
A new tyrosinase model system: formation of a phenoxy- and hydroxy-bridged copper(II) complex with...
5
ELSEVIER lnorganicaChimicaActa263 (1997) 133-137 A new tyrosinase model system: formation of a phenoxy- and hydroxy-bridged copper(H) complex with partial hydrolysis of a tetraaza macrocyclic Schiff base ligand Rajeev Gupta, Rabindranath Mukherjee * Departmentof Chemistry,IndianInstituteofTeclmology, Kanpur 208016, India Received15 January 1997; revised 12March 1997; accepted 19 May 1997 Abstract A new tetraaza macrocyclic ligand capable of providing two nitrogen coordination to each copper center has been utilized to model tyrosinase-like activity. Aromatic hydroxylation in one of the rings has been observed with concomitant partial hydrolysis of the macrocycle. The final dicopper(ll) complex has a/z-phenoxo/z-hydroxo bridged structure as evidenced from spectroscopic and variable-temperature magnetic st)~dies. © 1997 Elsevier Science S.A. Keywordv: Phenoxo-bridged complexes; Coppercomplexes; Hydroxylation; Tyrosinase model 1, Introduction Considerable progress has been made in the chemical mod- eling of tyrosinase-like monooxygenase activity (aromatic ring hydroxylation) [ 1-4]. Investigations of the chemistry of copper monooxygcnase biomimetic systems is of contin- ued interest [5,6]. Karlin et al. reported the first model [4] consisting of an N-donor binucleating ligand having an m- xylyl spacer between the two tridentate bis[2-(2-pyridyl- ethyl)amine] coordination units. Recently wehavedescribed a new m-xylyl-based tyrosinase model system [7] with a binucleating ligand that provides only two nitrogen coordi- nation sites: an aliphatic nitrogen and a pyridyl nitrogen at each copper center. Martell and co-workers [8] demonstrated the first suc- cessful use of a macrocyclic ligand to bring about aromatic ring hydroxylation. In fact, using a hexaaza 24-membered macrocyclic tetra Schiff base ligand (2+2 condensation between isophthalaldehyde and diethylenetriamine) they observed that one of the two aromatic rings becomes hydrox- ylated upon exposure of their dicopper(l) complex to diox- ygen. Prompted by our known success and that of Martell and co-workers we wished to examine possible steric con- sequences and dioxygen reactivity when designing a macro- cyclic ligand capable of providing only two nitrogen coordination to each copper center. This paper describes the * Correspondingaulhor. 0020-1693/97/$17.00 © 1997 Elsevier ScienceS.A. All rightsreserved PIi S0020- ! 693 ( 97 ) 05655- 7 synthesis of a new teuaaza 18-membered macrocyclic tetra Schiff base ligand (L) and our findings on the reactions of its dicopper(I) complex with dioxygen. 2. Experimental 2.1. General All reagents are commercially available and were used without further purification. Acetonitrile was dried by distil- lation over calcium hydride. Chloroform and dicloromethane were putrefiedby washing with 5% sodium carbonate solution followed by water and dried over anhydrous calcium chlo- fide, before a final reflux and distillation. Diethyl ether was dried first with anhydrous calcium chloride and then refluxed with, and stored over, sodium. Methanol was distilled from magnesium methoxide. The complex [Cu(MeCN)4] [CIO4] was prepared as before [7]. The synthesis of the dicopper(I) complex was performed under an argon atmosphere using standard Schlenk-line techniques. IR spectra were recorded using a Perkin-Elmer model 1320 spectropho~ometer. UV- Vis spectra were recorded using a Perkin-Elmer Lambda 2 spectrophotometer. ~H NMR spectra were recorded on a JEOL-PMX 60 (60 MHz) or a WP-80 BrOker (80 MHz) spectrometer, and chemical shifts (8) are repotted in ppm downfield from TMS. The El-MS and FAB-MS (positive ion) specwa were recorded in MeCN solution with JEOL
-
Upload
rajeev-gupta -
Category
Documents
-
view
212 -
download
0
Transcript of A new tyrosinase model system: formation of a phenoxy- and hydroxy-bridged copper(II) complex with...

