
A MICROSCOPIC EXAMINATION OF THE INTERACTION BETWEEN ANTIBODIES, DENGUE ... · 1.2.2 Dengue...
196
A MICROSCOPIC EXAMINATION OF THE INTERACTION BETWEEN ANTIBODIES, DENGUE VIRUS AND MONOCYTES ZHANG LIXIN (B.Sc, NUS) A THESIS SUBMITTED FOR THE DEGREE OF MASTER OF SCIENCE DEPARTMENT OF MICROBIOLOGY NATIONAL UNIVERSITY OF SINGAPORE 2010
Transcript of A MICROSCOPIC EXAMINATION OF THE INTERACTION BETWEEN ANTIBODIES, DENGUE ... · 1.2.2 Dengue...

A MICROSCOPIC EXAMINATION OF THE INTERACTION BETWEEN ANTIBODIES, DENGUE VIRUS
AND MONOCYTES
ZHANG LIXIN (B.Sc, NUS)
A THESIS SUBMITTED
FOR THE DEGREE OF MASTER OF SCIENCE DEPARTMENT OF MICROBIOLOGY
NATIONAL UNIVERSITY OF SINGAPORE
2010


Acknowledgements
I will like to extend my deepest gratitude to my main supervisor Associate Professor
Ooi Eng Eong for his constant guidance and many stimulating discussions. I will also
like to thank my co-supervisor Professor Mary Ng Mah Lee and her lab members for
their critical suggestions.
I am also very grateful to Tan Hwee Cheng, Chan Kuan Rong, Angelia Chow,
Angeline Lim and Dr Brendon Hanson for their kind support during the course of my
research.
Not to forget the fantastic groups of colleagues from both Duke-NUS and DMERI that
created a very cheerful and conducive environment to for research.
Lastly, I will also like to extend indebtedness to my beloved family and friends for
their continuous shower of concern, patience and understanding throughout the
whole course of graduate studies.


Contents
Summary ............................................................................................................................ i
List of Tables ................................................................................................................... iii
List of Figures ....................................................................................................................v
List of Publications ........................................................................................................ vii
List of Abbreviations ...................................................................................................... ix
Chapter 1: Introduction
1.1 Dengue Background ....................................................................................................3
1.1.1 Americas ..................................................................................................................7
1.1.2 Southeast Asia .......................................................................................................11
1.1.3 Singapore ...............................................................................................................15
1.2 Disease and management .........................................................................................19
1.2.1 Dengue Fever .......................................................................................................19
1.2.2 Dengue Hemorrhagic Fever/Dengue Shock Syndrome .......................................19
1.2.3 Current treatment/Dengue control ........................................................................22
1.2.4 Vaccine development and progress ......................................................................23
1.3 Life cycle of dengue virus .........................................................................................28
1.3.1 Structure and genome of DENV ..........................................................................28
1.3.2 Entry and exit of DENV .......................................................................................30

1.4 Role of host immune response in dengue ................................................................37
1.4.1 T cells ...................................................................................................................37
1.4.2 ADE ......................................................................................................................37
1.4.3 Other risk factors of disease severity ...................................................................39
1.4.4 Antibody neutralization of DENV .......................................................................39
1.4.5 Monocytes/macrophages ......................................................................................41
1.5 Discovery and use of fluorescent proteins in research ..........................................44
1.5.1 Discovery of GFP: short story of Aequorin, O Shimomura .................................44
1.5.2 From GFP to rainbow coloured fruit proteins ......................................................44
1.5.3 Fluorescent proteins in research ...........................................................................47
1.6 Gaps in knowledge and Hypothesis/Objectives of this study ................................48
Chapter 2: Materials and methods
2.1 Cell culture ................................................................................................................55
2.2 Primary monocytes culture ......................................................................................55
2.3 Antibodies ..................................................................................................................56
2.4 Virus culture and purification .................................................................................57
2.5 Plaque assay ...............................................................................................................57
2.6 Virus labelling ...........................................................................................................57
2.7 Immunofluorescence assay for viral infection ........................................................58

2.8 Flow cytometry determination of percentage of labelled dengue virus ...............59
2.9 Detection by SYBR green-based real-time PCR ....................................................59
2.10 Growth kinetics .......................................................................................................60
2.11 Humanization of 3H5 and 4G2 mouse monoclonal antibodies ...........................60
2.12 Binding affinity ELISA ..........................................................................................60
2.13 Titration of h3H5/h4G2 to determine neutralizing concentrations on monocytes
............................................................................................................................................61
2.14 DENV immune complex co-localization studies in monocytes ...........................61
2.15 Sucrose gradient analysis of DENV immune complex sizes ................................62
2.16 Dynamic light scattering (DLS) analysis of DENV immune complex sizes .......63
2.17 Statistical analysis ...................................................................................................63
Chapter 3: Results
3.1 Producing Fluorescent DENV .................................................................................67
3.1.1 Alexa Fluor labelling of DENV ...........................................................................67
3.1.2 Efficiency of Alexa Fluor dye labelling ...............................................................76
3.1.3 Reproducibility of labelling .................................................................................78
3.1.4 Growth kinetics of labelled DENV ......................................................................80
3.2 Visualizing the fate of antibody-DENV complexes in monocytes .........................82
3.2.1 Humanized 3H5 and 4G2 ......................................................................................82
3.2.2 Determining the neutralizing concentrations of h3H5 and h4G2 on monocytes .85
3.2.3 Visualizing the entry and endosomal trafficking of antibody-virus complexes in
THP-1 monocytic cell line ............................................................................................87

3.2.4 Primary monocytes ...............................................................................................93
3.2.5 Antibody-DENV immune complex interactions with primary monocytes ..........95
3.3 Fc receptor usage for internalization ....................................................................101
3.4 Inhibition of immune complex uptake ..................................................................109
3.4.1 Concentration dependence .................................................................................109
3.4.2 Competition for Fc receptors ..............................................................................109
3.4.3 Immune complex size and internalization ..........................................................115
3.4.3.1 Sucrose gradient separation of immune complex sizes ..........................115
3.4.3.2 Immune complex size by Dynamic Light Scattering (DLS) ..................116
Chapter 4: Discussion/Conclusion
4.1 Fluorescence labeling of DENV .............................................................................123
4.2 Cellular fate of DENV immune complexes in monocytes ....................................124
4.3 Antibody concentrations and complex size ...........................................................125
4.4 Conclusions ..............................................................................................................126
4.5 Future work .............................................................................................................127
Bibliography 131
Appendix 153

Abstract
Dengue is a significant disease globally. An estimated 50 to 100 million dengue
infections occur annually, and more are at risk of being infected with 2.5 billion people
living in dengue endemic countries. Although vector reduction programmes may limit
dengue virus (DENV) transmission, it has not been carried out at a scale sufficient to
control the disease globally. A tetravalent dengue vaccine is therefore needed to halt this
worldwide escalation in disease incidence. Serotype-specific antibodies generated in a
course of infection are thought to confer lifelong immunity to the same serotype of
DENV; whereas cross-reactive antibodies are more frequently associated with antibody-
mediated enhancement of infection, leading to more severe disease. Despite the fact that
antibody-DENV interactions can lead to immunity or immunopathogenesis, the factors
governing such outcomes of infection have not been well defined. This has thus led to
long delays in the development of a safe and effective vaccine. In this thesis, we sought
to understand the immunity end of the spectrum through early antibody-DENV
interactions with monocytes (the primary targets of dengue infection) that lead to
neutralization of the virus, using confocal microscopy.
A simplified method of labelling DENV with a fluorescent Alexa Fluor dye with minimal
modification to viral viability was developed in this study and subsequently used to
visualize the early cellular processes taking place when monocytes encounter antibody-
DENV complexes. Using two human-mouse chimeric antibodies, h3H5 and h4G2, as our
model for serotype-specific and cross-reactive antibodies, respectively, we observed
significantly different sub-cellular trafficking characteristics in human monocytes. At the
minimal antibody concentration to fully neutralize 10 multiplicity of infection (MOI) of
i

ii
DENV, immune complexes with 3µg/ml h3H5 were rapidly internalized through the
activatory FcγRI and transported to LAMP-1 positive compartments within 30min, while
that with 100µg/ml h4G2 bound to both FcγRI and FcγRII but internalization was
delayed. This delay in internalization appeared to be antibody concentration dependent as
increasing h3H5 concentration to 100 and 400µg/ml showed similar blockade of uptake.
These observations were also verified in primary monocyte cultures.
One possible explanation would be that larger viral aggregates were formed at higher
antibody concentrations and that inhibited efficient Fc receptor-mediated uptake by the
monocytes. Using a combination of sucrose gradient to separate the viral aggregates by
size and dynamic light scattering to estimate their diameter, the data indicates that viral
aggregates with average diameter of 192nm were formed with 100µg/ml of antibody,
which is significantly larger than virus only (49.1nm) or Fab only controls (57.7nm).
Taken collectively, increasing concentrations of antibody result in the formation of
DENV aggregates of different sizes, which appeared to inhibit internalization. The
mechanism for this is not through competition for FcR by free and unbound antibody.
Instead the data suggests that larger viral aggregates may enable antibodies to cross-link
FcR that are normally expressed at lower density. Lowering the antibody concentration
allowed for efficient internalization, followed rapidly by trafficking of the immune
complex to the late endosome. However, at these concentrations, viral replication was
only prevented with serotype-specific but not cross-reactive antibody.

List of Tables
Table Title Page 1-1 Current on-going vaccine development
26-27
1-2 Principle host factors in dengue
33-36
3-1 Viral RNA copy number to infectious particle ratio
79
3-2 Binding affinity of 3H5 and 4G2 to DENV-2 before and after humanization
83
3-3 Binding affinity of h3H5 and h4G2 to AF594-DENV and non-labelled DENV
84
iii

iv

List of Figures
Figure Title Page 1-1 Countries/areas at risk of dengue transmission
5
1-2 The change in global distribution of dengue serotypes from 1970 to 2004
6
1-3 Reinfestation of Aedes aegypti in the Americas post eradication
9
1-4 Incidences of DF/DHF cases in the Americas
10
1-5 Dengue situation in Southeast Asia
13-14
1-6 Dengue situation in Singapore
18
1-7 Range of dengue disease
21
1-8 Structure and proteome of DENV
29
1-9 Replication lifecycle of Flavivirus
32
1-10 mFruit fluorescent proteins derived from mRFP or somatic hypermutation (SHM)
46
3-1 Dengue virus viability post AF594 labelling
70-75
3-2 Efficiency of Alexa Fluor dye labelling as a function of percentage infected
77
3-3 Comparing the growth kinetics of purified non-labelled and labelled DENV
81
v

vi
3-4 Neutralizing concentrations of h3H5 and h4G2 on monocytes
86
3-5 Neutralizing h3H5-DENV complexes entry and transport in THP-1
89
3-6 Neutralizing h4G2-DENV complexes entry and transport in THP-1
91
3-7 Primary monocytes yield
94
3-8 Entry and endocytic trafficking of h3H5-opsonized DENV in primary monocytes
97
3-9 Entry and endocytic trafficking of h4G2-opsonized DENV in primary monocytes
99
3-10 Fc gamma receptors expression on monocytes
103
3-11 Fc receptor requirements by h3H5 or h4G2-opsonized DENV in THP-1 cells
105
3-12 Fc receptor requirements by h3H5 or h4G2-opsonized DENV in primary monocytes
107
3-13 Effect of increased antibody concentrations on endocytic trafficking of DENV
111
3-14 Competition for Fc receptors
113
3-15 Immune complex size analysis by sucrose gradient and dynamic light scattering (DLS)
118-119

vii
List of Publications
Published papers
1. Zhang SLX, Tan HC, Hanson BJ, Ooi EE. 2010. A simple method for Alexa
Fluor dye labelling of dengue virus. J Virol Methods 167(2):172-177
2. Zhang SLX, Tan HC, Ooi EE. 2011. Visualizing dengue virus through Alexa
Fluor labeling. Journal of Visualized Experiments Immunology and Infection.
(http://www.jove.com/main.php?SectionID=2) (Accepted, in press)
Manuscript in submission
Chan KR, Zhang SLX, Tan HC, Chan YK, Chow A, Lim PC, Vasudevan SG,
Hanson BJ, Ooi EE. Engagement of the inhibitory FcγRIIB neutralizes dengue
virus immune complexes in monocytic cells.
1. Manuscript in submission.
Conference presentations
1. Zhang SLX, Tan HC, Hanson BJ, Ooi EE. Direct fluorescent labelling dengue
virus. 4th Asian Dengue Reseach Network Meeting, December 2009, Singapore.

viii

List of abbreviations
ADE Antibody-dependent enhancement AF594/488 SE Alexa Fluor 594/488 succinimidyl ester AF594/488/647 Alexa Fluor 594/488/647 ATCC American Type Culture Collection BFP Blue fluorescence protein BHK-21 Baby hamster kidney cell line C6/36 Aedes albopictus cell line Cy3 Cyanine 3-bihexanoic acid DC Dendritic cells
DC-SIGN Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin
DENV Dengue virus DF Dengue fever DHF Dengue hemorrhagic fever
DiD
1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt
DLS Dynamic light scattering DMSO Dimethyl sulfoxide DSS Dengue shock syndrome E Envelope protein EEA-1 Early endosomal antigen 1 FACS Fluorescence activated cell sorting/flow cytometry FBS Fetal bovine serum FcR Fc receptor FcγR Fc gamma receptor FRET Förster resonance energy transfer G-CSF Granulocyte colony-stimulating factor GFP Green fluorescence HLA Human leukocyte antigen HLA-DM Human leukocyte antigen - DM HNE Buffer Hepes, sodium chloride and EDTA buffer hr hour IFNγ Interferon-gamma IgG Immunoglobulin G IL-6 Interleukin-6 IL-8 Interleukin-8 Imax Maximum fluorescence intensity LAMP-1 Lysosomal-associated membrane protein 1 LAV Live-attenuated vaccine
ix

x
mab monoclonal understanding MIIC Major histocompatibility complex class II molecules min minute MIP-1β Macrophage inflammatory protein -1β ml Milli liter mM Milli meter MOI Multiplicity of infection nm Nano meter NS Non-structural PAHO Pan American Health Organization PBMC Peripheral blood mononuclear cells PBS Phosphate-buffered saline PBST 1xPBS + 0.05% Tween PCR Polymerase chain reaction pFA Paraformaldehye pfu/ml plaque forming unit per milli liter prM pre-Membrane protein PRNT Plaque reduction neutralization test SBB Sodium bicarbonate buffer SHM Somatic hypermutation THP-1 Human monocytic cell line TMB 3,3’,5,5’-Tetramethylbenzidine TNFα Tumor necrosis factor-alpha Vero African green monkey kidney epithelial cell line WHO World Health Organization WNV West Nile virus μl Micro liter μM Micro meter

Chapter 1
Introduction
Zhang Lixin (HT080076A)


1.1. Dengue background
The earliest record of illnesses compatible with dengue fever found to date was first
published in a Chinese ‘encyclopedia of disease symptoms and remedies’ during the Jin
Dynasty (265-420 AD), and formally edited in 610 AD (Sui Dynasty) and again in 992
AD (Northern Song Dynasty) [Nobuchi, 1979]. In 1779-80, major epidemics of dengue-
like illness were reported in Asia, Africa and North America [Hirsch, 1883; Howe, 1977;
Pepper, 1941; Rush, 1789], indicating that dengue virus (DENV) had a wide
geographical distribution as early as the 18th century. This is likely a consequence of a
flourishing international sea trade. However, it was not until the World War II in the
1940s that the first of four DENV serotypes, designated DENV 1 (Hawaii strain) and 2
(NGC strain) were isolated [Hotta, 1952; Sabin and Schlesinger, 1945]. Two more
serotypes, DENV 3 and 4, were isolated from patients with a hemorrhagic disease during
an epidemic in Manila in 1956 [Hammon et al., 1960]. Since then, thousands of DENV
have been isolated from all parts of the tropics; all fitting into the four serotype
classification.
DENV belongs to the family Flaviviridae, which consists of 53 different viruses [Gubler
et al., 2007]. Among these are yellow fever, Japanese encephalitis, tick-borne
encephalitis and West Nile viruses. DENV is transmitted by Aedes mosquitoes, mainly
Aedes aegypti, and infects an estimated 50 million people annually with 2.5 billion more
people at risk of infection each year in the tropical and sub-tropical regions (Fig.1-1).
Hence, dengue is the most important mosquito-borne viral disease in the world [WHO,
2007]. Furthermore, these numbers only represent the tip of the iceberg as many dengue
infections are asymptomatic or present with non-specific febrile illness [Gubler, 1989b].
3

Nonetheless, its incidence has increased 30-fold over the past 50 years with increasing
geographical expansion to new countries [Gubler, 2002; Mackenzie et al., 2004].
Unprecedented global population growth and the associated unplanned and uncontrolled
urbanization, lack of effective mosquito control in dengue endemic areas, decay in public
health infrastructures in most countries, and increase in air travel which provides the ideal
mechanism for the transport of dengue between population centres of the world are the
major contributors to the re-emergence of the disease [Gubler, 1998]. With increased air
travel and exchange of viruses across borders, most endemic countries now have more
than one circulating dengue serotype (Fig. 1-2) [Mackenzie et al., 2004].
4

Figure 1-1 Countries/areas at risk of dengue transmission.
Dengue is the most important mosquito-borne viral disease in the world, infecting an estimated 50 million people annually with more at risk in countries within the tropical and sub-tropical regions. Figure shows the geographical distribution of countries/areas at risk of dengue transmission. Adapted from DengueNet, World Health Organization Map Production: Public Health Mapping and GIS World Health Organization.
5

Figure 1-2 The change in global distribution of dengue serotypes from 1970 to 2004.
Increased human travel over the decades led to a wider distribution of dengue viruses worldwide and these countries have become hyperendemic with more than 1 serotype reported. Adapted from Nat Med 10(12 Suppl): S98-109, 2004.
6

1.1.1. Americas
First records of dengue-like disease outbreaks in the Americas can be traced back to the
fifteenth century in French West Indies and Panama [Wilson and Chen, 2002]. This
coincides with the introduction of Aedes aegypti on slave ships arriving from West
Africa. Since then, the vector has become well established in tropical and temperate areas
of the Americas [Wilson and Chen, 2002].
Aedes aegypti not only transmits DENV, it also serves as an epidemic vector for yellow
fever virus. In an effort to control yellow fever transmission, the Pan American Health
Organization (PAHO) launched a large-scale intensive campaign that led to the
eradication of Aedes aegypti from almost all countries in the Americas by 1960s [Soper,
1963]. The programme not only controlled yellow fever, it also disrupted the dengue
transmission cycle. As a result, there was no recorded dengue epidemics from 1946 to
1963 [Wilson and Chen, 2002].
The support for vector control programmes waned with the decreased incidence of yellow
fever [Downs, 1969; Sencer, 1969; Soper, 1969]. Consequently, vector control activities
declined and dengue re-emerged in 1960s and 1970s as Aedes aegypti start to re-infest
areas where it was eliminated and subsequently spread to areas where it had never been
reported (Fig. 1-3) [Gubler, 1989a; Gubler, 1998; Wilson and Chen, 2002]. Dengue
haemorrhagic fever (DHF) made its first appearance in 1981 when a new strain of dengue
2 was introduced into Cuba and caused a massive epidemic with a total of 344,203 cases,
of which, 10,312 were severe and 158 were fatal [Kouri et al., 1986]. Since then, dengue
7

outbreaks continued to occur frequently and DHF has been reported in other parts of the
Caribbean and Central and South America (Fig. 1-4a and b) [Gubler, 1998].
Over a period of 30 years, many countries within the Americas (areas with previous
dengue, as well as new territories) have become endemic with multiple co-circulating
dengue serotypes [Gubler, 1998]. This corresponds to the expansion and establishment of
the Aedes mosquitoes in these areas. With increased human movement due to trade and
tourism activities, DENV was moved to new geographic areas by viremic individuals that
were either pre-symptomatic or who develop subclinical infection [Wilder-Smith and
Gubler, 2008].
It is predicted that global warming will increase the epidemic potential of vector-borne
transmission of DENV as fewer mosquitoes would be necessary to maintain or spread the
virus to vulnerable human populations [Patz et al., 1998]. It is possible that as the
temperature rises, mosquitoes can thrive in regions where it was previously too cold to,
hence expanding the geographic locations of dengue [Halstead, 2008a]. Increased
temperature also shorten the development time of mosquitoes, thus more mosquitoes can
reach sexual maturity earlier and spread the virus [Halstead, 2008a]. Higher ambient
temperature also shortens the extrinsic incubation time and hence it takes a shorter time
for the mosquitoes to become infective [Halstead, 2008a]. However, this remains
controversial as many researchers reasoned that the re-emergence and spread of dengue
epidemics are direct consequences of urbanization and globalization, which led to
increased movement of people and trade commodities, combined with inefficient public
health measures, rather than climate change alone [Halstead, 2008a; Ooi and Gubler,
2009b].
8

Figure 1-3 Reinfestation of Aedes aegypti in the Americas post eradication.
A large scale Aedes aegypti eradication programme launched to control yellow fever dramatically decreased the vector throughout most of the Americas by 1970. However as the support for the vector control programmes waned over time, Aedes aegypti regained the geographical distribution it held before the eradication was initiated and further spread to areas where it was not reported. Adapted from Pan American Health Organization (PAHO).
9
(a)
(b)
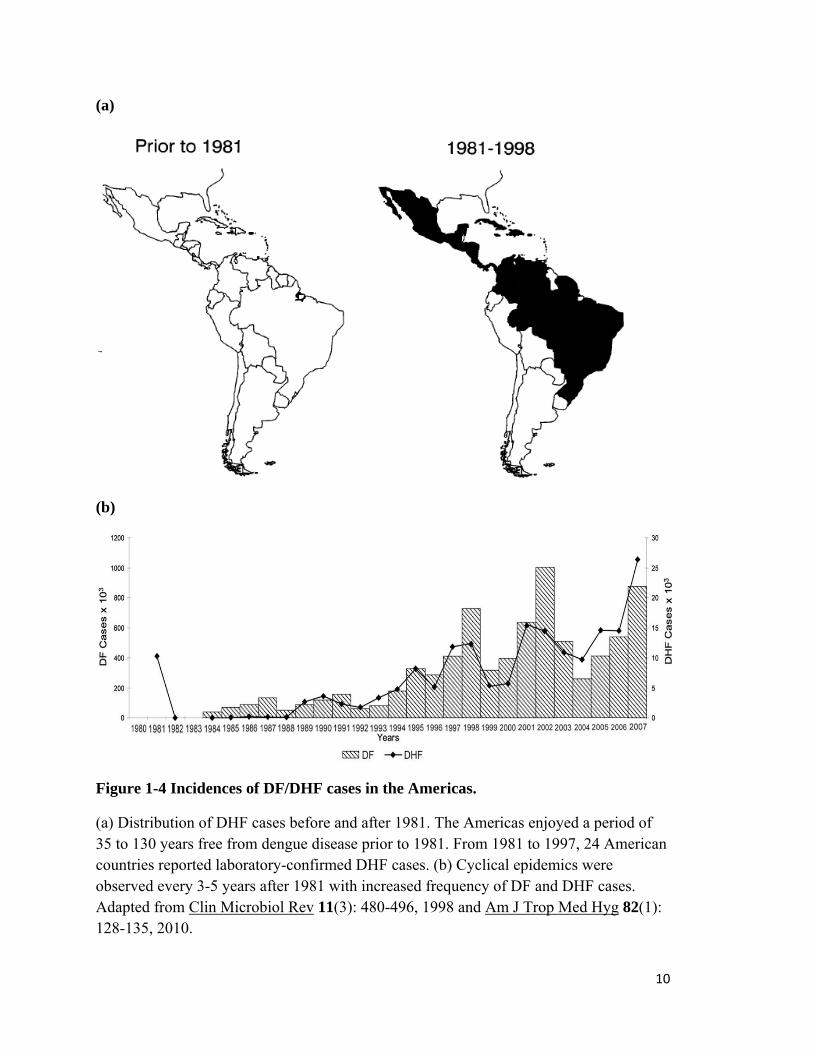
Figure 1-4 Incidences of DF/DHF cases in the Americas.
(a) Distribution of DHF cases before and after 1981. The Americas enjoyed a period of 35 to 130 years free from dengue disease prior to 1981. From 1981 to 1997, 24 American countries reported laboratory-confirmed DHF cases. (b) Cyclical epidemics were observed every 3-5 years after 1981 with increased frequency of DF and DHF cases. Adapted from Clin Microbiol Rev 11(3): 480-496, 1998 and Am J Trop Med Hyg 82(1): 128-135, 2010.
10

1.1.2. Southeast Asia
Multiple DENVs have been known to be transmitted in Southeast Asia for a long time
(Fig. 1-5) [Halstead, 2006]. There was no large-scale vector programmes against Aedes
aegypti, unlike in the Americas. However, adoption of effective anti-mosquito hygienic
practices in some areas of Asia during the colonial era helped to control mosquito
populations [Halstead, 1965].
The Second World War changed the epidemiology of dengue in Southeast Asia
permanently as the destruction of cities changed the landscape and places abandoned the
colonial system [Halstead, 2006]. Movement of troops during war aided the dispersal of
the DENVs between population centres of the Asia-Pacific regions and by the end of war,
most countries in Southeast Asia were hyperendemic and epidemic DHF emerged a few
years later [Gubler, 1997]. Urbanization happened quickly after the war as millions of
people moved into cities looking for jobs, which led to hurried but unplanned growth of
urban centres, and hence poor housing, water supply and sewerage system [Ooi and
Gubler, 2008]. As such, people tend to store water in households, and together with the
abandoned war equipment and rubbish, these became ideal mosquito breeding sites and
high density of susceptible human hosts in the cities provided ideal conditions for virus
propagation [Ooi and Gubler, 2009a]. Thus, the geographic distribution of the viruses and
vectors expanded, and the densities of Aedes aegypti increased, making many countries in
this region highly permissive for epidemic transmission [Wilder-Smith and Gubler,
2008].
11

Dengue soon emerged as the leading health burden in Southeast Asia. In 1954,
Philippines recorded its first DHF outbreak and a second outbreak 2 years after in 1956
[Gubler, 1997]. Since then, DHF cases have been reported yearly [Halstead, 1980].
Thailand also had similar dengue history as the Philippines with DHF/DSS (dengue
shock syndrome) documented as early as 1950s in Bangkok [Halstead, 1980; Halstead
and Yamarat, 1965]. Vietnam, Indonesia, Cambodia, Sri Lanka, Malaysia and Singapore,
all reported cases DHF/DSS in the period of 1956-1978 [Halstead, 1980].
12

(a)
13

(b)
Figure 1-5 Dengue in Southeast Asia
(a) Distribution of countries in South and Southeast Asia with records of mosquito-borne hemorrhagic fever outbreaks between 1950 and 1964. Adapted from Yale J Biol Med 37(6): 434-454, 1965. (b) Incidence rate of dengue in Southeast Asia, 2005. Adapted from WHO Denguenet.
14

