorigenicity in CRPC cells. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs as well as a control or IGF2 expression vector and following 72 hour treatment with DMSO, docetaxel or cabazitaxel. Data represent the mean ± SD. (b) Tumor incidence and latency of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs as well as a control or IGF2 expression vector. Data represent the mean ± SD.

58
Figure 20: Exogenous IGF2 rescues the defects instigated by GATA2 knockdown on chemotherapy resistance and tumorigenicity in CRPC cells. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs and following 72 hour treatment with vehicle or rIGF2 (100ng/ml) as well as DMSO, docetaxel or cabazitaxel. Data represent the mean ± SD. (b) Representative soft agar colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs and continuously treated with vehicle or rIGF2 (100ng/ml). Data represent the mean ± SD. *P<0.05.

59
3.2: GATA2 directly upregulates IGF2
3.2.1: GATA2 binds to and activates the IGF2 P4 promoter
We previously observed that IGF2 mRNA levels are strongly upregulated and responsive
to GATA2 knockdown in the chemotherapy resistant sublines (Figure 10). We first confirmed
these observations at the protein level (Figure 21), and we next sought to investigate the
mechanism whereby GATA2 regulates IGF2 expression in our models.
Figure 21: IGF2 protein levels are elevated and responsive to GATA2 knockdown in chemotherapy resistant CRPC sublines. (a) Representative immunoblots of IGF2 protein levels in DU145-DR, 22Rv1-DR and ARCaPM-DR relative to their parental counterparts. (b) Representative immunoblots of IGF2 protein levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs.
The human IGF2 gene is transcribed from as many as five promoters termed P0-P4
(Livingstone, 2013). Interestingly, probing the TRANSFAC database with these promoter
sequences indicated that P4 contains two predicted GATA2 binding sites that we termed GBE1
and GBE2 (Figure 22(a)). Indeed, chromatin immunoprecipitation followed by qPCR (ChIP-
qPCR) studies showed that GATA2 occupies the GBEs but not adjacent control regions in the
three chemotherapy resistant sublines (Figure 22(b)).
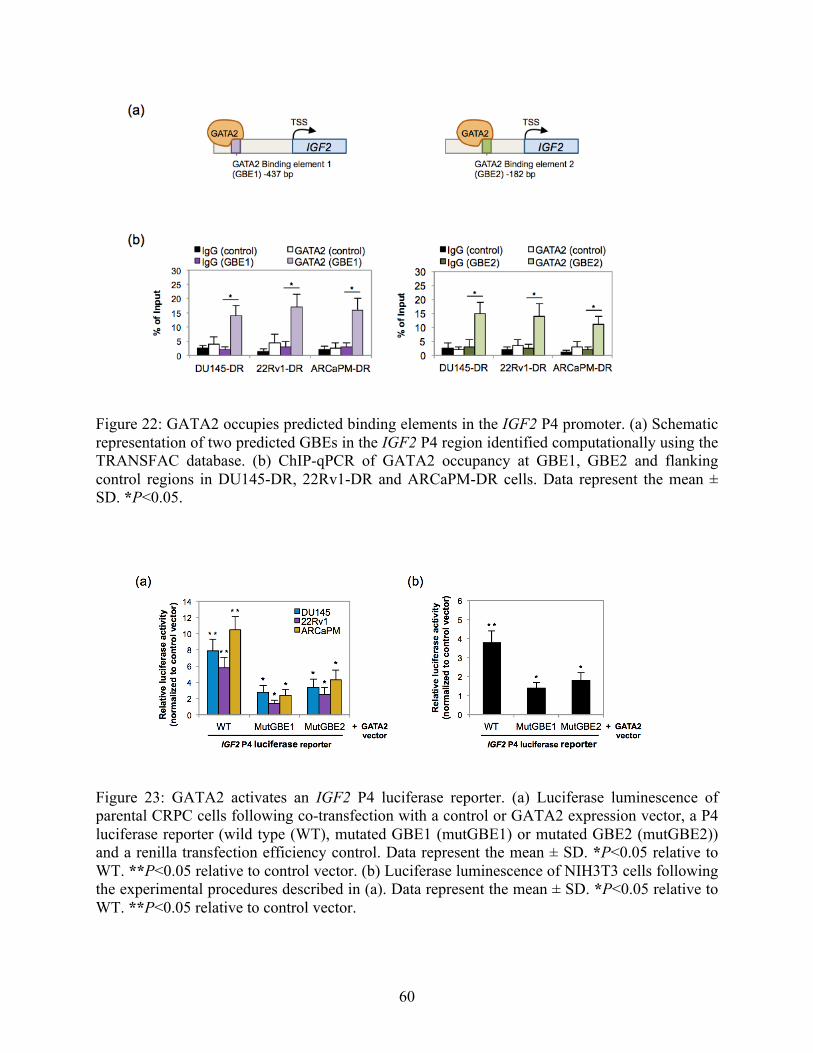
60
Figure 22: GATA2 occupies predicted binding elements in the IGF2 P4 promoter. (a) Schematic representation of two predicted GBEs in the IGF2 P4 region identified computationally using the TRANSFAC database. (b) ChIP-qPCR of GATA2 occupancy at GBE1, GBE2 and flanking control regions in DU145-DR, 22Rv1-DR and ARCaPM-DR cells. Data represent the mean ± SD. *P<0.05.
Figure 23: GATA2 activates an IGF2 P4 luciferase reporter. (a) Luciferase luminescence of parental CRPC cells following co-transfection with a control or GATA2 expression vector, a P4 luciferase reporter (wild type (WT), mutated GBE1 (mutGBE1) or mutated GBE2 (mutGBE2)) and a renilla transfection efficiency control. Data represent the mean ± SD. *P<0.05 relative to WT. **P<0.05 relative to control vector. (b) Luciferase luminescence of NIH3T3 cells following the experimental procedures described in (a). Data represent the mean ± SD. *P<0.05 relative to WT. **P<0.05 relative to control vector.

61
In addition, co-transfection assays showed that GATA2 expression can activate luciferase
transcription from a P4 reporter in the three parental cell lines, while mutation of the GBEs
reverses this effect (Figure 23(a)). Moreover, these observations were independently validated in
embryonic fibroblast cells (Figure 23(b)). Thus, we observed that GATA2 may occupy predicted
binding elements in the P4 sequence and thereby activate IGF2 transcription.
3.2.2: GATA2 and IGF2 are co-expressed during prostate cancer progression
Figure 24: IGF2 is co-expressed with GATA2 during prostate cancer progression. Representative immunohistochemistry images and quantifications of IGF2 protein levels during disease progression in a series of human paraffin embedded prostate cancer tissues. Line, median; box, 25th to 75th percentiles; bars, 1.5 times IQR; dots, outliers. n=124. Bar, 100µm. *P<0.05.
Finally, a corollary of our hypothesis that GATA2 transcriptionally upregulates IGF2 is
that these genes are co-expressed in clinical prostate cancer tissues. Indeed, we previously
observed that IGF2 is upregulated during disease progression in the two clinical transcriptome
datasets (Figure 10(b)). To confirm and extend this finding, we performed an
immunohistochemistry analysis with our cohort of 124 human paraffin embedded prostate cancer
tissues. We observed that IGF2 protein expression increases during the progression from primary
prostate cancer to disseminated CRPC, while the subset of patients treated with taxane
chemotherapy exhibits the highest levels (Figure 24).

62
3.3: IGF2 activates a polykinase program downstream of GATA2
3.3.1: Characterization of dephosphorylation patterns in knockdown models
We next investigated possible downstream mechanisms underlying the GATA2-IGF2
axis. IGF2 exhibits homology with insulin, and IGF2 ligand binding may activate both the
IGF1R and INSR tyrosine kinases (Livingstone, 2013). The receptors phosphorylate scaffolding
intermediates from the IRS and SHC families that in turn activate PI3K and MAPK signaling,
respectively. Accordingly, the downstream effectors of IGF pathway signaling principally
include AKT, JNK, ERK1/2 and p38 and are context dependent.
Immunoprecipitation studies revealed that IGF2 knockdown in the chemotherapy
resistant cultures results in dephosphorylation of both the IGF1R and INSR tyrosine kinases
(Figure 25(a)). We also noted dephosphorylation of IRS and SHC scaffolding intermediaries,
suggesting that IGF2 may signal through both the PI3K and MAPK pathways, respectively
(Figure 25(b)). Indeed, further studies showed that IGF2 knockdown consistently results in
dephosphorylation of AKT and JNK, but not p38, in the three chemotherapy resistant sublines.
In addition, ERK1/2 is dephosphorylated in DU145-DR, but not 22Rv1-DR or ARCaPM-DR.
Moreover, GATA2 knockdown in the chemotherapy resistant sublines recapitulates this
phosphorylation pattern, while an IGF2 expression vector results in a complete rescue (Figure
25(c)). These results suggest that GATA2 regulates a context dependent polykinase program
through IGF2.

63
Figure 25: IGF2 activates a polykinase program through IGF1R and INSR downstream of GATA2. (a) Representative immunoblots of phospho-tyrosine (pTyr) protein levels following immunoprecipitation of IGF1R or INSR in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing control or IGF2 shRNAs in serum free conditions. (b) Representative immunoblots of relevant IGF signaling protein levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or IGF2 shRNAs in serum free conditions. (c) Representative immunoblots of relevant IGF signaling protein levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells stably expressing indicated control or GATA2 shRNAs as well as a control or IGF2 expression vector in serum free conditions.

64
3.3.2: Chemical inhibition of the polykinase program is functionally significant
We next investigated whether the polykinase program regulated by GATA2 and IGF2 is
functionally significant. To this end, we evaluated the effect of two independent combinations of
well characterized chemical inhibitors of PI3K/AKT (LY294002 and MK2206), JNK (SP600125
and AS601245) and MEK (U0126 and PD98059) on chemotherapy resistance and soft agar
growth as a surrogate assay for tumorigenicity in our three in vitro models. We observed that
combined inhibition of PI3K/AKT and JNK signaling strongly diminishes the chemotherapy
resistance (Figures 26(a) and 27(a)) and soft agar growth (Figures 26(b) and 27(b)) of the three
models. Moreover, as predicted by our immunoblot results (Figure 25), additional MEK/ERK
signaling inhibition in the DU145-DR model further attenuates these properties. Therefore, our
data collectively suggest that GATA2 confers aggressive properties in part through a network of
kinases downstream of IGF2.

65
Figure 26: Chemicals inhibitors of the polykinase program reduce chemotherapy resistance and soft agar growth. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells following 72 hour treatment with PI3K (LY294002, 10µM), JNK (SP600125, 10µM) and MEK/ERK (U0126, 1µM) pathway inhibitors alone or in combination with DMSO, docetaxel (125nM) or cabazitaxel (DU145-DR and 22Rv1-DR, 25nM; ARCaPM-DR, 5nM). Data represent the mean ± SD. *P<0.05. (b) Representative soft agar colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells treated continuously with the cells and conditions described in (a). Data represent the mean ± SD. *P<0.05.

66
Figure 27: Alternative chemicals inhibitors of the polykinase program reduce chemotherapy resistance and soft agar growth. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells following 72 hour treatment with AKT (MK2206, 1µM), JNK (AS601245, 5µM) and MEK/ERK (PD98059, 10µM) pathway inhibitors alone or in combination with DMSO, docetaxel (125nM) or cabazitaxel (DU145-DR and 22Rv1-DR, 25nM; ARCaPM-DR, 5nM). Data represent the mean ± SD. *P<0.05. (b) Representative soft agar colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells treated continuously with the cells and conditions described in (a). Data represent the mean ± SD. *P<0.05.

67
3.4: Dual IGF1R/INSR inhibition improves the efficacy of chemotherapy and survival in
preclinical models
3.4.1: Dual IGF1R/INSR inhibitors improve the efficacy of chemotherapy in vitro
Prompted by our data implicating the GATA2-IGF2 axis in chemotherapy resistant
prostate cancer, we speculated that pharmacological disruption of this pathway may confer
therapeutic benefit. We studied dual inhibitors of the IGF1R and INSR tyrosine kinases, since
this class of drugs is in advanced clinical development. In particular, we focused on OSI-906, a
potent, selective and orally bioavailable dual IGF1R/INSR inhibitor that is currently in phase III
clinical testing for refractory adrenocortical carcinoma (Mulvihill et al., 2009).
We first performed colony formation and flow cytometry experiments with the
chemotherapy resistant cultures in vitro. We observed that monotherapy with OSI-906 or two
other dual IGF1R/INSR inhibitors exhibits negligible activity, while the inhibitors potently
sensitize the three chemotherapy resistant sublines to both docetaxel and cabazitaxel (Figures
28(a) and 28(b)). Moreover, immunoblots showed that exposure to OSI-906 results in the same
context dependent dephosphorylation pattern caused by IGF2 knockdown (Figure 28(c)).

68
Figure 28: Dual IGF1R/INSR inhibition improves the efficacy of chemotherapy in vitro. (a) Representative colony formation assays and quantifications of DU145-DR, 22Rv1-DR and ARCaPM-DR cells following 72 hour treatment with DMSO, docetaxel (125nM) or cabazitaxel (DU145-DR and 22Rv1-DR, 25nM; ARCaPM-DR, 5nM) as well as the dual IGF1R/INSR inhibitors BMS-536924 (1µM), GSK-1904529A (1µM) and OSI-906 (1µM) or vehicle control. Data represent the mean ± SD. (b) Flow cytometry detection of Annexin V and PI levels in cells and conditions described in (a). Data represent the mean ± SD. *P<0.05. (c) Representative immunoblots of relevant IGF signaling pathway protein levels in DU145-DR, 22Rv1-DR and ARCaPM-DR cells following 72 hour treatment with DMSO or OSI-906 (1µM).
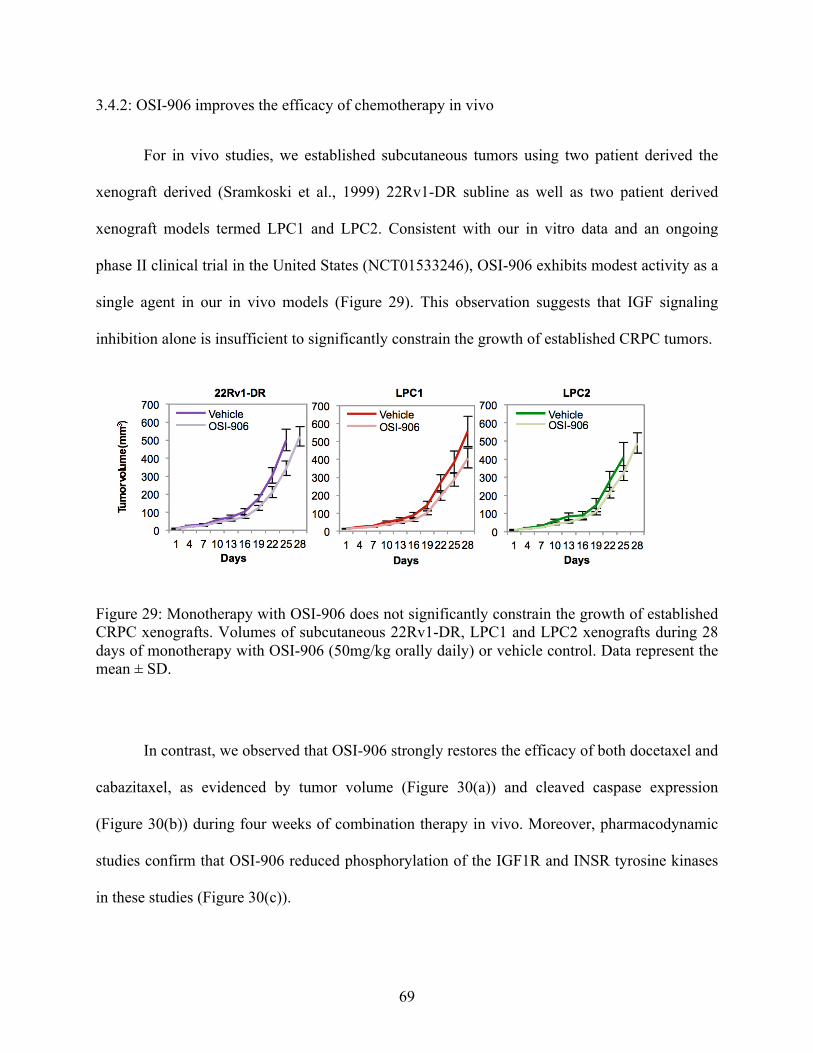
69
3.4.2: OSI-906 improves the efficacy of chemotherapy in vivo
For in vivo studies, we established subcutaneous tumors using two patient derived the
xenograft derived (Sramkoski et al., 1999) 22Rv1-DR subline as well as two patient derived
xenograft models termed LPC1 and LPC2. Consistent with our in vitro data and an ongoing
phase II clinical trial in the United States (NCT01533246), OSI-906 exhibits modest activity as a
single agent in our in vivo models (Figure 29). This observation suggests that IGF signaling
inhibition alone is insufficient to significantly constrain the growth of established CRPC tumors.
Figure 29: Monotherapy with OSI-906 does not significantly constrain the growth of established CRPC xenografts. Volumes of subcutaneous 22Rv1-DR, LPC1 and LPC2 xenografts during 28 days of monotherapy with OSI-906 (50mg/kg orally daily) or vehicle control. Data represent the mean ± SD.
In contrast, we observed that OSI-906 strongly restores the efficacy of both docetaxel and
cabazitaxel, as evidenced by tumor volume (Figure 30(a)) and cleaved caspase expression
(Figure 30(b)) during four weeks of combination therapy in vivo. Moreover, pharmacodynamic
studies confirm that OSI-906 reduced phosphorylation of the IGF1R and INSR tyrosine kinases
in these studies (Figure 30(c)).

70
Figure 30: OSI-906 restores the efficacy of chemotherapy in vivo. (a) Volumes of subcutaneous 22Rv1-DR, LPC1 and LPC2 xenografts during 28 days of combination therapy with vehicle, docetaxel (10mg/kg ip. weekly) or cabazitaxel (10mg/kg ip. weekly) as well as OSI-906 (25mg/kg orally four days weekly) or vehicle control. Data represent the mean ± SD. *P<0.05. (b) Representative immunohistochemistry images and quantifications of cleaved caspase 3 levels in subcutaneous 22Rv1-DR, LPC1, and LPC2 xenografts after 14 days of the combination therapy described in (a). Bar, 100µm. Data represent the mean ± SD. *P<0.05. (c) Representative immunohistochemistry images of pIGF1R and pINSR protein levels in subcutaneous 22Rv1-DR, LPC1, and LPC2 xenografts after 14 days of the combination therapy described in (a). Bar, 100µm.

71
3.4.3: OSI-906 improves the efficacy of chemotherapy and survival in preclinical models of
disseminated CRPC without added general drug toxicity
To extend the therapeutic benefit of combination therapy to preclinical models of
disseminated chemotherapy resistant disease, we performed intracardiac (IC) injections of
22Rv1-DR, LPC1 and LPC2 cells as well as toxicity profiles and survival analyses. Similar to a
previous report (Drake et al., 2005), IC injection of 22Rv1-DR, LPC1 and LPC2 cells resulted in
metastatic colonization of bone, liver and other organs, recapitulating disseminated human
disease in vivo (data not shown). We also exploited the in vitro nature of the 22Rv1-DR model to
generate a luciferase labeled subline suitable for in vivo bioluminescent imaging. Our studies
showed that addition of OSI-906 to standard chemotherapy confers therapeutic benefit, as
evidenced by reduced photon flux in 22Rv1-DR (Figure 31(a)) and improved overall survival in
all three independent preclinical models (Figure 31(b)). Notably, combining OSI-906 with
weekly taxane chemotherapy did not instigate a significant increase in general drug toxicity
(Figure 31(c)).