E L S E V I E R lnorganica Chimica Acta 263 (1997) 133-137
A new tyrosinase model system: formation of a phenoxy- and hydroxy-bridged copper(H) complex with partial hydrolysis of
a tetraaza macrocyclic Schiff base ligand
Rajeev Gupta, Rabindranath Mukherjee *
Department of Chemistry, Indian Institute ofTeclmology, Kanpur 208 016, India
Received 15 January 1997; revised 12 March 1997; accepted 19 May 1997
Abstract
A new tetraaza macrocyclic ligand capable of providing two nitrogen coordination to each copper center has been utilized to model tyrosinase-like activity. Aromatic hydroxylation in one of the rings has been observed with concomitant partial hydrolysis of the macrocycle. The final dicopper(ll) complex has a/z-phenoxo/z-hydroxo bridged structure as evidenced from spectroscopic and variable-temperature magnetic st)~dies. © 1997 Elsevier Science S.A.
Keywordv: Phenoxo-bridged complexes; Copper complexes; Hydroxylation; Tyrosinase model
1, Introduction
Considerable progress has been made in the chemical mod- eling of tyrosinase-like monooxygenase activity (aromatic ring hydroxylation) [ 1-4]. Investigations of the chemistry of copper monooxygcnase biomimetic systems is of contin- ued interest [5,6]. Karlin et al. reported the first model [4] consisting of an N-donor binucleating ligand having an m- xylyl spacer between the two tridentate bis[2-(2-pyridyl- ethyl)amine] coordination units. Recently wehavedescribed a new m-xylyl-based tyrosinase model system [7] with a binucleating ligand that provides only two nitrogen coordi- nation sites: an aliphatic nitrogen and a pyridyl nitrogen at each copper center.
Martell and co-workers [8] demonstrated the first suc- cessful use of a macrocyclic ligand to bring about aromatic ring hydroxylation. In fact, using a hexaaza 24-membered macrocyclic tetra Schiff base ligand ( 2 + 2 condensation between isophthalaldehyde and diethylenetriamine) they observed that one of the two aromatic rings becomes hydrox- ylated upon exposure of their dicopper(l) complex to diox- ygen. Prompted by our known success and that of Martell and co-workers we wished to examine possible steric con- sequences and dioxygen reactivity when designing a macro- cyclic ligand capable of providing only two nitrogen coordination to each copper center. This paper describes the
* Corresponding aulhor.
0020-1693/97/$17.00 © 1997 Elsevier Science S.A. All rights reserved PIi S0020- ! 693 ( 97 ) 05655- 7
synthesis of a new teuaaza 18-membered macrocyclic tetra Schiff base ligand (L) and our findings on the reactions of its dicopper(I) complex with dioxygen.
2. Experimental
2.1. General
All reagents are commercially available and were used without further purification. Acetonitrile was dried by distil- lation over calcium hydride. Chloroform and dicloromethane were putrefied by washing with 5% sodium carbonate solution followed by water and dried over anhydrous calcium chlo- fide, before a final reflux and distillation. Diethyl ether was dried first with anhydrous calcium chloride and then refluxed with, and stored over, sodium. Methanol was distilled from magnesium methoxide. The complex [Cu(MeCN)4] [CIO4] was prepared as before [7]. The synthesis of the dicopper(I) complex was performed under an argon atmosphere using standard Schlenk-line techniques. IR spectra were recorded using a Perkin-Elmer model 1320 spectropho~ometer. UV- Vis spectra were recorded using a Perkin-Elmer Lambda 2 spectrophotometer. ~H NMR spectra were recorded on a JEOL-PMX 60 (60 MHz) or a WP-80 BrOker (80 MHz) spectrometer, and chemical shifts (8) are repotted in ppm downfield from TMS. The El-MS and FAB-MS (positive ion) specwa were recorded in MeCN solution with JEOL

13a R. ¢Tupta, R. Mukherjee / lnorganica Chiraica Acta 263 (1997) 133-137
!)-300 and JEOL SX-102 spectrometers, respectively, at the Central Drug Research Institute (CDRI), Lucknow. The details of the magnetic measurements have already been described in the literature [9].