1.1.3. Singapore
DHF first appeared in Singapore in the 1960s and quickly became a major cause of
childhood death [Ooi et al., 2006]. A Vector Control Unit was set up in 1966 in response
to dengue and after a series of entomologic surveys [Chan et al., 1971a; Chan et al.,
1971b; Chan et al., 1971c; Chan et al., 1971d; Ho et al., 1971] and a pilot project was
started to control the Aedes vectors in an area with high incidence of DHF [Chan, 1967],
and a vector control system based on entomologic surveillance and larval source
reduction was launched in 1968 [Chan, 1985; Chan et al., 1977]. In 1966, DHF was made
a notifiable disease and in 1977, DF also became a notifiable disease [Chan, 1985].
Since the implementation of the vector control programme, the premises index fell
sharply from 16% and was maintained at approximately 2% till present day (Fig. 1-6a)
[Chan, 1967]. As with the reduction of vectors, the number of cases of dengue infections
dropped and Singapore enjoyed a 15-yr period of low dengue incidences [Ooi et al.,
2006]. However, despite having a successful vector control programme in place, dengue
incidences surged in the 1990s and the overt (symptomatic) attack rates were several
folds higher than in the 1960s (Fig. 1-6a) [Ooi et al., 2006].
Multiple factors appear to be associated with the re-emergence of dengue in Singapore
[Ooi et al., 2006]: (1) Lowered herd immunity. Reduced dengue transmission in the
1970s and 1980s resulted in lowered herd immunity to DENV [Egger et al., 2008; Goh,
1995]. Low levels of population immunity provided an ideal condition for dengue
transmission despite low Aedes mosquito density. (2) Transmission outside the home. A
seroprevalence study on 1,068 children ≤ 15 years of age indicated a shift of dengue
infection away from home [Ooi et al., 2001]. School-age children were 9 times more
15

likely to have antibodies to dengue than preschool children [Ooi et al., 2001]. Preschool
children spent most of their time at home or at pre-school care centres whereas school-
age children have formal half-day education in schools followed by extra-curriculum
activities, hence spend more time outside of home. This suggested that the risk of
acquiring dengue was higher when a person spent more time away from home [Ooi et al.,
2001]. This was supported by the lower premises index in residences compared to non-
residences in 1997 where schools (27.0%), construction sites (8.3%), factories (7.8%) and
vacant properties (14.6%), had much higher premises indexes than residential properties
(2.1% in landed premises and 0.6% in apartments) [Tan, 1997]. (3) Dengue in adults. The
proportion of patients ≤ 15 years of age had been on the decline while the proportion of
patients ≥ 25 years of age had been on the rise (Fig. 1-6b). Dengue infections in adults
tend to be more clinically overt than in children thereby contributing to the increase in
overall incidence of dengue [Ooi et al., 2003; Seet et al., 2005]. (4) Shift in surveillance
emphasis. Over time, the vector control programme evolved to emphasize on early case
detection and warning system to identify active virus transmission areas, rather than
active vector surveillance and elimination [Ooi et al., 2006]. The passive vector
surveillance was ineffective at stopping virus transmission, especially in people with
subclinical or mild undifferentiated fever, to the uninfected mosquitoes.
Countries embarking on Aedes control programmes may face similar epidemiological
problems as those encountered by Singapore [Ooi et al., 2006]. Hence an effective
tetravalent vaccine is the only sustainable solution for dengue. However, until a large-
scale immunization program for dengue becomes available, a community-based
16

integrated control of Aedes aegypti remains the key to prevention and control of DF/DHF
[Gubler and Clark, 1994].
17

(a)
(b)
Figure 1-6 Dengue situation in Singapore. (a) Annual incidences of DF and DHF, and the premises index, in Singapore between 1966 and 2005. Since the implementation of vector control programme in 1968, the premises index fell dramatically and was maintained at about 2% to 2005. DF/DHF cases also plummeted with the reduced premises index until their resurgence in 1990. (b) Proportion of dengue patients below 15 years of age is on the decline while the proportion of patients aged 25 years and above are on the rise. Adapted from Emerg Infect Dis 12(6): 887-893, 2006.
18

1.2. Disease and disease management
Dengue infections are mostly undetected or can present as a self-limiting febrile illness
(dengue fever, DF), which in some cases develop into more severe forms of the disease:
dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS). Figure 1-7 summarizes
the typical manifestations of dengue infection (a) and spectrum of DHF (b).
1.2.1. Dengue fever
Clinical presentations of DF often depend on the age of the patients. While dengue
infection in infants and young children may present as undifferentiated febrile disease
accompanied by maculopapular rash or mild DF, older children and adults may
experience sudden onset of high fever, severe headache, retro-orbital pain, myalgia and
arthralgia, nausea and vomiting, and rash (Fig. 1-7a) [George and Lum, 1997].
Leukopenia and thrombocytopenia may also be observed [George and Lum, 1997].
Recovery is often accompanied with fatigue and depression, especially in adults
[Halstead et al., 2007].
1.2.2. Dengue haemorrhagic fever/Dengue shock syndrome
The 1997 World Health Organization (WHO) guidelines describe DHF as being
characterized by fever lasting 2-7 days, plasma leakage, hemorrhagic tendency and
thrombocytopenia (Fig. 1-7a). Patients may experience similar symptoms as DF with the
addition of hemorrhagic phenomena, usually indicative by positive tourniquet test, easy
bruising and bleeding at venepuncture sites (Fig. 1-7b). The critical stage of the disease
typically occurs at the end of the febrile phase when the temperature declines rapidly, and
19

plasma leakage and hemoconcentration can be observed. The patient, if not properly
managed, can quickly progress into shock (DSS) and be in danger of death.
In addition to the DHF symptoms, a DSS patient could appear restless, have cold clammy
skin, weak but rapid pulse and narrow pulse pressure (< 20mmHg) [WHO, 1997]. The
duration of shock is usually short, lasting 12 to 24 hours, during which the patient
typically dies or recovers rapidly after appropriate volume replacement therapy [WHO,
1997]. Patients who survive the shock usually recover within 2 to 3 days [WHO, 1997].
20

Figure 1-7 Range of dengue disease.
(a) Dengue infections can have a wide range of clinical manifestations as shown in the generalized time course of events associated with DF, DHF and DSS. (b) WHO guidelines require each of the four criteria to be met for a diagnosis of DHF. The DHF patient can be further categorized to different grades (I-IV) if the corresponding criteria are met. Adapted from Nat Rev Microbiol 5(7): 518-528, 2007.
21

1.2.3. Current treatment/dengue control
Asymptomatic infections and undifferentiated febrile illnesses are the most common
outcomes of DENV infection in children and can represent more than 50% of cases
[Burke et al., 1988; Endy et al., 2002]; while DENV infection in adults are more likely to
be symptomatic [DeTulleo and Kirchhausen, 1998; Seet et al., 2005]. DF is frequently
self-limiting although it can be incapacitating. Currently, there is no vaccine or specific
therapy for dengue and all treatment is based on assessment by medical practitioner,
usually symptomatic and supportive. Severe disease (i.e., DHF or DSS) is rare but may
be fatal. Without proper, timely treatment, case fatality can exceed 30% in those with
severe disease; however, with intensive supportive fluid replacement therapy, case
fatality can be reduced to <1% [WHO, 2009]. Maintenance of the circulating fluid
volume and hemodynamic status is the central feature of severe case management.
Without an effective vaccine or anti-viral drug against DENV, the only way to control the
disease is to limit the exposure of susceptible people to the vectors. Many vector control
approaches are employed. These include the use of different larvicide, adulticide,
insecticide-treated materials and genetic strategies to reduce or block viral transmission
by mosquitoes. One potentially interesting and useful method is the use of Wolbachia, an
endosymbiotic bacterium, to deliver disease-resistance genes into mosquitoes, thereby
making them refractory to the human pathogens they transmit. Recently, two groups
independently demonstrated that introduction of Wolbachia into Aedes aegypti directly
inhibits DENV from infecting the mosquito and this may be due to the priming of
mosquito innate immune response and competition for the limited cellular resources
22

[Bian et al., 2010; Moreira et al., 2009]. This may serve as an eco-friendly option to
human pathogen disease control.
1.2.4. Vaccine development and progress
With dengue re-emerging and emerging in more than 100 countries, causing a burden to
the economy and claiming lives [Halstead, 2008b], a dengue vaccine is urgently needed.
Experiments to produce attenuated DENV as vaccines started way back in the early
twentieth century, where Sabin and Schlesinger reported that several passages of DENV
in mice changed the virus markedly that it can be used as vaccine to produce immunity
against dengue [Sabin and Schlesinger, 1945].
Now at the beginning of the 21st century, a pipeline of vaccine candidates does exist. The
leading candidate vaccine in clinical trials at present is the ChimeriVax dengue vaccine
(Sanofi Pasteur, Lyon, France). This live attenuated vaccine (LAV) uses the yellow fever
17D live attenuated vaccine strain as its genetic backbone with the yellow fever envelope
(E) and pre-membrane (prM) genes replaced with those from each of the 4 DENV
serotypes. LAVs most closely mimic a natural infection and thus are expected to induce
durable cellular and humoral immune responses. In order to be successful, they have to
be adequately attenuated yet maintaining high immunogenicity. The viruses must be able
to replicate well to evoke an immune response but sufficiently restricted in systemic
replication so that no transmission of the viruses by mosquitoes can occur. Importantly,
the attenuation has to be stable and should not revert to wild type or other virulent forms.
Antibody responses to the 4 components in the tetravalent formulation should not
interfere with each other as that could result in preferential antibody responses to the
23

better replicating components, leading to disease susceptibility [Blaney et al., 2006;
Blaney et al., 2005]. Unfortunately, these issues cannot be assessed accurately before
clinical trials due to the lack of a good animal model that reproduce the disease in human
beings. Nonetheless, these Sanofi chimeric viruses have been shown to be attenuated,
safe in non-human primates and phases I and II clinical trials, and elicit antibodies only to
dengue [McGee et al., 2008; Monath, 2007]. Other chimeric LAVs in clinical trials
include one that uses PDK-53 DENV 2 Mahidol vaccine candidate and another that uses
DENV-4 attenuated by 30-nucleotide deletion of the 3’ untranslated region as the genetic
backbone, with the E and prM genes replaced by that from the other DENVs [Blaney et
al., 2008; Blaney et al., 2007; Durbin et al., 2006; Durbin et al., 2005; Huang et al., 2003;
Rabablert et al., 2007].
Besides the LAVs, other vaccines in preclinical stages of development include subunit,
inactivated, DNA and vectored vaccines. Whole inactivated vaccines would have an
advantage over LAVs as it is impossible for the inactivated viruses to revert back to a
more pathogenic phenotype, thus making them safe for the immunocompromised. It
might be also be easier to attain a balanced antibody response against all 4 serotypes.
However, as they are also less likely to induce a robust and durable immune response,
multiple dosing or development of potent adjuvants would be required. These would
make the vaccines expensive and thus a less attractive option. A list of current vaccine
candidates is shown in Table 1-1.
As the pathogenic mechanism of DF/DHF remains incompletely understood, especially
with the possibility that cross-reacting antibodies could enhance DENV infection
(discussed below), these candidates still face many hurdles in safety and efficacy testing
24

25
before a safe, effective and affordable tetravalent dengue vaccine would be available on
the market.

26

27
Different approaches taken by various institutes and companies to develop a dengue vaccine. The list above shows the progress of the candidate vaccines. Adapted from Lancet Infect Dis 9(11): 678-687, 2009.
Table 1-1 Current ongoing vaccine developments.

1.3. Life cycle of DENV
1.3.1. Structure/genome of DENV
DENV is an enveloped, positive strand RNA virus, with a diameter of approximately
50nm. Its structure consists of an external icosahedral scaffold of 90 envelope
glycoprotein (E) dimers protecting the nucleocapsid shell, which contains the
approximately 10.7 kb RNA genome (Fig. 1-8a) [Kuhn et al., 2002]. The genome
translates into a large polyprotein encoding the three structural proteins (capsid,
envelope, pre-membrane/membrane) and 7 non-structural (NS) proteins (NS1, NS2A,
NS2B, NS3, NS4A, NS4B, NS5), that will be co- and post-translationally cleaved by
cellular and viral proteases [Lindenbach et al., 2005; Pastorino et al., 2010]. Figure 1-8b
summarizes the known functions of the different viral proteins.
28

(a)
(b)
Figure 1-8 Structure and proteome of DENV.
(a) Structure of whole virus showing each monomer of the E protein with domains I, II and III in red, yellow and blue respectively. (b) Flavivirus genome and polyprotein organization. The genome is translated into a single polyprotein which is co- and post-translationally cleaved by cellular and viral proteases to give 3 structural and 7 non-structural proteins with different functions. Adapted from Cell 108(5): 717-725, 2002 and Antiviral Res, 2010.
29

1.3.2. Entry and exit of DENV
Much of the knowledge on the mechanism of flavivirus entry into cells was built on work
done by Gollins and Porterfield with West Nile virus (WNV) on a murine macrophage-
like cell line, P388D1 [Gollins and Porterfield, 1985; Gollins and Porterfield, 1986a;
Gollins and Porterfield, 1986b; Kimura et al., 1986]. The virus first attaches itself to the
host cell via its attachment receptor and gets internalized through receptor-mediated
endocytosis using the same or another cellular receptor [Chu and Ng, 2004; Lindenbach
et al., 2005]. Once in the endosomes, the acidic environment triggers major conformation
changes in the E proteins, resulting in the fusion of viral membrane with the endosomal
membrane. The nucleocapsid containing the viral genome then exits the endosome into
the cytoplasm where viral replication follows. Newly formed, immature viral particles,
bud into the endoplasmic reticulum, travel through the trans-Golgi network and before
release from the cell by exocytosis, the prM protein is cleaved by host furin and the E
proteins are rearranged into homo-dimers to become mature virion (refer to Fig. 1-9).
Many molecules have been implicated in DENV entry process; however, none was
confirmed to be the cellular receptor for the virus (Table 1-2). Among them are putative
attachment/cellular receptors such as dendritic cell-specific intercellular adhesion
molecule (DC-SIGN, CD209), mannose receptor, heparin-sulphate, human monocyte-
derived macrophage membrane proteins of the following molecular weights: 27, 45, 67,
87kDa, heat shock proteins 70 and 90, CLEC5A and CD14-linked molecule [Cabrera-
Hernandez and Smtih, 2005; Chen et al., 2008; Lindenbach et al., 2005; Miller et al.,
2008; Moreno-Altamirano et al., 2002].
30

The primary targets of DENV infection in humans are the cells of mononuclear
phagocytic lineage including monocytes, macrophages and dendritic cells (DCs)
[Blackley et al., 2007; Halstead et al., 1977; Kou et al., 2008; Marovich et al., 2001;
Tassaneetrithep et al., 2003; Wu et al., 2000], although B cells, T cells, endothelial cells,
hepatocytes, and neuronal cells have also been reported to be permissive to DENV
[Clyde et al., 2006]. However, most of the studies on viral entry have been conducted on
cell lines of non-human origin or not from a mononuclear phagocytic lineage (e.g.
P338D1, murine macrophage-like cell line, Vero, African green monkey kidney epithelial
cell line). Hence, while these studies have provided valuable information on the
replication life cycle of flaviviruses, the in vivo relevance pertaining to human beings in
the event of natural dengue infection remains uncertain.
31

Figure 1-9 Replication lifecycle of Flavivirus.
The virus first enters the cell through receptor-mediated endocytosis and the acidic environment in the endosomes trigger major conformational change in the E proteins, resulting in fusion of viral membrane with endosomal membrane, hence the release of RNA genome into cytoplasm where replication begins. Virus assembly happens in the ER and the immature virion is then transported through the secretory pathway. The prM is cleaved by furin just before budding of the mature virion. Adapted from Antiviral Res 81(1): 6-15, 2009.
32

33

34

35

36
Table 1-2 Principle host factors in dengue. Many host proteins have been identified to play a role in flavivirus infection. The above table gives a summary of the host proteins, their known cellular functions and their possible roles in viral infection/replication. Adapted from Antiviral Res, 2010.

1.4. Role of host immune response in dengue
1.4.1. T cells
Findings from key epidemiological studies indicated that secondary infections of a
different serotype that is different from the primary infection in dengue-immune patients
often lead to more severe disease [Guzman et al., 2000; Sangkawibha et al., 1984]. A
possible explanation offered for the more severe disease outcome is the original antigenic
sin of the T cells [Mongkolsapaya et al., 2003]. Memory cells have a lower threshold for
activation compared to naive cells, hence, cross-reacting memory T cells from the
primary infection may be rapidly mobilized during the course of a secondary infection
[Veiga-Fernandes et al., 2000]. However, as they may be of lower affinity for the
secondary challenge antigen, they are less effective at clearing the secondary infection
[Mongkolsapaya et al., 2006]. They may instead promote immunopathology with
increased secretion of proinflammatory cytokines, leading to a more severe disease, but
die through apoptosis before clearing the infection [Dong et al., 2007; Mongkolsapaya et
al., 2006].
1.4.2. ADE
The more widely studied model to explain the association between secondary infection
and severe dengue is antibody-dependent enhancement (ADE) of DENV infection. Pre-
existing antibodies to a specific antigen have always been thought to be beneficial in
clearing an infection. This forms the basis of vaccination. However, in the case of
dengue, presence of heterotypic antibodies during a secondary infection different from
previous encounter or waning levels of maternally transferred anti-dengue antibodies in
37

infants born to dengue-immune mothers have also been associated with more severe
disease [Halstead and O'Rourke, 1977; Kliks et al., 1988; Simmons et al., 2007]. These
observations provided indirect but strong support for ADE. In this hypothesis, antibodies
generated from a previous infection may not neutralize a heterologous DENV serotype in
a secondary infection. Instead, they enhance viral infection by targeting the virus to Fc
receptor-bearing cells through the Fc portion of the antibody, thus bringing the virus
closer to the cell membrane where it can be internalized through Fc receptor-mediated
phagocytosis. As the antibody is non-neutralizing, it is incapable of stopping viral
replication, thus leading to increased viral burden and a more severe disease. Kliks et al
further tested pre-infection serum specimens collected during a prospective study of
dengue infections among school children in Bangkok for their ability to enhance DENV 2
growth in human monocytes in vitro and showed that greater enhancement of virus
growth correlated with symptomatic disease outcome in natural secondary infection
[Kliks et al., 1989], hence providing direct evidence that ADE in monocytes is central to
pathogenesis of DHF.
A recent prospective, nested, case-control study of dengue in infants in Philippines,
however, showed that infants exhibit a full range of disease severity after primary
infections and did not find any association between enhancing antibodies and the
development of DHF in the infants [Libraty et al., 2009]. This study even suggested that
rethinking or refinement of the current ADE pathogenesis model for infant DHF is
necessary and should stimulate new directions of research into mechanisms responsible
for the development of DHF. However, this difference in the ADE observations may be
due to differences DENV serotypes. Whereas the studies by Kliks et al (1988) and
38

Simmons et al (2007) involved DENV 2, DENV 3 was the predominant virus in the study
by Librarty et al (2009). Epidemiological observations have previously suggested that
DENV 2 and 4 cause more symptomatic disease in secondary infection compared to
DENV 1 and 3 [Henchal et al., 1982].
1.4.3. Others risk factors of disease severity?
It is also possible that both theories can work in conjunction in a secondary infection to
produce the increased viral load and heightened immune response that led to
pathogenesis of DHF/DSS. Beside these two well-received theories used to explain the
more severe disease experienced in certain individuals, recently more studies have
revealed many other factors that might influence the disease outcome. These include the
genetic background of an individual (e.g. race, HLA polymorphism and G6PD
deficiency), virus strain differences, levels of virus circulating in individual during acute
phase and nutritional status of the infected individual can all play a role in affecting the
severity of the infection [Chao et al., 2008; de la et al., 2007; Diamond et al., 2000;
Maron et al., 2010; Stephens, 2010; Thisyakorn and Nimmannitya, 1993; Vaughn et al.,
2000].
1.4.4. Antibody neutralization of DENV
The above review of the current state of knowledge of dengue vaccine development
underscores the need for mechanistic studies into the molecular basis for virus
neutralization. Antibodies generated against DENV are protective but are serotype-
specific. Individuals with previous dengue infection are protected against re-infection
with the same serotype and the anti-dengue antibodies can be detected in the serum even
39

after several decades post infection [Imrie et al., 2007; Russell et al., 1967; Sabin, 1952].
Homotypic immunity is thought to be lifelong with a brief period of cross-protection.
Hence it is important to understand the mechanistic difference between neutralization by
a homotypic antibody and a cross-reacting antibody, and steer the immune system away
from producing cross-reacting antibodies.
Antibody-mediated neutralization of animal viruses is generally thought to occur by a
‘multi-hit’ mechanism. In this model, it is postulated that virus inactivation is a function
of the number of antibodies bound to it [Della-Porta and Westaway, 1978; Pierson and
Diamond, 2008; Pierson et al., 2008]. Neutralization requires the engagement of the
virion by antibody with a stoichiometry that exceeds a particular threshold.
Concentrations of antibody that falls below this threshold or antibodies that can never
reach this threshold, even at maximal binding, will enhance infection in vitro in Fc
receptor bearing cells. Since all neutralizing antibodies can promote ADE in vitro at sub-
neutralizing concentrations, it is likely that DENV neutralization follows the ‘multi-hit’
model. Indeed, this has been demonstrated with the neutralization of DENV 2 by E
domain III specific antibody [Gromowski et al., 2008].
Many potent neutralizing monoclonal antibodies (mab) against flaviviruses have been
mapped to the lateral ridge of E domain III (e.g. 3H5: DENV 2 specific, E16: WNV)
[Oliphant et al., 2005; Sukupolvi-Petty et al., 2007]. However, these are murine
antibodies. Studies of human dengue immune sera demonstrated that E domain III
antibodies are present, but in low percentages and make only a minor contribution to the
total neutralizing capacity of human immune sera [Wahala et al., 2009]. Another study in
Taiwan revealed that antibodies to DENV during a natural course of infection are
40

predominantly cross-reactive and recognized epitopes containing residues at the fusion
loop of E domain II [Lai et al., 2008]. While an interrogation of the memory B cells from
7 individuals with previous DENV infection showed that 60% of the antibodies was
directed towards prM and the other 40% towards E [Dejnirattisai et al., 2010]. None of
the anti-prM antibodies showed high neutralization in this study, but can instead render
non-infectious immature or partially mature DENV infectious in Fc receptor-bearing
cells. These suggest that human antibody repertoire against DENV may be directed
towards the less neutralizing, more cross-reactive, but immunodominant epitope
involving the highly conserved fusion loop residues at domain II and prM, away from the
more potent type-specific neutralizing epitope at domain III. Eliminating or altering
specific DI/DII and prM epitopes may be a way to redirect antibody responses toward the
more protective neutralizing epitopes in DIII.
1.4.5. Monocytes/Macrophages
To mediate viral clearance, the Fc portion of the antibody-bound DENV immune
complexes presumably needs to interact with its Fc receptors (e.g. Immunoglobulin G,
IgG, interacts with Fc gamma receptors, FcγR). FcγRs comprise of multigene family of
integral membrane glycoproteins that upon aggregation by complexed IgG, exhibit
complex activation or inhibitory effects on cell function [Nimmerjahn and Ravetch,
2008]. Two activatory FcγRs are of particular concern in mediating immune complex
clearance: FcγRI (CD64) and FcγRIIa (CD32). FcγRI has high affinity for monomeric
and complexed IgG and is found exclusively on antigen-presenting
monocytes/macrophages and dendritic cells. FcγRIIa preferentially binds multimeric IgG
with at least 100 fold lower affinity than FcγRI, and is more broadly distributed among a
41

variety of myelogeneous cell types. FcγRI needs to associate with γ-chain subunits that
consist of immunoreceptor tyrosine-based activation motif (ITAM) for signaling whereas
FcγRIIa has its own ITAM constitutively expressed in the cytoplasmic tail.
Monocytes/macrophages bear both FcγRI and FcγRIIa and are the main mediators of
immune clearance. Macrophages are the first line of defence as they coat and monitor the
main body compartments. Monocytes circulate in the bloodstream and extravasate into
tissue to develop into macrophages or dendritic cells, in absence or presence of tissue
damage or infection, as part of their maturation process or part of an innate immune
response. Monocytes may also respond to pathogens they encounter while still in the
circulatory system. As sentinels of the immune system, they may encounter the virus
early and control viral entry into its target organ(s). However, the situation is made
complicated as they are also believed to be the primary sites of DENV replication
[Halstead, 1988; Kou et al., 2008]. Thus, it is important to understand their contribution
to pathogenesis as well as viral clearance.
Both FcRs have been shown to play a prominent role in mediating neutralization and
enhancement of DENV infection [Kou et al., 2008; Rodrigo et al., 2009; Rodrigo et al.,
2006; Schlesinger and Chapman, 1999]. Kou et al had an interesting finding that blocking
of either FcγRI or FcγRIIa on primary monocytes isolated from dengue-naive donors led
to approximately 40-60% reduction of ADE of dengue infection, while blocking both Fc
receptors had no significant additive effect. On the other hand, Rodrigo et al reported that
FcγRIIa is more efficient than FcγRI in enhancing dengue infection in COS cells
transiently transfected with either FcγRI or FcγRIIa. In this study, mutations of FcγRIIa
ITAM that abolish its signaling competency impaired phagocytosis, but unlike with
42

signaling-incompetent FcγRI (γ-chain ITAM mutants), immune enhancement appeared to
be unaffected. Ligand-clustered FcγRs are known to concentrate in cell membrane
regions rich in signaling molecules and potential virus receptor engagement sites. It is
thus possible that FcγRIIa, which preferentially binds immune complexes and exhibit a
high dissociation rate, is better equipped than FcγRI to utilize the alternative signaling
pathways and entry mechanisms made available by relocation to such sites where weakly
bound immune complexes might be more easily transferred to favorable entry pathways.
Because of their relative affinities to the two FcγRs, different subclasses of IgG also
modulate the neutralization of DENV in a different manner [Rodrigo et al., 2009].
There are also fundamental differences in the intracellular signaling pathways, receptor
trafficking and antigen processing/presentation stimulated by the FcγRI and FcγRIIa [Dai
et al., 2009] that can possibly lead to the differential neutralization/enhancement of
dengue infection. The two FcγRs utilizes different phospholipases for activation: FcγRI
uses phospholipase D1 and FcγRIIa uses phopholipase Cγ1. Immune complexes
internalized by FcγRI traffic to MIIC compartments (HLA-DM and LAMP-1 positive)
for processing whereas FcγRIIa traffics to compartments negative for HLA-DM and
LAMP-1 at equivalent time period, suggesting that they do not traffic to MIIC. It is also
noted that FcγRI activation elicits the production of higher levels of pro-inflammatory
cytokines, eg. TNF-α, IL-8, IL-6, IFN-γ, G-CSF and MIP-1β, which suggests that FcγRI
plays a dominant role over FcγRIIa in augmentation of inflammation and immunity.
43