72
Figure 31: OSI-906 improves the efficacy of chemotherapy and survival in preclinical models of disseminated CRPC without added general drug toxicity. (a) Bioluminescence of IC luciferase expressing and IC administered 22Rv1-DR cells during 28 days of combination therapy with vehicle, docetaxel (10mg/kg ip. weekly) or cabazitaxel (10mg/kg ip. weekly) as well as OSI-906 (25mg/kg orally four days weekly) or vehicle control. Data represent the mean ± SD. *P<0.05. (b) Kaplan-Meier survival analysis following IC administration of 22Rv1-DR, LPC1 and LPC2 cells and the combination strategy described (a). *P<0.05. (c) Weight loss in mice following IC administration of 22Rv1-DR, LPC1 and LPC2 cells and the combination strategy described in (a). Data represent the mean ± SD.

73
CHAPTER 4:
Discussion

74
4.1: Chemotherapy resistant CRPC remains an intractable clinical entity
Cancer is a leading cause of death in humans (Jemal et al., 2011). Among the estimated
7-8 million deaths worldwide in 2008, more than 250,000 resulted from prostate cancer. In the
United States, prostate cancer is the most common cancer diagnosis and the second leading cause
of cancer death in males (Siegel et al., 2014). Therefore, significant improvements in the
management of prostate cancer exercise a considerable demographic impact in the United States
and around the world.
The pioneering work of Charles Huggins in 1941 was the first breakthrough in the
management of prostate cancer (Huggins et al., 1941). Notably, Huggins demonstrated that
orchiectomy causes durable remissions in a large majority of prostate cancer patients nearly a
decade before Sydney Farber reported transient responses with antifolates for acute
lymphoblastic leukemia (Farber and Diamond, 1948). Moreover, the elaboration of the
hypothalamus-pituitary axis and the discovery of AR in the following decades led to the
development of GnRH agonists and AR antagonists, respectively, as medical alternatives to
surgical castration (Group, 1967; Liao et al., 1974; Schally et al., 2000). However, the
development of medical castration was marginally significant with respect to prostate cancer
survival (Seidenfeld et al., 2000). Indeed, both medical and surgical castration universally evolve
to CRPC within 1-3 years of therapy (Hellerstedt and Pienta, 2002).
Remarkably, the more than six decades spanning 1941 and 2004 produced no
improvement in the survival of patients with CRPC. During this time, the modern chemotherapy
age transformed the therapeutic landscape for an array of malignancies including leukemia,
lymphoma and testicular cancer, among others (Chabner and Roberts, 2005). However,

75
chemotherapy resistance in CRPC remained an intractable challenge. For example, a review of
twenty six trials conducted between 1987 and 1991 found that the average response rate to
cytotoxic chemotherapy was less than ten percent (Yagoda and Petrylak, 1993). It was therefore
a breakthrough when the two independent phase III clinical trials TAX-327 and SWOG-9916
demonstrated a survival benefit with the second generation taxane docetaxel in 2004 (Petrylak et
al., 2004; Tannock et al., 2004).
The decade since the pivotal docetaxel trials has introduced additional therapeutic options
for CRPC. The phase III TROPIC trial showed that the third generation taxane cabazitaxel
improves survival in CRPC patients progressing with docetaxel (de Bono et al., 2010). In
addition, the second generation androgen signaling inhibitors abiraterone and enzalutamide have
entered widespread clinical practice (Beer et al., 2014; de Bono et al., 2011; Ryan et al., 2013;
Scher et al., 2012). Finally, Sipuleucel-T and radium-223 improved survival in phase III trials
(Kantoff et al., 2010; Parker et al., 2013), although their widespread clinical application is
limited by several considerations including cost, pharmacodynamic markers and exclusion
criteria (Bishr and Saad, 2013). Despite these important advances of the last decade, CRPC
invariably progresses to a therapy resistant state that precedes lethality (Bishr and Saad, 2013).
Indeed, today lethal prostate cancer is a widespread phenomenon that is characterized by
resistance to chemotherapy as well as other treatments. Accordingly, the characterization of
mechanisms of resistance to these modalities is of considerable clinical significance.
4.2: The emerging role of GATA2 in cancer biology
4.2.1: Previous characterization of GATA2 in prostate cancer and other malignancies

76
The role of GATA2 in cancer biology received little interest until recently. A number of
studies have now shown that GATA2 mutations cause heritable disorders characterized
hematopoietic and vascular defects as well as predisposition to MDS and AML (Dickinson et al.,
2011; Hahn et al., 2011; Hsu et al., 2011; Ostergaard et al., 2011). Although sparingly studied,
these mutations most likely represent loss-of-function events (Hahn et al., 2011), leading to
speculation that stem cell exhaustion leads to bone marrow stress and transformation (Migliaccio
and Bieker, 2011). In contrast, gain-of-function mutations that repress differentiation occur in
BC CML (Zhang et al., 2008). Thus, the role of GATA2 in hematopoietic malignancy is
complex and merits further investigation.
A comprehensive study later showed that GATA2 is broadly required for RAS pathway
mutated NSCLC tumorigenesis, in vivo growth and overall survival (Kumar et al., 2012).
Moreover, detailed expression profiling and genome occupancy experiments showed that
GATA2 unexpectedly regulates the proteasome as well as IL1/NFκB and RHO signaling.
Surprisingly, this extensive in vitro and in vivo characterization was later questioned by a report
that GATA2 expression is strongly reduced in lung cancer relative normal tissues and that
GATA2 knockdown does not reduce the viability of cultured RAS pathway mutated NSCLC
cells (Tessema et al., 2014). Therefore, data regarding the role of GATA2 in RAS pathway
mutated NSCLC are conflicting at this time.
In prostate cancer, GATA2 has primarily been studied in the castration naïve context. An
initial study implicated GATA2 in androgen regulated expression of PSA (Perez-Stable et al.,
2000). Subsequent studies clearly established GATA2 as a pioneer factor for AR at several other
androgen regulated loci (Wang et al., 2007; Wang et al., 2009) as well as at the whole genome
level (Chen et al., 2013; Wu et al., 2014). Finally, a recent report suggests GATA2 may be

77
involved in metastatic progression (Chiang et al., 2014).
4.2.2: GATA2 confers aggressiveness in CRPC through a consensus signature
Our data both corroborate the emerging role of GATA2 in cancer and reveal new
insights. On the functional level, our finding that GATA2 is required for the tumorigenicity of
CRPC cells is highly reminiscent of the finding that GATA2 knockdown completely abrogates
the tumorigenicity of RAS pathway mutated A549 and PC9 NSCLC cells (Kumar et al., 2012).
On the other hand, our finding that GATA2 regulates chemotherapy resistance has not been
previously reported. Nonetheless, modifiers of survival signaling are known to broadly regulate
the efficacy of chemotherapeutics (Pommier et al., 2004), and our data therefore complement the
finding that GATA2 regulates survival in RAS pathway mutated NSCLC cells (Kumar et al.,
2012). Finally, a previous study of CML suggested that GATA2 contributes to BC by repressing
the differentiation potential of leukemic cells (Zhang et al., 2008). In this context, it is of interest
that we previously showed that DU145-DR and 22Rv1-DR exhibit loss of epithelial
differentiation markers (Domingo-Domenech et al., 2012), suggesting a possible conserved role
whereby GATA2 promotes a primitive cellular state in cancer.
The implementation of three independent cell line models combined with efficacious
shRNAs and whole genome expression profiling endowed our study with considerable power to
identify bona fide GATA2 regulated genes in CRPC. Indeed, these factors enabled the
identification of a consensus 28 member signature of GATA2 regulated genes that is potently
deregulated in patients with chemotherapy resistant CRPC. Moreover, the signature is rich in
cancer progression associated genes, many of which had not been previously associated with
prostate cancer. For example, TBC1D16 is a small G protein that was recently shown to promote
melanoma proliferation (Akavia et al., 2010) and is upregulated by GATA2 in our CRPC

78
models. Similarly, ADCY9 is an adenylate cyclase gene that was recently shown to promote
MAPK pathway inhibition resistance in melanoma (Johannessen et al., 2013) and is upregulated
by GATA2 in our CRPC models. On the other hand, PPP2R1B is a known lung and colorectal
cancer tumor suppressor gene (Wang et al., 1998) whose expression is derepressed by GATA2
knockdown in our CRPC models. Finally, DPYSL3 is a known metastasis suppressor gene in
prostate cancer (Gao et al., 2010) that is derepressed by GATA2 knockdown in our models.
Together with IGF2, these genes comprise a strong basis to explain the prominent biological
defects instigated by GATA2 knockdown in CRPC cells.
In order to distill the consensus signature to its clinically salient components, we focused
on genes that exhibited the most robust differential expression pattern consistent with regulation
by GATA2 in patient derived datasets. We thereby identified a core subset of 7 genes that
demonstrate both uniform deregulation by GATA2 knockdown in the three chemotherapy
resistant sublines as well as strongly complementary expression patterns during progression to
chemotherapy resistant CRPC in the Grasso et al. and Taylor et al. datasets.
In addition to its highly relevant expression pattern in our models and clinical prostate
cancer tissues, the seven member signature is also strongly enriched in cancer progression
associated genes. For example, PAK4 and FOXM1 are upregulated by GATA2 in our models
and upregulated during progression to chemotherapy resistant CRPC in clinical datasets. PAK4
is a serine/threonine kinase that promotes a number of cancer properties in a range of tumor
types (Radu et al., 2014). In prostate cancer, PAK4 promotes cellular motility (Ahmed et al.,
2008; Wells et al., 2010; Whale et al., 2013) as well as chemotherapy resistance and
tumorigenicity (Park et al., 2013). FOXM1 is a forkhead box transcription factor that promotes a
multitude of cancer properties in a large array of tumor types (Koo et al., 2012). Notably, the role

79
of FOXM1 in prostate cancer has attracted considerable interest. For example, FOXM1 regulates
the initiation and progression of prostate cancer in genetically engineered mouse models (Cai et
al., 2013; Kalin et al., 2006), and FOXM1 cooperates with AR to accelerate DNA replication and
proliferation (Liu et al., 2014). Moreover, a recent study showed that FOXM1 and CENPF
coordinately promote tumor growth and that co-expression of these genes is a robust predictor of
metastasis and survival in clinical databases (Aytes et al., 2014). Finally, our data are
complemented by a recent report that GATA2 regulates FOXM1 in the AR dependent LNCaP
cell line (Chiang et al., 2014).
Likewise, the seven member signature contains genes that are downregulated by GATA2
in our models, are downregulated during progression to chemotherapy resistant CRPC in clinical
datasets and are known to exhibit cancer inhibitory properties. For example, GALNT7 is a
galactosaminyltransferase that represses hepatocellular tumorigenesis (Shan et al., 2013) and
melanoma metastasis (Gaziel-Sovran et al., 2011). Similarly, ARRDC3 represses proliferation,
migration, invasion, soft agar growth and tumorigenicity in breast cancer (Draheim et al., 2010).
Importantly, a focused genetic screen of the seven member signature validated the roles
of PAK4, FOXM1 and ARRDC3 in chemotherapy resistance, soft agar growth or both in three
independent in vitro models. Moreover, GALNT7 regulated soft agar growth in 22Rv1-DR cells,
and the poorly characterized nuclear pore complex protein POM121 regulated both
chemotherapy resistance and soft agar growth in the three models. Finally, IGF2 regulated both
chemotherapy resistance and soft agar growth in the three models and was selected for detailed
mechanistic analysis owing to its potential therapeutic implications. Overall, our genetic screen
contributed to the identification of a discrete network of clinically and biologically validated
GATA2 effectors in CRPC.

80
Until now the investigation of GATA2 biology in prostate cancer has focused almost
exclusively on its role as a pioneer factor for androgen regulated genes. Indeed, studies have
shown that GATA2 is required for the recruitment of AR to several well characterized androgen
regulated genes (Wang et al., 2007; Wang et al., 2009), and this hierarchy likely operates at the
genome wide level (Chen et al., 2013; Wu et al., 2014). In this context, it is of considerable
interest that the DU145-DR and ARCaPM-DR cell lines do not exhibit detectable levels of AR
protein. Moreover, the consensus signature overlapped poorly with published signatures of AR
regulated genes in CRPC, and the core GATA2 regulated genes identified in our studies were
unresponsive to AR knockdown in the AR expressing 22Rv1-DR subline. Therefore, our data
support a prominent AR independent role for GATA2 in prostate cancer for the first time.
Nonetheless, we observed that GATA2 regulates subsets of androgen regulated genes in
the AR expressing 22Rv1-DR subline. Indeed, GATA2 knockdown in 22Rv1-DR cells resulted
in reduced levels of the canonical androgen regulated genes PSA and TMPRSS2. Moreover, our
finding that GATA2 regulates a multitude of cell cycle genes in 22Rv1-DR cells (e.g. CDK1,
CENPF, NUSAP1, PBK, PTTG1 and WEE1) is highly reminiscent of a recent report that AR
preferentially regulates cell cycle genes in an in vitro castrated LNCaP model (Wang et al.,
2009). Therefore, overall our data credential the hypothesis that GATA2 regulates gene
expression in prostate cancer through both AR dependent and independent mechanisms.
Interestingly, this hypothesis is consistent with a recent report conducted with the androgen
dependent LNCaP cell line wherein approximately half of genome wide GATA2 binding sites
did not overlap with those of AR (Wu et al., 2014).

81
4.2.3: The increasingly complex role of GATA2 in prostate cancer
Although the literature related to GATA2 in prostate physiology and disease remains
sparse, the currently published studies as well as ours strongly credential an increasingly central
and multifaceted role. For example, a constellation of data now strongly supports a significant
role for GATA2 in metastatic progression. In an initial study, protein levels of GATA2 in
prostatectomy samples were significantly predictive of biochemical relapse and distant
metastasis (Bohm et al., 2009). Moreover, a second study showed that GATA2 regulates focal
adhesion disassembly and cellular motility in the androgen dependent LNCaP cell line (Chiang
et al., 2014). Notably, our data complement these findings in at least two respects. First, we
found that GATA2 mRNA levels are elevated in disseminated chemotherapy resistant CRPC
tissues from the Grasso et al. and Taylor et al. datasets. Although this observation may be
explained in part by the castration and chemotherapy resistant nature of these tissues, the data are
nonetheless consistent with a role in metastatic progression. This notion is further supported by
our observation that GATA2 protein levels are elevated is paraffin embedded disseminated
CRPC tissues without previous chemotherapy. Second, our expression profiling studies showed
that GATA2 regulates a multitude of metastogenic genes including ARRDC3, DPYSL3,
FOXM1, GALNT7 and PAK4. Therefore, in vitro invasion assays and in vivo metastasis
experiments are strongly warranted at this time in order to definitively establish GATA2 as a
regulator of metastatic progression in prostate cancer.
In addition, several observations suggest that GATA2 promotes castration resistance.
Extensive laboratory and clinical evidence now supports the hypothesis that continued AR
signaling is a critical feature of castration resistance in prostate cancer (Zong and Goldstein,
2013). Moreover, it is now firmly established that GATA2 functions as a pioneer factor that

82
recruits AR to androgen regulated genes (Wang et al., 2009; Wu et al., 2014). These two
observations strongly support a likely role for GATA2 in castration resistance. Indeed, this
notion was indirectly supported by a rigorous study dissecting the role of AR in an in vitro model
of castrated LNCaP cells (Wang et al., 2009). In that study, the transcriptional program regulated
by AR changed significantly during the process of castration. Specifically, the authors reported
that AR predominantly regulates cell cycle related genes in the castrated setting and that AR
regulation of the anaphase promoting complex associated gene UBE2C in particular promotes
castration resistance. Notably, GATA2 binding to an androgen regulated enhancer was required
for AR mediated transcription of UBE2C. Interestingly, although GATA2 did not regulate
UBE2C in our 22Rv1-DR model, GATA2 did impinge on a multitude of cell cycle related genes
that are regulated by AR in castrated LNCaP cells (e.g. CDK1, CENPF, NUSAP1, PBK, PTTG1
and WEE1). Therefore, the published literature as well as our results invite a detailed
investigation of the likely role of GATA2 in castration resistance.
Finally, it is noteworthy that GATA2 has been implicated in both urogenital development
and adult prostate physiology. Although it did not specifically comment on the prostate, one
study used a Gata2 YAC in Gata2-/- embryos to demonstrate that GATA2 is indispensable for
normal urogenital development (Zhou et al., 1998). Furthermore, the authors showed that
GATA2 is expressed in the urogenital sinus, the embryonic antecedent of the adult prostate. In
addition, a recent study showed that GATA2 is expressed in adult prostate and may play a role in
adult prostate physiology (Rosa-Ribeiro et al., 2014).
Collectively, the published literature as well as our data highlight an intriguing
constellation of observations: GATA2 is likely involved in prostate development and physiology
as well as nearly every aspect of prostate cancer biology. These observations are highly

83
reminiscent of so called “lineage oncogene” transcription factors that have been characterized in
other tumor types. For example, ETV1 is expressed in and required for the development of
interstitial cells of Cajal, the putative cell-of-origin for gastrointestinal stromal tumors (GISTs),
and ETV1 is required for the in vivo growth of GIST cells (Chi et al., 2010b). Similarly, MITF is
required for development of the melanocytic lineage (Steingrimsson et al., 2004), and MITF
regulates the soft agar growth and chemotherapy resistance of melanoma cells (Garraway et al.,
2005). Studies in genetically engineered mouse models are needed to definitively establish the
role of GATA2 in prostate development and malignant transformation. Nonetheless, current
evidence strongly credentials the notion that GATA2 is a lineage specific transcription factor that
regulates prostate development, physiology and many facets of malignancy.
4.3: A novel role for IGF2 in prostate cancer
IGF2 has attracted considerable investigation over the decades, and today several aspects
of IGF2 biology in cancer are well described (Livingstone, 2013). First, IGF2 has emerged as a
classical oncofetal gene. Early studies showed that IGF2 is widely expressed in developing
embryos in rodents and humans (Han et al., 1987; Stylianopoulou et al., 1988), and the advent of
genetically engineered mouse models revealed its role as an important fetal growth hormone
(DeChiara et al., 1990). The discovery of imprinting stimulated a surge of interest in IGF2 and
the identification of IGF2 LOI in a wide range of malignancies (Cui, 2007). Interestingly, an
early whole genome expression profiling study found IGF2 to be the single most overexpressed
transcript in colorectal cancer (Zhang et al., 1997).
The discovery of widespread IGF2 overexpression in cancer stimulated interest in its
functional properties. Indeed, a multitude of genetically engineered mouse models revealed a