2.2. Syntheses
2.2.1. Macrocyclic ligand (L) The synthetic procedure followed for this new macrocyclic
ligand is adapted from a literature report [ 10]. 1,2-Diami- noethane (0.447 g, 7.46 retool) was dissolved in 280 ml of MeCN under magnetic stirring and to it isophthalaldehyde ( 1.0 g, 7.46 retool) dissolved in 160 ml of MeCN was added dropwise over a period of 4 h, at room temperature. The reaction mixture was allowed to stir for 2 days. The white powder thus formed was filtered and washed thoroughly with MeCN and dried in vacuo. Yield 1.04 g (88%). Anal. Calc. for C2oH2oN4: C, 75.92; H, 6.37; N, 17.71. Found: C, 75.43; H, 6.27; N, 18.13%. ~H NMR (CDCI£ 80 MHz): 8 3.97 (s, 4I-I, --CH2CHz-), 4.00 (s, 4H, --CH2CH2-), 7.19-8.44 (m, 12H, Ar-H and --C-H). MS spectrum: peak at 316 corre- sponding to C2oH2oN4 (intact maerocycle L).
L
2.2.2. The dicopper(1) complex (1) To a solution of L (0.1 g, 0.32 mmol) in dichloromethane
(15 ml) was added [Cu(MeCN)4] [004] (0.21 g, 0.64 mmol) dissolved in diehloromethane (15 ml), under mag- netic stirring. Immediately an intense yellow suspension formed. After stirring for 10 rain, solvent was removed to afford a bright yellow solid. Yield 0.21 g (89%). Anal. Calc. for C24H26NrOsCI2Cu2: C, 39.78; H, 3.62; N, 11.60. Found: C, 37.14, H, 3.73; N, 1 !.16%. (Unfortunately, all attempts to obtain better microanalytical data tile.n this have been unsuccessful so far due to its extreme moisture and air sen- sitivity. Anal. Calc. for CzsH2sNrOsCl,Cu2 (with one dich- ioromethane molecule): C, 37.06; H, 3.46; N, 10.38. Anal. Calc. for C~H3oNrO,oCl2Cu2 (with two molecules of water): C, 37.86; H, 3.94; N, I 1.04%. With the available experimen- tal evidence at hand (see below) we cannot rule out one possibility over the other.) FAB-MS spectrum: an intense peak at 461 (corresponding to ICu2(L)(HzO)}2+) and a peak at 661 (corresponding to {Cuz(L)(H20)(CIO4)2} +H +) are clearly observable. (Substitution of the water molecule in the latter species by an aeetonitrile molecule is
also seen in its spectrum.) This solid was used for dioxygen reactivity studies without further purification.
2.2.3. The dicopper(ll) complex (2) To the afore-mentioned dicopper(1) compound (0.21 g,
0.29 mmol) was added MeCN-MeOH ( 1:3 vol./vol., 60 mi) resulting in a yellow solution with some suspension. After stirring for 10 ,. "a, the reaction mixture was exposed to the atmosphere of dioxygen and allowed to stir for 24 h, at room temperature. Formation of a green solution with some yellow suspension was observed. The reaction mixture was then filtered to remove a yellow polymeric residue (0.032 g) (insoluble in common organic solvents) and a greenish-blue filtrate was obtained. On evaporation of the solvent a green- ish-blue crude solid product (0.155 g) resulted. This solid was then dissolved in MeCN (5 ml) and filtered. Diethyl ether ( 10 ml) diffusion to the filtrate at room temperature resulted in the formation of dark blue needle-shaped crystals in a day. The crystals were collected by filtration and washed with MeCN and dried in vacuo (on concentration the filtrate afforded some white solid, which was found to be isophthal- aldehyde by IH NMR). Yield 0.065 g (33%). Anal. Calc. for ClzH22N4Oi2Cu2C12: C, 23.55; H, 3.59; N, 9.15. Found: C, 23.93; H, 3.25; N, 8.97%. Absorption spectrum (in MeCN), ~ (nm) (e (din 3 reel -I cm- t ) ) : 591 (163), 352 (8957), 253 (38 152), 207 (27 472). FAB-MS spectrum: an intense peak at 612 (complex 2, see below).
3. Results and discussion
3.1. Synthesis and characterization of the macrocyclic ligand
The macrocyclic ligand L was prepared by the Schiffbase (2 + 2) condensation of 1,2-diaminocthane and isophthalal- dehyde in MeCN at room temperature. The IR spectrum of the ligand indicates that cyclization has occurred by the dis- appearance of bands associated with carbonyl and amine groups and the appearance of a band at 1640 cm- ' associated with imine stretching. A clean ~H NMR spectrum in CDCI3 adds credence to the macrocyclic structure of this product. The observed two singlets which we assign due to CH2CH2 sections of the ring deserve special mention. We believe that it is due to inherent ring strain present in this macrocycle, which renders it a non-planar structure and that is the reason for the observation of two singlcts. The final proof of the identity of the macrocyclie nature of L was obtained from its microanalytical data and mass spectrum. It is to be noted here that in its mass spectrum no peak corresponding to incomplete condensation products (possible non-macrocyclic products) could be identified. The non-template procedure followed here is a straightforward, efficient and high yield synthetic methodology.