1.5. Discovery and use of fluorescent proteins in research
1.5.1. Discovery of GFP: short story of Aequorin, O Shimomura
In 1960, Osamu Shimomura left Japan with a Fulbright Fellowship to embark on his
journey to understand the photophysics involved in Aequorean bioluminescence in the
laboratory of Prof. Frank Johnson at Princeton University, little knowing that this piece
of work which took more than twenty years of dedication, would transform into one of
the most useful tools in modern biology and medicine. He successfully isolated aequorin
and its companion protein, a fluorescent protein that absorbs the blue light emitted by
aequorin (lmax = 470 nm) through radiationless (Förster-type) energy transfer, and
fluoresces green (lmax = 509 nm), from the luminous jellyfish Aequorea. This protein is
later called green fluorescent protein (GFP). He also managed to solve the chromophore
structure of GFP. And it was this effort that won him the Nobel Prize in Chemistry for
2008, which he shares with Martin Chalfie and Roger Tsien, who first demonstrated that
it is possible to replace the original protein with GFP-fusion protein, thereby showing
where in the cell the protein resided in C. elegans and Drosophila oocytes, and expanded
the colour palette of fluorescent proteins by mutation, respectively [Chalfie et al., 1994;
Heim et al., 1994; Wang and Hazelrigg, 1994; Zimmer, 2009].
1.5.2. From GFP to rainbow coloured fruit proteins
In the same year Chalfie reported the work on GFP-fusion proteins, Roger Tsien reported
that the autocatalytic chromophore formation in GFP was oxygen dependent and
proposed the biosynthetic pathway for chromophore formation [Heim et al., 1994]. He
also described the creation of the first wavelength mutation of GFP and proposed the
44

possibility of utilizing fluorescence energy transfer (FRET) measurements between GFP
and its mutants, such as the newly created blue fluorescent protein (BFP = a Y66H GFP
mutant) [Heim et al., 1994]. And since then, the race has been on to produce better and
brighter fluorescent proteins to fill in every gap in the different spectral classes [Shaner et
al., 2007]. From a single green fluorescent protein in the 1970s, the colour palette has
expanded to rainbow colours of mFruit proteins (Fig. 1-10) and beyond. The search for
new fluorescent proteins is an endless quest as researchers look at isolating new
fluorescent proteins from marine animals or by mutating existing proteins to derive at
new proteins with improved properties such as brightness, photostability and Stokes shift.
A new generation of fluorophores, the Alexa Fluor dyes, are synthesized by Molecular
Probes, through the sulfonation of coumarin, rhodamine, xanthene (such as fluorescein),
and cyanine dyes. Sulfonation makes Alexa Fluor dyes negatively charged and
hydrophilic. Alexa Fluor dyes are generally more stable, brighter, and less pH-sensitive
than common dyes (e.g. fluorescein, rhodamine) of comparable excitation and emission
[Panchuk-Voloshina et al., 1999], and to some extent the newer cyanine series [Berlier et
al., 2003], making them more ideal for imaging studies.
45

Figure 1-10 mFruit fluorescent proteins derived from mRFP or somatic hypermutation (SHM). Adapted from Chem Soc Rev 38(10): 2823-2832, 2009.
46

1.5.3. Fluorescent proteins in research
Fluorescent proteins have gained increasing popularity over the years as they require no
additional substrate or conenzymes to fluoresce, and rarely causes phototoxicity in living
cells or whole organisms. Their applications as tools in biological researches are
numerous, including as minimally invasive markers to track and quantify individual or
multiple protein species, as probes to monitor protein-protein interactions, as photo-
modulatable proteins to highlight and follow the fate of specific protein populations
within a cell, and as biosensors to describe biological events and signals [Lippincott-
Schwartz and Patterson, 2003; Zhang et al., 2002].
Along with the continued advances in genetically fine-tuning the properties of fluorescent
protein variants to increase brightness levels, improve photostability, larger Stokes shift,
better expression and maturation in mammalian cells, it is important for the modern
imaging technology to keep pace with these developments. More sensitive and quicker
camera systems, filter systems for optimally detecting different fluorophores, superior
software for quantifying and discriminating fluorescent signals, and hardware for
photobleaching and photoactivating geometrically defined spatial patterns are desirable
[Lippincott-Schwartz and Patterson, 2003].
Greater understanding on how the fluorescent proteins work also led to their creative
usage in labelling by researchers, and with combination of different microscopy
techniques to reveal exciting new insights into the biology of living cells. Not only are
fluorescent proteins used in elucidating the protein dynamics of cells, they are also very
useful in visualizing pathogen-host interactions in cells. Van der Schaar et al interestingly
47

used a lipophilic dye, 1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine, 4-
chlorobenzenesulfonate salt (DiD), to fluorescently label DENV for tracking of the early
cellular entry process [van der Schaar et al., 2008; van der Schaar et al., 2007]. Using this
method, they were able to track single virus particles and characterize the dynamics of
viral transport in the cell.
1.6. Gaps in knowledge and Hypothesis/Objectives of this study
As most endemic countries have 4 circulating serotypes of DENV, the burden of
epidemic DF and DHF remains very high until a safe tetravalent dengue vaccine becomes
available. In order to overcome the challenges in vaccine production, a reliable surrogate
marker for immunity needs to be identified. Plaque reduction neutralization tests (PRNT)
have been the gold standard for determining the serotype that an individual was exposed
with previously in an uncomplicated scenario. However, distinguishing the actual
serotypes a person was infected with previously may not be that straightforward in the
event of secondary infection or immunization with Japanese encephalitis or yellow fever
virus, due to presence of cross-reactive antibodies. Greater insights into how homotypic
or cross-reacting antibody neutralizes the virus would provide the much needed correlates
of immune-related protection to the vaccines and would expedite the development of new
vaccines.
Although the exact pathogenesis of DENV remains unclear to date, monocytes appear to
have a central role in the clearance of antibody-virus immune complexes. It is thus to the
interest of our laboratory to understand antibody-mediated neutralization or enhancement
of DENV through the early interactions of antibody-DENV complexes with monocytes
48

and subsequently tease out the mechanisms that lead to the different outcomes of dengue
infection. Equipped with that knowledge, we can then develop an assay that allows us to
differentiate protective homotypic antibodies from the less protective cross-reacting
antibodies. This would provide the much needed test to accelerate vaccine development.
In the scope of this thesis, we will be focusing on antibody-mediated neutralization of
DENV. Preliminary findings from our laboratory using DiD labelled DENV and 2 well-
characterized mabs, namely 3H5 and 4G2 [Henchal et al., 1982; Henchal et al., 1983]
show that DENV complexes formed with minimally neutralizing amounts of homotypic
3H5 antibody interact with THP-1 cells (monocytic cell line) differently compared to that
of cross-reactive 4G2 antibody. As the minimally neutralizing concentrations of 3H5
(3µg/ml) and 4G2 (100µg/ml) are very different, we hypothesized that at these
concentrations of antibodies, the complexes formed interact differentially with the
monocytes, possibly through the engagement of different Fc receptors, and hence
different fates along the endocytic pathways. We also hypothesized the differential
interaction with the Fc receptors could be due to different complex sizes formed at the
different antibody concentrations.
To test our hypotheses, we have four specific aims.
Specific aims:
1) Fluorescent label DENV
In our previous study, a lipophilic fluorescent dye DiD labelled DENV was used to
visualize its trafficking into endosomal pathway. However, as the DiD dye is largely
quenched when in close-proximity to each other (e.g. on virus membrane), it is difficult
49

to look at early virus-cell interactions prior to fusion (where the lipids would then be
diluted out and fluorescence increases dramatically). Freezing and thawing of DiD-
labelled virus might also cause the leaking of the dye from the virus membrane, hence the
labelled virus can only be kept at 4°C for a short period of time. Freshly labelled virus
needs to be prepared for each experiment. Thus it is necessary to explore fluorescence-
labelling of DENV with another dye that is unquenched extracellularly and with stable
conjugation to the virus so that a large batch of labelled virus can be made and use in
multiple experiments for consistency.
Alexa Fluor dyes are a natural choice as they are more photostable and less sensitive to
low pH, thus making them ideal to study endocytic trafficking. They also come
conveniently in amine-reactive esters that conjugate permanently to the proteins.
However, as they bind to the protein part of the virus, most likely to the abundant E
proteins on the surface, careful optimization is necessary to ensure that labelled virions
remain infective. Hence the first part of the MSc project was aimed at fluorescence
labelling with Alexa Fluor dyes and characterizing the labelled virions.
2) Visualizing the fate of antibody-DENV complexes in monocytes
We next sought to visualize the early interactions of the complexes with monocytes and
their subsequent sub-cellular localization through confocal microscopy, using the two
mabs and the Alexa Fluor labelled DENV in THP-1 cell line. The fate of the antibody-
DENV complexes in THP-1 cells through the early endosomal and late endosomal
network, using EEA-1 and LAMP1 as their respective markers, would be tracked and
compared in a time course manner up to 2 hours. These experiments will be repeated on
50

primary monocytes to verify that the observations can also be reproduced in primary
cultures.
3) Fc receptor engagement
Monocytes normally express more than one Fc receptor, mainly FcγRI and FcγRII, on
their surfaces. Antibody-antigen complexes interaction with Fc receptor bearing cells is
most likely through the Fc receptors. As such, one possible explanation for any
differential interaction with the monocytes would be different Fc receptor engagement by
the complexes. Hence, we would next elucidate the Fc recptor utilization by complexes
formed with 3H5 or 4G2 mabs using confocal microscopy and this will shed light on any
preferential Fc receptor employment in an environment where both FcγRs are
simultaneously expressed.
4) Concentration and complex sizes
Being bivalent antibodies, IgGs are capable of binding to and cross-linking antigens into
aggregates. Therefore, to test whether different antibody concentrations bring about
DENV complexes of different sizes which could potentially influence the Fc receptor
interactions on monocytes surface, we would determine and compare the sizes of DENV
complexes formed at high (100µg/ml) and low (3µg/ml) of antibody.
51

52

Chapter 2
Materials and Methods
Zhang Lixin (HT080076A)


2. Materials and methods
2.1. Cell culture
All cells and hybridomas were obtained from American Type Culture Collection (ATCC,
Manassas, Va), and all cell culture media and supplements were purchased from Gibco
(Invitrogen, Singapore). Baby hamster kidney BHK-21 cells were maintained in Roswell
Park Memorial Institute 1640 medium (RPMI-1640), supplemented with 10% fetal
bovine serum, penicillin (100U/ml) and streptomycin (100µg/ml) at 37°C with 5% CO2.
African green monkey kidney Vero cells were maintained in Media-199 (M199)
supplemented with 10% fetal bovine serum and 4mM L-glutamine at 37°C with 5% CO2.
Raji B cells transfected with DC-SIGN was a kind gift from Timothy H. Burgess (Naval
Medical Research Centre, US) and maintained in RPMI medium supplemented with 10%
fetal bovine serum at 37°C with 5% CO2. THP-1 cells were maintained in RPMI-1640
supplemented with 0.05mM 2-mercaptoethanol and 10% fetal bovine serum at 37°C with
5% CO2. Mouse anti-dengue virus serotype 2 E protein monoclonal antibody hybridoma,
3H5 (ATCC: HB46) and mouse anti-flavivirus E protein antibody hybridoma, 4G2
(ATCC: HB112), were maintained in RPMI-1640 supplemented with 10% fetal bovine
serum.
2.2. Primary monocytes culture
Venous blood from the principal investigator was collected in BD sodium heparin
vacutainers (Biomed Diagnostics, Singapore). The blood was then diluted with 2 volumes
of 0.5% bovine serum albumin (BSA, Sigma Aldrich, Singapore) in phosphate buffered
solution (PBS, 1st Base, Singapore) (0.5% PBS/BSA), and carefully layered onto Ficoll-
55

hypaque (GE healthcare, Singapore). The blood was then subjected to centrifugation at
750 x g, without brakes. The interphase cells containing the peripheral blood
mononuclear cells (PBMCs) were aspirated and transferred to a clean tube. The PBMCs
were washed 3 times with 0.5% PBS/BSA and resuspended in growth medium (RPMI-
1640 supplemented with 10% fetal bovine serum, 100U/ml penicillin and 100µg/ml
streptomycin). The cells were then counted and seeded into T25 tissue culture flasks
(NUNC, Bio Laboratories, Singapore) at 1x107 per flask. The cells were incubated at
37°C, 5% CO2 for 2.5hrs to allow the monocytes to adhere to the flask surface. The
adhered cells were washed 5 times with PBS to remove the non-adherent lymphocytes,
and replenished with fresh growth medium. These monocytes were allowed to recover
overnight at 37°C, 5% CO2 before use in experiments.
2.3. Antibodies
Mouse anti-human EEA1 and LAMP1 antibodies (BD biosciences, Biomed Diagnostics,
Singapore) were used at 1:100 and 1:500 dilutions, respectively. Mouse anti-human
FcγRI (CD64, clone 10.1, eBioscience, Biomed Diagnostics, Singapore) and FcγRII
(CD32, clone IV.3, StemCell Technologies) antibodies were used at 1:1000 dilution.
Purified human IgG antibodies were purchased from Sigma Adrich, Singapore. Alexa
Fluor 488 (AF488) anti-mouse IgG and Alexa Fluor 647 (AF647) anti-human IgG
antibodies were purchased from Invitrogen and used at 1:100 dilution. Goat anti-human
IgG HRP antibody was purchased from Sigma Aldrich and used at 1:1000 dilution.
56

2.4. Virus culture and purification
Dengue 2 virus (ST) strain was first isolated from a clinical sample in Singapore General
Hospital using an Aedes albopictus mosquito cell line C6/36 and subsequently
propagated in Vero cells. The culture supernatant was harvested when 75% or more of
the cells showed cytopathic effect and clarified by centrifugation at 1000g for 30min at
4°C. Virus in the resulting supernatant was concentrated by centrifugation at 30,000g for
2 hours at 4°C. The pellet was resuspended in 5mM Hepes, 150mM NaCl, 0.1mM EDTA
(HNE buffer, pH7.4) and further purified over 30% sucrose cushion by
ultracentrifugation in Beckman SW41Ti rotor at 80,000g for 15 hours at 4°C. Purified
virus was resuspended in HNE buffer and stored in 100ul aliquots at -80°C. A limiting
dilution plaque assay was performed on BHK cell line to determine the viral titre in
plaque forming units per millilitre (pfu/ml).
2.5. Plaque assay
Ten-fold serial dilutions of virus were added to BHK-21 cells in 24 well plates and
incubated for 1hr at 37oC, with gentle rocking every 15 minutes. Media was then
aspirated and replaced with 0.8% methyl-cellulose in maintenance medium (RPMI-1640,
2% fetal bovine serum, penicillin and streptomycin). After 4 days at 37˚C, cells were
fixed with 25% formaldehyde and stained with 0.5% crystal violet. The plates were
washed, dried and the plaque forming units per ml (pfu/ml) calculated.
2.6. Virus labelling
For the initial experiment aimed at determining the optimal concentration of dye needed
to fluorescently label dengue virus, Alexa Fluor 594 succinimidyl ester (AF594SE,
57

Molecular Probes, Invitrogen) was reconstituted in dimethyl sulfoxide (DMSO) or 0.2M
sodium bicarbonate buffer, pH 8.3 (SBB) immediately before labelling, and added to
approximately 3x107 pfu dengue virus diluted in SBB at final concentrations of 10,
50,100, 200, 500 and 1000µM of dye, while stirring gently. The dye-virus mixture was
incubated at room temperature for 1 hour with gentle inversions every 15 minutes. The
labelling reaction was stopped by adding freshly prepared 1.5M hydroxylamine, pH 8.5
(Sigma-Aldrich, Singapore) and incubated at room temperature for 1 hour with gentle
inversions every 15 minutes. Labelled dengue virus was re-titrated after fluorescence
labelling and tested for fluorescence using immunofluorescence assay.
For subsequent AF594SE or AF488SE labelling involving a larger batch of virus, the dye
was dissolved directly in SBB and the same labelling approach was employed as above.
The labelled dengue virus was purified using Sephadex G-25 columns (Amersham, GE
Healthcare, Singapore) to remove the excess dye. The labelled virus was stored in 100ul
aliquots at -80°C, re-titrated and tested for fluorescence before use.
2.7. Immunofluorescence assay of viral infection
Equal volume of AF594-labelled dengue virus was incubated with Vero cells plated on
coverslips for 10 minutes at 37°C, washed, fixed with 3% paraformaldehyde (pFA,
Sigma-Aldrich, Singapore) and permeabilized with 0.1% saponin (Sigma-Aldrich,
Singapore). The cells were then incubated for 1 hr with centrifugation clarified
supernatant of 3H5 monoclonal antibody hybridoma culture, at room temperature. The
cells were washed three times in wash buffer (PBS containing 1mM calcium chloride,
1mM magnesium chloride and 0.1% saponin), followed by incubation with AF488 anti-
mouse IgG antibody for 45min at room temperature. Cells were then washed three times
58

in wash buffer, rinsed once in deionised water and mounted on to glass slides with
Mowiol 4-88 (Calbiochem, San Diego, CA) with 2.5% Dabco (Sigma-Aldrich,
Singapore). Cells were visualized at magnification x63 on a Leica Microsystem TCS SP5
confocal microscope and merged images exported for processing. Processing of the
images involved adjustment of the contrast on the images on a whole for clarity and
exported in individual colours for unmerged images using Adobe Photoshop CS3 version
10.
2.8. Flow cytometry determination of percentage of labelled dengue virus
Raji B cells transfected with DC-SIGN were counted, aliquoted, centrifuged and
resuspended in AF488-labelled dengue virus at multiplicity of infection (MOI) of 1 and
incubated for 10 minutes at 37°C, then fixed with 3% paraformaldehyde. The cells were
then washed 2 times with FACS buffer consisting of PBS with 0.1% fetal bovine serum
and permeabilised with 0.1% saponin in PBS. Cells were subsequently stained for the
presence of dengue virus using anti-E protein monoclonal antibody, 3H5, and PE anti-
mouse anti-IgG antibody before reading on BD FACS Calibur machine and analyzed
with CellQuest software, version 3.3 (Becton Dickinson).
2.9. Detection by SYBR green-based real-time PCR
Real-time detection of viral RNA was done as previously described ([Lai et al., 2007])
using pan-dengue forward and reverse primers with some modifications. Briefly, RNA
extraction was carried out using QIAamp viral RNA mini kit for purified non-labelled
and AF594-labelled dengue virus (AF594-DENV), or RNeasy mini kit (both from
QIAGEN, Hilden, Germany) for total RNA extraction from cells according to
59

manufacturer’s instructions. Complementary DNA was synthesized using SuperScript III
First Strand Synthesis System (Invitrogen, Singapore) following manufacturer’s
instructions for random hexamers. RNA copy number was then determined by SYBR
green-based real-time PCR on LightCycler 480 Real-Time PCR System (Roche
Diagnostics, Penzberg, Germany).
2.10. Growth kinetics
Vero cells are seeded at 2 x 105 per well in 24 well plates and allowed to adhere at 37°C
for 4 hours before infecting with MOI 0.1 of either purified dengue virus or AF594-
DENV for 1 hour at 37°C, with gentle rocking every 15 minutes. The cells were then
washed 2 times with 1ml per well PBS and replaced with maintenance medium (MM,
M199 with 2% FBS). Supernatant was harvested from wells and stored at -80°C for
plaque assay, and cells were washed 2 times in PBS and lysed with RLT buffer provided
in RNeasy mini kit at 0, 6, 8, 12, 24, 48 and 72 hours post adsorption. The cell lysates
were stored frozen until all samples were collected and subsequently extracted according
to kit’s protocol. Viral RNA was detected using real-time PCR.
2.11. Humanization of 3H5 and 4G2 mouse monoclonal antibodies
Chimeric forms of the mAb 3H5 (h3H5) and 4G2 (h4G2) containing mouse VH and VL
sequences and human γ1 and κ constant sequences were constructed as previously
described in Hanson et al [Hanson et al., 2006] by Angeline Lim.
2.12. Binding affinity ELISA
Nunc Maxisorp 96-well plate was coated with 100µl of 2x105 pfu/ml purified DENV
(labelled or non-labelled) overnight at 4°C. The plate was washed 5 times with PBS
60

containing 0.05% Tween-20 (PBST), with 1 min intervals, blotted dry and blocked for
1hr with 100ul of 1% skim milk in PBS at room temperature. The plate was washed 5
times with PBST, with 1 min intervals, before the addition of 50µl/well two-fold serial
dilutions of mouse 3H5/4G2 monoclonal antibodies or humanized 3H5/4G2 in 0.5% skim
milk in PBS (quadruplicates). The primary antibodies were incubated for 1hr at 37°C,
washed 5 times in PBST with 1 min intervals , before adding 25µl/well goat anti-human
IgG HRP for 1hr at 37°C. The plate was then washed 5 times in PBST and blotted dry.
100µl/well Tetramethylbenzidine (TMB) substrate was added and allowed colour to
develop in the dark before stopping with 50µl of 2M H2SO4. The OD was then read at
450nm.
2.13. Titration of h3H5/h4G2 to determine neutralizing concentrations on monocytes
Two-fold serial diluted h3H5 or h4G2 antibodies were allowed to complex with 2 x105
pfu of DENV 2 ST strain at 37°C before adding to 2 x 104 THP-1 cells or primary
monocytes (MOI 10). The culture supernatant was then harvested 72hrs post-infection for
plaque assay. The neutralizing concentrations of h3H5 or h4G2 on THP-1 cells or
primary monocytes were determined to be the highest dilution of antibodies that
produced no plaques.
2.14. DENV immune complex co-localization studies in monocytes
Neutralizing concentrations of h3H5 or h4G2, or neutralizing concentration of h3H5 with
the addition of purified human isotype control IgG to top up total antibody concentration
to 100 or 400µg/ml, were incubated with 10 MOI of AF594-DENV for 1hr at 37°C and
cooled on ice before adding to pre-cooled THP-1 cells or primary monocytes. The
61

mixture was further incubated for 15mins on ice to synchronize entry. The time course of
5, 10, 30, 60 and 120mins was started by transferring the mixture to 37°C, and ended by
transferring back to ice. The cells were then fixed in 3% pFA for 30mins on ice and
centrifuged on to positively-charged glass slides (Menzel-Gläser, Germany) at 800rpm
for 3min using a cytospin machine. The cells were then permeabilized with 0.1% saponin
and stained for early endosome marker (EEA1), late endosome/lysosome marker
(LAMP1), FcγRI or FcγRII at room temperature for 1hr. The slides were then washed 3
times with PBS containing 0.1% saponin and incubated with AF488 anti-mouse IgG and
AF647 anti-human IgG secondary antibodies for 45mins at room temperature in the dark.
The slides were washed 3 times and rinsed once in distilled water before mounting in
Mowiol 4-88 containing 2.5% Dabco. The cells were visualized at magnification x63 on
a Leica Microsystem TCS SP5 or Carl Zeiss LSM 710 confocal microscopes and merged
images exported for processing. Processing of the images involved adjustment of the
contrast on the images on a whole for clarity and exported in individual colours for
unmerged images using Adobe Photoshop CS3 version 10.
2.15. Sucrose gradient analysis of DENV immune complex sizes
Sucrose gradient was formed by careful layering of 10-60 sucrose solutions (in HNE
buffer) in 10% increments, starting with the densest at the bottom, in 13.5ml Ultra-Clear
tubes (Beckman Coulter, Singapore). The gradient was allowed to linearize overnight at
4°C.
Equal amounts of purified DENV were incubated with humanized antibodies at 3 or
100µg/ml h3H5 IgG or h3H5 Fab (h3H5 IgG that was enzymatically digested to give
62

only 1 arm of the Fab fragment) at 33.3µg/ml (mole equivalent amount of 100µg/ml
h3H5) or without antibodies (virus control) for 1hr at 37°C. The samples were then
carefully layered on top of the linearized 10-60% sucrose gradient, and centrifuged
overnight at 25,000rpm at 4°C, in SW41Ti rotor for 17hrs. Each gradient was then
harvested in 0.25ml fractions in a drop wise manner from the bottom of the tube.
Subsequently, the viral RNA was extracted from each fraction and quantified by real-
time PCR.
2.16. Dynamic light scattering (DLS) analysis of DENV immune complex sizes
Individual fractions from section 2.16 were also subjected to dynamic light scattering to
determine the diameter of the complexes formed by different concentrations of the
antibodies. 20µl of each fraction was loaded into Quartz cuvette for analysis by Zetasizer
Nano S machine (Malvern Instruments, UK) at 37°C using 50% sucrose in HNE buffer as
the dispersant. Data was then analysed using Zetasizer Nano software version 6.01. The
diameter reports generated were average of more than 10 readings.
2.17. Statistical analysis
Statistical analyses were performed using unpaired Student’s t test on Graphpad Prism 5
software. P value < 0.05 was considered significant.
63

64

Chapter 3
Results
Zhang Lixin (HT080076A)


3.1. Producing fluorescent DENV
Results reported in this section has been published in Zhang et al, J Virol Methods 2010;167:172-7.
3.1.1. Alexa Fluor labelling of DENV
To visualize the early interactions of DENV-antibody complexes with cells, we sought to
label DENV with a suitable fluorophore. DENV surface has several identical protein
subunits and can therefore be labelled at multiple sites using an amine reactive dye; the
resulting fluorescence intensity may be sufficient to track the virus under a fluorescence
microscope. Hence the goal here was to develop a simple procedure of labelling DENV
with a fluorescent Alexa Fluor succinimidyl ester that results in minimal loss of
infectivity post-labelling. Alexa Fluor dye was chosen as it has better photostability and
signal intensity compared to other common fluorophores such as fluorescein
isothiocyanate (FITC) or cyanine 3-bihexanoic acid (Cy3). In addition, Alexa Fluor dyes
are stable at a lower pH compared to the common fluorophores, which could be useful for
visualizing labelled viral particles as they move through the acidic compartments of
endosomes post-infection. Furthermore, the permanence of covalent conjugation of the
fluorophores to the surface proteins allows for storage of batch-labelled virus at -80°C,
providing uniformity across multiple experiments.
Labelling was initially carried out according to the protein labelling protocol supplied by
the manufacturer that requires the reconstitution of Alexa Fluor 594 succinimidyl ester
(AF594SE) in DMSO. To determine the optimum concentration of AF594SE to use for
labelling of DENV to obtain maximum fluorescence and minimum loss in viable virus
67

titre, a range of concentrations (10, 50, 100, 200, 500 and 1000μM) were tested. The
AF594 labelled-DENV (AF594-DENV) was then re-titrated by plaque assay (Fig. 3-1a).
Approximately 31- to 58-fold reductions in titre were obtained when DENV was labelled
with 10μM to 500μM of AF594SE, while DENV labelled with 1mM dye was reduced
approximately 796-fold compared to the virus control.
The infectivity of the labelled DENV was determined by immunofluorescence staining.
Vero cells seeded on coverslips were incubated with equal amounts of DENV labelled
with different concentrations of dye. The cells were then fixed, stained for E protein and
visualized using laser-scanning confocal microscope. The cells showed little to no
staining of E protein across the concentrations of AF594SE used (Fig. 3-1b). Taken
collectively, these data indicate that there was a significant drop in virus titre post
labelling. A plausible explanation is that DMSO has adverse effects on virus viability.
Hence, an alternative method to label DENV without the use of DMSO was sought.
Since the AF594SE dye is highly soluble in aqueous solution, we dissolved the dye
directly in the sodium bicarbonate labelling buffer (SBB), without DMSO. The
fluorescence labelling experiment was repeated with the same range of dye
concentrations, but this time with the AF594SE reconstituted in SBB. The titre of DENV
labelled with 10μM to 100μM of AF594SE dye showed approximately 15-fold reduction,
and DENV labelled with 500μM and 200μM AF594SE dye showed approximately 26 to
29-fold reductions, respectively, compared to equivalent number of non-labelled virus.
DENV labelled with 1mM of AF594SE dye showed 82-fold reduction in titre.
68

Using the modified labelling protocol, E protein can clearly be detected by
immunofluorescence staining in the cells inoculated with the AF594-DENV across the
range of dye concentrations used. However, its intensity diminished substantially for
DENV labelled with 500μM and 1mM of the dye, indicating a reduction in virus
infectivity post-labelling with higher concentrations of dye (Fig. 3-1c). This was
consistent with the plaque assay results. DENV labelled with 10μM and 50μM of dye
showed no discernable AF594 fluorescence, while DENV labelled with 100μM and
200μM of AF594SE dye displayed high AF594 fluorescence which co-localized well
with the E protein staining. Only weak AF594 fluorescence could be detected in cells
inoculated with DENV labelled with 500μM and 1mM of AF594SE dye.
The above results indicate that the modified protocol is able to produce labelled DENV
that retains its infectivity and can be visualized easily by fluorescence microscopy.
69