84
significant role in tumorigenesis. For example, IGF2 overexpression is sufficient to initiate breast
tumors (Bates et al., 1995; Pravtcheva and Wise, 1998) as well as several other malignancies in
mice (Moorehead et al., 2003; Rogler et al., 1994). Similarly, IGF2 modulates the penetrance of
large T antigen induced islet cell tumors (Christofori et al., 1994) and PTEN deficient breast
tumors (Church et al., 2012), and IGF2 is indispensable for the formation of PTCH deficient
medulloblastoma and rhabdomyosarcoma (Hahn et al., 2000). In addition, several studies
suggest that IGF2 may regulate angiogenesis (Kim et al., 1998; Yao et al., 2012) and metastasis
(Diaz et al., 2007; Pravtcheva and Wise, 1998; Sciacca et al., 2002). Finally, although modifiers
of survival signaling are known to broadly impinge on the efficacy of chemotherapeutics
(Pommier et al., 2004), studies implicating IGF2 in chemotherapy resistance have been
exceedingly scarce (Ogawa et al., 2010).
The role of IGF2 in prostate cancer has received little attention. An early study
implicated LOI in primary prostate cancer (Jarrard et al., 1995), and more recently it was shown
that IGF2 may contribute to steroidogenesis (Lubik et al., 2013). Therefore, our data break
considerable ground in characterizing the role of IGF2 in prostate cancer. First, we showed that
chemotherapy resistant CRPC models exhibit a strong and functionally significant IGF2
autocrine loop. Although IGF2 has scarcely been implicated in chemotherapy resistance, this
finding is highly consistent with a large literature implicating survival factors in this property.
Interestingly, soft agar growth and tumorigenicity are generally accepted as complementary
measures of cellular transformation (Shin et al., 1975). Our finding that IGF2 regulates these
properties is therefore consistent with genetically engineered mouse models that showed a role
for IGF2 in tumorigenesis. In addition, ChIP studies, reporter assays and functional genetic
experiments supported IGF2 as a mediator of GATA2 biology. Notably, our laboratory models

85
were complemented by results from both public domain datasets and paraffin embedded tissues,
which showed that IGF2 expression increases during prostate cancer progression and reaches its
highest levels in disseminated CRPC patients treated with chemotherapy. Therefore, overall our
data support a greatly expanded role for IGF2 in prostate cancer.
4.4: Therapeutic implications of the GATA2-IGF2 axis
Our studies revealed that GATA2 confers aggressiveness in CRPC through increased
chemotherapy resistance and tumorigenicity. Moreover, other reports as well data form our
studies further support significant roles in metastasis, castration resistance, and possibly
tumorigenesis. Overall, these data strongly credential GATA2 as a promising therapeutic target
in prostate cancer. In this context, it is noteworthy that whole body inducible knockout of Gata2
in adult mice provokes no detectable toxicity (Kumar et al., 2012).
Although the therapeutic targeting of transcription factors is generally considered
impracticable, is not without precedent. This notion is perhaps most strongly evidenced by
decades of clinical experience with breast and prostate cancer. For example, tamoxifen (Shiau et
al., 1998) and bicalutamide (Bohl et al., 2005) bind to the ligand binding domains of estrogen
receptor and AR, respectively, and both agents are mainstays of therapy today. Moreover,
mounting evidence suggests that inhibition of several other transcription factors, including the
STATs, NFκB and NOTCH1, among others, may enter clinical practice (Yeh et al., 2013). In
addition, characterizing in detail the protein complex that facilitates GATA2 transcription may
yield clinically relevant insights. For example, the histone acetyltransferase (HAT) EP300
acetylates GATA2 and thereby increases its transcriptional activity (Hayakawa et al., 2004), and
HATs are potentially viable therapeutic targets (Manzo et al., 2009).

86
Alternatively, focusing on the effectors of GATA2 biology may be a practicable strategy.
In this context, the serine/threonine kinase PAK4 is of particular interest. PAK4 is upregulated
and responsive to GATA2 knockdown in our chemotherapy resistant CRPC models. Moreover,
PAK4 is upregulated in clinical CRPC tissues treated with chemotherapy and PAK4 knockdown
in our models reduced chemotherapy resistance and soft agar growth. These results are highly
consistent with a recent study wherein stable PAK4 knockdown reduced the tumroigenicity and
chemotherapy resistance of PC3 cells (Park et al., 2013). Moreover, the small molecule PAK4
inhibitor PF-3758309 exhibits therapeutic activity with breast, colon, GIST and melanoma
xenografts (Murray et al., 2010). Although PF-3758309 did not enter advanced clinical
development owing to pharmacokinetic limitations (Dart and Wells, 2013), these data comprise
proof of principle that targeting PAK4 downstream of GATA2 may confer therapeutic benefit.
IGF2 was the most biologically and therapeutically meritorious candidate among the
GATA2 consensus signature genes. IGF2 was strikingly upregulated and responsive to GATA2
knockdown in our models and it exhibited a complementary expression pattern during
progression to chemotherapy resistant CRPC in multiple clinical databases. Moreover, functional
genetic experiments supported IGF2 as a significant mediator of GATA2 biology. Notably, these
data stimulated the preclinical characterization of a combination strategy involving OSI-906, a
dual IGF1R/INSR inhibitor currently in phase III testing. These findings have important
implications for both the interpretation of past clinical trials and the design of future studies.
The first generation of IGF signaling inhibitors to enter clinical development was IGF1R
antibodies, and these therapeutics unfortunately met with consistent failure in phase II and phase
III studies. For example, CP-751,871 was investigated for lung cancer in a phase III trial (Langer
et al., 2014), R1507 was investigated for lung cancer in a large randomized phase II trial

87
(Ramalingam et al., 2011) and AMG479 was investigated for breast (Robertson et al., 2013) and
pancreatic (Cohn et al., 2013) cancer in large randomized phase II trials. Notably, CP-751,871
failed to improve survival in combination with docetaxel for CRPC in a large randomized phase
II study (de Bono et al., 2014).
The disappointing results from these trials stimulated considerable interest in
understanding the mechanisms whereby cancer cells persist under IGF1R blockade (Pollak,
2012; Yee, 2012). Interestingly, IGF1R blockade in humans provokes increased levels of GH,
IGF1 and insulin (Atzori et al., 2011; Haluska et al., 2007; Tolcher et al., 2009). Elevated levels
of IGF1 may activate residual IGF1R and bind weakly to INSR, while elevated levels of insulin
in particular would be expected to robustly drive PI3K and MAPK signaling downstream of
INSR. Moreover, the high serum IGF2 levels physiologically present in humans would be
expected to efficiently activate INSR and downstream signaling irrespective of IGF1R blockade
(Pollak, 2012; Yee, 2012).
In addition to these general considerations, our data provide a further possible
explanation for the failure of IGF1R blockade, particularly in the setting of CRPC. Exposure to
docetaxel provoked a powerful IGF2 autocrine loop in our in vitro models. Moreover, we
showed for the first time that IGF2 is elevated in CRPC tissues treated with chemotherapy from
both public domain datasets and paraffin embedded tissues. Importantly, IGF2 efficiently
signaled through INSR in our three independent in vitro models, and this phenomenon would
also be expected in prostate cancer tissues. Therefore, our data predict that IGF1R blockade
would be ineffectual in the setting of docetaxel treatment for CRPC.
Overall, our results suggest that a detailed knowledge of the relevant molecular
pathophysiology may be instrumental in the design of future clinical trials, particularly those

88
involving targeted therapies. Indeed, it is possible that our characterization of IGF2 in CRPC
would have discouraged the design of a clinical trial with a combination of chemotherapy and
IGF1R blockade (de Bono et al., 2014). Fortunately, a second generation of IGF signaling
inhibitors has emerged which may circumvent IGF1R blockade resistance involving IGF2 and
INSR. Indeed, OSI-906 is a potent, selective and orally bioavailable small molecule inhibitor of
IGF1R and INSR that is in phase III development (Mulvihill et al., 2009), and OSI-906 improved
survival in combination with chemotherapy in our preclinical models. Moreover, several other
relevant therapeutics are currently in phase I and phase II development. For example, BMS-
754807 is a small molecule with similar properties to OSI-906 (Carboni et al., 2009; Wittman et
al., 2009), and MEDI-573 and BI-836845 are antibodies that neutralize IGF1 and IGF2
(Friedbichler et al., 2014; Gao et al., 2011). As we have shown, these therapeutics are better
suited to the molecular pathophysiology of CRPC, and they may succeed in improving survival
in a challenging clinical context in which previous strategies have failed.

89
MATERIALS AND METHODS

90
Taxanes, targeted pathway inhibitors and other reagents:
Docetaxel and was obtained from Sigma-Aldrich. Cabazitaxel as well as LY294002, MK2206,
SP00125, AS60125, U0126, PD98059, BMS-563924, GSK-1904529A and OSI-906 were
obtained from Selleck Chemicals. Anti-IGF2 (clone S1F2) was obtained from Millipore. IgG1
isotype control and human recombinant IGF2 were obtained from R&D.
Docetaxel resistant prostate cancer cell models:
The sublines DU145-DR and 22Rv1-DR were previously described (Domingo-Domenech et al.,
2012). ARCaPM cells were obtained and maintained in Prostate Epithelial Cell Medium (both
Novicure Biotechnology) supplemented with 10% FBS. Cells were grown at 37oC in a
humidified atmosphere with 5% CO2. Resistant clones were selected by culturing cells with
docetaxel dissolved in DMSO for 72 hours. A dose escalation strategy was implemented for 7
months until a concentration of 1µM was reached. In parallel, parental ARCaPM cells were
exposed to equal volumes of DMSO.
Colony formation and cell viability assays:
Clonogenic survival assays in response to drug treatment were performed by plating 103 cells in
35mm culture dishes. After 24 hours cells were treated with vehicle controls or with drugs for 72
hours. After 10-14 days cells were fixed with 4% paraformaldehyde in PBS, stained with crystal
violet solution and formed colonies of ≥50 cells were quantified microscopically. Cell viability
was analyzed using the Cell titer 96 Aquos Non-Reactive Cell Proliferation Assay (MTs) kit
(Promega). Cells were seeded at a density of 104 in 96 well culture dishes and 24 hours later
medium was removed and replaced with new medium containing vehicle control or drugs. After
72 hours, color absorbance was measured on a microplate spectrophotometer (Molecular
Dynamics) at 450 nm (test wavelength) and 620 nm (reference wavelength). The percentage of

91
surviving cells was estimated by dividing the A450nm-A620nm of treated cells by the A450nm-
A620nm of control cells.
Immunoblot and immunoprecipitation:
Whole cell extracts from cells grown with or without serum were prepared in sample buffer and
analyzed by immunoblot using standard procedures. Extracts from xenograft tumors were
obtained using a tissue homogenizer (Fisher Scientific). For immunoprecipitation, extracts were
incubated with the indicated antibodies overnight at 4°C. Following a 2 hour incubation with
protein A/G Dynabeads (Invitrogen), beads were washed four times, resuspended in 1x Laemmli
sample buffer and boiled for 5 minutes. SDS-PAGE resolved proteins were transferred to PVDF
membranes and incubated with primary antibodies. Secondary antibodies were used at 1:5000.
Antibodies:
Unless otherwise specified, the following antibodies were used for all reported applications.
Primary antibodies against GATA2, IGF2 and ß-Actin were obtained from Sigma-Aldrich.
Primary antibodies against cleaved PARP, pTyr, pIGF1R (immunoblot), IGF1R, pINSR
(immunoblot), INSR, pSHC1, SHC1, pAKT, AKT, pp38, p38, pERK1/2, ERK1/2, cleaved
Caspase 3 and AR were obtained from Cell Signaling Technology. Primary antibodies against
pIRS1, IRS1, pJNK1-3, JNK1-3 and pIGF1R (immunohistochemistry) were obtained from Santa
Cruz Biotechnology. Primary antibody against GATA2 was obtained from Novus Biologicals
(immunoprecipitation). Primary antibody against pINSR (immunohistochemistry) and PSMA
(immunohistochemistry) were obtained from Abcam.

92
Animal husbandry and surgical castration:
All mouse procedures were performed with NOD.Cg-Prkdcscid IL2rgtm1Wjl (NSG) mice obtained
from Jackson Laboratories. All protocols for mouse experiments were in accordance with
institutional guidelines and were approved by the Mount Sinai Medical Center Institutional
Animal Care and Use Committee (IACUC). Unless specified otherwise all mouse experiments
were performed using castrated male animals. For castration, anesthetized and surgically
prepared animals were placed in dorsal recumbency. Both testes were then pushed down into the
scrotal sacs by pressuring the abdomen. A 1cm incision was made in the scrotum to expose the
tunica. The tunica was pierced, the testes were pushed out one at a time and then raised to expose
the underlying blood vessels. The vas deferens with the prominent blood vessels running along
them were located using a forceps and the testis were dissected away from the fat and removed.
The vas deferens and ducts were then replaced back into the tunica, and skin incisions were
closed with stainless steel wound closures and removed after 10 days.
Tumorigenicity assay:
Limiting dilutions of cells were implanted subcutaneously in a 1:1 mixture of complete growth
medium and Matrigel (BD Biosciences). Tumor incidence (number of tumors per number of
injections) and tumor latency (time from injection to first tumor palpability) were evaluated
weekly. Tumors formed were confirmed histologically. When tumors became palpable at a
single injection site, they were surgically removed to allow continued evaluation of other sites.
Mice were monitored for up to 36 weeks, and animals with no sign of tumor formation were
examined at necropsy for confirmation.

93
Analysis of apoptosis by flow cytometry:
Adherent and detached cells were pooled, washed and labeled with Annexin-V-FITC and PI
using the Annexin-V-FLUOS Staining Kit (Roche) in accordance with manufacturer’s
instructions. Samples were acquired with a FACscan Flow Cytometer (BD Biosciences) and
analyzed with CellQuest Pro software (BD Biosciences) to determine the percentage of cells
displaying Annexin V and PI staining.
RNA extraction and qRT-PCR:
For cultured cells as well as explanted tumors total RNA was isolated using the RNeasy Mini kit
(Qiagen) in accordance with manufacturer’s instructions. Complementary DNA was synthesized
from equivalent concentrations of total RNA using the SuperScript III First-Strand Synthesis
SuperMix Kit (Invitrogen) in accordance with manufacturer’s instructions. Primer sequences
used for amplification experiments are shown below:
Gene Forward Sequence (5' to 3') Reverse Sequence (5' to 3') ACTB CATGTACGTTGCTATCCAGGC CTCCTTAATGTCACGCACGAT ARRDC3 ATGCAAGAGGACATGCGAAAG GTGGAAGCCTTCTTCGGAATTAT FOXM1 TGCAGCTAGGGATGTGAATCTTC GGAGCCCAGTCCATCAGAACT GALNT7 GGTTCATCTTACGCAGTTTGCT GTGGAAGCCTTCTTCGGAATTAT GATA2 CATCAAGCCCAAGCGAAGA TTTGACAGCTCCTCGAAGCA IGF2 CCGTGCTTCCGGACAACT GGACTGCTTCCAGGTGTCATATT PAK4 TAGGCCATTTGTCCTGGAGTTT GTTCATCCTGGTGTGGGTGAC
POM121 AGTGGCAGTGGACATTCAGC CGTAAGCGCCTGTCAAGGA NGRN ATGGCGGTTACCCTGAGTCT GGAATCGGATTGCTTGTTTCTGT CENPF CTCTCCCGTCAACAGCGTTC GTTGTGCATATTCTTGGCTTGC KLK3 CACAGGCCAGGTATTTCAGGT GAGGCTCATATCGTAGAGCGG TMPRSS2 GTCCCCACTGTCTACGAGGT CAGACGACGGGGTTGGAAG KRT 18 GAGACGTACAGTCCAGTCCTTGG CCACCTCCCTCAGGCTGTT KRT 19 CTGCGGGACAAGATTCTTGGT CCAGACGGGCATTGTCGAT HLA A AAAAGGAGGGAGTTACACTCAGG GCTGTGAGGGACACATCAGAG HLA B CAGTTCGTGAGGTTCGACAG CAGCCGTACATGCTCTGGA NOTCH2 GGCATTAATCGCTACAGTTGTGTCT GGAGGCACACTCATCAATGTCA HES1 CTGGAAATGACAGTGAAGCACCT ATTGATCTGGGTCATGCAGTTG

94
SMO ATGGATGGTGCCCGCCGAGAG ATGGTCTCGTTGATCTTGCTGG GLI1 CCCAACTCCACAGGCATAC ACACGAACTCCTTCCGCTCC GLI2 ACACCAACCAGAACAAGCAG AGTCTTCCCAGTGGCAGTTG
Coding sequences for genes of interest as well as a loading control (ACTB) were amplified from
500ng of complementary DNA using QuantiTect SYBR Green PCR Kit (Qiagen). Amplification
was carried out using an Mx3005P qPCR System (Agilent Technologies). Cycle threshold values
were determined and normalized to the loading control for each experiment. Fold changes for
experimental groups relative to respective controls were calculated using MX Pro software
(Agilent Technologies).
Clinical transcriptome dataset mining:
A gene was considered deregulated during the progression from primary to disseminated
chemotherapy resistant disease in GSE35988 (Grasso et al., 2012) and GSE21032 (Taylor et al.,
2010) if it exhibited multiple testing significance (FDR<0.05) using the significance analysis of
microarrays (SAM) algorithm (Tusher et al., 2001). For analyses of representation of the 28 gene
GATA2 signature and AR signatures in primary and disseminated chemotherapy resistant
disease, we calculated a Pearson correlation coefficient comparing the average expression of the
signatures and the expression data for each patient sample as previously described (Liu et al.,
2007). Briefly, the expression levels of genes in control shRNA expressing cells divided by the
expression levels of genes in their corresponding GATA2 shRNA expressing cells in the three
chemotherapy resistant cultures were averaged, log transformed and the resulting ratios were
used as expression levels for genes in the two signatures. Similarly, for the downloaded clinical
transcriptome datasets expression levels were calculated by log transformation of the signal
intensity for each probe divided by the average signal intensity of that probe in the entire dataset.
For generation of the seven gene core subset from the consensus 28 gene signature, genes

95
exhibiting significantly deregulated expression (FDR<0.1) during the progression to
disseminated chemotherapy resistant disease as well as the expression pattern predicted by
GATA2 knockdown in the chemotherapy resistant models were selected. For generation of the
CRPC AR signature from the Wang et al. study, datasets were downloaded from GEO
(GSE11428) and genes were considered significantly deregulated between ‘abl-siControl’ and
‘abl-siAR’ biological replicates if they exhibited multiple testing significance (FDR<0.05) as
well as fold change ≥1.5 using the SAM algorithm.
Human paraffin embedded prostate cancer tissues:
Human formalin fixed paraffin embedded primary (n=56), metastatic CRPC (n=35) and taxane
treated metastatic CRPC (n=33) tissue samples were collected from the Mount Sinai Medical
Center tumor biorepository under an Institutional Review Board (IRB) approved protocol. All
tissue sections were reviewed by a pathologist to confirm prostate cancer origin.
Immunohistochemistry:
Immunohistochemistry analyses were conducted on prostate cancer formalin fixed paraffin
embedded tissue sections from human samples and cell line (22Rv1-DR) or LPC xenografts.
Tissue sections (5µm) were deparaffinized and submitted to standard peroxidase based
immunohistochemistry procedures. Quantification of positive cells was determined by counting
the number of tumor cells in 10 contiguous high power fields in three different areas of each
section, and referred to the total number of counted cancer cells.
Generation of stably expressing shRNA sublines:
For shRNA mediated suppression of GATA2, two clones (GATA2_2668 and GATA2_2829)
and a non-targeting (Renilla) control were selected following a screen of a custom library.
Briefly, de novo predictions were obtained using the DSIR algorithm (Vert et al., 2006) and