R. Gupta. R. Mukherjee I lnorganica Chimica Acta 263 (1997) 133-137 135
3.2. Synthesis and characterization of the dicopper(1) complex
The dicopper(I) complex of this macrocyclic iigand was prepared under argon from the reaction between the ligand L and [Cu(MeCN)4] [C104] ( 1:2 ligand to metal mole ratio) in dichloromethane. The IR spectrum of this bright yellow solid in nujol mull shows the v(C=N) stretching band at I620 cm- ~. This compound exhibits a broad and strong per- chlorate absorption at 1085 cm- ' and also a medium intensity band at 620 cm- ', indicative of the presence of ionic per- chlorate. Additionally, the IR spectrum of this complex does show a weak, but clearly observable, set of niuile stretching vibrations of coordinated CH3CN at 2230-2260 cm- ~.
This yellow solid is sparingly soluble in CH3CH and is insoluble in most common organic solvents. Its ~H NMR spectrum (80 MHz) in deoxygenated CD3CN (dried over molecular sieves) closely resembles that of the free macro- cycle L. The peaks are comparatively broad and less struc- tured, though. In the low-field region, only two peaks are now observable: one at 8.34 ppm and the other at 7.61 ppm, cor- responding to a total of twelve protons, as expected. The bridging ethylene protons now appear as a broad singlet at 3.91 ppm. Moreover, a peak at 2.16 ppm, due to coordinated CH3CN, is clearly observable. Even though the *H NMR spectrum of this dicopper(1) complex in degassed deuterated N,N-dimethylformamide is not as clean as that in CDsCN, in this solvent more convincing evidence of the presence of coordinated CH3CN was obtained (2.19 ppm; integration corresponding to two CH3CN molecules). The broad nature of the peaks exhibited by this compound most probably is due to fast solvent exchange. The absence of any possible copper(If) impurity due to aerobic oxidation was confirmed by its yellow to orange color and lack of any absorption peak in the visible region. The authenticity of this complex was obtained from its microanalytical and FAB-MS spectrum. The chemical composition of this yellow solid is, beyond doubt, very close to that proposed below. Given the above specU'al results and that reported for a closely related struc- turaUy characterized dicopper(1) complex using a furan- based macrocyclic ligand [ 11 ], we tentatively assign the structure of this yellow solid as 1.
1 S :MeCN
3.3. Reaction of the dicopper(1) complex with dioxygen
On exposure to dry dioxygen, a suspension of dicopper complex 1 in CH3OH-MeCN (3:1, vol./vol.) develops a green coloration over a period of 24 h. After removal of a polymeric yellow solid, a bluish-green solution was obtained. Evaporation of solvent r-~ulted in a biuish-green solid. This solid was dissolved in 6 M HC1 and extraction with CHO3 produced a yellowish white solid. The tH NMR spectrmn of this solid in CDCI3 exhibits the spectral feature almost iden- tical to that reported by Martell and co-workers [llb]. The peak integration and mass spectral feature unambigtmmly reveal that this solid was a mixture of isophthalak~yde (rJ/z= 134) and 2,6-difonnyiphenoi (re~z= 150). The above results demonstrate that due to oxygenation of complex I oxygen insertion into the aromatic ring has occurred with only one of the arene rings hydroxylated. Based on tH NMR spectral integration it has been found that the yellow solid contains 63% isophthalaldehyde and 37% 2,6-difonnyl phe- nol and the efficiency of this hydroxylation reaction is 74%.
The bluish-green solid obtained from oxygenation of com- pound 1 was dissolved in MuCH and diffusion of diethyl ether afforded a blue crystalline solid.