(a)
70

(b) E protein AF594 labelling Merged
10µM
50µM
100µM
200µM
500µM
1mM
71

72
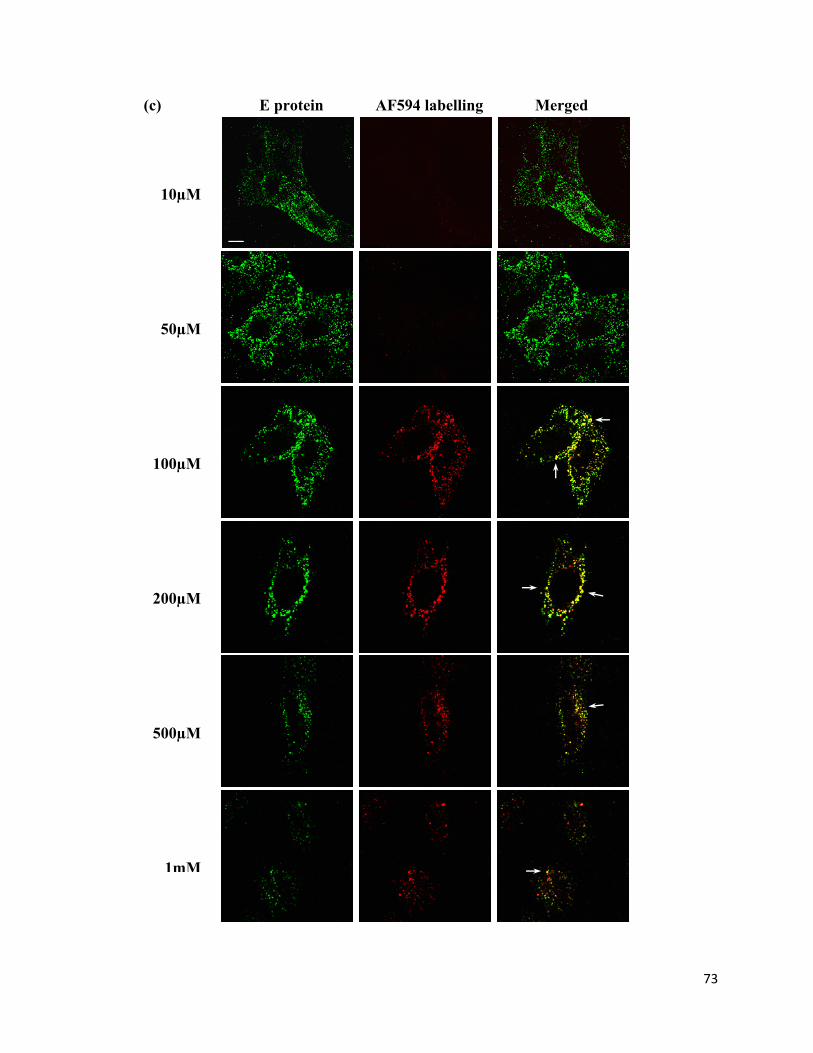
(c) E protein AF594 labelling Merged
10µM
50µM
100µM
200µM
500µM
1mM
73

74

Figure 3-1 Dengue virus viability post AF594 labelling
Purified dengue virus was labelled with various concentrations of AF594SE dye dissolved in DMSO or SBB for 1 hour at room temperature. (a) The labelled virus was then re-titrated post-labelling by plaque assay. Bar diagram shows average virus titre of four determinations, pre- and post-labelling. Error bars indicate standard deviation. The labelled virus with dye dissolved in DMSO (b) or SBB (c) was also tested for fluorescence intensity by immunofluorescence assay. Vero cells seeded on coverslips the day prior are infected with labelled DENV for 10 minutes at 37°C. The cells were subsequently fixed and labelled with anti-E antibody, and examined for co-localization of E protein (green) and AF594 labelling (red). Fluorescent signals are visualized under 63X magnification using Leica Microsystem TCS SP5 confocal microscope. Scale bar is 10µm. White arrows indicate areas of co-localization (yellow when merged).
75

3.1.2. Efficiency of Alexa Fluor dye labelling
To estimate the percentage of virus labelled with the fluorescence dye using this modified
method, flow cytometric studies were carried out using DENV labelled with AF488SE
dye (AF488-DENV). DC-SIGN transfected Raji B cells were infected with AF488-
DENV at a MOI of 1, stained for presence of E protein using 3H5 monoclonal antibody
and analysed by flow cytometry. Compared to the uninfected Raji B cells with DC-SIGN,
approximately 81.4% of the cells following infection were positive for the E protein. Of
these cells, 58.4% were positive for both E protein and AF488 (Fig. 3-3a), suggesting that
approximately 71% of the virions were labelled with the Alexa Fluor dye.
Although AF594-DENV could not be assessed for efficiency of labelling on the flow
cytometer used, due to the lack of appropriate excitation laser, Pearson’s correlation
values obtained from co-localization studies using confocal microscopy revealed that
similar labelling efficiency was achieved (Fig. 3-3b).
76

(a)
MOI 1 Uninfected
E pr
otei
n (P
E)
Dye-labelled dengue virus (AF488)
(b)
Figure 3-2 Efficiency of Alexa Fluor dye labelling as a function of percentage infected Raji B cells transfected with DC-SIGN were infected at a MOI of 1 with AF488-DENV for 10 minutes at 37°C, fixed and stained for presence of E protein. Fluorescence was then measured using BD FACS Calibur (a). Close to 100% of uninfected Raji B cells with DC-SIGN were double negative for both Alexa Fluor and anti-E antibody signals, whereas approximately 81% of the infected cells stained positive for E protein, and approximately 71% of which were also positive for Alexa Fluor 488 fluorescence, suggesting that approximately 71% of the virions were labelled with the dye. Confocal microscope analysis of Vero cells infected with AF594-DENV (red) and stained for E protein (green) showed a mean Pearson’s correlation of 0.677 ± 0.053 (Mean ± SD) (b), suggesting similar labelling efficiency (yellow indicates co-localization). Scale bar = 10nm.
77

3.1.3. Reproducibility of labelling
Three independent AF594SE labelling experiments were performed with similar starting
titre of 2x108 pfu of purified DENV using the modified protocol. The titre post-labelling
was then determined by plaque assay. All three experiments showed 3- to 8-fold
reduction in titre post-labelling and column purification, from a mean titre of 2.21x108
(SD = 3.68x107) to 3.78x107 (SD = 1.5x107). To exclude the possibility that the reduction
in titre post-labelling was due to a loss in infectivity of the virus, the ratio of RNA copy
number to plaque forming unit of DENV pre- and post-labelling was analysed from one
of these batches of labelled virus. No significant difference in the viral RNA copy
number to plaque forming unit ratio was observed between pre- and post-labelled virus
(Table 3-1, p > 0.5), indicating that the fall in titre was not due to inactivation of the
virions but rather due to loss during column purification.
78

Table 3-1
Viral RNA copy number to infectious particle ratio
Titre (pfu/ml) RNA copy number (/ml) RNA copy number/pfu
AF594-DENV 3.5 x 107 6.27 x 108 ± 2.61 x 108 17.9 ± 7.45
Purified DENV 1.0 x 109 1.67 x 1010 ± 1.30 x 1010 16.7 ± 12.96
Viral RNA was extracted from purified non-labelled DENV or AF594-DENV and RNA copy number quantitated by real-time PCR. The RNA copy number was then divided by the titre to obtain a ratio for both non-labelled DENV (16.7) and AF594-DENV (17.9). The similar ratios indicated that both labelled and non-labelled DENV have comparable infectious/non-infectious proportions, suggesting that the labelling process did not inactivate the virions.
79

3.1.4. Growth kinetics of labelled DENV
As the labelled virus would be used to track the progression of virus through cellular
compartments, we tested if labelling altered the efficiency of virus replication. Vero cells
were infected with either purified non-labelled or AF594-DENV at a MOI of 0.1. Culture
supernatant and cell lysates were collected up to 72 hours and assayed for virus titre in
supernatant as well as viral RNA in the cell lysates. No significant difference was
observed between labelled and unlabelled virus for both viral RNA copy number and the
number of infectious particles in the supernatant of the culture (Fig. 3-3) (P > 0.5). The
data indicates that the attachment of the fluorophores at the concentration used did not
alter the kinetics of DENV replication, in vitro.
80
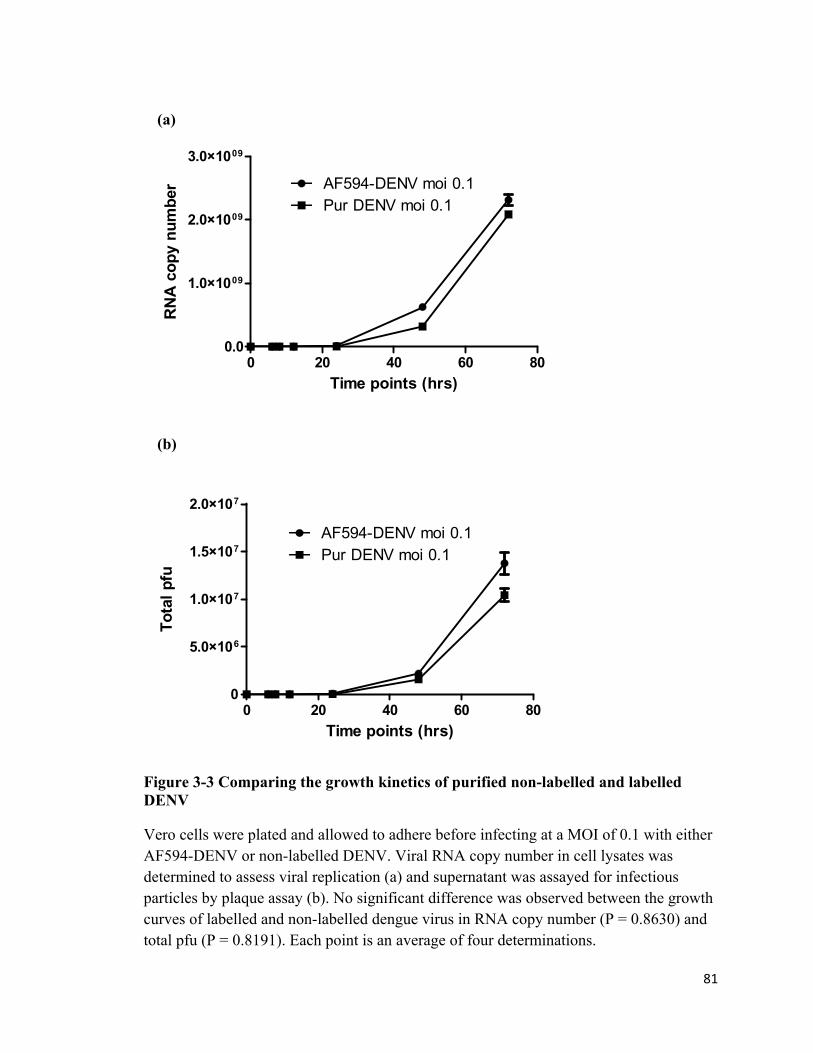
(a)
0 20 40 60 800.0
1.0×1009
2.0×1009
3.0×1009
AF594-DENV moi 0.1Pur DENV moi 0.1
Time points (hrs)
RN
A c
opy
num
ber
(b)
0 20 40 60 800
5.0×106
1.0×107
1.5×107
2.0×107
AF594-DENV moi 0.1Pur DENV moi 0.1
Time points (hrs)
Tota
l pfu
Figure 3-3 Comparing the growth kinetics of purified non-labelled and labelled DENV
Vero cells were plated and allowed to adhere before infecting at a MOI of 0.1 with either AF594-DENV or non-labelled DENV. Viral RNA copy number in cell lysates was determined to assess viral replication (a) and supernatant was assayed for infectious particles by plaque assay (b). No significant difference was observed between the growth curves of labelled and non-labelled dengue virus in RNA copy number (P = 0.8630) and total pfu (P = 0.8191). Each point is an average of four determinations.
81

3.2. Visualizing the fate of antibody-DENV complexes in monocytes
3.2.1. Humanized 3H5 and 4G2
To describe the early interactions of DENV-antibody complexes with human monocytes,
two mouse mabs, namely 3H5 and 4G2 [Henchal et al., 1982; Henchal et al., 1983] were
used. 3H5 mab is a DENV-2 type-specific and potently neutralizing antibody that binds
to an epitope in N-terminal region and FG loops of the lateral ridge of domain III on the
E protein [Trirawatanapong et al., 1992], while 4G2 mab is a flavivirus cross-reactive
antibody that recognises an epitope in the fusion peptide of domain II of E protein [Crill
and Chang, 2004]. However, the use of these mabs with human monocytes requires the
switch of the mouse Fc portion of the antibodies to that of human Fc region to ensure the
correct engagement of the Fc receptors on the human cells. Chimeric forms of the mAb
3H5 (h3H5) and 4G2 (h4G2) containing mouse VH and VL sequences and human γ1 and
κ constant sequences were constructed as previously described in Hanson et al [Hanson et
al., 2006]. These chimeric IgG1 antibodies retained similar binding characteristics to
DENV-2 as their parent mabs, indicated by the similarity of their respective Kd values
(p>0.05) (Table 3-2). The humanized antibodies were also tested for their binding
affinities to the AF594-DENV. There was no significant difference in binding of h3H5
between labelled and non-labelled virus (p>0.05); however, h4G2 has an increased
binding affinity to the labelled virus as compared to non-labelled virus (p=0.013) (Table
3-3).
82

Table 3-2
Binding affinity of 3H5 and 4G2 to DENV-2 before and after humanization
Antibodies Kd (±SE)/nM
p-value Before humanization After humanization
3H5 0.5189 ± 0.0935 0.3881 ± 0.0499 0.233
4G2 1.3560 ± 0.4630 1.5550 ± 0.2962 0.721
Kd values were determined by non-linear regression analysis. There were no differences in binding affinity to DENV-2 before and after humanization (P-value>0.05, not significant). (Data from Chan Kuan Rong)
83

Table 3-3
Binding affinity of h3H5 and h4G2 to AF594-DENV and non-labelled DENV
Antibodies Kd (±SE)/nM
p-value AF594-DENV Non-labelled DENV
h3H5 0.1124 ± 0.0111 0.0913 ± 0.0167 0.289
h4G2 0.2612 ± 0.0301 0.5434 ± 0.1052 0.013
Kd values were determined by non-linear regression analysis. There was no significant difference in binding of h3H5 to labelled and non-labelled DENV (P-value>0.05, not significant). Humanized 4G2 demonstrated an increased binding affinity to the labelled virus.
84

3.2.2. Determining the neutralizing concentrations of h3H5 and h4G2 on monocytes
Two-fold serial dilutions of h3H5 or h4G2 were allowed to complex with 10 MOI of
DENV before the mixture was added to the THP-1 cells or primary monocytes. The
supernatant was then harvested 72 hours later for plaque assay. The minimum
neutralizing concentrations of antibodies on THP-1 (Fig. 3-4a) or primary monocytes
(Fig. 3-4b) were determined to be the highest dilution that produced no plaques.
Humanized 3H5 fully neutralized DENV between the range of 3 to 400µg/ml antibodies,
with the minimum neutralizing concentration being 3µg/ml on THP-1 cells and 6µg/ml
on primary monocytes. Humanized 4G2 neutralized DENV at a concentration of
100µg/ml and above on both THP-1 and primary monocytes.
85

(a)
0.01 0.1 1 10 100 10000
20
40
60
80
100
120h3H5h4G2
h4G2 (100µg/ml)
Antibody concentration (µg/ml)
% N
eutr
aliz
atio
n
h3H5 (3µg/ml) (b)
0.01 0.1 1 10 100 10000
20
40
60
80
100
120h3H5h4G2
h4G2 (100µg/ml)
Antibody concentration (µg/ml)
% N
eutr
aliz
atio
n
h3H5 (6µg/ml)
Figure 3-4 Neutralizing concentrations of h3H5 and h4G2 on monocytes
Two-fold serial dilutions of h3H5 or h4G2 were incubated with DENV for 1hr before adding to (a) THP-1 cells, or (b) primary monocytes, and cultured for 72 hours. The supernatant was then harvested and plaque assay done to determine the titre. The graphs above show the percentage neutralization of DENV at various antibody concentrations compared to virus control. The highest dilution that resulted in no plaques was taken to be the minimum neutralizing concentration for each antibody. Each point is an average of at least 3 determinations. (Graph (a) from Tan Hwee Cheng)
86

3.2.3. Visualizing the entry and endosomal trafficking of antibody-virus complexes in THP-1 monocytic cell line
To understand the early events of antibody-DENV complexes (formed with h3H5 or
h4G2) interaction with monocytes that leads to subsequent neutralization of the virus, a
time course experiment was performed and the events visualized by confocal microscopy.
Neutralizing concentrations of h3H5 (3µg/ml) or h4G2 (100µg/ml) were complexed with
10 MOI of AF594-DENV for 1hr at 37°C. The immune complexes were incubated with
THP-1 cells on ice to allow synchronization of entry and time course started when
temperature was raised to 37°C. The cells were subsequently fixed and stained for early
endosome marker (EEA1) or late endosome/lysosome marker (LAMP-1) after 5, 10, 30,
60 or 120min incubation at 37°C, before viewing under 63x magnification on a confocal
microscope. The samples were also stained with anti-human IgG secondary antibodies to
confirm that the humanized antibodies co-localized with DENV.
With h3H5, the labelled virus and human antibody signals could be seen co-localizing
with LAMP-1 in approximately 2-3% of the cells after 5 and 10min incubation at 37oC
(Fig. 3-5). After 30min, the percentage of cells showing antibody-DENV complexes in
LAMP-1 positive compartments increased to 4-5%. The viral proteins and h3H5 could
still be observed in the LAMP-1 positive compartments at the 1 and 2hr time points.
With h4G2, however, significantly less immune complexes were attached to the cells and
they appeared to be trapped on the cell surface, indicated by the reduced number of
AF594-DENV spots and dimmer fluorescence, for all time points taken in the first hour
of observation (Fig. 3-6). Entry of the immune complexes into cells and co-localization
87

88
with LAMP-1 positive compartments were observed at the 2hr time point in 1-2% of the
cells.
As no productive replication of the virus occurred in THP-1 with these concentrations of
mabs, the observations suggest that the 2 mabs resulted in the formation of immune
complexes that interacted differently with THP-1. At the minimum concentration for
h3H5 to neutralize 10 MOI of DENV (3µg/ml), antibody-DENV complexes were
internalized early and extensive co-localization of the complexes in the LAMP-1 positive
compartments was observed. In contrast, no sub-cellular trafficking of the antibody-virus
complexes was observed with h4G2 at its minimum neutralizing concentration
(100µg/ml) till 2hr after elevation of temperature to 37°C.
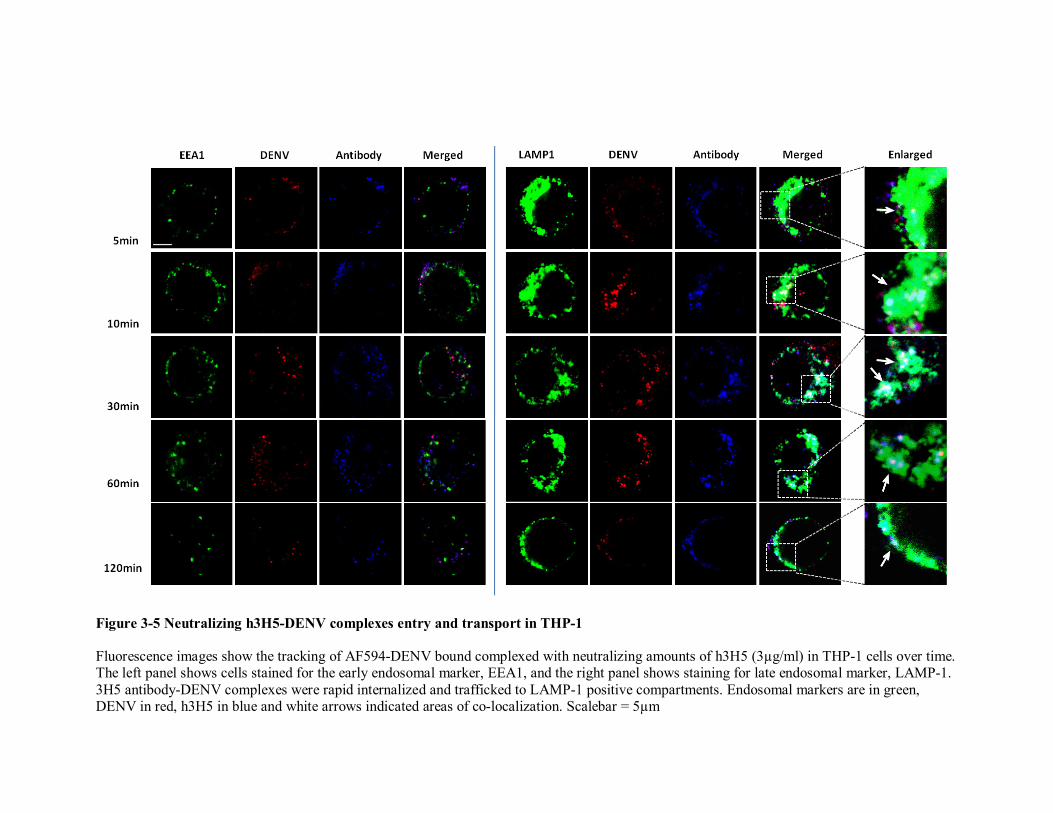
1
Figure 3-5 Neutralizing h3H5-DENV complexes entry and transport in THP-1
Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h3H5 (3µg/ml) in THP-1 cells over time. The left panel shows cells stained for the early endosomal marker, EEA1, and the right panel shows staining for late endosomal marker, LAMP-1. 3H5 antibody-DENV complexes were rapid internalized and trafficked to LAMP-1 positive compartments. Endosomal markers are in green, DENV in red, h3H5 in blue and white arrows indicated areas of co-localization. Scalebar = 5µm

90
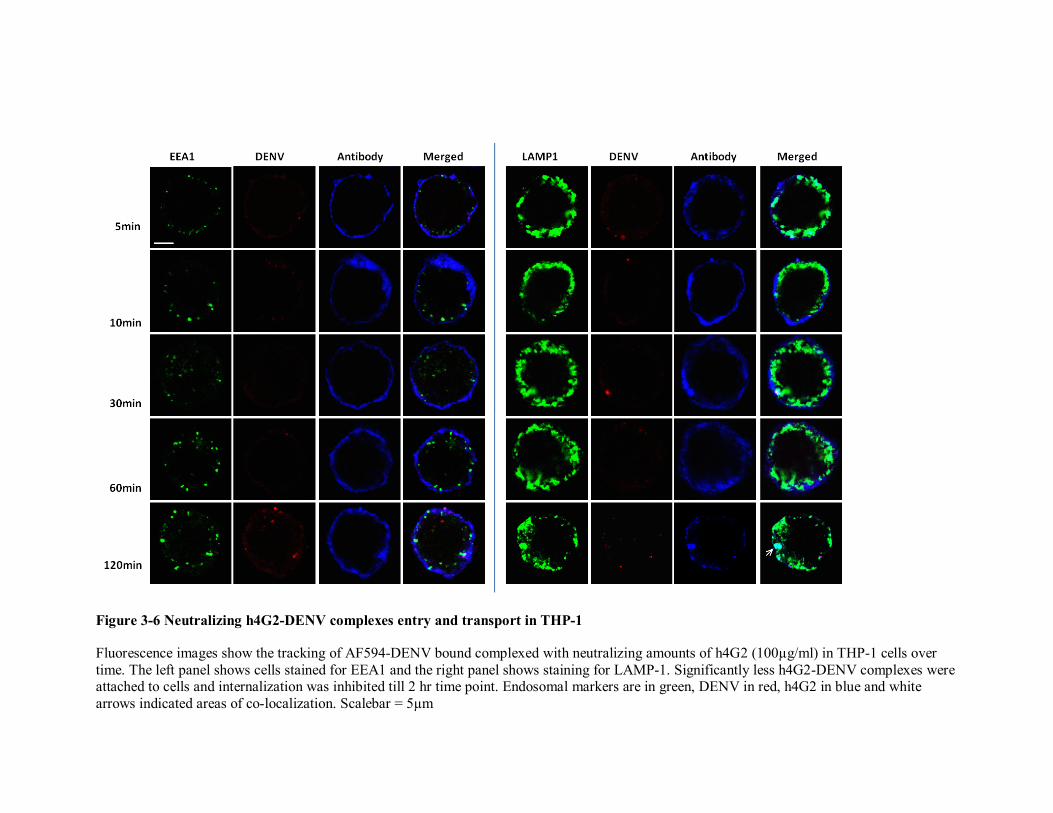
92
Figure 3-6 Neutralizing h4G2-DENV complexes entry and transport in THP-1
Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h4G2 (100µg/ml) in THP-1 cells over time. The left panel shows cells stained for EEA1 and the right panel shows staining for LAMP-1. Significantly less h4G2-DENV complexes were attached to cells and internalization was inhibited till 2 hr time point. Endosomal markers are in green, DENV in red, h4G2 in blue and white arrows indicated areas of co-localization. Scalebar = 5µm

92

3.2.4. Primary monocytes
The above observations were made using THP-1 cell line. To rule out any artefacts
due to the choice of cell line used, we repeated the experiments using primary
monocyte cultures.
Primary monocytes were isolated from peripheral blood using Ficoll density gradient
centrifugation and plastic adherence as described in the materials and methods. The
monocytes were then cultured overnight at 37°C, without any cytokine stimulation for
maturation, before use. This method of isolation yielded at least 80% monocytes (gate
R1), with a low percentage of lymphocytes contamination (gate R2), as confirmed by
FACS analysis (Fig. 3-7).
93

Figure 3-7 Primary monocytes yield
Primary monocytes were isolated from peripheral blood using Ficoll density centrifugation and plastic adherence. Each isolation yielded at least 80% purity by flow cytometry.
94

95
3.2.5. Antibody-DENV immune complex interactions with primary monocytes
The AF594-DENV was complexed with minimally neutralizing concentrations of
h3H5 (6µg/ml) or h4G2 (100µg/ml) and added to the primary monocytes. The fate of
the complexes was then followed at different time points using confocal microscopy.
Similar to what was observed with THP-1 cell line, DENV complexed with 6µg/ml of
h3H5 were internalized by the primary monocytes after incubation at 37°C. At the
5min time point, antibody-DENV complexes could be seen near the surface of the
cells, with an occasional co-localization of a DENV spot with EEA1 but no co-
localization with LAMP-1 (Fig. 3-8). However, at 10min time point, majority of the
complexes was already within LAMP-1 positive compartments. By 30, 60 and
120min time points, approximately 10% of the cells observed had the complexes co-
localizing strongly with LAMP-1 positive compartments.
Humanized 4G2 bound DENV, however, was able to attach to the primary monocytes
but entry into the cells was inhibited at the early time points. In approximately 5-10%
of the cells, the complexes were seen to line the surface at all time points observed up
to 60min. At the 2hr time point, h4G2 bound DENV can be detected in the LAMP-1
positive compartments in a small number of cells (Fig. 3-9).