96
guide strands were subsequently filtered using “sensor rules” to enrich for predictions harboring
sequence features associated with effective shRNAmir processing and potent knockdown
(Fellmann et al., 2011). The control and GATA2 shRNAs were cloned into the LMP (Dickins et
al., 2005) vector. Retroviral particles were generated by transient co-transfection of 2x106 Eco
Phoenix cells in 10cm dishes using FuGene (Roche) with 4µg of a VSVG packaging plasmid and
15µg of individual shRNA plasmids. For shRNA mediated suppression of IGF2, two clones
(V3LHS_400817 and V3LHS_382742) and a non-targeting (scrambled) control (RHS4346)
were selected following a screen of commercially available libraries (Thermo Fisher Scientific).
Lentiviral particles were generated by transient co-transfection of 2x106 HEK293T cells in 10cm
dishes using FuGene with 3µg each of pMD.G and CMVR8.9l packaging plasmids and 6µg of
individual shRNA plasmids. For both retrovirsus and lentiviruses, infectious supernatant was
collected 48 and 72 hours after transfection, concentrated by ultracentrifugation, resuspended in
full growth medium and added to cell cultures with 10µg/ml of polybrene (Sigma-Aldrich). 48
hours after infection at high MOI (> 95% infection efficiency), pools of shRNA expressing cells
were purified with a FACSAria II Cell Sorter (BD Biosciences) using GFP expression. First
passage stocks were validated by qRT-PCR and immunoblot before being stored in liquid
nitrogen. All studies with stably expressing shRNA sublines were performed with pools
passaged no more than five times.
RNA sequencing and expression analysis:
To characterize the transcriptional program regulated by GATA2, we performed RNA
sequencing of first passage chemotherapy resistant sublines stably expressing shRNAs. For each
chemotherapy resistant culture, we analyzed shControl-, shGATA2#1- and shGATA2#2-
expressing cells in biological triplicates. RNA was extracted and its integrity was checked by

97
either a 2100 Bioanalyzer using the RNA 6000 Nano assay or a 2200 TapeStation using the R6K
ScreenTape (both Agilent). All processed total RNA samples had RIN value of 7.7 or greater.
The sequencing library was prepared with the standard TruSeq RNA Sample Prep Kit v2
protocol (Illumina). Briefly, total RNA was poly-A-selected and then fragmented. The cDNA
was synthesized using random hexamers, end-repaired and ligated with appropriate adaptors for
sequencing. The library then underwent size selection and purification using AMPure XP beads
(Beckman Coulter). The recommended (Illumina) 6bp barcode bases were introduced at one end
of the adaptors during the PCR amplification step. The size and concentration of the RNAseq
libraries was measured by Bioanalyzer and Qubit fluorometry (Life Technologies) before
loading onto the sequencer. The mRNA libraries were sequenced on the HiSeq 2500 System
with 100 nucleotide single-end reads, according to the standard manufacturer's protocol
(Illumina). Obtained reads were then aligned to the UCSC hg19 human genome using Tophat
software (Trapnell et al., 2009) and gene level expression values obtained using Cufflinks
(Trapnell et al., 2010). For each chemotherapy resistant culture, the Cuffdiff utility was used to
identify differentially expressed genes. Genes were considered to be regulated by GATA2 if they
exhibited a fold change difference (FC≥1.5) as well as multiple testing significance (Padj<0.05)
in both shGATA2#1 and shGATA2#2 expressing cells. The GATA2 regulated genes from each
chemotherapy resistant culture were analyzed with the Ingenuity Pathways Analysis program
(http://www.ingenuity.com/index.html) to identify molecular and cellular functions enriched in
these gene lists. Categories with a significance of P<0.01 were considered.
siRNA knockdown:
Predesigned Silencer Select (Ambion) siRNAs were transfected into cells seeded overnight at a
density of 1x105 in six well plates. siRNAs and RNAiMAX lipofectamine (Life Technologies)

98
were diluted in OptiMEM (Life Technologies), combined and added dropwise to cells at a final
concentration of 10nM. Knockdown efficiency was assayed 24 hours later by qRT-PCR and for
each gene the two most independently efficacious siRNAs were selected for chemotherapy
resistance and soft agar colony formation assays.
Gene Clone Catalog # Control#1 - 4390843 FOXM1 S5248 4392420 FOXM1 S5250 4392420 PAK4 S20134 4390824 PAK4 S20135 4390824 POM121 S19145 4392420 POM121 S59623 4392420 IGF2 S7214 4392420 IGF2 S7115 4392420 SMO S13164 4392420 SMO S13165 4392420 NOTCH2 S9638 4392420 NOTCH2 S9639 4392420
Generation of stably expressing cDNA sublines:
GATA2 (PLOHS_ccsbBEn_10902), IGF2 (PLOHS_100010430), NGRN
(PLOHS_ccsbBEn_11979), GALNT7 (PLOHS_100074154) and ARRDC3
(PLOHS_100071659) clones from the ORFeome Collaboration and an RFP (OHS5832) control
were obtained in the Precision LentiORF lentiviral expression plasmid (Open Biosystems).
Lentiviral particles were generated by transient co-transfection of 2x106 HEK293T cells in 10cm
dishes using FuGene with 3µg each of pMD.G and CMVR8.9l packaging plasmids and 6µg of
cDNA plasmid. Supernatant was collected 48 and 72 hours after transfection, concentrated by
ultracentrifugation, resuspended in full growth medium and added to shRNA cultures with
10µg/ml of polybrene (Sigma-Aldrich). 48 hours after infection at high MOI (> 95% infection

99
efficiency), pools of cDNA expressing cells were selected with blasticidin for 5-7 days.
Retroviral particles driving the expression of myristoylated AKT (myrAKT) was a kind gift of
Dr. Adolfo Ferrando (Columbia University, New York). The vector containing a scramble
sequence that doesn’t target any human sequence was used as a control (Empty Vector), pools of
cells were selected with puromycin to generate stable cell lines. Low passage stocks were
validated by qRT-PCR and all studies with stably expressing shRNA-cDNA sublines were
performed with pools passaged no more than three times.
Soft agar assay:
Anchorage independent growth was determined by plating 1x103 cells in 35mm culture dishes in
0.33% Seaplaque agarose (Lonza) and cultured in RPMI supplemented with 10% fetal calf
serum. Colonies were counted under light microscopy after 21 days. For studies involving
continuous treatment with kinase inhibitors, antibodies and recombinant IGF2, growth medium
was replaced every 72 hours with fresh reagents.
ELISA:
Levels of secreted IGF2 were measured in cells cultured for 72 hours in serum free conditions
and using a human IGF2 ELISA kit (MyBiosource) in accordance with manufacturer’s
instructions. A microplate reader (Dynex Technologies) was used to detect the signal at 450nm.
Identification of predicted GBEs:
Evolutionarily conserved GBEs in the human IGF2 promoter were identified computationally
using multiple sequence alignment software (Ovcharenko et al., 2005). Multiple gene sequences
containing the IGF2 promoter regions were analyzed using the TRANSFAC database with
default settings.

100
Chromatin immunoprecipitation:
DU145-DR, 22Rv1-DR and ARCaPM-DR cells were crosslinked with paraformaldehyde and
nuclear extracts obtained using truChIP High Cell Chromatin Shearing Kit with SDS Shearing
Buffer (Covaris). Nuclear extracts were incubated with protein G magnetic beads (Millipore)
conjugated to anti-GATA2. The solution was then successively washed with low salt, high salt,
and LiCl buffers prior to elution. Following RNAse and proteinase K digestion and DNA
extraction, the immunoprecipitated and control (input) DNA was analyzed by qPCR using the
following primers:
Forward Sequence (5' to 3') Reverse Sequence (5' to 3') GBE1 GAGTTGGCAGGAGTCATGG GGGACTCCTGTTTGGAAATG Control 1 CAGAAGCCCACCCTGGTATGTT CATCCTGGCCCCGCCCACC GBE2 CACTGCCCTGTGCAGAGATG CAGCCAGTCTCTTCTCTCCTG Control 2 CACTGACCAGCCTGCAAACT GCTCAGCAGAAGGCTCGCTG
Luciferase reporter assay, constructs, and site directed mutagenesis:
The IGF2 P4 promoter sequence (-500 to +100 nucleotides) was amplified by PCR and cloned
into the pGL3 luciferase reporter vector (Promega). The resulting vector was used as a template
to mutate GATA2 binding elements by site directed mutagenesis using the following primers:
GBE Primer Sequence
1 Forward ACATTCGCGACAaaaaaaaaACACCCCCTCTGGCTCTGT Reverse ACAGAGCCAGAGGGGGTGTttttttttTGTCGCGAATGT
2 Forward ATGGCACTTCGCTAaaaaaaaaCAGGGCTCCCGGTTGGGGGTGCAG
GAGA
Reverse TCTCCTGCACCCCCAACCGGGAGCCCTGttttttttTAGCGAAGTGCCAT
After synthesizing the mutant strand by PCR, template sequence was digested with DpnI
restriction enzyme for 2 hours at 37°C. The mutant vector was transformed into competent cells
for nick repair, plasmid DNA was recovered and mutation of the binding site was confirmed by

101
Sanger sequencing. For the reporter assay, NIH3T3 cells were seeded into 6 well plates at a
density of 3x105 and allowed to attach overnight. Transfection mix was prepared by combining
3µg of pGL3, 1µg of pCMV-Renilla, and 3µg of a GATA2 expression vector
(PLOHS_ccsbBEn_10902) or RFP control (both Open Biosystems). Cells were lysed 48 hours
after transfection with 500µl of Luciferase Assay Buffer (Dual Glo, Promega) and 100µl of
lysate were analyzed in an automatic luminometer. 100µl of Stop & Glo reagent were then added
and Renilla luminescence measured after 10 minutes of incubation. Ratios of Firefly versus
Renilla luciferase were calculated to determine promoter activity.
Monitoring of subcutaneous xenograft growth:
For in vivo studies involving chemotherapy and/or OSI-906, subcutaneous xenografts were
generated by implantation of 106 indicated prostate cancer cells in a 1:1 mixture of growth
medium and Matrigel into the flanks of NSG mice. When subcutaneous tumors became palpable,
mice were randomly assigned to treatment groups containing four animals. The vehicles for
chemotherapy and OSI-906 were 10% DMSO in sterile 1xPBS and 25mM tartaric acid,
respectively. Tumor dimensions were monitored weekly using Vernier calipers. Tumor volume
was calculated according to the formula V=(a2xb)/2 where a and b are the minimal and maximal
diameter in millimeters, respectively. In accordance with institutional guidelines, mice bearing
subcutaneous xenografts greater than 500mm3 were sacrificed. Explanted tumors were weighed,
formalin fixed, and embedded in paraffin for pathological analysis.
IC injections:
IC injections were performed as previously described (Campbell et al., 2012). Briefly, the ventral
thorax of 3-4 week old castrated males was shaved prior to anesthesia with an isoflurane
vaporizer and nose cone. The thorax was sterilized with iodine and alcohol and a sterile marker

102
was used to mark a location half way between the sternal notch and the xyphoid process. 100µl
from a 1x106cell/ml suspension of 22Rv1-DR-luciferase, LPC1 or LPC2 cells in sterile 1xPBS
was drawn into a 30.5 guage needle. The upright syringe was gently inserted through the mark
and for each injection successful penetration into the left ventricle was confirmed visually by a
pulse of bright red blood into the syringe. Following each experiment a detailed necropsy was
performed to grossly and histologically confirm disseminated tumor burden.
In vivo bioluminescence imaging:
Imaging was performed using an IVIS Spectrum imager (Xenogen). Animals were anesthetized
using an isoflurane vaporizer and placed onto the warmed stage inside the camera box. Animals
next received intraperitoneal luciferin (200mg/kg) five minutes prior to imaging. For
quantification, rectangular regions of interest (ROIs) incorporating the entire animal were
measured. The signal was measured in photons per second using Living Image software.
General toxicity monitoring:
Body weights for every mouse were recorded every three days and fluctuations were computed
by the percentage of current body weight relative to baseline. When animals showed signs of
weight loss therapy was discontinued until resolution of the toxicity and reinitiated at a 50% of
the initial dose. In accordance with institutional guidelines all animals experiencing greater than
20% weight loss were sacrificed.
Statistical analyses:
Statistical analysis was carried out with SPSS software unless otherwise specified. Experimental
data expressed as mean ± SD were analyzed by Student’s t-test. All t-tests were conducted at the
two sided 0.05 level of significance. For genomic analyses, multiple testing significance was
calculated using Significance Analysis of Microarrays (SAM) or Tuxedo software. For

103
preclinical studies, survival analyses were performed using the Kaplan–Meier method and curves
were compared by the log rank test.

104
REFERENCES

105
Ahmed, T., Shea, K., Masters, J.R., Jones, G.E., and Wells, C.M. (2008). A PAK4-LIMK1 pathway drives prostate cancer cell migration downstream of HGF. Cellular signalling 20, 1320-1328.
Akavia, U.D., Litvin, O., Kim, J., Sanchez-Garcia, F., Kotliar, D., Causton, H.C., Pochanard, P., Mozes, E., Garraway, L.A., and Pe'er, D. (2010). An integrated approach to uncover drivers of cancer. Cell 143, 1005-1017.
Atzori, F., Tabernero, J., Cervantes, A., Prudkin, L., Andreu, J., Rodriguez-Braun, E., Domingo, A., Guijarro, J., Gamez, C., Rodon, J., et al. (2011). A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 6304-6312.
Awasthi, N., Zhang, C., Ruan, W., Schwarz, M.A., and Schwarz, R.E. (2012). BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Molecular cancer therapeutics 11, 2644-2653.
Aytes, A., Mitrofanova, A., Lefebvre, C., Alvarez, M.J., Castillo-Martin, M., Zheng, T., Eastham, J.A., Gopalan, A., Pienta, K.J., Shen, M.M., et al. (2014). Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer cell 25, 638-651.
Bae, S.K., Bae, M.H., Ahn, M.Y., Son, M.J., Lee, Y.M., Bae, M.K., Lee, O.H., Park, B.C., and Kim, K.W. (1999). Egr-1 mediates transcriptional activation of IGF-II gene in response to hypoxia. Cancer research 59, 5989-5994.
Banerjee, A., Roach, M.C., Trcka, P., and Luduena, R.F. (1990). Increased microtubule assembly in bovine brain tubulin lacking the type III isotype of beta-tubulin. The Journal of biological chemistry 265, 1794-1799.
Barlow, D.P., Stoger, R., Herrmann, B.G., Saito, K., and Schweifer, N. (1991). The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349, 84-87.
Bartolomei, M.S., Zemel, S., and Tilghman, S.M. (1991). Parental imprinting of the mouse H19 gene. Nature 351, 153-155.
Bates, P., Fisher, R., Ward, A., Richardson, L., Hill, D.J., and Graham, C.F. (1995). Mammary cancer in transgenic mice expressing insulin-like growth factor II (IGF-II). British journal of cancer 72, 1189-1193.
Beer, T.M., Armstrong, A.J., Rathkopf, D.E., Loriot, Y., Sternberg, C.N., Higano, C.S., Iversen, P., Bhattacharya, S., Carles, J., Chowdhury, S., et al. (2014). Enzalutamide in Metastatic Prostate Cancer before Chemotherapy. The New England journal of medicine.

106
Beer, T.M., Pierce, W.C., Lowe, B.A., and Henner, W.D. (2001). Phase II study of weekly docetaxel in symptomatic androgen-independent prostate cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 12, 1273-1279.
Belfiore, A., Frasca, F., Pandini, G., Sciacca, L., and Vigneri, R. (2009). Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine reviews 30, 586-623.
Belfiore, A., and Malaguarnera, R. (2011). Insulin receptor and cancer. Endocrine-related cancer 18, R125-147.
Bergman, D., Halje, M., Nordin, M., and Engstrom, W. (2013). Insulin-like growth factor 2 in development and disease: a mini-review. Gerontology 59, 240-249.
Berry, W., Dakhil, S., Gregurich, M.A., and Asmar, L. (2001). Phase II trial of single-agent weekly docetaxel in hormone-refractory, symptomatic, metastatic carcinoma of the prostate. Seminars in oncology 28, 8-15.
Berry, W., Dakhil, S., Modiano, M., Gregurich, M., and Asmar, L. (2002). Phase III study of mitoxantrone plus low dose prednisone versus low dose prednisone alone in patients with asymptomatic hormone refractory prostate cancer. The Journal of urology 168, 2439-2443.
Berthold, D.R., Pond, G.R., Soban, F., de Wit, R., Eisenberger, M., and Tannock, I.F. (2008). Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 26, 242-245.
Bill-Axelson, A., Holmberg, L., Ruutu, M., Garmo, H., Stark, J.R., Busch, C., Nordling, S., Haggman, M., Andersson, S.O., Bratell, S., et al. (2011). Radical prostatectomy versus watchful waiting in early prostate cancer. The New England journal of medicine 364, 1708-1717.
Bishr, M., and Saad, F. (2013). Overview of the latest treatments for castration-resistant prostate cancer. Nature reviews Urology 10, 522-528.
Blum, W.F., Ranke, M.B., and Bierich, J.R. (1988). A specific radioimmunoassay for insulin-like growth factor II: the interference of IGF binding proteins can be blocked by excess IGF-I. Acta endocrinologica 118, 374-380.
Bohl, C.E., Gao, W., Miller, D.D., Bell, C.E., and Dalton, J.T. (2005). Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 102, 6201-6206.
Bohm, M., Locke, W.J., Sutherland, R.L., Kench, J.G., and Henshall, S.M. (2009). A role for GATA-2 in transition to an aggressive phenotype in prostate cancer through modulation of key androgen-regulated genes. Oncogene 28, 3847-3856.
Bolla, M., Collette, L., Blank, L., Warde, P., Dubois, J.B., Mirimanoff, R.O., Storme, G., Bernier, J., Kuten, A., Sternberg, C., et al. (2002). Long-term results with immediate androgen