3.4. Synthesis and characterization of the blue solid
For the blue solid as KBr pellet the IR absorptions due to v(OH) are observed at 3500 an6 3410 cm -~. Additionally, several weak absorptions appear at 3320, 3220 and 3130 cm-t , due to symmetric and asymmetric N-H stretching vibrations [ 12]. This compound exhibits a strong band at 1645 cm- t, due to the ~(C=N) stretch. Absorptions are also observed due to ionic perchlorate. The positions and pattern are closely similar to those of 1. A characteristic feature of this compound is the appearance of a strong bead at 1560 cm- t, which does not exist in compound 1, but is typical for the t,(C~O) vibration oftbe phenol in the 2-hydrox'yplgn- ylimino chromophore [ 12]. This cmmbomtcs the tH NMR result described in the previous section. In this compound no absorptions due to aldehyde groups were observed.
This blue solid behaves as a 1:2 eleclrolyte in Me(2¢ solu- tion (AM=2(30 ~ -I CEll2 mol - I ) [13]. M i ~ I R spectral, solution electrical conductivity and FAB-MS results clearly correspond to the structure 2, implying a partial hydrolysis of the macrocyclic ligand L during oxygenation of complex L
2
lc~4122t~
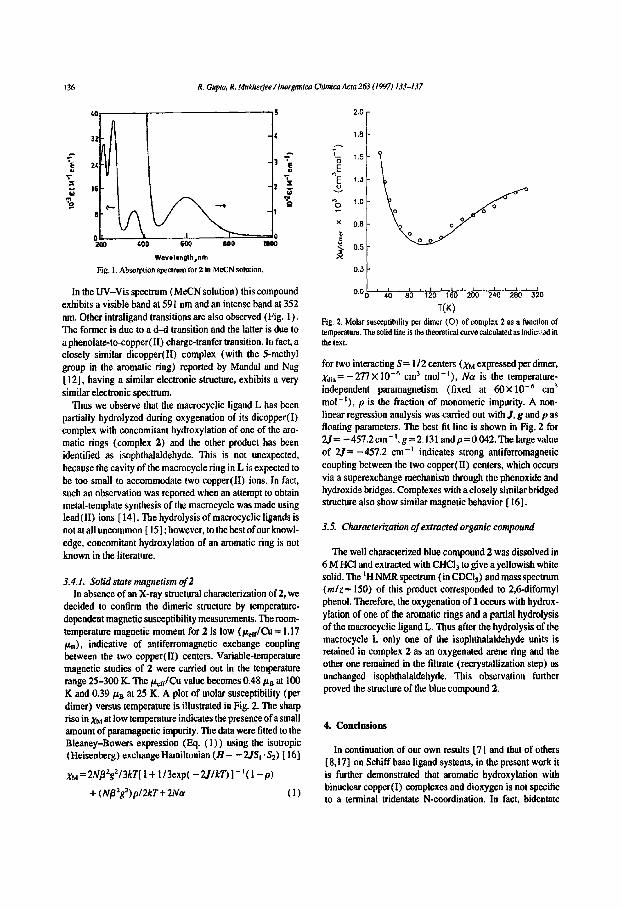
136 R. Gupta) R. Mu~erjee / lnorganica Chimica Acta 263 (1997) 133-137
40
3Z / 4
~o S 1
0 I 0 260 400 GO0 800 tO00
Wavelength,rim Fig. I. Absorption spectrum for2 in MeCN solution.
In the UV-Vis spectrum (MeCN solution) this compound exhibits a visible band at 591 nm and an intense band at 352 nm. Other intraligand transitions are also observed (Fig. 1). The former is due to a d-d transition and the latter is due to a phenolate-to-copper(II) charge-tranfer transition. In fact, a closely similar dicopper(II) complex (with the 5-methyl group in the aromatic ring) reported by Mandal and Nag [ 12], having a similar electronic structure, exhibits a very similar electronic spectrum.
Thus we observe that the macrocyclic ligand L has been partially hydrolyzed during oxygenation of its dicopper(I) complex with concomitant hydroxylation of one of the aro- matic rings (complex 2) and the other product has been identified as isophthalaldehyde. This is not unexpected, because the cavity of the macrocycle ring in L is expected to be too small to accommodate two copper(II) ions. In fact, such an observation was reported when an attempt to obtain metal-template synthesis of the macrocycle was made using lead(II) ions [ 14]. The hydrolysis of macrocyclic ligands is not at all uncommon [ 15 ]; however, to the best of our knowl- edge, concomitant hydroxylation of an aromatic ring is not known in the literature.