96

98
Figure 3-8 Entry and endocytic trafficking of h3H5-opsonized DENV in primary monocytes Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h3H5 in primary monocytes over time. The left panel shows cells stained for the early endosomal marker, EEA1, and the right panel shows staining for late endosomal/lysosome marker, LAMP-1. White arrows indicate areas of co-localization. Scalebar = 2µm

98

100
Figure 3-9 Entry and endocytic trafficking of h4G2-opsonized DENV in primary monocytes Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h4G2 in primary monocytes over time. The left panel shows cells stained for the early endosomal marker, EEA1, and the right panel shows staining for late endosomal marker, LAMP-1. White arrows indicate areas of co-localization. Scalebar = 2µm

100

3.3. Fc receptor requirement for internalization
Fc receptors (FcR) are important in mediating immune complex attachment and
internalization into monocytes. Two types of FcγRs, FcγRI and FcγRII, are present on
both THP-1 cells and primary monocytes (Fig. 3-10). Although both FcγRs have been
shown to mediate enhancement of DENV infection in monocytes [Kou et al., 2008;
Littaua et al., 1990], much less is known about their role in mediating viral clearance
in the presence of neutralizing antibodies in this cells. FcγRI has been shown to be
important in mediating clearance of neutralizing antibody-DENV complex in a recent
study using CV-1 cells transfected with FcγRI/γ [Rodrigo et al., 2009]. However, this
study cannot explain how antibody-DENV complexes interact with monocytes where
both FcγRs are naturally expressed simultaneously. Since FcγRI and FcγRIIa have
fundamental differences in their intracellular signaling pathways and antigen
processing/presentation when activated [Dai et al., 2009], differential receptor
engagement could explain the distinct fates of the DENV immune complexes in
monocytes. Hence, having a tool available to visualize this interaction, AF594-DENV
was used to shed some light on the FcγR engagement by the different complexes.
THP-1 cells were incubated with AF594-DENV complexed with either h3H5 at
3µg/ml or h4G2 at100µg/ml for 30 and/or 120min at 37°C, fixed and stained for
FcγRI or FcRγII before viewing with a confocal microscope. At 30min, DENV
opsonised by h3H5 interacts with both FcγRI and FcRγII on the surface of the cells,
but those complexes that were internalized, co-localized mainly with FcγRI (Fig. 3-
11). Although a few DENV spots can be seen to co-localize with FcRγII, they
constitute only a minority of the observation. This effect is much more pronounced on
primary monocytes (Fig. 3-12).
101

Humanized 4G2 bound DENV attached to THP-1 cell surface by both FcγRI and
FcRγII at 30min and remained on the outside of the cells (Fig. 3-11). The complexes
also appeared to cluster the Fc receptors as shown by the increased intensity of FcγRI
or FcRγII staining around the complexes. This was not observed with the h3H5-bound
DENV. The complexes that had entered the cells by 2hr after temperature shift to
37°C, appeared to co-localize to the same extent with both FcγRI and FcRγII. Again,
this effect is more evident with primary monocytes (Fig. 3-12).
These results indicated that DENV-h3H5 complexes can attach to either FcγRI or
FcRγII on the surface of the cells but internalization occurred primarily through
FcγRI; whereas for h4G2-bound virus, the immune complexes are able to bind to both
FcγRI and FcRγII on the cell surface but opsonization is inhibited until after 2 hours
of incubation. The subsequent uptake of the immune complex appeared to be
mediated by both FcγRI and FcRγII.
102

Figure 3-10 Fc gamma receptors expression on monocytes
Both FcγRI and FcRγII are expressed on THP-1 cell line and primary monocytes isolated from peripheral blood.
103

104

1
Figure 3-11 Fc receptor requirements by h3H5 or h4G2-opsonized DENV in THP-1 cells Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h3H5 (h3H5N) or h4G2 (h4G2N) in THP-1 cells after 30min and/or 120min incubation at 37°C. The left panel shows cells stained for FcγRI, and the right panel shows staining for FcRγII. White arrows indicate areas of co-localization. Scalebar = 5µm

106

108
Figure 3-12 Fc receptor requirements by h3H5 or h4G2-opsonized DENV in primary monocytes
Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h3H5 (h3H5N) or h4G2 (h4G2N) in primary monocytes after 30min and/or 120min incubation at 37°C. The left panel shows cells stained for FcγRI, and the right panel shows staining for FcRγII. White arrows indicate areas of co-localization. Scalebar = 2µm

108

3.4. Inhibition of immune complex uptake
3.4.1. Concentration dependence
We next evaluated if the concentration of antibodies influences how the complexes
interact with monocytes. Humanized 3H5 has a broad range of neutralizing activity on
DENV-2 (3 to 400µg/ml) (Figure 3-4a). To test the effect of high antibody
concentrations on the complexes interaction with monocytes, h3H5 concentration was
increased to 100 and 400µg/ml, similar to the neutralizing concentration of h4G2. At
these concentrations of h3H5, the proportion of cells positive for AF594-DENV
fluorescence reduced markedly from 4-5% to approximately 1-2% at 30min time
point, compared to h3H5 at 3µg/ml. Of these, majority of the antibody-DENV
complexes lined the surface of the cells with occasional entry of 1 or 2 complexes into
LAMP-1 positive compartments just below cell surface (Fig. 3-13a, c). Similarly,
complete inhibition of opsonisation can be observed in primary monocytes at
400µg/ml of 3H5 (Fig. 3-13d), although 100g/ml of the antibody did not stop uptake
of the complexes (Fig. 3-13b). Taken collectively, these results indicate that the
concentration of DENV antibody affects the efficiency of opsonization in monocytes.
3.4.2. Competition for Fc receptors
An obvious explanation for the above observation is that excess antibodies competed
with the immune complexes for the same Fc receptors. To test this possibility, we
compared the sub-cellular localization of antibody-DENV complexes neutralized with
100 or 400µg/ml of h3H5 to that of 3µg/ml but with an addition of isotype control
antibodies to give a total antibody concentration of 100 or 400µg/ml. The addition of
isotype control antibodies did not inhibit internalization of the virus in both THP-1
cells and primary monocytes even at 400µg/ml of total antibody concentration (Fig. 3-
109

110
14). These observations indicate that inhibition of FcγR-mediated internalization by
high concentration of h3H5 is likely to be a function of specific antibody-antigen
ratios and not competition for Fc receptors by the excess IgGs.

112
Figure 3-13 Effect of increased antibody concentrations on endocytic trafficking of DENV
Fluorescence images show the tracking of AF594-DENV bound complexed with 100 or 400µg/ml of h3H5 in THP-1 cells (left panel, a, c) or primary monocytes (right panel, b, d) at 30min. The cells were stained for LAMP-1 and viewed with confocal microscope. White arrows indicate areas of co-localization. Scalebar = 5µm (THP-1) or 2µm (primary monocytes)
(a) (b)
(d) (c)

112

114
Figure 3-14 Competition for Fc receptors Fluorescence images show the tracking of AF594-DENV bound complexed with neutralizing amounts of h3H5 with the addition of isotype control antibodies to a total concentration of 100 or 400µg/ml in THP-1 cells (left panel, a, c) or primary monocytes (right panel, b, d) at 30min time point. The cells were stained for LAMP-1 and viewed with confocal microscope. White arrows indicate areas of co-localization. Scalebar = 5µm (THP-1) or 2µm (primary monocytes)
(a) (b)
(d) (c)

114

3.4.3. Immune complex size and internalization
As competition for FcγR by free antibodies has been excluded as a possible
explanation (see section 3.4.2), we hypothesized that the inhibition of uptake of
DENV complexes formed at high concentrations of antibodies into monocytes might
be dependent on their size. Besides binding to epitopes on the DENV E protein and
thus inhibiting their ability to interact with the relevant cellular receptors or uncoating
of the virus post-uptake, antibodies can also aggregate viruses and how different
antibody concentrations affect the size of these viral aggregates as well as its
subsequent interaction with monocytes, is unknown. To test this hypothesis, the
immune complexes formed at 3 or 100µg/ml of h3H5 IgG or single Fab arm of h3H5
antibodies or virus only control were subjected to sucrose gradient and dynamic light
scattering (DLS) analyses.
3.4.3.1. Sucrose gradient separation of immune complex sizes
Being bivalent antibodies, h3H5 may form a wide variety of complexes with different
compositions of sizes and consequently, different sedimentation coefficients. Hence, a
sucrose gradient would be useful in separating such a heterogeneous mixture and
determining the most abundant complex size(s). Viral RNA were quantified from
each fraction and plotted against its fraction number to give a distribution curve (Fig.
3-15a). The distribution curves for the different conditions were then compared
against that of the virus control for any change brought about by the addition of
antibodies. A lower fraction number of the gradient would thus indicate higher
particle density.
Viral RNA distribution curve of the virus only control tube showed a peak at fraction
15, indicating the position where majority of the free virions would reside in the 10-
115

60% sucrose gradient. With the addition of h3H5 IgG, there was an obvious shift of
the curves to the left of that of the virus control, indicating that the antibodies
increased the sedimentation of the virus, suggesting the formation of aggregates. At
3µg/ml of h3H5 IgG, the peak shifted to fraction 12 and at 100µg/ml of h3H5 IgG,
there was a further shift of the peak to fraction 9. Unlike the virus control where the
spread of the curve is small with a sharp peak around fraction 15 (suggesting that the
virions were mostly uniformly sized), the spread of the distribution curves for DENV
with h3H5 IgG became wider (suggesting that the immune complexes formed were
heterogeneous in size). This shift in curves cannot be explained solely by the
attachment of antibodies to the surface of DENV as the addition of h3H5 Fab brought
about only a slight shift of 1 fraction to the left of virus control with little change to
the shape of the curve. Since the h3H5 Fab has the same binding affinity as the
parental h3H5 IgG but lacks both arms of the antigen-binding region, it is able to bind
to DENV but not cross-link them. Hence, it was expected that the shift due to the
attached h3H5 Fab on the surface of the virions should be minimal. This trend is
reproducible over three independent experiments.
3.4.3.2. Immune complex size by Dynamic Light Scattering (DLS)
To confirm that the shift in fraction numbers were indeed due to the increase in
complex sizes, individual fractions were analyzed using DLS. The DLS machine
works by shining a monochromatic light beam, such as a laser, onto a solution where
particles in Brownian motion causes a Doppler Shift when the light hits the moving
particle, thus changing the wavelength of the incoming light falling on the detector.
This change was then related to the size of the particle using Stokes-Einstein equation
to give a size distribution of the particles in suspension.
116

DLS analysis of virus only control showed that majority of the particles have an
average size of 49.1nm in diameter (fraction 15), corresponding to the size of a single
dengue virion (Fig. 3-15b). Addition of 3µg/ml of h3H5 increased the average
diameter of the DENV complex to 64.5nm (fraction 12). An IgG molecule is
approximately 15nm in length. Hence, it is likely that at this concentration of h3H5,
majority of the particles had antibody bound to them, but did not cross-link adjacent
virions to form aggregates. However, at 100µg/ml of h3H5, the average diameter of
the complexes increased to 192.0nm (fraction 9), indicating an aggregate of at least 3
virions in a row (Fig. 3-15c). With h3H5 Fab (approximately 7.5nm in length), the
majority of the complexes were 57.7nm in diameter (fraction 14), signifying that the
larger complex size formed with 100µg/ml of h3H5 IgG is specific action of antibody
cross-linking and not due to addition of exogenous proteins. A model displaying the
cross-sectional view of immune complexes is shown in Fig. 3-15c to illustrate the
possible configurations of antibody binding to DENV.
The above results demonstrated that 100µg/ml of h3H5 formed complexes with an
average of 3 dengue virions in diameter, which could be translated to a lattice
consisting of 27 virions in a 3-dimensional structure. These larger complexes may
account for the inhibition of entry of complexes at high concentrations of antibodies.
117

(a)
0 5 10 15 201.0×1006
1.0×1007
1.0×1008
1.0×1009
1.0×1010 Virus onlyVirus + 3µg/ml h3H5Virus + 100µg/ml h3H5Virus + h3H5 (Fab)
Bottom of tube Top of tube
Fraction no.
RN
A c
opy
num
ber/
frac
tion
(b)
(c)
118

Figure 3-15 Immune complex size analysis by sucrose gradient and dynamic light scattering (DLS)
Equal amounts of purified DENV were allowed to complex with 3µg/ml, 100µg/ml, 33.3µg/ml h3H5 Fab or no antibody (virus control) for 1hr at 37°C. The immune complexes were then loaded onto a linearized 10-60% sucrose gradient and spun overnight at 4°C. Fractions were collected from bottom of tube and analyzed for (a) viral RNA copy number per fraction, where each point is the mean of 3 independent experiments and error bars indicate the SEM, and (b) DLS for complex size. The size distribution of complexes in the corresponding peak fractions were shown as diameters in nanometers (d.nm). The proposed cross-sectional view of complexes based on the average complex sizes obtained from DLS analysis is shown in (c).
119

120

Chapter 4
Discussion
Zhang Lixin (HT080076A)


4.1. Fluorescence labelling of DENV
The early events of DENV infection in monocytes are not well understood and this has
led to long delays in the development of a safe and effective tetravalent dengue vaccine.
To improve this knowledge, we sought to visualize these interactions using fluorescently
labelled virus and cells. The first part of this study was focused on the development of a
DENV labelling method for AF594SE dye and characterizing the labelled virus to ensure
that the attached fluorophores did not impair the infectivity of the virus. An optimized
procedure for Alexa Fluor labelling of DENV was published [Zhang et al., 2010].
However, it should be noted that Alexa Fluor conjugation to DENV increased the binding
affinity of h4G2 antibody, but not h3H5, to the virions. The binding epitope of 3H5 had
been mapped to amino acids 386 to 397 (QLKLNWFKKGSS) of the DENV E protein
[Trirawatanapong et al., 1992], which consist of 3 lysine residues (K), where the amine-
reactive dye could potentially conjugate to. As the amine conjugation reaction is a
random event, the fluorophores should not target these lysine residues on all the E
proteins such that the epitopes of 3H5 are fully obscured. Hence, 3H5 binding to the
labelled virions was not affected. It is known that 4G2 binds to a conformational epitope
near the fusion loop, between amino acids 1-120 and 142-158 [Dejnirattisai et al., 2010],
but the precise amino acids that make up the epitope have not been mapped. There are
many arginine and lysine residues within these sequences, where the Alexa Fluor dye can
react to. It is not understood why 4G2 binds to the labelled virus with higher affinity, but
it can be speculated that the conjugation of the dye might have made the epitopes more
accessible to the antibody or, there might be some non-specific cross-reaction to the
fluorophore.
123

Notwithstanding these limitations, the Alexa Fluor labelled virions would serve as a very
useful tool for visualizing the early events of DENV complexes with monocytes.
4.2. Cellular fate of DENV immune complexes in monocytes
To identify the difference in the cellular fate of DENV complexes formed with either a
homotypic or heterotypic antibody in monocytes, two human-mouse chimeric antibodies,
h3H5 (homotypic) and h4G2 (heterotypic), were used. The visualization was first
demonstrated in THP-1 cells and then verified with primary monocytes.
Our observations suggested that the DENV immune complexes formed by the two
antibodies interact with monocytes differently. With h3H5 at 3µg/ml, almost all the
DENV complexes were rapidly internalized and co-localized to the late
endosomes/lysosomes within 30min, whereas DENV complexes formed with 100µg/ml
h4G2 stayed on the surface of cells and no sub-cellular trafficking was observed at this
time point. This phenomenon might be the result of differential ability of the complexes
to trigger Fc receptor-mediated phagocytosis. DENV complexes formed with 3µg/ml
h3H5 interacted with both FcγRI and FcγRII on the surface of cells but internalized
primarily through FcγRI at 30min time point. FcγRI is an activatory receptor. This
suggests that complexes formed at this concentration of h3H5 can activate Fc receptor-
mediated phagocytosis and presumably leads to rapid transport to the late
endosome/lysosomes for degradation of the cargo and thus stops productive infection of
DENV. On the other hand, DENV complexes formed with 100µg/ml h4G2 were able to
cluster both FcγRs on the surface of cells but unable to internalize at the 30min time
point. When the complexes were finally internalized to late endosomes/lysosomes at the
124

2hr time point, there were no preferential engagement of Fc receptor as the complexes
co-localized equally well to both FcγRI and FcγRII intracellularly. This suggests that the
attachment of DENV complexes formed with h4G2 had an inhibitory effect on
phagocytosis and this effect is only lifted after more than an hour of association with the
monocytes.
These observations suggest that the specific interaction of the complexes with monocytes
is dependent on the antibody-antigen ratios used. Indeed, the association of large
complexes with transient phagocytic defect in macrophages have been previously
reported in in vivo observations for both influenza and dengue [Astry and Jakab, 1984;
Chaturvedi et al., 1983], although a mechanistic explanation was not given for the
reported observations. This thesis explored the possibility that high concentration of
antibody aggregates viruses and thus forms immune complexes of a larger size.
4.3. Antibody concentrations and complex size
Immune complexes are the outcome of interactions of multivalent macromolecular
antigens, such as whole virions, with bivalent or pentavalent antibodies [Mannik, 1980].
Antibody concentrations in proportion to antigens affect the size of immune complexes
formed, which in turn influence the type and number of receptors they engage, and
subsequent pathways activated. A probable explanation for our observations above is that
with increasing antibody concentrations, the sizes of antibody-DENV complexes formed
increase correspondingly, and at 100g/ml of the antibody, the complexes are large enough
to interact with receptors that are normally expressed in lower density. As such, we next
125

questioned whether immune complex sizes might dictate the mechanism of DENV
neutralization by antibodies.
Sucrose gradient was used to separate the immune complex sizes formed by high
(100µg/ml) and low (3µg/ml) concentrations of h3H5 IgG antibody. Both concentrations
of h3H5 formed heterogeneous populations of immune complex sizes. However, analysis
of complex sizes using DLS at their respective peak fractions indicated that h3H5
at100µg/ml tends to form larger immune complexes (average diameter = 192nm) with
DENV than at 3µg/ml (average diameter = 64.5nm), thus suggesting the antibody cross-
link the virions at the higher concentration.
4.4. Conclusions
This thesis demonstrated that antibody concentrations have a direct impact on the size of
DENV immune complexes formed and their fates in monocytes. Higher antibody
concentration results in larger complexes that can associate with Fc receptors but were
inhibited from entry, whereas smaller complexes formed at lower antibody concentration
enter the monocytes through FcγRI. One possible explanation for the inhibition of entry
is the engagement of inhibitory Fc receptor. FcγRIIB is the only inhibitory Fc receptor. It
is a low affinity Fc gamma receptor that constitutively expresses immunoreceptor
tyrosine-based inhibitory motif (ITIM) in its cytoplasmic tail, normally present in much
lower density than activatory receptors on monocytes. Hence it is more likely to be cross-
linked and activated by large immune complexes. It is therefore postulated that
neutralization of DENV at high antibody concentrations engages FcγRIIB, which thus
inhibit internalization of the complex.
126

However, as the staining antibody used does not differentiate between the isoforms of
FcγRII, it was not possible to ascertain this. Nonetheless, a PhD student in our laboratory
is testing this hypothesis using biochemical approaches. Indeed, preliminary results
showed that knock down of FcγRIIB in THP-1 cells using specific siRNA significantly
increased uptake of complexes at high concentration of h3H5, compared to the non-
specific siRNA control; and conversely, over-expression of FcγRIIB significantly
reduced uptake of the complexes compared to mock transfected controls [Chan Kuan
Rong, personal communication].
These differential Fc receptor interactions led to discernable phenotypes in monocytes,
which could be used as a tool to distinguish between neutralization of homologous and
heterologous DENV serotypes.
4.5. Future work
From our data above, we propose that homotypic antibodies is able to neutralize DENV
at concentrations suitable for opsoniziation and directing of the complexes to late
endosomes/lysosomes for degradation while cross-reactive antibodies can only neutralize
DENV at a concentration that cross-links FcγRIIB where opsonization is inhibited. It is
tempting to speculate that cross reactive antibodies cannot neutralize DENV
intracellularly although more work will be needed to test this. However, this mechanism
is proposed largely based on study with the two mouse mabs. Although a small panel of
primary and secondary patient sera have been tested to showed similar observations
[Chan et al., manuscript in submission], further assessment needs to be done with larger
sample of well-characterized patients’ sera to confirm such a phenomenon. There is also
127

the possibility in the adaptation of our observations into an assay for screening vaccinees’
sera for presence of protective homotypic or more cross-reactive/less protective
heterotypic antibodies. If successful, this assay would be very valuable in production and
fine-tuning of dengue vaccines.
Our results also gave a preliminary insight to the correlation between immune complex
sizes and the proportion of activatory/inhibitory Fc receptors expression on monocytes.
The inhibitory receptor FcγRIIB counters cell activation and induces immune tolerance to
self-antigens and prevention of autoimmune diseases [Ravetch and Bolland, 2001]. Its
polymorphism or deficiency has been implicated with autoimmune diseases and their
severity [Clynes et al., 1999; Radstake et al., 2006; Smith and Clatworthy, 2010; Su et al.,
2007]. It would therefore be interesting to understand the contribution of FcγRIIB gene
polymorphism in dengue disease in the presence of other immune cells, such as B and T
cells.
In this thesis, we also suggested that neutralization of DENV by antibodies guides the
complexes into late endosomes/lysosomes through Fc receptor-mediated phagocytosis.
However, there are other routes whereby the virus can gain entry into the cells, e.g.
clathrin-mediated endocytosis, caveolin-dependent endocytosis [DeTulleo and
Kirchhausen, 1998; Pelkmans, 2005]. DENV complexes entry into monocytes can end up
in different pathways and thus different outcomes of infection (neutralization or
enhancement). However, DENV complexes entry into monocytes has been poorly
characterized. This information would be beneficial in specifically targeting the DENV
complexes to intended compartments by use of drugs to block undesired pathways such
128

that the DENV complexes, even at sub-neutralizing levels of antibodies, will be directed
to the ‘neutralizing’ pathways.
129

130

Bibliography
Zhang Lixin (HT080076A)


References
Anwar A, Leong KM, Ng ML, Chu JJ, Garcia-Blanco MA. 2009. The polypyrimidine tract-
binding protein is required for efficient dengue virus propagation and associates with
the viral replication machinery. J Biol Chem 284(25):17021-17029.
Astry CL, Jakab GJ. 1984. Influenza virus-induced immune complexes suppress alveolar
macrophage phagocytosis. J Virol 50(2):287-292.
Berlier JE, Rothe A, Buller G, Bradford J, Gray DR, Filanoski BJ, Telford WG, Yue S, Liu J,
Cheung CY, Chang W, Hirsch JD, Beechem JM, Haugland RP. 2003. Quantitative
comparison of long-wavelength Alexa Fluor dyes to Cy dyes: fluorescence of the
dyes and their bioconjugates. J Histochem Cytochem 51(12):1699-1712.
Bian G, Xu Y, Lu P, Xie Y, Xi Z. 2010. The endosymbiotic bacterium Wolbachia induces
resistance to dengue virus in Aedes aegypti. PLoS Pathog 6(4):e1000833.
Blackley S, Kou Z, Chen H, Quinn M, Rose RC, Schlesinger JJ, Coppage M, Jin X. 2007.
Primary human splenic macrophages, but not T or B cells, are the principal target
cells for dengue virus infection in vitro. J Virol 81(24):13325-13334.
Blackwell JL, Brinton MA. 1997. Translation elongation factor-1 alpha interacts with the 3'
stem-loop region of West Nile virus genomic RNA. J Virol 71(9):6433-6444.
Blaney JE, Jr., Durbin AP, Murphy BR, Whitehead SS. 2006. Development of a live
attenuated dengue virus vaccine using reverse genetics. Viral Immunol 19(1):10-32.
Blaney JE, Jr., Matro JM, Murphy BR, Whitehead SS. 2005. Recombinant, live-attenuated
tetravalent dengue virus vaccine formulations induce a balanced, broad, and
protective neutralizing antibody response against each of the four serotypes in rhesus
monkeys. J Virol 79(9):5516-5528.
Blaney JE, Jr., Sathe NS, Goddard L, Hanson CT, Romero TA, Hanley KA, Murphy BR,
Whitehead SS. 2008. Dengue virus type 3 vaccine candidates generated by
133

introduction of deletions in the 3' untranslated region (3'-UTR) or by exchange of the
DENV-3 3'-UTR with that of DENV-4. Vaccine 26(6):817-828.
Blaney JE, Jr., Sathe NS, Hanson CT, Firestone CY, Murphy BR, Whitehead SS. 2007.
Vaccine candidates for dengue virus type 1 (DEN1) generated by replacement of the
structural genes of rDEN4 and rDEN4Delta30 with those of DEN1. Virol J 4:23.
Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van
der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The
IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile
virus, and dengue virus. Cell 139(7):1243-1254.
Burke DS, Nisalak A, Johnson DE, Scott RM. 1988. A prospective study of dengue infections
in Bangkok. Am J Trop Med Hyg 38(1):172-180.
Cabrera-Hernandez A, Smtih DR. 2005. Mammalian dengue virus receptors. Dengue Bulletin
29:119-135.
Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. 1994. Green fluorescent protein as a
marker for gene expression. Science 263(5148):802-805.
Chan KL. 1967. A report on the pilot control scheme in mosquito eradication at the Geylang–
Tanjong Rhu area in Singapore. Singapore Public Health Bulletin 1:52-72.
Chan KL. 1985. Singapore’s dengue haemorrhagic fever control programme:a case study on
the successful control of Aedes aegypti and Aedes albopictus using mainly
environmental measures as a part of the integrated vector control. Southeast Asian
Medical Information Center, Tokyo.
Chan KL, Chan YC, Ho BC. 1971a. Aedes aegypti (L.) and Aedes albopictus (Skuse) in
Singapore City. 4. Competition between species. Bull World Health Organ 44(5):643-
649.
Chan KL, Ho BC, Chan YC. 1971b. Aedes aegypti (L.) and Aedes albopictus (Skuse) in
Singapore City. 2. Larval habitats. Bull World Health Organ 44(5):629-633.
Chan KL, Ng SK, Chew LM. 1977. The 1973 dengue haemorrhagic fever outbreak in
Singapore and its control. Singapore Med J 18(2):81-93.
134

Chan YC, Chan KL, Ho BC. 1971c. Aedes aegypti (L.) and Aedes albopictus (Skuse) in
Singapore City. 1. Distribution and density. Bull World Health Organ 44(5):617-627.
Chan YC, Ho BC, Chan KL. 1971d. Aedes aegypti (L.) and Aedes albopictus (Skuse) in
Singapore City. 5. Observations in relation to dengue haemorrhagic fever. Bull World
Health Organ 44(5):651-657.
Chao YC, Huang CS, Lee CN, Chang SY, King CC, Kao CL. 2008. Higher infection of
dengue virus serotype 2 in human monocytes of patients with G6PD deficiency. PLoS
One 3(2):e1557.
Chaturvedi UC, Nagar R, Mathur A. 1983. Effect of dengue virus infection on Fc-receptor
functions of mouse macrophages. J Gen Virol 64 (Pt 11):2399-2407.
Chavez-Salinas S, Ceballos-Olvera I, Reyes-Del Valle J, Medina F, Del Angel RM. 2008.
Heat shock effect upon dengue virus replication into U937 cells. Virus Res 138(1-
2):111-118.
Chen ST, Lin YL, Huang MT, Wu MF, Cheng SC, Lei HY, Lee CK, Chiou TW, Wong CH,
Hsieh SL. 2008. CLEC5A is critical for dengue-virus-induced lethal disease. Nature
453(7195):672-676.
Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM. 1997.
Dengue virus infectivity depends on envelope protein binding to target cell heparan
sulfate. Nat Med 3(8):866-871.
Chiou CT, Hu CC, Chen PH, Liao CL, Lin YL, Wang JJ. 2003. Association of Japanese
encephalitis virus NS3 protein with microtubules and tumour susceptibility gene 101
(TSG101) protein. J Gen Virol 84(Pt 10):2795-2805.
Chiu MW, Shih HM, Yang TH, Yang YL. 2007. The type 2 dengue virus envelope protein
interacts with small ubiquitin-like modifier-1 (SUMO-1) conjugating enzyme 9
(Ubc9). J Biomed Sci 14(3):429-444.
Chu JJ, Ng ML. 2004. Infectious entry of West Nile virus occurs through a clathrin-mediated
endocytic pathway. J Virol 78(19):10543-10555.
135