107
suppression and external irradiation in patients with locally advanced prostate cancer (an EORTC study): a phase III randomised trial. Lancet 360, 103-106.
Bolla, M., van Poppel, H., Tombal, B., Vekemans, K., Da Pozzo, L., de Reijke, T.M., Verbaeys, A., Bosset, J.F., van Velthoven, R., Colombel, M., et al. (2012). Postoperative radiotherapy after radical prostatectomy for high-risk prostate cancer: long-term results of a randomised controlled trial (EORTC trial 22911). Lancet 380, 2018-2027.
Boorjian, S.A., Karnes, R.J., Viterbo, R., Rangel, L.J., Bergstralh, E.J., Horwitz, E.M., Blute, M.L., and Buyyounouski, M.K. (2011). Long-term survival after radical prostatectomy versus external-beam radiotherapy for patients with high-risk prostate cancer. Cancer 117, 2883-2891.
Brown, J., Jones, E.Y., and Forbes, B.E. (2009). Keeping IGF-II under control: lessons from the IGF-II-IGF2R crystal structure. Trends in biochemical sciences 34, 612-619.
Buck, E., Gokhale, P.C., Koujak, S., Brown, E., Eyzaguirre, A., Tao, N., Rosenfeld-Franklin, M., Lerner, L., Chiu, M.I., Wild, R., et al. (2010). Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Molecular cancer therapeutics 9, 2652-2664.
Cai, Y., Balli, D., Ustiyan, V., Fulford, L., Hiller, A., Misetic, V., Zhang, Y., Paluch, A.M., Waltz, S.E., Kasper, S., et al. (2013). Foxm1 expression in prostate epithelial cells is essential for prostate carcinogenesis. The Journal of biological chemistry 288, 22527-22541.
Campbell, J.P., Merkel, A.R., Masood-Campbell, S.K., Elefteriou, F., and Sterling, J.A. (2012). Models of bone metastasis. Journal of visualized experiments : JoVE, e4260.
Cantor, A.B., Iwasaki, H., Arinobu, Y., Moran, T.B., Shigematsu, H., Sullivan, M.R., Akashi, K., and Orkin, S.H. (2008). Antagonism of FOG-1 and GATA factors in fate choice for the mast cell lineage. The Journal of experimental medicine 205, 611-624.
Carboni, J.M., Wittman, M., Yang, Z., Lee, F., Greer, A., Hurlburt, W., Hillerman, S., Cao, C., Cantor, G.H., Dell-John, J., et al. (2009). BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Molecular cancer therapeutics 8, 3341-3349.
Carver, B.S., Chapinski, C., Wongvipat, J., Hieronymus, H., Chen, Y., Chandarlapaty, S., Arora, V.K., Le, C., Koutcher, J., Scher, H., et al. (2011). Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer cell 19, 575-586.
Chabner, B.A., and Roberts, T.G., Jr. (2005). Timeline: Chemotherapy and the war on cancer. Nature reviews Cancer 5, 65-72.
Chang, A.J., Autio, K.A., Roach, M., 3rd, and Scher, H.I. (2014). High-risk prostate cancer-classification and therapy. Nature reviews Clinical oncology 11, 308-323.
Chang, A.N., Cantor, A.B., Fujiwara, Y., Lodish, M.B., Droho, S., Crispino, J.D., and Orkin, S.H. (2002). GATA-factor dependence of the multitype zinc-finger protein FOG-1 for its

108
essential role in megakaryopoiesis. Proceedings of the National Academy of Sciences of the United States of America 99, 9237-9242.
Chang, K.H., Li, R., Papari-Zareei, M., Watumull, L., Zhao, Y.D., Auchus, R.J., and Sharifi, N. (2011). Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 108, 13728-13733.
Chen, C.D., Welsbie, D.S., Tran, C., Baek, S.H., Chen, R., Vessella, R., Rosenfeld, M.G., and Sawyers, C.L. (2004). Molecular determinants of resistance to antiandrogen therapy. Nature medicine 10, 33-39.
Chen, Y., Chi, P., Rockowitz, S., Iaquinta, P.J., Shamu, T., Shukla, S., Gao, D., Sirota, I., Carver, B.S., Wongvipat, J., et al. (2013). ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nature medicine 19, 1023-1029.
Cheng, H., Snoek, R., Ghaidi, F., Cox, M.E., and Rennie, P.S. (2006). Short hairpin RNA knockdown of the androgen receptor attenuates ligand-independent activation and delays tumor progression. Cancer research 66, 10613-10620.
Chi, K.N., Hotte, S.J., Yu, E.Y., Tu, D., Eigl, B.J., Tannock, I., Saad, F., North, S., Powers, J., Gleave, M.E., et al. (2010a). Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 28, 4247-4254.
Chi, K.N., Siu, L.L., Hirte, H., Hotte, S.J., Knox, J., Kollmansberger, C., Gleave, M., Guns, E., Powers, J., Walsh, W., et al. (2008). A phase I study of OGX-011, a 2'-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 14, 833-839.
Chi, P., Chen, Y., Zhang, L., Guo, X., Wongvipat, J., Shamu, T., Fletcher, J.A., Dewell, S., Maki, R.G., Zheng, D., et al. (2010b). ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467, 849-853.
Chiang, Y.T., Wang, K., Fazli, L., Qi, R.Z., Gleave, M.E., Collins, C.C., Gout, P.W., and Wang, Y. (2014). GATA2 as a potential metastasis-driving gene in prostate cancer. Oncotarget 5, 451-461.
Chien, A.J., and Moasser, M.M. (2008). Cellular mechanisms of resistance to anthracyclines and taxanes in cancer: intrinsic and acquired. Seminars in oncology 35, S1-S14; quiz S39.
Christofori, G., Naik, P., and Hanahan, D. (1994). A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 369, 414-418.
Chun, T.H., Itoh, H., Subramanian, L., Iniguez-Lluhi, J.A., and Nakao, K. (2003). Modification of GATA-2 transcriptional activity in endothelial cells by the SUMO E3 ligase PIASy. Circulation research 92, 1201-1208.

109
Church, D.N., Phillips, B.R., Stuckey, D.J., Barnes, D.J., Buffa, F.M., Manek, S., Clarke, K., Harris, A.L., Carter, E.J., and Hassan, A.B. (2012). Igf2 ligand dependency of Pten(+/-) developmental and tumour phenotypes in the mouse. Oncogene 31, 3635-3646.
Cisternino, S., Bourasset, F., Archimbaud, Y., Semiond, D., Sanderink, G., and Scherrmann, J.M. (2003). Nonlinear accumulation in the brain of the new taxoid TXD258 following saturation of P-glycoprotein at the blood-brain barrier in mice and rats. British journal of pharmacology 138, 1367-1375.
Cohn, A.L., Tabernero, J., Maurel, J., Nowara, E., Sastre, J., Chuah, B.Y., Kopp, M.V., Sakaeva, D.D., Mitchell, E.P., Dubey, S., et al. (2013). A randomized, placebo-controlled phase 2 study of ganitumab or conatumumab in combination with FOLFIRI for second-line treatment of mutant KRAS metastatic colorectal cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 24, 1777-1785.
Coma, S., Allard-Ratick, M., Akino, T., van Meeteren, L.A., Mammoto, A., and Klagsbrun, M. (2013). GATA2 and Lmo2 control angiogenesis and lymphangiogenesis via direct transcriptional regulation of neuropilin-2. Angiogenesis 16, 939-952.
Constancia, M., Hemberger, M., Hughes, J., Dean, W., Ferguson-Smith, A., Fundele, R., Stewart, F., Kelsey, G., Fowden, A., Sibley, C., et al. (2002). Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 417, 945-948.
Cooper, C.R., Chay, C.H., and Pienta, K.J. (2002). The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia 4, 191-194.
Cooper, W.N., Luharia, A., Evans, G.A., Raza, H., Haire, A.C., Grundy, R., Bowdin, S.C., Riccio, A., Sebastio, G., Bliek, J., et al. (2005). Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. European journal of human genetics : EJHG 13, 1025-1032.
Cortes, J., Hochhaus, A., Hughes, T., and Kantarjian, H. (2011). Front-line and salvage therapies with tyrosine kinase inhibitors and other treatments in chronic myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29, 524-531.
Cui, H. (2007). Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Disease markers 23, 105-112.
Da Costa, T.H., Williamson, D.H., Ward, A., Bates, P., Fisher, R., Richardson, L., Hill, D.J., Robinson, I.C., and Graham, C.F. (1994). High plasma insulin-like growth factor-II and low lipid content in transgenic mice: measurements of lipid metabolism. The Journal of endocrinology 143, 433-439.
Dart, A.E., and Wells, C.M. (2013). P21-activated kinase 4--not just one of the PAK. European journal of cell biology 92, 129-138.
Daughaday, W.H., Hall, K., Raben, M.S., Salmon, W.D., Jr., van den Brande, J.L., and van Wyk, J.J. (1972). Somatomedin: proposed designation for sulphation factor. Nature 235, 107.

110
de Bono, J.S., Logothetis, C.J., Molina, A., Fizazi, K., North, S., Chu, L., Chi, K.N., Jones, R.J., Goodman, O.B., Jr., Saad, F., et al. (2011). Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine 364, 1995-2005.
de Bono, J.S., Oudard, S., Ozguroglu, M., Hansen, S., Machiels, J.P., Kocak, I., Gravis, G., Bodrogi, I., Mackenzie, M.J., Shen, L., et al. (2010). Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376, 1147-1154.
de Bono, J.S., Piulats, J.M., Pandha, H.S., Petrylak, D.P., Saad, F., Aparicio, L.M., Sandhu, S.K., Fong, P., Gillessen, S., Hudes, G.R., et al. (2014). Phase II randomized study of figitumumab plus docetaxel and docetaxel alone with crossover for metastatic castration-resistant prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 1925-1934.
de Pater, E., Kaimakis, P., Vink, C.S., Yokomizo, T., Yamada-Inagawa, T., van der Linden, R., Kartalaei, P.S., Camper, S.A., Speck, N., and Dzierzak, E. (2013). Gata2 is required for HSC generation and survival. The Journal of experimental medicine 210, 2843-2850.
DeChiara, T.M., Efstratiadis, A., and Robertson, E.J. (1990). A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345, 78-80.
DeChiara, T.M., Robertson, E.J., and Efstratiadis, A. (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849-859.
Dehm, S.M., Schmidt, L.J., Heemers, H.V., Vessella, R.L., and Tindall, D.J. (2008). Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer research 68, 5469-5477.
Derry, W.B., Wilson, L., Khan, I.A., Luduena, R.F., and Jordan, M.A. (1997). Taxol differentially modulates the dynamics of microtubules assembled from unfractionated and purified beta-tubulin isotypes. Biochemistry 36, 3554-3562.
Diaz, L.E., Chuan, Y.C., Lewitt, M., Fernandez-Perez, L., Carrasco-Rodriguez, S., Sanchez-Gomez, M., and Flores-Morales, A. (2007). IGF-II regulates metastatic properties of choriocarcinoma cells through the activation of the insulin receptor. Molecular human reproduction 13, 567-576.
Dickins, R.A., Hemann, M.T., Zilfou, J.T., Simpson, D.R., Ibarra, I., Hannon, G.J., and Lowe, S.W. (2005). Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nature genetics 37, 1289-1295.
Dickinson, R.E., Griffin, H., Bigley, V., Reynard, L.N., Hussain, R., Haniffa, M., Lakey, J.H., Rahman, T., Wang, X.N., McGovern, N., et al. (2011). Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 118, 2656-2658.

111
Dickinson, R.E., Milne, P., Jardine, L., Zandi, S., Swierczek, S.I., McGovern, N., Cookson, S., Ferozepurwalla, Z., Langridge, A., Pagan, S., et al. (2014). The evolution of cellular deficiency in GATA2 mutation. Blood 123, 863-874.
Dinchuk, J.E., Cao, C., Huang, F., Reeves, K.A., Wang, J., Myers, F., Cantor, G.H., Zhou, X., Attar, R.M., Gottardis, M., et al. (2010). Insulin receptor (IR) pathway hyperactivity in IGF-IR null cells and suppression of downstream growth signaling using the dual IGF-IR/IR inhibitor, BMS-754807. Endocrinology 151, 4123-4132.
Domingo-Domenech, J., Oliva, C., Rovira, A., Codony-Servat, J., Bosch, M., Filella, X., Montagut, C., Tapia, M., Campas, C., Dang, L., et al. (2006). Interleukin 6, a nuclear factor-kappaB target, predicts resistance to docetaxel in hormone-independent prostate cancer and nuclear factor-kappaB inhibition by PS-1145 enhances docetaxel antitumor activity. Clinical cancer research : an official journal of the American Association for Cancer Research 12, 5578-5586.
Domingo-Domenech, J., Vidal, S.J., Rodriguez-Bravo, V., Castillo-Martin, M., Quinn, S.A., Rodriguez-Barrueco, R., Bonal, D.M., Charytonowicz, E., Gladoun, N., de la Iglesia-Vicente, J., et al. (2012). Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer cell 22, 373-388.
Draheim, K.M., Chen, H.B., Tao, Q., Moore, N., Roche, M., and Lyle, S. (2010). ARRDC3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene 29, 5032-5047.
Drake, J.M., Gabriel, C.L., and Henry, M.D. (2005). Assessing tumor growth and distribution in a model of prostate cancer metastasis using bioluminescence imaging. Clinical & experimental metastasis 22, 674-684.
Drummond, I.A., Madden, S.L., Rohwer-Nutter, P., Bell, G.I., Sukhatme, V.P., and Rauscher, F.J., 3rd (1992). Repression of the insulin-like growth factor II gene by the Wilms tumor suppressor WT1. Science 257, 674-678.
Dumontet, C., and Sikic, B.I. (1999). Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 17, 1061-1070.
Dydensborg, A.B., Rose, A.A., Wilson, B.J., Grote, D., Paquet, M., Giguere, V., Siegel, P.M., and Bouchard, M. (2009). GATA3 inhibits breast cancer growth and pulmonary breast cancer metastasis. Oncogene 28, 2634-2642.
Eggener, S.E., Scardino, P.T., Walsh, P.C., Han, M., Partin, A.W., Trock, B.J., Feng, Z., Wood, D.P., Eastham, J.A., Yossepowitch, O., et al. (2011). Predicting 15-year prostate cancer specific mortality after radical prostatectomy. The Journal of urology 185, 869-875.
Evans, T., and Felsenfeld, G. (1989). The erythroid-specific transcription factor Eryf1: a new finger protein. Cell 58, 877-885.

112
Fabbri, F., Carloni, S., Brigliadori, G., Zoli, W., Lapalombella, R., and Marini, M. (2006). Sequential events of apoptosis involving docetaxel, a microtubule-interfering agent: a cytometric study. BMC cell biology 7, 6.
Fadilah, S.A., Cheong, S.K., Roslan, H., Rozie-Hanisa, M., and Yen, G.K. (2002). GATA-1 and GATA-2 gene expression is related to the severity of dysplasia in myelodysplastic syndrome. Leukemia 16, 1563-1565.
Farber, S., and Diamond, L.K. (1948). Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. The New England journal of medicine 238, 787-793.
Fellmann, C., Zuber, J., McJunkin, K., Chang, K., Malone, C.D., Dickins, R.A., Xu, Q., Hengartner, M.O., Elledge, S.J., Hannon, G.J., et al. (2011). Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Molecular cell 41, 733-746.
Ferguson-Smith, A.C. (2011). Genomic imprinting: the emergence of an epigenetic paradigm. Nature reviews Genetics 12, 565-575.
Ferlay, J., Shin, H.R., Bray, F., Forman, D., Mathers, C., and Parkin, D.M. (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. International journal of cancer Journal international du cancer 127, 2893-2917.
Fizazi, K., Yang, J., Peleg, S., Sikes, C.R., Kreimann, E.L., Daliani, D., Olive, M., Raymond, K.A., Janus, T.J., Logothetis, C.J., et al. (2003). Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clinical cancer research : an official journal of the American Association for Cancer Research 9, 2587-2597.
Flanigan, S.A., Pitts, T.M., Eckhardt, S.G., Tentler, J.J., Tan, A.C., Thorburn, A., and Leong, S. (2010). The insulin-like growth factor I receptor/insulin receptor tyrosine kinase inhibitor PQIP exhibits enhanced antitumor effects in combination with chemotherapy against colorectal cancer models. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 5436-5446.
Flanigan, S.A., Pitts, T.M., Newton, T.P., Kulikowski, G.N., Tan, A.C., McManus, M.C., Spreafico, A., Kachaeva, M.I., Selby, H.M., Tentler, J.J., et al. (2013). Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clinical cancer research : an official journal of the American Association for Cancer Research 19, 6219-6229.
Forbes, K., Westwood, M., Baker, P.N., and Aplin, J.D. (2008). Insulin-like growth factor I and II regulate the life cycle of trophoblast in the developing human placenta. American journal of physiology Cell physiology 294, C1313-1322.
Fox, E.M., Miller, T.W., Balko, J.M., Kuba, M.G., Sanchez, V., Smith, R.A., Liu, S., Gonzalez-Angulo, A.M., Mills, G.B., Ye, F., et al. (2011). A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer research 71, 6773-6784.

113
Frasca, F., Pandini, G., Scalia, P., Sciacca, L., Mineo, R., Costantino, A., Goldfine, I.D., Belfiore, A., and Vigneri, R. (1999). Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Molecular and cellular biology 19, 3278-3288.
Freedland, S.J., Humphreys, E.B., Mangold, L.A., Eisenberger, M., Dorey, F.J., Walsh, P.C., and Partin, A.W. (2005). Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA : the journal of the American Medical Association 294, 433-439.
Freedland, S.J., and Moul, J.W. (2007). Prostate specific antigen recurrence after definitive therapy. The Journal of urology 177, 1985-1991.
Friedbichler, K., Hofmann, M.H., Kroez, M., Ostermann, E., Lamche, H.R., Koessl, C., Borges, E., Pollak, M.N., Adolf, G., and Adam, P.J. (2014). Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand-neutralizing antibody, and mechanistic rationale for combination with rapamycin. Molecular cancer therapeutics 13, 399-409.
Froesch, E.R., Buergi, H., Ramseier, E.B., Bally, P., and Labhart, A. (1963). Antibody-Suppressible and Nonsuppressible Insulin-Like Activities in Human Serum and Their Physiologic Significance. An Insulin Assay with Adipose Tissue of Increased Precision and Specificity. The Journal of clinical investigation 42, 1816-1834.
Fu, V.X., Dobosy, J.R., Desotelle, J.A., Almassi, N., Ewald, J.A., Srinivasan, R., Berres, M., Svaren, J., Weindruch, R., and Jarrard, D.F. (2008). Aging and cancer-related loss of insulin-like growth factor 2 imprinting in the mouse and human prostate. Cancer research 68, 6797-6802.
Fujiwara, Y., Browne, C.P., Cunniff, K., Goff, S.C., and Orkin, S.H. (1996). Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proceedings of the National Academy of Sciences of the United States of America 93, 12355-12358.
Gao, H., Ouyang, X., Banach-Petrosky, W.A., Gerald, W.L., Shen, M.M., and Abate-Shen, C. (2006). Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 103, 14477-14482.
Gao, J., Chesebrough, J.W., Cartlidge, S.A., Ricketts, S.A., Incognito, L., Veldman-Jones, M., Blakey, D.C., Tabrizi, M., Jallal, B., Trail, P.A., et al. (2011). Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer research 71, 1029-1040.
Gao, X., Johnson, K.D., Chang, Y.I., Boyer, M.E., Dewey, C.N., Zhang, J., and Bresnick, E.H. (2013). Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. The Journal of experimental medicine 210, 2833-2842.