5 2.0
3.4,1. Solid state magnetism of 2 In absence of an X-ray structural characterization of 2, we
decided to confirm the dimeric structure by temperature- dependent magnetic susceptibility measurements, The room- temperature magnetic moment for 2 is low (/z¢~f/Cu = 1.17 /zn), indicative of antiferromagnetic exchange coupling between the two copper(II) centers. Variable-temperature magnetic studies of 2 were carried out in the temperature range 25-300 K. The/zcrf/Cu value becomes 0.48/Za at 100 K and 0.39/~n at 25 K, A plot of molar susceptibility (per dimer) versus temperature is illustrated in Fig. 2. The sharp rise in XM at low temperature indicates the presence era small amount of paramagnetic impurity. The data were fitted to the Bleaney-Bowers expression (Eq. (1)) using the isotropic (Heisenberg) exchangeHamiltonian (H =-- -2JSi" $2) [ 16]
XM = 2N~q2gZ/3kT[ 1 + 1/3exp( - 2J/kT) ] - ' ( 1 - p)
+ (N~2g:)p/2kT+ 2Na ( 1 )
L
m E
~0 ~.0
x 0.8
o.s
1.B
1,3,
0
0.3
0 % ' , ; ' 8b ' ~ o ' ~ ; o ' 2 ~ o ' 2 ~ o ' 2 ~ o ' 3 1 o T(K)
Fig. 2. Molar susceptibility per dimer (©) of complex 2 as a function of temperature. The solid line is the theoretical curve calculated as indic: .!~d in the text.
for two interacting S= 1/2 centers (AM expressed per dimer, X ~ = - 2 7 7 × 10 -6 cm 3 mol-~), Na is the temperature- independent paramagnetism (fixed at 60×10 -6 cm ~ mol- t ) , p is the fraction of monomeric impurity. A non- linear regression analysis was carried out with J, g and p as floating parameters. The best fit line is shown in Fig. 2 for 2,/= - 457.2 cm- i, g _ 2.131 and p = 0.042. The large value of 2 / = - 4 5 7 . 2 cm -~ indicates strong antiferromagnetic coupling between the two copper(H) centers, which occurs via a superexchange mechanism through the phenoxide and hydroxide bridges. Complexes with a closely similar bridged structure also show similar magnetic behavior [ I6].
3.5. Characterization of extracted organic compound
The well characterized blue compound 2 was dissolved in 6 M HCI and extracted with CHCI3 to give a yellowish white solid. The tH NMR spectrum (in CDCi 3) and mass spectrum (mlz = 150) of this product corresponded to 2,6-diformyl phenol. Therefore, the oxygenation of 1 occurs with hydrox- ylation of one of the aromatic rings and a partial hydrolysis of the macrocyclic ligand L. Thus after the hydrolysis of the macrocycl¢ L only one of the isophthalaldehyde units is retained in complex 2 as an oxygenated arene ring and the other one remained in the filtrate (recrystallization step) as unchanged isophthalaldehyde. This observation further proved the structure of the blue compound 2.
4. Conclusions
In continuation of our own results [7] and that of others [8,17] on Schiff base ligand systems, in the present work it is further demonstrated that aromatic hydroxylation with binuclear copper(I) complexes and dioxygen is not specific to a terminal tridentate N-coordination. In fact, bidentate

R. G~#pta, R. Mukherjee I lnorganica ChimK'a Acta 263 [1997) 133-137 137
N-coordination is sufficient enough, whether it is of the open chain type or part of a macrocyclic ligand system, to exhibit tyrosinasc-like activity. The present results show the feasi- bility ofa bidentate N-coordination of the latter type in chem- ical modeling of tyrosinase activity. Due to inherent strain associated with the present macrocyclic ligand, however, the oxygen insertion onto one of the arene rings is accompanied by the hydrolysis of the macrocyclic ligand.
5, Supplementary material
IR (L, complexes I and 2), 'H NMR (L and complex I) and MS (L, complexes 1 and 2 and extracted organic flag- mcnts) spectral data (l I pages) are available from the authors upon request.
Acknowledgements
This work was supported by the Department o f Science
and Technology (DST) and the Council of Scientific and
Industrial Research (CSIR), Government of India. R.G, acknowledges the award of a JRF by CSIR, We thank Mr Rahul Garg for his help in the synthesis of L and Dr Sreebrata Goswami (Department of Inorganic Chemistry, Indian Asso- ciation for the Cultivation of Science, Calcutta) for micro- analytical data.