Chu JJ, Yang PL. 2007. c-Src protein kinase inhibitors block assembly and maturation of
dengue virus. Proc Natl Acad Sci U S A 104(9):3520-3525.
Chua JJ, Ng MM, Chow VT. 2004. The non-structural 3 (NS3) protein of dengue virus type 2
interacts with human nuclear receptor binding protein and is associated with
alterations in membrane structure. Virus Res 102(2):151-163.
Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, Atkinson
JP, Diamond MS. 2006. West Nile virus nonstructural protein NS1 inhibits
complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci
U S A 103(50):19111-19116.
Clyde K, Kyle JL, Harris E. 2006. Recent advances in deciphering viral and host determinants
of dengue virus replication and pathogenesis. J Virol 80(23):11418-11431.
Clynes R, Maizes JS, Guinamard R, Ono M, Takai T, Ravetch JV. 1999. Modulation of
immune complex-induced inflammation in vivo by the coordinate expression of
activation and inhibitory Fc receptors. J Exp Med 189(1):179-185.
Crill WD, Chang GJ. 2004. Localization and characterization of flavivirus envelope
glycoprotein cross-reactive epitopes. J Virol 78(24):13975-13986.
Dai X, Jayapal M, Tay HK, Reghunathan R, Lin G, Too CT, Lim YT, Chan SH, Kemeny
DM, Floto RA, Smith KG, Melendez AJ, MacAry PA. 2009. Differential signal
transduction, membrane trafficking, and immune effector functions mediated by
FcgammaRI versus FcgammaRIIa. Blood 114(2):318-327.
Davis CW, Nguyen HY, Hanna SL, Sanchez MD, Doms RW, Pierson TC. 2006. West Nile
virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and
infection. J Virol 80(3):1290-1301.
Davis WG, Blackwell JL, Shi PY, Brinton MA. 2007. Interaction between the cellular protein
eEF1A and the 3'-terminal stem-loop of West Nile virus genomic RNA facilitates
viral minus-strand RNA synthesis. J Virol 81(18):10172-10187.
de la CSB, Kouri G, Guzman MG. 2007. Race: a risk factor for dengue hemorrhagic fever.
Arch Virol 152(3):533-542.
136

Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W,
Puttikhunt C, Edwards C, Duangchinda T, Supasa S, Chawansuntati K, Malasit P,
Mongkolsapaya J, Screaton G. 2010. Cross-reacting antibodies enhance dengue virus
infection in humans. Science 328(5979):745-748.
Della-Porta AJ, Westaway EG. 1978. A multi-hit model for the neutralization of animal
viruses. J Gen Virol 38(1):1-19.
DeTulleo L, Kirchhausen T. 1998. The clathrin endocytic pathway in viral infection. EMBO J
17(16):4585-4593.
Diamond MS, Edgil D, Roberts TG, Lu B, Harris E. 2000. Infection of human cells by
dengue virus is modulated by different cell types and viral strains. J Virol
74(17):7814-7823.
Dong T, Moran E, Vinh Chau N, Simmons C, Luhn K, Peng Y, Wills B, Phuong Dung N, Thi
Thu Thao L, Hien TT, McMichael A, Farrar J, Rowland-Jones S. 2007. High pro-
inflammatory cytokine secretion and loss of high avidity cross-reactive cytotoxic T-
cells during the course of secondary dengue virus infection. PLoS One 2(12):e1192.
Downs WG. 1969. Health protection in a shrinking world. Am J Trop Med Hyg 18(3):482.
Duan X, Lu X, Li J, Liu Y. 2008. Novel binding between pre-membrane protein and vacuolar
ATPase is required for efficient dengue virus secretion. Biochem Biophys Res
Commun 373(2):319-324.
Durbin AP, McArthur JH, Marron JA, Blaney JE, Thumar B, Wanionek K, Murphy BR,
Whitehead SS. 2006. rDEN2/4Delta30(ME), a live attenuated chimeric dengue
serotype 2 vaccine is safe and highly immunogenic in healthy dengue-naive adults.
Hum Vaccin 2(6):255-260.
Durbin AP, Whitehead SS, McArthur J, Perreault JR, Blaney JE, Jr., Thumar B, Murphy BR,
Karron RA. 2005. rDEN4delta30, a live attenuated dengue virus type 4 vaccine
candidate, is safe, immunogenic, and highly infectious in healthy adult volunteers. J
Infect Dis 191(5):710-718.
137

Egger JR, Ooi EE, Kelly DW, Woolhouse ME, Davies CR, Coleman PG. 2008.
Reconstructing historical changes in the force of infection of dengue fever in
Singapore: implications for surveillance and control. Bull World Health Organ
86(3):187-196.
Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus
products in infected cells interferes with stress granule formation and processing body
assembly. Proc Natl Acad Sci U S A 104(21):9041-9046.
Endy TP, Chunsuttiwat S, Nisalak A, Libraty DH, Green S, Rothman AL, Vaughn DW, Ennis
FA. 2002. Epidemiology of inapparent and symptomatic acute dengue virus infection:
a prospective study of primary school children in Kamphaeng Phet, Thailand. Am J
Epidemiol 156(1):40-51.
George R, Lum CS. 1997. Clinical spectrum of dengue infection. In: Gubler DJ, Kuno G,
editors. Dengue and dengue hemorrhagic fever. New York: CAB International. p 89-
113.
Goh KT. 1995. Changing epidemiology of dengue in Singapore. Lancet 346(8982):1098.
Gollins SW, Porterfield JS. 1985. Flavivirus infection enhancement in macrophages: an
electron microscopic study of viral cellular entry. J Gen Virol 66 ( Pt 9):1969-1982.
Gollins SW, Porterfield JS. 1986a. pH-dependent fusion between the flavivirus West Nile and
liposomal model membranes. J Gen Virol 67 ( Pt 1):157-166.
Gollins SW, Porterfield JS. 1986b. The uncoating and infectivity of the flavivirus West Nile
on interaction with cells: effects of pH and ammonium chloride. J Gen Virol 67 ( Pt
9):1941-1950.
Gromowski GD, Barrett ND, Barrett AD. 2008. Characterization of dengue virus complex-
specific neutralizing epitopes on envelope protein domain III of dengue 2 virus. J
Virol 82(17):8828-8837.
Gubler DJ. 1989a. Aedes aegypti and Aedes aegypti-borne disease control in the 1990s: top
down or bottom up. Charles Franklin Craig Lecture. Am J Trop Med Hyg 40(6):571-
578.
138

Gubler DJ. 1989b. Surveillance for dengue and dengue hemorrhagic fever. Bull Pan Am
Health Organ 23(4):397-404.
Gubler DJ. 1997. Dengue and dengue hemorrhagic fever: its history and resurgence as a
global public health problem. In: Gubler DJ, Kuno G, editors. Dengue and dengue
hemorrhagic fever. New York: CAB International. p 1-22.
Gubler DJ. 1998. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev 11(3):480-496.
Gubler DJ. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and
economic problem in the 21st century. Trends Microbiol 10(2):100-103.
Gubler DJ, Clark GG. 1994. Community-based integrated control of Aedes aegypti: a brief
overview of current programs. Am J Trop Med Hyg 50(6 Suppl):50-60.
Gubler DJ, Kuno G, Markoff L. 2007. Flaviviruses. In: Knipe DM, Howley PM, editors.
Field's Virology. 5th edition ed: Lippincott Williams & Wilkins.
Guzman MG, Kouri G, Valdes L, Bravo J, Alvarez M, Vazques S, Delgado I, Halstead SB.
2000. Epidemiologic studies on Dengue in Santiago de Cuba, 1997. Am J Epidemiol
152(9):793-799; discussion 804.
Halstead SB. 1965. Dengue and hemorrhagic fevers of Southeast Asia. Yale J Biol Med
37(6):434-454.
Halstead SB. 1980. Dengue haemorrhagic fever--a public health problem and a field for
research. Bull World Health Organ 58(1):1-21.
Halstead SB. 1988. Pathogenesis of dengue: challenges to molecular biology. Science
239(4839):476-481.
Halstead SB. 2006. Dengue in the Americas and Southeast Asia: do they differ? Rev Panam
Salud Publica 20(6):407-415.
Halstead SB. 2008a. Dengue virus-mosquito interactions. Annu Rev Entomol 53:273-291.
Halstead SB. 2008b. Dengue: Overview and History. Halstead SB, editor: Dengue. 508 p.
Halstead SB, O'Rourke EJ. 1977. Dengue viruses and mononuclear phagocytes. I. Infection
enhancement by non-neutralizing antibody. J Exp Med 146(1):201-217.
139

Halstead SB, O'Rourke EJ, Allison AC. 1977. Dengue viruses and mononuclear phagocytes.
II. Identity of blood and tissue leukocytes supporting in vitro infection. J Exp Med
146(1):218-229.
Halstead SB, Suaya JA, Shepard DS. 2007. The burden of dengue infection. Lancet
369(9571):1410-1411.
Halstead SB, Yamarat C. 1965. Recent Epidemics of Hemorrhagic Fever in Thailand.
Observations Related to Pathogenesis of a "New" Dengue Disease. Am J Public
Health Nations Health 55:1386-1395.
Hammon W, Rudnick A, Sather G. 1960. New hemorrhagic fevers of children in Philppines
and Thailand. Trans Assoc Am Physicians 73:140.
Hanson BJ, Boon AC, Lim AP, Webb A, Ooi EE, Webby RJ. 2006. Passive
immunoprophylaxis and therapy with humanized monoclonal antibody specific for
influenza A H5 hemagglutinin in mice. Respir Res 7:126.
Heim R, Prasher DC, Tsien RY. 1994. Wavelength mutations and posttranslational
autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A 91(26):12501-
12504.
Henchal EA, Gentry MK, McCown JM, Brandt WE. 1982. Dengue virus-specific and
flavivirus group determinants identified with monoclonal antibodies by indirect
immunofluorescence. Am J Trop Med Hyg 31(4):830-836.
Henchal EA, McCown JM, Seguin MC, Gentry MK, Brandt WE. 1983. Rapid identification
of dengue virus isolates by using monoclonal antibodies in an indirect
immunofluorescence assay. Am J Trop Med Hyg 32(1):164-169.
Hirsch AJ. 1883. Dengue, a comparatively new disease: Its symptoms. In: Creighton C,
editor. Handbook of Geographical and Historical Pathology, vol 1: London:
Sydenham Society.
Hirsch AJ, Medigeshi GR, Meyers HL, DeFilippis V, Fruh K, Briese T, Lipkin WI, Nelson
JA. 2005. The Src family kinase c-Yes is required for maturation of West Nile virus
particles. J Virol 79(18):11943-11951.
140

Ho BC, Chan KL, Chan YC. 1971. Aedes aegypti (L.) and Aedes albopictus (Skuse) in
Singapore City. 3. Population fluctuations. Bull World Health Organ 44(5):635-641.
Hotta S. 1952. Experimental studies on dengue. I. Isolation, identification and modification of
the virus. J Infect Dis 90(1):1-9.
Howe GM. 1977. A World Geography of Human Diseases. New York: Academic Press.
Huang CY, Butrapet S, Tsuchiya KR, Bhamarapravati N, Gubler DJ, Kinney RM. 2003.
Dengue 2 PDK-53 virus as a chimeric carrier for tetravalent dengue vaccine
development. J Virol 77(21):11436-11447.
Imrie A, Meeks J, Gurary A, Sukhbaatar M, Truong TT, Cropp CB, Effler P. 2007. Antibody
to dengue 1 detected more than 60 years after infection. Viral Immunol 20(4):672-
675.
Kimura T, Gollins SW, Porterfield JS. 1986. The effect of pH on the early interaction of West
Nile virus with P388D1 cells. J Gen Virol 67 ( Pt 11):2423-2433.
Kliks SC, Nimmanitya S, Nisalak A, Burke DS. 1988. Evidence that maternal dengue
antibodies are important in the development of dengue hemorrhagic fever in infants.
Am J Trop Med Hyg 38(2):411-419.
Kliks SC, Nisalak A, Brandt WE, Wahl L, Burke DS. 1989. Antibody-dependent
enhancement of dengue virus growth in human monocytes as a risk factor for dengue
hemorrhagic fever. Am J Trop Med Hyg 40(4):444-451.
Koh WL, Ng ML. 2005. Molecular mechanisms of West Nile virus pathogenesis in brain cell.
Emerg Infect Dis 11(4):629-632.
Kou Z, Quinn M, Chen H, Rodrigo WW, Rose RC, Schlesinger JJ, Jin X. 2008. Monocytes,
but not T or B cells, are the principal target cells for dengue virus (DV) infection
among human peripheral blood mononuclear cells. J Med Virol 80(1):134-146.
Kouri G, Guzman MG, Bravo J. 1986. Hemorrhagic dengue in Cuba: history of an epidemic.
Bull Pan Am Health Organ 20(1):24-30.
Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R,
Tsui M, Qian F, Montgomery RR, Lev S, Mason PW, Koski RA, Elledge SJ, Xavier
141

RJ, Agaisse H, Fikrig E. 2008. RNA interference screen for human genes associated
with West Nile virus infection. Nature 455(7210):242-245.
Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT,
Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. 2002. Structure
of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell
108(5):717-725.
Kurosu T, Chaichana P, Yamate M, Anantapreecha S, Ikuta K. 2007. Secreted complement
regulatory protein clusterin interacts with dengue virus nonstructural protein 1.
Biochem Biophys Res Commun 362(4):1051-1056.
Lai CY, Tsai WY, Lin SR, Kao CL, Hu HP, King CC, Wu HC, Chang GJ, Wang WK. 2008.
Antibodies to envelope glycoprotein of dengue virus during the natural course of
infection are predominantly cross-reactive and recognize epitopes containing highly
conserved residues at the fusion loop of domain II. J Virol 82(13):6631-6643.
Lai YL, Chung YK, Tan HC, Yap HF, Yap G, Ooi EE, Ng LC. 2007. Cost-effective real-time
reverse transcriptase PCR (RT-PCR) to screen for Dengue virus followed by rapid
single-tube multiplex RT-PCR for serotyping of the virus. J Clin Microbiol
45(3):935-941.
Liao CL, Lin YL, Wang JJ, Huang YL, Yeh CT, Ma SH, Chen LK. 1997. Effect of enforced
expression of human bcl-2 on Japanese encephalitis virus-induced apoptosis in
cultured cells. J Virol 71(8):5963-5971.
Libraty DH, Acosta LP, Tallo V, Segubre-Mercado E, Bautista A, Potts JA, Jarman RG,
Yoon IK, Gibbons RV, Brion JD, Capeding RZ. 2009. A prospective nested case-
control study of Dengue in infants: rethinking and refining the antibody-dependent
enhancement dengue hemorrhagic fever model. PLoS Med 6(10):e1000171.
Liew KJ, Chow VT. 2006. Microarray and real-time RT-PCR analyses of a novel set of
differentially expressed human genes in ECV304 endothelial-like cells infected with
dengue virus type 2. J Virol Methods 131(1):47-57.
142

Lin CW, Cheng CW, Yang TC, Li SW, Cheng MH, Wan L, Lin YJ, Lai CH, Lin WY, Kao
MC. 2008. Interferon antagonist function of Japanese encephalitis virus NS4A and its
interaction with DEAD-box RNA helicase DDX42. Virus Res 137(1):49-55.
Lindenbach BD, Thiel H-J, Rice CM. 2005. Flaviviridae: the viruses and their replication. In:
Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams
& Wilkins. p 1101-1152.
Lippincott-Schwartz J, Patterson GH. 2003. Development and use of fluorescent protein
markers in living cells. Science 300(5616):87-91.
Littaua R, Kurane I, Ennis FA. 1990. Human IgG Fc receptor II mediates antibody-dependent
enhancement of dengue virus infection. J Immunol 144(8):3183-3186.
Mackenzie JM, Khromykh AA, Parton RG. 2007. Cholesterol manipulation by West Nile
virus perturbs the cellular immune response. Cell Host Microbe 2(4):229-239.
Mackenzie JS, Gubler DJ, Petersen LR. 2004. Emerging flaviviruses: the spread and
resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med 10(12
Suppl):S98-109.
Mannik M. 1980. Physicochemical and functional relationships of immune complexes. J
Invest Dermatol 74(5):333-338.
Maron GM, Clara AW, Diddle JW, Pleites EB, Miller L, Macdonald G, Adderson EE. 2010.
Association between nutritional status and severity of dengue infection in children in
El Salvador. Am J Trop Med Hyg 82(2):324-329.
Marovich M, Grouard-Vogel G, Louder M, Eller M, Sun W, Wu SJ, Putvatana R, Murphy G,
Tassaneetrithep B, Burgess T, Birx D, Hayes C, Schlesinger-Frankel S, Mascola J.
2001. Human dendritic cells as targets of dengue virus infection. J Investig Dermatol
Symp Proc 6(3):219-224.
McGee CE, Lewis MG, Claire MS, Wagner W, Lang J, Guy B, Tsetsarkin K, Higgs S,
Decelle T. 2008. Recombinant chimeric virus with wild-type dengue 4 virus
premembrane and envelope and virulent yellow fever virus Asibi backbone sequences
is dramatically attenuated in nonhuman primates. J Infect Dis 197(5):693-697.
143

Miller JL, deWet BJ, Martinez-Pomares L, Radcliffe CM, Dwek RA, Rudd PM, Gordon S.
2008. The mannose receptor mediates dengue virus infection of macrophages. PLoS
Pathog 4(2):e17.
Monath TP. 2007. Dengue and yellow fever--challenges for the development and use of
vaccines. N Engl J Med 357(22):2222-2225.
Mongkolsapaya J, Dejnirattisai W, Xu XN, Vasanawathana S, Tangthawornchaikul N,
Chairunsri A, Sawasdivorn S, Duangchinda T, Dong T, Rowland-Jones S,
Yenchitsomanus PT, McMichael A, Malasit P, Screaton G. 2003. Original antigenic
sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med
9(7):921-927.
Mongkolsapaya J, Duangchinda T, Dejnirattisai W, Vasanawathana S, Avirutnan P, Jairungsri
A, Khemnu N, Tangthawornchaikul N, Chotiyarnwong P, Sae-Jang K, Koch M,
Jones Y, McMichael A, Xu X, Malasit P, Screaton G. 2006. T cell responses in
dengue hemorrhagic fever: are cross-reactive T cells suboptimal? J Immunol
176(6):3821-3829.
Moreira LA, Saig E, Turley AP, Ribeiro JM, O'Neill SL, McGraw EA. 2009. Human probing
behavior of Aedes aegypti when infected with a life-shortening strain of Wolbachia.
PLoS Negl Trop Dis 3(12):e568.
Moreno-Altamirano MM, Sanchez-Garcia FJ, Munoz ML. 2002. Non Fc receptor-mediated
infection of human macrophages by dengue virus serotype 2. J Gen Virol 83(Pt
5):1123-1130.
Nasirudeen AM, Liu DX. 2009. Gene expression profiling by microarray analysis reveals an
important role for caspase-1 in dengue virus-induced p53-mediated apoptosis. J Med
Virol 81(6):1069-1081.
Nimmerjahn F, Ravetch JV. 2008. Fcgamma receptors as regulators of immune responses.
Nat Rev Immunol 8(1):34-47.
Nobuchi H. 1979. The symptoms of a dengue-like illness recorded in a Chinese medical
encyclopedia (in Japanese). Kanpo no Rinsho 26:422.
144

Noisakran S, Perng GC. 2008. Alternate hypothesis on the pathogenesis of dengue
hemorrhagic fever (DHF)/dengue shock syndrome (DSS) in dengue virus infection.
Exp Biol Med (Maywood) 233(4):401-408.
Oh WK, Song J. 2006. Hsp70 functions as a negative regulator of West Nile virus capsid
protein through direct interaction. Biochem Biophys Res Commun 347(4):994-1000.
Oliphant T, Engle M, Nybakken GE, Doane C, Johnson S, Huang L, Gorlatov S, Mehlhop E,
Marri A, Chung KM, Ebel GD, Kramer LD, Fremont DH, Diamond MS. 2005.
Development of a humanized monoclonal antibody with therapeutic potential against
West Nile virus. Nat Med 11(5):522-530.
Ooi EE, Goh KT, Chee Wang DN. 2003. Effect of increasing age on the trend of dengue and
dengue hemorrhagic fever in Singapore. Int J Infect Dis 7(3):231-232.
Ooi EE, Goh KT, Gubler DJ. 2006. Dengue prevention and 35 years of vector control in
Singapore. Emerg Infect Dis 12(6):887-893.
Ooi EE, Gubler DJ. 2008. Dengue in Southeast Asia: epidemiological characteristics and
strategic challenges in disease prevention. Cad Saúde Pública, Rio de Janeiro 25(Sup
1):S115-S124.
Ooi EE, Gubler DJ. 2009a. Dengue in Southeast Asia: epidemiological characteristics and
strategic challenges in disease prevention. Cad Saude Publica 25 Suppl 1:S115-124.
Ooi EE, Gubler DJ. 2009b. Global spread of epidemic dengue: the influence of environmental
change. Future Virol 4(6).
Ooi EE, Hart TJ, Tan HC, Chan SH. 2001. Dengue seroepidemiology in Singapore. Lancet
357(9257):685-686.
Padwad YS, Mishra KP, Jain M, Chanda S, Karan D, Ganju L. 2009. RNA interference
mediated silencing of Hsp60 gene in human monocytic myeloma cell line U937
revealed decreased dengue virus multiplication. Immunobiology 214(6):422-429.
Panchuk-Voloshina N, Haugland RP, Bishop-Stewart J, Bhalgat MK, Millard PJ, Mao F,
Leung WY. 1999. Alexa dyes, a series of new fluorescent dyes that yield
145

exceptionally bright, photostable conjugates. J Histochem Cytochem 47(9):1179-
1188.
Paranjape SM, Harris E. 2007. Y box-binding protein-1 binds to the dengue virus 3'-
untranslated region and mediates antiviral effects. J Biol Chem 282(42):30497-30508.
Pastorino B, Boucomont-Chapeaublanc E, Peyrefitte CN, Belghazi M, Fusai T, Rogier C,
Tolou HJ, Almeras L. 2009. Identification of cellular proteome modifications in
response to West Nile virus infection. Mol Cell Proteomics 8(7):1623-1637.
Pastorino B, Nougairede A, Wurtz N, Gould E, de Lamballerie X. 2010. Role of host cell
factors in flavivirus infection: Implications for pathogenesis and development of
antiviral drugs. Antiviral Res.
Pattanakitsakul SN, Rungrojcharoenkit K, Kanlaya R, Sinchaikul S, Noisakran S, Chen ST,
Malasit P, Thongboonkerd V. 2007. Proteomic analysis of host responses in HepG2
cells during dengue virus infection. J Proteome Res 6(12):4592-4600.
Patz JA, Martens WJ, Focks DA, Jetten TH. 1998. Dengue fever epidemic potential as
projected by general circulation models of global climate change. Environ Health
Perspect 106(3):147-153.
Pelkmans L. 2005. Secrets of caveolae- and lipid raft-mediated endocytosis revealed by
mammalian viruses. Biochim Biophys Acta 1746(3):295-304.
Pepper OHP. 1941. A note on David Bylon and dengue. Ann Med Hist Ser 33:363.
Pierson TC, Diamond MS. 2008. Molecular mechanisms of antibody-mediated neutralisation
of flavivirus infection. Expert Rev Mol Med 10(10):e12.
Pierson TC, Fremont DH, Kuhn RJ, Diamond MS. 2008. Structural insights into the
mechanisms of antibody-mediated neutralization of flavivirus infection: implications
for vaccine development. Cell Host Microbe 4(3):229-238.
Polacek C, Friebe P, Harris E. 2009. Poly(A)-binding protein binds to the non-polyadenylated
3' untranslated region of dengue virus and modulates translation efficiency. J Gen
Virol 90(Pt 3):687-692.
146

Pryor MJ, Rawlinson SM, Butcher RE, Barton CL, Waterhouse TA, Vasudevan SG, Bardin
PG, Wright PJ, Jans DA, Davidson AD. 2007. Nuclear localization of dengue virus
nonstructural protein 5 through its importin alpha/beta-recognized nuclear
localization sequences is integral to viral infection. Traffic 8(7):795-807.
Rabablert J, Wasi C, Kinney R, Kasisith J, Pitidhammabhorn D, Ubol S. 2007. Attenuating
characteristics of DEN-2 PDK53 in flavivirus-naive peripheral blood mononuclear
cells. Vaccine 25(19):3896-3905.
Radstake TR, Franke B, Wenink MH, Nabbe KC, Coenen MJ, Welsing P, Bonvini E, Koenig
S, van den Berg WB, Barrera P, van Riel PL. 2006. The functional variant of the
inhibitory Fcgamma receptor IIb (CD32B) is associated with the rate of radiologic
joint damage and dendritic cell function in rheumatoid arthritis. Arthritis Rheum
54(12):3828-3837.
Ravetch JV, Bolland S. 2001. IgG Fc receptors. Annu Rev Immunol 19:275-290.
Rodrigo WW, Block OK, Lane C, Sukupolvi-Petty S, Goncalvez AP, Johnson S, Diamond
MS, Lai CJ, Rose RC, Jin X, Schlesinger JJ. 2009. Dengue virus neutralization is
modulated by IgG antibody subclass and Fcgamma receptor subtype. Virology 13:13.
Rodrigo WW, Jin X, Blackley SD, Rose RC, Schlesinger JJ. 2006. Differential enhancement
of dengue virus immune complex infectivity mediated by signaling-competent and
signaling-incompetent human Fcgamma RIA (CD64) or FcgammaRIIA (CD32). J
Virol 80(20):10128-10138.
Rush AB. 1789. An account of the bilious remitting fever, as it appeared in Philapelphia in
the summer and autumn of the year 1780. In: Rush AB, editor. Medical Inquiries and
Observations. Philadelphia: Prichard & Hall.
Russell PK, Udomsakdi S, Halstead SB. 1967. Antibody response in dengue and dengue
hemorrhagic fever. Jpn J Med Sci Biol 20 Suppl:103-108.
Sabin AB. 1952. Research on dengue during World War II. Am J Trop Med Hyg 1(1):30-50.
Sabin AB, Schlesinger RW. 1945. Production of Immunity to Dengue with Virus Modified by
Propagation in Mice. Science 101(2634):640-642.
147