114
Gao, X., Pang, J., Li, L.Y., Liu, W.P., Di, J.M., Sun, Q.P., Fang, Y.Q., Liu, X.P., Pu, X.Y., He, D., et al. (2010). Expression profiling identifies new function of collapsin response mediator protein 4 as a metastasis-suppressor in prostate cancer. Oncogene 29, 4555-4566.
Garraway, L.A., Widlund, H.R., Rubin, M.A., Getz, G., Berger, A.J., Ramaswamy, S., Beroukhim, R., Milner, D.A., Granter, S.R., Du, J., et al. (2005). Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117-122.
Gartrell, B.A., and Saad, F. (2014). Managing bone metastases and reducing skeletal related events in prostate cancer. Nature reviews Clinical oncology 11, 335-345.
Gaziel-Sovran, A., Segura, M.F., Di Micco, R., Collins, M.K., Hanniford, D., Vega-Saenz de Miera, E., Rakus, J.F., Dankert, J.F., Shang, S., Kerbel, R.S., et al. (2011). miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer cell 20, 104-118.
Gebeshuber, C.A., and Martinez, J. (2013). miR-100 suppresses IGF2 and inhibits breast tumorigenesis by interfering with proliferation and survival signaling. Oncogene 32, 3306-3310.
Giannakakou, P., Sackett, D.L., Kang, Y.K., Zhan, Z., Buters, J.T., Fojo, T., and Poruchynsky, M.S. (1997). Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. The Journal of biological chemistry 272, 17118-17125.
Giannoukakis, N., Deal, C., Paquette, J., Goodyer, C.G., and Polychronakos, C. (1993). Parental genomic imprinting of the human IGF2 gene. Nature genetics 4, 98-101.
Giddings, S.J., and Carnaghi, L.R. (1992). Insulin receptor gene expression during development: developmental regulation of insulin receptor mRNA abundance in embryonic rat liver and yolk sac, developmental regulation of insulin receptor gene splicing, and comparison to abundance of insulin-like growth factor 1 receptor mRNA. Molecular endocrinology 6, 1665-1672.
Goldman, J.M., and Melo, J.V. (2003). Chronic myeloid leukemia--advances in biology and new approaches to treatment. The New England journal of medicine 349, 1451-1464.
Gomella, L.G. (2009). Effective testosterone suppression for prostate cancer: is there a best castration therapy? Reviews in urology 11, 52-60.
Gonzalez-Garay, M.L., Chang, L., Blade, K., Menick, D.R., and Cabral, F. (1999). A beta-tubulin leucine cluster involved in microtubule assembly and paclitaxel resistance. The Journal of biological chemistry 274, 23875-23882.
Gottesman, M.M., Fojo, T., and Bates, S.E. (2002). Multidrug resistance in cancer: role of ATP-dependent transporters. Nature reviews Cancer 2, 48-58.
Grass, J.A., Boyer, M.E., Pal, S., Wu, J., Weiss, M.J., and Bresnick, E.H. (2003). GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and

115
domain-wide chromatin remodeling. Proceedings of the National Academy of Sciences of the United States of America 100, 8811-8816.
Grasso, C.S., Wu, Y.M., Robinson, D.R., Cao, X., Dhanasekaran, S.M., Khan, A.P., Quist, M.J., Jing, X., Lonigro, R.J., Brenner, J.C., et al. (2012). The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239-243.
Gravis, G., Fizazi, K., Joly, F., Oudard, S., Priou, F., Esterni, B., Latorzeff, I., Delva, R., Krakowski, I., Laguerre, B., et al. (2013). Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. The lancet oncology 14, 149-158.
Grote, D., Boualia, S.K., Souabni, A., Merkel, C., Chi, X., Costantini, F., Carroll, T., and Bouchard, M. (2008). Gata3 acts downstream of beta-catenin signaling to prevent ectopic metanephric kidney induction. PLoS genetics 4, e1000316.
Group, P.C.T.C. (2000). Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists' Collaborative Group. Lancet 355, 1491-1498.
Group, T.V.A.C.-o.U.R. (1967). Treatment and survival of patients with cancer of the prostate. The Veterans Administration Co-operative Urological Research Group. Surgery, gynecology & obstetrics 124, 1011-1017.
Guo, Z., Yang, X., Sun, F., Jiang, R., Linn, D.E., Chen, H., Chen, H., Kong, X., Melamed, J., Tepper, C.G., et al. (2009). A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research 69, 2305-2313.
Hahn, C.N., Chong, C.E., Carmichael, C.L., Wilkins, E.J., Brautigan, P.J., Li, X.C., Babic, M., Lin, M., Carmagnac, A., Lee, Y.K., et al. (2011). Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nature genetics 43, 1012-1017.
Hahn, H., Wojnowski, L., Specht, K., Kappler, R., Calzada-Wack, J., Potter, D., Zimmer, A., Muller, U., Samson, E., Quintanilla-Martinez, L., et al. (2000). Patched target Igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. The Journal of biological chemistry 275, 28341-28344.
Haldar, S., Chintapalli, J., and Croce, C.M. (1996). Taxol induces bcl-2 phosphorylation and death of prostate cancer cells. Cancer research 56, 1253-1255.
Haluska, P., Menefee, M., Plimack, E.R., Rosenberg, J., Northfelt, D., LaVallee, T., Shi, L., Yu, X.Q., Burke, P., Huang, J., et al. (2014). Phase I Dose-Escalation Study of MEDI-573, a Bispecific, Antiligand Monoclonal Antibody against IGFI and IGFII, in Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 4747-4757.
Haluska, P., Shaw, H.M., Batzel, G.N., Yin, D., Molina, J.R., Molife, L.R., Yap, T.A., Roberts, M.L., Sharma, A., Gualberto, A., et al. (2007). Phase I dose escalation study of the anti insulin-

116
like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 13, 5834-5840.
Hamilton, G.S., Lysiak, J.J., Han, V.K., and Lala, P.K. (1998). Autocrine-paracrine regulation of human trophoblast invasiveness by insulin-like growth factor (IGF)-II and IGF-binding protein (IGFBP)-1. Experimental cell research 244, 147-156.
Han, V.K., Bassett, N., Walton, J., and Challis, J.R. (1996). The expression of insulin-like growth factor (IGF) and IGF-binding protein (IGFBP) genes in the human placenta and membranes: evidence for IGF-IGFBP interactions at the feto-maternal interface. The Journal of clinical endocrinology and metabolism 81, 2680-2693.
Han, V.K., D'Ercole, A.J., and Lund, P.K. (1987). Cellular localization of somatomedin (insulin-like growth factor) messenger RNA in the human fetus. Science 236, 193-197.
Harris, L.K., and Westwood, M. (2012). Biology and significance of signalling pathways activated by IGF-II. Growth factors 30, 1-12.
Hayakawa, F., Towatari, M., Ozawa, Y., Tomita, A., Privalsky, M.L., and Saito, H. (2004). Functional regulation of GATA-2 by acetylation. Journal of leukocyte biology 75, 529-540.
Heilmann, A.M., Perera, R.M., Ecker, V., Nicolay, B.N., Bardeesy, N., Benes, C.H., and Dyson, N.J. (2014). CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer research 74, 3947-3958.
Hellerstedt, B.A., and Pienta, K.J. (2002). The current state of hormonal therapy for prostate cancer. CA: a cancer journal for clinicians 52, 154-179.
Holohan, C., Van Schaeybroeck, S., Longley, D.B., and Johnston, P.G. (2013). Cancer drug resistance: an evolving paradigm. Nature reviews Cancer 13, 714-726.
Holthuizen, P., Van Dijk, M.A., Rodenburg, R.J., Koonen-Reemst, A.M., and Sussenbach, J.S. (1993). Transcriptional regulation of the major promoters of the human IGF-II gene. Molecular reproduction and development 35, 391-393.
Hou, X., Huang, F., Macedo, L.F., Harrington, S.C., Reeves, K.A., Greer, A., Finckenstein, F.G., Brodie, A., Gottardis, M.M., Carboni, J.M., et al. (2011). Dual IGF-1R/InsR inhibitor BMS-754807 synergizes with hormonal agents in treatment of estrogen-dependent breast cancer. Cancer research 71, 7597-7607.
Hsu, A.P., Sampaio, E.P., Khan, J., Calvo, K.R., Lemieux, J.E., Patel, S.Y., Frucht, D.M., Vinh, D.C., Auth, R.D., Freeman, A.F., et al. (2011). Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 118, 2653-2655.
Hu, R., Dunn, T.A., Wei, S., Isharwal, S., Veltri, R.W., Humphreys, E., Han, M., Partin, A.W., Vessella, R.L., Isaacs, W.B., et al. (2009). Ligand-independent androgen receptor variants

117
derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer research 69, 16-22.
Huang, Z., Dore, L.C., Li, Z., Orkin, S.H., Feng, G., Lin, S., and Crispino, J.D. (2009). GATA-2 reinforces megakaryocyte development in the absence of GATA-1. Molecular and cellular biology 29, 5168-5180.
Huggins, C., Stevens, R.E., Jr, and Hodges, C.V. (1941). Studies on prostatic cancer: Ii. the effects of castration on advanced carcinoma of the prostate gland. Archives of Surgery 43, 209-223.
Ikonomi, P., Noguchi, C.T., Miller, W., Kassahun, H., Hardison, R., and Schechter, A.N. (2000). Levels of GATA-1/GATA-2 transcription factors modulate expression of embryonic and fetal hemoglobins. Gene 261, 277-287.
Irving, J.A., and Lala, P.K. (1995). Functional role of cell surface integrins on human trophoblast cell migration: regulation by TGF-beta, IGF-II, and IGFBP-1. Experimental cell research 217, 419-427.
Jarrard, D.F., Bussemakers, M.J., Bova, G.S., and Isaacs, W.B. (1995). Regional loss of imprinting of the insulin-like growth factor II gene occurs in human prostate tissues. Clinical cancer research : an official journal of the American Association for Cancer Research 1, 1471-1478.
Jemal, A., Bray, F., Center, M.M., Ferlay, J., Ward, E., and Forman, D. (2011). Global cancer statistics. CA: a cancer journal for clinicians 61, 69-90.
Jennbacken, K., Tesan, T., Wang, W., Gustavsson, H., Damber, J.E., and Welen, K. (2010). N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocrine-related cancer 17, 469-479.
Jiao, J., Wang, S., Qiao, R., Vivanco, I., Watson, P.A., Sawyers, C.L., and Wu, H. (2007). Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer research 67, 6083-6091.
Jin, J.K., Dayyani, F., and Gallick, G.E. (2011). Steps in prostate cancer progression that lead to bone metastasis. International journal of cancer Journal international du cancer 128, 2545-2561.
Johannessen, C.M., Johnson, L.A., Piccioni, F., Townes, A., Frederick, D.T., Donahue, M.K., Narayan, R., Flaherty, K.T., Wargo, J.A., Root, D.E., et al. (2013). A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504, 138-142.
Johansson, J.E., Andren, O., Andersson, S.O., Dickman, P.W., Holmberg, L., Magnuson, A., and Adami, H.O. (2004). Natural history of early, localized prostate cancer. JAMA : the journal of the American Medical Association 291, 2713-2719.
Johnson, K.D., Hsu, A.P., Ryu, M.J., Wang, J., Gao, X., Boyer, M.E., Liu, Y., Lee, Y., Calvo, K.R., Keles, S., et al. (2012). Cis-element mutated in GATA2-dependent immunodeficiency

118
governs hematopoiesis and vascular integrity. The Journal of clinical investigation 122, 3692-3704.
Jones, R.L., Kim, E.S., Nava-Parada, P., Alam, S., Johnson, F.M., Stephens, A.W., Simantov, R., Poondru, S., Gedrich, R., Lippman, S.M., et al. (2014). Phase I Study of Intermittent Oral Dosing of the Insulin-like Growth Factor-1 and Insulin Receptors Inhibitor OSI-906 in Patients With Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research.
Kalin, T.V., Wang, I.C., Ackerson, T.J., Major, M.L., Detrisac, C.J., Kalinichenko, V.V., Lyubimov, A., and Costa, R.H. (2006). Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer research 66, 1712-1720.
Kantoff, P.W., Halabi, S., Conaway, M., Picus, J., Kirshner, J., Hars, V., Trump, D., Winer, E.P., and Vogelzang, N.J. (1999). Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 17, 2506-2513.
Kantoff, P.W., Higano, C.S., Shore, N.D., Berger, E.R., Small, E.J., Penson, D.F., Redfern, C.H., Ferrari, A.C., Dreicer, R., Sims, R.B., et al. (2010). Sipuleucel-T immunotherapy for castration-resistant prostate cancer. The New England journal of medicine 363, 411-422.
Karl, P.I. (1995). Insulin-like growth factor-1 stimulates amino acid uptake by the cultured human placental trophoblast. Journal of cellular physiology 165, 83-88.
Kastner, P., and Chan, S. (2008). PU.1: a crucial and versatile player in hematopoiesis and leukemia. The international journal of biochemistry & cell biology 40, 22-27.
Kazenwadel, J., Secker, G.A., Liu, Y.J., Rosenfeld, J.A., Wildin, R.S., Cuellar-Rodriguez, J., Hsu, A.P., Dyack, S., Fernandez, C.V., Chong, C.E., et al. (2012). Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood 119, 1283-1291.
Kim, K.W., Bae, S.K., Lee, O.H., Bae, M.H., Lee, M.J., and Park, B.C. (1998). Insulin-like growth factor II induced by hypoxia may contribute to angiogenesis of human hepatocellular carcinoma. Cancer research 58, 348-351.
Klotz, L., and Emberton, M. (2014). Management of low risk prostate cancer-active surveillance and focal therapy. Nature reviews Clinical oncology 11, 324-334.
Kniss, D.A., Shubert, P.J., Zimmerman, P.D., Landon, M.B., and Gabbe, S.G. (1994). Insulinlike growth factors. Their regulation of glucose and amino acid transport in placental trophoblasts isolated from first-trimester chorionic villi. The Journal of reproductive medicine 39, 249-256.
Kobayashi-Osaki, M., Ohneda, O., Suzuki, N., Minegishi, N., Yokomizo, T., Takahashi, S., Lim, K.C., Engel, J.D., and Yamamoto, M. (2005). GATA motifs regulate early hematopoietic lineage-specific expression of the Gata2 gene. Molecular and cellular biology 25, 7005-7020.

119
Koga, S., Yamaguchi, N., Abe, T., Minegishi, M., Tsuchiya, S., Yamamoto, M., and Minegishi, N. (2007). Cell-cycle-dependent oscillation of GATA2 expression in hematopoietic cells. Blood 109, 4200-4208.
Koo, C.Y., Muir, K.W., and Lam, E.W. (2012). FOXM1: From cancer initiation to progression and treatment. Biochimica et biophysica acta 1819, 28-37.
Kooijman, R., van Buul-Offers, S.C., Scholtens, L.E., Schuurman, H.J., Van den Brande, L.J., and Zegers, B.J. (1995). T cell development in insulin-like growth factor-II transgenic mice. Journal of immunology 154, 5736-5745.
Kouros-Mehr, H., Bechis, S.K., Slorach, E.M., Littlepage, L.E., Egeblad, M., Ewald, A.J., Pai, S.Y., Ho, I.C., and Werb, Z. (2008). GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer cell 13, 141-152.
Kouros-Mehr, H., Slorach, E.M., Sternlicht, M.D., and Werb, Z. (2006). GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127, 1041-1055.
Kuhn, D.J., Berkova, Z., Jones, R.J., Woessner, R., Bjorklund, C.C., Ma, W., Davis, R.E., Lin, P., Wang, H., Madden, T.L., et al. (2012). Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 120, 3260-3270.
Kumar, M.S., Hancock, D.C., Molina-Arcas, M., Steckel, M., East, P., Diefenbacher, M., Armenteros-Monterroso, E., Lassailly, F., Matthews, N., Nye, E., et al. (2012). The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 149, 642-655.
Kyle, A.H., Huxham, L.A., Yeoman, D.M., and Minchinton, A.I. (2007). Limited tissue penetration of taxanes: a mechanism for resistance in solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 13, 2804-2810.
Langer, C.J., Novello, S., Park, K., Krzakowski, M., Karp, D.D., Mok, T., Benner, R.J., Scranton, J.R., Olszanski, A.J., and Jassem, J. (2014). Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 32, 2059-2066.
Lau, M.M., Stewart, C.E., Liu, Z., Bhatt, H., Rotwein, P., and Stewart, C.L. (1994). Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes & development 8, 2953-2963.
Lee, Y.I., Lee, S., Lee, Y., Bong, Y.S., Hyun, S.W., Yoo, Y.D., Kim, S.J., Kim, Y.W., and Poo, H.R. (1998). The human hepatitis B virus transactivator X gene product regulates Sp1 mediated transcription of an insulin-like growth factor II promoter 4. Oncogene 16, 2367-2380.
Lehr, J.E., and Pienta, K.J. (1998). Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. Journal of the National Cancer Institute 90, 118-123.

120
Li, B., Tsao, S.W., Chan, K.W., Ludwig, D.L., Novosyadlyy, R., Li, Y.Y., He, Q.Y., and Cheung, A.L. (2014). Id1-induced IGF-II and its autocrine/endocrine promotion of esophageal cancer progression and chemoresistance--implications for IGF-II and IGF-IR-targeted therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 20, 2651-2662.
Li, P., Cavallero, S., Gu, Y., Chen, T.H., Hughes, J., Hassan, A.B., Bruning, J.C., Pashmforoush, M., and Sucov, H.M. (2011). IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 138, 1795-1805.
Liao, S., Howell, D.K., and Chang, T.M. (1974). Action of a nonsteroidal antiandrogen, flutamide, on the receptor binding and nuclear retention of 5 alpha-dihydrotestosterone in rat ventral prostate. Endocrinology 94, 1205-1209.
Lim, K.C., Hosoya, T., Brandt, W., Ku, C.J., Hosoya-Ohmura, S., Camper, S.A., Yamamoto, M., and Engel, J.D. (2012). Conditional Gata2 inactivation results in HSC loss and lymphatic mispatterning. The Journal of clinical investigation 122, 3705-3717.
Ling, K.W., Ottersbach, K., van Hamburg, J.P., Oziemlak, A., Tsai, F.Y., Orkin, S.H., Ploemacher, R., Hendriks, R.W., and Dzierzak, E. (2004). GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. The Journal of experimental medicine 200, 871-882.
Litzenburger, B.C., Creighton, C.J., Tsimelzon, A., Chan, B.T., Hilsenbeck, S.G., Wang, T., Carboni, J.M., Gottardis, M.M., Huang, F., Chang, J.C., et al. (2011). High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 2314-2327.
Liu, R., Wang, X., Chen, G.Y., Dalerba, P., Gurney, A., Hoey, T., Sherlock, G., Lewicki, J., Shedden, K., and Clarke, M.F. (2007). The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England journal of medicine 356, 217-226.
Liu, Y., Gong, Z., Sun, L., and Li, X. (2014). FOXM1 and androgen receptor co-regulate CDC6 gene transcription and DNA replication in prostate cancer cells. Biochimica et biophysica acta 1839, 297-305.
Livingstone, C. (2013). IGF2 and cancer. Endocrine-related cancer 20, R321-339.
Locke, J.A., Guns, E.S., Lubik, A.A., Adomat, H.H., Hendy, S.C., Wood, C.A., Ettinger, S.L., Gleave, M.E., and Nelson, C.C. (2008). Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer research 68, 6407-6415.
Lubik, A.A., Gunter, J.H., Hollier, B.G., Ettinger, S., Fazli, L., Stylianou, N., Hendy, S.C., Adomat, H.H., Gleave, M.E., Pollak, M., et al. (2013). IGF2 increases de novo steroidogenesis in prostate cancer cells. Endocrine-related cancer 20, 173-186.