References
[ I ] (a) K.D. Km'lin and Y. Gulmoh, Prog. Inorg. Chent, 35 (1987) 219; (b) Z. Tyeldat and K.D. Kadin, Ace. Chem, Res., 22 (1989) 241; (c) K.D. Kadin and Z. Tyekla#, Adv. Inorg. Biocher~. 9 (1993) 123; (d) K.D. Katlin, Science. 261 (1993) 701.
[2] (a) N. Kitajima and Y. Mom-oka, Chem. Rev., 94 ( 1994} 737; (b) N. Kitajima, Adv. Inorg. Chem., 39 (1992) I; (c) T.N. Sotmll, Telrahedron. 45 (1989) 3.
[3] (a) E.I. Solomon, MJ. Baldwin aml M.D. Lowery, Chem. Rev., 92 (1992) 521; (b) E.I. Solomon, D.E. Root and CA. Brown, C/~m~ Rev., 94 ( 1994} 827: (c) E.I. Solomon and M.D. Lowery, Science, 259 (1993) 1575.
[4] (a) K.D. Katlin, P.L. Dahlstrom, S.N. Cozzene, P.M. S~'~rty and J. Zubiela, J. Chem. $oc.. Chem. Conunun., ( ! 981 ) 881; (b) K.D. K~i~ J.C. Hayes, Y. Gulmeh, R.W. Cmbe, J.W. MeKown, J.H. Hutchinson and J. Zebiela, J. Ant Chent Soc., 106 (1984) 2121.
[5] D.-H. Lee, N.N. Mu.qhy and K.D. IL~lin, lnorg. Chem., 35 (19961 804, and Refs. themn.
[6 ! J.A. Halfen, V.G. Young, Jr. and W.B. Tolman, J. Am. C?tem. ,f~c., i i8 (1996) 10920, and Refs. therein.
[7] D. Ghosh, T.K. Lal, S. Ghosh and R.N. Mekherjee, J. Chem. Sot., Chem. Commun., (1996) 13. and Refs. therein.
[8] (a) R. Menif and A.E. Martell, J. Chem. Soc.. Chem. CommmL, (1989) 1521; (b) R. Menif, A~E. Martell, Pj. S ~ o and A. Clearfield, lnorg. Chent, 29 (1990) 4723.
[9l M. Ray, D. Ghosh, Z Shirin and R.N. M u i ~ , lnorg. Chem., 36 (1997) in press.
[ 10l J. Jazwinski, J.-M. Lehn, R. Merit, J.-P. Vigneron. M. Cesario, J. Guilhem and C. Pasc.atd, Tetrahedron l.eZt., 28 (1987) 3489.
l I ! ] S.M. Nelson, F. Esho, A. Lave~y and M.G.B. Dww, J. Am. Chem. 5oc., 105 (I983) 5693.
[ 12] S.K. Mandai and K. Nag, J. Chem. Soc., Dalton Trans., (1984) 2141. [ 13] WJ. Geary, Coor,~ Chem. Rev.. 7 (1971) 81. [ 14] S.K. Mandal and K. Nag, J. Org. Chem., 51 (1986) 3900. [ 15] CJ. Harding, Q Lu, J.F. Malone, DJ. Marts, N. ~ V. McKee
and J. Nelson, J. Chem. Soc., Dahon Trans., (1995) 1739. [ 16] (a) JJ. GI2ybowski, P.H. Menell and F~L. Udxlch, lnorg. Chem.. 17
(1978) 3078; (b) N.A. Bailey, D-E. Femon, J. Lay, P.B. Roberts, J.- M. Lato~ and D. Limosin, J. Chem. See.. Dal~n Trans., (1986) 2681.
[ 17 ] (a) L. Casclla and L. Rigoni, J. Chem. $oc., Chem. Commun., { 1985 ) 1668; (b) OJ. Gelling, E van Bollmis, A. MeetsmaaadB.L. Feniaga, ]. Chem. Soc., Chem. Corrmm¢, (1988) 552; (c) L. Ca~ila, M. Gulloni. G. Pallasrza and L. Rigom, J. Ant Chem. Soc.. 110 (1988) 4221; (d) M.G. D~w, J. Trocha-Gdmshaw and K.P. McKillep, Polyhedron, 8 (1989) 2513; (el L. Casena, M. Gellotli, M. Barto~k, G. Pallanza and E. Lanmn0, J. Chem. Soc.. Chem. Cone*m~, (I991) 1235.


