Sangkawibha N, Rojanasuphot S, Ahandrik S, Viriyapongse S, Jatanasen S, Salitul V,
Phanthumachinda B, Halstead SB. 1984. Risk factors in dengue shock syndrome: a
prospective epidemiologic study in Rayong, Thailand. I. The 1980 outbreak. Am J
Epidemiol 120(5):653-669.
Schlesinger JJ, Chapman SE. 1999. Influence of the human high-affinity IgG receptor
FcgammaRI (CD64) on residual infectivity of neutralized dengue virus. Virology
260(1):84-88.
Seet RC, Ooi EE, Wong HB, Paton NI. 2005. An outbreak of primary dengue infection
among migrant Chinese workers in Singapore characterized by prominent
gastrointestinal symptoms and a high proportion of symptomatic cases. J Clin Virol
33(4):336-340.
Sencer DJ. 1969. Health protection in a shrinking world. Am J Trop Med Hyg 18(3):341-345.
Shaner NC, Patterson GH, Davidson MW. 2007. Advances in fluorescent protein technology.
J Cell Sci 120(Pt 24):4247-4260.
Simmons CP, Chau TN, Thuy TT, Tuan NM, Hoang DM, Thien NT, Lien le B, Quy NT,
Hieu NT, Hien TT, McElnea C, Young P, Whitehead S, Hung NT, Farrar J. 2007.
Maternal antibody and viral factors in the pathogenesis of dengue virus in infants. J
Infect Dis 196(3):416-424.
Smith KG, Clatworthy MR. 2010. FcgammaRIIB in autoimmunity and infection:
evolutionary and therapeutic implications. Nat Rev Immunol 10(5):328-343.
Soper FL. 1963. The elimination of urban yellow fever in the Americas through the
eradication of Aedes aegypti. Am J Public Health Nations Health 53:7-16.
Soper FL. 1969. Health protection in a shrinking world. Am J Trop Med Hyg 18(3):482-484.
Stadler K, Allison SL, Schalich J, Heinz FX. 1997. Proteolytic activation of tick-borne
encephalitis virus by furin. J Virol 71(11):8475-8481.
Stephens HA. 2010. HLA and other gene associations with dengue disease severity. Curr Top
Microbiol Immunol 338:99-114.
148

Su K, Yang H, Li X, Gibson AW, Cafardi JM, Zhou T, Edberg JC, Kimberly RP. 2007.
Expression profile of FcgammaRIIb on leukocytes and its dysregulation in systemic
lupus erythematosus. J Immunol 178(5):3272-3280.
Sukupolvi-Petty S, Austin SK, Purtha WE, Oliphant T, Nybakken GE, Schlesinger JJ,
Roehrig JT, Gromowski GD, Barrett AD, Fremont DH, Diamond MS. 2007. Type-
and subcomplex-specific neutralizing antibodies against domain III of dengue virus
type 2 envelope protein recognize adjacent epitopes. J Virol 81(23):12816-12826.
Tan BT. 1997. Control of dengue fever/dengue hemorrhagic fever in Singapore. Dengue
Bulletin 21:30-34.
Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, Eller
MA, Pattanapanyasat K, Sarasombath S, Birx DL, Steinman RM, Schlesinger S,
Marovich MA. 2003. DC-SIGN (CD209) mediates dengue virus infection of human
dendritic cells. J Exp Med 197(7):823-829.
Thayan R, Huat TL, See LL, Khairullah NS, Yusof R, Devi S. 2009. Differential expression
of aldolase, alpha tubulin and thioredoxin peroxidase in peripheral blood
mononuclear cells from dengue fever and dengue hemorrhagic fever patients.
Southeast Asian J Trop Med Public Health 40(1):56-65.
Thisyakorn U, Nimmannitya S. 1993. Nutritional status of children with dengue hemorrhagic
fever. Clin Infect Dis 16(2):295-297.
Trirawatanapong T, Chandran B, Putnak R, Padmanabhan R. 1992. Mapping of a region of
dengue virus type-2 glycoprotein required for binding by a neutralizing monoclonal
antibody. Gene 116(2):139-150.
Tsuda Y, Mori Y, Abe T, Yamashita T, Okamoto T, Ichimura T, Moriishi K, Matsuura Y.
2006. Nucleolar protein B23 interacts with Japanese encephalitis virus core protein
and participates in viral replication. Microbiol Immunol 50(3):225-234.
van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X,
Smit JM. 2008. Dissecting the cell entry pathway of dengue virus by single-particle
tracking in living cells. PLoS Pathog 4(12):e1000244.
149

van der Schaar HM, Rust MJ, Waarts BL, van der Ende-Metselaar H, Kuhn RJ, Wilschut J,
Zhuang X, Smit JM. 2007. Characterization of the early events in dengue virus cell
entry by biochemical assays and single-virus tracking. J Virol 81(21):12019-12028.
Vashist S, Anantpadma M, Sharma H, Vrati S. 2009. La protein binds the predicted loop
structures in the 3' non-coding region of Japanese encephalitis virus genome: role in
virus replication. J Gen Virol 90(Pt 6):1343-1352.
Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP,
Raengsakulrach B, Rothman AL, Ennis FA, Nisalak A. 2000. Dengue viremia titer,
antibody response pattern, and virus serotype correlate with disease severity. J Infect
Dis 181(1):2-9.
Veiga-Fernandes H, Walter U, Bourgeois C, McLean A, Rocha B. 2000. Response of naive
and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol 1(1):47-53.
Wahala WM, Kraus AA, Haymore LB, Accavitti-Loper MA, de Silva AM. 2009. Dengue
virus neutralization by human immune sera: role of envelope protein domain III-
reactive antibody. Virology 392(1):103-113.
Wang S, Hazelrigg T. 1994. Implications for bcd mRNA localization from spatial distribution
of exu protein in Drosophila oogenesis. Nature 369(6479):400-403.
WHO. 1997. Chapter 2: Clinical Diagnosis. Dengue haemorrhagic fever: diagnosis, treatment,
prevention and control 2nd edition Geneva: World Health Organization.
WHO. 2007. Report of Dengue Scientific Working Group. TDR⁄SWG⁄ 08, Geneva.
WHO. 2009. Dengue and dengue hemorrhagic fever.
Wilder-Smith A, Gubler DJ. 2008. Geographic expansion of dengue: the impact of
international travel. Med Clin North Am 92(6):1377-1390, x.
Wilson ME, Chen LH. 2002. Dengue in the Americas. Dengue Bulletin 26:44-61.
Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, Louder MK,
Filgueira L, Marovich MA, Wong HK, Blauvelt A, Murphy GS, Robb ML, Innes BL,
Birx DL, Hayes CG, Frankel SS. 2000. Human skin Langerhans cells are targets of
dengue virus infection. Nat Med 6(7):816-820.
150

Yang SH, Liu ML, Tien CF, Chou SJ, Chang RY. 2009. Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) interaction with 3' ends of Japanese encephalitis virus RNA
and colocalization with the viral NS5 protein. J Biomed Sci 16:40.
Yen YT, Chen HC, Lin YD, Shieh CC, Wu-Hsieh BA. 2008. Enhancement by tumor necrosis
factor alpha of dengue virus-induced endothelial cell production of reactive nitrogen
and oxygen species is key to hemorrhage development. J Virol 82(24):12312-12324.
Zhang J, Campbell RE, Ting AY, Tsien RY. 2002. Creating new fluorescent probes for cell
biology. Nat Rev Mol Cell Biol 3(12):906-918.
Zhang SL, Tan HC, Hanson BJ, Ooi EE. 2010. A simple method for Alexa Fluor dye
labelling of dengue virus. J Virol Methods 167(2):172-177.
Zimmer M. 2009. GFP: from jellyfish to the Nobel prize and beyond. Chem Soc Rev
38(10):2823-2832.
151

152

Appendix
Zhang Lixin (HT080076A)


1. Cell culture medium
1.1. To prepare fetal bovine serum (FBS)
Heat-inactivate at 56°C for 30mins and aliquot into 50ml Falcon tubes. Store at -20°C till
further use.
1.2. To prepare BHK-21 growth medium, 500ml
5ml of L-glutamine (200nM, Gibco)
5ml of sodium pyruvate (100mM, Gibco)
5ml of Penicillin/Streptomycin (100X liquid, Gibco)
50ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.3. To prepare BHK-21 maintenance medium, 500ml
5ml of L-glutamine (200nM, Gibco)
5ml of sodium pyruvate (100mM, Gibco)
5ml of Penicillin/Streptomycin (100X liquid, Gibco)
10ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.4. To prepare Vero growth medium, 500ml
5ml of sodium pyruvate (100mM, Gibco)
5ml of Non-essential amino acids (10mM, Gibco)
50ml FBS
Top up to 500ml with Medium-199 (Gibco) and filter-sterilize
1.5. To prepare Vero maintenance medium, 500ml
155

5ml of sodium pyruvate (100mM, Gibco)
5ml of Non-essential amino acids (10mM, Gibco)
10ml FBS
Top up to 500ml with Medium-199 (Gibco) and filter-sterilize
1.6. To prepare THP-1 growth medium, 500ml
5ml of L-glutamine (200nM, Gibco)
5ml of sodium pyruvate (100mM, Gibco)
5ml of HEPES (1M, Gibco)
454µl of β-mercaptoethanol (55mM, Gibco)
50ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.7. To prepare THP-1 maintenance medium, 500ml
5ml of L-glutamine (200nM, Gibco)
5ml of sodium pyruvate (100mM, Gibco)
5ml of HEPES (1M, Gibco)
454µl of β-mercaptoethanol (55mM, Gibco)
10ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.8. To prepare Raji-DC SIGN growth medium, 500ml
5ml of L-glutamine (200nM, Gibco)
50ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.9. To prepare Raji-DC SIGN maintenance medium, 500ml
156

5ml of L-glutamine (200nM, Gibco)
10ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.10. To prepare primary monocytes growth medium, 500ml
5ml of L-glutamine (200nM, Gibco)
5ml of sodium pyruvate (100mM, Gibco)
5ml of Non-essential amino acids (10mM, Gibco)
5ml of Penicillin/Streptomycin (100X liquid, Gibco)
50ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.11. To prepare primary monocytes maintenance medium, 500ml
5ml of L-glutamine (200nM, Gibco)
5ml of sodium pyruvate (100mM, Gibco)
5ml of Non-essential amino acids (10mM, Gibco)
5ml of Penicillin/Streptomycin (100X liquid, Gibco)
10ml FBS
Top up to 500ml with RPMI-1640 (Gibco) and filter-sterilize
1.12. To prepare 0.5% BSA/PBS
Dissolve 0.25g bovine serum albumin (BSA, Sigma) in 1XPBS and filter-sterilize.
157

2. Alexa Fluor labelling buffers
2.1. Sodium bicarbonate buffer, pH 8.3
Dissolve 0.84g of sodium bicarbonate powder in 50ml deionized water. Adjust pH, if
necessary, to 8.3 and filter-sterilize.
2.2. 1.5M Hydroxylamine, pH 8.5
Dissolve 1.05g of hydroxylamine in 5ml deionized water. Adjust pH to 8.5 with 5M
sodium hydroxide, top up to 10ml with deionized water and filter-sterilize.
2.3. HNE buffer, pH 7.4
0.651g HEPES (Sigma)
4.38g sodium chloride (Sigma)
0.019g EDTA (Sigma)
Top up with 500ml deionized water, adjust pH to 7.4 and filter-sterilize.
158

3. Plaque assay
3.1. 0.8% methycellulose overlay
A. 2X RPMI
Dissolve RPMI-1640 powder (Gibco) in 500ml deionized water
10ml sodium bicarbonate (7.5%, Gibco)
5ml sodium pyruvate (100mM, Gibco)
20ml FBS
Filter-sterilize
B. 1.6% carboxy-methyl cellulose (CMC)
Dissolve 8g of CMC (Calbiochem) in deionized water
Autoclave-sterilize at 121°C, 20min
Add 1 part of 2X RPMI (A) to 1 part of 1.6% CMC (B) in 50ml Falcon tubes. Shake
vigorously to mix. Store at 4°C.
3.2. 0.5% crystal violet in 25% formaldehyde
5g crystal violet (Sigma)
676ml 37% Formaldehyde
Top up to 1L with 1XPBS
159

4. Buffers for Immuno-assays
4.1. FACS wash buffer
10ml FBS
Top up to 1L with 1XPBS
4.2. FACS perm solution (0.1% saponin)
Dissolve 1g saponin (Sigma) in 1L of 1X PBS.
4.3. 12% paraformaldehyde (pFA)
Dissolve 12g of pFA (Sigma) in 90ml of deionized water. Adjust pH to 7.2 and top up to
100ml with 1X PBS. Filter-sterilize and store in -80°C in aliquots.
4.4. 3% pFA
Thaw a vial of 12% pFA and dilute 1 in 4 with 1X PBS.
4.5. Confocal wash buffer
Dissolve 1g saponin (Sigma) in 1L of 1X PBS.
4.6. Confocal perm buffer
1g of saponin (Sigma)
5g BSA (Sigma)
Top up to 100ml with 1X PBS
4.7. ELISA coating buffer
0.212g sodium carbonate (Sigma)
0.252g sodium bicarbonate (Sigma)
160

Dissolve in 50ml deionized water, adjust pH to 9.66 and filter-sterilize.
4.8. ELISA wash buffer (PBST)
Add 500µl of Tween-20 to 1L of 1X PBS.
4.9. ELISA blocking buffer (1%)
Dissolve 0.5g skim milk in 50ml of 1X PBS.
4.10. ELISA antibody diluents (0.5%)
Dissolve 0.25g skim milk in 50ml of 1X PBS
161

162
5. Sucrose gradient
5.1. 10% sucrose in HNE buffer
5g sucrose (Sigma)
Top up to 50ml with HNE buffer and filter-sterilize.
5.2. 20% sucrose in HNE buffer
10g sucrose (Sigma)
Top up to 50ml with HNE buffer and filter-sterilize.
5.3. 30% sucrose in HNE buffer
15g sucrose (Sigma)
Top up to 50ml with HNE buffer and filter-sterilize.
5.4. 40% sucrose in HNE buffer
20g sucrose (Sigma)
Top up to 50ml with HNE buffer and filter-sterilize.
5.5. 50% sucrose in HNE buffer
25g sucrose (Sigma)
Top up to 50ml with HNE buffer and filter-sterilize.
5.6. 60% sucrose in HNE buffer
30g sucrose (Sigma)
Top up to 50ml with HNE buffer and filter-sterilize.

Author's personal copy
Journal of Virological Methods 167 (2010) 172–177
Contents lists available at ScienceDirect
Journal of Virological Methods
journa l homepage: www.e lsev ier .com/ locate / jv i romet
A simple method for Alexa Fluor dye labelling of dengue virus
Summer Li-Xin Zhanga,b, Hwee-Cheng Tanb, Brendon J. Hansona, Eng Eong Ooia,b,∗
a Defence Medical and Environmental Research Institute, DSO National Laboratories, 27 Medical Drive, 117510 Singapore, Singaporeb Duke-NUS Graduate Medical School, 8 College Road, 169857 Singapore, Singapore
Article history:Received 6 March 2010Received in revised form 31 March 2010Accepted 8 April 2010Available online 23 April 2010
Keywords:Dengue virusAlexa FluorFluorescent virusLabellingFluorescence
a b s t r a c t
Dengue virus causes frequent and cyclical epidemics throughout the tropics, resulting in significant mor-bidity and mortality rates. There is neither a specific antiviral treatment nor a vaccine to prevent epidemictransmission. The lack of a detailed understanding of the pathogenesis of the disease complicates theseefforts. The development of methods to probe the interaction between the virus and host cells would thusbe useful. Direct fluorescence labelling of virus would facilitate the visualization of the early events invirus–cell interaction. This report describes a simple method of labelling of dengue virus with Alexa Fluorsuccinimidyl ester dye dissolved directly in the sodium bicarbonate buffer that yielded highly viable virusafter labelling. Alexa Fluor dyes have superior photostability and are less pH-sensitive than the commondyes, such as fluorescein and rhodamine, making them ideal for studies on cellular uptake and endosomaltransport of the virus. The conjugation of Alexa Fluor dye did not affect the recognition of labelled denguevirus by virus-specific antibody and its putative receptors in host cells. This method could have usefulapplications in virological studies.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
Dengue is a significant disease globally. With rapid urbaniza-tion, lack of vector control and increase in air travel, dengue hasbecome the most common and rapidly spreading arthropod-borneviral disease (Gubler, 1998). An estimated 50–100 million dengueinfections occur annually, and more are at risk of being infectedwith 2.5 billion people living in dengue endemic countries (WHO,2007). Despite research efforts, however, there remains an incom-plete understanding on the mechanism underlying pathogenesis,which limits antiviral and vaccine development. Among these gapsin knowledge are the specific cell-surface binding motifs and earlyviral entry processes. The availability of tools to visualize these earlyevents can accelerate this area of research.
Dengue virus (DENV) is an enveloped, positive strand RNA virusthat belongs to the family of Flaviviridae. Its structure consists ofan external icosahedral scaffold of 90 envelope glycoprotein (E)dimers protecting the nucleocapsid shell, which contains the RNAgenome (Kuhn et al., 2002). Dengue virus surface has several identi-cal protein subunits and can therefore be labelled at multiple sitesusing an amine reactive dye; the resulting fluorescence intensitymay be sufficient to track the virus under a fluorescence micro-scope.
∗ Corresponding author at: Duke-NUS Graduate Medical School, 8 College Road,169857 Singapore, Singapore. Tel.: +65 6516 8594; fax: +65 6221 2529.
E-mail address: [email protected] (E.E. Ooi).
A simple procedure of labelling dengue virus with a fluores-cent Alexa Fluor succinimidyl ester that results in minimal lossof infectivity post-labelling is described. Alexa Fluor dye was cho-sen as it has better photostability and signal intensity comparedto common fluorophores such as fluorescein isothiocyanate (FITC)or cyanine 3 bihexanoic acid (Cy3). In addition, Alexa Fluor dyesare stable at a lower pH compared to the common fluorophores,which could be useful for visualizing labelled viral particles asthey move through the acidic compartments of endosomes post-infection. Furthermore, the permanence of covalent conjugationof the fluorophores to the surface proteins allows for storage ofbatch-labelled virus at −80 ◦C, providing uniformity across multi-ple experiments. It also opens up the possibility of performing livecell imaging to compliment the ‘snapshot’ imaging of conventionalfluorescence microscopy.
2. Materials and methods
2.1. Cells and antibody
All cells and hybridomas were obtained from The AmericanType Culture Collection (ATCC, Manassas, VA), and all cell culturemedia and supplements were purchased from Gibco (Invitrogen,Singapore). Baby hamster kidney BHK-21 cells were maintained inRoswell Park Memorial Institute 1640 medium (RPMI-1640), sup-plemented with 10% fetal bovine serum, penicillin (100 U/ml) andstreptomycin (100 �g/ml) at 37 ◦C with 5% CO2. African green mon-key kidney Vero cells were maintained in Medium-199 (M199)
0166-0934/$ – see front matter © 2010 Elsevier B.V. All rights reserved.doi:10.1016/j.jviromet.2010.04.001

Author's personal copy
S.L.-X. Zhang et al. / Journal of Virological Methods 167 (2010) 172–177 173
supplemented with 10% fetal bovine serum and 4 mM l-glutamineat 37 ◦C with 5% CO2. Mouse anti-dengue virus serotype 2 E pro-tein monoclonal antibody hybridoma, 3H5 (ATCC: HB46), wasmaintained in RPMI-1640 supplemented with 10% fetal bovineserum. Dendritic cell-specific ICAM3-grabbing non-integrin (DC-SIGN) transfected-Raji B cell line was a kind gift from Timothy H.Burgess (Naval Medical Research Centre, US) and maintained inRPMI medium supplemented with 10% fetal bovine serum. AlexaFluor 488 (AF488) anti-mouse IgG antibody was purchased fromMolecular Probes, Invitrogen and used at 1:100 dilution.
2.2. Virus culture and purification
DENV serotype 2 (DENV-2) (ST) strain was first isolated from aclinical sample in the Singapore General Hospital using an Aedesalbopictus mosquito cell line C6/36 and subsequently propagatedin Vero cells. The culture supernatant was harvested when 75%of the cells showed cytopathic effect and clarified by centrifuga-tion at 1000 × g for 30 min at 4 ◦C. Virus in the supernatant wasconcentrated by centrifugation at 30,000 × g for 2 h at 4 ◦C. The pel-let was resuspended in 5 mM Hepes, 150 mM NaCl, 0.1 mM EDTA(HNE buffer, pH 7.4) and purified further on a 30% sucrose cush-ion by ultracentrifugation in a Beckman SW41Ti rotor at 80,000 × gfor 15 h at 4 ◦C. Purified virus was resuspended in HNE buffer andstored in 100 �l aliquots at −80 ◦C. A limiting dilution plaque assaywas performed on BHK cell line to determine the viral titre in plaqueforming units per millilitre (pfu/ml).
2.3. Plaque assay
Tenfold serial dilutions of virus were added to BHK-21 cells in24 well plates and incubated for 1 h at 37 ◦C, with gentle rock-ing every 15 min. The medium was then aspirated and replacedwith 0.8% methyl-cellulose in maintenance medium (RPMI-1640,2% fetal bovine serum, penicillin and streptomycin). After 4 days at37 ◦C, the cells were fixed with 25% formaldehyde and stained with0.5% crystal violet. The plates were washed, dried and the plaqueforming units per ml (pfu/ml) calculated.
2.4. Virus labelling
For the initial experiment aimed at determining the opti-mal concentration of dye needed to label DENV, Alexa Fluor594 succinimidyl ester (AF594SE, Molecular Probes, Invitrogen)was reconstituted in 0.2 M sodium bicarbonate buffer, pH 8.3(SBB) immediately before labelling, and added to approximately3 × 107 pfu DENV diluted in SBB at final concentrations of 10,50,100, 200, 500 and 1000 �M of dye, while stirring gently. Thedye-virus mixture was incubated at room temperature for 1 hwith gentle inversions every 15 min. The labelling reaction wasstopped by adding freshly prepared 1.5 M hydroxylamine, pH 8.5(Sigma–Aldrich) and incubated at room temperature for 1 h withgentle inversions every 15 min. Labelled DENV was re-titrated afterlabelling and tested for fluorescence using immunofluorescenceassay.
For subsequent AF594SE or AF488SE labelling involving largerbatches of virus, the same approach was employed and the labelledDENV was purified using Sephadex G-25 columns (Amersham, GEHealthcare, Singapore) to remove the excess dye. The labelled viruswas stored in 100 �l aliquots at −80 ◦C, re-titrated and tested forfluorescence before use.
2.5. Immunofluorescence assay
Equal volume of AF594-labelled DENV was incubated with Verocells plated on coverslips for 10 min at 37 ◦C, washed, fixed with 3%
paraformaldehyde (Sigma–Aldrich, Singapore) and permeabilizedwith 0.1% saponin (Sigma–Aldrich, Singapore). The cells were thenincubated for 1 h with undiluted centrifugation-clarified super-natant of 3H5 monoclonal antibody hybridoma culture, at roomtemperature. The cells were washed three times in wash buffer (PBScontaining 1 mM calcium chloride, 1 mM magnesium chloride and0.1% saponin), followed by incubation with AF488 anti-mouse IgGantibody for 45 min at room temperature. Cells were then washedthree times with the wash buffer, rinsed once with deionized waterand mounted on glass slides with Mowiol 4-88 (Calbiochem, SanDiego, CA) with 2.5% Dabco (Sigma–Aldrich, Singapore). Cells werevisualized at 63× magnification using a Leica Microsystem TCS SP5confocal microscope and merged images exported for processing.Processing of the images involved adjustment of the contrast of theimages on a whole for clarity and exported in individual colours forunmerged images using Adobe Photoshop CS3 version 10.
2.6. Flow cytometry determination of labelled DENV
Raji B cells transfected with DC-SIGN were counted, aliquoted,centrifuged and resuspended in AF488-labelled DENV at multiplic-ity of infection (MOI) of 1 and incubated for 10 min at 37 ◦C, thenfixed with 3% paraformaldehyde. The cells were then washed twicewith FACS buffer consisting of PBS with 0.1% fetal bovine serum andpermeabilized with 0.1% saponin in PBS. Cells were stained subse-quently for the presence of DENV using anti-E protein monoclonalantibody, 3H5, and PE anti-mouse anti-IgG antibody before readingon BD FACS Calibur machine and analysed with CellQuest software,version 3.3 (Becton Dickinson).
2.7. Detection by SYBR green-based real-time PCR
Real-time detection of viral RNA was conducted as describedpreviously (Lai et al., 2007) using pan-dengue forward and reverseprimers with some modifications. Briefly, RNA extraction was car-ried out using QIAamp viral RNA mini kit for purified non-labelledand AF594-labelled DENV (AF594-DENV), or RNeasy mini kit (bothfrom QIAGEN, Hilden, Germany) for total RNA extraction from cellsaccording to the manufacturer’s instructions. Complementary DNAwas synthesized using SuperScript III First Strand Synthesis Sys-tem (Invitrogen, Singapore) with random hexamers and accordingto the manufacturer’s instructions. RNA copy number was thendetermined by SYBR green-based real-time PCR on LightCycler 480Real-Time PCR System (Roche Diagnostics, Penzberg, Germany).
2.8. Growth kinetics
Vero cells were seeded at 2 × 105 per well in 24 well plates andincubated at 37 ◦C for 4 h before infecting at a MOI of 0.1 with eitherpurified DENV or AF594-DENV for 1 h at 37 ◦C, with gentle rockingevery 15 min. The cells were then washed twice with 1 ml per wellPBS and replaced with maintenance medium (M199 supplementedwith 2% FBS). Supernatant was harvested from wells and stored at−80 ◦C for plaque assay, and cells were washed twice in PBS andlysed with RLT buffer provided in RNeasy mini kit at 0, 6, 8, 12, 24, 48and 72 h post-adsorption. The cell lysates were stored frozen untilall samples were collected and subsequently extracted accordingto the kit’s protocol. Viral RNA was detected using real-time PCR.
2.9. Statistical analysis
Statistical analyses were performed using unpaired Student’s ttest (Graphpad Prism 5.0). P value < 0.05 was considered significant.