121
Luesink, M., Hollink, I.H., van der Velden, V.H., Knops, R.H., Boezeman, J.B., de Haas, V., Trka, J., Baruchel, A., Reinhardt, D., van der Reijden, B.A., et al. (2012). High GATA2 expression is a poor prognostic marker in pediatric acute myeloid leukemia. Blood 120, 2064-2075.
Lui, J.C., and Baron, J. (2013). Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proceedings of the National Academy of Sciences of the United States of America 110, 6181-6186.
Manzo, F., Tambaro, F.P., Mai, A., and Altucci, L. (2009). Histone acetyltransferase inhibitors and preclinical studies. Expert opinion on therapeutic patents 19, 761-774.
May, G., Soneji, S., Tipping, A.J., Teles, J., McGowan, S.J., Wu, M., Guo, Y., Fugazza, C., Brown, J., Karlsson, G., et al. (2013). Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell stem cell 13, 754-768.
McDonnell, T.J., Troncoso, P., Brisbay, S.M., Logothetis, C., Chung, L.W., Hsieh, J.T., Tu, S.M., and Campbell, M.L. (1992). Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer research 52, 6940-6944.
McGrath, K.E., Koniski, A.D., Malik, J., and Palis, J. (2003). Circulation is established in a stepwise pattern in the mammalian embryo. Blood 101, 1669-1676.
Mehra, R., Varambally, S., Ding, L., Shen, R., Sabel, M.S., Ghosh, D., Chinnaiyan, A.M., and Kleer, C.G. (2005). Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer research 65, 11259-11264.
Menghini, R., Marchetti, V., Cardellini, M., Hribal, M.L., Mauriello, A., Lauro, D., Sbraccia, P., Lauro, R., and Federici, M. (2005). Phosphorylation of GATA2 by Akt increases adipose tissue differentiation and reduces adipose tissue-related inflammation: a novel pathway linking obesity to atherosclerosis. Circulation 111, 1946-1953.
Merika, M., and Orkin, S.H. (1993). DNA-binding specificity of GATA family transcription factors. Molecular and cellular biology 13, 3999-4010.
Migliaccio, A.R., and Bieker, J.J. (2011). GATA2 finds its macrophage niche. Blood 118, 2647-2649.
Mimeault, M., Johansson, S.L., Vankatraman, G., Moore, E., Henichart, J.P., Depreux, P., Lin, M.F., and Batra, S.K. (2007). Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Molecular cancer therapeutics 6, 967-978.
Minegishi, N., Suzuki, N., Kawatani, Y., Shimizu, R., and Yamamoto, M. (2005). Rapid turnover of GATA-2 via ubiquitin-proteasome protein degradation pathway. Genes to cells : devoted to molecular & cellular mechanisms 10, 693-704.

122
Molkentin, J.D. (2000). The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. The Journal of biological chemistry 275, 38949-38952.
Moller, N., and Jorgensen, J.O. (2009). Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocrine reviews 30, 152-177.
Montgomery, R.B., Mostaghel, E.A., Vessella, R., Hess, D.L., Kalhorn, T.F., Higano, C.S., True, L.D., and Nelson, P.S. (2008). Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research 68, 4447-4454.
Moorehead, R.A., Sanchez, O.H., Baldwin, R.M., and Khokha, R. (2003). Transgenic overexpression of IGF-II induces spontaneous lung tumors: a model for human lung adenocarcinoma. Oncogene 22, 853-857.
Moulton, T., Crenshaw, T., Hao, Y., Moosikasuwan, J., Lin, N., Dembitzer, F., Hensle, T., Weiss, L., McMorrow, L., Loew, T., et al. (1994). Epigenetic lesions at the H19 locus in Wilms' tumour patients. Nature genetics 7, 440-447.
Mulholland, D.J., Tran, L.M., Li, Y., Cai, H., Morim, A., Wang, S., Plaisier, S., Garraway, I.P., Huang, J., Graeber, T.G., et al. (2011). Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer cell 19, 792-804.
Mulvihill, M.J., Cooke, A., Rosenfeld-Franklin, M., Buck, E., Foreman, K., Landfair, D., O'Connor, M., Pirritt, C., Sun, Y., Yao, Y., et al. (2009). Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future medicinal chemistry 1, 1153-1171.
Murphy, L.J., Bell, G.I., and Friesen, H.G. (1987). Tissue distribution of insulin-like growth factor I and II messenger ribonucleic acid in the adult rat. Endocrinology 120, 1279-1282.
Murray, B.W., Guo, C., Piraino, J., Westwick, J.K., Zhang, C., Lamerdin, J., Dagostino, E., Knighton, D., Loi, C.M., Zager, M., et al. (2010). Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proceedings of the National Academy of Sciences of the United States of America 107, 9446-9451.
Murray, S., Briasoulis, E., Linardou, H., Bafaloukos, D., and Papadimitriou, C. (2012). Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer treatment reviews 38, 890-903.
Nordin, M., Bergman, D., Halje, M., Engstrom, W., and Ward, A. (2014). Epigenetic regulation of the Igf2/H19 gene cluster. Cell proliferation 47, 189-199.
O'Dell, S.D., and Day, I.N. (1998). Insulin-like growth factor II (IGF-II). The international journal of biochemistry & cell biology 30, 767-771.

123
Ogawa, O., Eccles, M.R., Szeto, J., McNoe, L.A., Yun, K., Maw, M.A., Smith, P.J., and Reeve, A.E. (1993). Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms' tumour. Nature 362, 749-751.
Ogawa, T., Ogawa, K., Shiga, K., Furukawa, T., Nagase, H., Hashimoto, S., Kobayashi, T., and Horii, A. (2010). Upregulation of IGF2 is associated with an acquired resistance for cis-diamminedichloroplatinum in human head and neck squamous cell carcinoma. European archives of oto-rhino-laryngology : official journal of the European Federation of Oto-Rhino-Laryngological Societies 267, 1599-1606.
Ohlsson, R., Nystrom, A., Pfeifer-Ohlsson, S., Tohonen, V., Hedborg, F., Schofield, P., Flam, F., and Ekstrom, T.J. (1993). IGF2 is parentally imprinted during human embryogenesis and in the Beckwith-Wiedemann syndrome. Nature genetics 4, 94-97.
Orlic, D., Anderson, S., Biesecker, L.G., Sorrentino, B.P., and Bodine, D.M. (1995). Pluripotent hematopoietic stem cells contain high levels of mRNA for c-kit, GATA-2, p45 NF-E2, and c-myb and low levels or no mRNA for c-fms and the receptors for granulocyte colony-stimulating factor and interleukins 5 and 7. Proceedings of the National Academy of Sciences of the United States of America 92, 4601-4605.
Ostergaard, P., Simpson, M.A., Connell, F.C., Steward, C.G., Brice, G., Woollard, W.J., Dafou, D., Kilo, T., Smithson, S., Lunt, P., et al. (2011). Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nature genetics 43, 929-931.
Ovcharenko, I., Loots, G.G., Giardine, B.M., Hou, M., Ma, J., Hardison, R.C., Stubbs, L., and Miller, W. (2005). Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome research 15, 184-194.
Ozawa, Y., Towatari, M., Tsuzuki, S., Hayakawa, F., Maeda, T., Miyata, Y., Tanimoto, M., and Saito, H. (2001). Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood 98, 2116-2123.
Panda, D., Miller, H.P., Banerjee, A., Luduena, R.F., and Wilson, L. (1994). Microtubule dynamics in vitro are regulated by the tubulin isotype composition. Proceedings of the National Academy of Sciences of the United States of America 91, 11358-11362.
Pandini, G., Frasca, F., Mineo, R., Sciacca, L., Vigneri, R., and Belfiore, A. (2002). Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. The Journal of biological chemistry 277, 39684-39695.
Pandolfi, P.P., Roth, M.E., Karis, A., Leonard, M.W., Dzierzak, E., Grosveld, F.G., Engel, J.D., and Lindenbaum, M.H. (1995). Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nature genetics 11, 40-44.

124
Park, M.H., Lee, H.S., Lee, C.S., You, S.T., Kim, D.J., Park, B.H., Kang, M.J., Heo, W.D., Shin, E.Y., Schwartz, M.A., et al. (2013). p21-Activated kinase 4 promotes prostate cancer progression through CREB. Oncogene 32, 2475-2482.
Parker, C., Nilsson, S., Heinrich, D., Helle, S.I., O'Sullivan, J.M., Fossa, S.D., Chodacki, A., Wiechno, P., Logue, J., Seke, M., et al. (2013). Alpha emitter radium-223 and survival in metastatic prostate cancer. The New England journal of medicine 369, 213-223.
Pchejetski, D., Doumerc, N., Golzio, M., Naymark, M., Teissie, J., Kohama, T., Waxman, J., Malavaud, B., and Cuvillier, O. (2008). Chemosensitizing effects of sphingosine kinase-1 inhibition in prostate cancer cell and animal models. Molecular cancer therapeutics 7, 1836-1845.
Perez-Stable, C.M., Pozas, A., and Roos, B.A. (2000). A role for GATA transcription factors in the androgen regulation of the prostate-specific antigen gene enhancer. Molecular and cellular endocrinology 167, 43-53.
Peters, J. (2014). The role of genomic imprinting in biology and disease: an expanding view. Nature reviews Genetics 15, 517-530.
Petrylak, D.P., Tangen, C.M., Hussain, M.H., Lara, P.N., Jr., Jones, J.A., Taplin, M.E., Burch, P.A., Berry, D., Moinpour, C., Kohli, M., et al. (2004). Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. The New England journal of medicine 351, 1513-1520.
Pilepich, M.V., Winter, K., Lawton, C.A., Krisch, R.E., Wolkov, H.B., Movsas, B., Hug, E.B., Asbell, S.O., and Grignon, D. (2005). Androgen suppression adjuvant to definitive radiotherapy in prostate carcinoma--long-term results of phase III RTOG 85-31. International journal of radiation oncology, biology, physics 61, 1285-1290.
Pivot, X., Koralewski, P., Hidalgo, J.L., Chan, A., Goncalves, A., Schwartsmann, G., Assadourian, S., and Lotz, J.P. (2008). A multicenter phase II study of XRP6258 administered as a 1-h i.v. infusion every 3 weeks in taxane-resistant metastatic breast cancer patients. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 19, 1547-1552.
Ploussard, G., Terry, S., Maille, P., Allory, Y., Sirab, N., Kheuang, L., Soyeux, P., Nicolaiew, N., Coppolani, E., Paule, B., et al. (2010). Class III beta-tubulin expression predicts prostate tumor aggressiveness and patient response to docetaxel-based chemotherapy. Cancer research 70, 9253-9264.
Pollak, M. (2008). Insulin and insulin-like growth factor signalling in neoplasia. Nature reviews Cancer 8, 915-928.
Pollak, M. (2012). The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nature reviews Cancer 12, 159-169.

125
Pommier, Y., Sordet, O., Antony, S., Hayward, R.L., and Kohn, K.W. (2004). Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 23, 2934-2949.
Pound, C.R., Partin, A.W., Eisenberger, M.A., Chan, D.W., Pearson, J.D., and Walsh, P.C. (1999). Natural history of progression after PSA elevation following radical prostatectomy. JAMA : the journal of the American Medical Association 281, 1591-1597.
Pravtcheva, D.D., and Wise, T.L. (1998). Metastasizing mammary carcinomas in H19 enhancers-Igf2 transgenic mice. The Journal of experimental zoology 281, 43-57.
Puzanov, I., Lindsay, C.R., Goff, L.W., Sosman, J.A., Gilbert, J., Berlin, J., Poondru, S., Simantov, R., Gedrich, R., Stephens, A., et al. (2014). A Phase I Study of Continuous Oral Dosing of OSI-906, a Dual Inhibitor of Insulin-Like Growth Factor-1 and Insulin Receptors in Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research.
Qian, D.Z., Rademacher, B.L., Pittsenbarger, J., Huang, C.Y., Myrthue, A., Higano, C.S., Garzotto, M., Nelson, P.S., and Beer, T.M. (2010). CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity. The Prostate 70, 433-442.
Radu, M., Semenova, G., Kosoff, R., and Chernoff, J. (2014). PAK signalling during the development and progression of cancer. Nature reviews Cancer 14, 13-25.
Rainier, S., Johnson, L.A., Dobry, C.J., Ping, A.J., Grundy, P.E., and Feinberg, A.P. (1993). Relaxation of imprinted genes in human cancer. Nature 362, 747-749.
Ramalingam, S.S., Spigel, D.R., Chen, D., Steins, M.B., Engelman, J.A., Schneider, C.P., Novello, S., Eberhardt, W.E., Crino, L., Habben, K., et al. (2011). Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29, 4574-4580.
Ravenel, J.D., Broman, K.W., Perlman, E.J., Niemitz, E.L., Jayawardena, T.M., Bell, D.W., Haber, D.A., Uejima, H., and Feinberg, A.P. (2001). Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. Journal of the National Cancer Institute 93, 1698-1703.
Rinderknecht, E., and Humbel, R.E. (1976a). Amino-terminal sequences of two polypeptides from human serum with nonsuppressible insulin-like and cell-growth-promoting activities: evidence for structural homology with insulin B chain. Proceedings of the National Academy of Sciences of the United States of America 73, 4379-4381.
Rinderknecht, E., and Humbel, R.E. (1976b). Polypeptides with nonsuppressible insulin-like and cell-growth promoting activities in human serum: isolation, chemical characterization, and some biological properties of forms I and II. Proceedings of the National Academy of Sciences of the United States of America 73, 2365-2369.

126
Ritchie, C.K., Andrews, L.R., Thomas, K.G., Tindall, D.J., and Fitzpatrick, L.A. (1997). The effects of growth factors associated with osteoblasts on prostate carcinoma proliferation and chemotaxis: implications for the development of metastatic disease. Endocrinology 138, 1145-1150.
Robert-Moreno, A., Espinosa, L., de la Pompa, J.L., and Bigas, A. (2005). RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development 132, 1117-1126.
Robertson, J.F., Ferrero, J.M., Bourgeois, H., Kennecke, H., de Boer, R.H., Jacot, W., McGreivy, J., Suzuki, S., Zhu, M., McCaffery, I., et al. (2013). Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, phase 2 trial. The lancet oncology 14, 228-235.
Rodrigues, N.P., Janzen, V., Forkert, R., Dombkowski, D.M., Boyd, A.S., Orkin, S.H., Enver, T., Vyas, P., and Scadden, D.T. (2005). Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood 106, 477-484.
Rogler, C.E., Yang, D., Rossetti, L., Donohoe, J., Alt, E., Chang, C.J., Rosenfeld, R., Neely, K., and Hintz, R. (1994). Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. The Journal of biological chemistry 269, 13779-13784.
Rosa-Ribeiro, R., Nishan, U., Vidal, R.O., Barbosa, G.O., Reis, L.O., Cesar, C.L., and Carvalho, H.F. (2014). Transcription factors involved in prostate gland adaptation to androgen deprivation. PloS one 9, e97080.
Ross, H.M., Kryvenko, O.N., Cowan, J.E., Simko, J.P., Wheeler, T.M., and Epstein, J.I. (2012). Do adenocarcinomas of the prostate with Gleason score (GS) </=6 have the potential to metastasize to lymph nodes? The American journal of surgical pathology 36, 1346-1352.
Rossetti, L., Barzilai, N., Chen, W., Harris, T., Yang, D., and Rogler, C.E. (1996). Hepatic overexpression of insulin-like growth factor-II in adulthood increases basal and insulin-stimulated glucose disposal in conscious mice. The Journal of biological chemistry 271, 203-208.
Roth, B.J., Yeap, B.Y., Wilding, G., Kasimis, B., McLeod, D., and Loehrer, P.J. (1993). Taxol in advanced, hormone-refractory carcinoma of the prostate. A phase II trial of the Eastern Cooperative Oncology Group. Cancer 72, 2457-2460.
Ryan, C.J., Smith, M.R., de Bono, J.S., Molina, A., Logothetis, C.J., de Souza, P., Fizazi, K., Mainwaring, P., Piulats, J.M., Ng, S., et al. (2013). Abiraterone in metastatic prostate cancer without previous chemotherapy. The New England journal of medicine 368, 138-148.
Saad, F., Hotte, S., North, S., Eigl, B., Chi, K., Czaykowski, P., Wood, L., Pollak, M., Berry, S., Lattouf, J.B., et al. (2011). Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-

127
resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 5765-5773.
Sallman, D.A., Chen, X., Zhong, B., Gilvary, D.L., Zhou, J., Wei, S., and Djeu, J.Y. (2007). Clusterin mediates TRAIL resistance in prostate tumor cells. Molecular cancer therapeutics 6, 2938-2947.
Salmon, W.D., Jr., and Daughaday, W.H. (1957). A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. The Journal of laboratory and clinical medicine 49, 825-836.
Schally, A.V., Comaru-Schally, A.M., Plonowski, A., Nagy, A., Halmos, G., and Rekasi, Z. (2000). Peptide analogs in the therapy of prostate cancer. The Prostate 45, 158-166.
Scher, H.I., Fizazi, K., Saad, F., Taplin, M.E., Sternberg, C.N., Miller, K., de Wit, R., Mulders, P., Chi, K.N., Shore, N.D., et al. (2012). Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England journal of medicine 367, 1187-1197.
Schutz, F.A., Buzaid, A.C., and Sartor, O. (2014). Taxanes in the management of metastatic castration-resistant prostate cancer: Efficacy and management of toxicity. Critical reviews in oncology/hematology.
Sciacca, L., Mineo, R., Pandini, G., Murabito, A., Vigneri, R., and Belfiore, A. (2002). In IGF-I receptor-deficient leiomyosarcoma cells autocrine IGF-II induces cell invasion and protection from apoptosis via the insulin receptor isoform A. Oncogene 21, 8240-8250.
Seidenfeld, J., Samson, D.J., Hasselblad, V., Aronson, N., Albertsen, P.C., Bennett, C.L., and Wilt, T.J. (2000). Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. Annals of internal medicine 132, 566-577.
Seruga, B., Ocana, A., and Tannock, I.F. (2011). Drug resistance in metastatic castration-resistant prostate cancer. Nature reviews Clinical oncology 8, 12-23.
Sferruzzi-Perri, A.N., Owens, J.A., Pringle, K.G., Robinson, J.S., and Roberts, C.T. (2006). Maternal insulin-like growth factors-I and -II act via different pathways to promote fetal growth. Endocrinology 147, 3344-3355.
Shan, S.W., Fang, L., Shatseva, T., Rutnam, Z.J., Yang, X., Du, W., Lu, W.Y., Xuan, J.W., Deng, Z., and Yang, B.B. (2013). Mature miR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7 and vimentin in different signal pathways. Journal of cell science 126, 1517-1530.
Sharma, N.L., Massie, C.E., Ramos-Montoya, A., Zecchini, V., Scott, H.E., Lamb, A.D., MacArthur, S., Stark, R., Warren, A.Y., Mills, I.G., et al. (2013). The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer cell 23, 35-47.

128
Shiau, A.K., Barstad, D., Loria, P.M., Cheng, L., Kushner, P.J., Agard, D.A., and Greene, G.L. (1998). The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927-937.
Shin, S.I., Freedman, V.H., Risser, R., and Pollack, R. (1975). Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proceedings of the National Academy of Sciences of the United States of America 72, 4435-4439.
Shivdasani, R.A., Fujiwara, Y., McDevitt, M.A., and Orkin, S.H. (1997). A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. The EMBO journal 16, 3965-3973.
Siegel, R., Ma, J., Zou, Z., and Jemal, A. (2014). Cancer statistics, 2014. CA: a cancer journal for clinicians 64, 9-29.
Snoek, R., Cheng, H., Margiotti, K., Wafa, L.A., Wong, C.A., Wong, E.C., Fazli, L., Nelson, C.C., Gleave, M.E., and Rennie, P.S. (2009). In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 15, 39-47.
Soares, M.B., Ishii, D.N., and Efstratiadis, A. (1985). Developmental and tissue-specific expression of a family of transcripts related to rat insulin-like growth factor II mRNA. Nucleic acids research 13, 1119-1134.
Spinner, M.A., Sanchez, L.A., Hsu, A.P., Shaw, P.A., Zerbe, C.S., Calvo, K.R., Arthur, D.C., Gu, W., Gould, C.M., Brewer, C.C., et al. (2014). GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood 123, 809-821.
Springate, C.M., Jackson, J.K., Gleave, M.E., and Burt, H.M. (2005). Efficacy of an intratumoral controlled release formulation of clusterin antisense oligonucleotide complexed with chitosan containing paclitaxel or docetaxel in prostate cancer xenograft models. Cancer chemotherapy and pharmacology 56, 239-247.
Sramkoski, R.M., Pretlow, T.G., 2nd, Giaconia, J.M., Pretlow, T.P., Schwartz, S., Sy, M.S., Marengo, S.R., Rhim, J.S., Zhang, D., and Jacobberger, J.W. (1999). A new human prostate carcinoma cell line, 22Rv1. In vitro cellular & developmental biology Animal 35, 403-409.
Stanbrough, M., Bubley, G.J., Ross, K., Golub, T.R., Rubin, M.A., Penning, T.M., Febbo, P.G., and Balk, S.P. (2006). Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer research 66, 2815-2825.
Steenman, M.J., Rainier, S., Dobry, C.J., Grundy, P., Horon, I.L., and Feinberg, A.P. (1994). Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumour. Nature genetics 7, 433-439.

129
Steingrimsson, E., Copeland, N.G., and Jenkins, N.A. (2004). Melanocytes and the microphthalmia transcription factor network. Annual review of genetics 38, 365-411.
Stylianopoulou, F., Efstratiadis, A., Herbert, J., and Pintar, J. (1988). Pattern of the insulin-like growth factor II gene expression during rat embryogenesis. Development 103, 497-506.
Sudo, T., Nitta, M., Saya, H., and Ueno, N.T. (2004). Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer research 64, 2502-2508.
Sullivan, K.F., and Cleveland, D.W. (1986). Identification of conserved isotype-defining variable region sequences for four vertebrate beta tubulin polypeptide classes. Proceedings of the National Academy of Sciences of the United States of America 83, 4327-4331.
Sullivan, M.J., Taniguchi, T., Jhee, A., Kerr, N., and Reeve, A.E. (1999). Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation. Oncogene 18, 7527-7534.
Sun, S., Sprenger, C.C., Vessella, R.L., Haugk, K., Soriano, K., Mostaghel, E.A., Page, S.T., Coleman, I.M., Nguyen, H.M., Sun, H., et al. (2010). Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of clinical investigation 120, 2715-2730.
Sun, Y., Wang, B.E., Leong, K.G., Yue, P., Li, L., Jhunjhunwala, S., Chen, D., Seo, K., Modrusan, Z., Gao, W.Q., et al. (2012). Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer research 72, 527-536.
Sussenbach, J.S., Steenbergh, P.H., and Holthuizen, P. (1992). Structure and expression of the human insulin-like growth factor genes. Growth regulation 2, 1-9.
Suzuki, N., Ohneda, O., Minegishi, N., Nishikawa, M., Ohta, T., Takahashi, S., Engel, J.D., and Yamamoto, M. (2006). Combinatorial Gata2 and Sca1 expression defines hematopoietic stem cells in the bone marrow niche. Proceedings of the National Academy of Sciences of the United States of America 103, 2202-2207.
Sweeney, C.C., Y.H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Eisenberger, M.A.; Wong, Y.N.; Hahn, N.M.; Kohli, M.; Vogelzang, N.J.; Cooney, M.M.; Dreicer, R.; Picus, J.; Shevrin, D.H.; Hussain, M.; Garcia, J.A.; DiPaola R.S. (2014). Impact on overall survival (OS) with chemohormonal therapy versus hormonal therapy for hormone-sensitive newly metastatic prostate cancer (mPrCa): An ECOG-led phase III randomized trial. (2014 ASCO Annual Meeting).
Tada, Y., Yamaguchi, Y., Kinjo, T., Song, X., Akagi, T., Takamura, H., Ohta, T., Yokota, T., and Koide, H. (2014). The stem cell transcription factor ZFP57 induces IGF2 expression to promote anchorage-independent growth in cancer cells. Oncogene.

130
Taichman, R.S., Cooper, C., Keller, E.T., Pienta, K.J., Taichman, N.S., and McCauley, L.K. (2002). Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer research 62, 1832-1837.
Tamaki, A., Ierano, C., Szakacs, G., Robey, R.W., and Bates, S.E. (2011). The controversial role of ABC transporters in clinical oncology. Essays in biochemistry 50, 209-232.
Tanaka, H., Kono, E., Tran, C.P., Miyazaki, H., Yamashiro, J., Shimomura, T., Fazli, L., Wada, R., Huang, J., Vessella, R.L., et al. (2010). Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nature medicine 16, 1414-1420.
Taniguchi, T., Sullivan, M.J., Ogawa, O., and Reeve, A.E. (1995). Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proceedings of the National Academy of Sciences of the United States of America 92, 2159-2163.
Tannock, I.F., de Wit, R., Berry, W.R., Horti, J., Pluzanska, A., Chi, K.N., Oudard, S., Theodore, C., James, N.D., Turesson, I., et al. (2004). Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine 351, 1502-1512.
Tannock, I.F., Osoba, D., Stockler, M.R., Ernst, D.S., Neville, A.J., Moore, M.J., Armitage, G.R., Wilson, J.J., Venner, P.M., Coppin, C.M., et al. (1996). Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 14, 1756-1764.
Taylor, B.S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B.S., Arora, V.K., Kaushik, P., Cerami, E., Reva, B., et al. (2010). Integrative genomic profiling of human prostate cancer. Cancer cell 18, 11-22.
Terui, K., Takahashi, Y., Kitazawa, J., Toki, T., Yokoyama, M., and Ito, E. (2000). Expression of transcription factors during megakaryocytic differentiation of CD34+ cells from human cord blood induced by thrombopoietin. The Tohoku journal of experimental medicine 192, 259-273.
Tessema, M., Yingling, C.M., Snider, A.M., Do, K., Juri, D.E., Picchi, M.A., Zhang, X., Liu, Y., Leng, S., Tellez, C.S., et al. (2014). GATA2 is epigenetically repressed in human and mouse lung tumors and is not requisite for survival of KRAS mutant lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 9, 784-793.
Thompson, I., Thrasher, J.B., Aus, G., Burnett, A.L., Canby-Hagino, E.D., Cookson, M.S., D'Amico, A.V., Dmochowski, R.R., Eton, D.T., Forman, J.D., et al. (2007). Guideline for the management of clinically localized prostate cancer: 2007 update. The Journal of urology 177, 2106-2131.
Tognon, C.E., Somasiri, A.M., Evdokimova, V.E., Trigo, G., Uy, E.E., Melnyk, N., Carboni, J.M., Gottardis, M.M., Roskelley, C.D., Pollak, M., et al. (2011). ETV6-NTRK3-mediated breast

131
epithelial cell transformation is blocked by targeting the IGF1R signaling pathway. Cancer research 71, 1060-1070.
Tolcher, A.W., Sarantopoulos, J., Patnaik, A., Papadopoulos, K., Lin, C.C., Rodon, J., Murphy, B., Roth, B., McCaffery, I., Gorski, K.S., et al. (2009). Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 27, 5800-5807.
Trainor, C.D., Ghirlando, R., and Simpson, M.A. (2000). GATA zinc finger interactions modulate DNA binding and transactivation. The Journal of biological chemistry 275, 28157-28166.
Tran, C., Ouk, S., Clegg, N.J., Chen, Y., Watson, P.A., Arora, V., Wongvipat, J., Smith-Jones, P.M., Yoo, D., Kwon, A., et al. (2009). Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787-790.
Trapnell, C., Pachter, L., and Salzberg, S.L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105-1111.
Trapnell, C., Williams, B.A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M.J., Salzberg, S.L., Wold, B.J., and Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511-515.
Tsai, F.Y., Keller, G., Kuo, F.C., Weiss, M., Chen, J., Rosenblatt, M., Alt, F.W., and Orkin, S.H. (1994). An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature 371, 221-226.
Tsai, F.Y., and Orkin, S.H. (1997). Transcription factor GATA-2 is required for proliferation/survival of early hematopoietic cells and mast cell formation, but not for erythroid and myeloid terminal differentiation. Blood 89, 3636-3643.
Tsai, S.F., Martin, D.I., Zon, L.I., D'Andrea, A.D., Wong, G.G., and Orkin, S.H. (1989). Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 339, 446-451.
Tusher, V.G., Tibshirani, R., and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America 98, 5116-5121.
Udan, R.S., Culver, J.C., and Dickinson, M.E. (2013). Understanding vascular development. Wiley interdisciplinary reviews Developmental biology 2, 327-346.
Ueno, H., and Weissman, I.L. (2010). The origin and fate of yolk sac hematopoiesis: application of chimera analyses to developmental studies. The International journal of developmental biology 54, 1019-1031.

132
Underwood, L.E., Hintz, R.L., Voina, S.J., and Van Wyk, J.J. (1972). Human somatomedin, the growth hormone dependent sulfation factor, is antilipolytic. The Journal of clinical endocrinology and metabolism 35, 194-198.
van Bokhoven, A., Varella-Garcia, M., Korch, C., Johannes, W.U., Smith, E.E., Miller, H.L., Nordeen, S.K., Miller, G.J., and Lucia, M.S. (2003). Molecular characterization of human prostate carcinoma cell lines. The Prostate 57, 205-225.
van Buul-Offers, S.C., de Haan, K., Reijnen-Gresnigt, M.G., Meinsma, D., Jansen, M., Oei, S.L., Bonte, E.J., Sussenbach, J.S., and Van den Brande, J.L. (1995). Overexpression of human insulin-like growth factor-II in transgenic mice causes increased growth of the thymus. The Journal of endocrinology 144, 491-502.
van Dijk, M.A., Rodenburg, R.J., Holthuizen, P., and Sussenbach, J.S. (1992). The liver-specific promoter of the human insulin-like growth factor II gene is activated by CCAAT/enhancer binding protein (C/EBP). Nucleic acids research 20, 3099-3104.
Vasan, N., Boyer, J.L., and Herbst, R.S. (2014). A RAS Renaissance: Emerging Targeted Therapies for KRAS-Mutated Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research.
Verrills, N.M., Po'uha, S.T., Liu, M.L., Liaw, T.Y., Larsen, M.R., Ivery, M.T., Marshall, G.M., Gunning, P.W., and Kavallaris, M. (2006). Alterations in gamma-actin and tubulin-targeted drug resistance in childhood leukemia. Journal of the National Cancer Institute 98, 1363-1374.
Vert, J.P., Foveau, N., Lajaunie, C., and Vandenbrouck, Y. (2006). An accurate and interpretable model for siRNA efficacy prediction. BMC bioinformatics 7, 520.
Vicente, C., Conchillo, A., Garcia-Sanchez, M.A., and Odero, M.D. (2012a). The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Critical reviews in oncology/hematology 82, 1-17.
Vicente, C., Vazquez, I., Conchillo, A., Garcia-Sanchez, M.A., Marcotegui, N., Fuster, O., Gonzalez, M., Calasanz, M.J., Lahortiga, I., and Odero, M.D. (2012b). Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia 26, 550-554.
Visakorpi, T., Hyytinen, E., Koivisto, P., Tanner, M., Keinanen, R., Palmberg, C., Palotie, A., Tammela, T., Isola, J., and Kallioniemi, O.P. (1995). In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature genetics 9, 401-406.
Wang, Q., Li, W., Liu, X.S., Carroll, J.S., Janne, O.A., Keeton, E.K., Chinnaiyan, A.M., Pienta, K.J., and Brown, M. (2007). A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Molecular cell 27, 380-392.
Wang, Q., Li, W., Zhang, Y., Yuan, X., Xu, K., Yu, J., Chen, Z., Beroukhim, R., Wang, H., Lupien, M., et al. (2009). Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138, 245-256.

133
Wang, S., Wang, Z., Boise, L., Dent, P., and Grant, S. (1999). Loss of the bcl-2 phosphorylation loop domain increases resistance of human leukemia cells (U937) to paclitaxel-mediated mitochondrial dysfunction and apoptosis. Biochemical and biophysical research communications 259, 67-72.
Wang, S.S., Esplin, E.D., Li, J.L., Huang, L., Gazdar, A., Minna, J., and Evans, G.A. (1998). Alterations of the PPP2R1B gene in human lung and colon cancer. Science 282, 284-287.
Wang, Z.Q., Fung, M.R., Barlow, D.P., and Wagner, E.F. (1994). Regulation of embryonic growth and lysosomal targeting by the imprinted Igf2/Mpr gene. Nature 372, 464-467.
Ward, A., Bates, P., Fisher, R., Richardson, L., and Graham, C.F. (1994). Disproportionate growth in mice with Igf-2 transgenes. Proceedings of the National Academy of Sciences of the United States of America 91, 10365-10369.
Wells, C.M., Whale, A.D., Parsons, M., Masters, J.R., and Jones, G.E. (2010). PAK4: a pluripotent kinase that regulates prostate cancer cell adhesion. Journal of cell science 123, 1663-1673.
Weroha, S.J., and Haluska, P. (2012). The insulin-like growth factor system in cancer. Endocrinology and metabolism clinics of North America 41, 335-350, vi.
Whale, A.D., Dart, A., Holt, M., Jones, G.E., and Wells, C.M. (2013). PAK4 kinase activity and somatic mutation promote carcinoma cell motility and influence inhibitor sensitivity. Oncogene 32, 2114-2120.
Wittman, M.D., Carboni, J.M., Yang, Z., Lee, F.Y., Antman, M., Attar, R., Balimane, P., Chang, C., Chen, C., Discenza, L., et al. (2009). Discovery of a 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. Journal of medicinal chemistry 52, 7360-7363.
Wolf, E., Hoeflich, A., and Lahm, H. (1998). What is the function of IGF-II in postnatal life? Answers from transgenic mouse models. Growth hormone & IGF research : official journal of the Growth Hormone Research Society and the International IGF Research Society 8, 185-193.
Wolf, E., Kramer, R., Blum, W.F., Foll, J., and Brem, G. (1994). Consequences of postnatally elevated insulin-like growth factor-II in transgenic mice: endocrine changes and effects on body and organ growth. Endocrinology 135, 1877-1886.
Wong, Y.N., Ferraldeschi, R., Attard, G., and de Bono, J. (2014). Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nature reviews Clinical oncology 11, 365-376.
Wu, D., Sunkel, B., Chen, Z., Liu, X., Ye, Z., Li, Q., Grenade, C., Ke, J., Zhang, C., Chen, H., et al. (2014). Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic acids research 42, 3607-3622.

134
Wu, L., Birle, D.C., and Tannock, I.F. (2005). Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer research 65, 2825-2831.
Yagoda, A., and Petrylak, D. (1993). Cytotoxic chemotherapy for advanced hormone-resistant prostate cancer. Cancer 71, 1098-1109.
Yao, N., Yao, D., Wang, L., Dong, Z., Wu, W., Qiu, L., Yan, X., Yu, D., Chen, J., Sai, W., et al. (2012). Inhibition of autocrine IGF-II on effect of human HepG2 cell proliferation and angiogenesis factor expression. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine 33, 1767-1776.
Yee, D. (2012). Insulin-like growth factor receptor inhibitors: baby or the bathwater? Journal of the National Cancer Institute 104, 975-981.
Yeh, J.E., Toniolo, P.A., and Frank, D.A. (2013). Targeting transcription factors: promising new strategies for cancer therapy. Current opinion in oncology 25, 652-658.
Yerbury, J.J., Poon, S., Meehan, S., Thompson, B., Kumita, J.R., Dobson, C.M., and Wilson, M.R. (2007). The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 21, 2312-2322.
Yuasa, H., Oike, Y., Iwama, A., Nishikata, I., Sugiyama, D., Perkins, A., Mucenski, M.L., Suda, T., and Morishita, K. (2005). Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. The EMBO journal 24, 1976-1987.
Zapf, J., Walter, H., and Froesch, E.R. (1981). Radioimmunological determination of insulinlike growth factors I and II in normal subjects and in patients with growth disorders and extrapancreatic tumor hypoglycemia. The Journal of clinical investigation 68, 1321-1330.
Zegarra-Moro, O.L., Schmidt, L.J., Huang, H., and Tindall, D.J. (2002). Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer research 62, 1008-1013.
Zeng, X., Zhang, H., Oh, A., Zhang, Y., and Yee, D. (2012). Enhancement of doxorubicin cytotoxicity of human cancer cells by tyrosine kinase inhibition of insulin receptor and type I IGF receptor. Breast cancer research and treatment 133, 117-126.
Zhang, L., Zhou, W., Velculescu, V.E., Kern, S.E., Hruban, R.H., Hamilton, S.R., Vogelstein, B., and Kinzler, K.W. (1997). Gene expression profiles in normal and cancer cells. Science 276, 1268-1272.
Zhang, P., Behre, G., Pan, J., Iwama, A., Wara-Aswapati, N., Radomska, H.S., Auron, P.E., Tenen, D.G., and Sun, Z. (1999). Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proceedings of the National Academy of Sciences of the United States of America 96, 8705-8710.

135
Zhang, S.J., Ma, L.Y., Huang, Q.H., Li, G., Gu, B.W., Gao, X.D., Shi, J.Y., Wang, Y.Y., Gao, L., Cai, X., et al. (2008). Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America 105, 2076-2081.
Zhao, H., Desai, V., Wang, J., Epstein, D.M., Miglarese, M., and Buck, E. (2012). Epithelial-mesenchymal transition predicts sensitivity to the dual IGF-1R/IR inhibitor OSI-906 in hepatocellular carcinoma cell lines. Molecular cancer therapeutics 11, 503-513.
Zhau, H.Y., Chang, S.M., Chen, B.Q., Wang, Y., Zhang, H., Kao, C., Sang, Q.A., Pathak, S.J., and Chung, L.W. (1996). Androgen-repressed phenotype in human prostate cancer. Proceedings of the National Academy of Sciences of the United States of America 93, 15152-15157.
Zhong, H., Fazenbaker, C., Breen, S., Chen, C., Huang, J., Morehouse, C., Yao, Y., and Hollingsworth, R.E. (2014). MEDI-573 Alone or in Combination with Mammalian Target of Rapamycin Inhibitors, Targets the Insulin-like Growth Factor Pathway in Sarcomas. Molecular cancer therapeutics.
Zhou, Y., Lim, K.C., Onodera, K., Takahashi, S., Ohta, J., Minegishi, N., Tsai, F.Y., Orkin, S.H., Yamamoto, M., and Engel, J.D. (1998). Rescue of the embryonic lethal hematopoietic defect reveals a critical role for GATA-2 in urogenital development. The EMBO journal 17, 6689-6700.
Zhu, Y., Liu, C., Nadiminty, N., Lou, W., Tummala, R., Evans, C.P., and Gao, A.C. (2013). Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Molecular cancer therapeutics 12, 1829-1836.
Zingg, A.E., and Froesch, E.R. (1973). Effects of partially purified preparations with nonsuppressible insulin-like activity (NSILA-S) on sulfate incorporation into rat and chicken cartilage. Diabetologia 9, 472-476.
Zinn, R.L., Gardner, E.E., Marchionni, L., Murphy, S.C., Dobromilskaya, I., Hann, C.L., and Rudin, C.M. (2013). ERK phosphorylation is predictive of resistance to IGF-1R inhibition in small cell lung cancer. Molecular cancer therapeutics 12, 1131-1139.
Zong, Y., and Goldstein, A.S. (2013). Adaptation or selection--mechanisms of castration-resistant prostate cancer. Nature reviews Urology 10, 90-98.
Zoubeidi, A., Chi, K., and Gleave, M. (2010). Targeting the cytoprotective chaperone, clusterin, for treatment of advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 1088-1093.


