Author's personal copy
174 S.L.-X. Zhang et al. / Journal of Virological Methods 167 (2010) 172–177
3. Results
3.1. Infectivity of DENV post-labelling
DENV was re-titrated by plaque assay post-labelling withAF594SE (Fig. 1a). The virus titre of DENV labelled with 10–100 �Mof AF594SE dye showed approximately 15-fold reduction, andDENV labelled with 500 and 200 �M AF594SE dye showed approx-imately 26- to 29-fold reductions, respectively, compared toequivalent number of non-labelled virus. DENV labelled with 1 mMof AF594SE dye showed an 82-fold reduction.
3.2. Intensity of AF594 labelling
Equal volume of DENV labelled with various concentrations ofAF594SE dye was incubated with Vero cells seeded on coverslips,fixed in 3% paraformaldehyde and stained with mouse anti-E mon-oclonal antibody, 3H5, and AF488 anti-mouse IgG antibody. Thecoverslips were then mounted on glass slides and examined usinga confocal microscope.
Cells infected with DENV labelled with 10 �M to 1 mM AF594SEdye showed decreased staining for E protein with concentrationof dye above 200 �M, indicating a reduction in virus infectivitypost-labelling with higher concentrations of dye (Fig. 1b). This wasconsistent with the plaque assay results. DENV labelled with 10and 50 �M of dye showed no discernable AF594 fluorescence, whileDENV labelled with 100 and 200 �M of AF594SE dye displayed highAF594 fluorescence which co-localized well with the E protein ashighlighted by the 3H5 monoclonal antibody. Fluorescence for bothAF594 and E protein diminished with AF594SE dye concentrationsof 500 �M and 1 mM.
3.3. Efficiency of Alexa Fluor dye labelling
DC-SIGN transfected-Raji B cells were infected with AF488-labelled DENV at a MOI of 1, stained for the presence of E proteinusing 3H5 monoclonal antibody and analysed by flow cytometry.Compared to the uninfected Raji B cells with DC-SIGN, approxi-mately 81.4% of the cells following infection were positive for theE protein. Of these cells, 58.4% were positive for both E proteinand AF488 (Fig. 2a), suggesting that approximately 71% of the viri-ons were labelled with the Alexa Fluor dye. Similar conclusionscan be drawn from the Pearson’s correlation values obtained byanalysis of confocal microscopy images showing co-localization ofAF594-DENV with E protein (Fig. 2b).
3.4. Reproducibility of labelling
Three independent AF594SE labelling experiments were per-formed with the same approximate starting titre of 2 × 108 pfuof purified DENV using the same procedure as described in Sec-tion 2. The titre post-labelling was then determined by plaqueassay. All three experiments showed three- to eightfold reduc-tion in titre post-labelling and column purification, from a meantitre of 2.21 × 108 (SD = 3.68 × 107) to 3.78 × 107 (SD = 1.5 × 107). Toexclude the possibility that the reduction in titre post-labelling wasdue to a loss in infectivity of the virus, the ratio of RNA copy numberto plaque forming unit of DENV pre- and post-labelling was anal-ysed (the same batch of purified virus was used). No significantdifference in the viral RNA copy number to plaque forming unitratio was observed between pre- and post-labelled virus (Table 1,P = 0.9507), indicating that the fall in titre was not due to inactiva-tion of the virions but rather due to loss during column purification.
Fig. 1. Dengue virus viability post AF594 labelling. Purified dengue virus waslabelled with various concentrations of AF594SE dye for 1 h at room temperature(described in Section 2.4). The labelled virus was then re-titrated post-labelling byplaque assay. Bar diagram shows average virus titre of four determinations, pre- andpost-labelling (a). Error bars indicate standard deviation. The labelled virus was alsotested for fluorescence intensity by immunofluorescence assay (b). Vero cells seededon coverslips the day prior are infected with labelled DENV for 10 min at 37 ◦C. Thecells were subsequently fixed and labelled with anti-E antibody, and examined forco-localization of E protein (green) and AF594 labelling (red). Fluorescent signalsare visualized under 63× magnification using Leica Microsystem TCS SP5 confo-cal microscope. Scale bar is 10 �m. White arrows indicate areas of co-localization(yellow when merged). (For interpretation of the references to colour in this figurelegend, the reader is referred to the web version of the article.)

Author's personal copy
S.L.-X. Zhang et al. / Journal of Virological Methods 167 (2010) 172–177 175
Fig. 2. Efficiency of Alexa Fluor dye labelling as a function of percentage infected. Raji B cells transfected with DC-SIGN were infected at a MOI of 1 with AF488-DENV for 10 minat 37 ◦C, fixed and stained for the presence of E protein. Fluorescence was then measured using BD FACS Calibur (a). Close to 100% of uninfected Raji B cells with DC-SIGNwere double negative for both Alexa Fluor and anti-E antibody signals, whereas approximately 81% of the infected cells stained positive for E protein, and approximately71% of which are positive for Alexa Fluor fluorescence, suggesting that approximately 71% of the virions were labelled with the dye. Confocal microscope analysis of Verocells infected with AF594-DENV (red) and stained for E protein (green) showed a mean Pearson’s correlation of 0.677 ± 0.053 (mean ± SD) (b), suggesting similar labellingefficiency (yellow indicates co-localization). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of the article.)
3.5. Growth kinetics of labelled DENV
In order to ascertain further that labelling of the surface proteinsof DENV did not interfere with infectivity and growth kinetics of thevirus, Vero cells were infected with either purified non-labelled orAF594-DENV at a MOI of 0.1. Culture supernatant and cell lysateswere collected up to 72 h and assayed for virus titre in supernatantas well as viral RNA in the cell lysates. No significant differencewas observed between labelled and unlabelled virus for both viralRNA copy number and the number of infectious particles in thesupernatant of the culture (Fig. 3) (P > 0.8).
Table 1Viral RNA copy number to infectious particle ratio.
Titre (pfu/ml) RNA copynumber (/ml)
RNA copy numberto pfu ratio
AF594-DENV 3.5 × 107 6.27 × 108 17.9Purified DENV 1.0 × 109 1.67 × 1010 16.7
Viral RNA was extracted from purified non-labelled DENV or AF594-DENV and RNAcopy number quantitated by real-time PCR. The RNA copy number was then dividedby the titre to obtain a ratio for both non-labelled DENV (16.7) and AF594-DENV(17.9). The similar ratios indicated that both labelled and non-labelled DENV havecomparable infectious/non-infectious proportions, suggesting that the labellingprocess did not inactivate the virions.
4. Discussion
Direct fluorescent labelling of virus allows for real-time track-ing of post-internalization events and requires few labelling steps,removing the possibility of non-specific staining that could beintroduced with the use of indirect antiviral antibodies. The track-ing of single-virus entry into cell by labelling DENV with lipophilicDiD (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chorobenzenesulfonate salt) was described recently (van derSchaar et al., 2007, 2008). However, as DiD fluorescence is largelyquenched when the fluorophores are in close proximity to eachother (e.g. on virion surface) and will only fluoresce brightly fol-lowing fusion with cellular membranes, the DiD-labelled virionsare not traceable easily prior to fusion. This hampers the visualiza-tion of early virus–cell interaction. In addition, the DiD fluorescencepost-fusion tends to be diffused, possibly due to rapid lipid recy-cling, thus complicating co-localization studies. Furthermore, thelabelling is not stable and labelled virus can only be stored in thecold for up to 3 days. Hence, a fluorophore, such as Alexa Fluor, thatis unquenched extracellularly and stably conjugates to the virussurface proteins would enable the tracking of virus within a cellrather than visualizing lipid movement.
Currently, there is no standardized procedure for fluorescencelabelling of viruses and protein labelling protocols by manufac-

Author's personal copy
176 S.L.-X. Zhang et al. / Journal of Virological Methods 167 (2010) 172–177
Fig. 3. Comparing the growth kinetics of purified non-labelled and labelled DENV.Vero cells were plated and allowed to adhere before infecting at a MOI of 0.1 witheither AF594-DENV or non-labelled DENV. Viral RNA copy number in cell lysates wasdetermined to assess viral replication (a) and supernatant was assayed for infectiousparticles by plaque assay (b). There is no significant difference between the growthcurves of labelled and non-labelled dengue virus in RNA copy number (P = 0.8630)and total pfu (P = 0.8191). Each point is an average of four determinations.
turers often call for the use of dimethyl sufoxide (DMSO) toreconstitute the dye. DMSO can block productive infection ofvirus in cells, even when present in minute amounts, and mayinduce cell cytotoxicity (Aguilar et al., 2002) which could affectdownstream assays. An alternative method of producing infectivelabelled viruses without the use of DMSO would therefore be desir-able. This initiated the assessment of labelling of DENV with AlexaFluor succinimidyl ester dye dissolved directly in the sodium bicar-bonate buffer since the dye is highly soluble in aqueous solution.The results obtained indicate that this approach is able to produceviable labelled DENV that can be visualized easily by fluorescencemicroscopy. The findings were also reproducible and consistentlyyielded fluorescently labelled virus with less than tenfold drop intitre following column purification.
Alexa Fluor dyes are small molecules that react with free aminogroups, primarily arginine and lysine (Huang et al., 2004), usu-ally outward-facing amino acids. Despite its relatively small sizecompared to other fluorescent proteins, increasing the concen-tration of the dye used for labelling correlated with a reducedviral titre. This was observed with concentrations of AF594SE dyegreater than 200 �M. An optimal level of labelling should thusbalance between the degree of labelling and the functional abro-gation (Freistadt and Eberle, 2006). Consequently, although DENVlabelled with 200 �M of dye showed brighter fluorescence over100 �M of dye, 100 �M was chosen for subsequent experimentsas it provided adequate labelling with minimal loss of viability ofDENV.
The labelled DENV retained the ability to be recognized by anti-E antibody and DC-SIGN expressed on transfected-Raji B cells asdemonstrated by immunofluorescence assays performed on FACSand confocal microscope. Raji B cells are not normally suscepti-ble to DENV infection, but DC-SIGN transfection renders these cells
permissive to this virus (Martin et al., 2006; Tassaneetrithep et al.,2003; Wu et al., 2004). This suggests that conjugation of the AlexaFluor dye molecules to the surface of DENV did not alter nor blockimportant epitopes of the surface glycoproteins used for receptorrecognition. Additional assays were also conducted to determinewhether labelling of the viral proteins would interfere with itsinfection and replication in normally permissive cells. Study of thegrowth kinetics in Vero cells showed no significant difference in theinfectivity or replication rate between the labelled and the non-labelled virus as indicated by the viral RNA copy number in cellsand titre in the supernatant (P > 0.8).
To estimate the percentage of virus labelled with the fluores-cence dye using this method, flow cytometric studies were carriedout using DENV labelled with AF488SE dye. Approximately 58.4%of the cell population were positive for both AF488 and anti-Estaining, out of a total of 81.4% infected. This suggests that approx-imately 71.7% of the virus preparation was actually labelled withAF488 dye. Although AF594-DENV could not be assessed for effi-ciency of labelling on the flow cytometer used, due to the lack ofappropriate excitation laser, Pearson’s correlation values obtainedfrom co-localization studies using confocal microscopy revealedthat similar labelling efficiency can be achieved.
Even though it has not been demonstrated in this report, thisapproach of labelling DENV should apply to the other serotypes. Asthe labelling chemistry is the same, different fluorophores can alsobe used (AF594 and AF488 demonstrated here). This could increasethe range of applications of the labelled virus (e.g. flow cytometry,confocal microscopy). In addition, conjugation of dye to viral pro-teins is stable and the labelled virus could be stored frozen untiluse. Experience in this laboratory indicates that labelled virus canbe stored at −80 ◦C for up to 8 months without noticeable loss influorescence or viral titre. Thus, a large batch of virus can be labelledand used for consecutive experiments to minimize inter-assay vari-ations.
In conclusion, Alexa Fluor labelled virus provides a usefulmethod for tracking the early events in virus–cell interaction.
Competing interests
The authors declare that they have no competing interests.
Acknowledgement
This work was funded by the Duke-NUS Graduate MedicalSchool Program in Emerging Infectious Diseases.
References
Aguilar, J.S., Roy, D., Ghazal, P., Wagner, E.K., 2002. Dimethyl sulfoxide blocks her-pes simplex virus-1 productive infection in vitro acting at different stages withpositive cooperativity. Application of micro-array analysis. BMC Infect. Dis. 2, 9.
Freistadt, M.S., Eberle, K.E., 2006. Fluorescent poliovirus for flow cytometric cellsurface binding studies. J. Virol. Methods 134, 1–7.
Gubler, D.J., 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11,480–496.
Huang, S., Wang, H., Carroll, C.A., Hayes, S.J., Weintraub, S.T., Serwer, P., 2004. Anal-ysis of proteins stained by Alexa dyes. Electrophoresis 25, 779–784.
Kuhn, R.J., Zhang, W., Rossmann, M.G., Pletnev, S.V., Corver, J., Lenches, E., Jones, C.T.,Mukhopadhyay, S., Chipman, P.R., Strauss, E.G., Baker, T.S., Strauss, J.H., 2002.Structure of dengue virus: implications for flavivirus organization, maturation,and fusion. Cell 108, 717–725.
Lai, Y.L., Chung, Y.K., Tan, H.C., Yap, H.F., Yap, G., Ooi, E.E., Ng, L.C., 2007. Cost-effectivereal-time reverse transcriptase PCR (RT-PCR) to screen for Dengue virus fol-lowed by rapid single-tube multiplex RT-PCR for serotyping of the virus. J. Clin.Microbiol. 45, 935–941.
Martin, N.C., Pardo, J., Simmons, M., Tjaden, J.A., Widjaja, S., Marovich, M.A., Sun, W.,Porter, K.R., Burgess, T.H., 2006. An immunocytometric assay based on dengueinfection via DC-SIGN permits rapid measurement of anti-dengue neutralizingantibodies. J. Virol. Methods 134, 74–85.
Tassaneetrithep, B., Burgess, T.H., Granelli-Piperno, A., Trumpfheller, C., Finke, J., Sun,W., Eller, M.A., Pattanapanyasat, K., Sarasombath, S., Birx, D.L., Steinman, R.M.,

Author's personal copy
S.L.-X. Zhang et al. / Journal of Virological Methods 167 (2010) 172–177 177
Schlesinger, S., Marovich, M.A., 2003. DC-SIGN (CD209) mediates dengue virusinfection of human dendritic cells. J. Exp. Med. 197, 823–829.
van der Schaar, H.M., Rust, M.J., Chen, C., van der Ende-Metselaar, H., Wilschut, J.,Zhuang, X., Smit, J.M., 2008. Dissecting the cell entry pathway of dengue virusby single-particle tracking in living cells. PLoS Pathog. 4, e1000244.
van der Schaar, H.M., Rust, M.J., Waarts, B.L., van der Ende-Metselaar, H., Kuhn, R.J.,Wilschut, J., Zhuang, X., Smit, J.M., 2007. Characterization of the early events in
dengue virus cell entry by biochemical assays and single-virus tracking. J. Virol.81, 12019–12028.
WHO, 2007. Report of Dengue Scientific Working Group. TDR/SWG/08, Geneva.Wu, L., Martin, T.D., Carrington, M., KewalRamani, V.N., 2004. Raji B cells, misiden-
tified as THP-1 cells, stimulate DC-SIGN-mediated HIV transmission. Virology318, 17–23.

Visualizing Dengue virus through Alexa Fluor labeling
Authors: Summer Zhang, Hwee Cheng Tan, Eng Eong Ooi,
Authors: institution(s)/affiliation(s) for each author:
Summer Zhang Defence Medical and Environmental Research Institute DSO National Laboratories, Singapore [email protected]
Hwee Cheng Tan Program in Emerging Infectious Diseases Duke-NUS Graduate Medical School, Singapore [email protected]
Eng Eong Ooi Program in Emerging Infectious Diseases Duke-NUS Graduate Medical School, Singapore [email protected] Corresponding author:
Eng Eong Ooi
Keywords:
Dengue virus, Alexa Fluor, labeling, fluorescence
Short Abstract:
Taking advantage of the advancements in fluorophore development and imaging technology, a simple method of Alexa Fluor labeling of dengue virus was devised to visualize the early interactions between virus and cell.
Long Abstract:
The early events in the interaction between virus and cell can have profound influence on the outcome of infection. Determining the factors that influence this interaction could lead to improved understanding of disease pathogenesis and thus influence vaccine or therapeutic design. Hence, the development of methods to probe this interaction would be useful. Recent advancements in fluorophores development1-3 and imaging technology4 can be exploited to improve our current knowledge on dengue pathogenesis and thus pave the way to reduce the millions of dengue infections occurring annually.

The enveloped dengue virus has an external scaffold consisting of 90 envelope glycoprotein (E) dimers protecting the nucleocapsid shell, which contains a single positive strand RNA genome5. The identical protein subunits on the virus surface can thus be labeled with an amine reactive dye and visualized through immunofluorescent microscopy. Here, we present a simple method of labeling of dengue virus with Alexa Fluor succinimidyl ester dye dissolved directly in a sodium bicarbonate buffer that yielded highly viable virus after labeling. There is no standardized procedure for the labeling of live virus and existing manufacturer’s protocol for protein labeling usually requires the reconstitution of dye in dimethyl sulfoxide. The presence of dimethyl sulfoxide, even in minute quantities, can block productive infection of virus and also induce cell cytotoxicity6. The exclusion of the use of dimethyl sulfoxide in this protocol thus reduced this possibility. Alexa Fluor dyes have superior photostability and are less pH-sensitive than the common dyes, such as fluorescein and rhodamine2, making them ideal for studies on cellular uptake and endosomal transport of the virus. The conjugation of Alexa Fluor dye did not affect the recognition of labeled dengue virus by virus-specific antibody and its putative receptors in host cells7. This method could have useful applications in virological studies.
Protocol Text:
1.) Alexa Fluor labeling of dengue virus
1.1) Before the labeling reaction, purify dengue virus with sucrose cushion and prepare the necessary reagents and equipment as indicated in the protocol.
1.2) Prepare fresh 0.2M sodium bicarbonate buffer, pH 8.5 (labeling buffer), and 1.5M hydroxylamine buffer, pH 8.3 (stop reagent), just before labeling and filter sterilize with 0.2µm syringe filters.
1.3) Dilute approximately 3x108 plaque forming units (pfu) of purified dengue virus in 1ml of labeling buffer in a 2ml tube. This can be scaled up proportionally for batch labeling of virus.
1.4) Reconstitute the lyophilized Alexa Fluor 594 (AF594) succinimidyl esters to 1mM in labeling buffer immediately prior to the labeling reaction. Other fluorochromes from the Alexa Fluor dye series may be used according to one’s needs. Minimize exposure to light from this step onwards.
1.5) Add 100µl of the 1mM AF594 dye to the diluted virus while stirring gently with the pipette tip.
1.6) Incubate the labeling reaction mix at room temperature for 1hr in the dark. Mix by gentle inversions every 15mins.
1.7) Spin the tube briefly in a tabletop centrifuge and add 100µl of stop reagent to the reaction mix while stirring gently with the pipette tip.
1.8) Incubate at room temperature for an additional hour in the dark. Mix by gentle inversions every 15mins.

2.) Purifying Alexa Fluor labeled dengue virus
2.1) In the meantime, equilibrate the purification column with buffer of choice. In this experiment, a PD-10 column is equilibrated with 25ml of HNE buffer (5mM Hepes, 150mM NaCl, 0.1mM EDTA), pH 7.4, before use.
2.2) Apply the labeled virus to the top of the column and start collecting the flow-through once the labeled virus enters the matrix. Fill the column with HNE buffer once all the labeled virus has entered the matrix. Discard the first 2.5ml of flow-through and collect the next 2ml of labeled virus fraction.
2.3) Aliquot and store purified AF594 labeled dengue virus in -80°C, away from light source.
3.) Checking virus viability and fluorescence
3.1) Thaw one aliquot and determine the titer of the labeled virus by plaque assay before using the batch of labeled virus.
3.2) Seed 5x104 per well of Vero cells, grown in M-199 growth medium, in a 4-well plate with a coverslip on the bottom of well a day before infection.
3.3) Remove the culture supernatant in well and infect the cells with multiplicity of infection of 1 of the labeled virus in 100µl volume (diluted in M-199 maintenance medium as required) for 10min at 37°C.
3.4) Remove the inoculums and wash the coverslips twice in 1xPBS.
3.5) Fix the cells in 3% paraformalydehyde for 30mins.
3.6) Wash the coverslips 3 times in 1xPBS.
3.7) Permeabilize the cells with permeabilization solution containing 0.1% saponin and 5% BSA in 1xPBS for 30mins.
3.8) Incubate the cells with undiluted centrifugation-clarified supernatant of 3H5 monoclonal antibody hybridoma culture for 1hr in a humid chamber, protected from light.
3.9) Wash the coverslips 3 times in wash buffer (1xPBS containing 1mM calcium chloride, 1mM magnesium chloride and 0.1% saponin).
3.10) Incubate the cells with AF488 anti-mouse IgG antibody, 1:100 diluted in permeabilization solution, for 45mins in humid chamber, protected from light.
3.11) Wash the coverslips 3 times in wash buffer and rinse once in deionized water.

3.12) Dab the edge of coverslip against a paper towel to drain excess water and mount on to glass slide with 8μl Mowiol 4-88 containing 2.5% Dabco.
3.13) Allow the mounting solution to set overnight at 4°C before viewing using a Zeiss confocal microscope. Sequential acquisitions should be performed exciting one fluorophore at a time and switching between the detectors concomitantly.
3.14) Images are then analyzed for co-localization of the E protein antibody staining with the labeled virus using the Zeiss LSM Zen software to estimate the degree of labeling.
Representative Results:
An example of the yield of dengue virus labeled with AF594 dye is shown in Figure 2. Normally, less than 10-fold drop from the initial titer should be observed following successful labeling. However, it should be noted that all buffers have to be prepared fresh for the labeling to be successful and the Alexa Fluor succinimidyl esters should be used immediately upon reconstitution as they hydrolyze into nonreactive free acids in aqueous solutions8.
Next, the labeled virus has to be checked for sufficient fluorescence before use in experiments. A simple immunofluorescence assay was done on Vero cells and the degree of labeling can be estimated from the co-localization of the labeled virus with anti-E protein antibody staining. Several cells were examined and a typical confocal image is shown in Figure 3. Co-localization analysis of the images using the LSM Zen software demonstrated overlap coefficients ranging from 0.65 to 0.8, suggesting that approximately 65 to 80% of the virions were labeled with the dye.
Tables and Figures (Required):
Figure 1: Overall scheme depicting the Alexa Fluor dye labeling of dengue virus procedure. First, the relevant buffers and purified dengue virus are prepared. The Alexa Fluor dye is reconstituted and added to the dengue virus diluted in labeling buffer. The reaction is then stopped 1 hour later with the addition of stop reagent. Subsequently, the labeled virus is purified through a size exclusion column to remove free dye. Finally, the labeled virus is re-titrated by plaque assay and tested for fluorescence.
Figure 2: Mean number of viable virions (pfu/ml) as determined on a plaque assay before and after AF594 labeling. An aliquot of the AF594 labeled dengue virus is thawed and re-titrated by plaque assay and it typically shows less than 10-fold drop from the starting titer. Error bars indicate standard deviation of duplicates.
Figure 3: Co-localization of AF594 labels with dengue virus E proteins in Vero cells. Vero cells grown on coverslips the day prior were infected with AF594 labeled dengue at MOI of 1 for

10 minutes at 37°C. The cells were subsequently fixed and labeled with anti-E antibody, and examined for colocalization of E protein (green) and AF594 labeling (red). Fluorescent signals were visualized under 63X magnification using Zeiss LSM 710 confocal microscope. Scale bar is 10µm. Yellow indicate areas of colocalization, as shown in the inset.
Discussion:
Although AF594 dye was used in this report, a wide range of fluorophores in the Alexa Fluor succinimidyl esters series is available with similar labeling chemistry. This could extend the labeling application beyond imaging. Flow cytometry can be used as an alternative to confocal microscopy for estimating the degree of labeling for fluorophores that can be excited and detected by the FACS machine.
Alexa Fluor dyes are small molecules that react with free amino groups, primarily arginine and lysine9, normally outward facing residues of proteins. In our laboratory, conjugation of dengue virus with 100μM of Alexa Fluor 594 dye provided sufficient brightness for imaging with minimal loss in the viral titer. Different brightness may be achieved by varying the concentration of dye used. However, increasing the dye concentration can reduce virus viability7,10. One possible limitation is the interference in receptor binding due to the blockade of access by the fluorophores. Therefore, depending on application, an optimal level of labeling should be determined to ensure a balance between the degree of labeling and functional abrogation10. Do note that the concentration can also be affected by the post-packaging reactivity of the Alexa Fluor succinimidyl esters8.
The direct labeling of dengue virus with Alexa Fluor dye presented here does not require any additional labeling steps to visualize the virus, thus removing the possibility of non-specific staining from indirect antiviral antibodies. It also allows for real-time tracking of post-internalization events in live cell imaging. This method is relatively simple, and because the conjugation is stable, it can be used to produce and store batch-labeled virus for multiple experiments as opposed to lipophillic fluorescent dyes, such as long-chain carbocyanine1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine (DiD) or styryl dyes, which cannot be stored in the cold for more than 3 days.
Acknowledgments:
This work has been funded by the National Medical Research Council, Singapore.
Disclosures:
We have nothing to disclose.

Table of specific reagents and equipment:
Name of the reagent Company Catalogue number Comments (optional) Sodium bicarbonate Sigma-Aldrich S6297 Hydroxylamine Sigma-Aldrich 159417 Sodium hydroxide Merck 106498 AF594 succinimidyl esters
Molecular Probes, Invitrogen
A20004
PD-10 column GE Healthcare 17-0851-01 Hepes Sigma-Aldrich H6147 NaCl Sigma-Aldrich S3014 EDTA Sigma-Aldrich E9884 M-199 Invitrogen 11150 FBS Hyclone SH30070.03 4-well plate Nunc 176740 Coverslips Einst 0111520 Microscope slide Sail Brand 7105 3H5 hybridoma ATCC HB46 10x PBS 1st Base BUF-2040-10X1L Saponin Sigma-Aldrich S4521 BSA Sigma-Aldrich A7906 Magnesium chloride Sigma-Aldrich M2670 Calcium chloride Sigma-Aldrich C3306 Paraformaldehye Sigma-Aldrich 15,812-7 Mowiol 4-88 Calbiochem 475904 Dabco Sigma-Aldrich D27802 Tabletop centrifuge Eppendorf 5424 Confocal microscope Zeiss LSM 710 To prepare M-199 growth medium, add 50ml of FBS, 5ml of sodium pyruvate and 5ml of non-essential amino acids to 500ml of M-199, sterile filter. To prepare M-199 maintenance medium, add 15ml of FBS, 5ml of sodium pyruvate and 5ml of non-essential amino acids to 500ml of M-199, sterile filter.

References:
1 Olenych, S. G., Claxton, N. S., Ottenberg, G. K. & Davidson, M. W. The Fluorescent
Protein Color Palette. Vol. 21.5 1-34 (Current Protocols in Cell Biology, 2007). 2 Panchuk-Voloshina, N. et al. Alexa dyes, a series of new fluorescent dyes that yield
exceptionally bright, photostable conjugates. J Histochem Cytochem 47, 1179-1188 (1999).
3 Shaner, N. C., Patterson, G. H. & Davidson, M. W. Advances in fluorescent protein
technology. J Cell Sci 120, 4247-4260, doi:120/24/4247 [pii] 10.1242/jcs.005801 (2007).
4 Lippincott-Schwartz, J. & Patterson, G. H. Development and use of fluorescent protein
markers in living cells. Science 300, 87-91, doi:10.1126/science.1082520 300/5616/87 [pii] (2003).
5 Kuhn, R. J. et al. Structure of dengue virus: implications for flavivirus organization,
maturation, and fusion. Cell 108, 717-725, doi:S0092867402006608 [pii] (2002). 6 Aguilar, J. S., Roy, D., Ghazal, P. & Wagner, E. K. Dimethyl sulfoxide blocks herpes
simplex virus-1 productive infection in vitro acting at different stages with positive cooperativity. Application of micro-array analysis. BMC Infect Dis 2, 9 (2002).
7 Zhang, S. L., Tan, H. C., Hanson, B. J. & Ooi, E. E. A simple method for Alexa Fluor
dye labelling of dengue virus. J Virol Methods 167, 172-177, doi:S0166-0934(10)00130-8 [pii] 10.1016/j.jviromet.2010.04.001 (2010).
8 Alexa Fluor Succinimidyl Esters (Molecular Probes),
<probes.invitrogen.com/media/pis/mp10168.pdf> (2009). 9 Huang, S. et al. Analysis of proteins stained by Alexa dyes. Electrophoresis 25, 779-784,
doi:10.1002/elps.200305723 (2004). 10 Freistadt, M. S. & Eberle, K. E. Fluorescent poliovirus for flow cytometric cell surface
binding studies. J Virol Methods 134, 1-7, doi:S0166-0934(05)00272-7 [pii] 10.1016/j.jviromet.2005.08.011 (2006).

Figure 1

Figure 2

Figure 3

![Dengue Hemorrhagic Fever: A State-of-the-Art Review ......hemorrhagic fever (DHF)/dengue shock syndrome (DSS) or severe dengue (SD) [6, 7]. Symptomatic dengue virus infection can present](https://static.fdocuments.us/doc/165x107/613fd07cb44ffa75b8047786/dengue-hemorrhagic-fever-a-state-of-the-art-review-hemorrhagic-fever-dhfdengue.jpg)
















